

Abstract
The development of clinically actionable pharmaceuticals against coronavirus disease (COVID-19); an infectious disease caused by the SARS-CoV-2 virus is very important for ending the pandemic. Coronavirus spike glycoprotein (GP)-Receptor Binding Domain (RBD) and its interaction with host receptor angiotensin converting enzyme 2 (ACE2) is one of the most structurally understood but therapeutically untapped aspect of COVID-19 pathogenesis. Binding interface based on previous x-ray structure of RBD/ACE2 were virtually screened to identify fragments with high-binding score from 12,000 chemical building blocks. The hit compound was subjected to fingerprint-based similarity search to identify compounds within the FDA-approved drug library containing the same core scaffold. Identified compounds were then re-docked into of RBD/ACE2. The best ranked compound was validated for RBD/ACE2 inhibition using commercial kit. Molecular dynamics simulation was conducted to provide further insight into the mechanism of inhibition. From the original 12000 chemical building blocks, benzimidazole (BAZ) scaffold was identified. Fingerprint-based similarity search of the FDA-approved drug library for BAZ-containing compounds identified 12 drugs with the benzimidazole-like substructure. When these compounds were re—docked into GP/ACE2 interface, the consensus docking identified bazedoxifene as the hit. In vitro RBD/ACE2 inhibition kinetics showed micromolar IC50 value (1.237 μM) in the presence of bazedoxifene. Molecular dynamics simulation of RBD/ACE2 in the presence BAZ resulted in loss of contact and specific hydrogen-bond interaction required for RBD/ACE2 stability. Taken together, these findings identified benzimidazole scaffold as a building block for developing novel RBD/ACE2 complex inhibitor and provided mechanistic basis for the use of bazedoxifene as a repurposable drug for the treatment of COVID-19 acting at RBD/ACE2 interface.
Keywords: COVID-19, Receptor binding domain, ACE2, Bazedoxifene, SARS-COV2
Graphical abstract
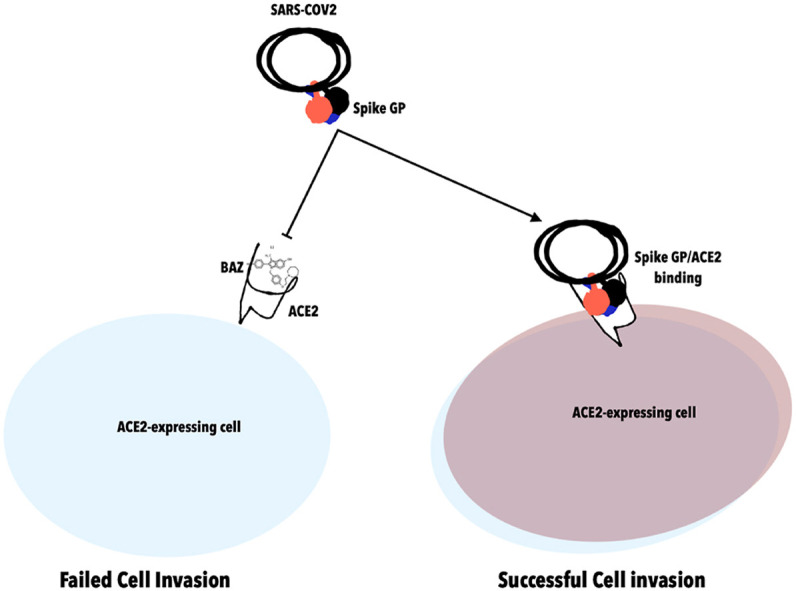
SARS-COV2 bears spike glycoprotein required for host ACE2 interaction; in the presence of Bazedoxifene (BAZ)RBD/ACE2 interaction is impossible; thus resulting in failed cell invasion.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of coronavirus disease 2019 (COVID-19); a pneumonia-like illness responsible for the COVID-19 pandemic infecting approximately 25 million people and causing more than 835,000 deaths (Johns Hopkins, Coronavirus resource center) and counting [1,2]. Two general approaches have been adopted for treating COVID-19 cases; vaccine and therapeutics development.
Small molecules such as remdesivir, hydroxychloroquine, ribavirin, and ritonavir were initially repurposed as therapeutics for COVID-19 [3] but have not performed satisfactorily clinically. New drugs specifically developed for COVID-19 by Merck (molnupiravir), Pfizer (ritonavir in combination with PF-07321332) [4] have been adjudged more efficacious [5]. Research efforts at developing plant-based therapeutic agents is also worthy of mention [6].
Although comparatively, vaccines have been more successful in combating COVID-19 than small molecular weight drugs within clinical settings, this success has been threatened by the fast-rising cases of mutations [7]. Thus, making discovery and development of novel anti-COVDI-19 agents highly desirable in the long run.
To this end, 12,000 chemical building blocks were computationally screened to identify possible virus entry inhibitor candidates acting at the spike glycoprotein-angiotensin (RBD) converting enzyme II (ACE2) interface [8]. Whilst most of the previous successful efforts have targeted SARS-COV-2 proteases [9], the current strategy provides a complementary approach such that a successful candidate will potentially reduce virus tissue reservoir as the distribution of ACE2 has been confirmed in most cells with the exception of matured erythrocytes [10]. Indeed, Pfizer/BioNTech's BNT162b2 and Moderna's mRNA-1273 platforms drive bioactivity through SARS-CoV-2 spike glycoprotein interaction with host (human) ACE2 receptor to gain entry into a cell to initiate tissue infection [11]. SARS-CoV-2 spike glycoprotein [12] is indeed a hot spot for mutation which clearly threatens vaccine efficacy. A few other research is also targeting RBD/ACE2 interface, of note is the discovery of bromelain [13], (−)-pipoxide, 2-(p-hydroxybenzyl)benzofuran-6-ol, 1-(4-hydroxy-3-methoxyphenyl)-2-4-[(E)-3-hydroxy-1-propenyl]-2-methoxyphenoxy-1,3-propanediol, and Rhein [14].
This study therefore identified benzimidazole fragment as particularly interesting, and when compounds bearing benzimidazole scaffold were searched from FDA-approved library, we have identified bazedoxifene as the most likely candidate inhibitor of RBD/ACE2 interaction through molecular docking, atomistic simulation and validation by inhibitory kinetics studies. The previous reports that benzimidazole is a potent entry inhibitor to hepatitis C virus [15] somewhat improves our overall confidence that bazedoxifene may represent a repurposable anti-COVID-19 agent.
2. Materials and methods
2.1. 2D fingerprint search, ligand library, and protein preparation
The database of FDA-approved drugs (open.fda.gov) were retrieved and filtered (Datawarrior Suite) using the molecular weight cutoff 200 ≥ 700 g/mol. The residual compounds were searched (open babel) for benzimidazole substructure using a path-based fingerprint (FP2). Each compound was scored using the Tanimoto coefficient and plotted as a function of the population size using Graphed prism.
Unless otherwise stated, all ligands for molecular docking were incorporated into Maestro and prepared using the Schrodinger suite version 2018–1. Employing Ligprep [16], Epik [17] with OPLS3 force field for protonation, stereoisomerization, tautomers generation, and to achieve biological conformers. Energy minimization was achieved for all tautomeric states at a pH of 7 ± 2.2019-nCoV RBD/ACE2 Complex (2.9 Å) was retrieved from PDB [18] with the PDB ID 6M17 [19]. The complex was prepared using the protein preparation wizard module in maestro 11.5 by default settings. The complex was optimized and then minimized using the OPLS3 force field by converging heavy atoms to RMSD of 0.3 Å. The complex interaction interface for docking was generated with the receptor grid generation tool in maestro 11.5. The interaction interface between RBD/ACE2 complex was defined by amino acid residues: K417/Q409/R403/D405/R408/Y505 (RBD) and D30/N33/T324/F356/(ACE2), with van der Waals scaling factor 1.00 and charge cutoff of 0.25 around the complex interaction interface residues.
2.2. Molecular docking
Molecular docking was conducted using Autodock Vina [20], and Glide module on maestro 11.5 [21]. The library of compounds was docked into the interface of the target using the standard precision (SP) and Extra precision (XP) algorithm [22], applying a scaling factor of 0.8 and partial charge cutoff of 0.15, the ligand was handled as flexible. 30 Å in all directions of the interface between RBD/ACE2 complex defined by amino acid residues: K417/Q409/R403/D405/R408/Y505 (RBD) and D30/N33/T324/F356/(ACE2) was used as dock grid dimension. Lastly, the binding affinity of the receptor-ligand complex was ranked according to Glide score (SP and XP) while Vina score was utilized for the Autodock vina program.
2.3. Molecular dynamics simulation
2.3.1. Biosystems setup for atomistic simulation
The input files for MD simulation for each of the biosystems (apo-RBD/ACE2, BAZ-RBD/ACE2; ACE2 = aa21-559; RBD = aa336-518) was generated using CHARMM-GUI webserver (www.charmm-gui.org). BAZ parametrization was performed using ParamChem service (https://cgenff.paramchem.org) as implemented on CHARMM-GUI webserver. During biosystem build-up, protein and ligand atoms were defined by CHARMM36 all-atom additive force field parameters, solvated in TIP3P explicit water model and neutralized with Na+/CL− at 0.15 M [23]. Details of biosystem setup using CHARMM-GUI have been previously published [[24], [25], [26]].
Molecular dynamics simulation was run on GROMACS (ver. 5) [27] software. During equilibration, the biosystems were subjected to constant pressure and temperature (NPT; 310K, 1 bar) conditions using Berendsen temperature and pressure coupling algorithms as implemented in GROMACS. van der Waals interactions were estimated at 10 Å, long-range electrostatic interactions were computed using particle mesh Ewald (PME) summation scheme while equation of atomic motion was integrated using the leap-frog algorithm at 2 fs time step for a total time of 50 ns with positional restraints imposed on the heavy atoms in all directions. Production simulations were performed at 100 ns with the removal of atomic restraints. All calculations were performed on Super-Micro workstations (32-E2600 Intel Xeon CPUs, M6000 GPUs Accelerator PCI-E ×16 Card/node) housed at Bio-Computing Research Unit (B-cRU), Mols and Sims, Ado Ekiti, Ekiti State, Nigeria.
2.3.2. Post-simulation trajectory analysis and MMPBSA calculation
Unless otherwise stated, 3D atomic representations were drawn using Visual Molecular Dynamics (VMD) software [28]. Interatomic distance was calculated using g_dist tool inbuilt in GROMACS software. Population plots from distance values were plotted using GraphPad-Prism software. Binding free energy was calculated GROMACS Tool for high-throughput MM-PBSA calculations (g_mmpbsa) algorithm as described [29]. All line graphs were plotted as mean of 2 independent runs using GraphPad prism (ver 6.0e, 2014).
2.4. RBD/ACE2 complex inhibition kinetics
Assessment of SARS-CoV2 RBD and hACE2 binding inhibition by Bazedoxifene (Cat: HY-A0031/CS-0932, Lot: 58414) was performed using COVID-19 Spike-ACE2 binding assay kit (RayBiotech, Inc, Cat #: CoV-SACE2-1, https://www.raybiotech.com/covid-19-spike-ace2-binding-assay-kit/) following the manufacturer's protocol) Bazedoxifene was dissolved in water and aliquoted at concentrations 0.01, 0.03, 0.1, 0.3, 1.0, 3.0 mM in triplicates for the study. The SARS-CoV2 RBD and hACE2 binding inhibitory capacity of each concentration was assessed in triplicate. Compounds at noted concentration were mixed with recombinant hACE2 protein orPBS (Control), added to ELISA plate pre-coated with recombinant SARS-Cov2 S-protein RBD and incubated overnight at 4 °C with shaking. Unbound ACE2 was removed by washing, and binding was assessed based on anti-ACE2 antibody- HRP-conjugated anti-goat IgG reaction with 3,3′,5,5′-tetramethylbenzidine (TMB). Absorbance at 450 nm was measured with a ELISA microplate reader. Dose-response graph was plotted using GraphPad Prism and IC50 value was calculated.
3. Results
3.1. Virtual screening identifies benzimidazole fragment as high-affinity binder at RBD/ACE2 interface
Fragment-based lead discovery (FBLD) represents an efficient approach to drug discovery, taking advantage of the highly diverse fragment libraries (12,000 compounds) available at the Chem Bridge Corporation (https://www.chembridge.com), a virtual screening performed to identify potential hits at the RBD/ACE2 interface. Whilst most of the fragments had docking scores between −1.0 and −6.0 kcal/mol, three compounds namely: 1-(5-Nitro-1H-benzimidazol-2-yl)ethanol, 1-(2-piperidin-1-ylethyl)-1H-pyrazol-4-amine and 5-(3-(aminoethyk)-1,2,4-oxadiazol-5-yl)pyrrolidine-2-one have docking scores of −7.0 kcal/mol (Fig. 1 A). Although, −7.0 kcal/mol docking score is weak, instead of subsequent growing and/or combining the lead fragments to produce other leads with a higher affinity and improved physicochemical properties, we rather screened the FDA-library for chemical compounds containing benzimidazole fragment as 1-(5-Nitro-1H-benzimidazol-2-yl)ethanol had the best docking score overall.
Fig. 1.

(A) Population count of docking score distribution of building block into RBD/ACE-2 interface; the inset show the best 3 ranked compounds 2D structures. Fig. 1B: (i) Population count of FDA-approved drugs with benzimidazole scaffold based on Tanimoto coefficient scores (inset shows 12 compounds with benzimidazole scaffold substructure). (ii) A snapshot of the placement of bazedoxifene (12) within the RBD/ACE2 interface and a detailed projection of the amino acids contributing to the binding interaction. Fig. 1B. (iii) Dose-response curve fitting of RBD/ACE2 binding in the presence of graded concentration of bazedoxifene (BAZ).
2D fingerprint search was conducted into the database of FDA-approved drugs to identify and rank (Tanimoto coefficient) compounds with benzimidazole scaffold which can be rapidly repurposed as entry inhibitors in the current pandemic. The best ranked compounds were twelve (12) including albendazole(1), etonitazene(2), galeterone(3), lansoprazole(4), mavatrep(5), mebendazole(6), omeprazole(7), pantoprazole(8), rabeprazole(9), tenatoprazole(10), triclabendazole(11), and bazedoxifene(12) (Fig. 1B, i, Table 1 ). When these compounds were docked into RBD/ACE2 interface using consensus scoring method [30], bazedoxifene (BAZ) was identified as exhibiting the lowest binding score (−6.24 kcal/mol) (Supplementary Table 1.0). An overview of the binding position within the RBD/ACE2 intercalates between a network of aromatic and charged amino acids at the RBD/ACE2 interface; some of the key residues proximal to BAZ binding pocket include: D30/N33/T324/F356/(ACE2) and K417/Q409/R403/D405/R408/Y505 (RBD) (Fig. 1B, ii). These amino acids have identified as key to successful initiation of cellular invasion [31]. When the finding was validated inSARS-COV-2 spike-ACE2 binding assay, a mean IC50 value of 1.237 μM was obtained (Fig. 1B, iii) thus, strongly suggesting that BAZ is a micromolar level inhibitor of RBD/ACE2 interaction and a potential drug in the current pandemic.
Table 1.0.
Compounds with high Tanimoto coefficient with benzimidazole.
| Compounds | Vina Dock | Glide XP | Glide SP | Mean Score |
|---|---|---|---|---|
| Albendazole (1) | −5.20 | −2.84 | −4.27 | −4.10 |
| etonitazene (2) | −6.00 | −2.48 | −4.78 | −4.42 |
| galeterone (3) | −6.80 | −2.52 | −4.03 | −4.45 |
| lansoprazole (4) | −6.30 | −5.23 | −5.83 | −5.79 |
| mavatrep (5) | −8.00 | −3.81 | −5.55 | −5.79 |
| mebendazole (6) | −6.20 | −3.41 | −3.85 | −4.49 |
| omeprazole (7) | −5.40 | −5.48 | −5.27 | −5.38 |
| pantoprazole (8) | −6.50 | −3.94 | −5.81 | −5.42 |
| rabeprazole (9) | −5.70 | −5.74 | −5.05 | −5.50 |
| tenatoprazole (10) | −6.00 | −4.03 | −3.91 | −4.65 |
| triclabendazole (11) | −5.70 | −4.18 | −4.69 | −4.86 |
| Bazedoxifene* (12) | −7.40 | −5.13 | −6.20 | −6.24 |
3.2. Molecular dynamics simulation identified the atomistic basis for RBD/ACE2 inhibition
Molecular dynamics (MD) simulation has become a routine in silico tools for drug discovery due to its key advantage of incorporating structural flexibility and entropic effects into target-drug interaction, which is limited in molecular docking. Thus, MD simulation is more accurate when monitoring atomistic motions and kinetics associated with drug–target recognition and binding [32]. Since our group has previous experience in deploying MD simulation for delineating mechanism of drug-target interaction in complexes such as lysophosphatidate-LPA1 [26], nefiracetam-NMDA receptors [33], we therefore deployed MD simulation for the current studies in order to provide better insight into the mechanism of inhibition.
First, investigation was conducted into the center of mass distance between residues constituting the interface (Fig. 2 A, i) showed that at approximately 40 ns, the distance reduced to 0.4 nm in apo state, indicating contact establishment; the presence of BAZ resulted in loss of contact with mean distance 0.8–1.2 nm starting at 40 ns till the end of the simulation. (Fig. 2A, ii). Three amino acid pairs were further studied providing insight into loss of contact. Glu23/Tyr473 (Fig. 2A, iii) and Asp30/Lys471 (Fig. 2A, iv) distance evolution with time strongly suggest loss of contact around 40 ns but Tyr41/T500 had already contact post equilibration in the presence of BAZ (Fig. 2A, v). Clearly, the presence of BAZ within the RBD/ACE2 interface promotes loss of contact and possibly dissociation as suggested by the in vitro data. Lastly, the details of the amino acid network required for BAZ binding and the energy of binding were worked out using MMPBSA method [29]. Several amino acids at the RBD/ACE2 interface participate in BAZ binding (Fig. 2B, i) but in terms of contribution, Glu329/Asp355/Asp350/D546 (ACE2) and Asp405/Glu406 and Asp420 (ACE2) have the most contribution using the energy −14.0 kj/mol energy cutoff, while the binding energy was estimated at −409.178 ± 21.114 kj/mol (Table 2 ) These data strongly suggest high affinity of RBD/ACE2 interface for BAZ.
Fig. 2.

(I) 3D representation of selected hydrogen-bond interaction at RBD (blue) and ACE2 (green) interface proximal to bazedoxifene (yellow stick) binding site. (ii-v)Time-dependent distance plots of RBD/ACE2, and selected residue pairs (see Y-axes). (D). (i) Energy contribution of amino acids (ACE2 = green points, RBD = blue points) to bazedoxifene binding based on MMPBSA energy calculations; a 3D projection of the most contributing amino acids based on 14.0 kj/mol energy cutoff.
Table 2.0.
MMPBSA Energy Estimation of Bazedoxifene at RBD/ACE2 interface.
| Energy Terms (Kj/mol) | |||||
|---|---|---|---|---|---|
| Compound | van der Waal energy | Electrostatic energy | Polar solvation energy | SASA energy | Binding energy |
| BAZ | −128.110 ± 20.285 | −431.479 ± 18.550 | 167.395 ± 39.644 | −16.984 ± 2.664 | −409.178 ± 21.114 |
4. Discussion
In addition to the challenges of developing and distributing safe and effective SARS-CoV-2 (COVID-19) vaccines [34], evidence for the presence of variants of concern (VOC) now mounts [35]. These VOCs in most cases accumulate mutations around the epitopes; causing viral escape or highly diminished vaccine efficacy [36].
It is therefore not unimaginable that cheaper, more effective therapeutic options must be investigated. One of the key successes here is the Pfizer-developed PF-07321332 4. PF-07321332 is an orally bioavailable SARS-CoV-2 main protease inhibitor with excellent off-target selectivity and in vivo safety profile [5].
In current study, Fragment-based lead discovery-aided fingerprint-based similarity search [37] of the FDA-approved chemical library identified benzimidazole scaffold-containing compounds as potential inhibitor of RBD/ACE2 interaction. Indeed, some previous reports have identified benzimidazole scaffold-containing compounds as potent anti-viral agents [38,39]. For instance, linear (dialkylamino)alkyl-derivatized 2-[(benzotriazol-1/2-yl)methyl]benzimidazoles exhibited nanomolar EC(50) activity on respiratory syncytial virus (RSV) [40].
The FDA-approved drug with the best docking result in this study is bazedoxifene. Bazedoxifene is a selective estrogen receptor modulator indicated for endometriosis [41]. It has also been indicated for postmenopausal osteoporosis [42]. The anti-SARS-COV-2 activity of bazedoxifene has been mentioned in some prior reports. For instance, in a study, it was demonstrated that bazedoxifene inhibits IL-6 signaling at therapeutic doses, leading to blockage of cytokine storm, ARDS and mortality in severe COVID-19 patients [43]. In another report, anti-SARS-CoV-2 activity with IC50 value of 3.44 μM, and CC50 value of 14.97 had been reported [44]. Here, we provide a mechanistic insight that bazedoxifene inhibits RBD/ACE2 complex. It is worthy of note that the computational data obtained by Yele et al. [45] and Mohapatra et al. [46] lend credence to our findings. Mudi et al. [47] also reported that benzimidazole preferentially inhibits SARS-COV-2-plagued VeroE6 cells while proposing main protease and non-structural as potential targets based on molecular docking and MD simulation studies. A further study by this group also confirmed that SARS-CoV-2 is susceptible to five-membered heterocycle-derivatives of benzimidazoles [48]. Indeed, in vitro conformation of bazedoxifene will encourage validation of gonadorelin, fondaparinux and atorvastatin which have been recently identified as RBD/ACE2 inhibitors through similar computational methods [13].
In conclusion, we have provided further evidence in support of bazedoxifene as a repurposable drug for the treatment of COVID-19 acting at RBD/ACE2 interface. These findings are of immediate importance as vaccine production, distribution and efficacy present a challenge in the global flight against COVID-19.
Ethics approval and consent to participate
The current study does not involve the use of human or animal subjects, therefore, ethical approval or consent to participate is not applicable.
Consent for publication
All authors have consented to publish this manuscript.
Authors’ contributions
Authors OO, and SF and ON conceived and designed the study. Authors OMO and BO performed molecular docking and complied the manuscript. OS and AI performed the kinetic assay studies. All authors prepared the final manuscript, and proofreading.
Funding
This research is supported by funds from the Nigerian Innovative Research Management Structures (NiReMaS) DAAD DIES ProGRANT, and Afe Babalola University Research Foundation.
Availability of data and material
All materials including primary data relating to this study will be made available upon request.
CRediT authorship contribution statement
Olaposi Omotuyi: Writing – original draft, Methodology, Funding acquisition, Conceptualization. Olusina M. Olatunji: Validation. Oyekanmi Nash: Conceptualization. Babatunji Oyinloye: Formal analysis. Opeyemi Soremekun: Writing – review & editing, Writing – original draft, Investigation. Ayodeji Ijagbuji: Visualization, Validation. Segun Fatumo: Supervision, Investigation, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The Authors acknowledge the contribution of University of Cologne, Germany in securing the NiReMaS grant; and also the staff of Mols and Sims, Nigeria for insightful discussion during the Bio-assay studies.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.micpath.2023.105994.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
Data availability
Data will be made available on request.
References
- 1.Li X., Zai J., Wang X., Li Y. Potential of large "first generation" human-to-human transmission of 2019-nCoV. J. Med. Virol. 2020;92(4):448–454. doi: 10.1002/jmv.25693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rehman S.U., Rehman S.U., Yoo H.H. COVID-19 challenges and its therapeutics. Biomed. Pharmacother. 2021;142 doi: 10.1016/j.biopha.2021.112015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., Dantonio A., Di L., Eng H., Ferre R., Gajiwala K.S., Gibson S.A., Greasley S.E., Hurst B.L., Kadar E.P., Kalgutkar A.S., Lee J.C., Lee J., Liu W., Mason S.W., Noell S., Novak J.J., Obach R.S., Ogilvie K., Patel N.C., Pettersson M., Rai D.K., Reese M.R., Sammons M.F., Sathish J.G., Singh R.S.P., Steppan C.M., Stewart A.E., Tuttle J.B., Updyke L., Verhoest P.R., Wei L., Yang Q., Zhu Y. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374(6575):1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 5.Heskin J., Pallett S.J.C., Mughal N., Davies G.W., Moore L.S.P., Rayment M., Jones R. Caution required with use of ritonavir-boosted PF-07321332 in COVID-19 management. Lancet. 2022;399(10319):21–22. doi: 10.1016/S0140-6736(21)02657-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omotuyi I.O., Nash O., Ajiboye B.O., Olumekun V.O., Oyinloye B.E., Osuntokun O.T., Olonisakin A., Ajayi A.O., Olusanya O., Akomolafe F.S., Adelakun N. Aframomum melegueta secondary metabolites exhibit polypharmacology against SARS-CoV-2 drug targets: in vitro validation of furin inhibition. Phytother Res. 2021;35(2):908–919. doi: 10.1002/ptr.6843. [DOI] [PubMed] [Google Scholar]
- 7.Aleem A., Akbar Samad A.B., Slenker A.K. StatPearls, Treasure Island. FL); 2022. Emerging variants of SARS-CoV-2 and novel therapeutics against coronavirus (COVID-19) [Google Scholar]
- 8.Choudhary S., Malik Y.S., Tomar S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol. 2020;11:1664. doi: 10.3389/fimmu.2020.01664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J., Lin C., Zhou X., Zhong F., Zeng P., Yang Y., Zhang Y., Yu B., Fan X., McCormick P.J., Fu R., Fu Y., Jiang H., Zhang J. Structural basis of the main proteases of coronavirus bound to drug candidate PF-07321332. J. Virol. 2022;96(8) doi: 10.1128/jvi.02013-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harmer D., Gilbert M., Borman R., Clark K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002;532(1–2):107–110. doi: 10.1016/s0014-5793(02)03640-2. [DOI] [PubMed] [Google Scholar]
- 11.Xia X. Domains and functions of spike protein in sars-Cov-2 in the context of vaccine design. Viruses. 2021;13(1) doi: 10.3390/v13010109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Omotuyi I.O., Nash O., Ajiboye O.B., Iwegbulam C.G., Oyinloye E.B., Oyedeji O.A., Kashim Z.A., Okaiyeto K. Atomistic simulation reveals structural mechanisms underlying D614G spike glycoprotein-enhanced fitness in SARS-COV-2. J. Comput. Chem. 2020;41(24):2158–2161. doi: 10.1002/jcc.26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tallei T.E., Fatimawali, Yelnetty A., Idroes R., Kusumawaty D., Emran T.B., Yesiloglu T.Z., Sippl W., Mahmud S., Alqahtani T., Alqahtani A.M., Asiri S., Rahmatullah M., Jahan R., Khan M.A., Celik I. An analysis based on molecular docking and molecular dynamics simulation study of bromelain as anti-SARS-CoV-2 variants. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.717757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan A., Randhawa A.W., Balouch A.R., Mukhtar N., Sayaf A.M., Suleman M., Khan T., Ali S., Ali S.S., Wang Y., Mohammad A., Wei D.Q. Blocking key mutated hotspot residues in the RBD of the omicron variant (B.1.1.529) with medicinal compounds to disrupt the RBD-hACE2 complex using molecular screening and simulation approaches. RSC Adv. 2022;12(12):7318–7327. doi: 10.1039/d2ra00277a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vausselin T., Seron K., Lavie M., Mesalam A.A., Lemasson M., Belouzard S., Feneant L., Danneels A., Rouille Y., Cocquerel L., Foquet L., Rosenberg A.R., Wychowski C., Meuleman P., Melnyk P., Dubuisson J. Identification of a new benzimidazole derivative as an antiviral against hepatitis C virus. J. Virol. 2016;90(19):8422–8434. doi: 10.1128/JVI.00404-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Release, S., Maestro, Schrödinger LLC, New York, NY, 2018-1, 2018.
- 17.Brooks W.H., Daniel K.G., Sung S.S., Guida W.C. Computational validation of the importance of absolute stereochemistry in virtual screening. J. Chem. Inf. Model. 2008;48(3):639–645. doi: 10.1021/ci700358r. [DOI] [PubMed] [Google Scholar]
- 18.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sastry G.M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 22.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 23.Huang J., MacKerell A.D., Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 2013;34(25):2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omotuyi I.O. Ebola virus envelope glycoprotein derived peptide in human Furin-bound state: computational studies. J. Biomol. Struct. Dyn. 2015;33(3):461–470. doi: 10.1080/07391102.2014.981207. [DOI] [PubMed] [Google Scholar]
- 25.Omotuyi O.I., Nash O., Safronetz D., Ojo A.A., Ogunwa T.H., Adelakun N.S. T-705-modified ssRNA in complex with Lassa virus nucleoprotein exhibits nucleotide splaying and increased water influx into the RNA-binding pocket. Chem. Biol. Drug Des. 2019;93(4):544–555. doi: 10.1111/cbdd.13451. [DOI] [PubMed] [Google Scholar]
- 26.Omotuyi O.I., Nagai J., Ueda H. Lys39-Lysophosphatidate carbonyl oxygen interaction locks LPA1 N-terminal cap to the orthosteric site and partners Arg124 during receptor activation. Sci. Rep. 2015;5 doi: 10.1038/srep13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 28.Hildebrand P.W., Rose A.S., Tiemann J.K.S. Bringing molecular dynamics simulation data into view. Trends Biochem. Sci. 2019;44(11):902–913. doi: 10.1016/j.tibs.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Kumari R., Kumar R., Open Source Drug Discovery C., Lynn A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 30.Ren X., Shi Y.S., Zhang Y., Liu B., Zhang L.H., Peng Y.B., Zeng R. Novel consensus docking strategy to improve ligand pose prediction. J. Chem. Inf. Model. 2018;58(8):1662–1668. doi: 10.1021/acs.jcim.8b00329. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y., Guo Y., Pan Y., Zhao Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys. Res. Commun. 2020;525(1):135–140. doi: 10.1016/j.bbrc.2020.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Vivo M., Masetti M., Bottegoni G., Cavalli A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016;59(9):4035–4061. doi: 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
- 33.Omotuyi O.I., Ueda H. Molecular dynamics study-based mechanism of nefiracetam-induced NMDA receptor potentiation. Comput. Biol. Chem. 2015;55:14–22. doi: 10.1016/j.compbiolchem.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Forman R., Shah S., Jeurissen P., Jit M., Mossialos E. COVID-19 vaccine challenges: what have we learned so far and what remains to be done? Health Pol. 2021;125(5):553–567. doi: 10.1016/j.healthpol.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Omotuyi O., Olubiyi O., Nash O., Afolabi E., Oyinloye B., Fatumo S., Femi-Oyewo M., Bogoro S. SARS-CoV-2 Omicron spike glycoprotein receptor binding domain exhibits super-binder ability with ACE2 but not convalescent monoclonal antibody. Comput. Biol. Med. 2022;142 doi: 10.1016/j.compbiomed.2022.105226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paul D., Pyne N., Paul S. Mutation profile of SARS-CoV-2 spike protein and identification of potential multiple epitopes within spike protein for vaccine development against SARS-CoV-2. Virusdisease. 2021;32(4):703–726. doi: 10.1007/s13337-021-00747-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bajusz D., Racz A., Heberger K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminf. 2015;7:20. doi: 10.1186/s13321-015-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tonelli M., Simone M., Tasso B., Novelli F., Boido V., Sparatore F., Paglietti G., Pricl S., Giliberti G., Blois S., Ibba C., Sanna G., Loddo R., La Colla P. Antiviral activity of benzimidazole derivatives. II. Antiviral activity of 2-phenylbenzimidazole derivatives. Bioorg. Med. Chem. 2010;18(8):2937–2953. doi: 10.1016/j.bmc.2010.02.037. [DOI] [PubMed] [Google Scholar]
- 39.Vitale G., Corona P., Loriga M., Carta A., Paglietti G., Giliberti G., Sanna G., Farci P., Marongiu M.E., La Colla P. 5-acetyl-2-arylbenzimidazoles as antiviral agents. Part 4. Eur. J. Med. Chem. 2012;53:83–97. doi: 10.1016/j.ejmech.2012.03.038. [DOI] [PubMed] [Google Scholar]
- 40.Tonelli M., Paglietti G., Boido V., Sparatore F., Marongiu F., Marongiu E., La Colla P., Loddo R. Antiviral activity of benzimidazole derivatives. I. Antiviral activity of 1-substituted-2-[(benzotriazol-1/2-yl)methyl]benzimidazoles. Chem. Biodivers. 2008;5(11):2386–2401. doi: 10.1002/cbdv.200890203. [DOI] [PubMed] [Google Scholar]
- 41.Kulak J., Jr., Fischer C., Komm B., Taylor H.S. Treatment with bazedoxifene, a selective estrogen receptor modulator, causes regression of endometriosis in a mouse model. Endocrinology. 2011;152(8):3226–3232. doi: 10.1210/en.2010-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palacios S. Bazedoxifene acetate for the management of postmenopausal osteoporosis. Drugs Today. 2011;47(3):187–195. doi: 10.1358/dot.2011.47.3.1587026. [DOI] [PubMed] [Google Scholar]
- 43.Smetana K., Jr., Rosel D., BrAbek J. vol. 34. 2020. Raloxifene and bazedoxifene could Be promising candidates for preventing the COVID-19 related cytokine storm, ARDS and mortality; pp. 3027–3028. (Vivo). 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeon S., Ko M., Lee J., Choi I., Byun S.Y., Park S., Shum D., Kim S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020;64(7) doi: 10.1128/AAC.00819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yele V., Sanapalli B.K.R., Mohammed A.A. Imidazoles and benzimidazoles as putative inhibitors of SARS-CoV-2 B.1.1.7 (Alpha) and P.1 (Gamma) variant spike glycoproteins: a computational approach. Chem. Zvesti. 2022;76(2):1107–1117. doi: 10.1007/s11696-021-01900-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohapatra R.K., Dhama K., El-Arabey A.A., Sarangi A.K., Tiwari R., Emran T.B., Azam M., Al-Resayes S.I., Raval M.K., Seidel V., Abdalla M. Repurposing benzimidazole and benzothiazole derivatives as potential inhibitors of SARS-CoV-2: DFT, QSAR, molecular docking, molecular dynamics simulation, and in-silico pharmacokinetic and toxicity studies. J. King Saud Univ. Sci. 2021;33(8) doi: 10.1016/j.jksus.2021.101637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mudi P.K., Mahanty A.K., Kotakonda M., Prasad S., Bhattacharyya S., Biswas B. A benzimidazole scaffold as a promising inhibitor against SARS-CoV-2. J. Biomol. Struct. Dyn. 2022:1–13. doi: 10.1080/07391102.2021.2024448. [DOI] [PubMed] [Google Scholar]
- 48.Mudi P.K., Mahato R.K., Verma H., Panda S.J., Purohit C.S., Silakari O., Biswas B. In silico anti-SARS-CoV-2 activities of five-membered heterocycle-substituted benzimidazoles. J. Mol. Struct. 2022;1261 doi: 10.1016/j.molstruc.2022.132869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All materials including primary data relating to this study will be made available upon request.
Data will be made available on request.