

Abstract
Renal cell carcinoma (RCC) is diagnosed through expensive cross-sectional imaging, frequently followed by renal mass biopsy, which is not only invasive but also prone to sampling errors. Hence, there is a critical need for a noninvasive diagnostic assay. RCC exhibits altered cellular metabolism combined with the close proximity of the tumor(s) to the urine in the kidney, suggesting that urine metabolomic profiling is an excellent choice for assay development. Here, we acquired liquid chromatography–mass spectrometry (LC–MS) and nuclear magnetic resonance (NMR) data followed by the use of machine learning (ML) to discover candidate metabolomic panels for RCC. The study cohort consisted of 105 RCC patients and 179 controls separated into two subcohorts: the model cohort and the test cohort. Univariate, wrapper, and embedded methods were used to select discriminatory features using the model cohort. Three ML techniques, each with different induction biases, were used for training and hyperparameter tuning. Assessment of RCC status prediction was evaluated using the test cohort with the selected biomarkers and the optimally tuned ML algorithms. A seven-metabolite panel predicted RCC in the test cohort with 88% accuracy, 94% sensitivity, 85% specificity, and 0.98 AUC. Metabolomics Workbench Study IDs are ST001705 and ST001706.
Keywords: renal cell carcinoma, metabolomics, machine learning, liquid chromatography mass spectrometry, nuclear magnetic resonance spectroscopy
Graphical Abstract
INTRODUCTION
In the United States, kidney cancer is one of the most lethal urinary cancers. In 2021, an estimated number of 76,080 patients will be diagnosed, with a death toll of 13,780.1 Approximately 90% of kidney and renal pelvis cancers are renal cell carcinomas (RCCs). RCC lacks specific symptoms in the early stages, and the latest statistics indicate that over 50% of patients are diagnosed incidentally.2,3 Diagnosis is typically performed via expensive imaging tests4,5 and biopsies, the latter being highly invasive and prone to sampling errors.2,6,7 Current treatments and early diagnosis, when tumors are localized, result in a 92.6% 5 year survival, while late diagnosis results in the decrease of 5 year survival to 13.0%.2 An improved, noninvasive, and cost-effective diagnostic test is urgently needed to diagnose RCC earlier in the course of the disease.
As early as in the Middle Ages, physical properties including taste, smell, and color of urine were used to diagnose diseases, and these properties are influenced by urine metabolites.8 Today, analytical chemistry platforms such as nuclear magnetic resonance (NMR) spectroscopy and liquid chromatography–mass spectrometry (LC–MS) can determine the chemical composition of urine in a high-throughput fashion for biomarker discovery and diagnostics.9,10 The metabolome closeness to the phenotype of biological systems supports its utility to investigate the biology of cancer, which is considered by many to effectively be a metabolic disease.11,12 The close proximity of the RCC tumor(s) to the urine suggests that metabolomic alterations may be ideally detected in this noninvasively collected biofluid.
The high-throughput nature of metabolomics experiments and the broad analyte coverage by both NMR and LC–MS often result in enormous datasets that frequently require machine learning approaches to investigate biological alterations.13 Machine learning is a branch of artificial intelligence that uses algorithms to uncover patterns in complex data without explicit programming.14 These models allow for the prediction of output(s) based on a set of inputs, such as the prediction of RCC status using a panel of metabolite abundances selected from the urine feature dataset.
Several previous studies have investigated urine metabolome changes associated with RCC.15–30 Kim et al. found 4-hydroxybenzoate, quinolinate, and gentisate to be differentially expressed at a false discovery rate of 0.28 between RCC (n = 29) and controls (n = 33) using ultra-high-performance liquid chromatography–mass spectrometry (UHPLC–MS) and gas chromatography–mass spectrometry (GC–MS).23 Monteiro et al. reported a 32-metabolite resonance signature from NMR urine metabolomics that discriminated RCC patients (n = 42) from controls (n = 49) using unsupervised learning.20 Urinary volatile metabolic profiling using GC–MS led to the discovery of a panel of 21 volatile organic compounds correlated with RCC when 30 RCC patients were compared to 37 controls, with 2,5,8-trimethyl-1,2,3,4-tetrahydronaphthalene-1-ol and 2-oxopropanal subsequently validated as potential RCC biomarkers in a small independent sample set.21 In 2019, Liu et al. used LC–MS to identify androstenedione, 7α-hydroxy-3-oxochol-4-en-24-oic acid, and lithocholyltaurine to be the most significantly altered metabolites between RCC (n = 100) and controls (n = 129).22 In 2020, Zhang et al. identified aminoadipic acid, 2-(formamido)-N1-(5-phospho-d-ribosyl) acetamidine, and α-N-phenylacetyl-l-glutamine to be predictive of RCC in a cohort of 68 healthy controls and 39 RCC patients using LC–MS.15 Unfortunately, none of these highlighted studies made their data widely available, complicating progress in the field.
To improve our understanding of metabolome alterations associated with RCC and to build on prior research conducted in the field, we here report a multiplatform (NMR + Hydrophilic Interaction Liquid Chromatography (HILIC) LC–MS) metabolomics study on a patient cohort of larger size than most previously published studies (healthy control = 179, RCC patients = 105). The use of custom-built machine learning models enabled us to investigate algorithms with different inductive biases and hyperparameter tuning. In addition, the dataset was not filtered to retain only endogenous metabolites, therefore allowing for the inclusion of xenobiotics and exposure metabolites such as 2-mercaptobenzothiazole and dibutylamine in the discriminatory panel. We have shown that seven MS-derived metabolites, which discriminated RCC patients from healthy controls with 88% accuracy in the test cohort, could be identified. In addition, four NMR-derived diagnostic markers discriminated RCC patients from healthy controls with an accuracy of 78%. These results underlie the promise of RCC detection using urine metabolomics, providing additional evidence for metabolic perturbations in RCC.
MATERIALS AND METHODS
Chemicals
Optima (ThermoFisher Scientific) LC–MS grade water and acetonitrile were used to prepare all mobile-phase components. Ammonium acetate (Sigma, molecular biology grade) and ammonium hydroxide 28–30% solution (Fisher Chemical) were used as additives for mobile phases. For NMR samples, D2O and 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) were obtained from Cambridge Isotope Laboratories (Andover, MA, USA).
Urine Collection
Patients at Emory University Hospital with a solid renal mass with potential for RCC and subsequently confirmed to be RCC following surgery were identified prospectively. Healthy controls were identified during an annual physical exam. All patients provided informed consents (Emory University approvals IRB00058903, IRB00054812, IRB00085068, and IRB00055316). Urine was collected at either a clinic appointment or at the time of surgery in a urine collection cup and placed on ice. Urine was mixed by turning the cup upside down five times, and 15 mL was transferred to a sterile tube followed by centrifugation at 1800g for 20 min at 4 °C. Ten milliliters of the supernatant was transferred to a clean, sterile tube, and one tablet of Complete Protease Inhibitor Cocktail (Sigma, St. Louis) was added to the tube. The tube was placed on ice for 10 min with periodic vortex mixing to dissolve the tablet. This urine was then transferred into 5 × 1.5 mL aliquots and stored at −80 °C.
Hydrophilic Interaction Liquid Chromatography–High-Resolution Mass Spectrometry
Urine samples were thawed on ice, and proteins were precipitated with addition of methanol in a 5:1 volume ratio to 50 μL of urine. Samples were vortex-mixed for 30 s, and after centrifugation at 21,100g for 5 min, the supernatant was transferred to a snap-on cap LC vial and stored at 4 °C until analysis. A sample preparation blank was analyzed jointly with the samples, and a pooled sample was created for use as quality control and to correct for instrument drift. Samples were analyzed in randomized order, and the pooled sample was included in approximately every tenth injection over the course of the batch.
Compounds were separated using an Ultimate3000 (ThermoFisher Scientific), fitted with a Waters Acquity UPLC BEH HILIC column (2.1 × 75 mm, 1.7 μm particle size). The compounds were eluted with the following gradient: 95:5 10 mM ammonium acetate with ~0.014% ammonium hydroxide: acetonitrile (mobile phase A) and acetonitrile with ~0.014% ammonium hydroxide (mobile phase B) using the following gradient program: 0 min 5% A; 3 min 63% A; 7 min 63% A; 7.1 min 5% A; 9.9 min 5%. The flow rate was set at 0.30 mL min−1 for 0–7.1 min, increased to 0.5 mL min−1 from 7.1 to 7.2 min, 7.2–9.5 min at 0.5 mL min−1, and decreased to 0.30 mL min−1 from 9.5 to 10.0 min. The column temperature was set to 50 °C, and the injection volume was 2 μL. A high-resolution accurate mass Q Exactive HF mass spectrometry system (ThermoFisher Scientific) was used for all measurements. The heated electrospray ionization (HESI) source was operated at a capillary temperature of 275 °C, a spray voltage of 3.5 kV, and sheath, auxiliary, and sweep gas flow rates of 48, 11, and 2 arbitrary units, respectively. MS data were acquired in the 70–1050 m/z range in both positive and negative ionization modes. MS/MS experiments were performed by acquiring mass spectra in a data-dependent acquisition fashion. Survey MS spectra were collected with a resolution setting of 120,000, and the top 10 dd-MS2 were collected at a resolution of 30,000 and an isolation window of 0.4 m/z. Stepped normalized collision energies of 10, 30, and 50 fragmented selected precursor ions in the HCD cell prior to combining all ions for Orbitrap analysis. Dynamic exclusion was set at 10 s, and ions with charges greater than 2 were omitted.
Data acquisition and processing were carried out using Xcalibur V4.0 (ThermoFisher Scientific) and Compound Discoverer V3.0 (ThermoFisher Scientific), respectively. Pooled QC injections were used to adjust for instrument drift using a LOESS algorithm. Background peaks were filtered from the dataset when signals were less than 5× of corresponding features in sample blank injections. A feature was filtered if it was present in less than 50% of the QC sample injections or if a relative standard deviation was observed to be greater than 30% in the QC injections.
Once a panel of discriminant features were selected, additional experiments were conducted with an Orbitrap IDX Tribrid mass spectrometer (ThermoFisher Scientific) using data-dependent acquisition methods to collect MS2 data for features that were missed during the original DDA data collection. For these experiments, a Waters Acquity UPLC BEH amide column (2.1 × 150 mm, 1.7 μm particle size) was used with the following mobile phases: 80:20 10 mM ammonium formate with 0.1% formic acid: acetonitrile (mobile phase A) and acetonitrile with 0.1% formic acid (mobile phase B). The gradient used was as follows: 0 min 5% A; 0.5 min 5% A; 8 min 60% A; 9.4 min 60% A; 11 min 5% A. The flow rate was set to 0.40 mL min−1, the column temperature was set to 40 °C, and the injection volume was 2 μL. Tandem MS spectra were collected for an inclusion list of precursors if they were above an intensity threshold of 6.0 × 103, using an isolation window of 0.8 m/z. Survey mass spectra were collected with a resolution of 60,000. Stepped normalized collision energies of 15, 30, and 45 fragmented the precursors in the HCD cell followed by Orbitrap analysis at a resolution of 30,000. Precursor ions were also sequentially fragmented with a CID collision energy of 45 and analyzed in the ion trap.
Data processing was performed with Compound Discoverer v3.0 (ThermoFisher Scientific), which included elemental formula prediction based on exact masses and isotope patterns. When elemental formula prediction was not achieved in the automated fashion via Compound Discoverer, the feature was manually analyzed using Xcalibur v3.0 to assign the elemental formula. Tentative annotations were assigned based on searches against literature and metabolomic databases, such as the Human Metabolome Database (HMDB), Metlin, mzCloud, and MassBank. Elemental formulas and exact masses with a mass error of 10 mDa were used in this case. Fragmentation patterns were also analyzed and matched against tandem MS databases such as mzCloud and locally built mzVault libraries in order to assign annotations.
Nuclear Magnetic Resonance Spectroscopy
Urine samples were thawed in a 4 °C cold room followed by centrifugation at 20,200 relative centrifugal force (rcf) for 20 min at 4 °C to remove any precipitated materials. A sample preparation robot (SamplePro, Bruker Biospin, Rheinstetten, Germany) was used to dispense 60 μL of NMR buffer into 5 mm SampleJet NMR tubes (Bruker Biospin, Billerica, MA, USA) followed by the transfer of 540 μL of urine sample and sample mixing. The NMR buffer used was 1.5 M KH2PO4/K2HPO4 buffer with a pH of 7.0 in D2O, containing 0.11 mM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS). DSS is used as a chemical shift reference (0.0 ppm). Quality assurance and quality control for this study are described in Supporting Information Section S1. NMR spectra were acquired using an Avance III HD 600 MHz Bruker NMR spectrometer with a Bruker SampleJet cooled to 5.6 °C. The following NMR experiments were conducted: one-dimensional nuclear Over-hauser effect pulse sequence with presaturation of water resonance (NOESYPR1D), two-dimensional (2D) 1H-13C heteronuclear single quantum correlation (HSQC), and HSQC-TOCSY (HSQC-total correlation spectroscopy). For 1D 1H NMR metabolomics spectra, phase and baseline correction and referencing were carried out with Bruker’s TopSpin software. Referencing to DSS was confirmed using the Edison laboratory in-house MATLAB scripts (https://github.com/artedison/Edison_Lab_Shared_Metabolomics_UGA). In addition, the ends of NMR spectra (less than −0.50 ppm, greater than 10.0 ppm) and water regions (between 4.89 and 4.68 ppm) were removed from all samples. Urine NMR spectra were aligned using constrained correlation optimized warping (CCOW)31 and normalized using probabilistic quotient normalization (PQN).32 NMRPipe was used to preprocess the 2D NMR Data (HSQC and HSQC-TOCSY).33 Metabolites were identified using the AssureNMR software (Bruker Biospin, USA) with the BBiorefcode metabolite database and COLMARm.34 Metabolites were assigned a confidence score from 1 to 5, with 5 as the highest confidence score. The scores were defined as follows: (1) putatively characterized compound classes or annotated compounds, (2) matches from 1D NMR to the literature and/or 1D BBiorefcode compound (AssureNMR) or other database libraries such as BMRB35 and HMDB,36 (3) matched to HSQC, (4) matched to HSQC and validated by HSQC-TOCSY (COLMARm), and (5) validated by spiking the authentic compound into the sample. Fifty metabolomic features in the aligned and normalized 1D 1H NMR spectra were quantified by taking spectral areas for integration and combined with MS features for downstream analysis (see Supporting Information Section S1 and Scheme S1 for NMR peak picking and integration details). Of the 50 metabolomic features quantified via NMR, 30 metabolites were identified with some metabolites having multiple resonances quantified, in addition to 11 unknown resonances.
Sample Cohort Selection
Propensity score matching37 was used to reduce the sample selection bias effect while balancing potential confounders among control and RCC patient groups. The covariates considered included age, gender, BMI, race, and smoking history. The propensity score was computed via a logistic regression model using the default parameters of the Scikit-learn38 linear model module in Python. A one-to-one propensity score matching with the caliper method, which allowed for a maximum distance of 1 × 10−5 between the propensity scores of matched pairs, resulted in the selection of 31 control subjects and 31 subjects with RCC to form the model cohort.
Feature Selection for RCC Prediction
Features were selected using the 62-model cohort. The normalized abundances of the 50 metabolomic features that were quantified by NMR and the 7097 normalized MS features were merged into one feature table in Python. The combined feature table was subjected to both filtering and wrapper feature selection methods.39,40 The features were filtered via the following sequential criteria: (1) features with a greater than 1-fold difference between the two groups were retained; (2) features with a q-value lower than 0.05 were retained (the q-value is defined as the p-value obtained from Student’s t test followed by Benjamini–Hochberg false discovery rate correction41); and (3) one of the two highly correlated features was removed, with a Pearson correlation coefficient cutoff of 0.8. The resulting features were autoscaled prior to further feature selection. A recursive feature elimination method under stratified fivefold cross validation conditions was implemented using random forests (RF-RFECV). The Scikit-learn38 default hyperparameters were used with the number of estimators set to 100 decision trees. In addition, a PLS regression method was applied on the same reduced feature set using the default PLS regression method in the cross-decomposition module in Scikit-learn. For each method, features were ranked based on importance for discriminating RCC patients from healthy controls. The Gini index was used in RF-RFECV, while variable importance in projection (VIP) scores were used in PLS regression. Finally, a voting-based system for potential biomarkers was used; the overlapping features among the top features from each method were selected as the final potential biomarkers. Variants of this method were used for selecting only upregulated biomarkers and NMR biomarkers in the study.
Machine Learning (ML) Methods for RCC Prediction
Random forest (RF), k-nearest neighbors (k-NN), linear kernel support vector machine (SVM-Lin), and radial basis kernel support vector machines (SVM-RBF) were used for predictions. Optimized hyperparameters for each ML method used the model cohort and the selected metabolite panel. A linear search for a single hyperparameter or a grid search for two (or more) hyperparameters was used under fivefold cross validation conditions. These tuned ML models were used to predict RCC status in the test cohort.
Random Forest.
Random forests are a collection of decision trees built using bootstrapped training samples, where decision trees are constructed using a random subset of metabolomic features as candidates for node splitting. The decision tree is an inverted tree starting with the root node at the top of the tree followed by internal nodes and finally leaf nodes. The root node and internal nodes are assigned specific metabolomic features, while the leaf nodes indicate the final prediction.
k-Nearest Neighbor.
k-NN classifiers are an instance-based learning algorithm that classifies samples via the vote of the majority of k (to be defined) closest neighbors. Distance measures considered for determining nearest neighbors during hyperparameter tuning include Euclidean (E) and Manhattan (M) distances.
Linear Kernel Support Vector Machine.
For binary classification, the goal of SVM-Lin is to generate a separating hyperplane that separates the classes in a j-dimensional space, where j is the number of features. Given n numbers of training samples x1, …xn ∈ Rj with a class membership of y1, …, yn ∈ (−1,1), where −1 represents controls and 1 represents RCC, the function of the separating hyperplane, defined here as the RCC metabolic score, is given by the following
where β0 and βj are the bias and the weight parameters, respectively, determined during training. The class membership of a new observation was defined by the sign of the RCC metabolic score (negative for control and positive for RCC). The function β0 + βx′ = 0 is the separating hyperplane that maximized the margin between the two classes, while the margin is defined as the following
The only hyperparameter to be tuned in the SVM-Lin is the non-negative regularization parameter cost (C), which allows for the flexibility of misclassification by the hyperplane margin. C in SVM controls the bias-variance tradeoff associated with statistical learning algorithms.
Radial Basis Function Kernel Support Vector Machine.
The RBF kernel is a kernel method that projects data in a higher-dimensional space for the purpose of linear separation, which is equivalent to a nonlinear decision boundary in the original feature space. SVM-RBF is defined by the following function
where xi are the training data, xi′ are the test data, and γ (gamma) is a positive tuning parameter. γ and C are the hyperparameters considered for tuning.
See Supporting Information Section S2 for model evaluation metrics.
Unsupervised Learning Methods
Hierarchical clustering analysis was conducted on 435 metabolic features, which were the top differential metabolites between RCC and controls, with greater than 1-fold change and q-value lower than 0.05. Of the 435 features, 433 were from LC–MS and 2 features were from NMR. The cluster map function in Seaborn was used.42 The linkage method for calculating clusters was weighted, while the distance metric was Euclidean. All features were autoscaled prior to analysis.
Data Availability and Implementation Environment
NMR data analysis was carried out using the Edison Lab in-house MATLAB scripts (https://github.com/artedison/Edison_Lab_Shared_Metabolomics_UGA, Matlab R2017b, The Mathworks, Inc.). Post metabolic features normalization computations were carried out in the Python 3.7.0 programming language using the following packages: Pandas for data handling,43 Matplotlib/Seaborn for data visualization,44 Numpy and Scipy for numerical computations,45,46 Statsmodel for statistical computations,47 and Sci-kit learn for machine learning.38 A Jupyter notebook was used as the integrated development environment (IDE).48 All Jupyter notebooks used in this study can be found here: https://github.com/artedison/RCC_MLprediction. The datasets collected in this work are available through the NIH Metabolomics Workbench49 with the project ID of PR001091 and study IDs ST001705 and ST001706. The dataset can be accessed via http://dx.doi.org/10.21228/M8P97V.
RESULTS AND DISCUSSION
Patient Selection
NMR measurements were conducted on 179 controls and 105 renal cell carcinoma (RCC) patient urine samples, while LC–MS measurements were conducted on 178 controls and 102 RCC patient urine samples. The subset of controls (n = 174) and RCC (n = 82) samples that were analyzed by both methods was selected for further investigation. While all control urine samples were collected in the clinic, RCC patient urine samples were collected both in the clinic and in the operating room. Preoperative procedures added cofounders to the samples collected in the operating room and were therefore not ideal for use in feature selection due to the potential for introducing bias in the RCC group. However, these operating room samples still had utility as part of the test cohort and were retained. The strategy for grouping of the samples into either model or test cohorts is presented in Figure 1. In the model cohort used for training purposes, 31 RCC urine samples collected in the clinic were matched via one-to-one propensity score matching (PSM)37 to 31 control urine samples. PSM seeks to balance the population characteristics of the case versus control samples in terms of characteristics, such as age, BMI, and smoking history, and is essential to obtain unbiased machine learning results and robust biomarker panels. In general, the model cohort consists of samples collected in the clinic. As such, features were selected, and models were trained solely using clinic samples. This addresses any sample collection bias concerns as model training was not carried out using the test cohort. Moreover, all discriminating features identified in the study were statistically insignificant (independent t test, BH-FDR q ≥ 0.05) when RCC samples collected in the clinic versus those collected in the operating room were compared (Figure S1).
Figure 1.
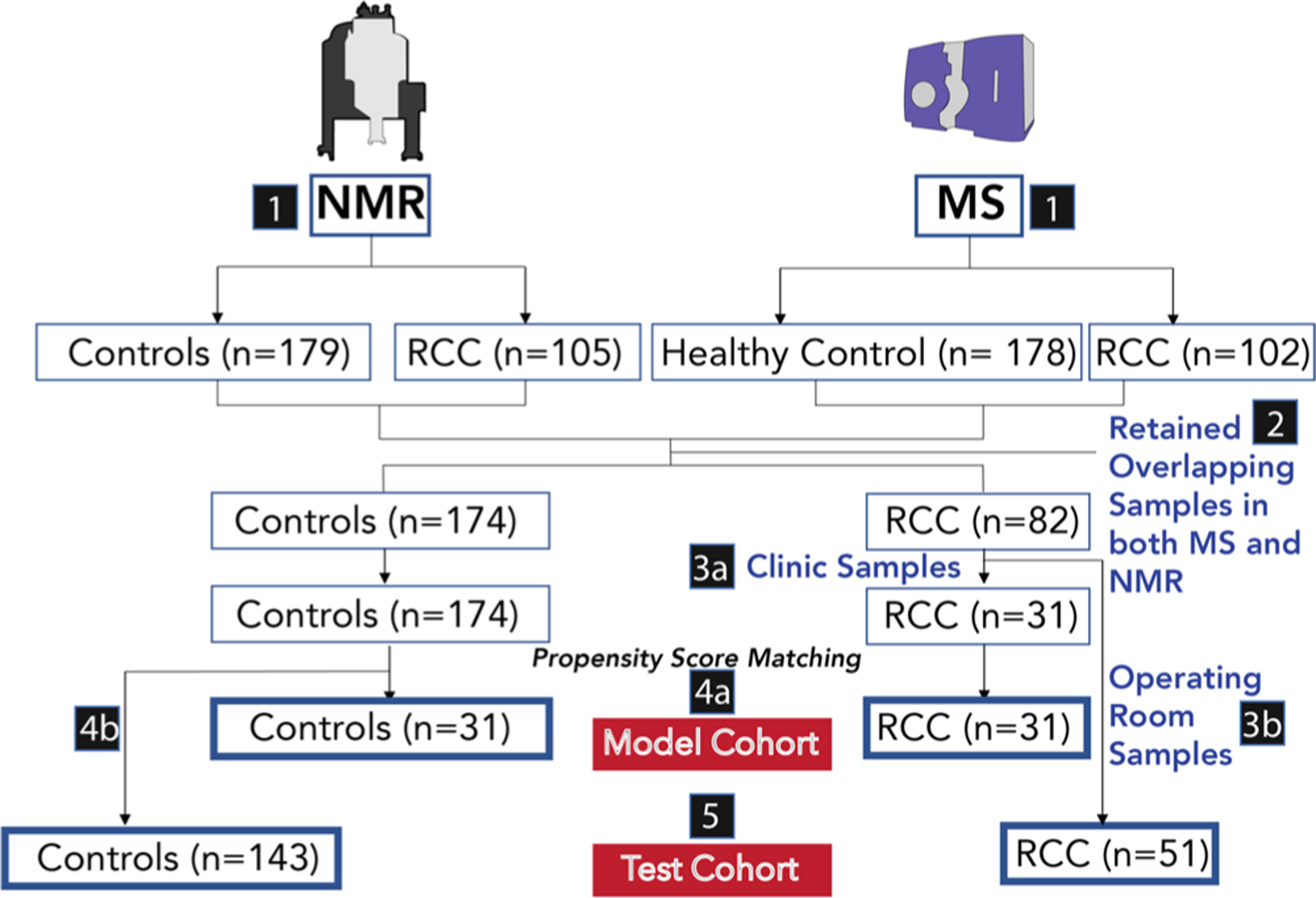
Flow chart for patient selection. Samples for which NMR and MS measurements were collected (1). A total of 284 samples, with 174 control individuals and 82 RCC patients, have their urine samples analyzed by both NMR and LC–MS methods (2). RCC samples collected in the clinic are selected for the model cohort (3a), while the operating room RCC samples are selected for the test cohort (3b). The model cohort was selected via propensity score matching from those samples collected in the clinic (31 RCC samples, 31 control samples) (4). The test cohort contained 51 RCC samples collected in the operating room and 143 controls collected in the clinic (5).
Figure 2a and Table S1 show the comparative statistics of the pre-PSM and post-PSM model cohorts. Adjusted covariates included gender, age, BMI, race, and smoking history. Following PSM, the cohorts were gender-matched (17 males, 14 females) and had statistically insignificant differences in age (p-value = 0.64) and BMI (p-value = 0.06). Smoking history and race statistics also improved considerably when compared to the prematched cohort. In addition, all RCC stages were represented in the model cohort: early-stage RCC (Stages I and II) represented 55% of the cohort, while late-stage RCC (Stages III and IV) represented 45% (Figure 2b, Table S2). The second subcohort in the study, the test cohort, was constructed from the remainder of the samples following removal of the model cohort. It was composed of 143 controls and 51 RCC patients (Figure 2c and Table S3.) The imbalance of gender, age, BMI, and smoking history in the test cohort made it a good candidate for a challenging test of the utility of the metabolic panel selected by modeling the PSM-adjusted model cohort.
Figure 2.
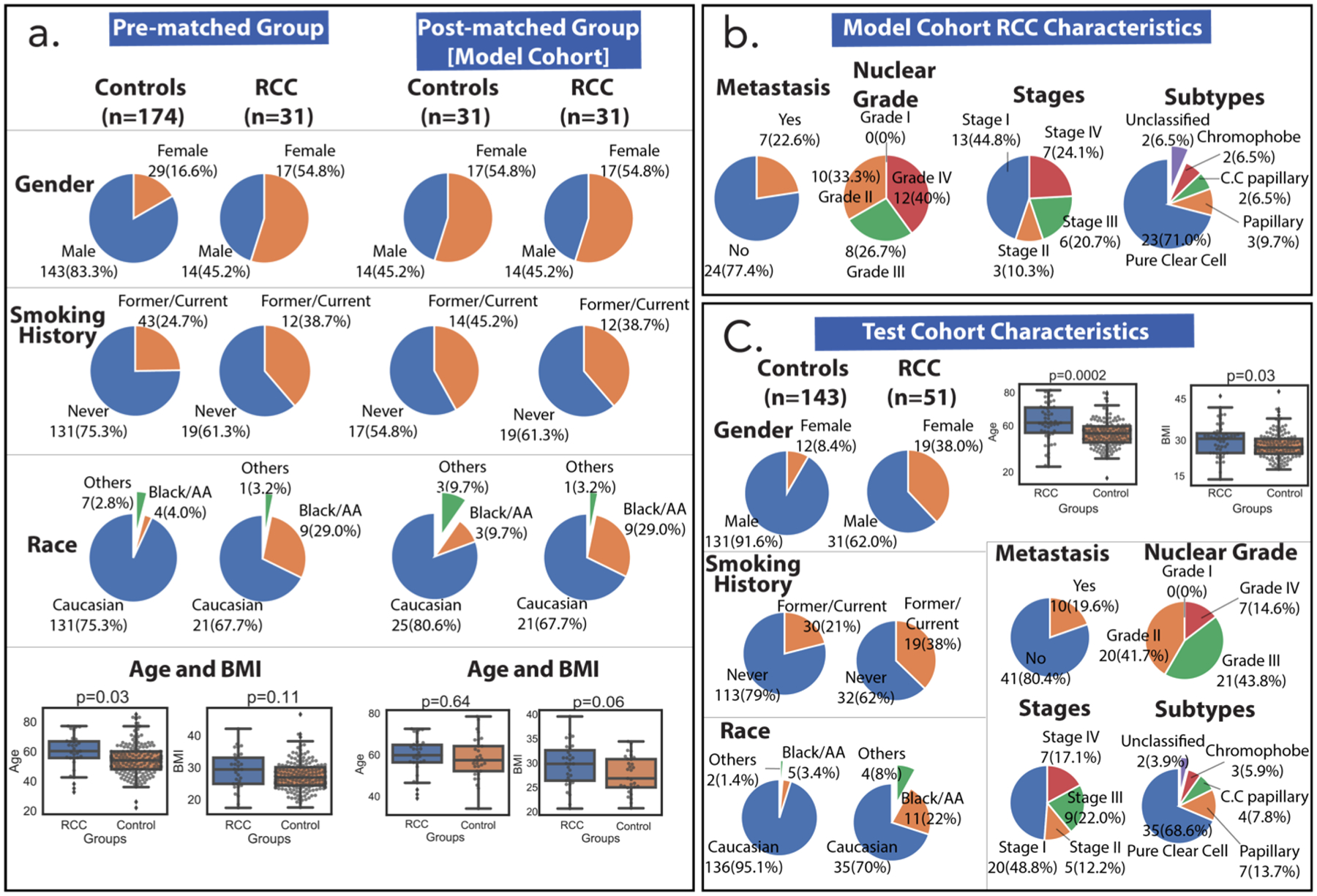
Cohort characteristics. (a) Model cohort characteristics (gender, smoking history, race, age, BMI in no particular order) are shown before and after propensity matching. p-Values were calculated for unequal and equal sample sizes using Welch’s and Student’s t tests, respectively. (b) Additional model cohort RCC characteristics (metastasis, nuclear grade, stage, and RCC subtype) show that the majority of the group was early-stage RCC and pure clear cell subtype. There was one nuclear grade datum that was unreported and two cancer stages that were not reported due to inconclusive TNM staging information. (c) Test cohort characteristics show differences that are useful in testing the feature panels selected using the model cohort. All p-values were calculated using Welch’s t test (unequal sample size). Three samples had unreported nuclear grades, and 10 samples did not have RCC staging due to inconclusive TNM staging. Abbreviations: AA: African American; BMI: body mass index; RCC: renal cell carcinoma; C.C. Papillary: clear cell papillary.
Metabolomics Analysis and Machine Learning Pipeline
After NMR data collection, ends of spectra and water regions were removed. Several alignment methods were attempted with CCOW31 giving the most reliable alignment, followed by data normalization. A total of 50 metabolic features were quantitated with NMR, and 30 metabolites were confirmed with 1H NMR and/or HSQC and HSQC-TOCSY as described in Materials and Methods (Table S4 and Figure 3a). A total of 7097 features were detected with LC–MS (4623 from positive mode and 2474 from negative mode), as described under Materials and Methods (Figure 3b).
Figure 3.
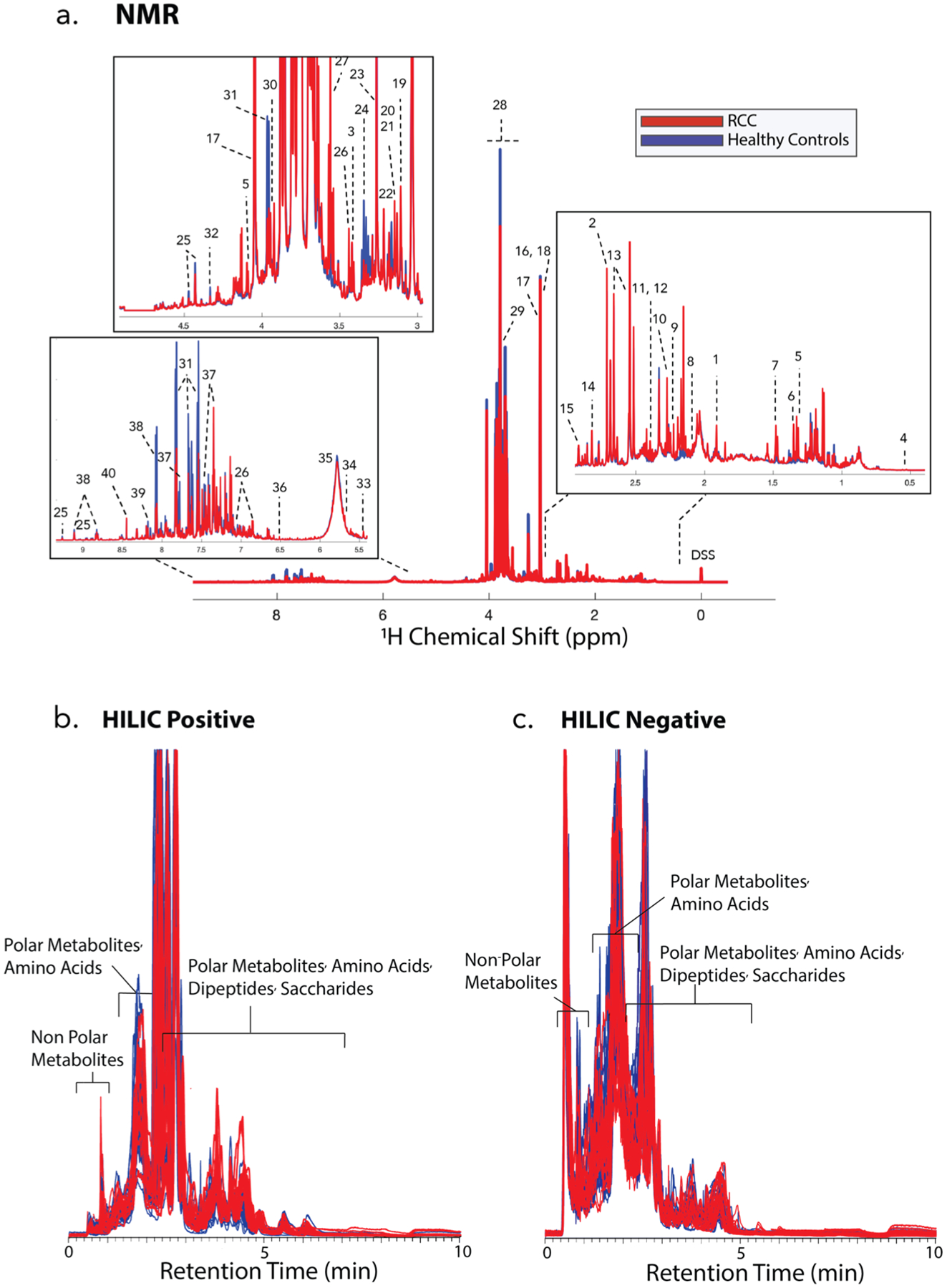
Raw data for various metabolomics platforms. (a) Average 600 MHz 1H 1D NOESY-PR NMR spectra of all urine samples tested in the study. (1) Acetate, (2) dimethylamine (DMA), (3) taurine, (4) bile acid (tentative assignment), (5) lactate, (6) α-hydroxyisobutyrate (HIBA), (7) alanine, (8) acetyl phosphate, (9) acetone, (10) acetoacetate, (11) succinate, (12) pyruvate, (13) citrate, (14) methylguanidine, (15) N,N-dimethylglycine (DMG), (16) creatine, (17) creatinine, (18) creatine phosphate, (19) cis-aconitate, (20) dimethylsulfone (DMS), (21) ethanolamine, (22) choline, (23) betaine, (24) syllo-inositol, (25) trigonellinamide, (26) 4-hydroxyphenylacetate (4-HPA), (27) glycine, (28) mannitol, (29) guadinoacetate, (30) glycolate, (31) hippurate, (32) tatrate, (33) allantoin, (34) cis-aconitate, (35) urea, (36) fumarate, (37) indoxyl sulfate, (38) trigonelline, (39) hypoxanthine, (40) formate, (41) 3-hydroxyisovaleric acid, (42) 4-aminohippuric acid, (43) 4-hydroxyhippuric acid, and (44) valine. (b) HILIC LC–MS positive ion mode data, displaying all samples. (c) HILIC LC–MS negative ion mode data, displaying all samples.
All 7147 metabolomic features from both platforms were merged, and data analysis proceeded according to the ML pipeline shown in Figure 4. The dataset was filtered to include 435 features with greater than 1-fold change between RCC and controls, and Student’s t test with Benjamini–Hochberg false discovery rate correction (q < 0.05) was performed. Figure 5 shows the hierarchical clustering of the 435 features selected in this analysis. To minimize the effect of feature multi-collinearity, one out of a pair of highly correlated features (Pearson correlation, r > 0.8) was retained resulting in 128 features for further analysis. The top 20 features with PLS-DA ranked by VIP scores and the top 20 features with RF-RFECV ranked by the Gini index were selected from this set of 128 features. Ten features were present on both feature lists selected by PLS-DA and RF-RFECV, leading to the 10-metabolite panel (Table S5 and Figure S2). This voting strategy was used to minimize bias from using only one machine learning algorithm for feature selection. Also, as a way of comparison with a more conventional workflow that relies less on machine learning, features with the top 10 highest q-values from the univariate analysis were selected, and a classification task was performed for the model cohort with logistic regression using the Metaboanalyst 5.0 biomarker analysis platform. Classification results showed an AUC of 0.86 and an accuracy of 83.3% (Figure S3). These were low performance scores compared to the 10 features selected via the voting-based feature selection methods and employed in the k-NN classifier (0.96 AUC and 95% accuracy) for the model cohort (Table S7). As such, we proceeded with the vote-based ML-derived features.
Figure 4.
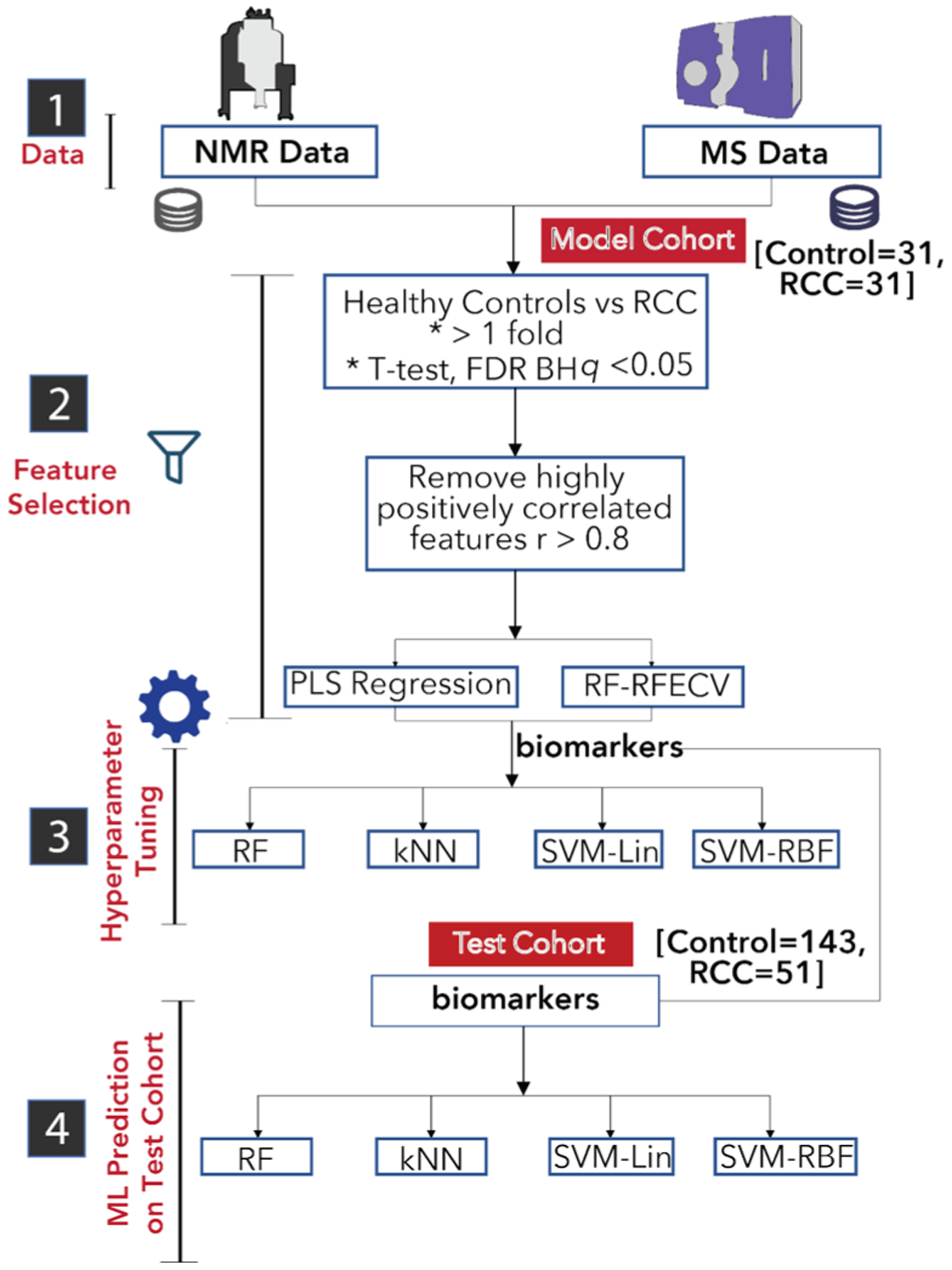
Machine learning pipeline. Using the model cohort, a hybrid method of feature selection resulted in a panel of 10 metabolites. Hyperparameters for four different machine learning models were tuned using the model cohort and the 10-metabolite panel. The RCC status of the test cohort was predicted with four models. PLS: partial least squares; RF-RFECV: random forest recursive feature elimination – cross validation; FDR-BH: false discovery rate Benjamini–Hochberg procedure; k-NN: k-nearest neighbors; SVM: support vector machines (Lin: linear, RBF: radial basis function).
Figure 5.
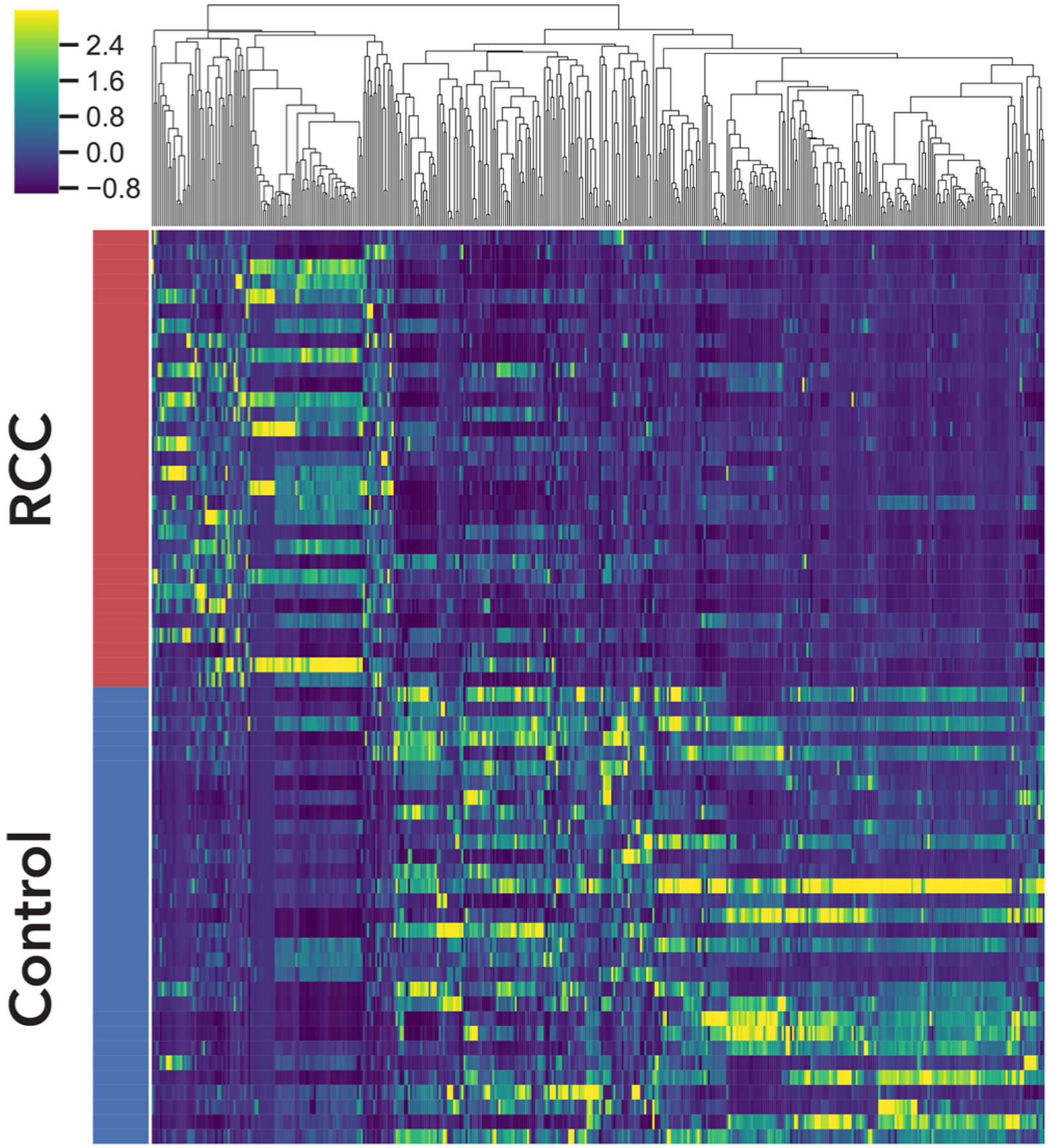
Hierarchical clustering of 435 metabolomic features with q-values <0.05 and >1-fold change in the model cohort. z-Scores are represented as shown in the color bar. Yellow represents higher abundances in RCC, while dark blue represents higher abundances in the controls. See Table S16 for details of metabolomic features.
For predicting RCC status, four machine learning (ML) algorithms were used: random forest (RF), k-nearest neighbor (k-NN), support vector machine with radial basis function (SVM-RBF), and linear kernel support vector machine (SVM-Lin). Selected hyperparameters were tuned using the 62-model cohort under fivefold cross validation conditions (Table S6). The tuned ML models were then used to predict RCC status in the test cohort. Overall, k-NN gave the best prediction with an AUC of 0.96, accuracy of 87%, specificity of 83%, and sensitivity of 96% (Table S7).
Eight of the 10 selected markers were in lower relative abundance in RCC samples (Figure S2) versus control samples, so we identified another panel containing features with higher relative abundance in the RCC patients’ urine versus control urine, as measuring increased abundance upon the appearance of disease is favored in clinical practice. Figure S4 describes the machine learning pipeline for upregulated metabolic features in RCC, which resulted in a five-metabolite panel (Table S8 and Figure S5). Again, selected hyperparameters were tuned using the 62-model cohort under fivefold cross validation conditions (Table S9). The tuned ML models were then used to predict RCC status in the test cohort. It was found that k-NN yielded the best prediction of the test cohort with an AUC of 0.92, accuracy of 81%, sensitivity of 86%, and specificity of 79% (Table S10), which was a slightly lower performance than for the 10-metabolite panel (Table S7).
High-resolution MS and tandem MS experiments were performed for metabolite annotation. Through standard procedures such as analyzing exact masses, isotopic relative ion abundances, and fragmentation patterns, five metabolites in the 10-metabolite panel (Table S5) and four of the five in the upregulated metabolite panel (Table S8) were annotated. A third metabolite panel was formed to include only annotated features from the first two panels. Table 1 and Figure 6 show the results using this last panel, namely, a seven-metabolite panel for RCC that included 2-phenylacetamide, Lys-Ile (or Lys-leu), dibutylamine, hippuric acid, mannitol hippurate, 2-mercaptobenzothiazole, and N-acetyl-glucosaminic acid (Table S11). ML hyperparameters were tuned using the 62-model cohort as described above (Table S12). ML models were used to predict RCC status in the test cohort with the most accurate model being linear SVM with an AUC of 0.98, accuracy of 88%, sensitivity of 94%, and specificity of 85% (Table 2).
Table 1.
Compound Annotation and Identification for the Seven-Metabolite Panel
| ID no. | retention time (min) | m/z | adduct type | mass error (ppm) | elemental formula | name | |
|---|---|---|---|---|---|---|---|
| theoretical | experimental | ||||||
| 720 | 5.68 | 136.0757 | 136.0755 | [M + H]+ | −1.47 | C8H9NO | 2-phenylacetamide |
| 1481 | 8.83 | 260.1969 | 260.1969 | [M + H]+ | 0.00 | C12H25N3O3 | Lys-Ile or Lys-leu |
| 2102 | 4.39 | 130.1590 | 130.1591 | [M + H]+ | 0.77 | C8H19N | dibutylamine (alkyl chain branching not determined, isomers possible) |
| 3804 | 2.59 | 202.0475 | 202.0478 | [M + Na]+ | 1.48 | C9H9NO3Na | hippuric acid |
| 6262 | 2.67 | 376.1249, 358.1143 | 376.1246, 358.1147 | [M + H2O−H]− [M−H]− | −0.68 | C15H21NO9 | hippurate-mannitol derivative |
| 6578 | 1.09 | 165.9790 | 165.9784 | [M−H]− | −3.61 | C7H5NS2 | 2-mercaptobenzothiazole |
| 6594 | 6.89 | 236.0776 | 236.0777 | [M + H]+ | 0.42 | C8H15NO7 | N-acetyl-glucosaminic acid |
Figure 6.
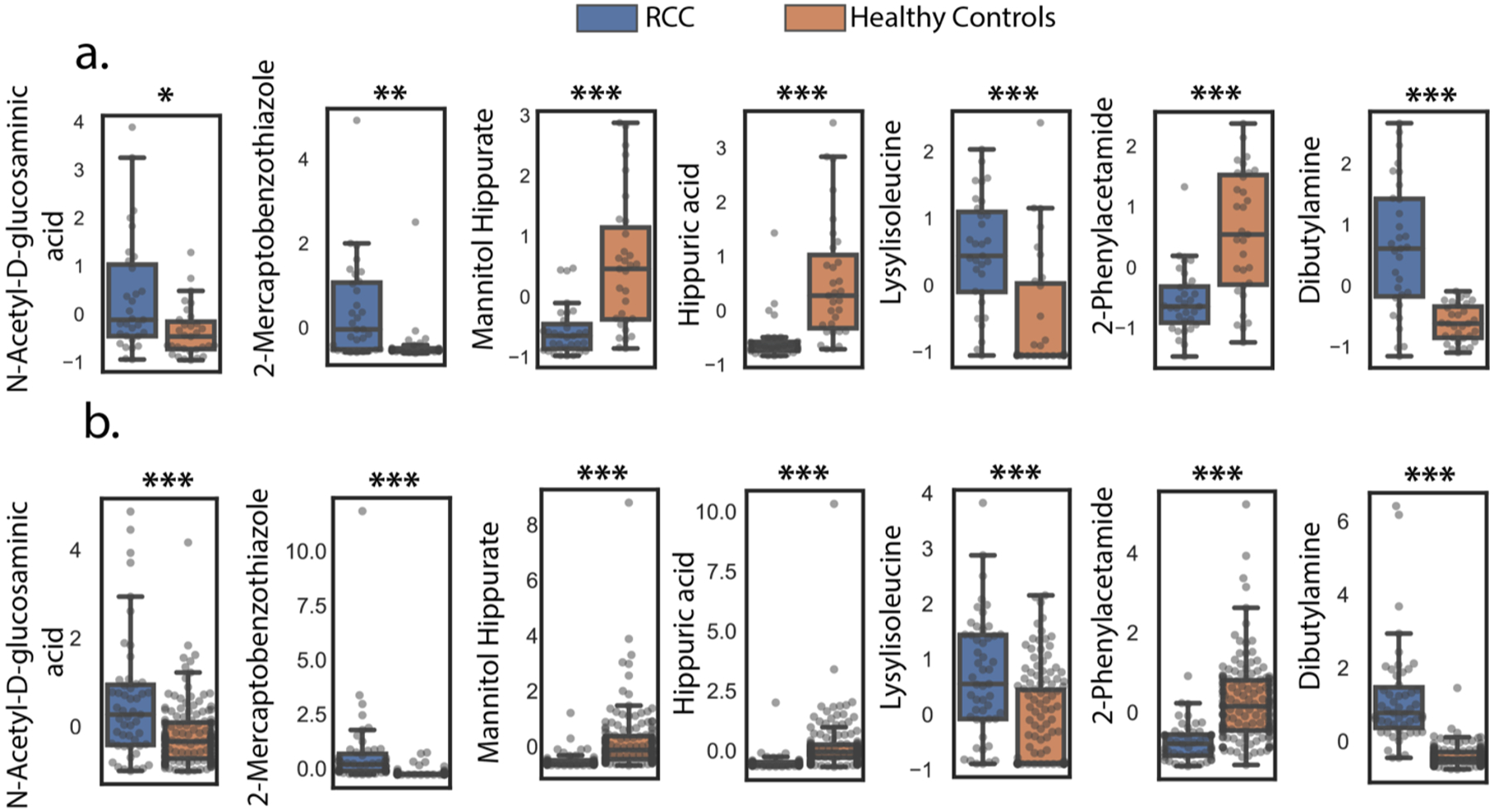
Relative abundances for the seven-metabolite panel in (a) the model cohort. After selecting features with greater than 1-fold changes between controls and RCC groups, q-values were computed by taking the FDR correction (Benjamini–Hochberg) after an independent t test. (*q ≤ 0.05, **q ≤ 0.01, ***q ≤ 0.001). (b) Relative abundances in the test cohort, p-values from Welch’s t test were reported (unequal sample size). (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). Raw data were transformed via autoscaling for visualization.
Table 2.
Machine Learning Performance for the Seven-Metabolite Biomarker Panel
| RF | k-NN | SVM-RBF | Linear SVM | |
|---|---|---|---|---|
| AUC | 0.96 ± 0.04 (0.99) | 0.96 ± 0.05 (0.94) | 0.97 ± 0.04 (0.94) | 0.97 ± 0.04 (0.98) |
| accuracy | 0.9 ± 0.06 (87%) | 0.92 ± 0.07 (80%) | 0.88 ± 0.09 (78%) | 0.87 ± 0.1 (88%) |
| sensitivity | 0.87 ± 0.12 (100%) | 0.83 ± 0.15 (92%) | 0.8 ± 0.19 (90%) | 0.77 ± 0.23 (94%) |
| specificity | 0.93 ± 0.08 (83%) | 1.0 ± 0.0 (76%) | 0.97 ± 0.07 (73%) | 0.97 ± 0.07 (85%) |
When combined with the 7097 LC–MS features, the 50 NMR features were not selected by machine learning procedures in any of the final panels. This is likely caused by the over-representation of MS features in the final feature list. To further investigate the utility of NMR features, the dataset was filtered with Student’s t test with Benjamini–Hochberg false discovery rate correction (q < 0.05). Following that, metabolomic features representing the same metabolites were removed via a Pearson correlation cutoff of 0.80 to retain only one feature representing a metabolite (Figure S6). This gave rise to a four-metabolite panel consisting of hippurate, trigonellinamide, lactate, and mannitol (Figure S7). As with other panels, selected hyperparameters were tuned using the 62-model cohort under fivefold cross validation conditions (Table S13). The tuned ML models were then used to predict RCC status in the test cohort. SVM-RBF yielded the best prediction in the test cohort with an AUC of 0.89, accuracy of 78%, sensitivity of 86%, and specificity of 76% (Table S14).
DISCUSSION
Machine learning enabled the accurate selection of metabolite markers that accurately distinguished urine samples from RCC patients to those from controls following propensity score matching of the cohorts. Because different machine learning techniques are driven by different induction biases, we used a variety of feature selection strategies to better down-select biomarkers. As initial feature filters, univariate statistical methods such as t tests, fold changes, and Pearson correlations were used for downsizing the metabolic feature set. The last few steps of the machine learning pipeline were based on two ML methods with differing inductive biases. PLS-DA assumes linear statistical relationships,50 while random forests can model more complex relationships in the dataset.51 This step was followed by voting for the top ranking overlapping metabolic features from the different methods tested. For the classification tasks, hyperparameter tuning of machine learning algorithms was carried out, culminating in excellent predictions of the test cohorts. These data analysis pipelines resulted in a 10-metabolite panel, a five-metabolite panel including only metabolites upregulated in RCC, and a four-metabolite marker containing only metabolites detected by NMR. The seven-identified metabolite biomarker proposals in the study gave an accuracy of 88% and an AUC of 0.98. This is likely a conservative assessment of the robustness of the biomarker given the small size of the training dataset versus a relatively large test cohort, given the constraint of patient selection. In general, many of the markers identified in these panels were novel, but a handful of markers have already been reported in the literature, validating the approach used in this study.
Examination of the biological role of the metabolites in the various panels constructed led to new insights into potential origins and mechanisms of disease progression in RCC. The metabolite 2-phenylacetamide decreased in RCC urine samples, indicating a downregulation of phenylalanine metabolism. Indeed, downregulation of phenylalanine metabolism has been reported in RCC cancer cells,52 while RCC urine metabolomics studies have also reported the downregulation of metabolites in the phenylalanine pathway such as 4-hydroxyphenylacetate and phenylacetyl-L-glutamine.15,20
The dipeptide lysyl-isoleucine/lysyl-leucine (Lys-Ile/Lys-leu) was observed to be increased in RCC urine samples. Upregulation of other types of dipeptides has been linked to RCC.22,53 For example, in a paired normal/clear cell renal cell carcinoma tissue metabolomics study by Hakimi and co-workers, numerous dipeptides were detected as being upregulated in RCC.53 In addition, dipeptides such as aspartyl-phenylalanine and glutamyl-threonine have been reported to be upregulated in a urine RCC metabolomics study.22 Increased dipeptide abundances are typically associated with the increased protein degradation/reutilization processes in tumors.53
Reduced levels of hippuric acid and feature C15H21NO9, likely a hippurate and mannitol derivative, in RCC patient urine were in line with the disrupted renal function that arises as a result of a disease, which may lead to the disruption of hippurate elimination or production.54 Hippurate is formed via the conjugation of glycine and benzoic acid, which takes place in the kidney, and this metabolite has been reported to have a strong association with diet and the gut microbiota.54 Reduced levels of hippurate in RCC patient urine were also reported in studies with smaller cohorts.20,28 In addition, reduced levels of hippurate have been reported in several RCC-predisposing conditions such as obesity55,56 and high blood pressure.57
N-Acetyl-d-glucosaminic acid, an acylaminosugar, was elevated in RCC in our study. Increased glucose uptake might be driving the elevation of the acylaminosugar via the hexosamine biosynthetic pathway (HBP). This increased HBP flux has been implicated in many cancer types58–62 as this pathway plays a central role in DNA repair, cellular signaling, and metastasis.63
In addition to endogenous metabolites, two exogenous metabolites were also selected as markers, 2-mercaptobenzothiazole (2-MBT) and dibutylamine. 2-MBT was found at higher levels in RCC patients’ urine. 2-MBT is used in acceleration of vulcanization; as such, it can be found in car tires. Other commodities that might contain 2-MBT include cables, rubber gloves, shoes, rubber bands, and toys.64,65 Humans are exposed to 2-MBT via inhalation and dermal or oral intake, and the compound has been detected in human urine.64,65 It has been identified as a marker for traffic intensity because of the tire tread wear linked to car usage, and calls were made for the revision of the risk assessment to 2-MBT.66 The International Agency for Research has classified it as “probably carcinogenic to humans”,66 while it has also been linked to an increased risk of bladder cancer.65
Higher levels of dibutylamine (DBA) were also present in RCC patient urine. Dibutylamine is a precursor to N-nitrosodibutylamine (NDBA), a nitrosamine.67 Nitrosamines are environmental carcinogens that can produce tumors in many organs in the body,68 with NBDA being one of the most potent bladder cancer carcinogens.69 Increased human urinary excretion of nitrosamines, including NBDA, has also been associated with esophageal cancer.70,71 Sources of amines and nitrosamines include drinking water67 and meat products.72,73
Hippuric acid, lactate, trigonellinamide, and mannitol were selected as markers in the NMR-only panel. Hippuric acid was also selected in the 10-metabolite panel, making the selection in the NMR-only panel unsurprising. The reduction in abundance of trigonellinamide (1-methylnicotinamide) in RCC patient urine could be indicative of a dysregulated nicotinate and nicotinamide metabolism,74 particularly considering that our study also identified a reduced level of trigonelline, a metabolite that showed a similar trend in a separate NMR study.20 Increased levels of lactate might reflect the activation of oncogenic aerobic glycolysis, the Warburg effect, which is a hallmark of cancerous cells.75 In addition, upregulation of lactate dehydrogenase A levels has been reported in RCC cells and tissues.76 Decreased mannitol excretion in RCC patients might be caused by dysregulations in energy metabolism, with this trend being reported in a separate urine metabolomics study.29
CONCLUSIONS
We have shown the potential utility of a urine assay in the clinical setting for RCC detection. This study, like others of its kind, has the limitation of numerous potential confounders that could impact biomarker discovery results. While randomized control trials (RCTs) are gold standards for epidemiology research, observational studies remain inescapable for studies like this as randomizing the intervention (RCC) is impossible. As such, to argue for the reduction in selection bias, we adjusted for five potential confounders in the study: age, BMI, gender, smoking history, and race. Of these, four adjustments were largely successful. Going forward, a much larger cohort, representing the diversity of race and geographical locations would be required for the validation of our biomarker proposals.
Supplementary Material
ACKNOWLEDGMENTS
Gonçalo J. Gouveia and Fariba Tayyari provided support in urine sample preparation and NMR data collection, respectively. In addition, we thank Dr. Jacquelyn Walejko and Sicong Zhang for NMR metabolomics computational advice. This work was supported by Georgia Institute of Technology’s Systems Mass Spectrometry Core Facility. The Georgia Research Alliance supported NMR data collection and analysis at UGA. Support of control samples was made available by the John and Mary Brock Discovery Fund. We also want to acknowledge the Cook Family Foundation, the Evans County Cares Foundation, and the Veterans Administration (1 I01 BX003367-01) for funding.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00213.
Section S1: NMR quality assurance and quality control and spectral binning; Scheme S1: NMR peak picking methods; Section S2: Model evaluation metrics; Figure S1: Relative quantification of all discriminating metabolomic features identified in the study, for RCC samples collected in the clinic versus operating room; Figure S2: Relative quantification of the 10-metabolite panel; Figure S3: Selection of metabolomic features with q-values and classification with logistic regression using the Metaboanalyst 5.0 biomarker analysis platform; Figure S4: Machine learning pipeline focused on upregulated features in RCC versus controls; Figure S5: Relative abundances for the panel of upregulated metabolites; Figure S6: Machine learning pipeline focused only on NMR features; Figure S7: Relative quantification of features in the NMR RCC metabolic panel; Figure S8: MS/MS annotation of 2-mercaptobenzothiazole and dibutylamine/n-butylisobutylamine/diisobutylamine; Table S1: Propensity score matching and study cohort characteristics; Table S2: Model cohort RCC characteristics; Table S3: Test cohort characteristics; Table S4: Quantified NMR features. ppm values, confidence score, fold changes, and q-values; Table S5: Chemical information of the 10-metabolite panel; Table S6: Machine learning hyperparameters used for binary classification using the MS-based 10-metabolite panel; Table S7: Machine learning performance using the MS RCC 10 panel biomarker; Table S8: Compound annotation and identification for the 5-metabolite panel upregulated in RCC; Table S9: Hyperparameters tuned for machine learning methods used for binary classification for the upregulated RCC biomarkers; Table S10: Machine learning performance using the upregulated MS RCC biomarkers; Table S11: Detailed MS/MS information for the panel of seven metabolites that distinguish RCC from control samples; Table S12: Machine learning hyperparameters tuned for binary classification using only the seven-identified metabolite panel; Table S13: Machine learning hyperparameters tuned for binary classification using only NMR RCC biomarkers; Table S14: Machine learning performance using NMR RCC biomarkers; Table S15: Review of some notable urine metabolomics studies comparing RCC to controls; Table S16: Metabolomic features with q-values <0.05 and >1-fold change (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.1c00213
The authors declare no competing financial interest.
Contributor Information
Olatomiwa O. Bifarin, Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia 30602, United States.
David A. Gaul, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
Samyukta Sah, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
Rebecca S. Arnold, Department of Urology, Emory University, Atlanta, Georgia 30308, United States
Kenneth Ogan, Department of Urology, Emory University, Atlanta, Georgia 30308, United States.
Viraj A. Master, Department of Urology, Emory University, Atlanta, Georgia 30308, United States; Winship Cancer Institute, Atlanta, Georgia 30302, United States
David L. Roberts, Department of Medicine, School of Medicine, Emory University, Atlanta, Georgia 30322, United States
Sharon H. Bergquist, Department of Medicine, School of Medicine, Emory University, Atlanta, Georgia 30322, United States
John A. Petros, Department of Urology, Emory University, Atlanta, Georgia 30308, United States; Atlanta VA Medical Center, Atlanta, Georgia 30033, United States
Facundo M. Fernández, School of Chemistry and Biochemistry and Petit Institute of Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States
Arthur S. Edison, Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center and Department of Genetics, Institute of Bioinformatics, University of Georgia, Athens, Georgia 30602, United States
REFERENCES
- (1).Siegel RL; Miller KD; Fuchs HE; Jemal A Cancer Statistics, 2021. CA Cancer J Clin 2021, 71, 7–33. [DOI] [PubMed] [Google Scholar]
- (2).Escudier B; Porta C; Schmidinger M; Rioux-Leclercq N; Bex A; Khoo V; Grunwald V; Gillessen S; Horwich A clinicalguidelines@esmo.org, E. G. C. E. a., Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2019, 30, 706–720. [DOI] [PubMed] [Google Scholar]
- (3).Gray RE; Harris GT Renal Cell Carcinoma: Diagnosis and Management. Am Fam Physician 2019, 99, 179–184. [PubMed] [Google Scholar]
- (4).Diaz de Leon A; Pedrosa I Imaging and Screening of Kidney Cancer. Radiol Clin North Am 2017, 55, 1235–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kang SK; Chandarana H Contemporary imaging of the renal mass. Urol Clin North Am 2012, 39, 161–170 vi. [DOI] [PubMed] [Google Scholar]
- (6).Patel HD; Johnson MH; Pierorazio PM; Sozio SM; Sharma R; Iyoha E; Bass EB; Allaf ME Diagnostic Accuracy and Risks of Biopsy in the Diagnosis of a Renal Mass Suspicious for Localized Renal Cell Carcinoma: Systematic Review of the Literature. J Urol 2016, 195, 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Haifler M; Kutikov A Update on Renal Mass Biopsy. Curr Urol Rep 2017, 18, 28. [DOI] [PubMed] [Google Scholar]
- (8).Nicholson JK; Lindon JC Systems biology: Metabonomics. Nature 2008, 455, 1054–1056. [DOI] [PubMed] [Google Scholar]
- (9).Zhang A; Sun H; Wu X; Wang X Urine metabolomics. Clin. Chim. Acta 2012, 414, 65–69. [DOI] [PubMed] [Google Scholar]
- (10).Fiehn O Combining genomics, metabolome analysis, and biochemical modelling to understand metabolic networks. Comp. Funct. Genomics 2001, 2, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Seyfried TN; Shelton LM Cancer as a metabolic disease. Nutr Metab 2010, 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Linehan WM; Srinivasan R; Schmidt LS The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 2010, 7, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xu C; Jackson SA Machine learning and complex biological data. Genome Biol. 2019, 20, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mitchell TM, Machine Learning. McGraw-Hill: New York, 1997; p xvii, 414 p. [Google Scholar]
- (15).Zhang M; Liu X; Liu X; Li H; Sun W; Zhang Y A pilot investigation of a urinary metabolic biomarker discovery in renal cell carcinoma. Int Urol Nephrol 2020, 52, 437–446. [DOI] [PubMed] [Google Scholar]
- (16).Wang Z; Liu X; Liu X; Sun H; Guo Z; Zheng G; Zhang Y; Sun W UPLC-MS based urine untargeted metabolomic analyses to differentiate bladder cancer from renal cell carcinoma. BMC Cancer 2019, 19, 1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rodrigues D; Monteiro M; Jeronimo C; Henrique R; Belo L; Bastos ML; Guedes de Pinho P; Carvalho M Renal cell carcinoma: a critical analysis of metabolomic biomarkers emerging from current model systems. Transl Res 2017, 180, 1–11. [DOI] [PubMed] [Google Scholar]
- (18).Oto J; Fernandez-Pardo A; Roca M; Plana E; Solmoirago MJ; Sanchez-Gonzalez JV; Vera-Donoso CD; Martinez-Sarmiento M; Espana F; Navarro S; Medina P Urine metabolomic analysis in clear cell and papillary renal cell carcinoma: A pilot study. J. Proteomics 2020, 218, 103723. [DOI] [PubMed] [Google Scholar]
- (19).Niziol J; Bonifay V; Ossolinski K; Ossolinski T; Ossolinska A; Sunner J; Beech I; Arendowski A; Ruman T Metabolomic study of human tissue and urine in clear cell renal carcinoma by LC-HRMS and PLS-DA. Anal. Bioanal. Chem 2018, 410, 3859–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Monteiro MS; Barros AS; Pinto J; Carvalho M; Pires-Luis AS; Henrique R; Jeronimo C; Bastos ML; Gil AM; Guedes de Pinho P Nuclear Magnetic Resonance metabolomics reveals an excretory metabolic signature of renal cell carcinoma. Sci. Rep 2016, 6, 37275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Monteiro M; Moreira N; Pinto J; Pires-Luis AS; Henrique R; Jeronimo C; Bastos ML; Gil AM; Carvalho M; Guedes de Pinho P GC-MS metabolomics-based approach for the identification of a potential VOC-biomarker panel in the urine of renal cell carcinoma patients. J Cell Mol Med 2017, 21, 2092–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Liu X; Zhang M; Liu X; Sun H; Guo Z; Tang X; Wang Z; Li J; Li H; Sun W; Zhang Y Urine Metabolomics for Renal Cell Carcinoma (RCC) Prediction: Tryptophan Metabolism as an Important Pathway in RCC. Front. Oncol 2019, 9, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kim K; Taylor SL; Ganti S; Guo L; Osier MV; Weiss RH Urine metabolomic analysis identifies potential biomarkers and pathogenic pathways in kidney cancer. OMICS 2011, 15, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kim K; Aronov P; Zakharkin SO; Anderson D; Perroud B; Thompson IM; Weiss RH Urine metabolomics analysis for kidney cancer detection and biomarker discovery. Mol. Cell. Proteomics 2009, 8, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ganti S; Weiss RH Urine metabolomics for kidney cancer detection and biomarker discovery. Urol Oncol 2011, 29, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ganti S; Taylor SL; Abu Aboud O; Yang J; Evans C; Osier MV; Alexander DC; Kim K; Weiss RH Kidney tumor biomarkers revealed by simultaneous multiple matrix metabolomics analysis. Cancer Res. 2012, 72, 3471–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kind T; Tolstikov V; Fiehn O; Weiss RH A comprehensive urinary metabolomic approach for identifying kidney cancerr. Anal. Biochem 2007, 363, 185–195. [DOI] [PubMed] [Google Scholar]
- (28).Ragone R; Sallustio F; Piccinonna S; Rutigliano M; Vanessa G; Palazzo S; Lucarelli G; Ditonno P; Battaglia M; Fanizzi FP; Schena FP Renal Cell Carcinoma: A Study Through NMR-Based Metabolomics Combined With Transcriptomics. Diseases 2016, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Oluyemi S; Falegan MWB; Shaykhutdinov RA; Pieroraio PM; Farshidfar F; Vogel HJ; Allaf ME; Hyndman ME Urine and Serum Metabolomics Analyses May Distinguish between Stages of Renal Cell Carcinoma. Metabolites 2017, 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Oluyemi S; Falegan SAAE; Andries Zijlstra M; Hyndman E; Vogel HJ Urinary Metabolomics Validates Metabolic Differentiation Between Renal Cell Carcinoma Stages and Reveals a Unique Metabolic Profile for Oncocytomas. Metabolites 2019, 9, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Niels-Peter VN; Carstensen JM; Smedsgaarda J Aligning of single and multiple wavelength chromatographic profiles for chemometric data analysis using correlation optimised warping. J. Chromatogr 1998, 805, 17–35. [Google Scholar]
- (32).Dieterle F; Ross A; Schlotterbeck G; Senn H Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem 2006, 78, 4281–4290. [DOI] [PubMed] [Google Scholar]
- (33).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfeifer J; Bax A NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (34).Bingol K; Li DW; Zhang B; Bruschweiler R Comprehensive Metabolite Identification Strategy Using Multiple Two-Dimensional NMR Spectra of a Complex Mixture Implemented in the COLMARm Web Server. Anal. Chem 2016, 88, 12411–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z; Nakatani E; Schulte CF; Tolmie DE; Kent Wenger R; Yao H; Markley JL BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wishart DS; Tzur D; Knox C; Eisner R; Guo AC; Young N; Cheng D; Jewell K; Arndt D; Sawhney S; Fung C; Nikolai L; Lewis M; Coutouly MA; Forsythe I; Tang P; Shrivastava S; Jeroncic K; Stothard P; Amegbey G; Block D; Hau DD; Wagner J; Miniaci J; Clements M; Gebremedhin M; Guo N; Zhang Y; Duggan GE; Macinnis GD; Weljie AM; Dowlatabadi R; Bamforth F; Clive D; Greiner R; Li L; Marrie T; Sykes BD; Vogel HJ; Querengesser L HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rosenbaum PR; Rubin DB The central role of the propensity score in observational studies for causal effects. Biometrika 1983, 70, 41–55. [Google Scholar]
- (38).Fabian Pedregosa GV; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; Vanderplas J; Passos A; Cournapeau D; Brucher M; Perrot M; Duchesnay É Scikit-learn: Machine Learning in Python. JMLR 2011, 12, 2825–2830. [Google Scholar]
- (39).Wang L; Wang Y; Chang Q Feature selection methods for big data bioinformatics: A survey from the search perspective. Methods 2016, 111, 21–31. [DOI] [PubMed] [Google Scholar]
- (40).Chandrashekar G; Sahin F A survey on feature selection methods. Computers and Electrical Engineering 2014, 40, 16–28. [Google Scholar]
- (41).Yoav Benjamini YH Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Statist. Soc. B 1995, 72, 12. [Google Scholar]
- (42).Waskom ML Seaborn: statistical data visualization. J. of Open Source Software 2021, 6, 3021. [Google Scholar]
- (43).McKinney W Data structures for statistical computing in python. Proceedings of the 9th Python in Science Conference 2010, 445, 56–61. [Google Scholar]
- (44).Hunter JD Matplotlib: A 2D graphics environment. Computing in Science & Engineering 2007, 9, 90–95. [Google Scholar]
- (45).van der Walt S; Colbert SC; Varoquaux G The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng 2011, 13, 22–30. [Google Scholar]
- (46).Oliphant TE Python for Scientific Computing. Computing in Science & Engineering 2007, 9, 10–20. [Google Scholar]
- (47).Seabold Skipper JP, Statsmodels: Econometric and statistical modeling with python. Proceedings of the 9th Python in Science Conference 2010. [Google Scholar]
- (48).Pérez F; Granger BE IPython: A System for Interactive Scientific Computing. Computing in Science and Engineering 2007, 9, 21–29. [Google Scholar]
- (49).Sud M; Fahy E; Cotter D; Azam K; Vadivelu I; Burant C; Edison A; Fiehn O; Higashi R; Nair KS; Sumner S; Subramaniam S Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Worley B; Powers R Multivariate Analysis in Metabolomics. Curr Metabolomics 2013, 1, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Boulesteix AL; Bender A; Lorenzo Bermejo J; Strobl C Random forest Gini importance favours SNPs with large minor allele frequency: impact, sources and recommendations. Brief Bioinform 2012, 13, 292–304. [DOI] [PubMed] [Google Scholar]
- (52).Pandey N; Lanke V; Vinod PK Network-based metabolic characterization of renal cell carcinoma. Sci. Rep 2020, 10, 5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hakimi AA; Reznik E; Lee CH; Creighton CJ; Brannon AR; Luna A; Aksoy BA; Liu EM; Shen R; Lee W; Chen Y; Stirdivant SM; Russo P; Chen YB; Tickoo SK; Reuter VE; Cheng EH; Sander C; Hsieh JJ An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lees HJ; Swann JR; Wilson ID; Nicholson JK; Holmes E Hippurate: the natural history of a mammalian-microbial cometabolite. J. Proteome Res 2013, 12, 1527–1546. [DOI] [PubMed] [Google Scholar]
- (55).Calvani R; Miccheli A; Capuani G; Tomassini Miccheli A; Puccetti C; Delfini M; Iaconelli A; Nanni G; Mingrone G Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int J Obes (Lond) 2010, 34, 1095–1098. [DOI] [PubMed] [Google Scholar]
- (56).Waldram A; Holmes E; Wang Y; Rantalainen M; Wilson ID; Tuohy KM; McCartney AL; Gibson GR; Nicholson JK Top-down systems biology modeling of host metabotype-microbiome associations in obese rodents. J. Proteome Res 2009, 8, 2361–2375. [DOI] [PubMed] [Google Scholar]
- (57).Holmes E; Loo RL; Stamler J; Bictash M; Yap IK; Chan Q; Ebbels T; De Iorio M; Brown IJ; Veselkov KA; Daviglus ML; Kesteloot H; Ueshima H; Zhao L; Nicholson JK; Elliott P Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 2008, 453, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zhu Q; Zhou L; Yang Z; Lai M; Xie H; Wu L; Xing C; Zhang F; Zheng S O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol 2012, 29, 985–993. [DOI] [PubMed] [Google Scholar]
- (59).Xu D; Wang W; Bian T; Yang W; Shao M; Yang H Increased expression of O-GlcNAc transferase (OGT) is a biomarker for poor prognosis and allows tumorigenesis and invasion in colon cancer. Int. J. Clin. Exp. Pathol 2019, 12, 1305–1314. [PMC free article] [PubMed] [Google Scholar]
- (60).Gu Y; Mi W; Ge Y; Liu H; Fan Q; Han C; Yang J; Han F; Lu X; Yu W GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res. 2010, 70, 6344–6351. [DOI] [PubMed] [Google Scholar]
- (61).Lynch TP; Ferrer CM; Jackson SR; Shahriari KS; Vosseller K; Reginato MJ Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem 2012, 287, 11070–11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).de Queiroz RM; Oliveira IA; Piva B; Bouchuid Catao F; da Costa Rodrigues B; da Costa Pascoal A; Diaz BL; Todeschini AR; Caarls MB; Dias WB Hexosamine Biosynthetic Pathway and Glycosylation Regulate Cell Migration in Melanoma Cells. Front. Oncol 2019, 9, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ma Z; Vosseller K Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J Biol Chem 2014, 289, 34457–34465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gries W; Kupper K; Leng G Rapid and sensitive LC-MS-MS determination of 2-mercaptobenzothiazole, a rubber additive, in human urine. Anal. Bioanal. Chem 2015, 407, 3417–3423. [DOI] [PubMed] [Google Scholar]
- (65).Murawski A; Schmied-Tobies MIH; Schwedler G; Rucic E; Gries W; Schmidtkunz C; Kupper K; Leng G; Conrad A; Kolossa-Gehring M 2-Mercaptobenzothiazole in urine of children and adolescents in Germany - Human biomonitoring results of the German Environmental Survey 2014–2017 (GerES V). Int J Hyg Environ Health 2020, 228, 113540. [DOI] [PubMed] [Google Scholar]
- (66).Avagyan R; Sadiktsis I; Bergvall C; Westerholm R Tire tread wear particles in ambient air–a previously unknown source of human exposure to the biocide 2-mercaptobenzothiazole. Environ Sci Pollut Res Int 2014, 21, 11580–11586. [DOI] [PubMed] [Google Scholar]
- (67).Wang W; Ren S; Zhang H; Yu J; An W; Hu J; Yang M Occurrence of nine nitrosamines and secondary amines in source water and drinking water: Potential of secondary amines as nitrosamine precursors. Water Res. 2011, 45, 4930–4938. [DOI] [PubMed] [Google Scholar]
- (68).Hecht SS Approaches to cancer prevention based on an understanding of N-nitrosamine carcinogenesis. Proc. Soc. Exp. Biol. Med 1997, 216, 181–191. [DOI] [PubMed] [Google Scholar]
- (69).Diana M; Felipe-Sotelo M; Bond T Disinfection byproducts potentially responsible for the association between chlorinated drinking water and bladder cancer: A review. Water Res. 2019, 162, 492–504. [DOI] [PubMed] [Google Scholar]
- (70).Zhao C; Lu Q; Gu Y; Pan E; Sun Z; Zhang H; Zhou J; Du Y; Zhang Y; Feng Y; Liu R; Pu Y; Yin L Distribution of N-nitrosamines in drinking water and human urinary excretions in high incidence area of esophageal cancer in Huai’an, China. Chemosphere 2019, 235, 288–296. [DOI] [PubMed] [Google Scholar]
- (71).Zhao C; Zhou J; Gu Y; Pan E; Sun Z; Zhang H; Lu Q; Zhang Y; Yu X; Liu R; Pu Y; Yin L Urinary exposure of N-nitrosamines and associated risk of esophageal cancer in a high incidence area in China. Sci. Total Environ 2020, 738, 139713. [DOI] [PubMed] [Google Scholar]
- (72).Bouma K; Schothorst RC Identification of extractable substances from rubber nettings used to package meat products. Food Addit. Contam 2003, 20, 300–307. [DOI] [PubMed] [Google Scholar]
- (73).Fiddler W; Doerr RC Gas chromatographic/chemiluminescence detection (thermal energy analyzer-nitrogen mode) method for the determination of dibutylamine in hams. J. AOAC Int 1993, 76, 578–581. [PubMed] [Google Scholar]
- (74).Slominska EM; Smolenski RT; Szolkiewicz M; Leaver N; Rutkowski B; Simmonds HA; Swierczynski J Accumulation of plasma N-methyl-2-pyridone-5-carboxamide in patients with chronic renal failure. Mol. Cell. Biochem 2002, 231, 83–88. [DOI] [PubMed] [Google Scholar]
- (75).Warburg O On the origin of cancer cells. Science 1956, 123, 309–314. [DOI] [PubMed] [Google Scholar]
- (76).Perroud B; Ishimaru T; Borowsky AD; Weiss RH Grade-dependent proteomics characterization of kidney cancer. Mol. Cell. Proteomics 2009, 8, 971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NMR data analysis was carried out using the Edison Lab in-house MATLAB scripts (https://github.com/artedison/Edison_Lab_Shared_Metabolomics_UGA, Matlab R2017b, The Mathworks, Inc.). Post metabolic features normalization computations were carried out in the Python 3.7.0 programming language using the following packages: Pandas for data handling,43 Matplotlib/Seaborn for data visualization,44 Numpy and Scipy for numerical computations,45,46 Statsmodel for statistical computations,47 and Sci-kit learn for machine learning.38 A Jupyter notebook was used as the integrated development environment (IDE).48 All Jupyter notebooks used in this study can be found here: https://github.com/artedison/RCC_MLprediction. The datasets collected in this work are available through the NIH Metabolomics Workbench49 with the project ID of PR001091 and study IDs ST001705 and ST001706. The dataset can be accessed via http://dx.doi.org/10.21228/M8P97V.