

Abstract
Messenger RNA-based therapeutics have shown tremendous potential, as demonstrated by the rapid development of messenger RNA based vaccines for COVID-19. Nevertheless, distribution of mRNA vaccines worldwide has been hampered by mRNA’s inherent thermal instability due to in-line hydrolysis, a chemical degradation reaction. Therefore, predicting and understanding RNA degradation is a crucial and urgent task. Here we present RNAdegformer, an effective and interpretable model architecture that excels in predicting RNA degradation. RNAdegformer processes RNA sequences with self-attention and convolutions, two deep learning techniques that have proved dominant in the fields of computer vision and natural language processing, while utilizing biophysical features of RNA. We demonstrate that RNAdegformer outperforms previous best methods at predicting degradation properties at nucleotide resolution for COVID-19 mRNA vaccines. RNAdegformer predictions also exhibit improved correlation with RNA in vitro half-life compared with previous best methods. Additionally, we showcase how direct visualization of self-attention maps assists informed decision-making. Further, our model reveals important features in determining mRNA degradation rates via leave-one-feature-out analysis.
Keywords: mRNA vaccine degradation, deep learning, bioinformatics, COVID-19 mRNA
Introduction
Messenger RNA therapeutics have emerged as a highly promising platform that provides modularity and potentially allows any protein to be delivered and translated [1, 2]. In comparison to recombinant proteins expressed in mammalian cell lines, mRNA is faster to produce with more flexibility using in vitro transcription; the rapid deployment mRNA-based vaccines against COVID-19 is a testament to the potential mRNA therapeutics [3, 4]. Nevertheless, mRNA-based therapeutics faces a fundamental limit, which is the inherent instability of mRNA molecules. As a result, mRNA vaccines still suffer from decreased efficacy because of RNA instability in vitro and in vivo. The degradation of RNA is dependent on how prone the molecule is to in-line hydrolytic cleavage, but currently not much is known about where in the backbone of a given mRNA is prone to hydrolysis and where is safe from degradation. Understanding mRNA degradation can help us design more thermostable RNA therapeutics that would allow increased equitability of distribution, cost reduction and even increased potency [5]. It has even been shown that it is possible to design more thermostable mRNA sequences that code for the same protein with equal amounts of translation via stochastic optimization [6].
Secondary structures of mRNA have been shown to be positively correlated with mRNA stability and translation and optimizing secondary structure to increase half-lives and translation efficiency has been shown to be viable [6, 7]. Many dynamic programming-based RNA secondary structure packages [8–12] can predict secondary structures of mRNA sequences with reasonable accuracies. However, RNA secondary structure packages are quite idealized and degradation of mRNA may depend on more than just secondary structure but also local and global context of mRNA sequences. Therefore, it is important to explore alternative avenues to study mRNA degradation.
Deep learning is a class of data-driven modeling approaches that has found much success in many fields including image recognition [13], natural language processing [14, 15], genomics [16] and computational biology [17]. It has allowed researchers to use recurrent neural networks such as Recurrent Neural Network/Long Short Term Memory/Gated Recurrent Unit to efficiently predict the function, origin, and properties of DNA/RNA sequences by training neural networks on large datasets [18–29], but these sequential computational approaches are difficult to parallelize and objects at long distances suffer from vanishing gradients. Convolutional neural networks are adept at motif recognition and works in the literature have adopted convolution-based architectures that offer better performance than previous non-deep learning approaches [30–38]; they still struggle to capture long-range dependencies, which are essential for DNA/RNA tasks. Graph-based models have also been applied to study RNA binding [39]. Transformers, on the other hand, is a recently proposed architecture that solely relies on attention mechanisms that can model dependencies regardless of the distance in the input or output sequences [14]. Transformers have been adopted in many natural language processing tasks and seen massive success [14, 15, 40], and very recently transformers have been used to classify DNA sequences [41–45]; however, we have found few studies using transformers to study the biophysical properties of RNA sequences.
In this study, we present a neural network model RNAdegformer that utilizes convolution and self-attention to capture both local and global dependencies, which enable the model to achieve high accuracy and provide interpretability in predicting degradation properties of mRNA sequences. We participated in the 21-day OpenVaccine challenge and used the dataset to train our RNAdegformer [46]. While double-stranded DNA forms hydrogen bonds between its complementary bases, single-stranded RNA forms secondary structures by itself, which have been known to stabilize RNA molecules. Therefore, we use existing biophysical models to inject knowledge of RNA secondary structure into our deep learning models to predict RNA degradation. Combining advanced learning techniques (supervised,unsupervised and semi-supervised), we demonstrate that the RNAdegformer outperforms previous best methods at predicting RNA degradation rates at each position of a given sequence, a task of great importance to predict and produce stable mRNA vaccines and therapeutics. Further, we show RNAdegformer generalizes better to predict half-lives of sequences much longer than those in the training dataset compared with other machine learning and dynamic programming algorithms. Last but not least, RNAdegformer also reveals feature importance in predicting mRNA degradation through the usage of leave-one-feature-out (LOFO) test, advancing our understanding of RNA degradation.
Methods
OpenVaccine challenge dataset
The OpenVaccine challenge [46] hosted by the Eterna community sought to rally the data science expertise of Kaggle competitors to develop models that could accurately predict degradation of mRNA. During the 21-day challenge, competitors were provided with 2400 107-bp mRNA sequences with the first 68 base pairs labels with five degradation properties at each position. These properties are reactivity, deg_pH10, deg_Mg_pH10, deg_50C and deg_Mg_50C. More details on these properties can be found at https://www.kaggle.com/c/stanford-covid-vaccine/data.
Like most Kaggle competitions, the test set was divided into a public test set and a private test set. Results on the public test set was available during the competition, whereas private test set results were hidden (Figure 1A). The experimental procedures to obtain the training and public test set are detailed in [7]. The final evaluation was done on a portion of the private test set consisting of 3005 130-bp mRNA sequences, whose degradation measurements were conducted during the 21-day challenge and revealed at the end. The test set was subjected to screening based on three criteria:
Figure 1.
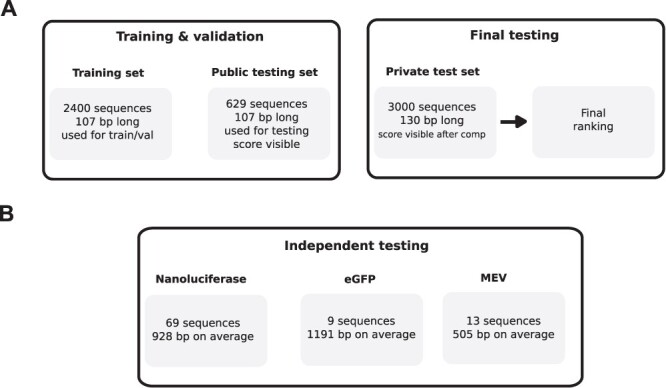
Datasets used in this study A. Visulization of the OpenVaccine dataset B. Visulization of the in vitro half-life dataset consisting of eGFP, nanoluciferase, and MEV sequences.
Minimum value across all five degradation properties must be greater than -0.5
Mean signal/noise across all five degradation properties must be greater than 1.0 [signal/noise is defined as mean( measurement value over 68 nts )/mean( statistical error in measurement value over 68 nts)].
Sequences were clustered into clusters with less than 50% sequence similarity and chosen from clusters with 3 or fewer members
After screening, only 1172 sequences remained on the test set. Final evaluation was done on 3 of the 5 properties (reactivity, deg_Mg_pH10 and deg_Mg_50C). Unlike the training set, the test set has longer mRNA sequences with more sequence diversity and more measurements (first 91 positions) per sequence; in fact, more predictions had to be made for the test set than there were training samples. The metric used for ranking in the competition is mean columnwise root mean squared error (MCRMSE):
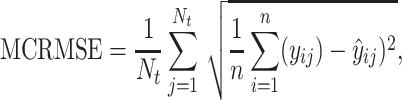 |
(1) |
where 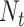 is the number of columns,
is the number of columns, 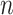 the number of positions predicted,
the number of positions predicted, 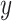 the ground truth and
the ground truth and 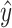 the predicted value. In addition, we also use
the predicted value. In addition, we also use 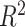 score (coefficient of determination) during further analysis.
score (coefficient of determination) during further analysis.
In vitro half-life dataset
In addition to the OpenVaccine dataset, we also use a recently available dataset of in vitro half-lives of three CDS groups: eGFP, Nanoluciferase and a short multi-epitope vaccine (MEV) for independent testing [7]. This dataset consists of 69 Nanoluc CDS variants, 13 eGFP variants and 9 MEV variants (Figure 1B). Note that these sequences are full-length mRNA sequences with UTR regions. Since our model does not directly output half-lives, we sum up reactivity predictions of the CDS region as a proxy for half-life and calculate Pearson R correlation of reactivity sums and half-lives to evaluate performance of our model.
K-mers with 1-D convolutions
RNAdegformer captures local dependencies by extracting k-mers with 1-D convolutions, after embedding each nucleotides. The extraction of k-mers from a RNA sequence can be considered a sliding window of size 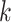 taking snapshots of the sequence while moving one position at a time from one end of the sequence to the other, which is conceptually identical to the convolution operation used in deep learning. Consider a simple example of convolution involving a vector
taking snapshots of the sequence while moving one position at a time from one end of the sequence to the other, which is conceptually identical to the convolution operation used in deep learning. Consider a simple example of convolution involving a vector 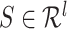 , where
, where 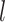 is the length of the vector, and a convolution kernel
is the length of the vector, and a convolution kernel 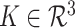 , which convolves over the vector
, which convolves over the vector 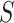 . If the convolutional kernel strides one position at a time, an output vector of dot products
. If the convolutional kernel strides one position at a time, an output vector of dot products 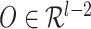 is computed:
is computed:
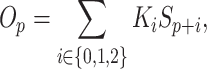 |
(2) |
where 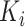 is the
is the 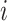 th element of the convolution weight matrix,
th element of the convolution weight matrix, 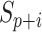 is the
is the 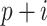 th element in the input vector and
th element in the input vector and 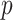 denotes the position in the output vector
denotes the position in the output vector  . In this case, the convolution operation aggregates local information with three positions at a time, so if
. In this case, the convolution operation aggregates local information with three positions at a time, so if 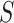 is a sequence of nucleotides, the convolution operation is essentially extracting 3-mers from the DNA sequence
is a sequence of nucleotides, the convolution operation is essentially extracting 3-mers from the DNA sequence 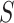 .
.
Since our model takes sequences of RNA nucleotides as input, we first transform each nucleotide into embeddings of fixed size 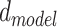 . So now for each sequence we have a tensor
. So now for each sequence we have a tensor 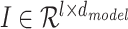 , where
, where 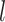 is the length of the sequence. Now to create k-mers we perform convolutions on the tensor
is the length of the sequence. Now to create k-mers we perform convolutions on the tensor 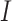 without padding and stride = 1. When a convolution operation with kernel size
without padding and stride = 1. When a convolution operation with kernel size 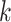 is performed over
is performed over 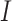 , a new tensor
, a new tensor 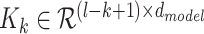 representing the sequence of k-mers is generated. Finally each k-mer is represented by a feature vector of size
representing the sequence of k-mers is generated. Finally each k-mer is represented by a feature vector of size  . The 1D convolution layers are always followed by a layer normalization layer [47].
. The 1D convolution layers are always followed by a layer normalization layer [47].
Indeed our representation of k-mers deviates from conventional representation of words in deep learning, where each word in the vocabulary directly corresponds to a feature vector in a look up table. The disadvantage of using look up tables for k-mers is that a very small percentage of all possible k-mers are present in the OpenVaccine dataset, and it is nearly impossible for the network to generalize to unseen k-mers. Additionally, embeddings of k-mers of larger sizes require a prohibitively large amount of parameters, since the total possible amount of k-mers for a given 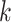 is
is 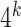 . For example, the biggest k-mer we created with convolution is 9 nucleotides long and using embeddings to represent would need
. For example, the biggest k-mer we created with convolution is 9 nucleotides long and using embeddings to represent would need 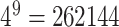 embeddings of size (in our case) 256.
embeddings of size (in our case) 256.
Transformer encoder
RNAdegformer understands global dependencies with self-attention that operates on k-mer representations. For self-attention, we implement the vanilla transformer encoder [14], which uses the multi-head self-attention mechanism. First, the k-mer representations (each k-mer is represented by a feature vector of size 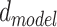 ) are linearly projected into lower dimensional (
) are linearly projected into lower dimensional (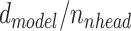 ) keys, values and queries for
) keys, values and queries for 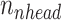 times. Next, the self-attention function is computed with the lower dimensional keys, values and queries for
times. Next, the self-attention function is computed with the lower dimensional keys, values and queries for 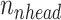 times independently. In this case, the self-attention function essentially computes a pairwise interaction matrix relating every k-mer to every k-mer (including self to self interaction) and computes a weighted sum of the values. It has been posited that the multi-head mechanism allows different heads to learn different hidden representations of the input, leading to better performance. The multi-head self-attention mechanism can be summarized in a few equations:
times independently. In this case, the self-attention function essentially computes a pairwise interaction matrix relating every k-mer to every k-mer (including self to self interaction) and computes a weighted sum of the values. It has been posited that the multi-head mechanism allows different heads to learn different hidden representations of the input, leading to better performance. The multi-head self-attention mechanism can be summarized in a few equations:
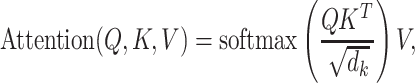 |
(3) |
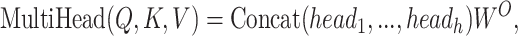 |
(4) |
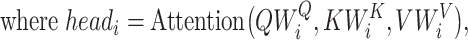 |
(5) |
where 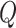 ,
, 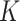 and
and 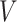 are the query, key, and vector,
are the query, key, and vector, 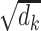 is the size of the attention head,
is the size of the attention head,  is the linear transformation matrix for the concatenated attention head outputs and
is the linear transformation matrix for the concatenated attention head outputs and 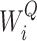 ,
, 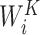 and
and 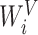 are the linear transformation matrices for
are the linear transformation matrices for 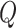 ,
, 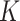 and
and 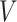 before the
before the 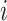 th attention head, respectively.
th attention head, respectively.
Since we are only using the transformer encoder, 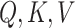 come from the same sequence of feature vectors (hence the name self-attention), each of which represents a k-mer with positional encoding.
come from the same sequence of feature vectors (hence the name self-attention), each of which represents a k-mer with positional encoding.
The self-attention mechanism enables each k-mer to attend to all k-mers (including itself), so global dependencies can be drawn between k-mers at any distance. Contrary to recurrence and convolutions, both of which enforce sparse local connectivity, transformers allow for dense or complete global connectivity. The ability to draw global dependencies of transformers is a huge advantage over recurrence and convolutions, both of which struggle with long sequences.
The self-attention function is followed by a position-wise feedforward network applied separately and identically to each position:
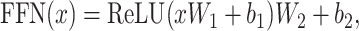 |
(6) |
where 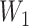 and
and 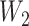 are linear transformation matrices, and
are linear transformation matrices, and 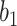 and
and 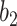 are bias matrices for the two linear transformations, respectively. The position-wise feedforward network is basically two linear transforms with a ReLU (Rectified Linear Unit) activation in between. Conventionally, the combination of self-attention and position-wise feedforward network is referred to as the transformer encoder layer, and a stack of transformer encoder layers is referred to the transformer encoder.
are bias matrices for the two linear transformations, respectively. The position-wise feedforward network is basically two linear transforms with a ReLU (Rectified Linear Unit) activation in between. Conventionally, the combination of self-attention and position-wise feedforward network is referred to as the transformer encoder layer, and a stack of transformer encoder layers is referred to the transformer encoder.
Incorporating biophysical models to predict RNA degradation
Accurate prediction of RNA degradation requires more than just sequence information, and here we detail how we utilize biophysical models as additional features when training RNAdegformer. Biophysical models such as Viennafold predicts RNA secondary structure using dynamic programming and thermodynamic scoring functions; they cannot directly predict RNA degradation but the secondary structure predictions are very useful when training a neural network to directly predict degradation. Firstly, we include the predicted structure per position (by folding algorithms), which describes whether a nucleotide is paired or unpaired with another one via hydrogen bonding. The predicted structure is generated using arnie with log_gamma set to 0. Also, we include the predicted loop type assigned by bpRNA from folding algorithm predictions [48]. Two embedding layers are added to represent structure and loop type, and the resulting feature vectors are concatenated and the dimensionality reduced with a linear transformation. Additionally, we directly add a modified version of the base-pairing probability matrix into the attention function (note that this is simply a modified version of 3 with 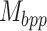 as an additional input):
as an additional input):
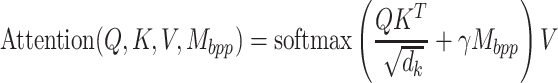 |
(7) |
where 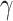 is a learnable parameter and
is a learnable parameter and 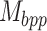 is the modified base-pairing probability matrix (Algorithm 1) . The original base-pairing probability matrix contains the probabilities for every possible base pair in an RNA sequence and has been used for many RNA informatics tasks. Here in addition to base-pairing probabilities, we also stack inverse, inverse squared and inverse cubed pairwise distance matrices on top of the original base-pairing probability matrix, where the distance is the the number of covalent bonds between the pair of nucleotides (this can also be considered the path length in an RNA graph where the only edges are the covalent bonds). The inverse distance matrices encode some information about the relative distance between pairs of nucleotides, since pairs of nucleotides with a small number of covalent bonds in between are likely to be closer to each other spatially. Because the distance matrix already encodes information about position, we do not use positional encoding for mRNA.
is the modified base-pairing probability matrix (Algorithm 1) . The original base-pairing probability matrix contains the probabilities for every possible base pair in an RNA sequence and has been used for many RNA informatics tasks. Here in addition to base-pairing probabilities, we also stack inverse, inverse squared and inverse cubed pairwise distance matrices on top of the original base-pairing probability matrix, where the distance is the the number of covalent bonds between the pair of nucleotides (this can also be considered the path length in an RNA graph where the only edges are the covalent bonds). The inverse distance matrices encode some information about the relative distance between pairs of nucleotides, since pairs of nucleotides with a small number of covalent bonds in between are likely to be closer to each other spatially. Because the distance matrix already encodes information about position, we do not use positional encoding for mRNA.
|
|

Because 1-D convolution operation used in the RNAdegformer does not use padding, the convolution product ends up with reduced dimensionality in the 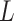 dimension when the convolution kernel size is bigger than 1. As a result, the base pairing probability matrix cannot be directly added to self-attention matrix. To circumvent this, we do 2D convolution with the same kernel size as the 1D convolution on the modified base pairing probability matrix without padding, so the dimensionality of the feature map becomes
dimension when the convolution kernel size is bigger than 1. As a result, the base pairing probability matrix cannot be directly added to self-attention matrix. To circumvent this, we do 2D convolution with the same kernel size as the 1D convolution on the modified base pairing probability matrix without padding, so the dimensionality of the feature map becomes 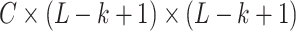 . The attention function now is:
. The attention function now is:
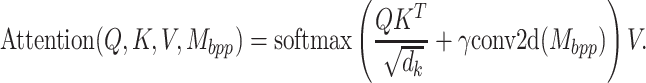 |
(8) |
Conceptually, instead of a base pair to base pair interaction mapping, the 2D convolution product of the modified base pairing probability matrix can be seen as a k-mer to k-mer pairwise interaction mapping with matching dimensionality to the 1D convolution k-mer products. Aside from matching dimensionality, the 2D convolution operation also makes up for some missing information regarding the geometry of mRNA folding. To illustrate this, we visualize an mRNA sequence in the OpenVaccine dataset to explain the physical and mathematical reasoning behind the 2D convolution operation (Figure 2A). While inspecting the interaction between A-20 (A at position 20), G-21, C-40 and U-41, we can visually see that A-20 and C-40 are quite close to each other and imagine that there is some degree of interaction between them, despite A-20 and C-40 not forming hydrogen bonds. However, looking at the portion of base pairing probability (BPP) matrix and distance matrix corresponding to the 2 2 connection between A-20 (A at position 20), G-21, C-40 and U-41, we see that neither the BPP matrix nor the distance matrix convey this information, as the component (40,20) has zero or close to zero values on both the BPP matrix and the distance matrix. When a 2 2 convolution kernel operates on the BPP matrix and distance matrix (for illustration purposes here we simply draw a kernel with all values set to unity), it essentially fuses the four connections between A-20, G-21, C-40 and U-41 and creates a strong connection between the 2 2mers (A-20, G-21 and C-40, U-41). Now it becomes much easier for the network to learn the interaction between A-20 and G-40 (as well as for G-21 and U-41).
Figure 2.
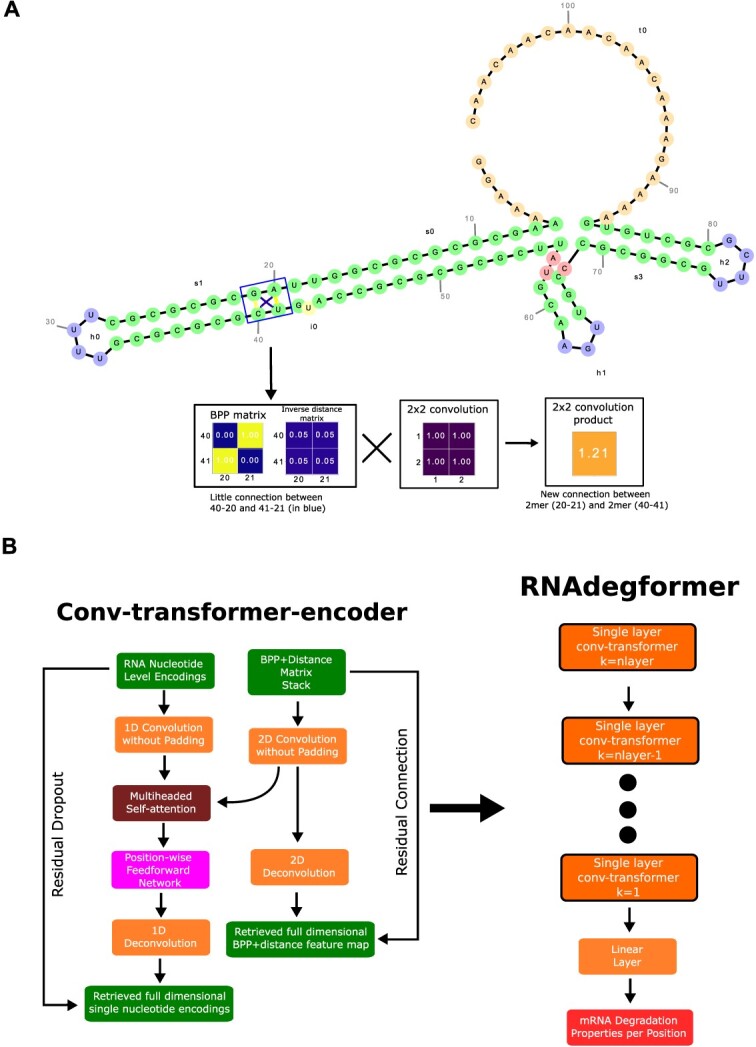
RNAdegformer combines convolution and self-attention to predict RNA degradation. A. RNAdegformer architecture which takes advantage of additional input information from biophysical models. B. Visualization of BPP+distance matrix, attention weights of a non-pretrained RNAdegformer, and attention weights of a pretrained RNAdegformer
The combination of convolution and self-attention cannot produce nucleotide position wise predictions, since it generates k-mer encodings instead of single base pair encoding. In order to make predictions per nucleotide position, we introduce additional deconvolution layers to retrieve full dimensional encodings, which allow residual connections of both 1D and 2D encodings before and after the transformer encoder. We name these blocks as Conv-transformer-encoders. As a result, both the single nucleotide embeddings and the modified BPP matrix go through deep transforms before outputting predictions.
Now we can summarize the RNAdegformer architecture used for the RNA task (Figure 2B), which can be seen as a special case of a series of multiple Conv-Transformer-encoders, each with a single transformer encoder layer followed by a deconvolution layer. Also, because the OpenVaccine challenge requires making predictions at each position of the RNA sequence, it is important for the last transformer encoder layer right before outputting predictions to operate on single nucleotide encodings instead of k-mer encodings. With these considerations in mind, we choose a simple strategy to construct the stack of RNAdegformers with two main hyperparameters 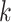 and
and 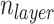 set equal to each other. The first single layer Conv-transformer-encoder has
set equal to each other. The first single layer Conv-transformer-encoder has 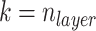 and we decrease the size of the convolution kernel by 1 for the next Conv-Transformer-encoder. Therefore, when we get to the last Conv-Transformer-encoder in the stack,
and we decrease the size of the convolution kernel by 1 for the next Conv-Transformer-encoder. Therefore, when we get to the last Conv-Transformer-encoder in the stack, 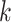 becomes 1 and the last Conv-Transformer-encoder is simply a transformer encoder layer with an added bias from the BPP feature map.
becomes 1 and the last Conv-Transformer-encoder is simply a transformer encoder layer with an added bias from the BPP feature map.
Optimizer and training schedule
We started with Adam but found it to be underfitting. Therefore, we switched to a more recent and powerful optimizer, Ranger, which uses gradient centralization from https://github.com/lessw2020/Ranger-Deep-Learning-Optimizer [49]. As for the training schedule, we used flat and anneal, where training starts with a flat learning rate of 1e-3 and then 75% through all the epochs training proceeds with cosine annealing schedule reducing learning rate down to 0 at the end of training. Weight decay is set to 0.1.
Learning approaches
In the OpenVaccine challenge, the number of samples in the test set exceeds that in the training set, so we use a combination of learning methods (Figure 3). Here we describe those learning methods.
Figure 3.

The RNAdegformer is first pretrained in unsupervised settings, trained with ground truth labels in supervised settings, and finally trained with pseudo labels and ground truth labels in semi-supervise settings.
Multitasking learning: during pretraining, mutated/masked sequence, structure and predicted loop type are inputted into the RNAdegformer and then the RNAdegformer is trained with crossentropy loss to retrieve the correct sequence, structure and predicted loop type simultaneously at each position of the RNA sequence. During training on ground truth labels and pseudo labels, the RNAdegformer is trained to predict five different degradation properties simultaneously at each measured position of the RNA sequence.
Unsupervised learning (pretraining): we use all available sequences in the OpenVaccine challenge dataset to pretrain our network on randomly mutated and masked (with NULL token) sequence retrieval loss (basically softmax to retrieve correct nucleotide/structure/loop). During pretraining, the RNAdegformer learns the rules of RNA structure, guided by biophysical knowledge provided by biophysical models.
Supervised learning: during supervised learning, the RNAdegformer is trained on target values of RNA degradation properties.
Semi-supervised learning: Following RNA supervised learning, the RNAdegformer is retrained in semi-supervised fashion on pseudo labels generated by an ensemble of RNAdegformer with different depths. Similar to previous work with semi-supervised learning [50], we retrain the models first using pseudo labels at a flat learning rate and then finetune with ground truth labels in the training set with cosine anneal schedule.
In practice, we first pretrain our neural networks with unsupervised learning, then train our neural networks on ground truth labels with supervised learning, and lastly use labeled and unlabeled data in conjunction with semi-supervised learning.
Error weighted loss
We used a form of RMSE loss, which is also the same mathematically as the competition metric:
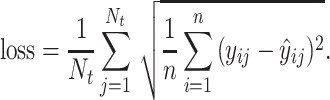 |
(9) |
where 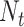 is the number of columns,
is the number of columns, 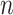 is the number of positions predicted,
is the number of positions predicted, 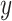 is the ground truth and
is the ground truth and 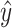 is the predicted value. Because the OpenVaccine dataset came from experimental measurements that had errors, we adjusted the losses based on the error for each measurement during supervised training:
is the predicted value. Because the OpenVaccine dataset came from experimental measurements that had errors, we adjusted the losses based on the error for each measurement during supervised training:
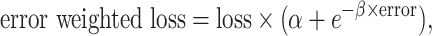 |
(10) |
where loss is the per position loss to be back propagated, error is the experimental error for that position and 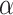 and
and 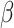 are tunable hyperparameters to control how loss is weighted based on experimental errors . If
are tunable hyperparameters to control how loss is weighted based on experimental errors . If 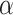 is set to 1 and
is set to 1 and 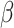 to infinity, the loss values stay the same; otherwise gradients from measurements with large errors would be lowered to prevent the neural network from overfitting to experimental errors. We find the best setup to be
to infinity, the loss values stay the same; otherwise gradients from measurements with large errors would be lowered to prevent the neural network from overfitting to experimental errors. We find the best setup to be 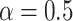 and
and 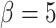 .
.
Usage of biophysical models during training
We used secondary structures predicted by an ensemble of biophysical models including RNAsoft [11], rnastructure [12], CONTRAfold [9], EternaFold [51], NUPACK [10] and Vienna [8]. Arnie (https://github.com/DasLab/arnie) is used as a wrapper to generate secondary structure predictions. For each sequence, we generated secondary structure predictions at 37 and 50C, since two of the scored degradation properties were measured at different temperatures. Although we also need to make predictions for a degradation property at pH10, none of the biophysical models used could generate predictions at different pH’s. With six packages, we ended with up with 12 secondary structure predictions for each sequence. During training, we randomly select one of the 12 secondary structure predictions for each sample during a forward and backward propagation pass. During validation and testing, we use the averaged predictions made with all 12 secondary structure predictions.
Best hyperparameters
For models trained during the OpenVaccine competition, 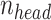 is set to 32,
is set to 32, 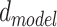 is set to 256, dropout is set to 0.1 and conv2d filter size is set to 32,
is set to 256, dropout is set to 0.1 and conv2d filter size is set to 32, 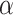 and
and 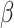 are both 1. Half of the models were trained with sequences with signal to noise greater than 1 and half were trained with signal to noise greater than 0.5. During post competition experiments, we found that results are better when penalizing measurements with high error more by reducing
are both 1. Half of the models were trained with sequences with signal to noise greater than 1 and half were trained with signal to noise greater than 0.5. During post competition experiments, we found that results are better when penalizing measurements with high error more by reducing 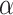 to 0.5 and increasing
to 0.5 and increasing 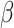 to 5. Although using more models at more different conditions can give better results, for better reproducibility we only trained five models with
to 5. Although using more models at more different conditions can give better results, for better reproducibility we only trained five models with 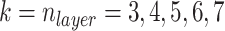 for each experiment hereinafter. Here
for each experiment hereinafter. Here 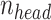 is set to 32,
is set to 32, 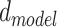 is 256, dropout is set to 0.1 and conv2d filter size is set to 8. We also only use sequences with signal to noise greater than 0.25 for training and signal to noise greater than 1 for 10-fold cross validation.
is 256, dropout is set to 0.1 and conv2d filter size is set to 8. We also only use sequences with signal to noise greater than 0.25 for training and signal to noise greater than 1 for 10-fold cross validation.
Results
Interpretablility of self-attention assists decision-making
By the end of the competition, we had trained two sets of models to use in the final submissions, one that was trained directly on short sequences with labels and one that was pretrained with all available sequences (including unlabeled test sequences) before training on short sequences with labels. Note that test sequences appear to have a different distribution via t-sne analysis (Figure 4A). In order to robustly select submissions, we visualized and evaluated the learned attention weights from the transformer encoder (Figure 4B). Since we added the BPP matrix and distance matrix as a bias, both learned attention distributions of pretrained and non-pretrained models resembled the BPP and distance matrix, but there were also some key differences. The non-pretrained model paid heavy attention indiscriminately to pairs of positionally close nucleotides, as indicated by the bright stripes parallel and close to the diagonal of the attention matrix. This indicates the non-pretrained model thought that positionally close nucleotides were always important when making predictions on mRNA degradation properties, which seemed highly unlikely. On the other hand, the pretrained model did not show the same bias towards pairs of positionally close nucleotides and was able to recognize the weak BPP connections that were barely visible on the original BPP matrix. In this case, the model made more effective use of the BPP matrix generated by biophysical models. Because of these considerations and given the short timeframe of the competition, we had to make a prompt decision to focus more on experimenting with pretrained models, and we favored pretrained models in our final submissions. Results on the private test set validated our selection based on visual inspection of attention weights (Figure 4B). Pretrained models performed much better than non-pretrained models on both public test set and private test set. The non-pretrained models would have placed us at 39th/1636 instead of 7th/1636. Notably, the predictions on the private test set have much higher error, likely both due to longer sequence length and more sequence diversity in the private test set.
Figure 4.

RNAdegformer accurately predicts RNA degradation at nucleotide level. A. T-sne plot of RNA sequences in the training set (blue), test set (orange), and randomly generated set (green) B. Visualization of BPP+distance matrix, attention weights of a non-pretrained RNAdegformer, and attention weights of a pretrained RNAdegformer C. Comparison of pretrained and non-pretrained models trained during the OpenVaccine competition and post competition experiments.
Semi-supervised learning leads to more accurate predictions in RNA degradation
Following the competition, we found that using predictions generated by an ensemble predictions of the private test set as pseudo-labels in semi-supervised settings, we could reduce the test set MCRMSE to 0.33722, compared with the top solution’s 0.34198 (Figure 4B). This is somewhat surprising given the that the predictions used as pseudo-labels could only score 0.3438 on the private test set. When we compare the 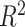 scores of supervised only, pretrained and semi-supervised RNAdegformer, we see that pretraining and semi-supervised learning led to sizable improvements over the supervised only (Figure 4C, Figure 5). Also, we notice that predictions for deg_Mg_pH10 have significantly larger errors than the other two properties, which is expected because the biophysical models used are incapable of generating different secondary structure predictions at different pHs. It is also important to note that the semi-supervised learning approach requires pseudo-labeling and knowing the test set distribution before hand. Therefore, while pseudo-labeling is effective under the competition setting, it is a risky approach and the performance gain may not transfer in real life applications. In addition, we also report performance of our models using different packages and ensembled together ( 1). Overall, while RNAsoft and Vienna consistently provide the best performance, ensembling results from different biophysical models still provides an advantage over any individual biophysical model.
scores of supervised only, pretrained and semi-supervised RNAdegformer, we see that pretraining and semi-supervised learning led to sizable improvements over the supervised only (Figure 4C, Figure 5). Also, we notice that predictions for deg_Mg_pH10 have significantly larger errors than the other two properties, which is expected because the biophysical models used are incapable of generating different secondary structure predictions at different pHs. It is also important to note that the semi-supervised learning approach requires pseudo-labeling and knowing the test set distribution before hand. Therefore, while pseudo-labeling is effective under the competition setting, it is a risky approach and the performance gain may not transfer in real life applications. In addition, we also report performance of our models using different packages and ensembled together ( 1). Overall, while RNAsoft and Vienna consistently provide the best performance, ensembling results from different biophysical models still provides an advantage over any individual biophysical model.
Figure 5.

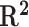 score on the OpenVaccine private test set of the RNAdegformer.
A. supervised only, B. pretrained, C. semi-supervised.
score on the OpenVaccine private test set of the RNAdegformer.
A. supervised only, B. pretrained, C. semi-supervised.
Table 1.
Performance using inputs generated by different biophysical models to predict mRNA degradation in the OpenVaccine dataset
| Package | Public MCRMSE | Private MCRMSE | Public MCRMSE semi-supervised | Private MCRMSE semi-supervised |
|---|---|---|---|---|
| RNAsoft | 0.23154 | 0.34521 | 0.23101 | 0.33841 |
| rnastructure | 0.23637 | 0.34639 | 0.23502 | 0.33989 |
| EternaFold | 0.23158 | 0.34613 | 0.23081 | 0.33878 |
| Contrafold_2 | 0.23245 | 0.34858 | 0.23123 | 0.34111 |
| NUPACK | 0.23679 | 0.34955 | 0.23587 | 0.34292 |
| Vienna | 0.23492 | 0.34515 | 0.2337 | 0.33864 |
| avg of all | 0.22976 | 0.34375 | 0.22914 | 0.33722 |
Figure 7.

RNAdegformer webapp A. Home page of the RNAdegformer webapp B. Page where the user can visualize secondary structure and predict degradation rates of any RNA sequence.
RNAdegformer is a strong predictor of mRNA in vitro half-life
Comparing predictions of RNAdegformer on groups of CDS variants mRNA design sequences to half-life data [7], we see that the RNAdegformer provides improved correlation (Pearson R=-0.655) with half-lives of the largest group of nanoluciferase CDS variants (928 bp on average) compared previous models quantifying mRNA degradation such as Degscore (Pearson R=-0.637) and sum unpaired probability (SUP) (Pearson R=-0.58) (Figure 4D). On smaller groups of CDS variants of eGFP (1191 bp on average) and Multi-Epitope-Vaccine (MEV) (505 bp on average), RNAdegformer shows larger improvement (-0.499/-0.578) over Degscore (-0.288/-0.240) and SUP (-0.103/-0.130). Compared with top solutions in OpenVaccine, we find the RNAdegformer generalizes better to longer CDS variants (Nanoluciferase and GFP) (2), both of which are much longer than sequences in the training data. Overall, RNAdegformer is the only method that provides correlation equal to or better than -0.5 across all 3 CDS groups. It has been shown that computational design of CDS regions of mRNA sequences can lead to close to 3 time increase in in vitro half-life using Degscore [7], a ridge regression model, following dynamic programming optimization of Gibbs free energy using linear design [52]; we believe RNAdegformer can guide computational design of CDS regions to produce even more stable sequences.
Table 2.
Pearson correlation of RNAdegformer predictions with in vitro half-life compared with other top methods
| NLuc Eterna | eGFP | MEV | |
|---|---|---|---|
| RNAdegformer | -0.655 | -0.499 | -0.578 |
| nullrecurrent (1st place) | -0.601 | -0.220 | -0.685 |
| kazuki (2nd place) | -0.623 | -0.376 | -0.597 |
| Degscore | -0.637 | -0.288 | -0.240 |
| Vienna SUP | -0.590 | -0.103 | -0.130 |
RNAdegformer reveals most important features in RNA stability predictions
To understand what features are the most important in predicting RNA stability, we took a simple LOFO approach. We retrained our models from scratch with the best settings but left out one of the available features (sequence, predicted structure, predicted loop type or predicted base pair probabilities), and then evaluated model performance with the absence of one feature (Figure 4G). We found that our models took the biggest performance hit when sequence information was left out. This indicates that aside from secondary structure, stable raw sequence motifs/motif pairs could be extracted from experimental data to enable design of more stable RNA molecules [7]; additionally, this also demonstrates the powerful capabilities of the RNAdegformer in terms of motif/pairwise motif recognition. Performance hit when leaving out sequence information is followed by predicted base pair probabilities, which describe the ensemble of possible folds a particular RNA sequence can adopt. This shows that base pairing probabilities are a rich, detailed and holistic feature that describe RNA secondary structure well. It has also been shown experimentally that base pairing probabilities are highly correlated with RNA half lives [7], and designing RNA coding sequences to maximize pairing has been proposed as a strategy to produce more stable RNA molecules [53]; our LOFO results support that proposal. Further, RNAdegformer’s ability to directly and effectively incorporate 2D features like base pairing probabilities also demonstrates its flexibility. Surprisingly, leaving out structure and loop features had little impact on model performance as the MCRMSE remained almost the same. However, the minimal impact of structure and loop features is actually consistent with the previous findings regarding importance of base pairing probabilities. The structure and loop features only represent the singular most likely fold based on predicted base pair probabilities; in other words, they do not describe RNA secondary structure in the same holistic fashion as base pairing probabilities. While Degscore and linear design leverage either structure information or thermodynamics, our RNAdegformer can simultaneously and effectively utilize the most important features in regards to mRNA degradation: sequence and BPP, as revealed by our LOFO ablation studies.
RNAdegformer webapp
We created a web application (Figure 7A) developed using H2O.ai’s wave and made it available at https://github.com/Shujun-He/RNAdegformer-Webapp, so users can utilize our RNAdegformer to predict and visualize RNA degradation without any need to code. To use the webapp, the user simply needs to type in the RNA sequence of interest (Figure 7B) and RNA degradation predictions will be generated as a downloadale CSV file, along with relevant features such as BPP and secondary structure. In addition, the secondary structure, attention weight and degradation properties of the RNA sequence will also be visualized, allowing the user to visually inspect the degradation predictions.
Figure 6.
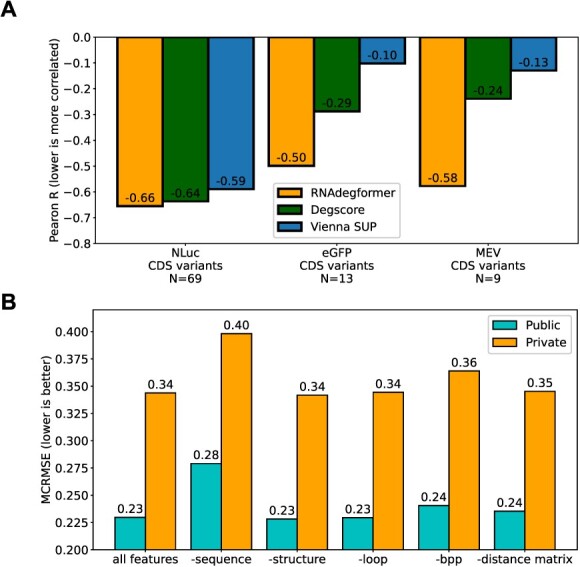
RNAdegformer generalizes well to longer mRNA sequences and reveals important features in determininh mRNA degradation A. Half-life correlation of RNAdegformer predictions compared to previous best methods. B. LOFO feature importance on the OpenVaccine dataset.
Discussion
In this work, we present the RNAdegformer, an effective neural network architecture that is capable of accurately predicting degradation properties of mRNA COVID-19 vaccine candidates per nucleotide. We participated in the recent 21-day OpenVaccine challenge with an adaptation of the RNAdegformer and placed 7th out of 1636 teams of top machine learning experts from all over the globe. We also demonstrate with semi-supervised learning, the RNAdegformer outperforms even the top solution in the OpenVaccine challenge by a considerable margin; RNAdegformer also generalizes well to predict half-lives of unseen mRNA sequences, with better correlation than previous best methods. Further, RNAdegformer reveals the most important features in predicting RNA degradation. Our results show that self-attention and convolution are a powerful combination enabling learning both global and local dependencies effectively to predict RNA degradation and even half-life . It has long been posited that the transformer architecture can excel beyond natural language processing, and our work demonstrates that.
Although there has been many dynamic programming algorithms that predict mRNA secondary structure, there is no precedence on predictions of mRNA degradation properties per nucleotide. After a chaotic 2020 caused by COVID-19, mRNA vaccines have emerged as a fast and effective solution to the COVID problem, with companies like Pfizer and Moderna rolling out mRNA vaccines at unprecedented speeds. However, storage and transport remain a challenge with fragile mRNA vaccines (Pfizer-BioNtech’s vaccine has to be stored as 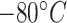 and Moderna’s at
and Moderna’s at 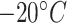 . One strategy to reduce mRNA hydrolysis is to redesign RNAs to code for the same proteins but form double-stranded regions, which are protected from these degradative processes [7, 53]. Our work can provide guidance and trained models can act as a screening tool in development of more stable mRNA vaccines in the future. Messenger RNA vaccines and therapeutics have many applications towards infectious diseases and cancers [54–57], and it is our hope the RNAdegformer will aid design of more stable mRNA vaccines that can withstand harsher conditions than current ones. It is important to note, however, that there is still significant gap between errors on the 107 bp mRNA OpenVaccine public set sequences and the 130 bp mRNA OpenVaccine private set sequences, both due to difference in sequence length and diversity. Actual COVID-19 candidates are even longer and modeling those remains a challenge in the future. For these long sequences, one key challenge is the quadratic computational complexity of self-attention, which prohibits training on long sequences. Notably, much work has been done in very recent times on reducing the quadratic computational complexity of self-attention to linear to enable training of transformer like self-attention on much longer sequences [58–61], i.e. linear transformers. Whole genomes and COVID-19 mRNA vaccines both greatly exceed the length limit of full self-attention, and COVID-19 vaccine candidates, in particular, are around 4000 bp long, so these new approaches may serve as effective tools to solve the challenges.
. One strategy to reduce mRNA hydrolysis is to redesign RNAs to code for the same proteins but form double-stranded regions, which are protected from these degradative processes [7, 53]. Our work can provide guidance and trained models can act as a screening tool in development of more stable mRNA vaccines in the future. Messenger RNA vaccines and therapeutics have many applications towards infectious diseases and cancers [54–57], and it is our hope the RNAdegformer will aid design of more stable mRNA vaccines that can withstand harsher conditions than current ones. It is important to note, however, that there is still significant gap between errors on the 107 bp mRNA OpenVaccine public set sequences and the 130 bp mRNA OpenVaccine private set sequences, both due to difference in sequence length and diversity. Actual COVID-19 candidates are even longer and modeling those remains a challenge in the future. For these long sequences, one key challenge is the quadratic computational complexity of self-attention, which prohibits training on long sequences. Notably, much work has been done in very recent times on reducing the quadratic computational complexity of self-attention to linear to enable training of transformer like self-attention on much longer sequences [58–61], i.e. linear transformers. Whole genomes and COVID-19 mRNA vaccines both greatly exceed the length limit of full self-attention, and COVID-19 vaccine candidates, in particular, are around 4000 bp long, so these new approaches may serve as effective tools to solve the challenges.
In summary, we have developed a convolution and transformer-based deep learning platform toward prediction of mRNA degradation and half-lives. Our work has demonstrated success in RNA stability and half-life predictions. We believe that by further development and optimization, we will solve many challenges including understanding RNA degradation and structure relationships, aiding next-generation mRNA therapy development, and more.
Key Points
Messenger RNA therapeutics have emerged as a highly promising platform that provides modularity and potentially allows any protein to be delivered and translated. Messenger RNA can produced rapidly and flexibly with in vitro transcription, but it suffers from chemical instability due to in-line hydrolysis.
We present a model architecture RNAdegformer that utilizes convolution and self-attention to capture both local and global dependencies, which enable the model to achieve high accuracy and provide interpretability in predicting degradation properties of mRNA sequences
Using unsupervised (pretraining), supervised and semi-supervised learning in conjunction with each other, we demonstrate that RNAdegformer outperforms the top solution in OpenVaccine at predicting RNA degradation rates at each position of a given RNA sequence, a task of great importance to predict and produce stable mRNA vaccines and therapeutics.
RNAdegformer generalizes better to predict half-lives of sequences much longer than those in the training dataset compared with other machine learning and dynamic programming algorithms.
RNAdegformer also reveals feature importance in predicting mRNA degradation through the usage of leave-one-feature-out (LOFO) test, advancing our understanding of RNA degradation
Author contributions statement
S.H. and B.G conceived the project. R.S.provided critical feedback on the analysis of RNA sequences and biological context. S.H. implemented the deep learning algorithms (in Pytorch) and participated in the OpenVaccine challenge. Q.S. supervised the project and provided guidance. S.H., B.G. and R.S. wrote the manuscript in consultation with Q.S.
Data availability
OpenVaccine dataset is available at https://www.kaggle.com/c/stanford-covid-vaccine/data. Pretrained models can be accessed from Kaggle notebooks: https://www.kaggle.com/shujun717/rnadegformer-inference.
Code availability
All training code to fully reproduce results is released at https://github.com/Shujun-He/RNAdegformer, and a web application (Figure 7) developed using H2O.ai’s wave is available at https://github.com/Shujun-He/RNAdegformer-Webapp.
Supplementary Material
Acknowledgments
We would like to thank Das Lab for providing the data on RNA degradation and hosting the OpenVaccine competition. We would also like to thank Dr Yang Shen, Dr Shuiwang Ji and his student Hao Yuan for proofreading the manuscript.
Shujun He is a PhD student at the Artie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, Texas, United States. His research interests include deep learning and mRNA stability.
Baizhen Gao is a PhD student at the Artie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, Texas, United States. His research interests include gene circuit design and protein engineering.
Rushant Sabnis is a Ph.D. student at the Artie McFerrin Department of Chemical Engineering,Texas A&M University, College Station, Texas. His research interest include protein engineering and synthetic biology.
Qing Sun is an Assistant Professor at the Artie Mcferrin Department of Chemical Engineering, Texas A&M University, College Station, Texas, United States. Her research interests include synthetic biology for biomedical and environmental applications.
Contributor Information
Shujun He, Department of Chemical Engineering, Texas A&M University, 100 Spence St, 77843, Texas, United States.
Baizhen Gao, Department of Chemical Engineering, Texas A&M University, 100 Spence St, 77843, Texas, United States.
Rushant Sabnis, Department of Chemical Engineering, Texas A&M University, 100 Spence St, 77843, Texas, United States.
Qing Sun, Department of Chemical Engineering, Texas A&M University, 100 Spence St, 77843, Texas, United States.
Funding
National Institutes of Health (R01AI165433); Texas A&M University X-grants.
References
- 1. Kramps T, Elbers K. Introduction to RNA vaccines. RNA Vaccines 2016;1499:1–11. [DOI] [PubMed] [Google Scholar]
- 2. Kaczmarek JC, Kowalski PS, Anderson DG. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med 2017;9(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waickman AT, Victor K, Newell K, et al. MRNA-1273 vaccination protects against SARS-COV-elicited lung inflammation in nonhuman primates. JCI Insight 2022;7(13):e160039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baden LR, El Sahly HM, Essink B, et al. Efficacy and safety of the mRNA-1273 sars-cov-2 vaccine. N Engl J Med 2021;384(5):403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schoenmaker L, Witzigmann D, Kulkarni JA, et al. MRNA-lipid nanoparticle covid-19 vaccines: structure and stability. Int J Pharm 2021;601:120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mauger DM, Cabral BJ, Presnyak V, et al. MRNA structure regulates protein expression through changes in functional half-life. Proc Natl Acad Sci 2019;116(48):24075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leppek K, Byeon GW, Kladwang W, et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat Commun 2022;13(1):1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lorenz R, Bernhart SH, Siederdissen CHZ, et al. Viennarna package 2.0. Algorithm Mol Biol 2011;6(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Do CB, Woods DA, Batzoglou S. Contrafold: RNA secondary structure prediction without physics-based models. Bioinformatics 2006;22(14):90–98. [DOI] [PubMed] [Google Scholar]
- 10. Dirks RM, Pierce NA. An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. J Comput Chem 2004;25(10):1295–304. [DOI] [PubMed] [Google Scholar]
- 11. Andronescu M. Rnasoft: a suite of RNA secondary structure prediction and design software tools. Nucleic Acids Res 2003;31(13):3416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reuter JS, Mathews DH. Rnastructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 2010;11(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. He K, Zhang X, Ren S, et al. Deep residual learning for image recognition. IEEE Conference on Computer Vision and Pattern Recognition 2016;770–78.
- 14. Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. CoRR. abs/1706.037622017. [Google Scholar]
- 15. Devlin J, Chang M-W, Lee K, et al. BERT: pre-training of deep bidirectional transformers for language understanding. CoRR. abs/1810.048052018. [Google Scholar]
- 16. Schreiber J, Singh R. Machine learning for profile prediction in genomics. Curr Opin Chem Biol 2021;65:35–41. [DOI] [PubMed] [Google Scholar]
- 17. Angermueller C, Pärnamaa T, Parts L, et al. Deep learning for computational biology. Mol Syst Biol 2016;12(7):878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quang D, Xie X. Danq: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res 2016;44:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nielsen AAK, Voigt CA. Deep learning to predict the lab-of-origin of engineered DNA. Nat Commun 2018;3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trabelsi A, Chaabane M, Ben-Hur A. Comprehensive evaluation of deep learning architectures for prediction of DNA/RNA sequence binding specificities. Bioinformatics 2019;35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Angenent-Mari NM, Garruss AS, Soenksen LR, et al. A deep learning approach to programmable RNA switches. Nat Commun 2020;5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lam JH, Li Y, Zhu L, et al. A deep learning framework to predict binding preference of RNA constituents on protein surface. Nat Commun 2019;4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amin N, McGrath A, Chen YP. Evaluation of deep learning in non-coding RNA classification. Nat Mach Intell 2019;1:246–56. [Google Scholar]
- 24. He Y, Shen Z, Zhang Q, et al. A survey on deep learning in DNA/RNA motif mining. Brief Bioinform 2020;10:bbaa229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Qiao S, Ji S, et al. Deepsite: bidirectional LSTM and CNN models for predicting DNA-protein binding. Int J Mach Learn & Cyber 2020;11:841–51. [Google Scholar]
- 26. Liu Q, Fang L, Yu G, et al. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat Commun 2019:2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H. Identifying centromeric satellites with dna-brnn. Bioinformatics 2019;35(21):4408–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Angermueller C, Lee HJ, Reik W, et al. Deepcpg: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol 2017;18:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oubounyt M, Louadi Z, Tayara H, et al. Deepromoter: robust promoter predictor using deep learning. Front Genet 2019;10:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren J, Song K, Deng C, et al. Identifying viruses from metagenomic data using deep learning. Quant Biol 2020;8(1):64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tampuu A, Bzhalava Z, Dillner J, et al. Viraminer: deep learning on raw DNA sequences for identifying viral genomes in human samples. PLoS One 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Le NQK, Yapp EKY, Nagasundaram N, et al. Classifying promoters by interpreting the hidden information of DNA sequences via deep learning and combination of continuous fasttext n-grams. Front Bioeng Biotechnol 2019;7:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu F, Miao Y, Liu Y, et al. Rnn-virseeker: a deep learning method for identification of short viral sequences from metagenomes. IEEE/ACM Trans Comput Biol Bioinform 2020;19:1. [DOI] [PubMed] [Google Scholar]
- 34. Zhou J, Theesfeld CL, Yao K, et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat Genet 2018;50(8):1171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alipanahi B, Delong A, Weirauch MT, et al. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 2015;33(8):831–8. [DOI] [PubMed] [Google Scholar]
- 36. Zhang S, Hailin H, Zhou J, et al. Analysis of ribosome stalling and translation elongation dynamics by deep learning. Cell Syst 2017;5(3):212–20. [DOI] [PubMed] [Google Scholar]
- 37. Zhang S, Hailin H, Jiang T, et al. Titer: predicting translation initiation sites by deep learning. Bioinformatics 2017;33(14):i234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 2015;12(10):931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang S, Zhou J, Hailin H, et al. A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res 2015;44(4):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Radford A, Wu J, Child R, et al. Language models are unsupervised multitask learners. OpenAI Blog 2019;1(8):9. [Google Scholar]
- 41. Avsec Ž, Agarwal V, Visentin D, et al. Effective gene expression prediction from sequence by integrating long-range interactions. 2021. [DOI] [PMC free article] [PubMed]
- 42. Ji Y, Zhou Z, Liu H, et al. Dnabert: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 2021;37(15):2112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Clauwaert J, Waegeman W. Novel transformer networks for improved sequence labeling in genomics. IEEE/ACM Trans Comput Biol Bioinform 2020;19:1. [DOI] [PubMed] [Google Scholar]
- 44. Le NQK, Ho Q-T, Nguyen T-T-D, et al. A transformer architecture based on Bert and 2D convolutional neural network to identify DNA enhancers from sequence information. Brief Bioinform 2021;22(5):bbab005. [DOI] [PubMed] [Google Scholar]
- 45. Ullah F, Ben-Hur A. A self-attention model for inferring cooperativity between regulatory features. Nucleic Acids Res 2021;49:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wayment-Steele HK, Kladwang W, Watkins AM, et al. Deep learning models for predicting RNA degradation via dual crowdsourcing. Nat Mach Intell 2022. [DOI] [PMC free article] [PubMed]
- 47. Ba JL, Kiros JR, Hinton GE. Layer normalization, 2016. Retrieved from https://arxiv.org/abs/1607.06450.
- 48. Danaee P, Rouches M, Wiley M, et al. bpRNA: large-scale automated annotation and analysis of RNA secondary structure. Nucleic Acids Res 2018;46(11):5381–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yong H, Huang J, Hua X, et al. Gradient centralization: a new optimization technique for deep neural networks. European Conference on Computer Vision 2020.
- 50. Zeki Yalniz I, Jégou H, Chen K, et al. Billion-scale semi-supervised learning for image classification. CoRR. abs/1905.005462019. [Google Scholar]
- 51. Wayment-Steele HK, Kladwang W, Das R. RNA secondary structure packages ranked and improved by high-throughput experiments. Nat Methods 2022;19:1234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. He Z, Liang Z, Li Z, et al. Lineardesign: efficient algorithms for optimized mRNA sequence design. 2020. Retrieved from https://arxiv.org/abs/2004.10177.
- 53. Wayment-Steele HK, Kim DS, Choe CA, et al. Theoretical basis for stabilizing messenger RNA through secondary structure design. Nucleic Acids Research 2021;49:10604–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pardi N, Hogan MJ, Porter FW, et al. MRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov 2018;17(4):261–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jeong DE, McCoy M, Artiles K, et al. Assemblies of putative sars-cov2-spike-encoding mRNA sequences for vaccines bnt162b2 and mRNA-1273. Retrieved from https://github.com/NAalytics/Assemblies-of-putative-SARS-CoV2-spike-encoding-mRNA-sequences-for-vaccines-BNT-162b2-and-mRNA-1273.
- 56. Zhang NN, Li XF, Deng YQ, et al. A thermostable mRNA vaccine against covid-19. Cell 2020;182:1271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Y, Zhang Z, Luo J, et al. mRNA vaccine: a potential therapeutic strategy. Mol Cancer 2021;20:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beltagy I, Peters ME, Cohan A. Longformer: the long-document transformer, 2020. Retrieved from https://arxiv.org/abs/2004.05150.
- 59. Wang S, Li BZ, Khabsa M, et al. Linformer: self-attention with linear complexity, 2020.
- 60. Choromanski K, Likhosherstov V, Dohan D, et al. Rethinking attention with performers, 2020.
- 61. Zaheer M, Guruganesh G, Dubey A, et al. Big bird: transformers for longer sequences. Proceedings of NeurIPS. 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
OpenVaccine dataset is available at https://www.kaggle.com/c/stanford-covid-vaccine/data. Pretrained models can be accessed from Kaggle notebooks: https://www.kaggle.com/shujun717/rnadegformer-inference.