

Keywords: aldosterone, aldosterone-sensitive distal nephron, epithelial Na+ channel, mineralocorticoid receptor, 11β-hydroxysteroid dehydrogenase 2
Abstract
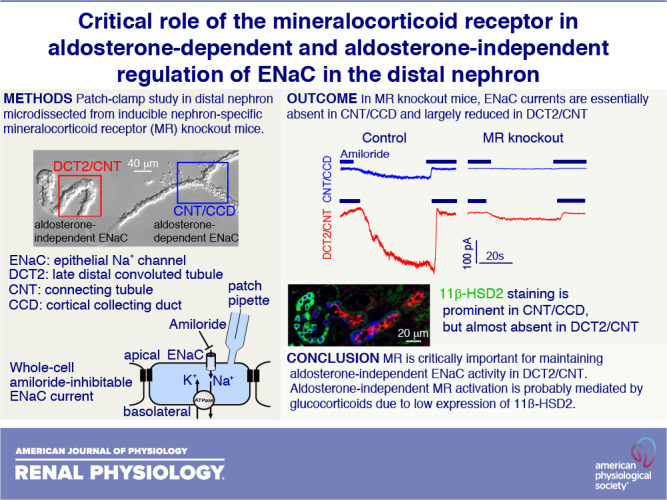
The epithelial Na+ channel (ENaC) constitutes the rate-limiting step for Na+ absorption in the aldosterone-sensitive distal nephron (ASDN) comprising the late distal convoluted tubule (DCT2), connecting tubule (CNT), and collecting duct (CD). Previously, we demonstrated that ENaC activity in the DCT2/CNT transition zone is constitutively high and independent of aldosterone, in contrast to its aldosterone dependence in the late CNT/initial cortical CD (CCD). The mineralocorticoid receptor (MR) is expressed in the entire ASDN. Its activation by glucocorticoids is prevented through 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2) abundantly expressed in the late but probably not early part of the ASDN. We hypothesized that ENaC function in the early part of the ASDN is aldosterone independent but may depend on MR activated by glucocorticoids due to low 11β-HSD2 abundance. To test this hypothesis, we used doxycycline-inducible nephron-specific MR-deficient [MR knockout (KO)] mice. Whole cell ENaC currents were investigated in isolated nephron fragments from the DCT2/CNT or CNT/CCD transition zones using the patch-clamp technique. ENaC activity was detectable in the CNT/CCD of control mice but absent or barely detectable in the majority of CNT/CCD preparations from MR KO mice. Importantly, ENaC currents in the DCT2/CNT were greatly reduced in MR KO mice compared with ENaC currents in the DCT2/CNT of control mice. Immunofluorescence for 11β-HSD2 was abundant in the CCD, less prominent in the CNT, and very low in the DCT2. We conclude that MR is critically important for maintaining aldosterone-independent ENaC activity in the DCT2/CNT. Aldosterone-independent MR activation is probably mediated by glucocorticoids due to low expression of 11β-HSD2.
NEW & NOTEWORTHY Using a mouse model with inducible nephron-specific mineralocorticoid receptor (MR) deficiency, we demonstrated that MR is not only critical for maintaining aldosterone-dependent ENaC activity in CNT/CCD but also for aldosterone-independent ENaC activity in DCT2/CNT. Furthermore, we demonstrated that cells of this latter nephron segment express little 11β-HSD2, which probably allows glucocorticoids to stimulate MR, resulting in aldosterone-independent ENaC activity in DCT2/CNT. This site-specific ENaC regulation has physiologically relevant implications for renal sodium and potassium homeostasis.
INTRODUCTION
Fine tuning of renal Na+ and K+ excretion occurs in the distal part of the nephron where the epithelial Na+ channel (ENaC) is expressed. This nephron segment is usually called the aldosterone-sensitive distal nephron (ASDN) and comprises the late distal convoluted tubule (DCT2), connecting tubule (CNT), and entire collecting duct (CD) (1–3). In the cortical CD (CCD), ENaC activity is tightly regulated by the mineralocorticoid hormone aldosterone. ENaC activity is absent or very low in the CCD of rats and mice maintained on a standard salt diet but increases with increasing aldosterone levels, for example, in response to a low-salt diet (4, 5). In microdissected mouse tubules, we confirmed the aldosterone dependence of ENaC in the transition zone comprising the late part of the CNT and early part of the CCD (CNT/CCD). In contrast, we demonstrated that ENaC was largely aldosterone independent in the transition zone comprising the DCT2 and early part of the CNT (DCT2/CNT) (6, 7). This finding has recently been confirmed by another group reporting that angiotensin II plays an important role in maintaining ENaC activity in the DCT2/CNT by a mechanism independent of the mineralocorticoid receptor (MR) (8). Moreover, aldosterone-independent ENaC stimulation has been observed in response to an acute dietary K+ load (9). However, the cellular and molecular mechanisms involved in aldosterone-independent ENaC regulation in the DCT2/CNT region of the ASDN are not yet fully understood.
Results from global and tissue-specific knockout (KO) mouse models have provided some important clues regarding the site-specific roles of aldosterone and MR in controlling ENaC function in the ASDN. Importantly, global MR deficiency leads to a much more severe renal phenotype than deficiency of aldosterone or deficiency of MR restricted to CD principal cells (10–12). Similarly, CD-specific KO of the α-subunit of ENaC results in a relatively mild phenotype (13), comparable to the phenotype of MR KO in CD principal cells (12). In contrast, global knockout of β-ENaC results in a severe salt-losing syndrome with hyperkalemia and neonatal death, which is similar to the phenotype caused by global MR KO (14). Based on these findings, we hypothesized that, albeit aldosterone independent, ENaC function in the DCT2/CNT is MR dependent.
MR is equally sensitive to mineralocorticoids and glucocorticoids, whereas the concentration of free glucocorticoids in blood plasma is approximately two orders of magnitude higher than that of mineralocorticoids. This would imply that under physiological conditions, MR is completely occupied and activated by glucocorticoids and that aldosterone has no capacity to regulate cell functions through MR (15). The ability of cells to discriminate between mineralocorticoids and glucocorticoids largely depends on expression of the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which converts glucocorticoids into inactive metabolites (15, 16). Thus, in cells expressing high levels of 11β-HSD2, MR is protected from being activated by glucocorticoids and becomes aldosterone responsive. Indeed, genetic deficiency of this enzyme leads to a rare form of pseudohyperaldosteronism, the so-called apparent mineralocorticoid excess syndrome (17). Interestingly, there is some evidence that expression of 11β-HSD2 is lower in the proximal portion of the ASDN than in its distal portion (18–20). In contrast, expression of MR appears to be more uniform along the ASDN. This raises the possibility that protection of MR by 11β-HSD2 may be less complete in the early than late ASDN. If the DCT2 and early CNT expressed MR but only little 11β-HSD2, MR activation by glucocorticoids may stimulate ENaC activity in this part of the ASDN. Thus, aldosterone-independent ENaC activity in the DCT2/CNT may be MR dependent. To test this hypothesis, we used mice with nephron-wide, doxycycline-inducible MR deficiency (21). Whole cell and outside-out ENaC currents in the CNT/CCD transition zone versus the DCT2/CNT transition zone were systematically studied and compared using microdissected tubules, as previously described (6, 7, 22). Furthermore, expression of 11β-HSD2 was assessed in the DCT, CNT, and CCD using immunofluorescence.
MATERIALS AND METHODS
Animals
The doxycycline-inducible nephron-specific MR KO mice (MRPax8/LC1) used in this study were generated as previously described (21). Mice were bred and maintained in the animal facility of Friedrich-Alexander-Universität Erlangen-Nürnberg under the governance of the responsible regulatory local authorities, and all procedures were approved by the animal welfare officer of Friedrich-Alexander-Universität Erlangen-Nürnberg (TS-1/03 ZellPhys and TS-11/2017 ZellPhys). To induce nuclear receptor subfamily 3 group C member 2 (Nr2c3) deletion in renal tubular cells, MR-floxed (lox, lox) mice, being transgenic for Pax8-rtTA and TRE-LC1, were treated with doxycycline and constituted the experimental group, termed “MR KO mice” in the present study. Transgenes that did not lead to the excision of MR (MR-floxed mice being transgenic for either Pax8-rtTA or TRE-LC1) were used as the control group. Breeding pairs or triplets were composed of heterozygous animals. The genotypes of the offspring were distributed as expected for Mendelian inheritance. All MR KO and control mice of a particular litter were used. This resulted in a random allocation of animals to the experimental groups. In total, 77 mice were used for this study. Eleven MR KO animals had to be excluded from the experiments due to critical loss of body weight and two MR KO mice due to anatomic abnormalities (single kidney). In nine animals, no electrophysiological recordings were obtained due to technical reasons. Six animals were excluded from data analysis, because their genotype could not be confirmed after the experiment. Results from the remaining 49 mice (19 females and 30 males) were included in the analysis. In the initial publication describing this mouse model, no differences between male and female animals were reported for the parameters investigated (21). Therefore, data from male and female animals were pooled. To avoid overlooking potential sex effects, different symbols were used to distinguish between data points obtained from female versus male mice in figures summarizing pooled data. Figure legends clarify the sex of the animals used for representative traces. For electrophysiological recordings, a single animal was used per experimental day, and genotypes were systematically alternated to balance age in experimental and control groups. Blinding was not used at any of the experimental steps.
MR KO and control mice received standard chow (3.2 g of Na+ per kg of food; Altromin, Spezialfutter, Lage, Germany) and had free access to drinking water. The latter was replaced at the age of 26–28 days after birth by drinking water containing 2 mg/mL doxycycline hydrochloride (Santa Cruz Biotechnology, Heidelberg, Germany). In addition, this doxycycline solution contained 2% sucrose and 0.45% NaCl to make it more palatable and to compensate for renal Na+ losses due to nephron-specific MR KO. Control and MR KO mice received this sucrose- and NaCl-enriched doxycycline drinking solution ad libitum for at least 14 days and on average for 28 ± 2.7 days (means ± SE).
Genotyping
Genotyping was performed as previously described (21). In brief, DNA was extracted from mouse biopsies (tail tip or ear), and genotyping was performed by PCR analysis. After the experiment, genotyping was repeated from cortical kidney tissue. The KO band corresponding to the recombination of the floxed Nr2c3 allele was detected by PCR. Only mice with confirmed recombination of the floxed Nr2c3 allele were included in the analysis of the MR KO group.
Preparation of Tubules
Tubules were prepared essentially as previously described (6, 7, 22). Briefly, after terminal anesthesia with ketamine and xylazine overdosage (300 mg/kg ketamine and 60 mg/kg xylazine), the thorax and abdomen were opened. The animal was euthanized by bleeding from the vena cava inferior expedited by perfusing the circulatory system with Ringer’s solution via the left ventricle. This was followed by perfusion with Leibowitz medium (Thermo Fisher Scientific, Schwerte, Germany) containing 350 U/mL collagenase type IV (Biochrom). Kidneys were harvested, and coronary slices were prepared and incubated in Leibowitz medium with collagenase at 37°C for 20–30 min for electrophysiological experiments or for 40–45 min for immunohistochemistry.
Electrophysiology
Patch-clamp experiments were performed essentially as previously described (6, 7, 22). After incubation with collagenase, slices were kept in ice-cold Leibowitz medium. Tubules were isolated manually, and nephron segments were identified using morphological criteria as previously described (6). We identified and isolated tubular segments with characteristic branching indicative of the merging of the CNT into the CCD and extended the dissection toward the DCT. Morphologically, the DCT can be recognized by extensive convolutions and large cells with visible basal membrane infoldings. The transitional zone between the DCT2 and early CNT is characterized by a decreasing tubular diameter and smaller rounded cells. The zone comprising the late CNT and early CCD is defined by its proximity to the tubular branching points. In this segment, cells are even smaller than in the early CNT and the tubular diameter is further reduced. Under the dissecting microscope, there are no clear-cut boundaries between the DCT2 and early CNT or between the late CNT and initial CCD. In particular, after transfer of the tubular segments to the perfusion chamber and after opening the tubules, it is difficult to distinguish between the DCT2 and early CNT or between the late CNT and initial CCD. Therefore, in our patch-clamp experiments, we distinguished only between two regions of the ASDN: 1) the DCT2 and initial CNT (DCT2/CNT) and 2) the late CNT and initial CCD (CNT/CCD). An isolated ASDN fragment was placed on a small piece of a glass coverslip coated with Cell-Tak (Corning, Kaiserslautern, Germany) and transferred to a flow chamber on the stage of an inverted microscope. To gain access to the apical membrane, tubules were cut open with a broken glass pipette attached to a micromanipulator. In the exposed epithelial layer of the opened tubule, principal cells were identified according to their characteristic shape and were approached with a patch pipette as previously described (6). Bath solution contained Na gluconate (140 mM), K gluconate (5 mM), CaCl2 (2 mM), Ba acetate (10 mM), MgCl2 (1 mM), and HEPES (10 mM) and was titrated to pH 7.4 with Tris. Pipette solution contained K gluconate (85 mM), Na gluconate (5 mM), Mg ATP (2 mM), MgCl2 (2 mM), CsOH (40 mM), TEA-OH (20 mM), and HEPES (10 mM) and was titrated to pH 7.2 with gluconic acid. Experiments were started in the presence of the ENaC inhibitor amiloride (2 µM) purchased from Sigma-Aldrich (Steinheim, Germany). Whole cell and outside-out currents were recorded at a holding potential of −60 mV, favoring inward Na+ currents. The amiloride-sensitive whole-cell current (ΔIami) was determined by subtracting the current measured in the presence of amiloride from that measured in its absence. In most cases, two recordings were made in the CNT/CCD and two recordings in the DCT2/CNT for each mouse. For technical reasons, this was not achieved in all animals. Thus, in a few animals, only one recording was obtained from one or both segments. Moreover, in five mice, recordings were obtained only from the CNT/CCD and in one animal, only from the DCT2/CNT. Throughout the text, N values correspond to the number of animals used and n values indicate the number of individual recordings.
Estimate of Channel Open Probability
After recordings in whole cell mode, outside-out patches were excised. Recordings with resolvable single-channel current levels were used for the analysis of current level distribution (7, 23). Levels were determined as maxima of binned current amplitude histograms and were numbered. The “all closed” level (level 0) was determined by application of amiloride. Levels 1, 2, …, n correspond to simultaneous opening of 1, 2, …, n channels. Single-channel current amplitude was determined as the interval between adjacent levels. Current values at each timepoint were attributed to the current level to which they were nearest. The probability of a current level to occur was determined by dividing the number of data points attributed to this level by the total number of data points in an analyzed recording. The channel activity was estimated as NPo, which was determined as follows:
where “level” is the current level (level 0, 1, 2, …), “MaxLevels” is the maximal number of observed levels, and “plevel” is the probability of each level. To estimate the number of channels in the patch, the observed probability for n channels being open simultaneously was fitted with a binomial distribution; the best fit was determined by maximal-likelihood estimate. Open probability (Po) was estimated as the ratio of NPo to the best N estimate. Original Nest-o-Patch software was used for the analysis (24).
Immunofluorescence Staining of MR in Isolated Tubules
Microdissection of tubules was essentially performed as described above Preparation of Tubules and Electrophysiology. Incubation time in collagenase was increased to 40–45 min to obtain more complete ASDN fragments. Segments were attached to a slide covered with Cell-Tak and fixed with 4% paraformaldehyde in PBS (pH 7.4) for 10 min on ice. After fixation, tubular cells were permeabilized with 0.1% Triton X-100 (Sigma, Taufkirchen, Germany) in PBS for 15 min on ice. Unspecific binding sites were blocked with 1× Roti-Immunoblock (Roth, Karlsruhe, Germany) for 10 min at room temperature. For MR detection, a primary monoclonal antibody (monoclonal mouse anti-rat-MR rMR1-18 6G1) was kindly provided by C. E. Gomez-Sanchez (25) and was used in a dilution of 1:40 in 0.5% BSA-0.04% sodium azide with overnight incubation at 4°C. Staining was visualized with goat anti-mouse DyLight488-conjugated secondary antibody (Pierce Biotechnology, Rockford, IL) used in a 1:100 dilution. Slides were covered with Fluoroshield including DAPI for nuclear counterstaining (Sigma). MR staining in microdissected tubules was evaluated with a Zeiss Axiovert 200 M microscope using structured illumination technology (ApoTome).
Immunofluorescence Staining of 11β-HSD2 in Renal Tissue
Anesthetized mice were fixed by perfusion with 2% paraformaldehyde in PBS via the left ventricle for 5 min at room temperature. These experiments were approved by the University of Maryland-Baltimore Animal Care and Use Committee. The kidneys were then removed and fixed (24 h at 4°C), rinsed in PBS, and embedded in paraffin. Three-micrometer-thick cross sections, cut at the level of the papilla, were placed on chrome-alum gelatin-coated glass coverslips and dried on a warming plate. Sections were then deparaffinized in two xylene baths and two absolute ethanol baths, 5 min each, and rehydrated in a graded ethanol series to distilled water. For epitope retrieval, coverslips were placed in pH 8 solution (1 mM Tris, 0.5 mM EDTA, and 0.02% SDS). The retrieval solution and sections were heated to boiling in a microwave oven, transferred to a boiling water bath (15 min), and then cooled to room temperature before the sections were thoroughly washed in distilled water to remove SDS. Sections were preincubated for 30 min with 2% BSA, 0.2% fish gelatin, and 0.2% sodium azide in PBS. Incubations with primary antibodies diluted in PBS containing 1% BSA, 0.2% fish gelatin, 0.1% Tween 20, and 0.2% sodium azide took place overnight in a humid chamber at 4°C. After a thorough wash in high-salt solution (incubation medium plus added sodium chloride at 0.5 M), sheep anti-11β-HSD2 (Chemicon, now Millpore Sigma, AB1296) was detected with Alexa Fluor 488-conjugated donkey anti-sheep IgG (Jackson Laboratories). Guinea pig anti-NaCl cotransporter (NCC) (26) was detected with Alexa Fluor 568-conjugated donkey anti-guinea pig IgG (Jackson Laboratories), and mouse anti-calbindin D28 (Swant CB38) was detected with Alexa Fluor 633-conjugated donkey anti-mouse IgG (Invitrogen). Unconjugated secondary antibodies from Jackson Laboratories were coupled to the respective fluorophores using kits from Invitrogen.
Quantitative Analysis of Images
Nephron segments were identified in coronal kidney sections by confocal microscopy using a Zeiss LSM 510, ×40 objective. The early distal convoluted tubule (DCT1) was defined as the NCC++, calbindin−+ staining tubule, the DCT2 was defined as an adjacent NCC−+, calbindin++ staining tubule, and the CNT was defined as an NCC−−, calbindin++ staining tubule. Cytoplasmic 11β-HSD2 pixel intensity was measured using Volocity 5 3D Image Analysis Software (Perkin-Elmer). Cytoplasmic regions of interest were measured from at least five cells/tubule in at least five adjacent DCT1-DCT2-CNT tubules/mouse in eight mice.
Statistics
Observed distributions of data were in many cases clearly different from a Gaussian distribution. Therefore, data are presented in the following form: median (lower quartile, upper quartile), and a nonparametric Wilcoxon rank-sum test was used to estimate the statistical significance of differences between groups. A P value below 0.05 was considered a statistically significant difference. In each experimental group, the variability of current values from one animal was similar to that of current values from different animals. Therefore, each individual current recording was considered to be an independent observation. Statistical analysis was done using R software version 4.0 (The R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
In MR KO Mice, MR Deficiency in the DCT2/CNT Is Similar to That in the CNT/CCD
In the original description of the MRPax8/LC1 mouse line (21), it has been reported that in the whole kidney of KO animals, MR expression was reduced by ∼80% at the mRNA level and by ∼90% at the protein level. Moreover, it was shown that MR protein expression in the isolated DCT/CNT and CCD from MR KO mice could not be detected by Western Blot analysis. We used an immunohistochemical approach to confirm MR KO in this mouse model and to investigate whether the degree of MR KO in the DCT2/CNT is similar to that in the CNT/CCD. For this purpose, nuclear MR staining was analyzed in microdissected fragments of the ASDN from control mice and from MR KO mice. Microdissected tubular fragments consisted on average of 160 cells and included both the CNT/CCD and DCT2/CNT portions. Four to eight tubular fragments from every experimental animal were investigated. In control mice, prominent nuclear MR staining was observed (Fig. 1, A and B). In contrast, in the majority of MR KO mice, nuclei were MR negative (Fig. 1, C and D). To quantify MR expression, we performed costaining experiments with the nuclear dye DAPI (Fig. 1, B and D) to determine the percentage of MR-positive nuclei. In control mice, MR costaining with DAPI was found in 68% (63%, 72%) of nuclei. In contrast, in MR KO mice, the percentage of MR-positive nuclei was overall significantly lower, with a median of 4.16% (0.55%, 25%) (Fig. 1E). Interestingly, MR KO efficiency varied between individual animals, and substantial nuclear MR staining was preserved in some mice (e.g., animals 4 and 5 in Fig. 1E). Nevertheless, our results indicate that overall MR KO in the ASDN was highly effective in the majority of doxycycline-treated and genetically confirmed MR KO animals.
Figure 1.

Mineralocorticoid receptor (MR) staining in isolated fragments of the aldosterone-sensitive distal nephron (ASDN) from control (A and B) and MR knockout (KO) mice (C and D). In the control mouse, the MR antibody mainly caused nuclear staining (red) in the connecting tubule (CNT)/cortical collecting duct (CCD) as well as in the late distal convoluted tubule (DCT2)/CNT. This nuclear staining was essentially absent in the MR KO mouse. Areas indicated in A and C are shown on an enlarged scale in B and D, respectively. DAPI costaining (blue) confirmed the nuclear localization of MR staining in the control mouse and its absence in the MR KO mouse. E: summary data from similar experiments as shown in A– D. The percentage of MR-positive nuclei in tubules isolated from control (N = 7) and MR KO mice (N = 8) was determined in several ASDN fragments from each animal. Each point corresponds to an individual tubular fragment. Data points connected by a vertical line are from the same animal identified by a number. Median and quartiles are shown. Mouse 1 in the control group and mouse 4 in the MR KO group were females; all other animals were males. The age of the animals ranged from 45 to 104 days in the control group and from 53 to 102 days in the MR KO group.
To rule out segment-specific differences in MR KO efficiency, we calculated the percentage of MR-positive cells separately in the DCT2/CNT and CNT/CCD in the same preparations as shown in Fig. 1. In control mice, 66% (61%, 73%) of cells were MR positive in the CNT/CCD and 71% (63%, 73%) in the DCT2/CNT (Fig. 2A). In contrast, in MR KO mice, the percentage of MR-positive cells was significantly reduced to 3.8% (0%, 16.2%) in the CNT/CCD and to 1.96% (0%, 48%) in the DCT2/CNT (Fig. 2B). Thus, the degree of MR KO in the DCT2/CNT was similar to that in the CNT/CCD. This was also true for individual animals despite considerable interanimal variability of overall KO efficiency (Fig. 2B).
Figure 2.

In control and mineralocorticoid receptor (MR) knockout (KO) mice, the percentage of MR-positive cells in the connecting tubule (CNT)/cortical collecting duct (CCD) is similar to that in the late distal convoluted tubule (DCT2)/CNT. A and B: percentage of MR-positive cells in the CNT/CCD (left) and DCT2/CNT (right) from control (A) and MR KO mice (B). Each point represents the analysis of a corresponding segment from an individual isolated tubule. The same immunohistochemical preparations were used as in Fig. 1, but the CNT/CCD and DCT2/CNT regions were analyzed separately. To indicate the origin of each tubular segment, the dots are color coded using the same colors as used for the individual mice in Fig. 1. Data are presented as medians and quartiles.
ENaC Activity Is MR Dependent in the CNT/CCD
Using aldosterone synthase-deficient mice, we have previously established that ENaC is aldosterone dependent in the CNT/CCD but aldosterone independent in the DCT2/CNT (6). This implies that ENaC activity in the CNT/CCD depends on the presence of MR. To confirm this, we investigated ENaC activity in the microdissected CNT/CCD from control and nephron-specific MR KO mice using patch-clamp experiments as previously described (6, 7). To assess ENaC activity, ΔIami was determined by washout and reapplication of amiloride (2 µM), as shown in Fig. 3. The current traces on the top of Fig. 3, A and B, were recorded in the CNT/CCD of a control mouse or a MR KO mouse, respectively. As expected, sizeable ΔIami was detectable in the control CNT/CCD but not in the CNT/CCD from the MR KO mouse. Data from similar recordings are shown in Fig. 4A. In the control CNT/CCD, median ΔIami was 40 pA (27 pA, 107 pA) (n = 19), which is in good agreement with previously reported data for mice maintained on a standard salt diet (6, 7). Moreover, our finding that ΔIami was detectable in essentially all control recordings from the CNT/CCD (Fig. 4A, control) confirmed that we reliably selected principal cells for our recordings. In contrast, in doxycycline-treated and genetically verified MR KO mice, ΔIami in the CNT/CCD was overall significantly lower, with a median of 0 pA and an upper quartile of 185 pA (n = 44, P = 0.01). In the majority of recordings (27 of 44 recordings) from the CNT/CCD of MR KO mice, ΔIami was undetectable or barely detectable (<10 pA), indicating effective MR KO. Interestingly, in several recordings from the CNT/CCD of MR KO mice, ΔIami values were similar to those in the control CNT/CCD or even higher, reaching values in the nanoampere range (Fig. 4A). These findings suggest that MR expression was preserved at least in some cells in the CNT/CCD of doxycycline-treated and genetically verified MR KO mice, which is consistent with our immunohistochemical findings.
Figure 3.

Mineralocorticoid receptor (MR) knockout (KO) nearly abolishes amiloride-sensitive currents (ΔIami) in the connecting tubule (CNT)/cortical collecting duct (CCD) and substantially reduces ΔIami in the late distal convoluted tubule (DCT2)/CNT. Representative whole cell current traces are shown from the CNT/CCD (top traces) and DCT2/CNT (bottom traces) isolated from a female control mouse (A) and a male MR KO mouse (B). Black bars indicate the presence of amiloride (2 µM) in the bath solution; holding potential = −60 mV. Amiloride washout resulted in a gradual increase of the inward current to a plateau level. Reapplication of amiloride returned the current to its baseline level and was used to determine ΔIami.
Figure 4.

Summary of amiloride-sensitive currents (ΔIami) values from similar experiments as shown in Fig. 3. Connecting tubule (CNT)/cortical collecting duct (CCD) and late distal convoluted tubule (DCT2)/CNT data are summarized in A and B, respectively. Individual measurements, medians, and quartiles are shown. In both diagrams, the left and middle columns show all ΔIami values obtained from control mice and mineralocorticoid receptor (MR) knockout (KO) mice (MR KO overall), respectively. Data points from female and male animals are depicted with circles and triangles, respectively. No obvious sex effect was noticed in any experimental group. Individual ΔIami values from control mice are depicted with black symbols. Red symbols are used for ΔIami values from MR KO mice in which ΔIami in the CNT/CCD did not exceed 10 pA in any measurement. Blue symbols indicate ΔIami values obtained from mice not meeting this criterion. For clarity, the red ΔIami values are shown separately in the right columns called “MR KO max. < 10 pA” in A and “MR KO max. < 10 pA in CNT/CCD” in B. Numbers of mice (N) and individual measurements (n) are given below each group of mice. The age of control mice (5 females and 6 males) ranged from 36 to 68 days, and the age of MR KO mice (12 females and 11 males) ranged from 35 to 65 days.
MR Deficiency Decreases ENaC Activity in the DCT2/CNT
In the recordings shown on the bottom of Fig. 3A and summarized in Fig. 4B, we confirmed that ENaC activity in the DCT2/CNT was substantially higher than in the CNT/CCD. Indeed, median ΔIami in the DCT2/CNT of control mice was 231 pA (162 pA, 357 pA) (n = 15). This ΔIami value was significantly higher than that in the CNT/CCD of control mice (P < 0.001), which is consistent with our previous findings (6, 7). Importantly, in the DCT2/CNT of MR KO mice, ΔIami with a median of 57 pA (29 pA, 129 pA) (n = 32) was significantly smaller than ΔIami in the DCT2/CNT of control mice (P < 0.001; Fig. 4B, MR KO overall). This inhibitory effect of MR deficiency on ΔIami in the DCT2/CNT became even more prominent when we included in the analysis only those MR KO mice in which ΔIami measurements in the CNT/CCD did not exceed 10 pA. The aim of this selection criterion was to produce a bias toward MR KO mice with a high degree of MR KO efficiency. Indeed, in this group of MR KO mice (Fig. 4B, MR KO maximum < 10 pA in the CNT/CCD), median ΔIami in the DCT2/CNT was 35 pA (21 pA, 95 pA) (n = 17). Thus, in mice with functional evidence for MR deficiency, the activity of ENaC in the DCT2/CNT was reduced by ∼85% compared with control mice with intact MR.
Po in the DCT2/CNT From MR KO Mice Is Not Reduced Compared With Control
In the DCT2/CNT of MR KO mice, reduced ENaC activity may be due to lower channel density at the cell surface, lower channel Po, or a combination of both effects. To distinguish between these possibilities, we performed single-channel recordings in outside-out patches to compare Po of ENaC in the DCT2/CNT of control and MR KO mice. Typical single-channel current traces from successful outside-out patches obtained from a control DCT2/CNT and a DCT2/CNT from an MR KO mouse are shown in Fig. 5, A and B, respectively. Median ensemble ΔIami values determined in the corresponding whole-cell recordings before achievement of the outside-out configuration were 206 pA (180 pA, 373 pA) in the DCT2/CNT from control mice and 52 pA (38 pA, 119 pA) in the DCT2/CNT from MR KO mice, consistent with the whole-cell current data shown in Fig. 4B. In recordings with resolvable single-channel activity, amiloride was applied to determine the current level at which all channels are closed. NPo was derived from amplitude histograms, and Po was estimated by analyzing the current level probability distribution as previously described (Fig. 5, C and D) (7, 23). ENaC Po was not manifestly reduced in the DCT2/CNT of MR KO mice compared with control. If anything, there was a small trend toward an increased Po in MR KO mice with a median Po of 0.33 (0.18, 0.43) versus 0.25 (0.10, 0.33) in control animals (Fig. 5E). These findings suggest that the reduced ΔIami values in whole-cell recordings cannot be attributed to a lower single-channel Po but are mainly due to decreased channel expression at the cell surface. The representative traces shown in Fig. 5, A and B, seem to support this conclusion, with seven channels estimated to be present in the control patch (Fig. 5C) but only two channels in the patch from the MR KO animal (Fig. 5D). On average, the estimated number of channels present in outside-out patches from the DCT2/CNT of MR KO animals was not significantly different from that of control mice (Fig. 5F). This is possibly due to an experimental bias, because in outside-out patches, individual channel levels can be resolved only when active channels are present in the patch and when their number is relatively small. Thus, with our experimental approach, we are likely to underestimate the number of channels in control mice and overestimate the number of channels in MR KO mice. In addition, small differences in pipette tip diameter and patch size may hamper the quantification of the number of channels by this method. In contrast, the Po estimates are less affected by these confounding factors. On balance, our single-channel data suggest that MR deficiency reduces cell surface expression of ENaC in the DCT2/CNT.
Figure 5.
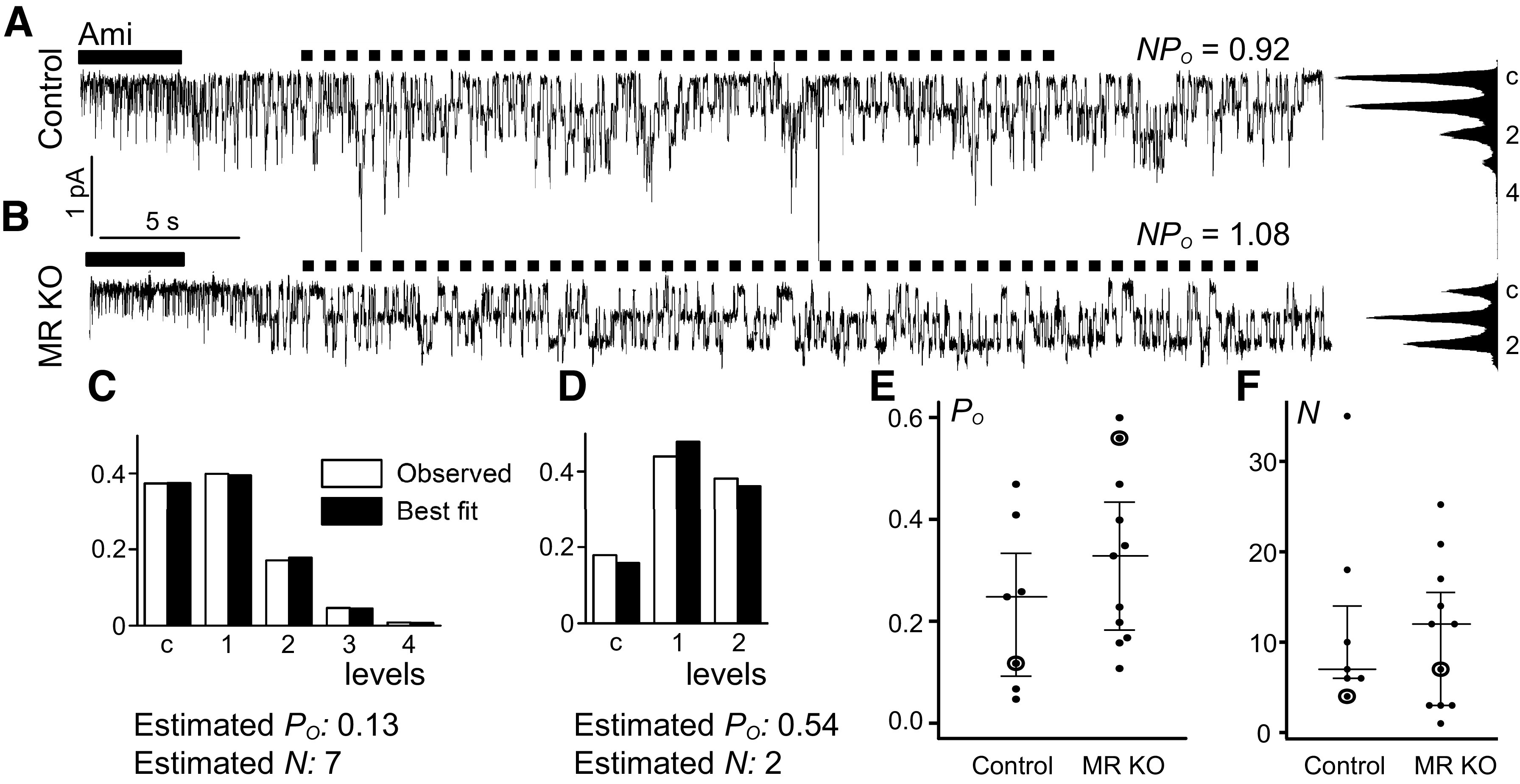
Single-channel open probability (Po) of the epithelial Na+ channel (ENaC) in the late distal convoluted tubule (DCT2)/connecting tubule (CNT) from mineralocorticoid receptor (MR) knockout (KO) mice is similar to that in control mice. A and B: representative outside-out patch-clamp recordings from the DCT2/CNT of a male control mouse (A) and a female MR KO mouse (B). Black bars indicate amiloride (Ami) application; dotted lines delineate intervals used for the analysis of the distribution of current levels, the construction of amplitude histograms, and the calculation of channel activity (NPo). C and D: observed (open bars) and best-fit (black bars) probability distribution for the current levels of the traces shown in A and B, respectively; corresponding estimated Po values and estimated numbers of channels (N) are given. E: summary of estimated Po values in control (N = 5 animals, n = 7 individual recordings) and MR KO mice (N = 9 animals, n = 11 individual recordings). F: summary of estimated number of channel values from the same experiments. Individual observations with medians and quartiles are shown. The data points from the representative traces shown in A and B are encircled.
Expression of 11β-HSD2 Is Essentially Absent in the DCT1, Rather Weak in the DCT2, and Progressively Increases Along the CNT and CCD
Expression of 11β-HSD2 is probably lower in the proximal portion of the ASDN than in its distal portion (18–20). To confirm this, kidney sections were costained for 11β-HSD2 (Fig. 6A), for NCC as a marker of the DCT (Fig. 6B), and for calbindin as a marker of the DCT2, CNT, and CCD (Fig. 6C) (27). Using this approach, and by selecting transition segments containing the contiguous DCT-CNT-CCD for analysis, each of the segments can be identified. The DCT1 can be recognized as an NCC-positive tubular fragment with weak or negative staining for calbindin. In contrast, the DCT2 stains strongly positive for calbindin and NCC, whereas the CNT and CCD are calbindin positive and NCC negative (Fig. 6D). We observed cytosolic and nuclear calbindin staining, which is consistent with previously reported findings (28–30). Calbindin is a soluble protein and likely to be sensitive to fixation, which may explain its variable nuclear localization. Fluorescence intensity was quantified to estimate the relative expression level of 11β-HSD2 in different tubular segments. In good agreement with previous studies, we found no specific 11β-HSD2 staining in the DCT1. 11β-HSD2 staining was detectable but very weak in the DCT2 and abruptly increased at the transition from the DCT2 to CNT with a further increase in the CCD (Fig. 6E).
Figure 6.
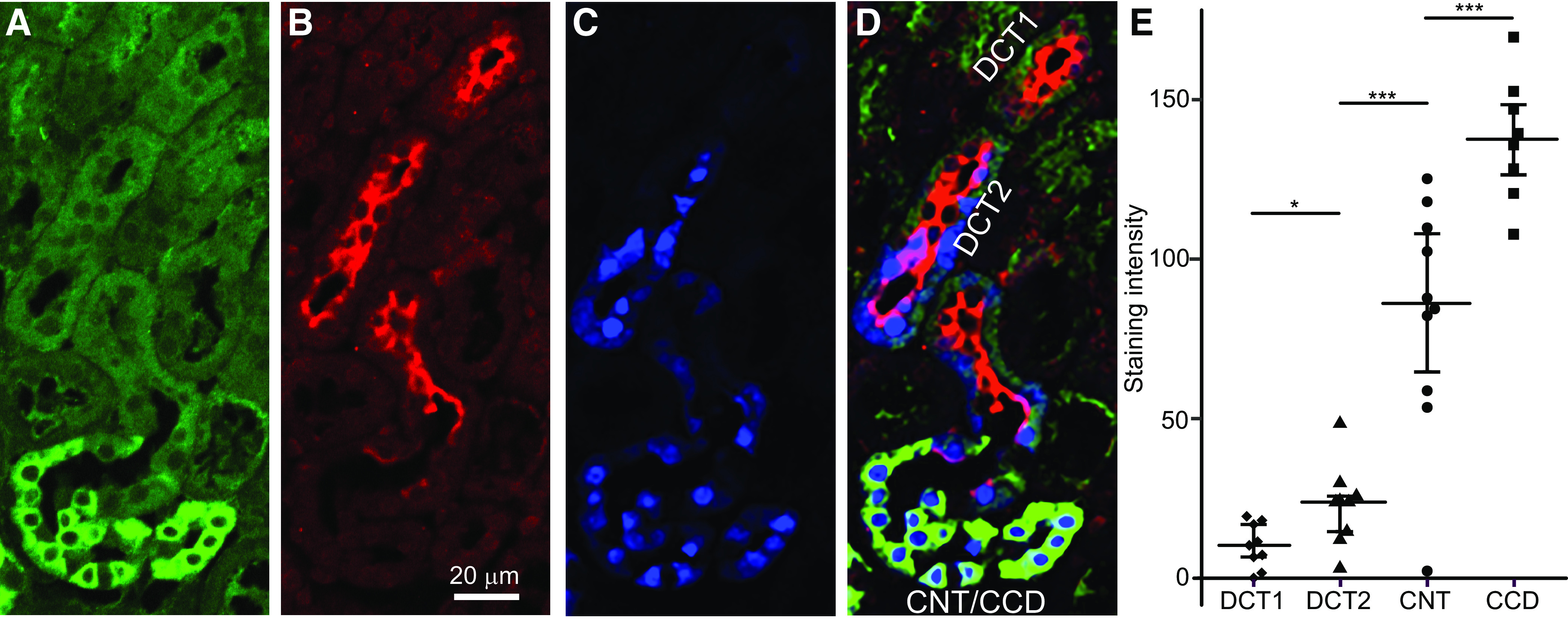
11β-Hydroxysteroid dehydrogenase 2 (11β-HSD2) expression along the distal nephron (A–D). Parallel staining of 11β-HSD2 with different nephron-specific markers is shown. A: immunofluorescence staining for 11β-HSD2. B: staining for NaCl cotransporter (NCC) as a marker of the distal convoluted tubule (DCT). C: staining of calbindin as a marker of the late DCT (DCT2), connecting tubule (CNT), and cortical collecting duct (CCD). D: merged image. Green indicates 11β-HSD2, red indicates NCC, and blue indicates calbindin. E: staining intensity for 11β-HSD2 in different parts of the nephron. DCT1, early DCT. *P < 0.05; ***P < 0.001.
DISCUSSION
In a previous study, we demonstrated that ENaC activity is fully preserved in the DCT2/CNT of aldosterone synthase-deficient mice. Thus, ENaC is aldosterone independent in the DCT2/CNT, unlike in the CNT/CCD (6). In the present study, we used a mouse model with inducible nephron-specific MR deficiency (21) to demonstrate that aldosterone-independent ENaC activity in the DCT2/CNT is largely MR dependent. Moreover, we provide immunohistochemical evidence showing that expression of 11β-HSD2 is low or even absent in the DCT2/CNT. Thus, in the DCT2/CNT, conversion of glucocorticoids into inactive metabolites is likely to be incomplete. Consequently, circulating glucocorticoids could activate MR, which probably explains the substantial MR-dependent but aldosterone-independent component of ENaC activity in this part of the ASDN.
In MR KO Mice, Expression of MR Is Equally Reduced in the DCT2/CNT and CNT/CCD With Considerable Interanimal Variability of KO Efficiency
Our immunohistochemical experiments confirmed that in MR KO mice, expression of MR was significantly suppressed in the entire ASDN, as expected from previously reported findings that MR expression in MR KO mice was reduced by ∼80% and ∼90% at the mRNA and protein level, respectively (21). Moreover, we demonstrated that the degree of MR KO in the DCT2/CNT was similar to that in the CNT/CCD. Our immunohistochemical data also demonstrate that MR KO was not always complete, with considerable interanimal variability regarding the degree of KO efficiency. This latter observation is reflected by our functional data demonstrating that ENaC-mediated whole-cell currents were minimal (<10 pA) in most, but not in all, recordings from the CNT/CCD microdissected from MR KO mice. Previously, we have shown that aldosterone is essential for ENaC activity in the CNT/CCD (6). This justifies the conclusion that in CNT/CCD cells with largely suppressed ENaC activity (<10 pA), MR KO was probably complete. On the other hand, we have to conclude that in CNT/CCD cells with larger ENaC currents, MR expression was at least partially preserved.
Several factors may contribute to incomplete MR KO. Pax8 is a developmental gene, and its expression decreases after birth (31, 32). In newborn mice, MR deficiency is known to lead to death due to salt loss (10). Therefore, to induce MR KO, we started doxycycline treatment 26–28 days after birth. This made it possible to compensate for renal salt wasting due to MR deletion by adding NaCl to the drinking solution. The Pax8-rtTAtg system has been previously shown to produce reliable gene KO in mice treated with doxycycline at the age of 8 wk (33) and has been used successfully for nephron-specific deletion of serum and glucocorticoid-inducible kinase (34) and neural precursor cell expressed developmentally downregulated 4-2 (35). Nevertheless, we cannot rule out that Pax8 promotor activity may have been too low in some MR KO mice to induce effective MR KO in all cells in the ASDN. In addition, Cre recombinase activity depends on the specific DNA structure in proximity to the loxP sites (36), which may explain why deletion of some genes is more effective than that of others, possibly including the MR gene. Moreover, MR promotes cell survival and proliferation (37). Therefore, even very few cells escaping MR gene deletion may reduce overall KO efficiency substantially due to the selective advantage of MR-expressing cells regarding cell survival and proliferation.
ENaC Currents in Some CNT/CCD Cells From MR KO Mice Were Higher Than Control Currents Probably Due to Incomplete MR KO and Compensatory Upregulation of Plasma Aldosterone
Interestingly, in a minority of recordings in the CNT/CCD from MR KO mice, we observed ENaC currents that were as high as those observed in the CNT/CCD from control mice or even higher. As discussed in the preceding section, it is safe to conclude that CNT/CCD cells with sizeable ENaC currents express functional MR. To compensate for their overall reduced MR expression, MR KO mice increase their plasma aldosterone to very high levels (21). This probably explains why in MR KO mice ENaC currents were surprisingly high in those CNT/CCD cells that escaped MR deletion. Indeed, in some cases, ENaC currents in the CNT/CCD from MR KO mice were 10 times higher than the maximal currents observed in the CNT/CCD of control mice and comparable to currents previously observed in the CNT from rats after infusion of pharmacological doses of aldosterone (38). Mosaic gene expression of Cre recombinase is not uncommon (39) and may be the reason why some cells escaped MR deletion. Moreover, it is conceivable that cells with preserved MR expression have an advantage to remain viable in microdissected split-open tubular fragments and to be selected in our patch-clamp experiments. Thus, some of the recordings with very high ENaC currents in the CNT/CCD from MR KO mice may be due to a selection bias.
Even in MR KO mice with low KO efficiency (e.g., mouse 4 and mouse 5 in Fig. 1), overall MR-positive nuclear staining appeared to be slightly reduced. Thus, plasma aldosterone levels are likely to be elevated also in mice with partially preserved MR function in the ASDN. In addition, it has been previously reported that Pax8 activity in adult kidneys is higher in the renal medulla than in the cortex (31). Therefore, incomplete MR KO in the ASDN does not rule out pronounced MR deficiency in the medullary CD. Overall ENaC activity in the outer medullary CD has been reported to be comparable to that in the CCD (40). Thus, reduced ENaC activity due to MR KO in the medullary CD alone may be sufficient to elicit an increase in plasma aldosterone, which could explain the higher than normal ENaC currents in CNT/CCD cells of MR KO mice with preserved MR. Our functional data and immunohistochemical analysis highlight the need to consider cell-to-cell and animal-to-animal variability when using nephron-specific and inducible KO mouse models. Importantly, our data confirm the suitability of the mouse model used in the present study to explore the effect of nephron-specific MR KO on ENaC currents in the DCT2/CNT.
Aldosterone-Independent ENaC Activity in the DCT2/CNT Largely Depends on the Presence of MR
In contrast to aldosterone synthase deficiency (6), nephron-wide MR KO caused a significant and substantial reduction of ENaC currents in the DCT2/CNT to ∼25% of control. An even more pronounced reduction to ∼15% was observed when we considered only those MR KO mice with ΔIami values in the CNT/CCD not exceeding 10 pA, which suggests effective MR deletion in these animals. According to our single-channel recordings in outside-out patches, ENaC Po in the DCT2/CNT was not significantly different in MR-deficient mice compared with control mice. Thus, the reduced ENaC whole-cell currents in the DCT2/CNT from MR-deficient mice is most likely due to decreased channel expression at the cell surface. This is consistent with the commonly held view that MR signaling is critically important for increasing ENaC abundance in the apical membrane. Taken together, our findings clearly indicate that MR is not only critical for aldosterone-dependent ENaC activity in the CNT/CCD but also plays a major role in maintaining aldosterone-independent ENaC activity in the DCT2/CNT.
Qualitatively, our findings are in agreement with a recent study that also reported a significant reduction of ENaC currents in the DCT2/CNT obtained from another mouse model with doxycycline-inducible nephron-specific MR deficiency (8). In this kidney-specific MR knockout (KS-MR-KO) mouse model, MR deficiency also significantly reduced ENaC currents in the DCT2/CNT, albeit only by ∼40% (8). Moreover, application of losartan, an inhibitor of the angiotensin II type 1 receptor, strongly inhibited ENaC in the DCT2/CNT of both control mice and KS-MR-KO mice but not in the CCD. Therefore, it was concluded that the angiotensin II type 1 receptor plays an important role in maintaining ENaC activity in the DCT2/CNT by an MR-independent manner. This latter conclusion is consistent with previously reported findings that treatment with losartan prevented apical translocation of ENaC in the DCT/CNT of aldosterone synthase-deficient mice (41). We have no clear explanation for the relatively weak effect of MR deletion in the KS-MR-KO mouse model compared with the pronounced effect observed in our mouse model. One possibility is a different protocol for the induction of MR deletion in the KS-MR-KO model, which was also generated through use of the Pax8-rtTA system (42). Treatment with doxycycline for 2 wk was started at the age of 8 wk and was followed by 2 wk without doxycycline before the electrophysiological experiments (8). This protocol may induce incomplete MR deletion at least in some animals with time for partial recovery of MR expression when doxycycline is discontinued. This may explain why the MR-dependent component of ENaC activity in the DCT2/CNT appeared to be less prominent in the KS-MR-KO mouse model. Interestingly, the plasma K+ concentration in these mice was ∼4 mM (42), which is in contrast to the more pronounced hyperkalemia of ∼8 mM observed under the normal-salt diet in MR KO mice from the same strain as used in the present study (21). This supports the conclusion that overall MR deficiency in KS-MR-KO mice may have been less severe than in the model used in the present study. In our view, both studies support the conclusion that MR plays a significant role in maintaining aldosterone-independent ENaC activity in the DCT2/CNT. Obviously, this does not rule out that other factors contribute to ENaC regulation in the DCT2/CNT, with angiotensin II being a strong candidate. Indeed, the finding that MR deletion essentially abolished ENaC currents in the CNT/CCD but only partially inhibited ENaC currents in the DCT2/CNT indicates that the MR dependence of ENaC is less complete in this latter part of the ASDN. Future studies are needed to elucidate additional components involved in maintaining and regulating ENaC activity in the DCT2/CNT.
11β-HSD2 Is Highly Expressed in the CCD and CNT but Not in the DCT
The observation that MR expression, but not aldosterone, is critically important for ENaC activity in the DCT2/CNT raises the question as to how MR may be stimulated in this nephron segment in an aldosterone-independent manner. MR has equal affinity to mineralocorticoids and glucocorticoids. This implies that in aldosterone target tissues, the specificity of aldosterone action largely depends on the presence of 11β-HSD2, which converts cortisol and corticosterone into inactive metabolites (15, 16). It is well established that 11β-HSD2 is highly expressed in the CD. In contrast, conflicting data have been reported about its expression in more proximal parts of the nephron. In the human kidney, high expression of 11β-HSD2 has been described beginning in the thick ascending limb (TAL) and continuing through the entire DCT and CNT into the CD (43). Using different antibodies, 11β-HSD2 expression was detected in the human CD and TAL (44). In the rat kidney, 11β-HSD2 expression was observed in the DCT2, CNT, and CCD (45, 46). Similar results were reported for the mouse kidney, with high 11β-HSD2 expression in the DCT2, lower expression in the DCT1, and no detectable expression in the TAL (19). Interestingly, another study reported that 11β-HSD2 and NCC do not colocalize in the mouse kidney (20). This latter finding suggests that 11β-HSD2 is absent or barely expressed in the DCT. In the present study, we used immunofluorescence and costaining experiments to reinvestigate this issue. We detected the highest 11β-HSD2 staining in the CCD with a slightly reduced expression in the CNT and a largely reduced 11β-HSD2 signal in the DCT2/CNT transition zone with essentially no specific signal in the DCT1. Thus, our data support the hypothesis that 11β-HSD2 expression in the DCT2 and possibly in the initial part of the CNT is sufficiently low to permit MR activation by glucocorticoids and consequently aldosterone-independent stimulation of ENaC. In this sense, our findings challenge the view that the DCT2 is functionally part of the ASDN, at least regarding the aldosterone-sensitivity of ENaC.
Perspectives and Significance
Using a mouse model with inducible nephron-specific MR deficiency, we demonstrated a key role of MR for maintaining ENaC activity not only in the CNT/CCD but also in the DCT2/CNT. Furthermore, we demonstrated that cells of this latter nephron segment express little 11β-HSD2, which probably allows glucocorticoids to stimulate MR, resulting in aldosterone-independent ENaC activity in the DCT2/CNT (Fig. 7). However, unlike in the CNT/CCD, MR deficiency does not completely abolish ENaC activity in the DCT2/CNT, which is consistent with the concept that additional mechanisms, for example, angiotensin II (8), contribute to ENaC function and regulation in this nephron segment. Our findings have (patho)physiological implications, because appropriate ENaC regulation in the DCT2/CNT through MR signaling is likely to play an important role in the long-term control of arterial blood pressure. The specific relevance of ENaC regulation in the DCT2/CNT for blood pressure control is highlighted by the finding that in Liddle syndrome, a severe form of hereditary salt-sensitive hypertension, ENaC is hyperactive mainly in this early part of the ASDN (7). Moreover, there is good evidence to suggest that the early part of the ASDN, including the DTC2/CNT region, contributes substantially to renal K+ secretion (2). This concept is also supported by the previously reported finding that aldosterone synthase-deficient mice adapt to a high-K+ diet by upregulating the secretory K+ channel, the renal outer medullary K+ channel, in the DCT2/CNT (41). Thus, aldosterone-independent but MR-dependent ENaC activity in the DCT2/CNT may be critical for renal K+ excretion because it maintains a lumen-negative transepithelial potential difference as a driving force for K+ secretion even when plasma aldosterone levels are low and ENaC activity is negligible in more distal parts of the ASDN.
Figure 7.

In the absence of aldosterone, glucocorticoids stimulate the epithelial Na+ channel (ENaC) in the late distal convoluted tubule (DCT2)/connecting tubule (CNT) but not in the CNT/cortical collecting duct (CCD). Low expression of 11β-hydroxysteroid dehydrogenase 2 (11β-HSD2) in the DCT2/CNT probably allows glucocorticoids to stimulate mineralocorticoid receptor (MR), resulting in aldosterone-independent ENaC activity in this nephron segment. Shown is a schematic diagram of a distal tubule with the early distal convoluted tubule (DCT1), DCT2, CNT, and CCD. The convergence of two CNTs marks the beginning of the CCD. The expression pattern of 11β-HSD2, MR, and apically localized ENaC is illustrated in green, red, and blue, respectively, with color intensity reflecting the level of expression. MR-mediated ENaC activation (red arrows) can only occur in those parts of the nephron where 11β-HSD2 does not prevent (X) glucocorticoid action (black arrows). Please note that the diagram corresponds to a situation in which the circulating aldosterone level is negligible and apical ENaC expression is essentially absent in the late CNT and CCD where ENaC activity is known to be aldosterone dependent (6).
GRANTS
This work was supported by Grant KO 1057/10-1 from the Deutsche Forschungsgemeinschaft (German Research Foundation) to C.K.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.N., M.B., and C.K. conceived and designed research; V.N., M.B., J.C., and R.C. performed experiments; V.N., M.B., J.C., E.H., R.C., P.A.W., and C.K. analyzed data; V.N., M.B., E.H., P.A.W., and C.K. interpreted results of experiments; V.N., M.B., E.H., and P.A.W. prepared figures; V.N., M.B., and C.K. drafted manuscript; V.N., M.B., J.C., E.H., P.A.W., and C.K. edited and revised manuscript; V.N., M.B., J.C., E.H., R.C., P.A.W., and C.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Jessica Rinke and Lorenz Mc Cargo for providing expert technical assistance.
REFERENCES
- 1.Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflugers Arch 458: 111–135, 2009. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- 2.Meneton P, Loffing J, Warnock DG. Sodium and potassium handling by the aldosterone-sensitive distal nephron: the pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004. doi: 10.1152/ajprenal.00454.2003. [DOI] [PubMed] [Google Scholar]
- 3.Loffing J, Zecevic M, Féraille E, Kaissling B, Asher C, Rossier BC, Firestone GL, Pearce D, Verrey F. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol 280: F675–F682, 2001. doi: 10.1152/ajprenal.2001.280.4.F675. [DOI] [PubMed] [Google Scholar]
- 4.PáCha J, Frindt G, Antonian L, Silver RB, Palmer LG. Regulation of Na channels of the rat cortical collecting tubule by aldosterone. J Gen Physiol 102: 25–42, 1993. doi: 10.1085/jgp.102.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frindt G, Sackin H, Palmer LG. Whole-cell currents in rat cortical collecting tubule: low-Na diet increases amiloride-sensitive conductance. Am J Physiol Renal Physiol 258: F562–F567, 1990. doi: 10.1152/ajprenal.1990.258.3.F562. [DOI] [PubMed] [Google Scholar]
- 6.Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol 303: F1289–F1299, 2012. doi: 10.1152/ajprenal.00247.2012. [DOI] [PubMed] [Google Scholar]
- 7.Nesterov V, Krueger B, Bertog M, Dahlmann A, Palmisano R, Korbmacher C. In Liddle syndrome, epithelial sodium channel is hyperactive mainly in the early part of the aldosterone-sensitive distal nephron. Hypertension 67: 1256–1262, 2016. doi: 10.1161/HYPERTENSIONAHA.115.07061. [DOI] [PubMed] [Google Scholar]
- 8.Wu P, Gao ZX, Zhang DD, Duan XP, Terker AS, Lin DH, Ellison DH, Wang WH. Effect of angiotensin II on ENaC in the distal convoluted tubule and in the cortical collecting duct of mineralocorticoid receptor deficient mice. J Am Heart Assoc 9: e014996, 2020. doi: 10.1161/JAHA.119.014996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Frindt G, Xu Y, Uchida S, Palmer LG. Aldosterone-dependent and -independent regulation of Na+ and K+ excretion and ENaC in mouse kidneys. Am J Physiol Renal Physiol 319: F323–F334, 2020. doi: 10.1152/ajprenal.00204.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, Kriz W, Warth R, Greger R, Schütz G. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci USA 95: 9424–9429, 1998. doi: 10.1073/pnas.95.16.9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makhanova N, Lee G, Takahashi N, Lopez MLS, Gomez R A, Kim H-S, Smithies O, Sequeira Lopez ML, Gomez RA, Kim H, Smithies O. Kidney function in mice lacking aldosterone. Am J Physiol Renal Physiol 290: F61–F69, 2006. doi: 10.1152/ajprenal.00257.2005. [DOI] [PubMed] [Google Scholar]
- 12.Ronzaud C, Loffing J, Bleich M, Gretz N, Gröne H-J, Schütz G, Berger S. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 18: 1679–1687, 2007. doi: 10.1681/ASN.2006090975. [DOI] [PubMed] [Google Scholar]
- 13.Rubera I, Loffing J, Palmer LG, Frindt G, Fowler-Jaeger N, Sauter D, Carroll T, Mcmahon A, Hummler E, Rossier BC. Collecting duct-specific gene inactivation of αENaC in the mouse kidney does not impair sodium and potassium balance. J Clin Invest 112: 554–565, 2003. doi: 10.1172/JCI16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDonald FJ, Yang B, Hrstka RF, Drummond HA, Tarr DE, McCray PB Jr, Stokes JB, Welsh MJ, Williamson RA. Disruption of the β subunit of the epithelial Na+ channel in mice: hyperkalemia and neonatal death associated with a pseudohypoaldosteronism phenotype. Proc Natl Acad Sci USA 96: 1727–1731, 1999. doi: 10.1073/pnas.96.4.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funder J. 30 years of the mineralocorticoid receptor: mineralocorticoid receptor activation and specificity-conferring mechanisms: a brief history. J Endocrinol 234: T17–T21, 2017. doi: 10.1530/JOE-17-0119. [DOI] [PubMed] [Google Scholar]
- 16.Hunter RW, Bailey MA. Glucocorticoids and 11β-hydroxysteroid dehydrogenases: mechanisms for hypertension. Curr Opin Pharmacol 21: 105–114, 2015. doi: 10.1016/j.coph.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Funder JW. Apparent mineralocorticoid excess. J Steroid Biochem Mol Biol 165: 151–153, 2017. doi: 10.1016/j.jsbmb.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 18.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, Ellison DH. 11β-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998. [Erratum in J Am Soc Nephrol 9: 2179, 1998]. doi: 10.1681/ASN.V981347. [DOI] [PubMed] [Google Scholar]
- 19.Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC, Loffing J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol 299: F1473–F1485, 2010. doi: 10.1152/ajprenal.00437.2010. [DOI] [PubMed] [Google Scholar]
- 20.Hunter RW, Ivy JR, Flatman PW, Kenyon CJ, Craigie E, Mullins LJ, Bailey MA, Mullins JJ. Hypertrophy in the distal convoluted tubule of an 11β-hydroxysteroid dehydrogenase type 2 knockout model. J Am Soc Nephrol 26: 1537–1548, 2015. doi: 10.1681/ASN.2013060634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canonica J, Sergi C, Maillard M, Klusonova P, Odermatt A, Koesters R, Loffing-Cueni D, Loffing J, Rossier B, Frateschi S, Hummler E. Adult nephron-specific MR-deficient mice develop a severe renal PHA-1 phenotype. Pflugers Arch Eur J Physiol 468: 895–908, 2016. doi: 10.1007/s00424-015-1785-2. [DOI] [PubMed] [Google Scholar]
- 22.Nesterov V, Dahlmann A, Bertog M, Korbmacher C. Trypsin can activate the epithelial sodium channel (ENaC) in microdissected mouse distal nephron. Am J Physiol Renal Physiol 295: F1052–F1062, 2008. doi: 10.1152/ajprenal.00031.2008. [DOI] [PubMed] [Google Scholar]
- 23.Nesterov V. Nest-O-Patch 2.1. User’s Guide. https://www.researchgate.net/publication/326450502_Nest-O-Patch_21_User%27s_guide. doi: 10.13140/RG.2.2.16080.00001. [DOI]
- 24.Nesterov V. Nest-o-Patch ver. 2.1:2017 [Computer software]. Publisher: Sourceforge. https://sourceforge.net/projects/nestopatch/.
- 25.Gomez-Sanchez CE, de Rodriguez AF, Romero DG, Estess J, Warden MP, Gomez-Sanchez MT, Gomez-Sanchez EP. Development of a panel of monoclonal antibodies against the mineralocorticoid receptor. Endocrinology 147: 1343–1348, 2006. doi: 10.1210/en.2005-0860. [DOI] [PubMed] [Google Scholar]
- 26.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2597–2606, 2017. doi: 10.1681/ASN.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loffing J, Loffing-Cueni D, Valderrabano V, Kläusli L, Hebert SC, Rossier BC, Hoenderop JG, Bindels RJ, Kaissling B. Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol Renal Physiol 281: F1021–F1027, 2001. doi: 10.1152/ajprenal.0085.2001. [DOI] [PubMed] [Google Scholar]
- 28.Mounier F, Hinglais N, Brehier A, Thomasset M, Lacoste M, Sich M, Gubler MC. Ontogenesis of 28 kDa vitamin D-induced calcium-binding protein in human kidney. Kidney Int 31: 121–129, 1987. doi: 10.1038/ki.1987.18. [DOI] [PubMed] [Google Scholar]
- 29.Roth J, Brown D, Norman AW, Orci L. Localization of the vitamin D-dependent calcium-binding protein in mammalian kidney. Am J Physiol Renal Physiol 243: F243–F252, 1982. doi: 10.1152/ajprenal.1982.243.3.F243. [DOI] [PubMed] [Google Scholar]
- 30.German DC, Ng MC, Liang CL, McMahon A, Iacopino AM. Calbindin-D28k in nerve cell nuclei. Neuroscience 81: 735–743, 1997. doi: 10.1016/S0306-4522(97)00206-6. [DOI] [PubMed] [Google Scholar]
- 31.Sharma R, Sanchez-Ferras O, Bouchard M. Pax genes in renal development, disease and regeneration. Semin Cell Dev Biol 44: 97–106, 2015. doi: 10.1016/j.semcdb.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 32.Strachan T, Read AP. PAX genes. Curr Opin Genet Dev 4: 427–438, 1994. doi: 10.1016/0959-437x(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 33.Traykova-Brauch M, Schönig K, Greiner O, Miloud T, Jauch A, Bode M, Felsher DW, Glick AB, Kwiatkowski DJ, Bujard H, Horst J, von Knebel Doeberitz M, Niggli FK, Kriz W, Gröne H-J, Koesters R. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 14: 979–984, 2008. doi: 10.1038/nm.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faresse N, Lagnaz D, Debonneville A, Ismailji A, Maillard M, Fejes-Tóth G, Naray-Fejes-Toth A, Staub O. Inducible kidney-specific Sgk1 knockout mice show a salt-losing phenotype. Am J Physiol Renal Physiol 302: F977–F985, 2012. doi: 10.1152/ajprenal.00535.2011. [DOI] [PubMed] [Google Scholar]
- 35.Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler-Jaeger N, Boase NA, Perrier R, Maillard M, Yang B, Stokes JB, Koesters R, Kumar S, Hummler E, Loffing J, Staub O. Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest 123: 657–665, 2013. doi: 10.1172/JCI61110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohan DE. Progress in gene targeting: using mutant mice to study renal function and disease. Kidney Int 74: 427–437, 2008. doi: 10.1038/ki.2008.146. [DOI] [PubMed] [Google Scholar]
- 37.Dooley R, Harvey BJ, Thomas W. The regulation of cell growth and survival by aldosterone. Front Biosci (Landmark Ed) 16: 440–457, 2011. doi: 10.2741/3697. [DOI] [PubMed] [Google Scholar]
- 38.Frindt G, Palmer LG. Na channels in the rat connecting tubule. Am J Physiol Renal Physiol 286: F669–F674, 2004. doi: 10.1152/ajprenal.00381.2003. [DOI] [PubMed] [Google Scholar]
- 39.Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods 14: 381–392, 1998. doi: 10.1006/meth.1998.0593. [DOI] [PubMed] [Google Scholar]
- 40.Frindt G, Ergonul Z, Palmer LG. Na channel expression and activity in the medullary collecting duct of rat kidney. Am J Physiol Renal Physiol 292: F1190–F1196, 2007. doi: 10.1152/ajprenal.00399.2006. [DOI] [PubMed] [Google Scholar]
- 41.Todkar A, Picard N, Loffing-Cueni D, Sorensen MV, Mihailova M, Nesterov V, Makhanova N, Korbmacher C, Wagner CA, Loffing J. Mechanisms of renal control of potassium homeostasis in complete aldosterone deficiency. J Am Soc Nephrol 26: 425–438, 2015. doi: 10.1681/ASN.2013111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, Mccormick JA, Yang C-L, Ellison DH. Direct and indirect mineralocorticoid effects determine distal salt transport. J Am Soc Nephrol 27: 2436–2445, 2016. doi: 10.1681/ASN.2015070815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krozowski Z, MaGuire JA, Stein-Oakley AN, Dowling J, Smith RE, Andrews RK. Immunohistochemical localization of the 11β-hydroxysteroid dehydrogenase type II enzyme in human kidney and placenta. J Clin Endocrinol Metab 80: 2203–2209, 1995. doi: 10.1210/jcem.80.7.7608280. [DOI] [PubMed] [Google Scholar]
- 44.Kyossev Z, Walker PD, Reeves WB. Immunolocalization of NAD-dependent 11β-hydroxysteroid dehydrogenase in human kidney and colon. Kidney Int 49: 271–281, 1996. doi: 10.1038/ki.1996.39. [DOI] [PubMed] [Google Scholar]
- 45.Kim SW, de Seigneux S, Sassen MC, Lee J, Kim J, Knepper MA, Frøkiær J, Nielsen S. Increased apical targeting of renal ENaC subunits and decreased expression of 11βHSD2 in HgCl2-induced nephrotic syndrome in rats. Am J Physiol Renal Physiol 290: F674–F687, 2006. doi: 10.1152/ajprenal.00084.2005. [DOI] [PubMed] [Google Scholar]
- 46.Bachmann S, Bostanjoglo M, Schmitt R, Ellison DH. Sodium transport-related proteins in the mammalian distal nephron – distribution, ontogeny and functional aspects. Anat Embryol (Berl) 200: 447–468, 1999. doi: 10.1007/s004290050294. [DOI] [PubMed] [Google Scholar]