

ABSTRACT
Cancer-associated fibroblasts (CAFs) are considered one of the most critical stromal cells that interact with pancreatic ductal adenocarcinoma (PDAC) and promote tumor growth, metastasis, and treatment resistance. Previous studies illustrated macroautophagy/autophagy contributes to CAF activation during tumor progression. Here in our study, we found that autophagy deficiency in CAFs impedes CAF activation by inhibiting proline biosynthesis and collagen production. Furthermore, we uncovered that autophagy promotes proline biosynthesis through mitophagy-mediated regulation of NADK2 (NAD kinase 2, mitochondrial), an enzyme responsible for production of mitochondrial NADP(H). Using an orthotopic mouse model of PDAC, we found that inhibiting mitophagy by targeting PRKN (parkin RBR E3 ubiquitin protein ligase) in the stroma reduced tumor weight. Thus, inhibition of CAFs mitophagy might be an attractive strategy for stroma-focused anti-cancer intervention. Abbreviations: ACTA2/α-SMA: actin alpha 2, smooth muscle, aorta; ACTB/β-actin: actin, beta; ALDH18A1/P5CS: aldehyde dehydrogenase 18 family, member A1; ATG3: autophagy related 3; ATG5: autophagy related 5; BNIP3L: BCL2/adenovirus E1B interacting protein 3-like; CAFs:cancer-associated fibroblasts; COL1A1: collagen, type I, alpha 1; DES: desmin; ECM: extracellular matrix; FABP4: fatty acid binding protein 4, adipocyte; FAP/FAPα: fibroblast activation protein; IHC: immunohistochemical staining; LAMP1: lysosomal-associated membrane protein 1; NADK2: NAD kinase 2, mitochondrial; PC1: pro-collagen 1; PDAC: pancreatic ductal adenocarcinoma; PDGFR: platelet derived growth factor receptor; PDPN: podoplanin; PRKN: parkin RBR E3 ubiquitin protein ligase; PSCs: pancreatic stellate cells; VIM: vimentin; WT: wild-type
KEYWORDS: Autophagy, cancer-associated fibroblasts (CAFs), mitophagy, NADK2, proline biosynthesis
Introduction
Pancreatic cancer (PC) is one of the most malignant tumors which is characterized by its early metastasis and poor prognosis with 5-year survival rate less than 9% [1]. The tumor microenvironment of pancreatic ductal adenocarcinoma (PDAC) is composed of extracellular matrix (ECM) proteins, tumor vasculature, fibroblasts and immune cells [2]. The microenvironment of tumor has long been considered as a contributor to malignant development [3]. Stroma-associated pancreatic stellate cells (PSCs) contribute in cancer progression as they are of great abundance in PDAC and can cause intense fibrotic stromal response [4–6]. Notably, there is a transition from quiescent PSCs to cancer-associated fibroblasts (CAFs), which is a hyper-activated myofibroblast-like state [7] that exhibits increased deposition of collagens [8] and secretes excessive amounts of ECM proteins, cytokines, chemokines, and growth factors to support tumor metabolism [2,9,10]. These activated CAFs are characterized by increased expression of some markers, including ACTA2/αSMA (actin alpha 2, smooth muscle, aorta), FAP/FAPα (fibroblast activation protein), and PDGFR (platelet derived growth factor receptor) [11].
Autophagy, a conserved process of lysosome-dependent protein degradation [12,13], has been reported to regulate the transition process from PSCs to CAFs, during which IL6 and ECM molecules are produced to promote tumor proliferation and metastasis [14]. Similarly, in stroma of squamous cell carcinoma, autophagy causes the downregulation of RBPJ protein which negative control the normal fibroblast conversing to CAFs [15]. Recently, a study using polyoma middle T antigen-driven (PyMT) mammary tumorigenesis model demonstrated that genetic ablation of stromal fibroblast autophagy significantly impedes the stromal desmoplastic response [16].
These findings illustrate that autophagy in stromal fibroblasts contributes to desmoplasia during tumor progression, however, further exploration is still needed to elucidate the mechanism of CAF activation. Upon nutrition starvation, autophagy is required for maintenance of amino acid levels through constitutive turnover of cytopolasmic components [17]. Here in our study, we found that autophagy deficiency in CAFs disrupts collagen production by inhibiting proline biosynthesis. Furthermore, we uncovered that autophagy in CAFs promotes proline biosynthesis through mitophagy, which supports CAF activation and tumor growth in vivo.
Results and discussion
Genetic inhibition of autophagy changes the phenotype of CAFs
It has been reported that autophagy is required for the transition process from PSCs to CAFs [14]. An important question is whether control of fibroblast autophagy and CAF activation can be genetically associated. To validate whether autophagy promotes CAF activation in vivo, we orthotopically transplanted 15,376 T (mouse PDAC, from C57Bl/6 J isogenic tetO_LKRASG12D TRP53/p53 L/+PTF1A/p48-Cre, termed 15,376 T) cells into C57Bl/6 J mice without (Atg5+/+) or with (atg5Δ/Δ) conditional whole-body ATG5 deficiency. (Orthotopic 15,376 T tumors faithfully recapitulated histological and molecular features of the primary tumors, suggesting that the cultured tumor cell line may serve to complement tumor studies in vivo [18].) Consistent with previous reports [4], tumors were significantly smaller when grown in autophagy-deficient hosts compared to Atg5+/+ hosts (Figure 1(a,b)), demonstrating that host autophagy promoted tumor growth. Next, we investigated histological sections of the resected orthotopic tumors. We found that the number of ACTA2/α-SMA- or COL1A1 (collagen, type I, alpha 1)-positive cells was significantly decreased by autophagy deficiency (Figure 1(c-f)). A recent study reported that pancreatic stellate cells (PSCs) show very high expression of the adipocyte marker FABP4 (fatty acid binding protein 4, adipocyte) [19]. By double immunofluorescent staining of FABP4 and ACTA2/α-SMA in wild-type (WT) mice and autophagy-deficient (Ubc-Cre-Esr/ERT2; atg5fl/fl) mice, we found that the decreased expression of α-SMA and induction of FABP4 in autophagy-deficient hosts (Figure 1(g)), indicating that the transition from quiescent PSCs to CAFs is inhibited in autophagy-deficient hosts. Consistently with the in vivo data, both the mRNA and protein levels of FABP4 were increased in both atg3-KO and atg5-KO CAFs (Figure 1(h,i), Figure S1A). It has been reported that the level of collagen is decreased in quiescent PSCs compared with activated CAFs [20,21]. Therefore, we speculated that autophagy inhibition attenuates CAF activation, leading to reduced collagen deposition. In addition, in both WT hosts and autophagy-deficient hosts, CASP3 (caspase 3) was negative in most ACTA2/α-SMA-positive CAFs and FABP4-positive PSCs [19] indicating that loss of autophagy did not affect cell death in the tumor stroma (Figure S1B).
Figure 1.
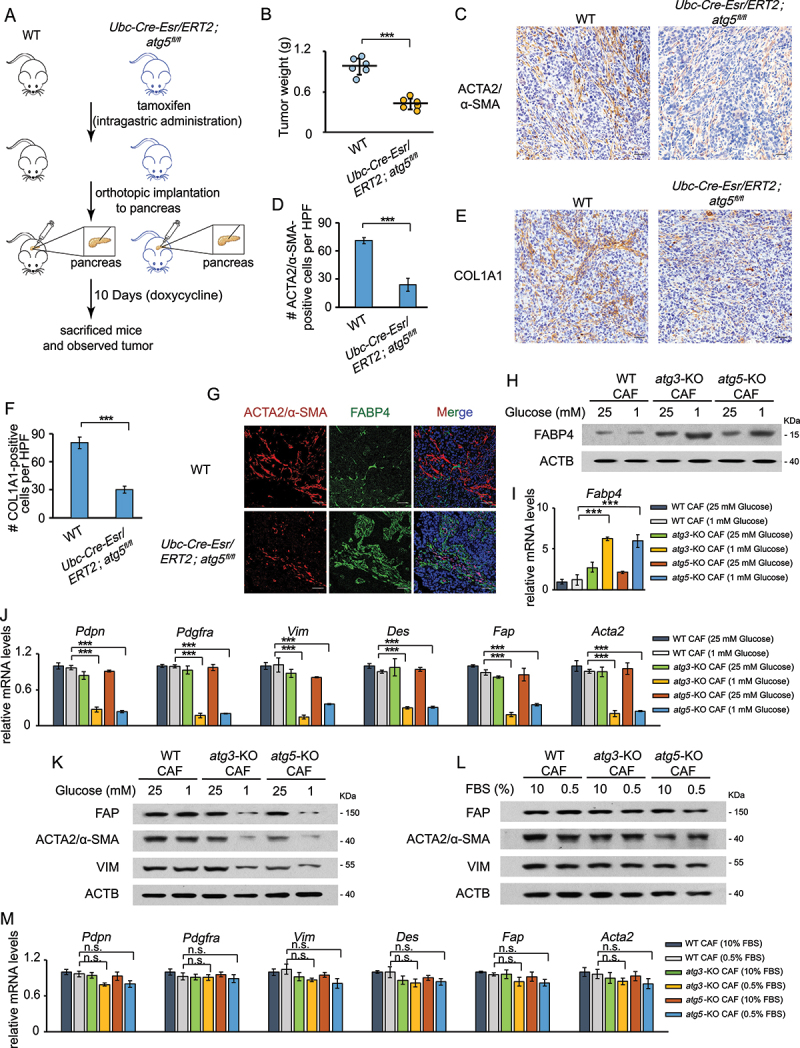
CAF activation depends on autophagy. (a and b) 15,376 T cells were orthotopically injected into WT mice and autophagy-deficient (Ubc-Cre-Esr/ERT2; atg5fl/fl) mice (a). After 10 days, the tumors were weighed (b). (c, d) ACTA2/α-SMA immunohistochemical (IHC) staining in the tumor sections from WT mice and autophagy-deficient (Ubc-Cre-Esr/ERT2; atg5fl/fl) mice after orthotopic implantation of 15,376 T cells. Representative images from n = 6 mice per group are illustrated (C), Scale bars: 20 μm. Quantification of ACTA2-positive cells in WT and Ubc-Cre-Esr/ERT2; atg fl/fl mice (d). HFP = 40x field, n = 15 fields from 6 mice. (e and f) COL1A1 IHC staining in the tumor sections from Atg5+/+or atg5Δ/Δ mice after orthotopically implantation of 15,376 T cells. Representative images from n = 6 mice per group are illustrated (e), Scale bars: 20 μm. Quantification of COL1A1-positive cells n WT and Ubc-Cre-Esr/ERT2; atg5fl/fl mice (f). HFP = 40x field, n = 15 fields from 6 mice. (g) 15,376 T cells were orthotopically injected into WT mice and autophagy-deficient (Ubc-Cre-Esr/ERT2; atg5fl/fl) mice. After 10 days, the tumors were analyzed by FABP4 and ACTA2/α-SMA immunofluorescence staining. Scale bars: 30 μm. (h and i) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. Western blotting was performed to determine FABP4 protein levels (h). RNA was then extracted and RT-PCR was performed to analyze the mRNA level of FABP4 (i). (j) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. RNA was then extracted and RT-PCR was performed to analyze the mRNA level of Pdpn, Pdgfra, Vim, Des, Fap, and Acta2. (k) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. Western blotting was performed to determine VIM, FAP and ACTA2/α-SMA protein levels. (l) WT CAFs and atg3-KO CAFs were cultured in complete media (10% FBS) or low serum media (0.5% FBS) for 48 h. Western blotting was performed to analyze the protein levels of activated CAFs markers. (m) WT CAFs and atg3-KO CAFs were cultured in complete media (10% FBS) or low serum media (0.5% FBS) for 48 h. RNA was then extracted and RT-PCR was performed to analyze the mRNA level of Pdpn, Pdgfra, Vim, Des, Fap, and Acta2.
It is well-known that cancer cells primarily rely on glucose and therefore take up large amount of glucose, leading to a competition with surrounding nonmalignant cells (such as CAFs and immune cells) for glucose [22]. To mimic situations that might occur in vivo, we subjected CAFs to low-glucose treatment in vitro. We then tested the mRNA levels of activated CAFs markers including Pdpn (podoplanin), Pdgfra, Vim (vimentin), Des (desmin), Fap and Acta2. As shown in Figure 1(j), compared to WT CAFs, these markers expressions were further reduced in both atg3-KO and atg5-KO CAFs under low glucose starvation. Consistently, the protein level of VIM, FAP and ACTA2/α-SMA were also reduced in atg3-KO or atg5-KO CAFs (Figure 1(k)). However, no obverse change was detected in response to low serum treatment (Figure 1(l-m)), suggesting that autophagy is critical for CAF activation, particularly under glucose starvation.
Autophagy supports proline biosynthesis and collagen production in CAFs
Tumor stroma generally exhibit increased deposition of collagens compared with quiescent fibroblasts [8]. Therefore we performed Masson’s Trichrome staining in orthotopical samples from WT and autophagy-deficient mice and the results demonstrated that there was less collagen content in autophagy-deficient hosts (Figure 2(a,b)). Consistently, decreased expression of COL1A1 and collagen secretion in atg3-KO CAFs were also observed upon glucose deprivation ((Figure 2(c,d)). Glycine and proline are highly abundant in collagen, comprising approximately 44% of the collagen molecule [23]. As we know, autophagy is an important process of replenishing amino acids when cells faced nutrient-deprived environments [17]. Therefore, we next tested the amino acid levels in WT CAFs and autophagy-deficient CAFs. As shown in Figure 2(e), several amino acids, particularly proline, were lower in atg3-KO CAFs compared with that in WT CAFs under glucose deprivation condition. Interestedly, we also observed that ornithine and citrulline, which are closely associated to proline synthesis, were also declined in autophagy-deficient CAFs after glucose depletion (Figure 2(f–h)). Therefore, we predicted that the decrease of proline induced by autophagy-deficiency may partially result from reduced proline synthesis. To test this hypothesis, we performed [U-13C]-glutamine tracing experiments to assess whether autophagy affects diversion of glutamine carbons into proline biosynthesis (Figure 2(i)). As shown in Figure 2(j), glutamine-derived proline was reduced in atg3-KO CAFs upon glucose deprivation. In addition, the fractions of ornithine and putrescine derived from [U-13C]-glutamine were also decreased in autophagy-deficient CAFs (Figure 2(k,l)). Collectively, these data suggested that autophagy has an impact on proline biosynthesis in CAFs.
Figure 2.
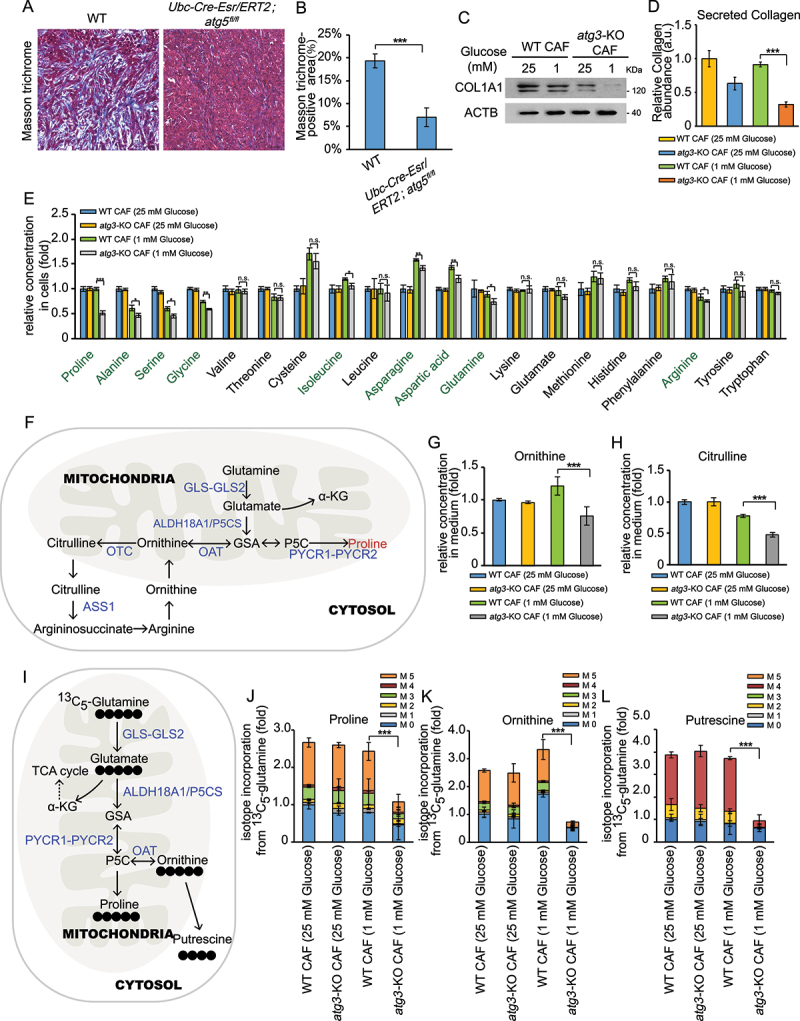
Proline synthesis depends on autophagy in CAFs. (A and B) Masson’s trichrome staining in the tumor sections from Atg5+/+or atg5Δ/Δ mice after orthotopic implantation of 15,376 T cells (a). Quantification of Masson trichrome-positive area in Atg5+/+or atg5Δ/Δ mice (b). (c) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. Western blotting was performed to determine COL1A1 protein levels. (d) Secreted collagen levels quantified by Sirius red staining in extracellular matrix (ECM) derived from WT CAFs and atg3-KO CAFs, cultured for 48 h in complete media (25 mM glucose) or low glucose media (1 mM glucose), in the presence of 50 μM LAA2P. (E) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. The levels of intracellular free amino acids were quantified using mass spectrometry. (f) Schematic of proline biosynthesis pathway. (g and h) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. The levels of ornithine (g) and citrulline (h) were quantified using mass spectrometry. (i) Schematic of proline biosynthesis pathway traced by glutamine labeled with [13C] at all five carbons ([U-13C] glutamine). (j and l) WT CAFs and atg3-KO CAFs were cultured in DMEM (no glutamine) supplemented with 2 mM [U-13C] glutamine for 24 h. The levels of labeled amino acids were quantified using mass spectrometry.
Autophagy deficiency reduces proline biosynthesis by repressing NADK2
We next investigate how autophagy influences proline biosynthesis in CAFs. Recent studies have shown that mitochondrial NADPH pool is essential to enable proline biosynthesis [24,25]. Therefore, we examined the role of autophagy in regulating NADPH levels in whole cells and immunopurified mitochondria in CAFs. As shown in Figure 3(a,b), although the total NADP(H) abundance was slightly changed at whole cell level upon autophagy deficiency, mitochondrial NADP(H) abundance was obviously reduced in atg3-KO CAFs. Moreover, NADK2, an enzyme responsible for production of mitochondrial NADP(H) and proline biosynthetic [24,25] (Figure 3(c)), was decreased in autophagy-deficient CAFs after glucose depletion (Figure 3(d)).
Figure 3.
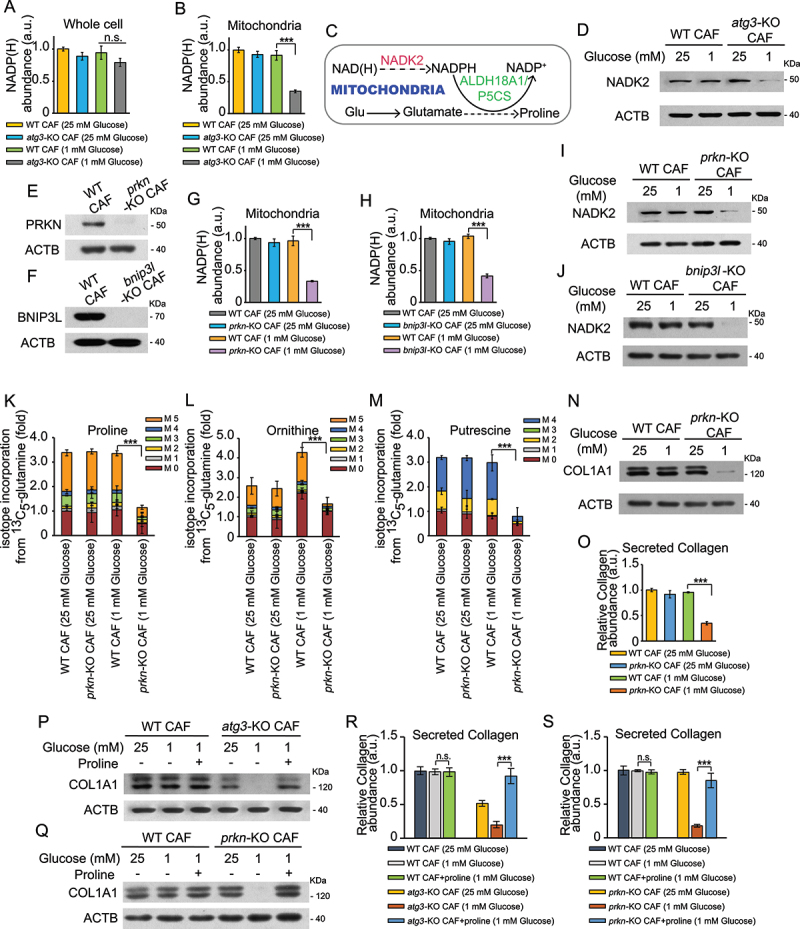
Autophagy deficiency reduces NADP(h) poor by repressing NADK2 in CAFs. (a and b) Measurement of total NADP(H) abundance in whole cells (a) or immunopurified mitochondria (b) of WT CAFs and atg3-KO CAFs by culturing with complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. (c) Schematic of the role of NADK2 controlling proline biosynthesis in the mitochondria. (d) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. Western blotting was performed to determine NADK2 protein levels. (e and f) Western blotting was performed to determine PRKN (e) and BNIP3L (f) protein levels. (g) Measurement of total NADP(H) abundance in immunopurified mitochondria of WT CAFs and prkn-KO CAFs by culturing with complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. (h) Measurement of total NADP(H) abundance in immunopurified mitochondria of WT CAFs and bnip3l-KO CAFs by culturing with complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. (I) WT CAFs and prkn-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. Western blotting was performed to determine NADK2 protein levels. (J) WT CAFs and bnip3l-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. Western blotting was performed to determine NADK2 protein levels. (k and m) WT CAFs and prkn-KO CAFs were cultured in DMEM (no glutamine) supplemented with 2 mM [U-13C] glutamine for 24 h. The levels of labeled amino acids were quantified using mass spectrometry. (n) WT CAFs and PRKN-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. Western blotting was performed to determine COL1A1 protein levels. (o) Secreted collagen levels quantified by Sirius red staining in ECM derived from WT CAFs and prkn-KO CAFs, cultured for 48 h in complete media (25 mM glucose) or low glucose media (1 mM glucose), in the presence of 50 μM LAA2P. (p) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) with or without proline (300 μM each) for 24 h. Western blotting was performed to determine COL1A1 protein level. (q) WT CAFs and atg3-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) with or without proline (300 μM each) for 24 h. Western blotting was performed to determine COL1A1 protein level. (r) Secreted collagen levels quantified by Sirius red staining in ECM derived from WT CAFs and atg3-KO CAFs, cultured for 48 h in complete media (25 mM glucose) or low glucose media (1 mM glucose) supplemented with or without 300 μM proline), in the presence of 50 μM LAA2P. (s) Secreted collagen levels quantified by Sirius red staining in ECM derived from WT CAFs and prkn-KO CAFs, cultured for 48 h in complete media (25 mM glucose) or low glucose media (1 mM glucose) supplemented with or without 300 μM proline), in the presence of 50 μM LAA2P.
Mitophagy is a selective form of autophagy targeting dysfunctional or redundant mitochondria for degradation [26], we then test whether mitophagy is involved in regulating mitochondrial NADP(H) abundance. We generated mitophagy-deficient CAFs by using CRISPR-Cas9 technology to knock-out PRKN or BNIP3L (BCL2/adenovirus E1B interacting protein 3-like), which are essential for mitophagy [27]. Consistently, mitochondrial NADP(H) abundance and NADK2 level were also reduced in mitophagy-deficient CAFs under glucose depletion (Figure 3(e-j)). These results demonstrated that autophagy deficiency reduces mitochondrial NADPH poor in a mitophagy-dependent manner.
Since mitochondrial NADPH poor is essential for proline biosynthesis [24,25], we next performed [U-13C]-glutamine tracing experiments to assess whether mitophagy affects the diversion of glutamine carbons into proline biosynthesis. As shown in Figure 3(k-m), glutamine-derived proline, ornithine, and putrescine were reduced in prkn-KO CAFs upon glucose deprivation, suggesting that mitophagy has an impact on proline biosynthesis in CAFs. In addition, we found that both the protein level of COL1A1 and collagen secretion were also decreased in prkn-KO CAFs after glucose depletion (Figure 3(n-o)), indicating that loss of mitophagy could lead to collagen secretion deficiency. Previous work demonstrated that stroma derived from autophagy-deficient CAFs exhibited reduced levels of multiple immune and inflammatory populations in addition to reduced collagen deposition [16]. Consistently, IHC staining revealed that ADGRE1/F4/80-, CD3-, and CD4-positive cells were decreased in autophagy-deficient (Ubc-Cre-Esr/ERT2; atg5fl/fl) mice compared with WT mice (Figure S2A,B), suggesting that stroma autophagy influences the recruitment of certain immune cell populations in vivo. However, no obverse change was detected in the mitophagy-deficient (prkn−/−) hosts (Figure S2A,B).
To determine whether proline played a role in autophagy or mitophagy deficiency-reduced collagen production, we cultured atg3-KO and prkn-KO CAFs in the low-glucose media with or without exogenous proline. CAFs lacking ATG3 or PRKN showed decreased COL1A1 expression and collagen secretion, which were rescued by proline supplementation to the low-glucose medium (Figure 3(p-s)). NADK2 or ALDH18A1/P5CS (aldehyde dehydrogenase 18 family, member A1) are two important enzymes in the mitochondrial proline biosynthetic pathway [24,25,28]. To further validate the role of proline, we stably knocked out NADK2 or ALDH18A1/P5CS in CAFs (Figure 4(a,b)). Cell viability was not decreased in nadk2-KO or aldh18a1/p5cs-KO CAFs compared with WT CAFs (Figure S2C), indicating that reduced proline could not cause cell death. Next, we orthotopically implanted PDAC cells with the same amount of WT CAFs, nadk2-KO CAFs or aldh18a1/p5cs-KO CAFs into the pancreas of C57Bl/6 J mice. As shown in Figure 4(c-f), both desmoplastic reaction and COL1A1-positive cell populations were reduced in tumor samples which were co-injected with nadk2-KO or aldh18a1/p5cs-KO CAFs. Consistent with the in vivo results, the protein level of COL1A1 and collagen secretion was also reduced in nadk2-KO or aldh18a1/p5cs-KO CAFs, further demonstrating that proline synthesis plays a critical role in collagen production (Figure 4(g-h)). We next tested whether impaired synthesis of proline in CAFs influenced tumor cell proliferation. As shown in Figure 4(i-j), tumor-promoting activity of CM from CAFs was impaired when proline biosynthesis was inhibited upon glucose depletion. Then we tested the amino acid levels in CAFs CM and found that, besides proline, alanine was also declined in nadk2-KO or aldh18a1/p5cs-KO CAFs after glucose depletion (Figure 4(k-l)), consistent with a previous report [4]. Therefore, we speculated that impaired proline synthesis has an impact on alanine secretion from CAFs, which might result from CAFs inactivation.
Figure 4.
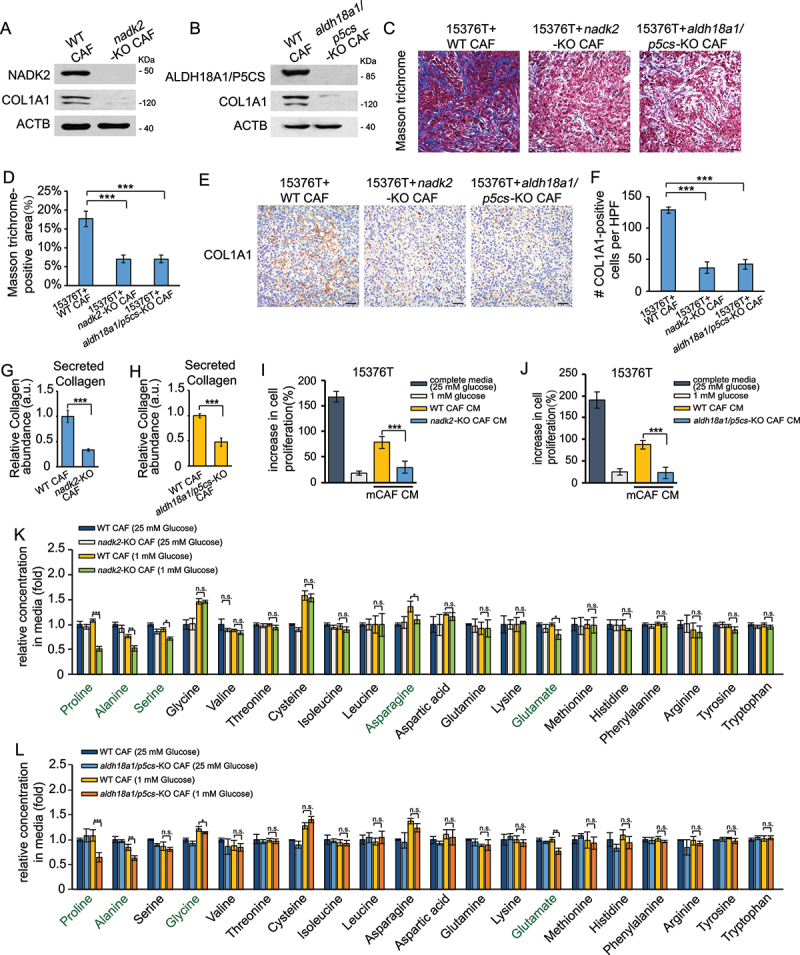
The effect of proline synthesis on collagen production. (a and b) Western blotting was performed to determine COL1A1 protein levels in WT CAFs, nadk2-KO CAFs (a) and aldh18a1/p5cs-KO CAFs (b) cultured for 24 h in complete media (25 mM glucose). (c and d) 15,376 T with nadk2-KO CAFs (1:1), WT CAFs (1:1) or aldh18a1/p5cs-KO CAFs (1:1) were orthotopically injected into C57BL/6 J mice. After 10 days, the tumors were removed and analyzed by Masson’s trichrome staining (c). Scale bars: 20 μm. Quantification of Masson trichrome-positive area in tumor samples which were co-injected with WT, nadk2-KO or aldh18a1/p5cs-KO CAFs (d). (E and F) 15,376 T with nadk2-KO CAFs (1:1), WT CAFs (1:1) or aldh18a1/p5cs-KO CAFs (1:1) were orthotopically injected into C57BL/6 J mice. After 10 days, the tumors were removed and analyzed by COL1A1 immunohistochemical staining (e). Scale bars: 20 μm. Quantification of COL1A1-positive cells in tumor samples which were co-injected with WT, nadk2-KO or aldh18a1/p5cs-KO CAFs (f). HFP = 40x field, n = 15 fields from 6 mice. (g) Secreted collagen levels quantified by Sirius red staining in ECM derived from WT CAFs and nadk2-KO CAFs cultured for 48 h in complete media, in the presence of 50 μM LAA2P. (h) Secreted collagen levels quantified by Sirius red staining in ECM derived from WT CAFs and aldh18a1/p5cs-KO CAFs cultured for 48 h in complete media, in the presence of 50 μM LAA2P. (i) The cells were counted to calculate the cell proliferation. 15,376 T cells were incubated in complete media or conditioned medium from different cell lines for 24 h. The conditioned medium was collected from the nadk2-KO CAFs with 70%-80% confluences cultured in low glucose media (1 mM glucose) or complete media (25 mM glucose) for 24 h. (j) The cells were counted to calculate the cell proliferation. 15,376 T cells were incubated in complete media or conditioned medium from different cell lines for 24 h. The conditioned medium was collected from the aldh18a1/p5cs-KO CAFs with 70%-80% confluences cultured in low glucose media (1 mM glucose) or complete media (25 mM glucose) for 24 h. (k) WT CAFs and nadk2-KO CAFs (Nadk2 knocked-out) were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. The levels of intracellular free amino acids were quantified using mass spectrometry. (l) WT CAFs and aldh18a1/p5cs-KO CAFs (Nadk2 knocked out) were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. The levels of intracellular free amino acids were quantified using mass spectrometry.
Mitophagy is required to maintain the CAFs phenotype
Next, we investigated whether mitophagy deficiency changes the phenotype of CAFs. As shown in Figure 5(a,b), the mRNA levels of activated CAFs markers including Pdpn, Pdgfra, Vim, Des, Fap, Acta2 were reduced in prkn-KO and bnip3l-KO mCAFs upon low-glucose depletion. To further confirm the effect of mitophagy on CAF activation in vivo, we orthotopically transplanted 15,376 T cells into WT or mitophagy-deficient prkn−/− mice. Consistently with the in vitro data, there was less collagen content in mitophagy-deficient hosts (Figure 5(c,d)). In addition, both ACTA2/α-SMA-positive and COL1A1-positive cell populations were reduced in prkn-KO mice, demonstrating that mitophagy in stromal promoted CAF activation (Figure 5(e-h)). We then tested the effect of mitophagy-deficient CAFs on supporting PDAC growth under glucose starvation. As shown in Figure 5(i,j), tumor-promoting activity of conditioned medium from CAFs was impaired when mitophagy was inhibited under glucose starvation. Moreover, in the orthotopic model of PDAC, tumors were significantly smaller when grown in prkn−/− mice compared to Prkn+/+ mice (Figure 5(k)), which highlights the importance of stromal mitophagy in PDAC.
Figure 5.
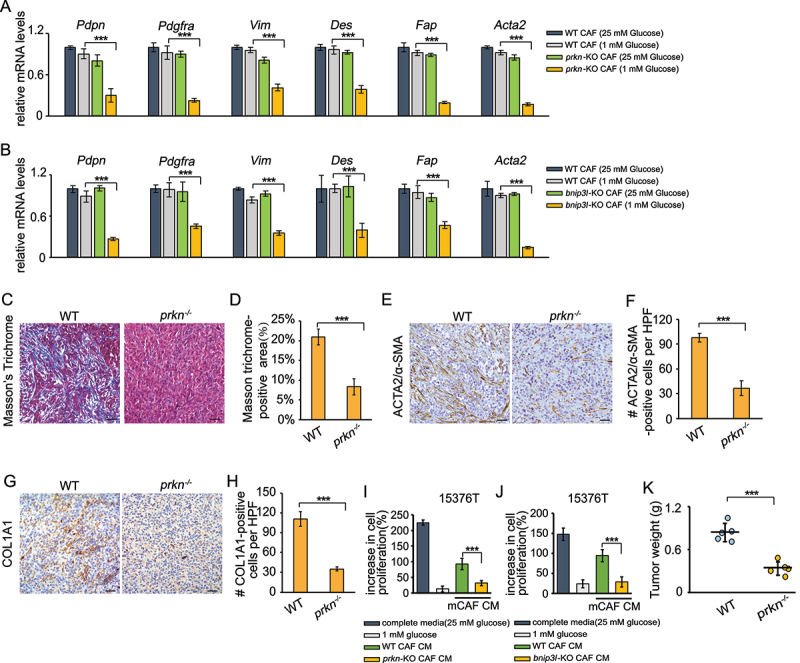
Mitophagy is required to maintain the CAFs phenotype. (a) WT CAFs and prkn-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. RNA was then extracted and RT-PCR was performed to analyze analyzed the mRNA level of Pdpn, Pdgfra, Vim, Des, Fap, and Acta2. (b) WT CAFs and bnip3l-KO CAFs were cultured in complete media (25 mM glucose) or low glucose media (1 mM glucose) for 24 h. RNA was then extracted and RT-PCR was performed to analyze analyzed the mRNA level of Pdpn, Pdgfra, Vim, Des, Fap, and Acta2. (c and d) Masson’s trichrome staining in the tumor sections from WT and prkn−/− mice after orthotopically implantation of 15,376 T cells (c). Quantification of Masson trichrome-positive area in WT and prkn−/− mice (d). (e and f) ACTA2/α-SMA immunohistochemical staining in the tumor sections from WT and prkn−/− mice after orthotopically implantation of 15,376 T cells. Representative images from n = 5 mice per group are illustrated (E). Quantification of ACTA2/α-SMA-positive cells in WT and prkn−/− mice (F). HFP = 40x field, n = 15 fields from 5 mice. (g and h) COL1A1 immunohistochemical staining in the tumor sections from WT and prkn−/− mice after orthotopically implantation of 15,376 T cells. Representative images from n = 5 mice per group are illustrated (g). Quantification of COL1A1-positive cells in WT and prkn−/− mice (h). HFP = 40x field, n = 15 fields from 5 mice. (i) 15,376 T was cultured in conditioned medium (1 mM glucose) from WT CAFs and nadk2-KO CAFs for 24 h. The cells were counted to calculate the cell proliferation. (j) 15,376 T was cultured in conditioned medium (1 mM glucose) from WT CAFs and aldh18a1/p5cs-KO CAFs for 24 h. The cells were counted to calculate the cell proliferation. (k) 15,376 T cells were orthotopically injected into WT and prkn−/− mice. After 10 days, the tumors were weighed.
In summary, the findings we present in this report provide mechanism insights into the role of autophagy in CAF activation. It has been reported that impaired collagen secretion in autophagy-deficient cells due to defects in procollagen degradation via the lysosome [16,29,30]. Consistently, our data indicated that autophagy, but not mitophagy, is required for the pro-collagen 1 (PC1) degradation in CAFs (Figure S3A,B). As shown in Figure S3A, B, LAMP1-PC1 colocalization was increased in prkn-KO CAFs upon treatment with CHQ (at 25 mM glucose). However, the colocalization was not increased in prkn-KO CAFs upon CHQ treatment after glucose starvation (at 1 mM glucose) (Figure S3C-E). Especially, our study showed that mitophagy in CAFs could promote collagen secretion by regulating the synthesis of proline, but not degradation of procollagen (Figure S3C-E). Therefore, our findings uncover a novel pathway that autophagy in CAFs has an impact on desmoplasia by influencing proline biosynthesis.
Materials and methods
Transgenic mice cell line. Primary mouse PDAC lines 15,376 T used in this study was isolated as described [31] from genetically engineered C57BL/6 mice [18] (tetO_LKRASG12D TRP53/p53 L/+PTF1A/p48-Cre), and maintained in DMEM medium containing 10% FBS and 1 μg/mL doxycycline.
Mouse models. prkn KO mice (prkn−/−) were purchased from Cyagen Biosciences (Suzhou, China). The nuclear background of the mice was C57BL/6 N. Heterozygous mice were then interbred to obtain homozygous WT control and KO mice. Mice carrying two conditional alleles of ATG5 (Atg5fl/fl) were crossbred with UBC-Cre Esr/ERT2 mice (Model Organisms Center, Shanghai). Cre-mediated recombination was induced with intragastric administration of tamoxifen in corn oil (2 mg/day for 5 days). Control groups were intragastric administrated with corn oil. All genotyping was done by PCR.
Cell culture. All cell lines were grown in DMEM (MACGENE, CM15023) supplemented with 10% (vol:vol) fetal bovine serum and the appropriate amount of penicillin-streptomycin in a 37°C incubator with a humidified, 5% CO2 atmosphere. CAFs were generated from C57BL/6 J harboring mouse PDAC. These animals were pre-treated with doxycycline (Selleck, S5159) drinking water (2 g/L) and kept in doxycycline regimen for the duration of the experiment, and then were injected with10515376T cells into the pancreas. Pancreatic tumors were extracted at 10 days, digested in collagenase and dispase (Roche, 10,269,638,001) and mechanically minced. Neoplastic tissues were dissociated at 37°C for 1 h. Cells were collected in cell culture dishes in DMEM with 10% FBS in the absence of doxycycline to limit the growth of iKRAS mouse PDAC cells. After 24 h, mCAFs were immortalized by transfection with SV40 (Addgene, 22,298; deposited by Dr. Eric Campeau) retro-viruses.
CAFs were verified by measuring ACTA2 expression.
Antibodies and reagents. The antibodies used were anti-ATG3 (Medical & Biological Laboratories, M133-3), anti-ACTB/ACTIN (Santa Cruz Biotechnology, sc-7210), anti-ATG5 (Novus, NB110-53,818), anti-MAP1LC3B/LC3 for western blot (Cell Signaling Technology, 2775S), anti-NADK2 (Abcam, ab181028), anti-BNIP3L (Proteintech, 12,986-1-AP), anti-PRKN/PARKIN (Cell Signaling Technology, 2132S), anti-ACTA2/α-SMA for IHC and IF (GeneTech, GM085129), anti- ACTA2/α-SMA for western blot (Proteintech, 55,135-1-AP), anti-COL1A1 (Proteintech, 67,288-1-Ig), anti-FAP (Biodragon, BD-PT6136), anti-VIM/vimentin (Santa Cruz Biotechnology, sc-6260), anti-CASP3 (caspase 3) (Santa Cruz Biotechnology, sc-7148), anti-CD3 (Proteintech, 12,986-1-Ig), anti-CD4 (Biodragon, BD-PM3070), anti-ADGRE1/F4/80 (Proteintech, 28,463-1-Ig), anti-FABP4 (Proteintech, 15,872-1-AP), anti-LAMP1 (lysosomal-associated membrane protein 1) for IF (Sino Biological, 11,215-MM07), anti-Procollagen 1 (Bioss, bs-4482 R), anti-ALDH18A1/P5CS (Proteintech, 17,719-1-AP).
Doxycycline (S5159) and E-64 (S7379) were purchased from Selleck. Stable isotope-labeled glutamine, 13C5-glutamine (CLM-1822-H-0.1) was produced by Cambridge Isotope Laboratories. Gelatin (0.1% gelatin; CC052) was purchased from M&C GENE TECHNOLOGY, L-Proline (HY-Y0252) and L-ascorbic acid 2-phosphate triodium (LAA2P; HY-107837) were purchased from MedChemExpress (MCE), chloroquine (CHQ, C6628) was purchased from Sigma-Aldrich.
Western Blotting. Cells were lysed in lysis buffer supplemented with protease inhibitor (Roche, 4,693,132,001) and NP40 on ice. Equal amounts of proteins (20–40 μg) were separated by 6–15% SDS polyacrylamide gel electrophoresis. Equal loading of proteins was confirmed by quantification of ACTB/β-actin.
Gene expression analysis by real-time PCR (qPCR). Total RNA was isolated with TRIzol (Applygen, 1030) and reverse transcripted with 2 μg. Relative gene expression was determined by realtime PCR using an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, CA, USA), per the manufacturer’s recommended instructions. Each analysis was performed in triplicate. Sequences for qPCR primers are as follows: Actb (mouse): ctaaggccaaccgtgaaaag, accagaggcatacagggaca; Rna5s (mouse): ggccataccaccctgaacgc, cagcacccggtattcccagg; Pdpn (mouse): accgtgccagtgttgttctg, agcacctgtggttgttattttgt; Pdgfra (mouse): agagttacacgtttgagctgtc, gtccctccacggtactcct; Vim (mouse): cgtccacacgcacctacag, gggggatgaggaatagaggct; Des (mouse): gtggatgcagccactctagc, ttagccgcgatggtctcatac; Fap (mouse): gtcacctgatcggcaatttgt, ccccattctgaaggtcgtagat; Acta2 (mouse): gtcccagacatcagggagtaa, tcggatacttcagcgtcagga.
Generation of CRISPR-Cas9 knockout (KO) cell lines. The Cas9 knockout CAFs cell lines were established using CRISPR-Cas9 methods. We used the lentiCRISPRv2 puro vector purchased from Addgene (98,290; deposited by Dr. Brett Stringer). The sgRNA was designed by online software (http://crispr.mit.edu). Lentiviruses were produced using pMDLg/pRRE (Addgene, 12,251; deposited by Dr. Didier Trono), pRSV-REV (Addgene, 12,253; deposited by Dr. Didier Trono), and pCMV-VSV-G (Addgene, 8454; deposited by Dr. Bob Weinberg) vectors together with lentiCRISPRv2. These plasmids were transfected into 293 T cells and viruses were collected 48 h after transfection by filtering through 0.45-μm Steriflip filter (Millipore, SLHV033 N) and concentrated. Cells were infected with the complete medium containing corresponding virus and 4 μg/ml polybrene (MACGENE, MC032). After 48 h infection, the puromycin selection (2.5 μg/mL) started and was continued for two weeks to generate stable cell pools. The sgRNA sequences are as follows: Atg3 (mouse): sense: CACCGAATGTGATCAACACGGTGAA; sense:AAACTTCACCGTGTTGATCACATTC; Atg5 (mouse): sense:CACCGAAGAGTCAGCTATTTGACGT; antisense: AAACACGTCAAATAGCTGACTCTTC; Prkn (mouse): sense: CACCGACATAGTACAGAGACCACGG; antisense: AAACCCGTGGTCTCTGTACTATGTC; Bnip3l (mouse): sense: CACCGTCGTCCAACAGGTTCCTGGG; antisense: AAACCCCAGGAACCTGTTGGACGAC; Aldh18a1/P5cs (mouse): sense:CACCGTGGACTGATGGCCTTGTACG; antisense: AAACCGTACAAGGCCATCAGTCCAC; Nadk2 (mouse): sense: CACCGCTGCTCGAACTCGTAGCGGG; antisense: AAACCCCGCTACGAGTTCGAGCAGC.
Cell proliferation assay. Cells were seeded in 12-well plates at a density of 5 × 104 cells/well and treated with different treated medium after culturing for 24 h. Cell proliferation was evaluated after 24 h, the cells were collected, diluted with a trypan blue working solution and counted.
Immunofluorescence analysis. Cells were cultured in confocal plates. After appropriate treatment, cells were fixed with 4% paraformaldehyde and permeabilized with methanol. The confocal plates were incubated with blocking solution (0.8% BSA [DingGuo, BF-0011] in PBS [HARVEYBIO, HZB1859]) for 1 h and exposed overnight to primary antibody (1:50 dilution for all antibodies) at 4°C. After incubation for 2 h with secondary antibodies (ZSGB-BIO, ZB-2305, ZB-2301), samples were incubated with DAPI. Immunofluorescent images were obtained using confocal microscope (FV1000-IX81).
De-paraffined sections with 3-μm thickness were repaired by microwave. The sections were then treated with 0.5% Triton X-100 (Solarbio, T8200) for 30 min and blocked with goat serum (ZSGB-BIO, ZLI-9022) for 1 h at room temperature. Washing with PBS 3 times between each step. The sections were incubated with antibodies, overnight at 4°C in a humidified chamber, followed by incubation with secondary antibodies and DAPI. Anti-Fade Mounting Medium (Solarbio, S2100) was used to seal the sections. Immunofluorescent images were obtained using confocal microscope.
Cell death. Post-treatment cell viability was determined by staining with propidium iodide (PI, 2 μg/ml; MedChemExpress, HY-D0815) and flow cytometric analysis on a FACScan.
Rapid isolation of mitochondria. Rapid isolation of mitochondria was performed following the published methodology [32]. WT cells or cells with atg3 or prkn or bnip3l knockout were engineered to express the HA-tagged SYNJ2BP/OMP25 protein. 3XHA-EGFP-SYNJ2BP/OMP25 was amplified and cloned into pcDNA 3.1. For transfecting CAFs, Neon transfection system (Invitrogen, MPK5000) was used following the manufacturer’s instructions. Cells were washed on ice and dounce homogenized in KPBS (136 mM KCl, 10 mM KH2PO4, pH 7.25). The homogenate was then centrifuged at 1000 g for 2 min at 4°C and the supernatant from the centrifuged sample was applied to anti-HA beads (Thermo Fisher Scientific, 88,837) and incubated with end-over-end rotator for 8 h. The resultant beads were then washed with KPBS. Then 80:20 methanol:water was used to elute mitochondria for NADP(H) measurements.
Measurement of NADP(H). NADP(H) measurements were performed using an NADP+/NADPH Assay Kit (Beyotime, S0179), with modifications as described in published study [33]. Briefly, metabolites from whole cells and isolation of mitochondria samples were extracted with 80:20 methanol:water. Then the metabolites was dried down in a SpeedVac concentrator for 2 h. Metabolites were then resuspended with NADPH extraction buffer and centrifuged for 5 min at 600 g. The supernatant was split in half after centrifued. One half was kept on ice for 30 min, the other half was exacted to 60°C incubation for 30 min to decompose NADP+. 100 μL of G6PDH mix (98 uL buffer and 2 μL G6PDH enzyme mix from the manufacturer) was added to each sample and incubated for 10 min at 37°C. 10 μL of manufacturer’s NADPH developer was then added into each well and incubated for one hour at 37°C. Values were recorded with a microplate reader at 450 nm. The amount of NADPH was calculated from the standard curves and normalized for cell counts.
Extracellular matrix (ECM) extraction and collagen staining. ECM extraction and collagen staining were performed as previously described [28]. In brief, confluent CAFs were grown for two days on plates coated with 0.1% gelatin in the presence of 50 μM L-ascorbic acid 2-phosphate in the different media. Plates were decellularized with 0.5% Triton X-100 for 5 min on a rotating platform. Three times the volume of PBS was added, and ECM was equilibrated overnight at 4°C. To measure collagen abundance, extracted ECM was stained with picrosirius red (Freemore, D00041). The stain was extracted with 0.1 M NaOH, and collagen staining was measured at 550 nm using a microplate reader. Values were normalized for cell counts.
Xenografts. All experimental uses of mice in this study were housed and supplied by the Experimental Animal Center of Peking University Health Science Center (Beijing, China). Five-to-six-week-old C57BL/6 J mice were used for orthotopic tumorigenesis. These animals were given doxycycline-containing diet and kept in doxycycline regimen for the duration of the experiment for induction of tumor development. Mice were injected with10515376T cells into the pancreas. Briefly, mice were fixed in a supine position and performed an abdominal incision. The spleen was identified and gently ripped out through the incision to expose the pancreas. Twenty μL of 15,376 T cells were injected into the pancreas. The injection site was clamped for about 30s after removing the syringe. The tissues were put back carefully in its original position and the wound was sutured with absorbable sutures. Ten days after inoculation, the mice were sacrificed when the tumors have grown. Then the tumor volume was measured with a vernier caliper. The drug was injected intraperitoneally, starting from the fifth day. All animal experiments were performed in accordance with a protocol approved by the Department of Laboratory Animal Science of Peking University Health Science Center and supervised by the institutional review board of Peking University.
Metabolite extraction and stable isotope labeling. Cells were washed with ice-cold PBS, quenched with 80% methanol and subjected to three rapid freeze-thaw cycles. The cell debris was removed by centrifugation (12,000 g, 15 min) at 4°C. After sample normalization based on total protein levels, the supernatant containing aqueous metabolites was collected and evaporated to dryness using a SpeedVac concentrator. For glutamine tracing experiments, cells were seed in 10-well plates and cultured for 24 h in DMEM supplemented with 10% FBS. And then medium was replaced with DMEM without l-glutamine and glucose (Gibco, A1443001) supplemented with 2 mM [U-13C] l-glutamine (Cambridge Isotope Laboratories, CLM-1822-H-0.1), 10% dialyzed FBS and 25 mM glucose or 1 mM glucose. After 24 h, metabolism was quenched and metabolites were extracted using the above method.
Immunohistochemical (IHC) staining. For IHC analysis, 3 mm sample sections were deparaffinized, rehydrated and then blocked endogenous peroxidases by incubation in 3% H2O2 for 10 min. After retrieving antigen with citric acid buffer (pH6.0), sections were incubated with anti-ACTA2/α-SMA (1:100), anti-COL1A1 (1:100), respectively, overnight at 4°C. Tissue sections followed by incubation with the HRP-conjugated secondary antibodies (ZSGB-BIO, PV-6000) at 37°C for 30 min. Staining was completed by 1–2 min incubation with diaminobenzidine (DAB) substrate (ZSGB-BIO, ZLI-9018). Counterstaining was conducted for 2 min using hematoxylin. The tissue sections were visualized using a BX53 upright microscope (Olympus) and the mean staining intensity was calculated by Image-Pro Plus software.
Masson’s trichrome staining. Masson’s trichrome staining was used to detect collagen distribution. De-paraffined sections with 5-μm thickness were stained according to instructions (Applygen, B1130). Images were taken under upright microscopy for analysis.
Statistical analysis. Statistical analysis was performed by using the SPSS statistical software package (standard version 22; SPSS Inc., Chicago, IL, USA). For two groups statistical analyses, unpaired Student’s t-test was used. Error bars represent standard error of standard deviation (SD), as indicated in the legends. n.s.: non-significant, *: p < 0.05, **: p < 0.01, ***: p < 0.001.
Supplementary Material
Funding Statement
This study was supported by the National Key R&D Program of China (2019YFC1005200, 2017YFA0503900), National Natural Science Foundation of China (81874145, 82173020 and 82073057), Peking University (PKU2020LCXQ024, BMU2022XKQ004), Beijing Natural Science Foundation (7,202,228; Clinical Medicine plus X Project of Peking UniversityClinical Medicine plus X Project of Peking University [PKU2020LCXQ024]
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2093026
References
- [1].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2020. CA Cancer J Clin. 2020. Jan;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- [2].Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020. Aug;17(8):487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Katheder NS, Khezri R, O’Farrell F, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017 Jan 19 541(7637):417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016 Aug 25 536(7617):479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Feig C, Gopinathan A, Neesse A, et al. The pancreas cancer microenvironment. Clin Cancer Res. 2012 Aug 15 18(16):4266–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kamphorst JJ, Nofal M, Commisso C, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015 Feb 1 75(3):544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Calvo F, Ege N, Grande-Garcia A, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013. Jun;15(6):637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016 Aug 23; 16(9):582–598. [DOI] [PubMed] [Google Scholar]
- [9].Pereira BA, Vennin C, Papanicolaou M, et al. CAF subpopulations: a new reservoir of stromal targets in pancreatic cancer. Trends Cancer. 2019. Nov;5(11):724–741. [DOI] [PubMed] [Google Scholar]
- [10].Neuzillet C, Tijeras-Raballand A, Ragulan C, et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J Pathol. 2019. May;248(1):51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Whittle MC, Hingorani SR. Fibroblasts in pancreatic ductal adenocarcinoma: biological mechanisms and therapeutic targets. Gastroenterology. 2019. May;156(7):2085–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007. Nov;8(11):931–937. [DOI] [PubMed] [Google Scholar]
- [13].Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol. 2011 Aug 3. 10.1101/sqb.2011.76.011023 [DOI] [PubMed] [Google Scholar]
- [14].Endo S, Nakata K, Ohuchida K, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017. May;152(6):1492–1506 e24. [DOI] [PubMed] [Google Scholar]
- [15].Goruppi S, Jo SH, Laszlo C, et al. Autophagy controls CSL/RBPJ kappa stability through a p62/SQSTM1-Dependent mechanism. Cell Rep. 2018 Sep 18 24(12):3108–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rudnick JA, Monkkonen T, Mar FA, et al. Autophagy in stromal fibroblasts promotes tumor desmoplasia and mammary tumorigenesis. Genes Dev. 2021 Jul 1 35(13–14):963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 2010. Sep;21(7):683–690. [DOI] [PubMed] [Google Scholar]
- [18].Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012 Apr 27 149(3):656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Helms EJ, Berry MW, Chaw RC, et al. Mesenchymal lineage heterogeneity underlies nonredundant functions of pancreatic cancer-associated fibroblasts. Cancer Discov. 2022. Feb;12(2):484–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sherman MH, Yu RT, Engle DD, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014 Sep 25 159(1):80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ohlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017 Mar 6 214(3):579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yan L, Raj P, Yao W, et al. Glucose metabolism in pancreatic cancer. Cancers (Basel). 2019 Sep 29;11(10). 10.3390/cancers11101460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li P, Wu G. Roles of dietary glycine, proline, and hydroxyproline in collagen synthesis and animal growth. Amino Acids. 2018. Jan;50(1):29–38. [DOI] [PubMed] [Google Scholar]
- [24].Zhu J, Schworer S, Berisa M, et al. Mitochondrial NADP(H) generation is essential for proline biosynthesis. Science. 2021 May 28 372(6545):968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tran DH, Kesavan R, Rion H, et al. Mitochondrial NADP(+) is essential for proline biosynthesis during cell growth. Nat Metab. 2021. Apr;3(4):571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011. Mar;7(3):297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Springer MZ, Macleod KF. In brief: mitophagy: mechanisms and role in human disease. J Pathol. 2016. Nov;240(3):253–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schworer S, Berisa M, Violante S, et al. Proline biosynthesis is a vent for TGFbeta-induced mitochondrial redox stress. EMBO J. 2020 Apr 15 39(8):e103334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cinque L, Forrester A, Bartolomeo R, et al. FGF signalling regulates bone growth through autophagy. Nature. 2015 Dec 10 528(7581):272–275. [DOI] [PubMed] [Google Scholar]
- [30].Forrester A, De Leonibus C, Grumati P, et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019 Jan 15;38(2). 10.15252/embj.201899847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003 Dec 15 17(24):3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen WW, Freinkman E, Sabatini DM. Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat Protoc. 2017. Oct;12(10):2215–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hoxhaj G, Ben-Sahra I, Lockwood SE, et al. Direct stimulation of NADP(+) synthesis through Akt-mediated phosphorylation of NAD kinase. Science. 2019 Mar 8 363(6431):1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.