

Abstract
Motivation
Compound–protein interaction (CPI) plays an essential role in drug discovery and is performed via expensive molecular docking simulations. Many artificial intelligence-based approaches have been proposed in this regard. Recently, two types of models have accomplished promising results in exploiting molecular information: graph convolutional neural networks that construct a learned molecular representation from a graph structure (atoms and bonds), and neural networks that can be applied to compute on descriptors or fingerprints of molecules. However, the superiority of one method over the other is yet to be determined. Modern studies have endeavored to aggregate information that is extracted from compounds and proteins to form the CPI task. Nonetheless, these approaches have used a simple concatenation to combine them, which cannot fully capture the interaction between such information.
Results
We propose the Perceiver CPI network, which adopts a cross-attention mechanism to improve the learning ability of the representation of drug and target interactions and exploits the rich information obtained from extended-connectivity fingerprints to improve the performance. We evaluated Perceiver CPI on three main datasets, Davis, KIBA and Metz, to compare the performance of our proposed model with that of state-of-the-art methods. The proposed method achieved satisfactory performance and exhibited significant improvements over previous approaches in all experiments.
Availability and implementation
Perceiver CPI is available at https://github.com/dmis-lab/PerceiverCPI.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Drug development is a high-cost low-efficient process. New drug approval typically requires 10–15 years and costs 2.8 billion dollars on an average (Wouters et al., 2020). Various approaches based on artificial intelligence have been introduced to alleviate this problem. In recent years, traditional machine learning (ML) algorithms have been deployed to model the prediction of the interaction between compounds and proteins as a binary classification problem (Bahi and Batouche, 2021). However, binding affinity, which indicates the interaction strength of the drug–target pair, is a continuum value; hence, considering compound–protein interaction (CPI) as a regression problem is both effective and sufficient.
Through the binding mechanism, drugs can have a positive or negative influence on the functions carried out by proteins, which affect the targeted disease conditions (You et al., 2018). Understanding drug–target binding affinity makes it possible to identify candidate drugs that can inhibit or stimulate a given protein. Researchers have attempted to exploit meaningful information from given proteins and compounds. Notably, in terms of protein information extraction, most previous approaches consider the protein sequence as a plain text and then use a 1D convolutional neural network (1DCNN) with different methods of protein sequence numbering. Nevertheless, two types of models have shown excellent performance in terms of obtaining information from chemical compounds: deep neural networks (DNNs) such as a multiple layer perceptron (MLP) neural network, and 1DCNN performing on descriptors or fingerprints, and graph neural networks (GNNs) and their variants for extracting knowledge from a graph-structured dataset (Yang et al., 2019).
With respect to the first approach for molecular descriptors, Öztürk et al. (2018) proposed DeepDTA, which adopts two 1DCNNs to perform on raw sequences and the simplified molecular-input line-entry system (SMILES) (Weininger, 1988) as one-hot vectors. Using 1DCNN, the authors aimed to extract local residues and atomic features to predict binding affinity. DeepConv-DTI (Lee et al., 2019) followed a similar idea of DeepDTA by introducing a deep learning (DL) model to predict CPIs using raw protein sequences with Morgan/circular fingerprints (Morgan, 1965) as a compound representation. They used a 1DCNN on entire sequences of proteins to capture local residue patterns, while applying MLP neural network on molecular fingerprints to get drug features. Subsequently, Lee et al. concatenated aforementioned features, then transmitted them to a fully connected layer and predicted the property.
Regarding the second method, GNNs that follow a neighborhood aggregation scheme have become increasingly popular for graph-structured data (Scarselli et al., 2009). Numerous variants of GNN models have been proposed to achieve state-of-the-art (SOTA) performances in graph-based tasks in various fields of deep learning. Aware of the strength of GNNs, Nguyen et al. (2021) converted a compound representation into a graph represented by nodes (atoms) and edges (bonds); they, then, used four types of GNNs, graph convolutional networks (GCNs), graph attention networks (GATs), graph isomorphism networks (GINs) and a combination of GCNs and GATs, to capture molecular information. The knowledge extracted from atoms and bonds was then concatenated with the output of three 1DCNNs, which were used to learn different levels of abstract features from raw protein sequences.
Transformers (Vaswani et al., 2017) have shown a good performance in many AI fields, such as computer vision and natural language processing. Inspired by their potential to capture features between two sequences, Chen et al. (2020) proposed TransformerCPI, which is based on the architecture of an autoregressive encoder–decoder, using a combination of multiheaded attention and positional feed-forward to perform the CPI task. In their approach, molecular graphs were propagated to a GCN to obtain atomic features. Meanwhile, protein sequences were converted into sequential representations by separating a protein sequence into an overlapping 3-g amino acid sequence. Then, all words were translated into real-valued embeddings using the pretraining approach. The output was processed through 1DCNNs to obtain the final representation of the protein. Subsequently, these two representations were combined using a modified self-attention mechanism followed by MLPs. Motivated by the effectiveness of the self-attention mechanism, HyperattentionDTI was created (Zhao et al., 2022). The model was designed to input both compounds and proteins as plain sequences to two stacked 1DCNNs. In contrast to previous attention-based models, HyperattentionDTI inferred an attention vector by using a Sigmoid activation function rather than using a Softmax activation function.
The drawbacks of the existing approaches can be summarized as follows:
Because molecular descriptor vectors or fingerprints [such as extended-connectivity fingerprint (ECFP)] contain useful chemical knowledge from the start, the use of molecular fingerprints and molecular descriptors might lead to a better performance than using complex graphs on small datasets. However, owing to the representation’s simplification, models deploying them may underfit larger datasets.
On the other hand, GNNs must always learn a meaningful chemical space embedding from scratch. In addition, because of the global pooling step, which is simply chosen as the sum or average of all atomic features, over-smoothing and information loss are also crucial issues for GNNs.
Integration of the compound network’s and protein network’s representation is often performed by a simple concatenation, which is practically unsuitable for revealing the relationship between these molecules in practice.
Obtaining informative messages from protein sequences is a focus of research not only in CPI tasks but also in the general bioinformatics. Most current approaches consider protein sequences as plain texts, which cannot sufficiently reveal the real 3D structures of proteins.
In this study, we developed Perceiver CPI, a deep-learning model that addresses three of the abovementioned challenges (1, 2 and 3). Our approach is mainly inspired by that of Perceiver IO (Jaegle et al., 2021b) and a directed message-passing neural network (D-MPNN) (Yang et al., 2019). The contributions of this study are summarized as follows:
To avoid over-smoothing and information loss problems, we propose a novel method to enrich the representation of compounds by combining the information from both ECFPs and graph information.
To the best of our knowledge, Perceiver CPI is the first approach to use nested cross-attention for capturing the relations between protein and molecule representations.
Experimental results show that Perceiver CPI can achieve SOTA performance in novel pair and novel compound settings, and is competitive or slightly better than the baseline models in a novel protein setting.
2 Materials and methods
2.1 Feature encoding
2.1.1 Compound information encoding
Unlike previous approaches, which have commonly used either ECFP or molecular graph information constructed from SMILES, our proposed approach adopts ECFP to enrich the information extracted from the compound using D-MPNN (Yang et al., 2019). More specifically, we represent a molecule s using two forms:
A Morgan/circular fingerprint vector Ms as a binary vector, which indicates the existence of specific substructures. The Morgan algorithm searches each atom of the molecule and obtains all possible paths through the atom with a specific radius. Then, each unique path is hashed into a number based on a maximum of bit number.
A molecular graph , which represents the interactions between a set of atoms A by a set of bonds B.
We then process Ms and Gs through a MLP neural network and D-MPNN, respectively. Owing to its ability to approximate any continuous mapping, the MLP neural network is used to capture complex non-linear relationship features from Ms to yield OMs as the output. D-MPNN operates on hidden states and messages associated with directed edges (bonds) instead of messages associated with vertices (atoms). Each bond in the graph has a hidden state (i.e. feature vector) that contains atomic features (atomic number, number of bonds for each atom, formal charge, chirality, number of bonded hydrogens, hybridization, aromaticity and atomic mass) and bond features [bond type (single/double/triple/aromatic), conjugation, ring membership and stereochemistry] (Stokes et al., 2020). For each bond Bvw, we aggregate the function of the hidden states of all arriving neighboring bonds with the hidden state itself. Then, the hidden state of edge is updated using the obtained message and the previous hidden state of the atom. In other words, the hidden state of bond Bvw is obtained by updating the old hidden state with the newly obtained message. The corresponding message-passing update equations from atom Av to atom Aw are as follows:
| (1) |
where x is the feature of the corresponding atom A, ft is a MLP layer.
Specifically, in the message-passing phase, all messages arriving at bond Bvw are aggregated using a permutation-invariant aggregation function Average. Rather than using the summation function as suggested by the original D-MPNN, which caused model instability when training on small datasets, we adopt the average function Average to help the model update gradually. The aggregated representation is then combined with the existing hidden state via the MLP ft, resulting in an updated node feature vector hvw. Notably, all hidden states are initially set to (with evw as the feature of bond Bvw, is ReLU activation function). The main idea behind the message-passing technique is to prevent the distortion of messages between atoms. For example, the message from An to will only be propagated to and in the next iteration, whereas, in a conventional MPNN, it will be sent to node An, creating an unnecessary loop in the message-passing process. In the readout phase, we use one more average function to construct a final representation OGs.
Finally, after having two outputs OMs from MLP net and OGs from D-MPNN, we combine this information by adopting a cross-attention mechanism:
| (2) |
| (3) |
| (4) |
where Q is created from the output of the Morgan fingerprint MLP OMs, and K and V are generated from the output OGs of the D-MPNN by the projection functions (where and b are weight and bias, respectively). C and d are the embedding dimensions and number of heads, respectively. Figure 1 visualizes the attention module. Note that, OMs and OGs are 1D arrays. In our experiments, one more projection function f0 was used for a dimension reduction purpose at the end of the block. We found that a single-head cross-attention outperformed other multi-head cross-attentions.
Fig. 1.
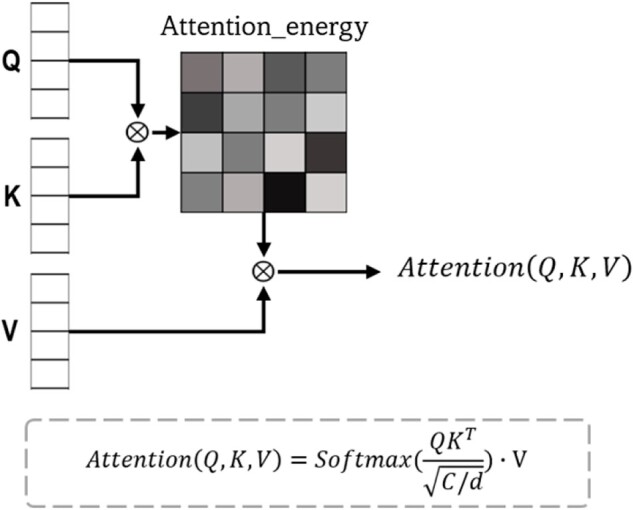
Demonstration of attention blocks. While the self-attention block accepts inputs from only single source, cross-attention blocks receive information from two sources
Using two modalities, we provide multiple views from compounds to the model; hence, Perceiver CPI is able to learn comprehensive patterns precisely. The ECFP provides information on the existence of substructures, whereas the graph representation considers the carrying knowledge that shows how they connect to one another.
2.1.2 Protein information encoding
The protein t was encoded using the tasks assessing protein embeddings (TAPE) tokenizer, where the initial feature of each residue was represented by the corresponding number following the UniRep Vocabulary (Rao et al., 2019). We used this one-hot encoding scheme for protein sequences, mainly because it is the simplest method to construct a unified representation (UniRep), which is broadly applicable and generalized to unseen regions of sequence space (Alley et al., 2019). The input is zero-padded to ensure that the number of output features remains fixed and then propagated into the blocks of 1DCNNs. Finally, we obtain the final output features. To help the model learn more deeply, we use the skip connection type to gradually change the weight of the network (He et al., 2016). Skip connections suggest skipping some of the layers in the neural network and feeding the output of one layer as the input to the next layers, thereby ensuring feature reusability to avoid the shattered gradient problem. The shattered gradient problem occurs in DNNs when the gradients resemble white noise and negatively impact the training (Balduzzi et al., 2017). Residual connection resolves this by introducing a spatial structure to the gradients, thus stabilizing the training process. Eventually, the output of the 1D convolution block can be expressed as follows:
Algorithm 1.
An algorithm for residual block of 1DCNN
Require: M,
Result:
for M do
( + embin * λ);
;
end for
where M is the number of 1DCNN layers, λ is fixed to isolate the effect of scaling and is a layer normalization function. emb represents the protein embedding with initialization emb0. Motivated by transformers, a combination of normalization and skip connection is observed to be helpful in facilitating the model’s capacity to learn of the model to protein information. Furthermore, the use of is intended to normalize the distributions of intermediate layers that might mitigate the gradient malformation to enable smoother gradients, faster training and better generalization accuracy. In particular, we use gated linear unit activation, a finite context approach through stacked convolutions, which can efficiently extract information from a sequence because it allows parallelization over sequential token features (Dauphin et al., 2017).
2.2 Compound–protein interaction
After obtaining two output representations from the three-element networks, we need to precisely integrate them to ultimately teach the model to capture valuable information that reveals CPI properties. The effective fusion of these multiple input sources is becoming increasingly important, as these multi-modality features have been shown to generate highly accurate performances in various tasks. A significant fusion method synergistically combines the two modalities and guarantees that the resultant product reflects the binding features of the input modalities (Mohla et al., 2020; Chen et al., 2021). Inspired by Perceiver IO and Perceiver (Jaegle et al., 2021a,b), we propose a novel method that leverages a highly asymmetric attention mechanism to distill compound information iteratively and then structure the final interaction representation using the protein information from a single cross-attention module. In the cross-attention block, we aim to force the model to capture patterns that show the effect of information from the compound on the protein information. In other words, we use this method because we primarily intend to determine how the protein reacts with the compound. More characteristically, after representing a compound, we process the compound latents by applying a series of self-attention modules to refine the compound representation. Finally, we combine Comps and Prott by applying a cross-attention module that maps latent arrays to the protein representation. The final interaction representation can be expressed as follows:
| (5) |
| (6) |
Using the cross-attention mechanism, we can model the semantic relevance between the protein and compound features, thus drawing attention to significant interaction information and benefiting the binding affinity prediction task. The cross-attention module generates cross-attention energy (also known as an attention map), which is then used to weight the feature map to achieve informative and discriminative feature representation. Moreover, the computation and memory complexity of generating attention energy in cross-attention are linear rather than quadratic, making the entire process more efficient.
2.3 Loss function and optimizer
In our experiment, we used the mean squared error (MSE) loss function with LAMB optimizer, which stands for ‘layer-wise adaptive moments optimizer for batch training’ (You et al., 2019). As can be seen, the training may become unstable if this ratio is too high. However, the weights do not change rapidly enough if the ratio is too small. Using the trust ratio, LAMB enables the model to be more confident in each step and scale much larger batch sizes without causing divergence. The hyperparameters in our neural network are searched using Bayesian optimization algorithms.
2.4 Benchmark datasets
To compare SOTA models with the proposed Perceiver CPI model and analyze its performance, we used three well-known benchmark datasets. To make use of the complementary information captured by the various bioactivity types, including dissociation constant (Kd), inhibition constant (Ki) or the half maximal inhibitory concentration (IC50), Tang et al. (2014) introduced a model-based integration approach called KIBA to generate an integrated drug–target bioactivity matrix. KIBA scores were created to optimize the consistency of the three measurements. The Davis dataset contains the interactions of 68 kinase inhibitors with 442 kinases covering >80% of the human catalytic protein kinome without missing interactions (Davis et al., 2011). The original unit of the dataset is Kd values; however, normalization of the label helps improve the performance. Hence, log transformation was applied to scale the label in the smaller range pKd = . Specifically, we used KIBA and Davis from the open-source software named DeepPurpose (Huang et al., 2021). For Metz data, Metz et al. (2011) presented a critical statistical analysis of kinomics screening data across 170 different protein kinases and establishing rigorous criteria. The PDBbind dataset contained 16 151 interactions (Wang et al., 2005). After filtering and processing to qualify the dataset, 6689 unique pairs were retained (Li et al., 2020). Table 1 shows the summary statistics for all datasets.
Table 1.
Statistics of the benchmark datasets
| Dataset | Proteins | Drugs | Interactions | Density (%) |
|---|---|---|---|---|
| Davis (Davis et al., 2011) | 442 | 68 | 30 056 | 100 |
| KIBA (Tang et al., 2014) | 229 | 2068 | 117 657 | 24.84 |
| Metz (Metz et al., 2011) | 170 | 1423 | 35 259 | 14.57 |
| PDBbind (Wang et al., 2005) | 2079 | 5535 | 6989 | 0.06 |
Furthermore, the density of all four datasets is shown in Figure 2. We employed kernel density estimation, a fundamental data smoothing problem where inferences about the population are made based on a finite data sample to reveal the dataset density. Figure 2 indicates that almost all the Davis dataset binding affinity values were highly concentrated around five. In particular, 69.64% of the Davis dataset had affinity binding values of five, whereas 71.96% of the KIBA dataset were in the range from 11.1 to 12. Due to the skew distribution of Davis dataset and KIBA dataset, we empirically forced the model to perform a larger weight update for data points, which did not belong to the high density area 10 times larger than the others. For instance, in Davis dataset, the data points have binding affinity in the range from 0 to 5 were discounted by 0.5, while the out-ranged data points multiplied by 5. Conversely, the Metz and PDBbind datasets exhibited well-balanced distributions with fewer outliers than the others; however, the sparsity of these datasets is extremely high.
Fig. 2.

Visualization of benchmark datasets with kernel density estimation
For a fair comparison, we also used the GPCR classification dataset to evaluate the enrichment factor (EF) from the Directory of Useful Decoys-Enhanced database (DUD-E) database. EF is used to show the performance of the model in finding true positives throughout the background database compared to random selection (Huang et al., 2006). Moreover, EF reveals the concentration of the annotated ligands among the top-scoring compounds compared to their concentrations throughout the entire dataset. For instance, the enrichment factor at 1% is the percentage of ligands found when 1% of decoys were found (Jain, 2008). Decoys from the DUD-E database were extracted from the ZINC database and were commercially available compounds for virtual screening (Mysinger et al., 2012). The 2D-similarity between the active compounds and decoy compounds are measured by calculating the Tanimoto distance. The statistics of the GPCR dataset are shown in Table 2 for training obtained from TransformerCPI, which was extracted from the GLASS database (Chan et al., 2015). The dataset provides experimentally validated GPCR–ligand associations. A threshold of 6.0 was set to divide the original dataset into positive and negative sets. Finally, Table 3 presents the test sets collected from the GPCR and Diverse subsets in the DUD-E database.
Table 2.
Statistic of GPCR dataset
| Proteins | Compounds | Positive pairs | Negative pairs | Density (%) |
|---|---|---|---|---|
| 356 | 5359 | 7989 | 7354 | 0.8 |
Table 3.
Statistics of GPCR and diverse subsets from DUD-E database
| Subset | Number of target | Actives | Decoys |
|---|---|---|---|
| GPCR subset | 5 | 1480 | 99 856 |
| Diverse subset | 7 | 1759 | 107 591 |
3 Experimental results and discussion
3.1 Experimental results
Model conversion: As mentioned above, we considered the CPI task as a regression problem. Nevertheless, only a few methods have used a similar concept; we transformed binary classification models, such as TransformerCPI, DeepconvDTI and HyperattentionDTI into regression models by modifying their final layers. To maintain the original performance, the output of the last layer was transformed to a single perceptron node, and the loss function was altered to the MSE loss function.
Experimental procedure: Owing to the density of the Davis dataset and because the dataset approximately covers 80% of the human catalytic protein kinomes, we decided to perform three experiments: novel pair setting, novel compound setting and novel protein setting; only novel pair setting was applied on the KIBA and Metz datasets. Finally, the PDBbind dataset and GPCR dataset with the GPCR and the Diverse subsets from the DUD-E database were used for an additional analysis. To calculate the similarities, the protein similarity is measured by the percentage of the number of aligned amino acids over the total length (in Perceiver CPI, the length of the proteins was fixed to 500). Meanwhile, the compound similarity was calculated using the Tanimoto similarity function.
Novel pair (Davis, KIBA and Metz): There were no overlaps between the training and test datasets. Neither the training compound nor the training protein appeared in the test set.
Novel-hard pair (Davis): There were no overlaps between the training and test datasets. The testing interactions were highly selective for similarities less than 0.3 by comparing to training interactions.
Novel compound (Davis): There were no intersections of compounds in the training set and compounds in the test set.
Novel protein (Davis): There were no intersections of proteins in the training set and proteins in the test set.
Cross-domain experiment (Davis and PDBbind): There were no overlaps between the training and test datasets. We trained the model with the Davis dataset and tested it with the PDBbind dataset.
Enrichment factor analysis [GPCR, GPCR subset (DUD-E dataset), Diverse subset (DUD-E dataset)]: There were no overlaps between the training and test datasets. We trained the model with the GPCR dataset and tested it with subsets from the DUD-E dataset (the duplicated target ‘CXCR4’ was removed from the Diverse subset).
Evaluation metric: To evaluate the performance on the regression task, we used the mean squared error (MSE) metric to measure the performance of the models and the concordance index (CI) metric to evaluate the proportion of concordant prediction pairs per the total number of label pairs, which tells us whether the predicted binding affinity values of two random drug–target pairs were predicted in the same order as their truth values. In the enrichment factor analysis, we adopted an EF score at fraction 1% (EF) to show the performance of the models in determining the annotated ligands among the top binding affinity compounds and the Boltzmann-enhanced discrimination of the receiver operating characteristic score to focus more on early enrichment with α = 80.5 (BEDROC).
Cross-validation: We applied five-fold cross-validation to calculate the performance of baseline models and Perceiver CPI in four experiments: novel pair, novel-hard pair, novel compound, novel protein. The validation set was taken arbitrarily from the training set following the ratio training: validation = 80%: 20% for all experiments.
Table 4 compares the performance of Perceiver CPI with five SOTA deep-learning baseline models for the three types of separations. Regarding the two principal tasks (novel pair and novel compound), Perceiver CPI showed remarkable performances. With reference to the novel pair setting, our proposed model achieved an MSE of 0.463(±0.013) and CI of 0.638(±0.028), whereas the competitors performed poorly. In the novel compound experiment, Perceiver CPI reached the lowest MSE (0.378(±0.010)) and had the highest value in CI (0.726(±0.017)). We discovered no significant difference between the previous approaches and our model in the novel protein tasks. Nonetheless, Perceiver CPI performed better than the others in terms of MSE and was competitive with the first-placed model in terms of CI metric. As shown in Table 5, in the most challenging setting, when the test set was significantly different from what the model trained on, the proposed method outperformed baseline compactors by providing precise predictions, resulting in the lowest MSE. In practice, the number of proteins is finite, and most of them will eventually be annotated, which means that the CPI task is mainly about finding a new compound with existing proteins in the real world. Interestingly, our model was also more stable than the others as indicated by its lower standard deviation among the validations.
Table 4.
Comparison of the proposed method with SOTA model in terms of three settings from the Davis dataset with 5-fold cross-validation
| Model | Novel pair |
Novel compound |
Novel protein |
|||
|---|---|---|---|---|---|---|
| MSE | CI | MSE | CI | MSE | CI | |
| DeepDTA (Öztürk et al., 2018) | 0.631(±0.059) | 0.533(±0.027) | 0.482(±0.034) | 0.613(±0.029) | 0.701(±0.045) | 0.759(±0.015) |
| DeepConvDTI (Lee et al., 2019) | 0.598(±0.057) | 0.546(±0.043) | 0.512(±0.046) | 0.681(±0.012) | 0.789(±0.109) | 0.714(±0.034) |
| TransformerCPI (Chen et al., 2020) | 0.549(±0.038) | 0.490(±0.032) | 0.522(±0.027) | 0.592(±0.026) | 0.708(±0.032) | 0.676(±0.005) |
| GraphDTA (GINs) (Nguyen et al., 2021) | 0.846(±0.058) | 0.459(±0.032) | 0.452(±0.051) | 0.670(±0.018) | 0.970(±0.061) | 0.660(±0.016) |
| HyperattentionDTI (Zhao et al., 2022) | 0.671(±0.045) | 0.517(±0.013) | 0.506(±0.015) | 0.578(±0.019) | 0.784(±0.063) | 0.674(±0.020) |
| Perceiver CPI (ours) | 0.463(±0.013) | 0.638(±0.028) | 0.378(±0.010) | 0.726(±0.017) | 0.667(±0.018) | 0.758(±0.010) |
Note: The metrics are MSE (the lower, the better) and CI (the higher, the better) (± standard deviation)
Table 5.
Comparison of Perceiver CPI and other SOTA competitors on novel-hard pair setting
| Model | MSE | CI |
|---|---|---|
| DeepDTA (Öztürk et al., 2018) | 0.948(±0.218) | 0.565(±0.040) |
| DeepConvDTI (Lee et al., 2019) | 0.768(±0.290) | 0.571(±0.052) |
| TransformerCPI (Chen et al., 2020) | 0.806(±0.254) | 0.508(±0.071) |
| GraphDTA (GINs) (Nguyen et al., 2021) | 0.931(±0.314) | 0.542(±0.070) |
| HyperattentionDTI (Zhao et al., 2022) | 0.873(±0.246) | 0.600(±0.049) |
| Perceiver CPI (ours) | 0.701(±0.244) | 0.609(±0.072) |
Considering the most challenging setting, the novel pair split settings with KIBA and Metz datasets, as shown in Table 6, DeepConvDTI achieved inferior performance using ECFP representation for compounds. Although ECFP captures useful information for CPI prediction, owing to its simplicity, the knowledge from compounds is still not fully used. Therefore, instead of using the ECFP independently, a combination of ECFP and graph representations of the compound was utilized to further improve performance. The two datasets contained many missing interactions, resulting in the underperformance of all models. In the KIBA dataset, Perceiver CPI attained a lower MSE than the baseline by 0.028 and a higher CI. In particular, it was extremely difficult to obtain correct predictions using the Metz dataset, which has a 14.57% density.
Table 6.
Comparison of Perceiver CPI performance to SOTA baseline models in novel pair task from on KIBA and Metz datasets
| Model | KIBA |
Metz |
||
|---|---|---|---|---|
| MSE | CI | MSE | CI | |
| DeepDTA (Öztürk et al., 2018) | 0.668(±0.055) | 0.600(±0.011) | 0.781(±0.060) | 0.627(±0.011) |
| DeepConvDTI (Lee et al., 2019) | 0.550(±0.009) | 0.635(±0.007) | 0.703(±0.027) | 0.671(±0.016) |
| TransformerCPI (Chen et al., 2020) | 0.630(±0.057) | 0.563(±0.014) | 1.081(±0.125) | 0.557(±0.016) |
| GraphDTA (GINs) (Nguyen et al., 2021) | 0.698(±0.042) | 0.591(±0.013) | 1.232(±0.094) | 0.615(±0.010) |
| HyperattentionDTI (Zhao et al., 2022) | 1.022(±0.062) | 0.590(±0.015) | 1.064(±0.080) | 0.630(±0.013) |
| Perceiver CPI (ours) | 0.522(±0.010) | 0.638(±0.013) | 0.658(±0.016) | 0.675(±0.012) |
Moreover, we performed a cross-domain experiment to determine the adaptability of our method to an unseen domain dataset. We chose two datasets (Davis and PDBbind) owing to their overlapping properties and measurements. First, we eliminated all overlapping interactions from the PDBbind dataset to the Davis dataset. Second, while we divided the Davis dataset into training and validation sets at a ratio of 80%:20%, the processed PDBbind dataset was used as a test set. The results in Table 7 show that Perceiver CPI significantly outperformed the baselines. The proposed approach achieved a higher performance on CI metrics than SOTA models, while exhibited a lower MSE. In other words, Perceiver CPI provides more precise predictions than the compared models. In particular, all models, including ours, performed poorly in the cross-domain experiment because of the quantity and quality of the training dataset.
Table 7.
Results of the cross-domain experiment (trained on Davis and tested on PDBbind)
| Model | MSE | CI |
|---|---|---|
| DeepDTA (Öztürk et al., 2018) | 4.716 | 0.500 |
| DeepConvDTI (Lee et al., 2019) | 5.400 | 0.477 |
| TransformerCPI (Chen et al., 2020) | 4.962 | 0.497 |
| GraphDTA (GINs) (Nguyen et al., 2021) | 6.323 | 0.516 |
| HyperattentionCPI (Zhao et al., 2022) | 5.946 | 0.410 |
| Perceiver CPI (ours) | 4.612 | 0.532 |
We tested the model and other classifiers and five docking-based programs [Gold (Jones et al., 1997), Glide (Friesner et al., 2004), Surflex (Jain, 2003), FlexX (Rarey et al., 1996) and Blaster (Irwin et al., 2009)] on subsets from the DUD-E database. We converted Perceiver CPI architecture into a classifier by changing the loss function from MSELoss to CrossEntropyLoss, as well as by transforming the last layer into a sigmoid function. Ligand enrichment among top-ranking hits is an important criterion for molecular docking and drug–target interactions. Table 8 reveals that Perceiver CPI achieved a better performance for multiple targets in an EF and BEDROC than the other deep-learning models. However, the docking-based method outperformed the data-driven method for most protein targets on both metrics (Supplementary Table S7). Perhaps the combination of the two good methods might lead to an excellent performance. Moreover, the accumulation of extended datasets may enhance the predictions of the ML/DL models.
Table 8.
Enrichment factor analysis results for subsets in the DUD-E database (UP: EF, DOWN: BEDROC)
| Family | DeepConvDTI | TransformerCPI | HyperattentionDTI | Perceiver CPI (ours) | Gold | Glide | Surflex | FlexX | Blaster |
|---|---|---|---|---|---|---|---|---|---|
| GPCR (DUD-E) | 9.728(±11.534) | 0.814(±1.178) | 3.982(±3.119) | 16.366(±15.921) | N/a | N/a | N/a | N/a | 11.8(±8.136) |
| (DUD-E) | 0.152(±0.174) | 0.018(±0.040) | 0.071(±0.058) | 0.236(±0.177) | 0.282(±0.154) | 0.198(±0.205) | 0.284(±0.098) | 0.156(±0.135) | N/a |
| Diverse | 0.292(±0.774) | 0.922(±0.819) | 1.075(0.876) | 1.88(±1.297) | N/a | N/a | N/a | N/a | 13.571(±12.908) |
| (DUD-E) | 0.005(±0.015) | 0.021(0.016) | 0.023(±0.018) | 0.031(±0.022) | 0.295(±0.180) | 0.258(±0.170) | 0.118(±0.093) | 0.104(±0.059) | N/a |
In summary, the proposed Perceiver CPI achieves a competitive or better performance than SOTA deep-learning baselines in all settings, due to the fact that our model adopts the strength of a attention mechanism to dynamically adjust the features of drugs and proteins in different combinations.
3.2 Discussion
3.2.1 Difference between perceiver CPI and perceiver IO
Perceiver IO is an updated version of Perceiver, which uses an asymmetric attention mechanism to accept input information into a tight latent space. Subsequently, the output of Perceiver is merged with the query system using an additional cross-attention. The key insight is to produce each output by attending to the latent array using a specific output query associated with that output. Therefore, the target of Perceiver IO is the input compound. However, the purpose of Perceiver CPI is to seek a change in the protein caused by the effect of a compound; hence, our target is the input protein. As shown in Figure 3, we take the key (K) and value (V) from the protein information, contrary to Perceiver IO, which considers the protein information as an output query array (Q). Besides, empirical experiment results with the original structure of Perceiver IO showed poorer performance on the CPI task when compared to Perceiver CPI.
Fig. 3.

Overview of perceiver CPI. The model is a combination of three-element neural networks that take information from three sources: molecular graphs, Morgan fingerprints for constructing compound patterns and protein sequences for extracting protein knowledge
4 Conclusion and future work
In this study, we deployed cross-attention mechanisms to address the CPI task. We proposed a novel attention mechanism to not only enrich the information extracted from a compound using ECFP knowledge but also to capture CPI information effectively. The proposed Perceiver CPI model exhibited a significantly improved performance on three benchmark datasets when compared with SOTA baselines in terms of MSE and CI.
Although Perceiver CPI has demonstrated excellent performance, much work remains to improve the performance of CPI prediction tasks in the future.
Finding and extracting meaningful features from proteins remains a difficult but worthwhile task. For instance, AlphaFold2 from DeepMind can be used to predict the 3D structure of proteins (Jumper et al., 2021).
The information taken from compounds can still be cultivated more profitably, such as by using the META-Learning method to construct a better representation from small datasets.
Utilizing information from 3D structures produced from SMILES, as GeoMol attempts to do, is also a promising method because of its high information capacity (Ganea et al., 2021).
Adopting the transfer learning method for individual neural networks (compound and protein networks) to generate improved representations from the beginning with the help of prior knowledge should also be considered.
The interpretability of Perceiver CPI is limited by the dimensionality reduction of MLP from the hidden state update process in the message-passing step and from the attention blocks. Addressing such useful features would form a valuable part of future work.
Supplementary Material
Acknowledgements
We sincerely thank Dr. Sungjoon Park (Department of Medicine, University of California San Diego, La Jolla, CA, USA) for assistance with the methodology discussion and experimental design comments, which greatly improved the quality of our work. We greatly thank the anonymous reviewers for their careful of reading of our manuscript and their many insightful comments and suggestions.
Contributor Information
Ngoc-Quang Nguyen, Department of Computer Science and Engineering, Korea University, Seoul 02841, Republic of Korea.
Gwanghoon Jang, Department of Computer Science and Engineering, Korea University, Seoul 02841, Republic of Korea.
Hajung Kim, Interdisciplinary Graduate Program in Bioinformatics, Korea University, Seoul 02841, Republic of Korea.
Jaewoo Kang, Department of Computer Science and Engineering, Korea University, Seoul 02841, Republic of Korea; Interdisciplinary Graduate Program in Bioinformatics, Korea University, Seoul 02841, Republic of Korea; AIGEN Sciences, Seoul 04778, Republic of Korea.
Data availability
Our study used open-access datasets, and the data-related links are available in the Data availability section in the supplementary document.
Funding
This work was supported by the National Research Foundation of Korea [NRF-2020R1A2C3010638, NRF-2014M3C9A3063541]; the Ministry of Health & Welfare, Republic of Korea [HR20C0021]; and the ICT Creative Consilience program [IITP-2021-0-01819] supervised by the IITP (Institute for Information & communications Technology Planning & Evaluation).
Conflict of Interest: none declared.
References
- Alley E.C. et al. (2019) Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods, 16, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi M., Batouche M. (2021) Convolutional neural network with stacked autoencoders for predicting drug-target interaction and binding affinity. Int. J. Data Mining Model. Manag., 13, 81–113. [Google Scholar]
- Balduzzi D. et al. (2017) The shattered gradients problem: if resnets are the answer, then what is the question? In: Proceedings of the 34 th International Conference on Machine Learning, Sydney, Australia. pp. 342–350. PMLR.
- Chan W.K. et al. (2015) GLASS: a comprehensive database for experimentally validated GPCR-ligand associations. Bioinformatics, 31, 3035–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-F.R. et al. (2021) CrossViT: cross-attention multi-scale vision transformer for image classification. In: ICCV 2021 took place virtually from October 11 to October 17, 2021. pp. 357–366.
- Chen L. et al. (2020) TransformerCPI: improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics, 36, 4406–4414. [DOI] [PubMed] [Google Scholar]
- Dauphin Y.N. et al. (2017) Language modeling with gated convolutional networks. In: Proceedings of the 34 th International Conference on Machine Learning, Sydney, Australia. pp. 933–941. PMLR.
- Davis M.I. et al. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol., 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Friesner R.A. et al. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem., 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Ganea O. et al. (2021) GeoMol: torsional geometric generation of molecular 3d conformer ensembles. In: Advances in Neural Information Processing Systems, Virtual, Vol. 34.
- He K. et al. (2016) Deep residual learning for image recognition. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Las Vegas, Nevada, USA. pp. 770–778.
- Huang K. et al. (2021) DeepPurpose: a deep learning library for drug–target interaction prediction. Bioinformatics, 36, 5545–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N. et al. (2006) Benchmarking sets for molecular docking. J. Med. Chem., 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J.J. et al. (2009) Automated docking screens: a feasibility study. J. Med. Chem., 52, 5712–5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaegle A. et al. (2021a) Perceiver: general perception with iterative attention. In: IInternational Conference on Machine Learning, 18-24 July 2021, Virtual. pp. 4651–4664. PMLR.
- Jaegle A. et al. (2021b) Perceiver IO: A General Architecture for Structured Inputs & Outputs. In: International Conference on Learning Representations.
- Jain A.N. (2003) Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem., 46, 499–511. [DOI] [PubMed] [Google Scholar]
- Jain A.N. (2008) Bias, reporting, and sharing: computational evaluations of docking methods. J. Comput. Aided Mol. Des., 22, 201–212. [DOI] [PubMed] [Google Scholar]
- Jones G. et al. (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol., 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Jumper J. et al. (2021) Highly accurate protein structure prediction with alphafold. Nature, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. et al. (2019) DeepConv-DTI: prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol., 15, e1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. et al. (2020) MONN: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Systems, 10, 308–322.e11. [Google Scholar]
- Metz J.T. et al. (2011) Navigating the kinome. Nat. Chem. Biol., 7, 200–202. [DOI] [PubMed] [Google Scholar]
- Mohla S. et al. (2020) FusAtNet: dual attention based spectrospatial multimodal fusion network for hyperspectral and LiDAR classification. In: IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops, CVPR 2020, virtually. pp. 92–93.
- Morgan H.L. (1965) The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. J. Chem. Doc., 5, 107–113. [Google Scholar]
- Mysinger M.M. et al. (2012) Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem., 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. et al. (2021) GraphDTA: predicting drug–target binding affinity with graph neural networks. Bioinformatics, 37, 1140–1147. [DOI] [PubMed] [Google Scholar]
- Öztürk H. et al. (2018) DeepDTA: deep drug–target binding affinity prediction. Bioinformatics, 34, i821–i829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R. et al. (2019) Evaluating protein transfer learning with tape. In; Advances in Neural Information Processing Systems, Vancouver Convention Center, Vancouver Canada, Vol. 32. [PMC free article] [PubMed] [Google Scholar]
- Rarey M. et al. (1996) A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol., 261, 470–489. [DOI] [PubMed] [Google Scholar]
- Scarselli F. et al. (2009) The graph neural network model. IEEE Trans. Neural Netw., 20, 61–80. [DOI] [PubMed] [Google Scholar]
- Stokes J.M. et al. (2020) A deep learning approach to antibiotic discovery. Cell, 180, 688–702.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. et al. (2014) Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. J. Chem. Inf. Model., 54, 735–743. [DOI] [PubMed] [Google Scholar]
- Vaswani A. et al. (2017) Attention is all you need. In: Advances in Neural Information Processing Systems, Long Beach Convention Center (300 E Ocean Blvd, Long Beach, CA 90802, United States), Vol. 30. [Google Scholar]
- Wang R. et al. (2005) The PDBbind database: methodologies and updates. J. Med. Chem., 48, 4111–4119. [DOI] [PubMed] [Google Scholar]
- Weininger D. (1988) Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J. Chem. Inf. Model., 28, 31–36. [Google Scholar]
- Wouters O.J. et al. (2020) Research and development costs of new drugs—reply. JAMA, 324, 518–518. [DOI] [PubMed] [Google Scholar]
- Yang K. et al. (2019) Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model., 59, 3370–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J. et al. (2018) Graph convolutional policy network for goal-directed molecular graph generation. In: Advances in Neural Information Processing Systems, Palais des Congrès de Montréal, Montréal Canada, Vol. 31. [Google Scholar]
- You Y. et al. (2019) Large batch optimization for deep learning: training Bert in 76 minutes. In: International Conference on Learning Representations, Virtual Conference.
- Zhao Q. et al. (2022) HyperAttentionDTI: improving drug–protein interaction prediction by sequence-based deep learning with attention mechanism. Bioinformatics, 38, 655–662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our study used open-access datasets, and the data-related links are available in the Data availability section in the supplementary document.