

Abstract
Abiraterone is a standard treatment for metastatic castrate-resistant prostate cancer (mCRPC) that slows disease progression by abrogating androgen synthesis and antagonizing the androgen receptor (AR). Here we report that inhibitors of the mitotic regulator polo-like kinase-1 (Plk1), including the clinically active third-generation Plk1 inhibitor onvansertib, synergizes with abiraterone in vitro and in vivo to kill a subset of cancer cells from a wide variety of tumor types in an androgen-independent manner. Gene expression analysis identified an AR-independent synergy-specific gene set signature upregulated upon abiraterone treatment that is dominated by pathways related to mitosis and the mitotic spindle. Abiraterone treatment alone caused defects in mitotic spindle orientation, failure of complete chromosome condensation, and improper cell division independently of its effects on AR signaling. These effects, while mild following abiraterone monotherapy, resulted in profound sensitization to the anti-mitotic effects of Plk1 inhibition, leading to spindle assembly checkpoint-dependent mitotic cancer cell death and entosis. In a murine patient-derived xenograft model of abiraterone-resistant metastatic castration resistant prostate cancer (mCRPC), combined onvansertib and abiraterone resulted in enhanced mitotic arrest and dramatic inhibition of tumor cell growth compared to either agent alone. Overall, this work establishes a mechanistic basis for the phase 2 clinical trial (NCT03414034) testing combined onvansertib and abiraterone in mCRPC patients and indicates this combination may have broad utility for cancer treatment.
Keywords: Plk1, castrate-resistant prostate cancer, abiraterone, onvansertib, drug synergy, spindle orientation, mitosis
INTRODUCTION
Prostate cancer is largely driven by signaling through the AR, a member of the nuclear receptor superfamily (1). Accordingly, for prostate cancer patients for whom surgical resection or radiotherapy is contraindicated, first-line therapy targets AR signaling by suppressing androgen production within the testes (androgen deprivation therapy; ADT). While ADT is initially effective for the vast majority of men, patients invariably develop mCRPC. This occurs through multiple mechanisms including AR gene amplification, expression of AR splice variants (AR-v7), increased crosstalk between the AR and other signaling pathways, and upregulation of intratumoral androgen synthesis (2). Treatment of mCRPC involves further suppressing AR signaling using the antiandrogens abiraterone acetate or enzalutamide. Abiraterone, a synthetic derivative of pregnenolone, abrogates residual adrenal and intratumoral androgen synthesis by inhibiting Cyp17A1 (Supplementary Fig. S1A) (3,4). In addition, abiraterone directly antagonizes the AR (5). Enzalutamide is a direct AR antagonist that inhibits AR signaling irrespective of androgen synthesis (6). Treatment of CRPC patients with abiraterone or enzalutamide transiently arrests tumor progression; however, most mCRPC patients acquire resistance to these antiandrogens with median progression-free survival of 1–2 years (7,8).
Several signaling pathways cross-talk with the AR, including the PI3-kinase/Akt pathway, the MAPK pathway, and WNT/β-catenin signaling, modulating tumor cell survival, invasion, and metastasis, contributing to prostate cancer development and drug resistance (9,10). In addition, Polo-like kinase 1 (Plk1) (11,12), and components of the DNA damage response (DDR) contribute to prostate cancer susceptibility and progression (13), including BRCA1, BRCA2, MSH2, MSH6, PMS2, NBS1, RAD51, ATM, CHEK2, and PALB2 (14–16).
Plk1 is a serine/threonine kinase that plays a critical role in nearly every stage of mitosis (17,18). Plk1 is overexpressed and associated with poor prognosis in many cancers including prostate cancer (19,20), and is thought to be particularly important for mitotic cell division in cancer cells due to elevated replicative stress and chromosomal instability (21,22). Overexpression of Plk1 in prostate epithelial cells results in malignant transformation, enhanced migration and epithelial to mesenchymal transition (23) and is clinically associated with higher grade disease (20). Furthermore, Plk1 suppresses the pro-apoptotic function of the FOXO1 transcription factor (24) and postulated to cross-talk with the Wnt/β-catenin pathway in CRPC (25).
Synergistic combination therapies are of particular interest due to their potential for enhancing efficacy and cancer cell selectivity, overcoming drug resistance, and lowering toxicity though decreased individual drug dosage (26). Given the emerging importance of the Plk1 and DDR signaling pathways in prostate cancer, we sought to investigate whether Plk1 inhibitors and other anti-mitotic drugs, or the induction of genotoxic stress, in combination with inhibition of AR signaling, could be used to synergistically enhance anti-tumor responses in mCRPC cells. Here we report the surprising discovery that synergistic tumor-cell killing by combinations of Plk1 inhibitors and abiraterone was completely independent of AR signaling, occurs in a variety of cancer cell types extending far beyond prostate cancer, and was not recapitulated by combining abiraterone with inhibitors of other mitotic kinases. We show that abiraterone treatment alone causes defects in mitotic spindle orientation, chromosome condensation, and bipolar mitotic division, and results in synergistic mitotic cancer cell death and entosis in a subset of cancer cells when combined with inhibitors of Plk1.
MATERIALS AND METHODS
Culturing of cells
All cell lines were cultured in a 37°C humidified incubator supplied with 5% CO2, were maintained subconfluent, and used for no more than 20 passages. All media was supplemented with fetal bovine serum (FBS) unless explicitly stated as charcoal-stripped FBS (csFBS), contained 2 mM glutamine, and lacked antibiotics. C4-2 (MD Anderson; RRID: CVCL_4782), LNCaP (ATCC; RRID: CVCL_1379), 22Rv1 (ATCC; RRID: CVCL_1045), PSN-1 (KI High Throughput Sciences Core; RRID:CVCL_1644), AU565 (ATCC; RRID: CVCL_1074), HCC1954 (KI High Throughput Sciences Core; RRID:CVCL_1259), and NCIH661 (ATCC; RRID:CVCL_1577) were grown in RPMI-1640 with 10% FBS. LNCaP95 (Steven Balk Lab; RRID:CVCL_ZC87) was grown RPMI without phenol red and with 10% csFBS (Gibco). Panc 10.05 (ATCC; RRID:CVCL_1639) was grown in RPMI-1640 with 15% FBS, and 10 IU/ml human recombinant insulin (Gibco). OAW28 (Sigma Aldrich; RRID:CVCL_1614) was grown in Advanced DMEM (Gibco) with 10% FBS and supplemented with insulin. PC-3 (ATCC; RRID: CVCL_0035) cells were grown in F-K12 media with 10% FBS. DU145 (ATCC; RRID:CVCL_0105) and Cal33 (DSMZ; RRID: CVCL_1108) cells were grown in DMEM media with 10% FBS. SK-OV-3 (ATCC; RRID:CVCL_0532) cells were grown in McCoy’s 5A with 10% FBS. MV-4-11 (DSMZ; RRID:CVCL_0064) cells were grown in IMDM with 10% FBS. Calu3 (ATCC; RRID:CVCL_0609) cells were grown in MEM with 10% FBS. OCI-AML-3 (DSMZ; RRID:CVCL_1844) cells were grown in MEM with 20% FBS. KYSE450 (DSMZ; RRID:CVCL_1353) cells were grown in 1:1 RPMI:Ham’s F12 with 2% FBS. G418 was used at a concentration of 400 and 500 μg/ml for C4-2 and SK-OV-3 cells, respectively. Puromycin was used at a concentration of 2 μg/ml for C4-2 and SK-OV-3 cells. All cell lines used in this study were authenticated using short tandem repeat genotyping (Labcorp), except for LNCaP95 which lack a reference STR profile, and confirmed to be negative for mycoplasma contamination using MycoAlert™ PLUS Assay (Lonza).
Measurements of drug sensitivity and drug combination synergy
Cells were plated in 96- or 384-well plates at a density of 4000 or 1000 cells per well in 22.5 or 95 μl media, respectively. For experiments using the suspension AML cell lines, MV-4-11 and OCI-AML-3, 10,000 cells were plated in 384-well plates in 22.5 μl. The following day drugs, diluted in 5 μl or 2.5 μl media for 96- or 384-well plates, respectively, were added to the wells. A constant amount of DMSO vehicle was maintained in all wells. Unless otherwise stated, after 72-hour incubation, viability relative to control was measured using CellTiter-Glo™ (Promega) according to the manufacturer’s recommendations. Luminescence in individual wells in plates with opaque walls was measured using and Infinite™ M200 Pro plate reader (Tecan Group Ltd.). Relative viability was calculated by dividing the luminescent signal from each well by that measured in the vehicle-control well on an individual replicate basis. All abiraterone used in this study is specifically abiraterone acetate (Selleck Chemicals). For experiments examining cell number and relative confluence, drug-treated cells were washed with PBS and fixed with 4% formaldehyde for 1 hour, stained with 1 μM SYTO™ 60 (Molecular Probes) and imaged on an Odyssey™ CLx scanner (LiCOR Biosciences).
Flow cytometry analysis of cell cycle distribution and apoptosis
The indicated cells were treated as described and then collected by centrifugation or trypsinization for suspension or adherent cells, respectively. The media, trypsin, and PBS wash were collected together to avoid loss of loosely attached or detached cells. Cells were fixed in 4% formaldehyde in PBS for 15 minutes, washed with PBS containing 1% bovine serum albumin (PBS-BSA), and then stored in methanol at −20°C overnight. Cells were then washed twice in PBS-BSA 0.1% Tween-20, incubated with primary antibodies overnight at 4°C, washed with PBS-BSA 0.1% Tween-20, and incubated for 1 hour with fluorescent-dye conjugated secondary antibodies (diluted 1:200, Alexa Fluor, Molecular Probes) at room temperature for 1 hour. The fixed cells were then washed with PBS-BSA 0.1% Tween-20, resuspended in PBS containing 1 μg/ml 4,6-diamidino-2-phenylindole (DAPI, Molecular Probes) to stain DNA, and analyzed using a BD™ LSRII flow cytometer (Becton Dickinson) and the FlowJo™ software package. Primary antibodies used included anti-phospho serine 10 histone H3 (pHH3; EMD Millipore, 3H10; RRID:AB_310016) and anti-cleaved capase-3 (BD Pharmingen, 599565; RRID:AB_397274). For cell-cycle analysis post Cyp17A1 knockdown, prostate cancer cells were detached by treatment with Accutase (Gibco), fixed in ethanol, resuspended in propidium iodide (PI) staining solution (50 μg/mL PI, 0.1 mg/mL RNase A, and 0.05% Triton X-100), and analyzed on a FACSCanto II (BD Biosciences).
Time-lapse live-cell microscopy
Live-cell microscopy for analysis of mitotic spindles, duration of mitosis, and phenotypes associated with drug treatment was performed using C4-2, LNCaP, and SK-OV-3 cells transduced with pLNCX2-H2B-mCherry and pLVX-Puro-mEmerald-TUBA1B (27). Cells were plated in 12-well plates using 1.5 ml phenol red-free media. The following day drugs were added as indicated and the cells were imaged on an EVOS™ FL Auto Cell Imaging System (Invitrogen) equipped with an onstage incubator to maintain 37°C, adequate humidity, and 5% CO2. Images were acquired at 15-minute intervals for 72 hours using a 20X objective lens. Images were compiled into movies and analyzed using the Fiji distribution of ImageJ v2.1.0 (RRID:SCR_002285) (28). For image analysis of spindle rotation, and cell fate post mitosis, cells to be analyzed per condition were picked at random using only the first frame of the time lapse. Once the cell entered its first mitosis, the angle of the spindle’s major axis was tracked starting from when it was first apparent, prometaphase, until telophase. For each frame transition we calculated both the clockwise and counterclockwise deviations and assumed that the actual rotation was the lesser of the two (the shortest rotational path). Given the modest amount of rotation that occurred in vehicle only treated cells, we believe this assumption would rarely, if ever, provide us with the wrong conclusion. Clockwise rotation was considered positive rotation, whereas counterclockwise rotation was negative. The net angular displacement is the total change in angle of the spindle during mitosis from start until finish and is the absolute value of the sum of all stepwise rotations. The cumulative angular distance is the total amount of rotation regardless of direction that occurred during mitosis and is the sum of the absolute value of all rotations.
Indirect immunofluorescence of fixed cells and tumor sections
For examining mitotic spindles in fixed cells, C4-2 cells were plated in 12-well plates containing poly-L-lysine coated glass coverslips (Corning-BioCoat #354085) and subjected to the indicated drugs the following day. After the indicated amount of time cells were washed for 1 minute with microtubule stabilization buffer (MTSB: 4 M glycerol, 100 mM PIPES pH 6.9, 1 mM EGTA, 5 mM MgCl2), 2 minutes with MTSB 0.5% Triton X-100, 2 minutes with MTSB, 5 minutes with calcium buffer (100 mM PIPES, pH 6.8, 1 mM MgCl2, 1 mM CaCl2, 0.5% Triton X-100) and then fixed for 10 minutes with 1% glutaraldehyde (Electron Microscopy Sciences) in PBS. Residual glutaraldehyde was quenched with NaBH4 (0.5 mg/ml in ddH2O) for 12 minutes twice and then washed twice with TBS-BSA (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% BSA) for 5 minutes. Fixed cells were blocked for 1 hour in TBS-BSA containing 0.5% Triton X-100. TBS-BSA was used for anti-α-Tubulin (Novus Biologicals, YL1/2; RRID:AB_10078394) and anti-CENP-A (Thermo Fisher, PA5-17194; RRID:AB_10987425) antibody incubation overnight at 4°C, followed by a 5-minute wash, fluorescent secondary antibody (1:200, Alexa Fluor, Molecular Probes) incubation 1 hour at room temperature, and an additional 5-minute wash. Cells were stained using 5 μg/ml DAPI, washed in PBS for 3 minutes, and coverslips mounted on glass slides using the antifade reagent ProLong Gold™ (Molecular Probes). Images were acquired using a Deltavision™ Ultimate Focus microscope (Applied Precision) using a 100X 1.40 NA objective. Z-stacks were deconvolved and maximum intensity Z-stack projections were generated using softWoRx™ software (Applied Precision; RRID:SCR_019157). The Fiji distribution of ImageJ (RRID:SCR_002285) was used for image analysis. For immunofluorescence analysis of DU145 cancer cells, 1 – 1.5 × 104 were seeded in each well of a 4-chamber slide in 500 mL of culture medium and treated as indicated for 12 hours. Cell culture medium was removed and cells were rinsed with PBS three times. Cell fixation was done using 4% paraformaldehyde (pH 7.4) for 10 minutes at 37°C followed by 400 μL of ice-cold methanol for 5 minutes at −20°C. Fixed cells were permeabilized with 0.1% Triton X-100 in PBS (room temperature for 15 minutes), and blocking was done with 2% BSA in PBS (room temperature for 60 minutes). The anti-α-Tubulin primary antibodies incubation was performed overnight. Fluorescent dye–labeled secondary antibody along with DAPI diluted in 500 μL of 0.1% BSA was added for 45 minutes in room temperature. Negative and positive controls were used. Z-stacks were acquired and deconvolved as above but using a 60X 1.4 NA objective. The analysis using ImageJ was done by two independent investigators.
For immunohistochemical staining of tumor samples, formalin-fixed paraffin-embedded tumors were sectioned and mounted on charged slides. Sections (4 μm) were quenched with 3% H2O2 (UltraVision Hydrogen Peroxide Block, Epredia), blocked with Rodent Block M (BioCare Medical), labeled with anti-phospho histone H3 antibody (pHH3; Cell Signaling Technology, 9701S; RRID:AB_331535) and an Alexa Fluor® 488 conjugated secondary antibody (Molecular Probes), and then stained with DAPI. Images were acquired using a Deltavision™ Ultimate Focus microscope using a 40X 0.85 NA objective. Randomly selected fields of cells were analyzed using QuPath v0.2.3 (RRID:SCR_018257) (29). Images that contained very few cells or large necrotic regions were replaced with a new randomly selected image. Cells were identified in DAPI images and then pHH3 positive cells were quantified. Automated cell identification and pHH3 quantification was manually inspected for accuracy to ensure accurate cell counting and avoidance of false positive and negative pHH3 assignment to cells.
Preparation of lysates and immunoblotting
Cells were lysed directly on the plate after washing with PBS using lysis buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 5% glycerol, 5 mM EDTA, 1 mM NaF, 10 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, cOmplete™ EDTA-free protease inhibitors, and PhosSTOP™ phosphatase inhibitors). The media and PBS wash were reserved, centrifuged, and any cells present were combined with the lysate to prevent loss of loose or unattached cells. After sonication and protein concentration normalization, 6X sodium dodecyl sulfate - polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer (208 mM Tris-HCl pH 6.8, 42% glycerol, 3 M β-mercaptoethanol, 10% SDS, 5 mg/ml bromophenol blue) was added and lysates were boiled for 5 minutes. Following SDS-PAGE, immunoblots were blocked with Odyssey™ blocking buffer (LiCOR Biosciences) and incubated with primary antibody overnight at 4°C and then secondary antibody for 1 hour at room temperature. Immunoblots were scanned on an Odyssey™ CLx scanner (LiCOR Biosciences). Antibodies used included anti-AR (Cell Signaling Technologies, D6F11; RRID:AB_10691711), anti-MAD2 (Bethyl Labs, A300-300A; RRID:AB_309443), anti-Cyp17A1 (Cell Signaling Technologies, E6Y3S; RRID:AB_2797724), anti-Vinculin (Santa Cruz Biotechnology, 7F9; RRID:AB_1131294), anti-Plk1 (Cell Signaling Technologies, 208G4; RRID:AB_2167409), anti-TCTP (Cell Signaling Technologies, D10F2; RRID:AB_11220419), anti-phospho serine 46 TCTP (Cell Signaling Technologies, #5251; RRID:AB_10547143), and anti-β-actin (Sigma Aldrich, AC-15; RRID:AB_476744).
siRNA knockdown and drug sensitivity analysis
Silencer® Select siRNAs (s8392 and s8393, Invitrogen) targeting Mad2 (gene name MAD2L1) and the Negative Control No. 1 siRNA (#4390844, Invitrogen) were used for Lipofectamine™ RNAiMAX (Thermo Fisher Scientific) based transfection at 10 nM concentration according to the manufacturer’s recommendations. After 24 hours, cells were replated into both 6- and 384-well plates for preparation of cell lysates and synergy experiments, respectively. The following day cells in the 6-well plates were lysed for immunoblots, whereas cells in the 384-well plates were treated with a combination drug matrix that was assessed for synergy after 72 hours. For siCYP17A1 experiments Dharmacon On-TARGETplus™ human CYP17A1 siRNA (LQ-008469-02-0005) were used.
Gene expression analysis by RNA sequencing
Cells were plated in 6-well plates, and 24 hours later treated with the indicated drugs. After 16 hours cells were lysed and total RNA was isolated using NucleoSpin® RNA Plus Mini Kit (Macherey-Nagel) according to the manufacturer’s recommendations. For RNA sequencing of C4-2, LNCaP, LNCaP95, and 22Rv1 prostate cancer cells biological triplicate samples were collected. For the non-prostate 12 cancer cell line panel single samples were collected and results analyzed in a paired manner. Samples were submitted to the MIT BioMicro Center for library preparation and sequencing. RNA quality was assessed using a Fragment Analyzer (Agilent Technologies) and RNA sequencing libraries were prepared using 400 ng of total RNA using the Kapa mRNA Hyperprep kit (Roche) at 1/3rd reaction volume using 14 cycles of PCR. Libraries were analyzed using the Fragment Analyzer and quantified by qPCR prior to pooling and sequencing on a NextSeq® 500 System (Illumina) using 75 nt single end reads. Sequences were aligned to the human transcriptome (Gencode v29 GRCh38.p12) followed by transcript quantification using Salmon v0.14 (RRID:SCR_017036) (30). Transcript abundance was transformed into gene set enrichment scores using GSVA v1.36 (RRID:SCR_021058) (31) and MSigDB v7.0 (RRID:SCR_016863), the relevant contrasts performed using limma v3.44 (RRID:SCR_010943) (32), and differentially expressed gene sets identified based on an ≤ 0.01 FDR cut off.
Organoid generation, culture, and drug sensitivity measurements
Prostate cancer 3D organoids were derived from PDXs grown in castrated male NOD scid mice (Taconic; RRID:IMSR_TAC:nodsc). Following euthanasia tumors were extracted. Minced tumor fragments for organoid culturing were digested in Accumax™ - Cell Aggregate Dissociation Medium (Thermo Fisher Scientific), resuspended in DMEM/F12 plus 10% FBS and passed through a 250-μm cell strainer (Thermo Fisher Scientific, Pierce Tissue Strainers, #87791) to remove tissue debris and obtain smaller cell clusters. Cells were plated on Matrigel covered tissue cultured plates (Corning Matrigel®, Growth Factor Reduced Basement Membrane Matrix, LDEV-free, # 354230) and prostate-organoid specific media (33). After 7 days, organoids were transferred to 96-well plates and treated with onvansertib, abiraterone, and the combination for 6 days. Cell viability was assessed with CellTiter-Glo® 3D Cell Viability Assay (Promega). Six biological replicates were assessed per treatment group.
In vivo studies using a tumor-implantable microwell device
C4-2 CRPC and SK-OV-3 ovarian cancer xenograft tumors were grown in four- to six-week old NCR nude mice (Taconic; RRID:IMSR_TAC:ncrnu), male or female, respectively. Five million C4-2 or two million SK-OV-3 cells in serum free media were mixed one to one with growth factor reduced Matrigel® (Invitrogen) in a total volume of 200 μl and injected in the hind flank using a 23- or 27-gauge needle, respectively. Cells were found to be free of murine pathogens by IMPACT rodent pathogen testing (IDEXX BioAnalytics) prior to injection. Tumors took four to eight weeks to grow.
Microdose containing tumor-implantable devices were manufactured, implanted, and in vivo drug responses analyzed as previously described (34). The cylindrical microdevices (4 mm x 820 μm) micromachined from medical-grade Delrin® acetal resin blocks (DuPont) each contain eighteen 200 μm (diameter) x 250 μm (depth) reservoirs. Abiraterone acetate and BI2536 were mixed with PEG 1450 at 12.5% by weight, and 1 μg of the dry powder mixture then packed into a reservoir. Wells containing the combination of drugs contained 12.5% of each drug by weight. Implantation of the devices was accomplished using a 19-gauge spinal biopsy needle (Angiotech) with a retractable needle obturator. Tumors were excised 24 to 36 hours after device implantation, fixed in 10% formalin for 24 hours, and embedded in paraffin. Sections were stained with cleaved caspase-3 antibody (Cell Signaling Technologies, 9664; RRID:AB_2070042) followed by detection with horseradish peroxidase conjugated secondary antibody and diaminobenzidine with hematoxylin used as a counterstain, following standard immunohistochemistry techniques. Images were acquired using an EVOS® Cell Imaging System (Invitrogen) microscope, and scored using ImageJ (RRID:SCR_002285) (28) in a blinded manner. If upon implantation a well was adjacent to necrotic tissue then the response to the drugs could not be assessed. Because of this the number of measurements is not the same between treatment groups and ranged from 5 to 13. The apoptotic index was calculated as the percentage of cells that stained positive for cleaved caspase-3 within a 400 μm radius of the microwell-tissue interface, as described in (35).
All animal studies were approved by the Massachusetts Institute of Technology Committee for Animal Care or Beth Israel Deaconess Institutional Animal Care and Use Committee, conducted in compliance with both the Animal Welfare Act Regulations and other federal statutes relating to animals and experiments involving animals, and adhered to the principles set forth in the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996 (Institutional Animal Welfare Assurance #A-3125-01).
In vivo studies using LVCaP-2CR PDX
LVCaP-2CR PDXs were grown in castrated male NOD scid gamma mice (RRID:BCBC_1262) obtained from an in-house colony at the Sidney Kimmel Comprehensive Cancer Center (SKCCC) at Johns Hopkins as described previously (36). Twenty milligrams of LVCaP-2CR contained in 200 μl of 50% Matrigel® (Invitrogen) was subcutaneously injected in the hind flank of the mice. After 4 weeks, 0.1 cm3 tumors had formed and the animals were randomized into 4 groups. Onvansertib was suspended in 0.5% methylcellulose 0.1% Tween 80 and administered at 60 mg/kg. Abiraterone acetate (Selleck Chem) was administered at 0.5 mmole/kg (i.e. 196 mg/kg) in 200 μl of 5% benzl alcohol 95% safflower oil. Both were administered by oral gavage. Animals were treated in cycles consisting of 5 consecutive days on treatment, followed by a 24 to 48-hour drug holiday. Tumor size was measured at the indicated days and volume was calculated according to the ellipsoid volume formula (length × width × height × π/6). All animal procedures for the LVCaP-2CR PDX study were approved by the Johns Hopkins University School of Medicine Institutional Animal Care and Use Committee.
Measurement of tubulin polymerization in vitro
In vitro tubulin polymerization was measured using a fluorescence-based porcine tubulin polymerization kit (Cytoskeleton, Inc., BK011P) according to the manufacturer’s instructions. Tubulin polymerization is initiated by addition of GTP in the presence of the indicated drugs while maintaining a constant final concentration of DMSO. Polymerization was tracked by fluorescence at 1-minute intervals over the course of 100 minutes using an Infinite™ M200 pro plate reader (Tecan Group Ltd.).
Data availability
The data generated in this study are available within the article and its Supplementary data files. The RNA sequencing data generated in this study are publicly available in the Gene Expression Omnibus (GEO) at GSE217537.
RESULTS
The antiandrogen abiraterone acetate in combination with inhibitors of Polo-like kinase 1 synergistically kills CRPC cells
To explore pathways in CRPC cells that could be targeted to create abiraterone-based synergistic drug combinations, we performed a combinatorial dose-response study with abiraterone focused on anti-mitotic and DNA damaging agents that could be rapidly translated into the clinic. The microtubule-stabilizing taxane docetaxel was investigated because it is an alternative first-line treatment for mCRPC, and is a second-line agent after development of antiandrogen resistance (37). Doxorubicin was explored because it synergizes with docetaxel in prostate cancer (38). Furthermore, Polkinghorn et al. reported that the AR regulates DDR pathways promoting resistance to DNA damage (39). A Plk1 inhibitor (BI2536) was explored based on our lab’s long-standing interest in how this kinase controls mitotic progression and silences the G2/M checkpoint after DNA damage through phospho-priming and Polo-box dependent interactions (40–45). In addition, during CRPC development the AR selectively activates genes involved in mitosis, CRPC cells are particularly sensitive to Plk1 inhibitors, and AR splice variants associated with antiandrogen resistance upregulate genes involved in mitosis including Plk1 (11,46,47). Based on our previous work indicating that the timing of drug addition may be a critical parameter (48), we examined the effect of combinatorial dose matrices when abiraterone was added simultaneously with a second drug, and when drugs were administered in a time-staggered manner (Fig. 1A). To mimic androgen deprivation, C4-2 CRPC cells were screened in media containing charcoal-stripped fetal bovine serum (csFBS).
Figure 1. The combination of Plk1 inhibitors and abiraterone synergistically kill CRPC cells.
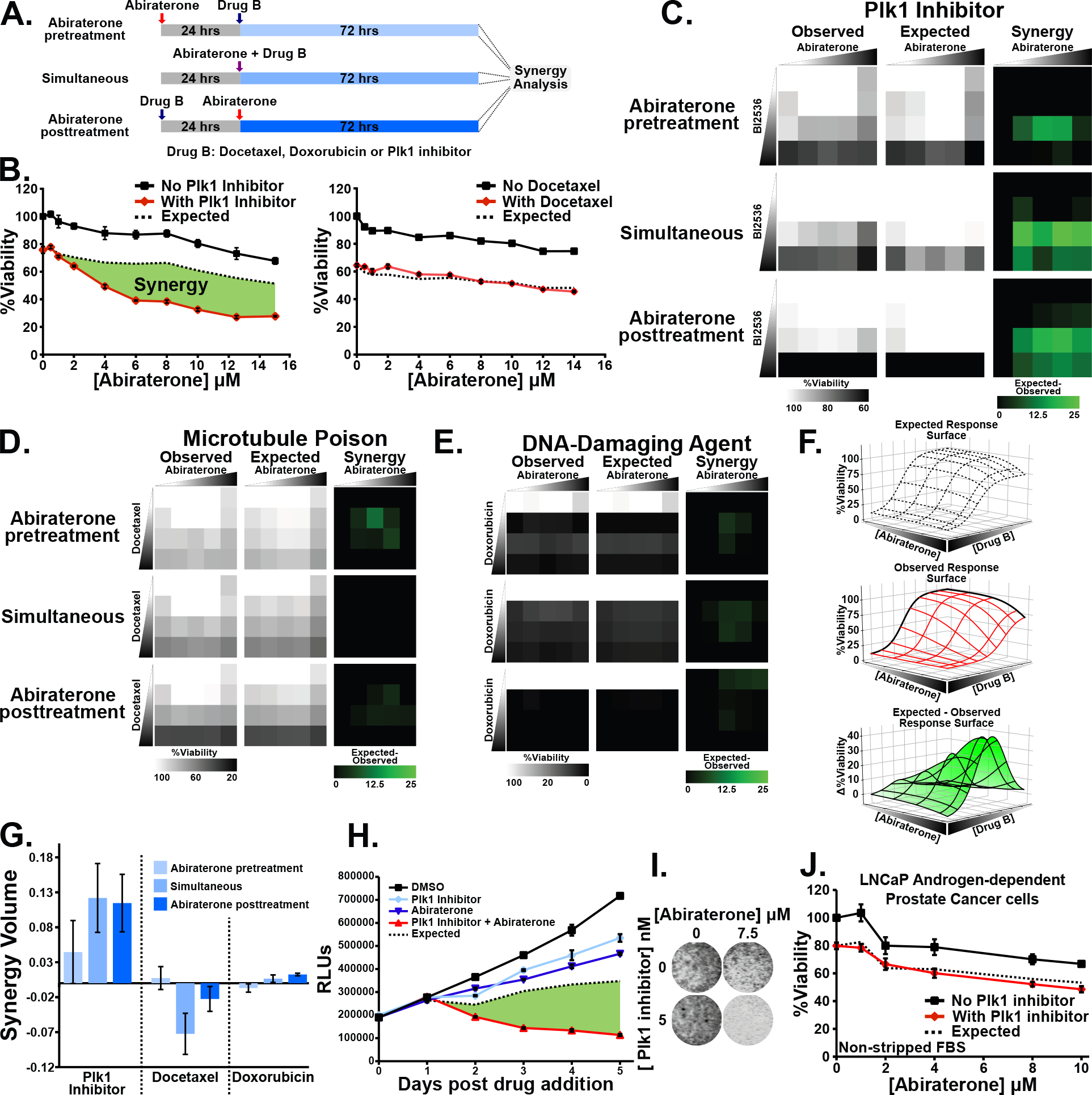
(A) Schematic indicating abiraterone-based synergy analysis in a time-staggered and non-staggered manner.
(B) Assessing synergy between abiraterone and a Plk1 inhibitor BI2536 (4 nM) or docetaxel (5 nM). C4-2 CRPC cells growing in media containing csFBS were subjected to abiraterone in the absence (black line) or presence (red line) of the second drug for 72 hours. The concentration of the second drug was chosen based on a 20–30% decrease in relative viability when used alone. Mean ± SEM (n = 3) is shown. Expected viability (dashed black line) is calculated according to the Bliss independence model of drug additivity. Synergy, depicted by the green area, is a decrease in viability beyond the expected additive effect.
(C-E) C4-2 cells grown using csFBS were subjected to dose matrices of abiraterone (0, 1, 5, 10, and 20 μM) and either BI2536, or docetaxel (0, 1, 10, and 50 nM ), or doxorubicin (0, 1, 10, and 50 μM) as depicted in (A). Observed viability, expected viability, and synergy (expected minus observed) are shown.
(F) Graphical representation of volumetric measurement of synergy. The observed relative viability response matrix is plotted as a dose response surface (middle). Deviation from the expected response surface (top) generates an expected minus observed response surface (bottom). The total synergy present is calculated as the integrated volume beneath the expected minus observed response surface.
(G) Synergy volume measurements calculated as in (F) from dose-response matrices in (C-E). Mean ± SEM (n = 3).
(H) C4-2 cells in media containing csFBS were treated with 5 nM BI2536, 10 μM abiraterone, and the combination. Shown is relative viability, expected viability, and synergy in green. Mean ± SEM (n = 3).
(I) C4-2 cells were grown using csFBS, subjected to the indicated drugs for 72 hours, and confluence assessed by SYTO 60 staining.
(J) LNCaP androgen-dependent cells were grown using non-stripped FBS to assess synergy between BI2536 (5 nM) and abiraterone as in (B).
Observed cell viability was compared to the results expected from drug additivity using the Bliss independence model (27,49). These experiments revealed dramatic synergy between the Plk1 inhibitor BI2536 and abiraterone (Fig. 1B), particularly when the entire dose-response matrix is analyzed (Fig. 1C). By comparison, expected and observed responses following abiraterone co-treatment with docetaxel or doxorubicin essentially overlap (Fig. 1B, D, and E). Each dose matrix was quantified by plotting the observed and expected response surfaces and synergy defined as the integrated volume between the observed and expected surfaces (Fig. 1F) (27). Synergy was clearly observed when abiraterone was combined with the Plk1 inhibitor, but not with docetaxel or doxorubicin, in these matrices regardless of drug sequencing (Fig. 1G). A detailed time course readily confirmed synergy between Plk1 inhibitors and abiraterone and this was further validated by direct measurement of cell confluence (Fig. 1H and 1I). The C4-2 cell line, a widely used model for CRPC, was derived by passage of LNCaP androgen-dependent prostate cancer cells through castrated mice (50). In contrast to C4-2 cells, co-treatment resulted in additivity in the parental LNCaP cells (Fig. 1J).
Plk1 inhibitor-abiraterone synergistic killing of CPRC cells is independent of abiraterone’s effects on AR signaling but specific to inhibition of the mitotic kinase Plk1
To assess the role of AR signaling and androgen levels in this synergistic response, we tested whether abiraterone and Plk1 inhibitors synergistically killed C4-2 cells in the presence of exogenous androgens. Unexpectedly, the observed synergy between abiraterone and Plk1 inhibitors appeared even more robust in non-charcoal-stripped FBS (Fig. 2A). In contrast, the presence or absence of androgens did not alter the non-synergistic response of LNCaP cells to this drug combination (Fig. 1J and Supplementary Fig S1B). We next tested whether Plk1 inhibitors would synergize with another AR antagonist enzalutamide (Fig. 2B, Supplementary Fig. S1A). Surprisingly, no synergy was observed between enzalutamide and Plk1 inhibitors in C4-2 cells regardless of androgen levels (Fig. 2C and Supplementary Fig. S1C). Under these conditions, abiraterone and enzalutamide inhibit AR transcriptional activity to equivalent extents (see below), suggesting that AR inhibition does not underlie synergistic killing by the abiraterone-Plk1 inhibitor combination. Because abiraterone inhibits Cyp17A1, preventing androgen, glucocorticoid and estrogen synthesis, abiraterone’s AR-independent effects could be due to changes in other steroid hormones (Supplementary Fig. S1A). However, the steroidal and non-steroidal Cyp17A1 inhibitors galeterone and orteronel (Fig. 2B) did not synergize with Plk1 inhibitors (Fig. 2D and E). Plk1 inhibition can induce AR protein degradation (51), suggesting an alternative AR-dependent synergistic mechanism. We confirmed that Plk1 inhibition decreased AR abundance, however, this occurred in both C4-2 and LNCaP cells (Supplementary Fig. S1D) while only the former showed synergistic killing by this combination. To further demonstrate no AR role in this synergy, we employed an AR-degrading PROTAC, ARV-110. Plk1 inhibition did not sensitize C4-2 CRPC cells to ARV-110 despite rapid and robust AR protein loss (Fig. 2F and G).
Figure 2. Plk1 inhibitor-abiraterone synergy is independent of AR signaling, occurs in multiple prostate cancer cell lines, and is specific to Plk1 activity.
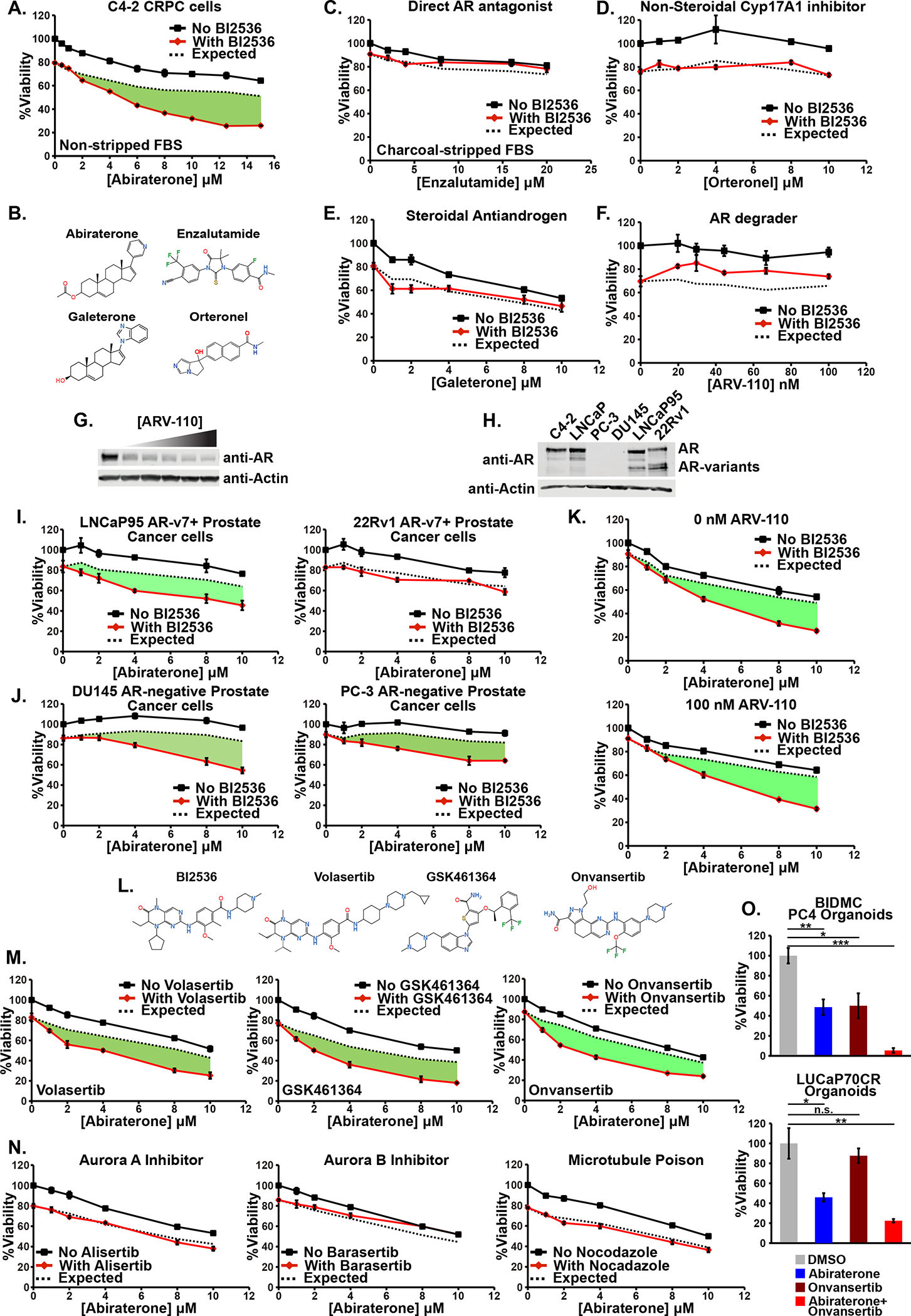
(A) C4-2 cells were grown in media containing FBS and subjected to increasing concentrations of abiraterone in the presence (red line) or absence (black line) of the Plk1 inhibitor BI2536 (3 nM) for 72 hours. Mean ± SEM (n = 3). Expected viability (dashed black line) was calculated according to the Bliss independence model of drug additivity. Synergy, depicted by the green area, is a decrease in viability beyond the expected additive effect.
(B) Comparison of the chemical structures of the antiandrogens abiraterone acetate, enzalutamide, galeterone, and orteronel.
(C) C4-2 cells were grown using csFBS and synergy between the antiandrogen enzalutamide and BI2536 (2.5 nM) was assessed and plotted as in (A).
(D-F) Synergy experiments between the antiandrogens orteronel, Galeterone, and ARV-110, and BI2536 (6 nM) performed and plotted as in (A).
(G) AR protein immunoblot of lysates from C4-2 cells treated with ARV-110 for 24 hours. The highest dose was 100 nM followed by four 1.5-fold serial dilutions and are the same doses used in (F).
(H) AR protein immunoblot of lysates from the indicated cell. The lower bands are AR splice variants, including AR-v7.
(I-J) LNCaP95 and 22Rv1 AR-v7+ CRPC, and DU145 and PC-3 AR-negative prostate cancer cells were assessed for synergy between abiraterone and BI2536 (4, 3, 5, and 7.5 nM, respectively) as in (A).
(K) C4-2 cells were assessed for synergy between abiraterone and BI2536 (4 nM) in the absence (top) or presence (bottom) of ARV-110 as in (A).
(L) Chemical structures of the panel of Plk1 inhibitors used.
(M) Synergy between abiraterone and volasertib (5 nM), GSK461364 (5 nM), or onvansertib (10 nM) was assessed as in (A).
(N) C4-2 cells were subjected abiraterone in the presence or absence of alisertib (20 nM, left), barasertib (200 nM, middle), or nocodazole (200 nM, right). Doses were chosen based on a ~20% decrease in viability when these antimitotic drugs were used in isolation. Synergy between these drugs was assessed as in (A).
(O) BIDMC PC4 CRPC organoids were treated with onvansertib (200 nM) and abiraterone (10 μM) for 8 days and then relative viability was assessed. LUCaP70CR CRPC organoids were treated with onvansertib (40 nM) and abiraterone (10 μM) for 5 days and then relative viability assessed. Mean ± SEM (n = 3) is shown. n.s. not significant, * p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001 by Student’s two-tailed t-test.
Resistance to antiandrogens in CRPC can be caused by expression of AR-v7, a constitutively active splice variant lacking the ligand binding domain that signals even in the presence of antiandrogens (36,52). LNCaP95 and 22Rv1, two AR-v7+ CRPC cell lines (Fig. 2H), were therefore tested. LNCaP95 demonstrated synergistic killing by the abiraterone- Plk1 inhibitor combination while 22Rv1 did not (Fig. 2I). Similar to full-length AR, the protein abundance of AR splice variants decreased in response to the combination regardless of the cell’s synergistic phenotype (Supplementary Fig. S1D). We next examined AR-negative DU145 and PC-3 prostate cancer cell lines (Fig. 2H). Both DU145 and PC-3 cells demonstrated synergistic killing by combined abiraterone and Plk1 inhibitor treatment (Fig. 2J) despite the lack of AR expression. Finally, to specifically address whether, in AR-expressing CRPC cells, this synergy could be partially dependent on AR signaling, abiraterone and Plk1 inhibitors were added to C4-2 cells in the presence ARV-110 (at a dose that reduced AR protein 92%). The synergistic response was unaltered (Fig. 2G and K). Taken together, these data firmly establish that Plk1 inhibitor-abiraterone synergy cannot be ascribed to suppression of AR signaling or androgen synthesis, a conclusion that is reinforced by multiple experiments described below.
We next explored whether this synergy was truly dependent on Plk1 using a panel of Plk1 inhibitors (Fig. 2L). While BI2536 and volasertib have structural similarities and can inhibit Plk2 and Plk3, GSK461364 and onvansertib are structurally disparate and Plk1-specific inhibitors (53). All three Plk1 inhibitors sensitized C4-2 cells to abiraterone regardless of androgen levels (Fig. 2M and Supplementary Fig. S1E). We further confirmed lack of synergy between the Plk1-specific inhibitor onvansertib and other antiandrogens (Supplementary Fig. S1F) as well as AR independence by demonstrating onvansertib-abiraterone synergy in the presence ARV-110 (Supplementary Fig. S1G).
Plk1 is intimately involved in multiple stages of mitosis, and mitotic arrest is a well-documented effect of Plk1 inhibition (17). It was therefore plausible that any anti-mitotic agent would synergize with abiraterone. However, co-treatment with docetaxel, which causes prometaphase and metaphase arrest, did not sensitize C4-2 cells to abiraterone (Fig. 1B). Furthermore, abiraterone in combination with inhibitors of the mitotic kinases Aurora A (alisertib), Aurora B (barasertib), or a microtubule polymerization inhibitor (nocodazole) did not cause synergistic tumor cell killing (Fig. 2N). These data argue that synergy between abiraterone and Plk1 inhibitors involves some unique aspect of Plk1 function that is not a generic effect of mitotic arrest. However, neither Plk1 abundance nor kinase activity alone were biomarkers for prostate cancer cell line synergistic responses to combined Plk1 inhibition and abiraterone based on western blotting for Plk1 or phosphorylation of a canonical Plk1 substrate, TCTP (Supplementary Fig. S1H) (54).
We next evaluated the effect of combined onvansertib and abiraterone in BIDMC PC4 and LuCaP70CR 3-D organoids generated from CRPC PDXs (55), which express high levels of AR and grow in androgen-deprived media. Onvansertib or abiraterone monotherapy inhibited cell growth compared to control, but the combination treatment elicited a synergistic response (Bliss independence CI of 0.82 and 0.77, respectively) demonstrating that this synergy is not confined to 2-D cell line models (Figure 2O).
Abiraterone treatment upregulates mitosis and mitotic spindle related gene sets in a synergy-specific and AR-independent manner
To gain insight into the mechanism of abiraterone-Plk1 inhibitor synergy, we designed a comprehensive RNA sequencing experiment (Fig. 3A) using C4-2 and LNCaP cells following treatment with abiraterone or enzalutamide alone, and in combination with onvansertib, a highly Plk1-specific inhibitor being investigated in human cancer patients. Enzalutamide served as a control to identify AR-independent transcriptional effects of abiraterone, while LNCaP cells allowed identification of transcriptional signatures specific to a synergistic phenotype. Gene set variation analysis (GSVA) was then used to generate pathway-level quantification so that biologically meaningful gene sets could be readily compared between conditions (31). First, the effects of abiraterone and enzalutamide on AR-dependent transcription was determined using a curated list of androgen responsive genes (Hallmark Androgen Response gene set) (56). Both antiandrogens suppressed AR-dependent transcription equivalently in both cell lines (Fig. 3B), yet only abiraterone synergizes with onvansertib, and only in C4-2 cells (Fig. 1B and J), supporting the notion that this synergy is not due AR inhibition. Importantly, combined abiraterone and onvansertib did not synergistically repress AR-dependent transcription, and the small effect that onvansertib treatment had on AR-dependent genes (Fig. 3B) and AR transcript levels (Supplementary Fig. S2A) occurred in both cell lines. In addition, abiraterone treatment did not alter Plk1 transcript levels (Supplementary Fig. S2B) indicating that synergy is not due to transcriptional inter-regulation between these pathways.
Figure 3. Abiraterone treatment induces an AR-independent synergy-specific gene expression signature dominated by mitosis and mitotic spindle related gene sets.
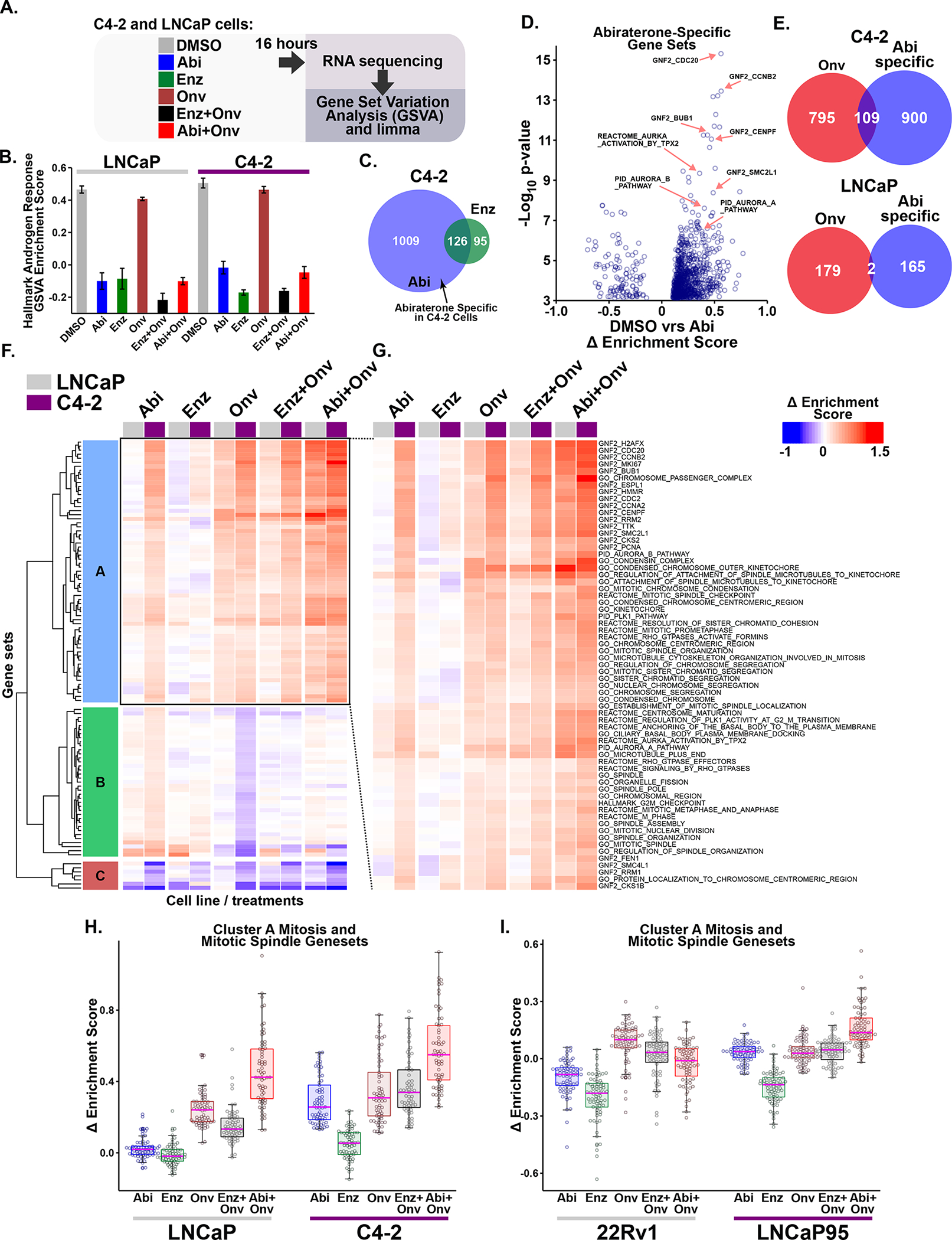
(A) Diagram of the RNA sequencing experiment. C4-2 and LNCaP cells were grown in FBS-containing media, treated for 16 hours with 5 μM abiraterone (Abi), 10 μM enzalutamide (Enz), onvansertib (15 or 30 nM, respectively; Onv), or the indicated combinations.
(B) AR-dependent transcription was investigated using the Hallmark Androgen Response gene set. Bars indicate mean enrichment score ± SEM.
(C) Identification of abiraterone-specific gene sets. Venn diagram depicts overlap of differentially expressed gene sets (FDR ≤ 0.01) in the two conditions.
(D) Volcano plot of abiraterone-specific gene sets in C4-2 cells. The Δ enrichment score represents the difference in mean enrichment scores between drug and control treatment. Several gene sets related to mitosis and the SAC are indicated by red arrows. GNF2 gene sets represent cancer gene neighborhoods (57). Members of these gene sets have expression patterns highly covariant with the seed gene listed in the name.
(E) Venn Diagram comparing abiraterone-specific and onvansertib differential gene set expression (FDR ≤ 0.01). Onvansertib and abiraterone share a large number of significant gene sets in C4-2, but not LNCaP, cells.
(F) Unsupervised hierarchical clustering of Δ enrichment scores using all treatments (columns) and the 109 gene sets (rows) identified in (E) using C4-2 cells. Columns depicting LNCaP and C4-2 gene set expression are marked at the top with grey and purple boxes, respectively. For labelled heatmap see Supplementary Fig. S2C.
(G) Expanded view of cluster A that is induced by abiraterone but not enzalutamide in C4-2 but not LNCaP cells, organized and displayed as in (F). Nearly all of the gene sets in cluster A are related to mitosis or the mitotic spindle.
(H) Box and whisker plot of cluster A gene set expression in C4-2 and LNCaP cells. Boxes depict the median and upper and lower quartiles. Whiskers extend to the most extreme value located less than 1.5 times the interquartile range from the upper or lower quartile. Individual gene sets are overlaid as circles.
(I) RNA sequencing analysis of LNCaP95 and 22Rv1 CPRC cells performed and analyzed as in A-H. Doses used were 10 μM abiraterone, 20 μM enzalutamide, and 15 nM onvansertib for LNCaP95 cells, and 5 μM abiraterone, 10 μM enzalutamide, and 20 nM onvansertib for 22Rv1 cells. The 109 gene set signature was isolated and Δ enrichment scores were hierarchically clustered (Supplementary Fig. S3B). Shown is a box and whisker plot of gene sets contained in cluster A, which contains many gene sets related to mitosis and the mitotic spindle.
AR-independent transcriptional effects of abiraterone in C4-2 cells that might contribute to synergy were identified by isolating gene sets that were regulated by abiraterone but not enzalutamide, thereby defining an abiraterone-specific gene set signature. While the majority of gene sets altered by enzalutamide treatment were also altered by abiraterone treatment, the converse was not true (Fig. 3C). Notably, among the most statistically significant upregulated abiraterone-specific gene sets, many were related to mitosis and Plk1 function or were gene neighborhoods (57) of mitotic genes (Fig. 3D), indicating that the abiraterone-specific effects likely overlap with the consequences of Plk1 inhibition. We therefore identified gene sets that were both abiraterone-specific and differentially regulated by onvansertib treatment (Fig. 3E). In C4-2 cells, among 904 gene sets that were differentially expressed after onvansertib treatment, 109 were in common with the abiraterone-specific gene set signature, in sharp contrast to LNCaP cells where only two gene sets met these criteria. When unsupervised hierarchical clustering was performed on these 109 gene sets (Fig. 3E), three clusters emerged (Clusters A-C, Fig. 3F and Supplementary Fig. S2C). The largest of the three, cluster A, was composed almost entirely of gene sets related to mitosis or mitotic spindle assembly (Fig. 3G). Cluster A gene sets were then mapped back onto the LNCaP and C4-2 expression data revealing upregulation by both abiraterone and onvansertib treatment in C4-2 cells, but only by onvansertib in the LNCaP cells (Fig. 3H). These gene sets were not significantly upregulated by enzalutamide in either cell line. Interestingly, combination treatment with abiraterone and onvansertib very strongly upregulated the Cluster A mitotic gene set, while the enzalutamide and onvansertib combination did not (Fig. 2H).
To confirm that this AR-independent synergy-specific mitotic gene set signature was not limited to C4-2 cells, similar RNA sequencing analysis was performed using LNCaP95 and 22Rv1 AR-v7+ CPRC cells (Fig. 2I). Due to AR-v7 expression, the repression of the Hallmark Androgen Response gene set by abiraterone or enzalutamide treatment was muted in 22Rv1 cells and entirely absent in LNCaP95 cells. Nonetheless, combined abiraterone-onvansertib treatment synergistically killed LNCaP95 cells despite having no effect on AR-dependent gene expression. In contrast no synergistic killing was observed in 22Rv1 cells that showed mild repression of the Hallmark Androgen Response gene set (Supplementary Fig. S3A). Similar to that seen in C4-2 and LNCaP cells, the mitotic gene set signature was upregulated by abiraterone, but not enzalutamide, and increased synergistically by combined abiraterone and onvansertib, but not combined enzalutamide and onvansertib treatment, only in LNCaP95 cells (Fig. 3I and Supplementary Fig. S3B). Taken together, this abiraterone- and synergy-specific gene set signature indicates that abiraterone has AR-independent effects resulting in upregulation of gene sets related to mitosis, suggesting that abiraterone itself may directly affect one or more mitotic processes.
Abiraterone treatment causes multiple mitotic defects in a manner that is distinct from its AR blockade effects
Abiraterone’s potential AR-independent effects on mitosis and mitotic spindle morphology were investigated using indirect immunofluorescence. C4-2 and LNCaP cells were treated with DMSO, abiraterone, or enzalutamide, fixed and stained for tubulin, the centromeric histone CENP-A, and DNA (DAPI). In contrast to Plk1 inhibitors or mitotic spindle poisons (27), abiraterone treatment did not grossly alter spindle organization. Close examination, however, revealed an altered chromatin morphology in C4-2 cells, but not in LNCaP cells, after abiraterone treatment. In DMSO- and enzalutamide-treated cells, metaphase chromosomes displayed highly condensed chromatin with discrete easily observable chromosome arms. Following abiraterone treatment, the majority of metaphase C4-2 cells, but not LNCaP cells, had insufficiently condensed chromatin to define individual chromosome arms (Fig. 4A and B; Supplementary Fig. S4A and B). This is most clearly seen in Supplementary Movies S1, S2, and S3 comparing Z-stacked images of treated C4-2 cells. To quantify reduced chromatin condensation, intercentromeric distance between sister chromatids was measured using CENP-A staining (Supplementary Movie S4) (58,59). This revealed that abiraterone, but not enzalutamide treatment, increased intercentromeric distance in C4-2 but not LNCaP cells (Fig. 4C and D and Supplementary Fig. S4C), consistent with chromosome condensation defects.
Figure 4. Abiraterone treatment prevents stable mitotic spindle orientation and impairs chromosome condensation independently of effects on AR signaling.
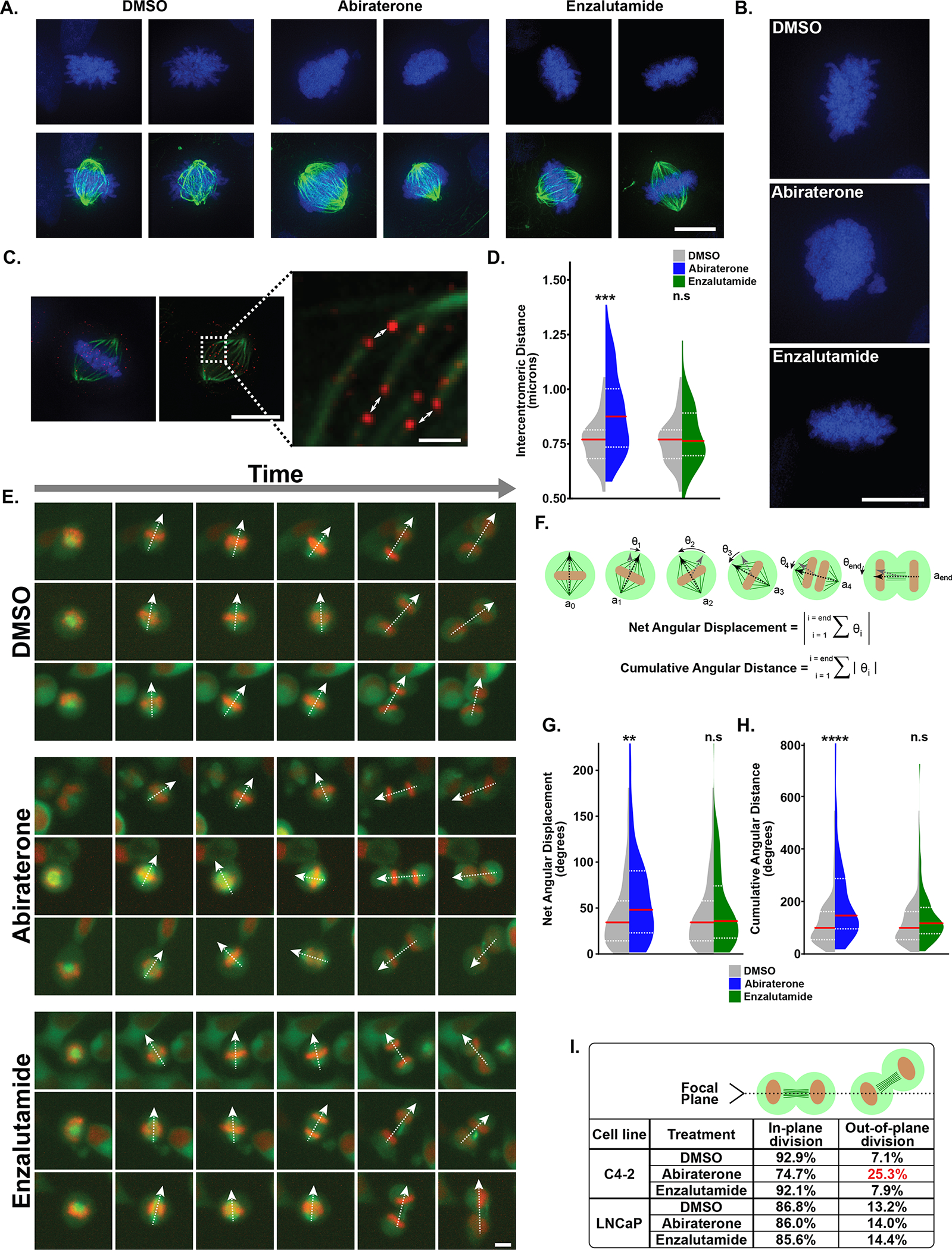
(A) C4-2 cells were treated with DMSO, abiraterone (5 μM), or enzalutamide (10 μM) for 24 hours prior to fixation and staining with DAPI (blue) and antibodies against tubulin (green). Additional examples in Supplementary Fig. S4A. DMSO and enzalutamide treated cells had well-defined chromosome arms in contrast to decondensed chromosome arms seen in abiraterone-treated cells. Scale bar lower right 10 μm. See Supplementary Movies S1–S3 for deconvolved Z-stacked images.
(B) Enlarged micrographs of cells treated and analyzed as in (A). Scale bar lower right 10 μm.
(C) C4-2 cells treated as in (A) stained for CENP-A (red). Centromeres on sister chromatids were identified by their paired orientation in three-dimensional space and microtubules emanating from opposite spindle poles. See Supplementary Movie S4 for more information. Intercentromeric distance was measured between paired centromeres (white arrows). Scale bar lower right of top and bottom images 10 and 1 μm, respectively.
(D) Violin plots depicting intercentromeric distances observed in DMSO, abiraterone and enzalutamide treated cells. The median (red line) and upper and lower quartiles (dotted white lines) are shown. N=50, *** p ≤ 0.001, n.s. not significant using a two-tailed Mann-Whitney U test.
(E) C4-2 cells expressing H2B-mCherry and mEmerald-tubulin were treated with DMSO, abiraterone (5 μM), or enzalutamide (10 μM), and analyzed by live-cell microscopy at 15-minute intervals. Individual cells (rows) were tracked as they progressed from prophase (a single cell with condensed chromatin), to cytokinesis (two daughter cells containing decondensed chromatin). The dashed white arrows represent the major axis of the mitotic spindle. Scale bottom right 10 μm. Related to Supplementary Movie S5.
(F) The vector axis (ai) of the mitotic spindle was tracked from when it was first apparent until telophase. Frames numbers (starting at zero) are denoted by the subscript i. The rotation between each frame was calculated (grey to black arrows; θi). Clockwise rotation was positive, counterclockwise rotation negative. Net angular displacement defined as the absolute value of the total rotation that occurred between the initial and final angle. Cumulative angular distance is the sum of the absolute values of all stepwise rotations - the total spindle rotation that occurred throughout mitosis.
(G, H) Violin plots comparing net angular displacement and cumulative angular distance in DMSO versus abiraterone or enzalutamide treated cells, with distributions plotted and analyzed as in (D). N ≥ 92, ** p ≤ 0.01, **** p ≤ 0.0001, n.s. not significant.
(I) The frequency of mitotic divisions that occurred nonparallel to the growth surface. Nonparallel division was apparent when one daughter cell left the focal plane during cytokinesis. See Supplementary Movies S6 and S7 for examples.
Time-lapse microscopy was then used to analyze impacts of abiraterone treatment on the dynamic process of mitosis in C4-2 and LNCaP cells expressing histone H2B-mCherry and mEmerald-tubulin. Abiraterone, but not enzalutamide, caused a statistically significant but small increase in the duration of mitosis in both cell types (Supplementary Fig. S4D). Furthermore, abiraterone, but not enzalutamide, caused significant defects in mitotic spindle orientation in both cell types. Spindles in abiraterone-treated cells failed to form a stable axis with respect to the cell cortex, and displayed continual rotation during progression from prometaphase through telophase (Fig. 4E, Supplementary Movie S5). The angle of the spindle’s major axis was tracked from when it was first apparent until telophase. This allowed calculation of the net displacement and cumulative spindle rotation that occurred throughout mitosis (Fig. 4F). A clear increase in the net displacement and cumulative rotational angle was observed throughout mitosis in abiraterone-treated, but not enzalutamide-treated, C4-2 and LNCaP cells (Fig. 4G–H and Supplementary Fig. S4E and S4F), indicating that this is not a consequence of AR inhibition or prolonged mitotic arrest (Supplementary Fig. S4G).
Abiraterone treatment did not result in the formation of multipolar spindles in either cell type (Supplementary Fig. S4H). However, C4-2, but not LNCaP, cells demonstrated a substantial increase in out-of-plane cell division (i.e. non-parallel to the plane of growth) following abiraterone, but not enzalutamide, treatment (Fig. 4I, Supplementary Movies S6 and S7). Taken together, we interpret this as evidence that abiraterone treatment induces multiple mitotic defects independent of its effects on AR signaling, and that these mitotic defects were more pronounced in a cell line that showed synergistic killing by the abiraterone-Plk1 inhibitor combination.
Abiraterone in combination with Plk1 inhibition causes synergistic mitotic arrest followed by cell death in a SAC-dependent manner
The above findings, indicate that abiraterone’s effects on mitosis, while mild in isolation, could render a subset of cancer cells more susceptible to the anti-mitotic effects of Plk1 inhibition. To examine this, C4-2 cells were treated with abiraterone, onvansertib, or the combination, and cell cycle distribution analyzed by flow cytometry using DAPI, and antibodies against phospho-histone H3 (pHH3), a marker of mitotic cells. Abiraterone caused a small but statistically significant increase in the percentage of mitotic cells, while onvansertib markedly increased pHH3+ population consistent with mitotic arrest (Fig. 5A–C). Interestingly, combined onvansertib plus abiraterone induced a two-fold increase in mitotic arrest compared to onvansertib monotherapy.
Figure 5. The combination of abiraterone and Plk1 inhibitors causes synergistic mitotic arrest followed by tumor cell death in a SAC-dependent manner.
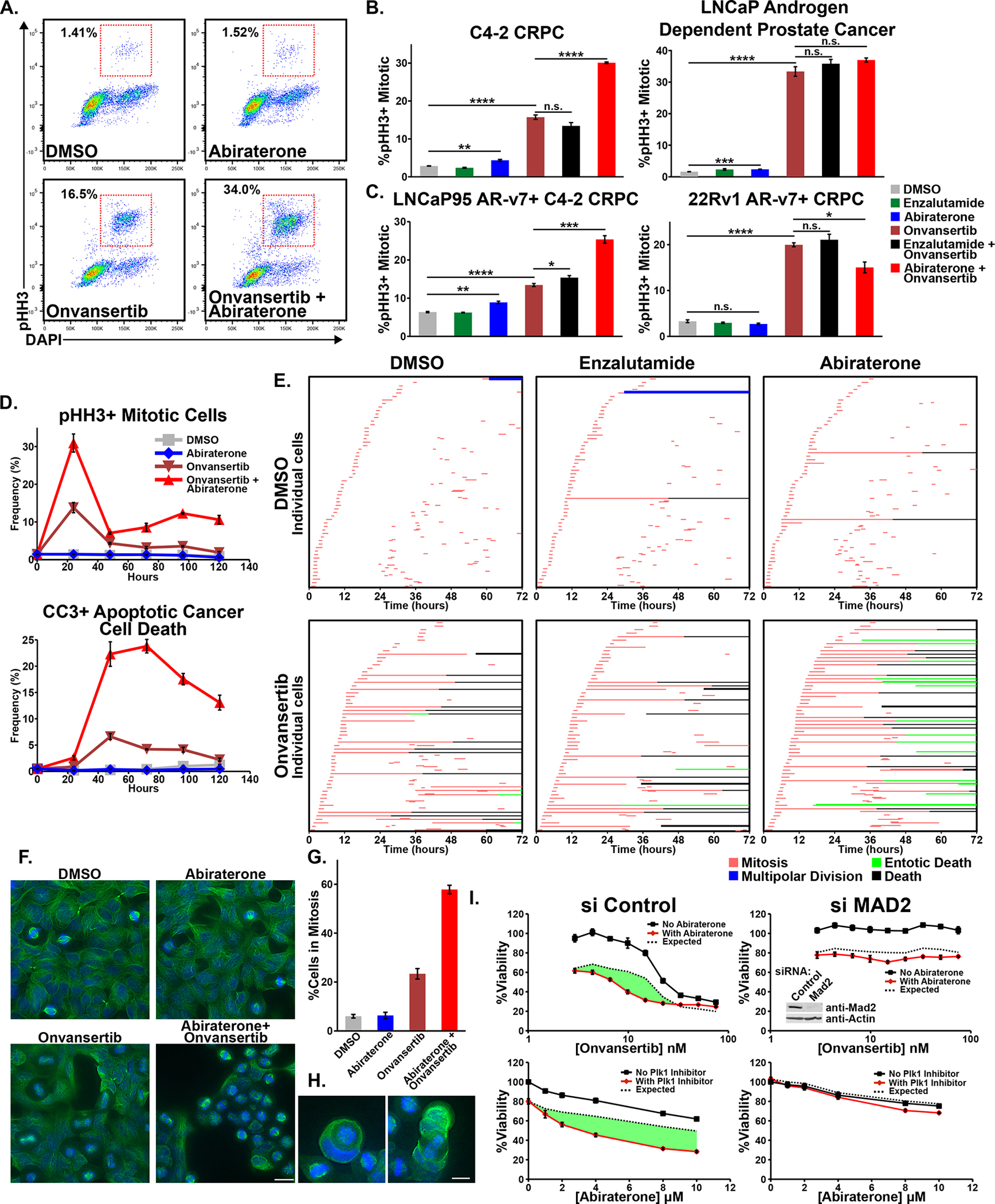
(A) C4-2 cells were subjected to DMSO, abiraterone (5 μM), onvansertib (15 nM), or the combination for 24 hours, cell cycle distribution analyzed by flow cytometry. Red boxes indicate mitotic cells.
(B) Mitotic arrest, measured as in (A), in C4-2 and LNCaP cells after 16-hour treatment with abiraterone (5 μM), enzalutamide (10 μM), or onvansertib (15 nM and 30 nM, respectively) or the indicated combinations. Mean ± SEM (n = 3). n.s. not significant, * p ≤ 0.05, ** p ≤ 0.01, and **** p ≤ 0.0001 by Student’s two-tailed t-test.
(C) LNCaP95 cells were treated with 10 μM abiraterone, 20 μM enzalutamide, 15 nM onvansertib, or the indicated combinations. 22Rv1 cells were treated with 5 μM abiraterone, 10 μM enzalutamide, 20 nM onvansertib, or the indicated combinations. Data was collected and analyzed as in (B).
(D) C4-2 cells were treated as in (A) over a 120-hour time course. The percentage of mitotic and CC3-positive apoptotic cells was measured by flow cytometry. Mean ± SEM (n = 3). See Supplementary Fig. S5C for other cell-cycle stages.
(E) Time-lapse microscopy of C4-2 cells treated with DMSO, abiraterone (5 μM), and enzalutamide (10 μM); alone and in combination with onvansertib (15 nM). Sixty cells were analyzed per condition Each row begins by tracking a cell until initiation of mitosis (red), daughter cells were then tracked possibly initiating mitosis (red) to create up to four cells. Prolonged mitotic arrest was often followed by cell death (black) or entosis (green). See Supplementary Movies S6, S8, and S9 for examples.
(F) AR-negative DU145 prostate cancer cells were treated with onvansertib (10 nM), abiraterone (10 μM), or the combination for 12 hours, then fixed. Tubulin (green) and DNA (DAPI, blue) were examined by microscopy. Scale bar at bottom right 20 μm.
(G) Quantification of mitotic cells from microscopy presented in (F). Mean ± SEM.
(H) Entosis of DU145 mitotic cells after cotreatment with abiraterone and onvansertib. Scale bar at bottom right 10 μm.
(I) C4-2 cells were transfected with a control or Mad2-targeting siRNA and 48 hours later assessed for synergy. Shown is onvansertib sensitivity in the presence of 8 μM abiraterone (top) and abiraterone sensitivity in the presence of 15 nM onvansertib (bottom). Viability relative to control 72 hours after drug addition, mean ± SEM (n = 3). The dotted black line represents the Bliss independence expected response and synergy is shown in green. Immunoblot inset (upper right panel) confirmed Mad2 knockdown 48 hours after siRNA transfection. For an independent Mad2-targeting siRNA see Supplementary Fig. S5G.
To determine whether this synergistic mitotic arrest was independent of abiraterone’s effect on AR signaling, C4-2 cells were treated with vehicle, abiraterone, or enzalutamide; alone or in combination with onvansertib, and analyzed as above. As shown in Fig. 5B, the combination of enzalutamide and onvansertib showed essentially the same extent of mitotic arrest as that obtained with onvansertib treatment alone, whereas the abiraterone plus onvansertib combination resulted in strong synergistic mitotic arrest. Importantly, in non-synergistic LNCaP cells, onvansertib-induced mitotic arrest was not enhanced by co-treatment with either enzalutamide or abiraterone (Fig. 5B). Similar results were obtained in the two AR-v7+ CPRC cell lines, with a synergistic accumulation of mitotically-arrested LNCaP95 cells, but not 22Rv1 cells, following abiraterone, but not enzalutamide, co-treatment with onvansertib (Fig. 5C). Furthermore, siRNA knockdown of Cyp17A1 failed to enhance mitotic arrest caused by onvansertib, indicating synergistic mitotic arrest was not due to androgen synthesis inhibition or altered steroidal hormone levels (Supplementary Fig. S5A and B). When assessed over time, the combination treatment-induced mitotic arrest peaked at 24 hours, followed by synergistic apoptotic cancer cell death as judged by cleaved caspase-3 (CC3) staining (Fig. 5D and Supplementary Fig. S5C). These data indicate that AR-independent effects of abiraterone synergize with Plk1 inhibition to cause enhanced mitotic arrest, followed by markedly increased apoptosis consistent with mitotic catastrophe (60).
To acquire a more granular understanding of this synergistic mitotic phenotype and assess overall cell fate, time-lapse microscopy of C4-2 cells expressing H2B-mCherry and mEmerald-tubulin was performed. Individual cells were tracked following treatment with vehicle, abiraterone, or enzalutamide; alone and in combination with onvansertib (Fig. 5E). Onvansertib at this dose elicited a heterogeneous response, with a subset of cells displaying prolonged mitotic arrest (~24 hours) while others progressed through mitosis unabated. Prolonged mitotic arrest was frequently followed by cell death (Supplementary Movie S8). Combined abiraterone-onvansertib markedly increased the frequency of severe mitotic arrest compared to onvansertib treatment alone, whereas enzalutamide treatment did not enhance onvansertib induced mitotic arrest.
While prolonged mitotic arrest caused by combined abiraterone-onvansertib was frequently followed by cell death, we additionally observed a marked increase in entosis (Fig. 5E and Supplementary Movie S9). Entosis is a form of cell death that involves one cell invading another, and has been reported to be a consequence of mitotic arrest in certain situations (60,61). While entosis occurred occasionally among C4-2 cells treated with onvansertib alone, it was commonly seen in cells co-treated with abiraterone and onvansertib. This was not seen following combined enzalutamide and onvansertib treatment. Rock1 kinase activity is required for mitotic entosis (61), an observation we confirmed in this context (Supplementary Fig. S5D). Notably, Rock1 inhibition did not reduce the total synergistic cancer cell killing following combined abiraterone and onvansertib treatment (Supplementary Fig. S5E). Thus, while the observed entosis may be a consequence of abiraterone-onvansertib induced mitotic arrest, it is not causal for synergistic cell killing.
To further confirm that combined abiraterone and Plk1 inhibitors synergistically disrupt mitosis in prostate cancer cells in an AR-independent manner, DU145 AR-negative cells were treated with onvansertib, abiraterone, or the combination. Despite lacking detectable AR protein (Fig. 2H), these cells demonstrated synergistic G2/M arrest upon combination treatment when analyzed by flow cytometry (Supplementary Fig. S5F) or quantification of mitotic figures by microscopy (Fig. 5F and G). Moreover, entotic figures were observed among co-treated cells indicating that induction of entosis is AR-independent (Fig. 5H).
Our observation that abiraterone impairs mitosis, rendering some cancer cells more susceptible to mitotic arrest following Plk1 inhibition implies that this synergy should depend on a functional SAC. The SAC inhibits anaphase progression by inhibiting APC/C-Cdc20 until chromosome biorientation is achieved though proper microtubule-kinetochore attachments and tension between opposite spindle poles (62). Mad2 is an essential component of the SAC, and loss of this protein prevents mitotic arrest caused by spindle assembly defects. When we examined onvansertib-abiraterone synergy in C4-2 cells after Mad2 knockdown by two distinct siRNAs, synergy was eliminated (Fig. 5I and Supplementary Fig. S5G). Mps1 is a dual-specificity kinase required for kinetochore localization of multiple SAC components, including Mad2, and Mps1 inhibition abrogates SAC activity (63). Loss of SAC activity through Mps1 inhibition eliminated synergy between abiraterone and onvansertib (Supplementary Fig. S5H). Tubulin is a common off-target binding partner of drugs (64), and we have previously shown that microtubule polymerization inhibitors synergize with Plk1 inhibition causing SAC-dependent synergistic mitotic arrest and cancer cell death (27). However, in vitro tubulin polymerization was unaffected by excessive concentrations of abiraterone (Supplementary Fig. S5I) indicating that synergy with Plk1 inhibitors was not due to abiraterone binding directly to tubulin to inhibit microtubule polymerization.
The abiraterone/Plk1 inhibitor combination synergistically kill cancer cells from diverse tumor types in culture and in vivo
The observation that AR-independent abiraterone-induced mitotic defects sensitize a subset of prostate cancer cells to Plk1 inhibition raised the possibility that the efficacy of this combination was not limited to AR-driven cancers. We therefore examined a set of acute myeloid leukemia (AML), pancreatic, and ovarian cancer cell lines for synergy between abiraterone and onvansertib. As shown in Fig. 6A and B, MV-4-11 AML cells, PSN-1 pancreatic cancer cells, and SK-OV-3 ovarian cancer cells were synergistically killed by the combination, while OCI-AML-3 AML, Panc 10.05 pancreatic cancer cells, and OAW28 ovarian cancer cells were not. In general, synergistic cell lines displayed a greater fold change in mitotic arrest as a result of combination treatment compared to onvansertib alone (purple bars in Fig. 6C and see Supplementary Fig. S6A). In addition, similar to the prostate cancer cell lines, there was no correspondence between a synergistic phenotype and Plk1 protein abundance or activity (Supplementary Fig. S6B). Importantly, none of the non-prostate cancer cell lines expressed detectable levels of the AR (Fig. 6D), clearly demonstrating that synergistic mitotic arrest and cell death caused by the combination of abiraterone and Plk1 inhibition is AR independent.
Figure 6. Synergistic mitotic arrest and cancer cell killing by combined abiraterone and Plk1 inhibitor treatment occurs in multiple AR-negative non-prostate cancer cell lines in vitro and in vivo.
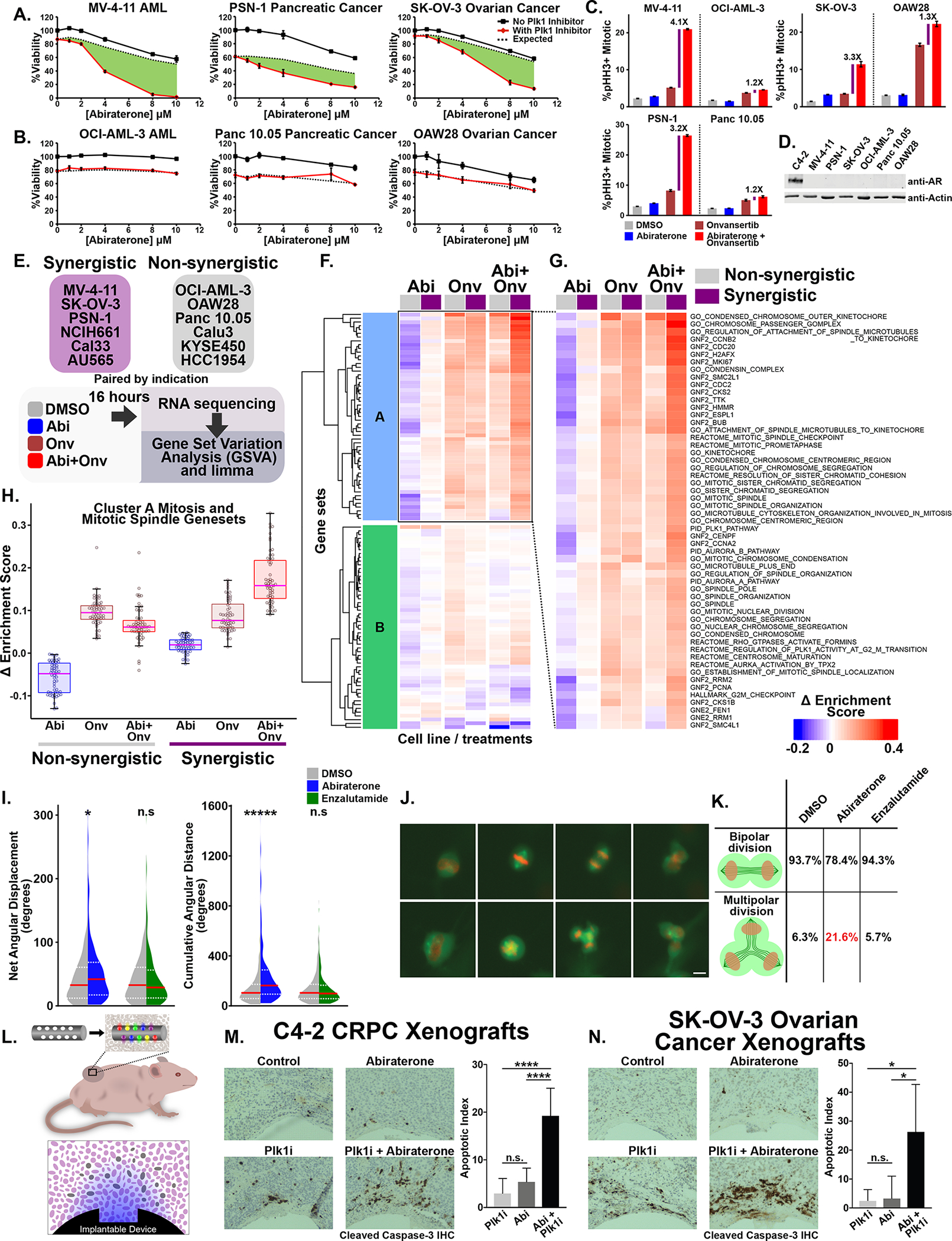
(A, B) Six tissue-paired non-prostate cancer cell lines were tested for abiraterone-onvansertib synergy. Shown is relative viability in response to abiraterone in the absence (black lines) or presence (red lines) of onvansertib (MV-4-11, OCI-AML-3, and PSN-1 22.2 nM; Panc 10.05, and SK-OV-3 33.3 nM; OAW28 15 nM) after 72 hours. Mean ± SEM (n = 3). Dotted line indicates the Bliss independence expected result and green areas indicate synergy.
(C) Shown for six cell lines, paired by tissue, are the percentage of mitotic cells after 16-hours drug treatment and cell-cycle analysis by flow cytometry. Purple bars indicate the fold-change in %pHH3+/mitotic cells between onvansertib monotherapy and the abiraterone-onvansertib combination. See Supplementary Fig. S6A for other cell cycle stages. Abiraterone treatment was 10 μM for all cell lines except OAW28, which was 5 μM. Onvansertib concentrations used were 20 nM for MV-4-11, OCI-AML-3, and PSN-1; 30 nM for Panc 10.05 and SK-OV-3; 15 nM for OAW28. Mean ± SEM (n = 3).
(D) AR protein immunoblot using the indicated cell lysates.
(E) RNA sequencing experimental design among six non-prostate cancer cell lines that show synergy and six that do not (See panel A, B and Supplementary Fig. S6C). For drug doses used see Supplementary Fig. S6D.
(F) The AR-independent synergy-specific gene set signature derived in Fig. 3 was applied to the non-prostate cancer RNA-seq data. The mean Δ enrichment scores among synergistic (purple boxes) and nonsynergistic (grey boxes) cancer cell lines were used for unsupervised hierarchical clustering. For labelled heatmap see Supplementary Fig. S6E.
(G) Cluster A mitosis- and spindle-related gene sets that are induced by abiraterone only in the synergistic cell lines
(H) Box and whisker plot comparing expression of cluster A gene sets in synergistic and non-synergistic cell lines plotted as in Fig. 3H.
(I) SK-OV-3 ovarian cancer cells expressing H2B-mCherry and mEmerald-tubulin were treated with DMSO, abiraterone (10 μM), or enzalutamide (20 μM), and imaged at 15-minute intervals for 72 hours. As in Fig. 4E–H, the vector axis of the mitotic spindle was tracked. Violin plots comparing the distribution of net angular displacement (left) and cumulative angular distance (right) in DMSO versus drug treated cells. The red line represents the median and dotted white lines are upper and lower quartiles (n ≥ 141). * p ≤ 0.05, ***** p ≤ 0.00001, n.s. not significant using a two-tailed Mann-Whitney U test.
(J) Micrographs from live-cell microscopy of SK-OV-3 cells demonstrating normal/bipolar division (top row) and multipolar division (bottom row). Scale bar bottom right 10 μm.
(K) Quantification of bipolar and multipolar division of SK-OV-3 cells treated as in (I).
(L) Diagram of the tumor-implantable device used to examine synergy between abiraterone and Plk1 inhibitors in vivo. Colored gradients represent drug concentration gradients created upon device implantation. On the top is the implantable device with respect to the tumor bearing mouse, on the bottom is a depiction of a microwell and drug gradient corresponding to the tumor sections shown in (M) and (N).
(M) C4-2 CRPC tumors in non-castrated male mice were implanted with drug-loaded devices. Microwells were loaded with drug at 12.5% by weight in isolation or 12.5% by weight each when combined. Tumor sections adjacent to the microwells containing the indicated drugs were stained with anti-CC3 antibodies to detect apoptotic cells (left). Clear sections at the bottom of each image are where the implantable device was previously located. On the right is the percentage of apoptotic cells within a 400 μm radius of wells. Mean ± SEM; n ≥ 9; n.s. not significant and **** p ≤ 0.0001 by a Student’s two-tailed t-test.
(N) SK-OV-3 ovarian cancer tumors in female mice were implanted with devices, processed, analyzed, and quantified as in (M). N ≥ 5; n.s. not significant and * p ≤ 0.05 by Student’s two-tailed t-test.
We next evaluated our AR-independent synergy-specific mitotic gene set signature defined in Fig. 3 in a variety of disparate non-prostate cancer cell lines. We identified additional pairs of cells lines (derived from Her2+ breast cancer, lung adenocarcinoma, and head and neck tumors), in which one member of the pair responded synergistically to the abiraterone-onvansertib combination and one did not (Supplementary Fig. S6C). This 12-cell line panel was then used for RNA sequencing analysis after treatment with abiraterone, onvansertib, or the combination (Fig. 6E and Supplementary Fig. S6D). Expression data was transformed into gene set enrichment scores using GSVA, and the previously defined 109 gene set signature used for unsupervised hierarchical clustering (Fig. 6F and Supplementary Fig. S6E), resulting in two clusters (A and B). Cluster A contained a multitude of gene sets related to mitosis and the mitotic spindle (Fig. 6G). As seen in the prostate cancer cell lines, abiraterone treatment resulted in upregulation of cluster A, and combined abiraterone-onvansertib treatment further enhanced their expression only in the synergistic cell lines (Fig. 6H). This confirmation of AR-independent upregulation of a mitosis- and mitotic spindle-related gene set signature following abiraterone monotherapy and combined abiraterone-onvansertib treatment in non-prostate cancer cells demonstrates its specificity for a synergistic phenotype.
We next examined effects of abiraterone treatment on mitotic phenotypes in AR-negative SK-OV-3 ovarian cancer cells that had demonstrated pronounced synergy (Fig. 6A). SK-OV-3 cells were analyzed by live-cell microscopy and indirect immunofluorescence in an identical manner to Fig. 4C–H. Similar to C4-2 and LNCaP cells, abiraterone but not enzalutamide treatment caused a mild increase in the duration of mitosis (Supplementary Fig. S6F) as well as increased spindle rotation (Fig. 6I), albeit less pronounced. However, in these cells abiraterone treatment did not cause defects in chromosome condensation or out-of-plane cell division (Supplementary Fig. S7A and B). Instead abiraterone treatment alone resulted in a marked increase in multipolar spindle formation and cell division (Fig. 6J and K), consistent with abiraterone’s induction of multiple cell type-specific AR-independent mitotic defects.
To examine the potentially greater-than-additive effects of these drugs on apoptotic cancer cell death in vivo, we examined C4-2 CRPC xenografts grown in non-castrated mice using a tumor-implantable device capable of achieving multiplexed drug sensitivity testing within tumors (Fig. 6L) (27,34). The device consists of a small cylinder containing multiple drug-loaded microwells that is implanted into a tumor, generating spatially distinct concentration gradients in the adjacent tissue. Following incubation, tumors were excised, and stained for CC3 as a marker of apoptotic cancer cell death. As shown in Fig. 6M, little apoptotic cell death was observed in C4-2 tumor sections adjacent to wells containing abiraterone or the Plk1 inhibitor alone. In contrast, significant apoptotic cell death was seen in tumor sections adjacent to wells containing the abiraterone/Plk1 inhibitor combination (3.6 fold higher relative to abiraterone alone). We next used this implantable device in AR-negative SK-OV-3 ovarian cancer xenografts which showed little apoptosis in response to treatment with either monotherapy, but substantial apoptosis in response to combined abiraterone-Plk1 inhibitor treatment (8.3-fold relative to abiraterone alone) (Fig. 6N).
Plk1 inhibition in combination with abiraterone acts synergistically in patient-derived CRPC tumor xenografts.
To more closely model the pharmacokinetics, tumor biology, and antiandrogen resistance of mCRPC patients, an AR-v7+ PDX model of CRPC, LVCaP-2CR was utilized (36). As shown in Fig. 7A, relative to vehicle control, abiraterone and onvansertib monotherapy had modest effects on tumor growth. The combined abiraterone and onvansertib, however, reduced tumor growth to a much greater extent (see Supplementary Fig. S7C for animal weights). These responses were analyzed using a version of Bliss independence adapted to tumor volume data (65), and abiraterone-onvansertib synergy confirmed in vivo (Fig. 7B).
Figure 7. Abiraterone and Plk1 inhibitors synergize in vivo and cause enhanced mitotic arrest in a murine PDX model of CRPC.
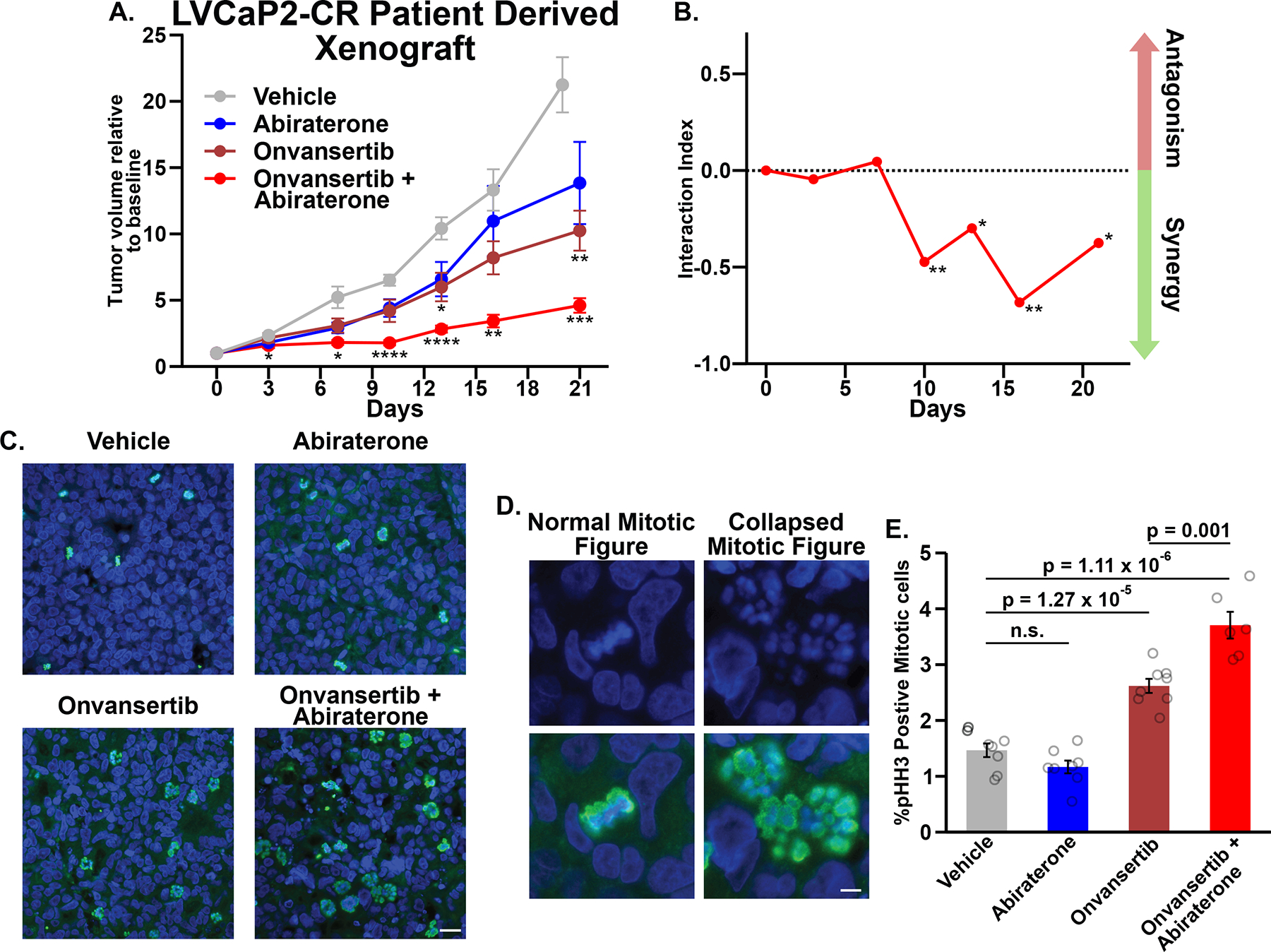
(A) Castrated male mice carrying LVCaP-2CR PDXs were administered the indicated drugs. Each tumor’s volume was normalized to day 0. Mean ± SEM where n ≥ 6; *, **, and *** indicate p ≤ 0.05, 0.01, and 0.001, respectively, relative to vehicle control using Tukey’s multiple comparisons test.
(B) Drug combination interaction indices according to (65) where values < 0 indicate synergy. * p ≤ 0.05 and ** p ≤ 0.01, using a bootstrap-t method with 10,000 random samples.
(C) End-of-study tumors were fixed and sections were stained with DAPI (blue) and anti-pHH3 antibodies (green). Scale bar bottom right 20 μm.
(D) Examples of mitotic figures in tumor sections stained with DAPI (blue). Bottom micrographs include pHH3 staining (green). Scale bar bottom right 5 μm.
(E) Quantification of pHH3+/mitotic cells in tissue sections. Plotted is the mean ± SEM, n ≥ 6 tumors per condition and ≥ 6724 cells per tumor. Data from individual tumors are overlaid as circles. P values were calculated using a Student’s two-tailed t-test; n.s. not significant.
Finally, we sought to determine if combined abiraterone and onvansertib causes synergistic mitotic arrest in vivo. End-of-study tumor sections were stained with DAPI and anti-pHH3 antibodies (Fig. 7C). Onvansertib and the combination treatment clearly increased the abundance of mitotic cells. Moreover, mitotic cells in these tumor sections contained decondensed and dispersed chromatin (Fig. 7D), resembling collapsed/monopolar spindles observed after prolonged Plk1 inhibition (27,66). We quantified mitotic cells in tissue sections from all tumors. As expected, onvansertib-treated tumors had increased frequency of mitotic cells. Despite no effect from abiraterone monotherapy, co-treatment resulted in ~30% increase in pHH3-positive cells compared to onvansertib alone (Fig. 7E). These CRPC PDX data confirm that abiraterone enhances the antimitotic effects of Plk1 inhibition resulting in synergistic mitotic arrest in vivo. Taken together, these organoid and human PDX data demonstrate clear synergistic anti-tumor activity of combined abiraterone and Plk1 inhibition, and provides strong preclinical support for the clinical trial (NCT03414034) testing the combination of onvansertib and abiraterone in mCRPC patients.
DISCUSSION
Our early finding of synergistic CPRC cell death in response to combined abiraterone and Plk1 inhibition, and those of Liu and colleagues (67,68), led to a phase 2 clinical trial (NCT03414034) in mCRPC patients (69). However, the mechanism of action of the combination has remained unknown until now. Here we report a cell-biological mechanism for synergistic cancer cell killing, demonstrate it is AR-independent, and applicable to a variety of AR-negative cancer types. Consistent with our findings, Zhang et al. showed strong synergy between abiraterone and Plk1 inhibitors in prostate cancer cells, especially with regard to in vivo efficacy of combined Plk1 inhibitor volasertib with abiraterone in the LuCAP35CR AR-v7+ CRPC PDX. In that study the authors concluded that synergy was AR-dependent, and resulted from Plk1-dependent potentiation of androgen signaling and synthesis. In contrast, our work shows this drug combination causes AR-independent synergistic mitotic arrest and cancer cell death both in culture and in vivo, through a process that appears to involve abiraterone-induced mitotic defects. The fact that abiraterone-Plk1 inhibitor synergy was observed in two AR-v7+ PDX models (LuCAP35CR: Zhang et al. and LVCaP2-CR: Fig. 7), supports the AR-independence of this synergy in vivo since AR-v7-expressing tumors are poorly responsive to abiraterone (36,52), and suggests this combination could remain useful in the context of antiandrogen resistance, a clinical setting with significant and increasing unmet need (70).
Abiraterone’s mitotic effects beyond AR antagonism and Cyp17A1 inhibition likely stem from its resemblance to endogenous steroids. Additional abiraterone binding partners may be protein(s) that bind steroidal molecules, and this will be investigated in future studies. These AR-independent effects likely do not contribute significantly to abiraterone’s monotherapy efficacy, since there is fairly strong cross-resistance between abiraterone and enzalutamide in mCRPC patients (71). Nonetheless, the AR-independent target(s) specifically synergize with Plk1 inhibition (Fig. 2M and N) in a subset of cancer cells. Plk1 is known to play important roles in centrosome maturation, spindle assembly, and kinetochore-microtubule interactions (72–74). Interestingly, among gene sets in our AR-independent synergy-specific signature were numerous related to centromeres/kinetochores, microtubules and pathways that regulate kinetochore-microtubule attachments (Fig. 3F–G). Together with our data showing that synergistic killing by this combination occurs in metaphase and is SAC-dependent (Fig. 5I; Supplementary Fig. S5G and H), these findings argue that stabilization of kinetochore-microtubule attachments is one of the key Plk1 functions whose compromise contributes to synergistic cell death.
Notably, we found that that AR-independent effects of abiraterone result in multiple cell-type specific mitotic defects. In both synergistic and non-synergistic cell lines abiraterone treatment impaired stable spindle orientation. Additional abiraterone-induced mitotic defects were seen in synergistic cell lines: chromatin decondensation and out-of-plane cell division in C4-2 cells, and multipolar spindle formation and cell division in SK-OV-3 cells. Because only synergistic cell lines had aberrant abiraterone-induced cell division, their synergistic response may stem from an inability to cope with these more severe mitotic events prior to cell division. Together with the mitotic spindle-enriched AR-independent synergy-specific gene set signature, we interpret this as evidence that abiraterone induces more severe mitotic phenotypes in a subset of cancer cells that enhance their response to Plk1 inhibition. The direct target of abiraterone responsible for these mitotic defects, and further elucidating the manner of SAC-dependent synergistic cell death will be the subject of future investigation.
In summary, our data demonstrates a cellular mitotic mechanism rationalizing the use of combined abiraterone and onvansertib in patients with mCRPC, and provides a compelling opportunity for repurposing of abiraterone as a novel antimitotic agent, that can be combined with Plk1 inhibitors to synergistically kill a subset of cancer cell types. Importantly, our finding of AR-independent effects of abiraterone and significant synergy in non-prostate cancer tumor types provides scientific justification for this combination outside the context of mCRPC. Abiraterone is an exceptionally well-tolerated drug in the field of oncology (7). Co-treatment with abiraterone could be an attractive way to enhance the utility of Plk1 inhibitors as anti-cancer agents in a wide variety of tumor types.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Abiraterone treatment induces mitotic defects that sensitize cancer cells to Plk1 inhibition, revealing an AR-independent mechanism for this synergistic combination that is applicable to a variety of cancer types.
ACKNOWLEDGEMENTS
We thank past and present members of the Yaffe lab, particularly Drs. Yi-Wen Kong, Daniel Lim, and Brian Joughin; Ian Hickson (Newcastle University); Jasmin Krüll and Catherine Henry (Koehler lab, MIT). We acknowledge the Koch Institute Genomics Core, MIT BioMicro Center, and the Robert A. Swanson (1969) Biotechnology Center for technical support, specifically the Flow Cytometry, Microscopy, and the Hope Babette Tang (1983) Histology Core Facilities. This work was supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute and P30-ES002109 from the National Institute of Environmental Health Sciences. We wish to thank the SKCCC at Hopkins Animal Core Facility supported by the SKCCC CCSG (P30-CA006973) for their services and assistance. This research was supported by grants from the National Institutes of Health (R35-ES028374 and R01-CA226898 to M.B. Yaffe.; P01-CA163227, and P50-CA090381 to S.P. Balk), awards from the Charles and Marjorie Holloway Foundation, the Ovarian Cancer Research Fund, the Prostate Cancer Foundation and the MIT Center for Precision Cancer Medicine to M.B. Yaffe, two Koch Institute - Dana-Farber/Harvard Cancer Center Bridge Project Grants to M.B. Yaffe, S.P. Balk, and D.J. Einstein, an American Cancer Society Post-Doctoral Fellowship PF-13-355-01-TBE to J.C. Patterson, a Prostate Cancer Foundation Young Investigator Award to D.J. Einstein, the Janssen/TRANSCEND Program, and a Sponsored Research Agreement from Cardiff Oncology Inc. to M.B. Yaffe.
Footnotes
DISCLOSURE OF POTENTIAL CONFLICT OF INTERESTS
Both Michael B. Yaffe and Jesse C. Patterson are co-inventors on a patent for the use of antiandrogens in combination with inhibitors of Plk1 (U.S. Patent and Trademark Office: 9,566,280). That patent has been licensed by Cardiff Oncology Inc. that owns the rights to onvansertib. Peter J.P. Croucher, Maya Ridinger, and Mark G. Erlander are employees and shareholders of Cardiff Oncology, Inc. All other authors declare no potential conflicts of interest.
REFERENCES
- 1.Augello MA, Den RB, Knudsen KE. AR function in promoting metastatic prostate cancer. Cancer and Metastasis Reviews 2014;33:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014;33:2815–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attard G, Belldegrun AS, de Bono JS. Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer. BJU Int 2005;96:1241–6. [DOI] [PubMed] [Google Scholar]
- 4.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel E a, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 2011;71:6503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards J, Lim AC, Hay CW, Taylor AE, Wingate A, Nowakowska K, et al. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: A rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res 2012;72:2176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung ME, Ouk S, Yoo D, Sawyers CL, Chen C, Tran C, et al. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J Med Chem 2010;53:2779–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. New England Journal of Medicine 2013;368:138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. New England Journal of Medicine 2014;371:424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lonergan P, Tindall D. Androgen receptor signaling in prostate cancer development and progression. J Carcinog 2011;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015;349:1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deeraksa a, Pan J, Sha Y, Liu X-D, Eissa NT, Lin S-H, et al. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene 2013;32:2973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu XS, Song B, Elzey BD, Ratliff TL, Konieczny SF, Cheng L, et al. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem 2011;286:35795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ta HQ, Gioeli D. The convergence of DNA damage checkpoint pathways and androgen receptor signaling in prostate cancer. Endocr Relat Cancer 2014;21:R395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541:359–64. [DOI] [PubMed] [Google Scholar]
- 15.Robinson D, van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wokołorczyk D, Kluźniak W, Stempa K, Rusak B, Huzarski T, Gronwald J, et al. PALB2 mutations and prostate cancer risk and survival. Br J Cancer 2021;125:569–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joukov V, de Nicolo A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci Signal 2018;11:1–26. [DOI] [PubMed] [Google Scholar]
- 18.Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol 2009;10:265–75. [DOI] [PubMed] [Google Scholar]
- 19.Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene 2005;24:287–91. [DOI] [PubMed] [Google Scholar]
- 20.Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, et al. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate 2004;60:240–5. [DOI] [PubMed] [Google Scholar]
- 21.Witkiewicz AK, Chung S, Brough R, Vail P, Franco J, Lord CJ, et al. Targeting the vulnerability of RB tumor suppressor loss in triple-negative breast cancer. Cell Rep 2018;22:1185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, et al. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol Cancer Ther 2010;9:2079–89. [DOI] [PubMed] [Google Scholar]
- 23.Wu J, Ivanov AI, Fisher PB, Fu Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife 2016;5:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gheghiani L, Shang S, Fu Z. Targeting the PLK1-FOXO1 pathway as a novel therapeutic approach for treating advanced prostate cancer. Sci Rep 2020;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cristóbal I, Rojo F, Madoz-Gúrpide J, García-Foncillas J. Cross talk between Wnt/β-Catenin and CIP2A/Plk1 signaling in prostate cancer: promising therapeutic implications. Mol Cell Biol 2016;36:1734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehár J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol 2009;27:659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patterson JC, Joughin BA, Prota AE, Mühlethaler T, Jonas OH, Whitman MA, et al. VISAGE reveals a targetable mitotic spindle vulnerability in cancer cells. Cell Syst 2019;9:74–92.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: ppen source software for digital pathology image analysis. Sci Rep 2017;7:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14:417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puca L, Bareja R, Prandi D, Shaw R, Benelli M, Karthaus WR, et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat Commun 2018;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jonas O, Landry HM, Fuller JE, Santini JT, Baselga J, Tepper RI, et al. An implantable microdevice to perform high-throughput in vivo drug sensitivity testing in tumors. Sci Transl Med 2015;7:284ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahn SW, Ferland B, Jonas OH. An interactive pipeline for quantitative histopathological analysis of spatially defined drug effects in tumors. J Pathol Inform 2021;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Y, Dalrymple SL, Coleman I, Zheng SL, Xu J, Hooper JE, et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene 2020;39:6935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lorente D, Mateo J, Perez-Lopez R, de Bono JS, Attard G. Sequencing of agents in castration-resistant prostate cancer. Lancet Oncol 2015;16:e279–92. [DOI] [PubMed] [Google Scholar]
- 38.Li K, Zhan W, Chen Y, Jha RK, Chen X. Docetaxel and doxorubicin codelivery by nanocarriers for synergistic treatment of prostate cancer. Front Pharmacol 2019;10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov 2013;3:1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kishi K, van Vugt MATM, Okamoto K, Hayashi Y, Yaffe MB. Functional dynamics of polo-like kinase 1 at the centrosome. Mol Cell Biol 2009;29:3134–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vugt MATM van, Gardino AK, Linding R, Ostheimer GJ, Reinhardt C, Ong S, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G2/M DNA damage checkpoint. PLoS Biol 2010;8:e1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008;455:119–23. [DOI] [PubMed] [Google Scholar]
- 43.Elia AEH, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, et al. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the polo-box domain. Cell 2003;115:83–95. [DOI] [PubMed] [Google Scholar]
- 44.Elia AEH, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003;299:1228–31. [DOI] [PubMed] [Google Scholar]
- 45.Lowery DM, Clauser KR, Hjerrild M, Lim D, Alexander J, Kishi K, et al. Proteomic screen defines the polo-box domain interactome and identifies Rock2 as a Plk1 substrate. EMBO Journal 2007;26:2262–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009;138:245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res 2012;72:3457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012;149:780–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bliss CI. The toxicity of poisons applied jointly. Annals of Applied Biology 1939;26:585–615. [Google Scholar]
- 50.Wu H-C, Hsieh J-T, Gleave ME, Brown NM, Pathak S, Chung LWK. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int J Cancer 1994;57:406–12. [DOI] [PubMed] [Google Scholar]
- 51.Hou X, Li Z, Huang W, Li J, Staiger C, Kuang S, et al. Plk1-dependent microtubule dynamics promotes androgen receptor signaling in prostate cancer. Prostate 2013;73:1352–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. New England Journal of Medicine 2014;371:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutteridge REA, Ndiaye MA, Liu X, Ahmad N. Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol Cancer Ther 2016;15:1427–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cucchi U, Gianellini LM, de Ponti A, Sola F, Alzani R, Patton V, et al. Phosphorylation of TCTP as a marker for polo-like kinase-1 activity in vivo. Anticancer Res 2010;30:4973–86. [PubMed] [Google Scholar]
- 55.Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Higano CS, et al. LuCaP prostate cancer patient-derived xenografts reflect the molecular heterogeneity of advanced disease and serve as models for evaluating cancer therapeutics. Prostate 2017;77:654–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database hallmark gene set collection Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samejima K, Samejima I, Vagnarelli P, Ogawa H, Vargiu G, Kelly DA, et al. Mitotic chromosomes are compacted laterally by KIF4 and condensin and axially by topoisomerase IIα. Journal of Cell Biology 2012;199:755–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samoshkin A, Arnaoutov A, Jansen LET, Ouspenski I, Dye L, Karpova T, et al. Human condensin function is essential for centromeric chromatin assembly and proper sister kinetochore orientation. PLoS One 2009;4:e6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell ceath 2018. Cell Death Differ 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Durgan J, Tseng YY, Hamann JC, Domart MC, Collinson L, Hall A, et al. Mitosis can drive cell cannibalism through entosis. Elife 2017;6:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Musacchio A. The molecular biology of spindle assembly checkpoint signaling dynamics. Current Biology 2015;25:R1002–18. [DOI] [PubMed] [Google Scholar]
- 63.Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, et al. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. Journal of Cell Biology 2010;190:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanabe K. Microtubule depolymerization by kinase inhibitors: unexpected findings of dual inhibitors. Int J Mol Sci 2017;18:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu J, Tracey L, Davidoff AM. Assessing interactions for fixed-dose drug combinations in tumor xenograft studies. J Biopharm Stat 2012;22:535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steegmaier M, Hoffmann M, Baum A, Garin-chesa P, Lieb S, Adolf R, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol 2007;17:316–22. [DOI] [PubMed] [Google Scholar]
- 67.Yaffe M, Patterson J. Combination therapies and methods of use thereof for treating cancer. United States Patent Office; 2014;9,566,280. [Google Scholar]
- 68.Zhang Z, Hou X, Shao C, Li J, Cheng JX, Kuang S, et al. PIk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res 2014;74:6635–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Einstein DJ, Choudhury AD, Saylor PJ, Patterson JC, Croucher P, Ridinger M, et al. A phase 2 study of onvansertib in combination with abiraterone and prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology 2022;40:TPS219. [Google Scholar]
- 70.Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. New England Journal of Medicine 2017;377:352–60. [DOI] [PubMed] [Google Scholar]
- 71.Buck SAJ, Koolen SLW, Mathijssen RHJ, de Wit R, van Soest RJ. Cross-resistance and drug sequence in prostate cancer. Drug Resistance Updates 2021;56:100761. [DOI] [PubMed] [Google Scholar]
- 72.van Vugt MATM, van de Weerdt BCM, Vader G, Janssen H, Calafat J, Klompmaker R, et al. Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for anaphase promoting complex/Cdc20 activation and initiation of cytokinesis. Journal of Biological Chemistry 2004;279:36841–54. [DOI] [PubMed] [Google Scholar]
- 73.Elowe S, Hümmer S, Uldschmid A, Li X, Nigg EA. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore-microtubule interactions. Genes Dev 2007;21:2205–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee K, Rhee K. PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. Journal of Cell Biology 2011;195:1093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its Supplementary data files. The RNA sequencing data generated in this study are publicly available in the Gene Expression Omnibus (GEO) at GSE217537.