

ABSTRACT
Herpes simplex encephalitis (HSE), mainly caused by herpes simplex virus type 1 (HSV-1), is a severe central nervous system disease commonly followed by cognitive impairment, behavioral changes, and focal neurological signs. Although increasing evidence implicates the central role of microglia in HSE progression, the intrinsic restrictors or the acquired environmental factors that balance the beneficial or detrimental immune responses in microglia remain unclear. In a recent study, we find that a gut microbial metabolite activates mitophagy to regulate microglia-mediated neuroinflammation and to mitigate HSE progression. HSV-1 neurotropic infection causes gut microbiota dysbiosis and microglial antiviral immune response, whereas depletion of gut microbiota by oral antibiotics treatment further results in hyperactivated microglia and exacerbated HSE pathology. Notably, exogenous administration of nicotinamide n-oxide (NAMO), an oxidative product of nicotinamide mainly produced by intestinal neomycin-sensitive bacteria, especially Lactobacillus gasseri and Lactobacillus reuteri, can significantly suppress HSE progression. Mechanistically, HSV-1 infection causes mitochondrial dysfunction and impairs mitophagy to activate microglia and promote proinflammatory cytokine production, whereas NAMO restores NAD+-dependent mitophagy to restrain microglial over-activation and to prevent HSV-1 early infection in neuronal cells. This work reveals a novel function of gut microbial metabolites as intrinsic regulators of microglia homeostasis and neuroinflammation via mitophagy.
Abbreviations: AD: Alzheimer disease; ABX: antibiotics; HSE: herpes simplex encephalitis; HSV-1: herpes simplex virus type 1; NAD+: nicotinamide adenine dinucleotide; NAMO: nicotinamide n-oxide; SCFAs: short-chain fatty acids.
KEYWORDS: Gut microbes, herpes simplex encephalitis, microbial metabolite, microglia activation, mitophagy, neuroinflammation, nicotinamide n-oxide
A considerable body of research has been conducted to explore the association between HSV-1 neurotropic infection and neurodegenerative diseases, such as Alzheimer disease (AD). Notably, microglia control and coordinate immune reaction to HSV-1 infection in the central nervous system, and unbridled microglial activity facilitates HSE progression. Microglia-mediated neuroinflammation and impaired microglial mitophagy are also tightly associated with AD progression, resulting in β-amyloid accumulation and MAPT/tau pathology. Therefore, the deep understanding of the complex interaction among microglia homeostasis, mitophagy and HSV-1 neurotropic infection will aid in the development of potential therapeutic strategies against HSV-1-related diseases.
Microglial maturation and function are modulated by host gut microbes, which orchestrate an optimal innate and adaptive immune response to control virus infection, such as influenza virus and hepatitis B virus. Specific microbial constituents, metabolites or microbe-associated molecular patterns serve as messengers to communicate with antiviral type I interferon signaling and to prime various immune cell subsets during systemic virus infection. In our recent study, we investigated whether gut microbes influence microglia homeostasis and the consequential antiviral responses during HSV-1 infection and HSE progression (Figure 1) [1].
Figure 1.
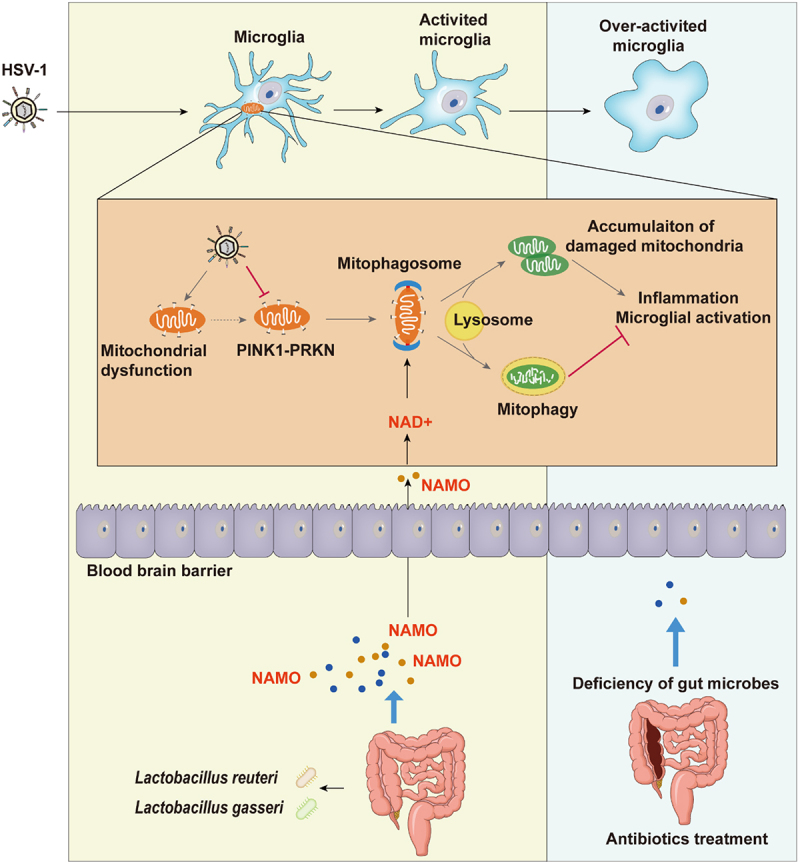
The gut microbial metabolite NAMO activates mitophagy to restrict microglia activation and HSE progression. HSV-1 neurotropic infection causes mitochondria dysfunction and inhibits PINK1-PRKN-dependent mitophagy, resulting in the accumulation of damaged mitochondria and microglia activation. On the contrary, the intestinal microbial metabolite NAMO, mainly derived from bacteria Lactobacillus gasseri and Lactobacillus reuteri, crosses the blood brain barrier to activate NAD+-dependent mitophagy to restrain microglia-mediated neuroinflammation and to suppress HSE progression. Deficiency of gut microbes further exaggerates microglial hyperactivation and HSE pathology.
We first observed that neurotropic HSV-1 infection significantly alters the composition and diversity of gut microbes, with high abundance of the genus Bacteroides, Odoribacter, Rikenellaceae RC9 gut group, and Ruminococcaceae UCG-014. The altered microbiota exhibit virus-specific tropisms that are different from that caused by other viruses, such as SARS-CoV-2 and influenza virus. Besides, HSV-1 significantly upregulates the production of inflammatory cytokines in the gut, suggesting that the enhanced intestinal inflammation might provide optimal environmental conditions for the growth of pathological bacteria, promoting their bidirectional regulation of immune responses in the gut and brain via the gut-brain-axis. Consistently, we depleted the intestinal microbiota of mice by administrating antibiotics (ABX) and found that ABX treatment significantly enhances cellular damage, exaggerates immune response to HSV-1 infection and promotes viral production and distribution, leading to the reduced survival rate of HSE mice. Collectively, these data clearly indicate a continuous protective contribution of intestinal microbes against HSE development.
By histopathological and RNA sequencing, we found that HSV-1 infection triggers the transformation from resting microglia (homeostatic M0 type) to activated microglia (pro-inflammatory M1 type), with changed immune properties and cell numbers, to stimulate the antiviral immune response (Figure 1). ABX treatment further primes activated microglia to a form with increased cytokine production (hyper-activated proinflammatory state), resulting in aggravated neuroinflammation and enhanced susceptibility to HSE-associated death. These results indicate that excessive activation of the microglial immune response leads to pathology, and gut microbiota act as a negative brake of unbridled microglia activation. Similarly, another previous study demonstrated that the gut microbiota restricts microglia-mediated pro-inflammatory and neurotoxic activities via AHR (aryl-hydrocarbon receptor) signaling.
Next, we focused on altered microbial metabolites linking gut microbiota to microglia activation and neuroinflammation. By using a non-targeted metabolomics technique, we found that HSV-1 infection significantly dysregulates amino acid metabolism and selectively switches tryptophan-nicotinamide transformation to tryptophan-phenylalanine transformation, leading to the increased abundance of phenylalanine, a typical feature associated with AD progression. In addition, we found that most microbiota-derived metabolites inhibit the interferon-based innate antiviral response in microglia. Among these differential regulated metabolites, we identified nicotinamide nitrogen oxide (NAMO), an oxidative product of nicotinamide derived from gut neomycin-sensitive bacteria, especially Lactobacillus reuteri and Lactobacillus gasseri, acting as a crucial mediator restricting microglia activation (Figure 1). Exogenous administration of NAMO to ABX-treated HSE mice diminishes microglia-mediated proinflammatory responses and restricts HSV-1 infection. NAMO also prevents neuron damage via inhibiting HSV-1 early infection of neuronal cells and reducing the microglia-mediated neuronal cytotoxic microenvironment. Importantly, gut microbiota-derived short-chain fatty acids (SCFAs) have been reported to positively modulate microglia maturation, morphology and function, whereas we confirmed NAMO as negative regulator restricting microglia activation, implying the cooperation of NAMO and SCFAs to maintain microglia homeostasis and immune status.
Mitophagy refers to the degradation of damaged mitochondria by the autophagic machinery, and a deficiency of mitophagy is a critical mediator for immune activation and inflammation in neuropathogenesis. Although the relationship between HSV-1 infection and macroautophagy has been extensively clarified, the role of mitophagy is currently unknown. In our study, we found that HSV-1 infection causes mitochondria dysfunction and downregulates PINK1-PRKN to inhibit mitophagy, resulting in the accumulation of damaged mitochondria and microglia activation (Figure 1). Modulation of mitophagy by the chemical inducer CCCP or inhibitor Mdivi-1 also significantly affect microglia activation and inflammatory cytokine release.
On the contrary, the gut metabolite NAMO restores mitophagy to remove damaged mitochondria and thereby inhibits microglia activation and inflammatory response. However, the complete and functional mitophagic flux activated by NAMO is not depended on PRKN and PINK1. Instead, NAMO promotes the production of nicotinamide adenine dinucleotide (NAD+) and activates NAD+- and ULK1-dependent mitophagy (Figure 1). Interestingly, both NAMO precursors, nicotinamide and nicotinic acid, are also able to reduce mitochondrial damage and enhance NAD+. Considering that the level of NAD+ declines in AD and the supplement of NAD+ can alleviate AD with lessened MAPT/tau levels, decreased DNA damage and improved cognitive function, the gut metabolite NAMO therefore represents an intrinsic defender to ameliorate neuroinflammation and AD symptoms via manipulating microglial mitophagy.
In summary, our study uncovers an unappreciated role of mitophagy bridging gut microbial regulatory effects and microglial activation to maintain immune homeostasis and avoid immunopathology. The altered microbiota composition and metabolite configuration, as well as the negative control of NAMO on microglial immune response, may enable therapeutic approaches for mitigating HSE and HSV-1 causally linked neurodegenerative disorders.
Acknowledgments
We thank Dr. Daniel J. Klionsky for editing this paper.
Funding Statement
This work was supported by the National Natural Science Foundation of China (No. 81573471, 81872908, 82104238); the Key Laboratory of Virology of Guangzhou (No. 201705030003); the Guangzhou Major program of the Industry-University-Research collaborative innovation (201704030087, 201604020178), and the Basic and Applied Basic Research Foundation of Guangdong Province (2021B1515120088, 2020A151511056601).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Li F, Wang Y, Song X, et al. The intestinal microbial metabolite nicotinamide n-oxide prevents herpes simplex encephalitis via activating mitophagy in microglia. Gut Microbes. 2022;14(1):2096989. [DOI] [PMC free article] [PubMed] [Google Scholar]