

Abstract
Para C—H borylations (CHB) of tetraalkylammonium sulfates and sulfamates have been achieved using bipyridine-ligated Ir boryl catalysts. Selectivities can be modulated by both the length of the alkyl groups in the tetraalkylammonium cations and the substituents on the bipyridine ligands. Ion pairing, where the alkyl groups of the cation shield the meta C—H bonds in the counter-anions, is proposed to account for para selectivity. The 4,4′-dimethoxy-2,2′-bipyridine ligand gave superior selectivities.
For two competing pathways, a difference in barrier heights of 2.5 kcal·mol−1 is sufficient for 99% of the reactants to follow the favored pathway in a chemical reaction. This is lower than the barrier for converting the anticonformer of butane to the gauche form.1 In transition-metal mediated reactions, a classic mode for selectively functionalizing bonds in a substrate relies on the coordination of an atom in a reactant functional group to the metal center of a compound or catalytic intermediate. The magnitudes of the ligand–metal interactions are at least an order of magnitude greater than the difference in barrier heights necessary for 99:1 selectivity. Consequently, design of catalysts where selectivity is conferred by weakly coordinating groups,2 as well as catalysts that leverage even weaker interactions (e.g., hydrogen-bonding, ion-pairing, dipole–dipole, etc.) for selective transformations,3,4 is attracting significant attention.
C–H functionalizations offer both atom and step economical means of converting ubiquitous C–H bonds to a range of functional groups.5,6 C–H borylations (CHBs) convert C–H bonds to C–B bonds and are mediated by both metal and metal-free catalysts.7–9 CHB reactions are valuable due to (i) facile substitution of the boron moiety by numerous functional groups and (ii) functional group tolerance of CHB catalysts, particularly those containing Ir.
The first Ir CHB catalysts enabled C(sp2)–H functionalizations with high selectivity for the most sterically accessible C–H bonds.10–12 In substrates where multiple C–H bonds are sterically accessible, early generation catalysts often give isomer mixtures, as well as multiply borylated products. To overcome these limitations, more selective Ir catalysts have been designed. The first examples were ortho-selective, relying on strongly coordinating functional groups in the substrate,13,14 while later reports exploited weaker interactions for ortho selectivity.15,16
By comparison, meta and para CHBs pose different challenges because their C–H bonds are farther from the functional group. One meta-selective CHB has been ascribed to a classical chelate-directed mechanism,17 while others rely on Ir ligands bearing groups that engage in noncovalent interactions with substrate functional groups to effect meta CHB.18–20
Figure 1 depicts approaches for para-selective CHB. The first CHBs with high para selectivity involved electrophilic additions of borenium cations to arenes bearing ortho, para-directors.21 Sterically directed CHBs rely on hindered phosphine ligands and substrates with large substituents.22 More recently, para borylations of esters and amides have been achieved through noncovalent interactions with K ions or coordination of the amide oxygen to hindered Lewis acids.23,24
Figure 1.
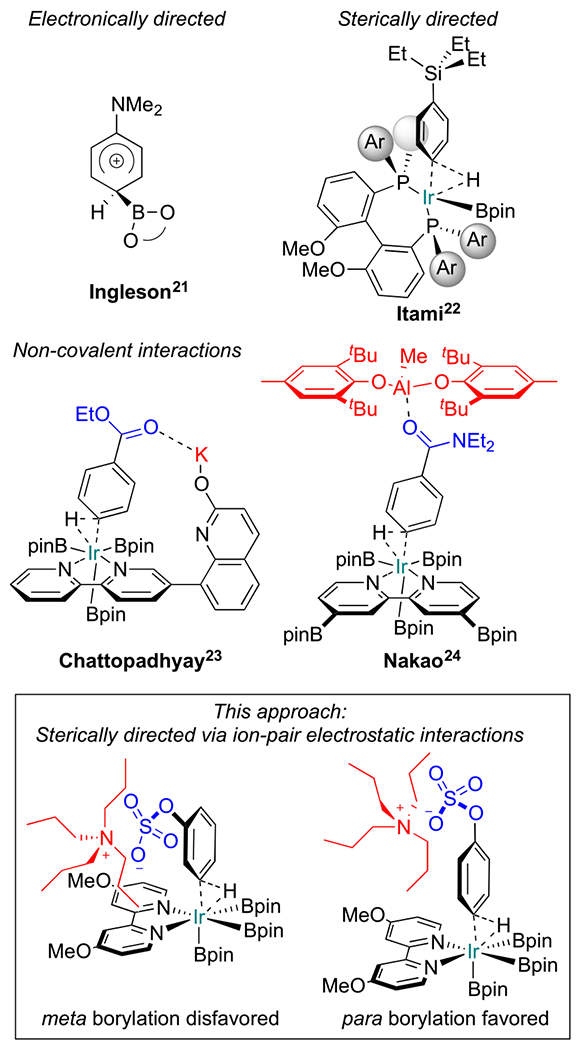
Para C–H borylations.
Our inspiration was based on the Phipps’ group ion-pair directed CHBs with one key difference.19 Instead of using oppositely charged groups on the ligand and substrate, combinations where groups on the ligand and substrate had the same charge were surveyed with the expectation that para borylation would be favored because electrostatic repulsions between the ligand and substrate would be minimized. However, a control experiment where tetrabutylammonium 2-chlorophenyl sulfate (1a′) was subjected to standard borylation conditions with a neutral bipyridine ligand (sealed tube with B2pin2 as the boron source, 1.5 mol % [Ir(cod)-OMe]2 as the precatalyst and 3 mol % dtbpy as the ligand, in THF at 80 °C) showed promising results with 6:1 para to meta regioselectivity (Table 1, entry 1). We hypothesized that substrate ion-pairing interactions, where the n-butyl groups of the cation shield the meta C–H bonds of the counter-anions, accounted for the para selectivity.
Table 1.
Ligand Effect on Para CHB of 1a′
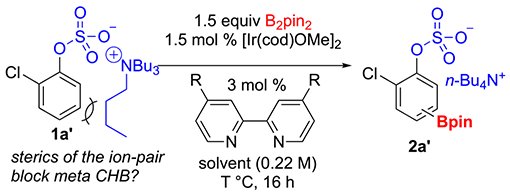
| |||||
|---|---|---|---|---|---|
| entry | ligand (R =) | solvent | T (°C) | conva | p/ma |
| 1 | t-Bu (L1) | THF | 80 | 100 | 6:1 |
| 2 | CN (L2) | THF | 80 | 0 | ND |
| 3 | H (L3) | THF | 80 | 89 | 7:1 |
| 4 | Me (L4) | THF | 80 | 96 | 10:1 |
| 5 | OMe (L5) | THF | 80 | 100 | 13:1 |
| 6 | tmphen (L6) | THF | 80 | 100 | 6:1 |
| 7 | phen (L7) | THF | 80 | 45 | 9:1 |
| 8 | NMe2 (L8) | dioxane | 80 | 100 | 12:1 |
| 9 | OMe (L5) | dioxane | 80 | 100 | 14:1 |
| 10 | OMe (L5) | dioxane | 60 | 100 | 16:1 |
| 11 | OMe (L5) | dioxane | 40 | 100 | 17:1 |
| 12b | OMe (L5) | dioxane | rt | 83 | 21:1 |
Determined by 1H NMR analysis.
Reaction carried out for 50 h
In Nakao’s study, ligand geometry played a key role in enhancement of the para selectivity.24 Similarly, we have observed that ligand choice can impact CHB regiochemistry where there is little steric differentiation between different arene C–H bonds.25
Therefore, we tested commercially available substituted bipyridine and phenanthroline ligands (Table 1) in CHB reactions run at 80 °C. The reactivity of the ligands is in accordance with previously noted electronic effects,26 with electron-rich ligands affording a more active system relative to electron-poor ligands. The borylation in THF with 4,4′-dimethoxy-2,2′-bipyridine (L5) as the ligand went to 100% conversion and afforded the best para selectivity (13:1). This observation is notable in that L5 is a nontraditional CHB ligand. Switching the solvent to dioxane slightly increased the para selectivity, whereas other apolar solvents worsened regioselectivity (see Supporting Information (SI) for details). Running the reactions at lower temperature (60 °C (entry 10) and 40 °C (entry 11)) further improved the para selectivity while still allowing for full conversion. The reaction at room temperature (entry 12) afforded 21:1 para selectivity, but starting material remained even after 50 h.
With the experiments in Table 1 establishing 4,4′-dimethoxy-2,2′-bipyridine and dioxane as our ligand and solvent of choice, we next investigated the effect of the tetraalkylammonium salt on para selectivity. DFT geometry optimization of 1a′ (Figure 2), using the B3LYP functional and 6-31G* basis set for all the atoms suggested to us that a slightly shorter alkyl chain would still block the meta position but leave the para position more exposed, thus potentially leading to improved para selectivity. As shown in Scheme 1, the CHB of tetrapropylammonium 2-chlorophenyl sulfate (1a) validated this hypothesis, as running the reaction with ligand L5 in dioxane at 40 °C pushed the para selectivity to 22:1. We also examined tetraethylammonium 2-chlorophenyl sulfate (1a′′) as a substrate. In terms of chain shortening, clearly diminishing returns had set in as the para selectivity decreased to 6:1.
Figure 2.
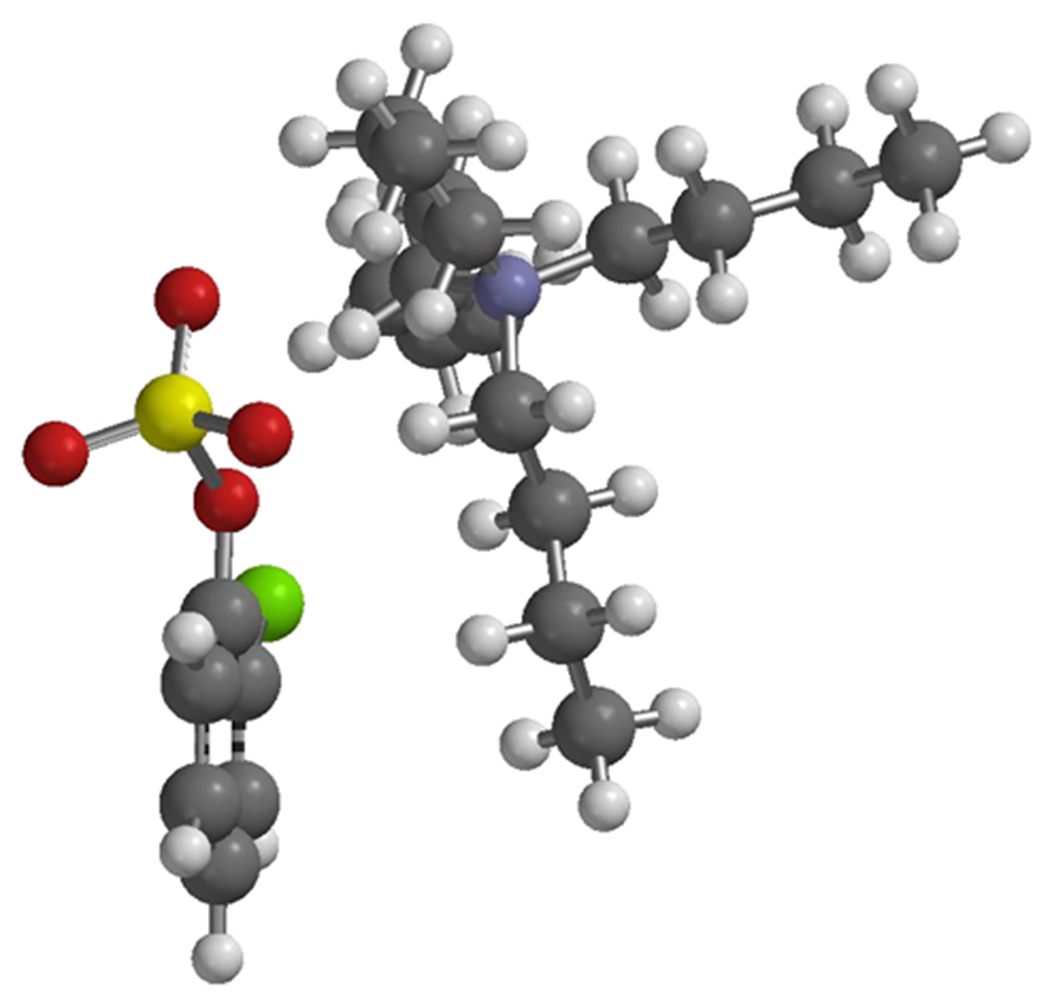
Lowest energy conformation geometry of 1a′.
Scheme 1.
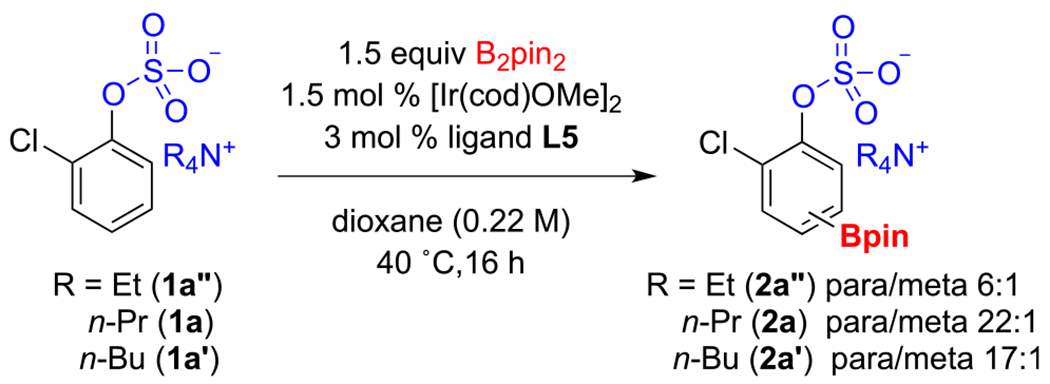
Effect of Alkyl Chain Length on Regioselectivity
Based on our results, we chose to test a series of phenol derived sulfates with n-Pr4N+ as the counterion to determine substrate scope. As illustrated in Scheme 2, borylations of a series of 2-substituted phenol derived sulfates produced the para regioisomer as the major isomer, often with >20:1 selectivity. Most isolated yields were in the 70–80% range. Notably upon isolation the para to meta isomer was enhanced, in some cases to >50:1.
Scheme 2. Borylation of Phenol Derived Sulfatesa.
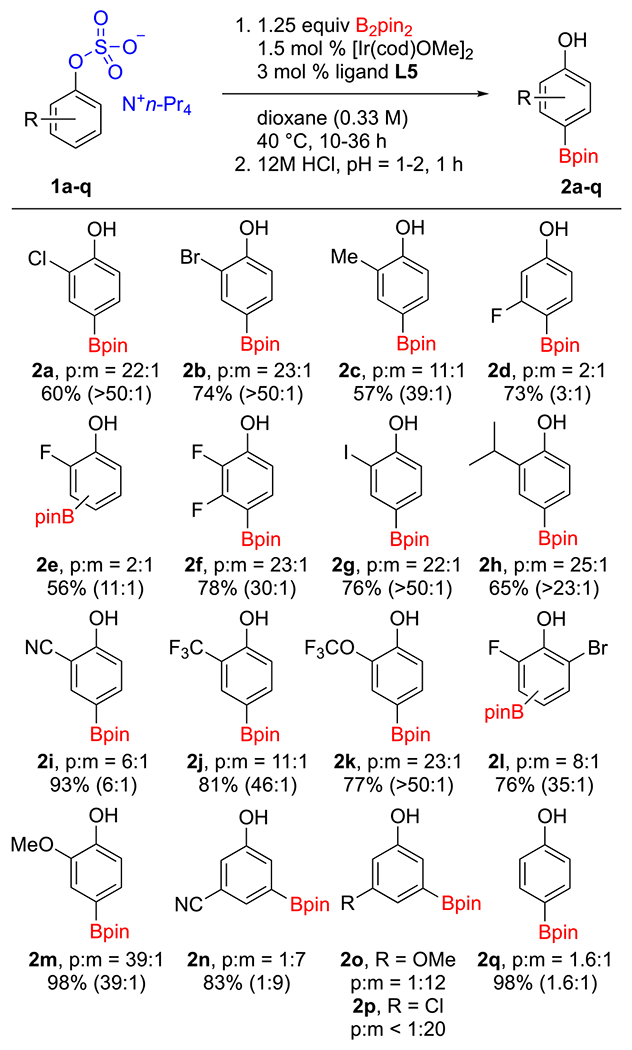
aPara/meta ratios were measured by 1H NMR on crude reaction mixtures of the borylated sulfates. Yields refer to isolated material with the para/meta ratio of the isolated material given in parentheses (2o and 2p were not isolated). See the SI for experimental details, including preparations of the starting sulfates.
Given that CHB ortho to small substituents is common,27 borylation at the C-3 and C-5 meta CH bonds of 2-fluoro-(1e) and 2-cyanophenol sulfate (1i) are possible. Indeed, analysis of the crude reaction mixture indicated that 1i gave a mixture of the para to 5-Bpin to 3,5-diBpin products in a ratio of approximately 7.5:1:0.4. For substrates 1e and 1l, the observed minor isomer was that with the Bpin ortho to the fluoro group and no diborylation was observed. Given this preference, it was perhaps somewhat surprising that 3-fluorophenol sulfate (1d) produced a relatively large amount of the meta regioisomer.
Not surprising was that the CHB of 3-substituted phenol sulfates (1n–p) gave the meta isomer as the major product, showing that such ion-pair interactions are limited in their ability to overcome steric crowding of the para CH position. Last, we borylated the sulfate of phenol and observed the para, meta, and 3,5-dimeta borylated products in a ratio of 4.4:1:1.8, or a para/meta ratio of 1.6:1. This result is consistent with the assumption that the ion pairing can only block one meta site and thus the reactions need a 2-substituent to sterically block the second meta CH bond.
Turning to anilines (Scheme 3), we subjected tetrapropylammonium 2-chlorophenylsulfamate (3a) to our now standard conditions. The para selectivity (40:1) was even better than that observed for 1a. Questioning if the chain length of the tetraalkylammonium salt would also impact the para selectivity for aniline derivatives, we prepared and reacted the tetrabutylammonium salt (3a′). In contrast to the phenol sulfates, employing this counterion met with 43:1 para selectivity and a higher isolated yield. Owing to this result and that the tetrabutylammonium salts are somewhat easier to prepare and isolate, we chose n-Bu4N+ as the counterion for CHBs of a series of aniline sulfamates. The para selectivities for 3b′ and 3d′ were excellent, while again selectivity for a 3-fluoro substrate (3c′) suffered, giving only a 2.4:1 para/meta ratio. Isolation of the hydrolyzed aniline following borylation of 3c′ proved difficult. Therefore, the reaction was quenched with acetyl chloride, facilitating the isolation of 4c′.
Scheme 3. Borylation of Aniline Derived Sulfamatesa.
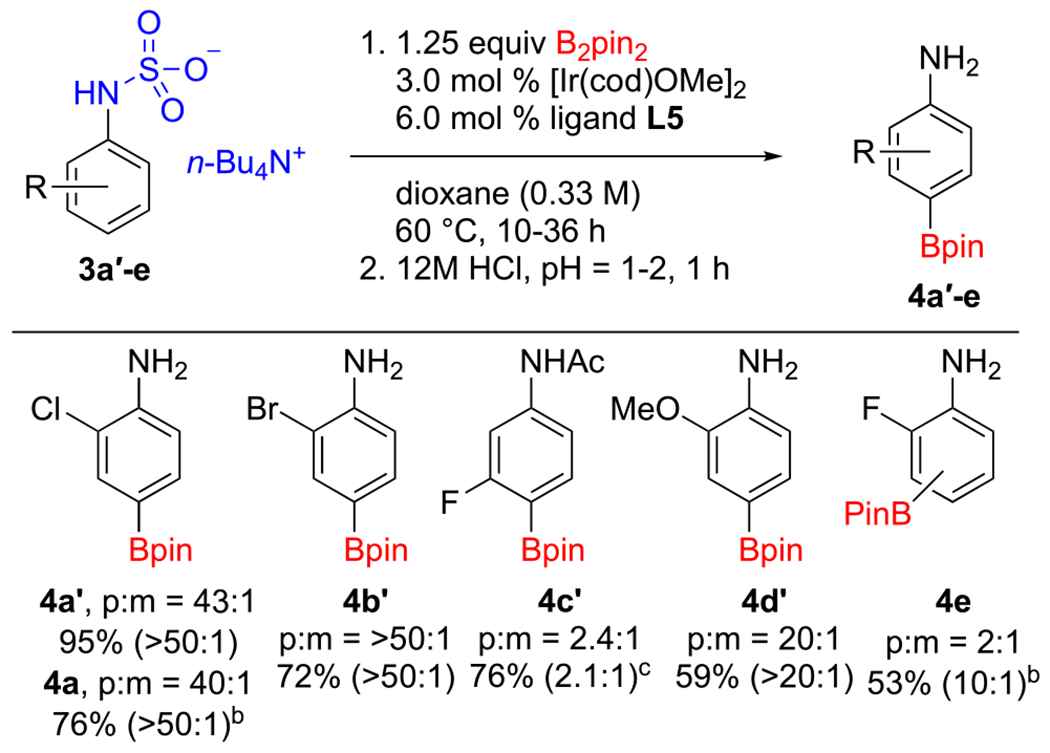
aPara/meta ratios were measured by 1H NMR (or 19F NMR for 4c′) on crude reaction mixtures of the borylated sulfamates. Yields refer to isolated material with the para/meta ratio of the isolated material given in parentheses. See the SI for experimental details, including preparations of the starting sulfamates. bRun with the n-Pr4N+ counterion. cProduct isolated as the acetamide.
Finally, we surveyed benzyl alcohol derived sulfates (Scheme 4). Generally, these substrates reacted with somewhat diminished para selectivity relative to their phenol and aniline counterparts. Products 6b and 6d were generated in lower yields owing in part to lower conversions and, for 6d, loss of the meta isomer upon isolation. Again, borylation of a substrate with fluorine in the 2-position (5g) afforded a significant amount of product with the Bpin ortho to the fluorine. By applying the CHB conditions to the n-Bu4N+ counterion, 5a–5c revealed that the counterion has a similar influence on the regioselectivity as observed for the phenols.
Scheme 4. Borylation of Benzyl Alcohol Derived Sulfatesa.
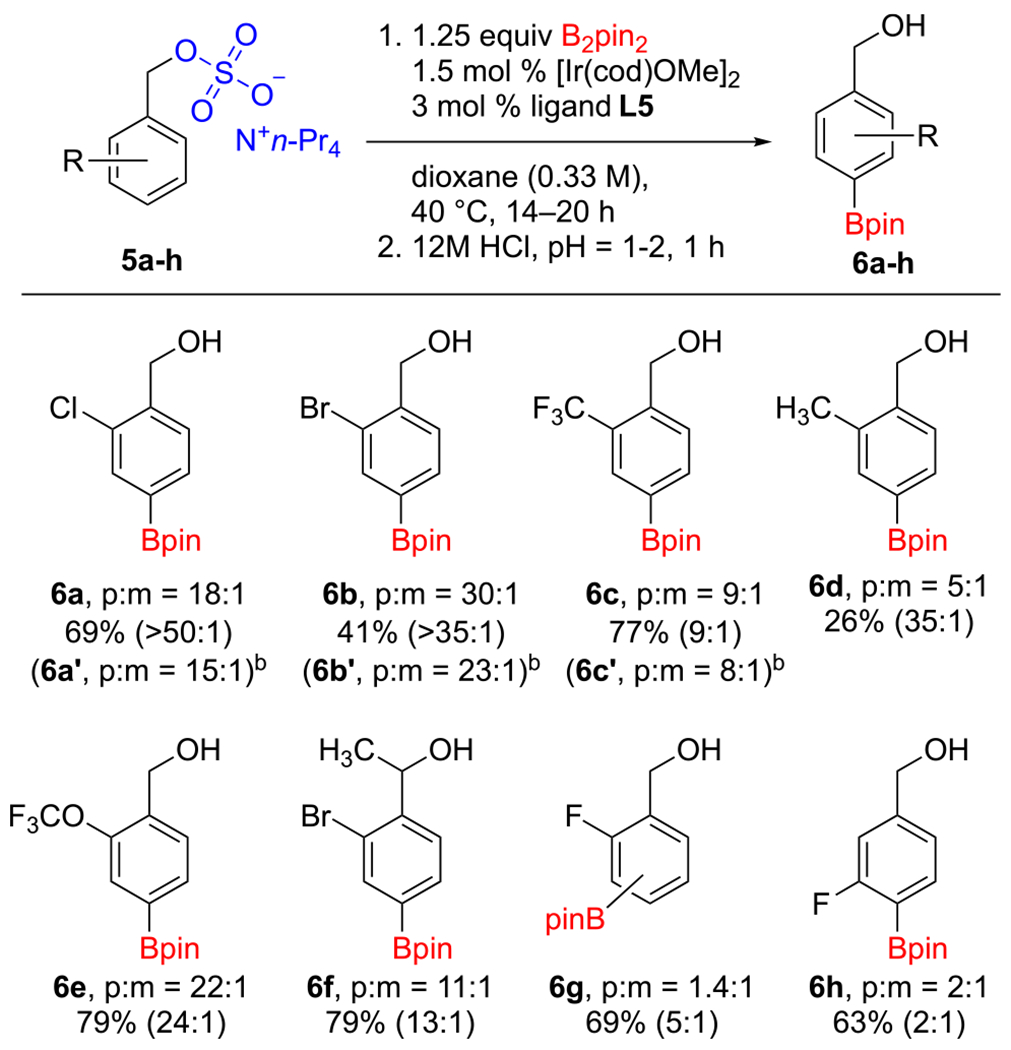
aPara/meta ratios were measured by 1H NMR on crude reaction mixtures of the borylated sulfates. Yields refer to isolated material with the para/meta ratio of the isolated material given in parentheses. See the SI for experimental details, including preparations of the starting sulfates. bRun with the n-Bu4N+ counterion.
In summary, ion-pair electrostatic interactions can be used to direct Ir-catalyzed borylation to the para position of sulfates and sulfamates derived from phenols, anilines, and benzyl alcohols. We hypothesize that the source of the para selectivity is a steric block created by the carbon chain of the tetrabutylammonium counterion. For sulfates derived from phenols and benzyl alcohols, n-Pr4N+ salts gave better selectivity than their n-Bu4N+ counterparts. The chain length of tetralkylammonium salt was not as influential on the borylation of the sulfamates derived from anilines. Notably, optimal results were observed with the nontraditional CHB ligand 4,4′-dimethoxy-2,2′-bipyridine. This serves to remind the community to look beyond dtbpy or tmphen when optimizing CHB reactions.28
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Daniel Holmes and Li Xie of the Max. T. Rogers NMR Facility and the MSU RTSF Mass Spectrometry Core for their assistance in compound characterization. We thank the NIH (GM63188) for financial support and BoroPharm, Inc. for a gift of B2pin2.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08464.
Experimental procedures, including preparation of starting materials, and compound characterization data (PDF)
The authors declare the following competing financial interest(s): S.L.M., M.R.S., and R.E.M. own a percentage of BoroPharm, Inc.
REFERENCES
- (1).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books: 2006. [Google Scholar]
- (2).Engle KM; Mei T-S; Wasa M; Yu J-Q Weak Coordination as a Powerful Means for Developing Broadly Useful C-H Functionalization Reactions. Acc. Chem. Res 2012, 45, 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Raynal M; Ballester P; Vidal-Ferran A; van Leeuwen PWNM Supramolecular Catalysis. Part 1: Non-Covalent Interactions as a Tool for Building and Modifying Homogeneous Catalysts. Chem. Soc. Rev 2014, 43, 1660–1733. [DOI] [PubMed] [Google Scholar]
- (4).Davis HJ; Phipps RJ Harnessing Non-Covalent Interactions to Exert Control over Regioselectivity and Site-Selectivity in Catalytic Reactions. Chem. Sci 2017, 8, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dixneuf PH; Doucet H C–H Bond Activation and Catalytic Functionalization I; Springer: 2015. [Google Scholar]
- (6).Dixneuf PH; Doucet H C–H Bond Activation and Catalytic Functionalization II; Springer: 2016. [Google Scholar]
- (7).Mkhalid IAI; Barnard JH; Marder TB; Murphy JM; Hartwig JF. C-H Activation for the Construction of C-B Bonds. Chem. Rev 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- (8).Xu L; Wang G; Zhang S; Wang H; Wang L; Liu L; Jiao J; Li P Recent Advances in Catalytic C–H Borylation Reactions. Tetrahedron 2017, 73, 7123–7157. [Google Scholar]
- (9).Fontaine F-G; Rochette É Ambiphilic Molecules: From Organometallic Curiosity to Metal-Free Catalysts. Acc. Chem. Res 2018, 51, 454–464. [DOI] [PubMed] [Google Scholar]
- (10).Cho JY; Iverson CN; Smith MR III. Steric and Chelate Directing Effects in Aromatic Borylation. J. Am. Chem. Soc 2000, 122, 12868–12869. [Google Scholar]
- (11).Cho J-Y; Tse MK; Holmes D; Maleczka RE Jr; Smith MR III. Remarkably Selective Iridium Catalysts for the Elaboration of Aromatic C-H Bonds. Science 2002, 295, 305–308. [DOI] [PubMed] [Google Scholar]
- (12).Ishiyama T; Takagi J; Ishida K; Miyaura N; Anastasi NR; Hartwig JF Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc 2002, 124, 390–391. [DOI] [PubMed] [Google Scholar]
- (13).Kawamorita S; Ohmiya H; Hara K; Fukuoka A; Sawamura M Directed Ortho Borylation of Functionalized Arenes Catalyzed by a Silica-Supported Compact Phosphine-Iridium System. J. Am. Chem. Soc 2009, 131, 5058–5059. [DOI] [PubMed] [Google Scholar]
- (14).Ishiyama T; Isou H; Kikuchi T; Miyaura N Ortho-C-H Borylation of Benzoate Esters with Bis(pinacolato)diboron Catalyzed by Iridium-Phosphine Complexes. Chem. Commun 2010, 46, 159–161. [DOI] [PubMed] [Google Scholar]
- (15).Roosen PC; Kallepalli VA; Chattopadhyay B; Singleton DA; Maleczka RE Jr.; Smith MR III. Outer-Sphere Direction in Iridium C-H Borylation. J. Am. Chem. Soc 2012, 134, 11350–11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chattopadhyay B; Dannatt JE; Andujar-De Sanctis IL; Gore KA; Maleczka RE Jr; Singleton DA; Smith MR III. Ir-Catalyzed Ortho-Borylation of Phenols Directed by Substrate-Ligand Electrostatic Interactions: A Combined Experimental/in Silico Strategy for Optimizing Weak Interactions. J. Am. Chem. Soc 2017, 139, 7864–7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bisht R; Chattopadhyay B Formal Ir-Catalyzed Ligand-Enabled Ortho and Meta Borylation of Aromatic Aldehydes via in Situ-Generated Imines. J. Am. Chem. Soc 2016, 138, 84–87. [DOI] [PubMed] [Google Scholar]
- (18).Kuninobu Y; Ida H; Nishi M; Kanai M A Meta-Selective C–H Borylation Directed by a Secondary Interaction between Ligand and Substrate. Nat. Chem 2015, 7, 712–717. [DOI] [PubMed] [Google Scholar]
- (19).Davis HJ; Mihai MT; Phipps RJ Ion Pair-Directed Regiocontrol in Transition-Metal Catalysis: A Meta-Selective C-H Borylation of Aromatic Quaternary Ammonium Salts. J. Am. Chem. Soc 2016, 138, 12759–12762. [DOI] [PubMed] [Google Scholar]
- (20).Yang L; Uemura N; Nakao Y Meta-Selective C–H Borylation of Benzamides and Pyridines by an Iridium–Lewis Acid Bifunctional Catalyst. J. Am. Chem. Soc 2019, 141, 7972–7979. [DOI] [PubMed] [Google Scholar]
- (21).Del Grosso A; Singleton PJ; Muryn CA; Ingleson MJ Pinacol Boronates by Direct Arene Borylation with Borenium Cations. Angew. Chem., Int. Ed 2011, 50, 2102–2106. [DOI] [PubMed] [Google Scholar]
- (22).Saito Y; Segawa Y; Itami K Para-C–H Borylation of Benzene Derivatives by a Bulky Iridium Catalyst. J. Am. Chem. Soc 2015, 137, 5193–5198. [DOI] [PubMed] [Google Scholar]
- (23).Hoque ME; Bisht R; Haldar C; Chattopadhyay B Noncovalent Interactions in Ir-Catalyzed C-H Activation: L-Shaped Ligand for Para-Selective Borylation of Aromatic Esters. J. Am. Chem. Soc 2017, 139, 7745–7748. [DOI] [PubMed] [Google Scholar]
- (24).Yang L; Semba K; Nakao Y Para-Selective C-H Borylation of (Hetero) Arenes by Cooperative Iridium/Aluminum Catalysis. Angew. Chem., Int. Ed 2017, 56, 4853–4857. [DOI] [PubMed] [Google Scholar]
- (25).Miller SL; Chotana GA; Fritz JA; Chattopadhyay B; Maleczka RE Jr; Smith MR III C-H Borylation Catalysts That Distinguish Between Similarly Sized Substituents Like Fluorine and Hydrogen. Org. Lett 2019, 21, 6388–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ishiyama T; Takagi J; Hartwig JF; Miyaura N A Stoichiometric Aromatic C-H Borylation Catalyzed by iridium(I)/2,2’-Bipyridine Complexes at Room Temperature. Angew. Chem, Int. Ed 2002, 41, 3056–3058. [DOI] [PubMed] [Google Scholar]
- (27).Chotana GA; Rak MA; Smith MR III. Sterically Directed Functionalization of Aromatic C-H Bonds: Selective Borylation Ortho to Cyano Groups in Arenes and Heterocycles. J. Am. Chem. Soc 2005, 127, 10539–10544. [DOI] [PubMed] [Google Scholar]
- (28).During preparation of our manuscript, we became aware that the Phipps group had developed a similar approach to para-selective borylation. We are grateful to them for agreeing to publish their results in a back-to-back fashion with our own: Mihai MT; Williams BD; Phipps RJ Para-Selective C–H Borylation of Common Arene Building Blocks Enabled by Ion-Pairing with a Bulky Countercation. J. Am. Chem. Soc 2019, DOI: 10.1021/jacs.9b07267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.