

Summary
In direct lineage conversion, transcription factor (TF) overexpression reconfigures gene regulatory networks (GRNs) to reprogram cell identity. We previously developed CellOracle, a computational method to infer GRNs from single-cell transcriptome and epigenome data. Using inferred GRNs, CellOracle simulates gene expression changes in response to TF perturbation, enabling in silico interrogation of network reconfiguration. Here, we combine CellOracle analysis with lineage tracing of fibroblast to induced endoderm progenitor (iEP) conversion, a prototypical direct reprogramming paradigm. By linking early network state to reprogramming outcome, we reveal distinct network configurations underlying successful and failed fate conversion. Via in silico simulation of TF perturbation, we identify new factors to coax cells into successfully converting their identity, uncovering a central role for the AP-1 subunit Fos with the Hippo signaling effector, Yap1. Together, these results demonstrate the efficacy of CellOracle to infer and interpret cell-type-specific GRN configurations, providing new mechanistic insights into lineage reprogramming.
Keywords: gene perturbation simulation, cell fate prediction, gene regulatory networks, machine learning, direct lineage reprogramming, single-cell analysis
Graphical abstract
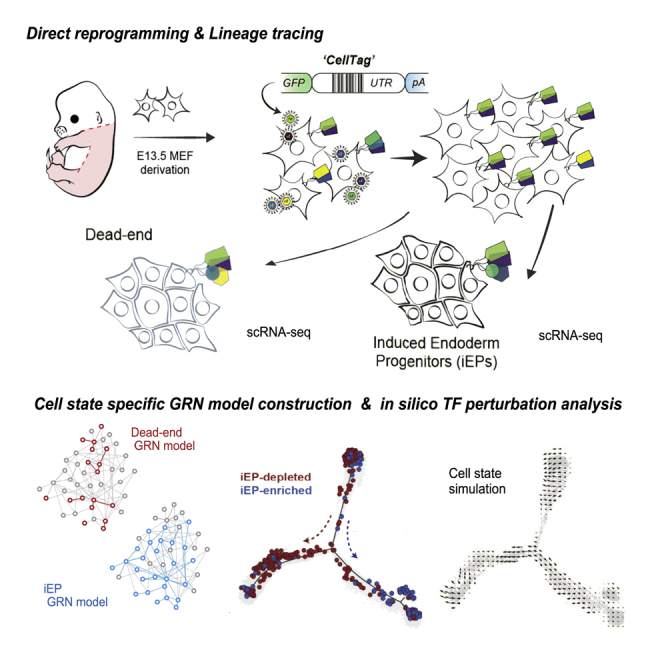
Highlights
-
•
CellOracle dissects gene regulatory network reconfiguration in direct lineage reprogramming
-
•
Lineage tracing fibroblast to endoderm progenitor reprogramming reveals early network changes
-
•
In silico interrogation of network reconfiguration identifies new reprogramming regulators
-
•
These analyses reveal a role for Fos with Hippo signaling effector, Yap1, in reprogramming
In this article, Morris and colleagues combine gene regulatory network (GRN) analysis and single-cell lineage tracing to dissect changes in cell identity during fibroblast to induced endoderm progenitor reprogramming. In silico perturbation simulation using CellOracle-inferred GRNs reveals new regulators of reprogramming and a role for Fos and Hippo signaling effector, Yap1, in the conversion and maintenance of reprogrammed cell identity.
Introduction
Direct lineage reprogramming aims to transform cell identity between fully differentiated somatic states via the forced expression of select transcription factors (TFs). Using this approach, fibroblasts have been directly converted into many clinically valuable cell types (Cohen and Melton, 2011). These protocols are currently limited, though, because only a fraction of cells convert to the target cell type and remain developmentally immature or incompletely specified (Morris and Daley, 2013). Therefore, the resulting cells are generally unsuitable for therapeutic application and have limited utility for disease modeling and drug screening in vitro.
A comprehensive characterization of cell identity is crucial to improve reprogramming methods. Gene regulatory networks (GRNs) represent the complex, dynamic molecular interactions that act as critical determinants of cell identity. These networks describe the intricate interplay between transcriptional regulators and multiple cis-regulatory DNA sequences, resulting in the precise spatial and temporal regulation of gene expression (Davidson and Erwin, 2006). Systematically delineating GRN structures enables a logic map of regulatory factor cause-effect relationships to be mapped. In turn, this knowledge supports a better understanding of how cell identity is determined and maintained, informing new strategies for cellular reprogramming.
We previously described CellOracle, a computational pipeline for GRN inference via integrating different single-cell data modalities (Kamimoto et al., 2020). CellOracle overcomes current challenges in GRN inference by using single-cell transcriptomic and chromatin accessibility profiles, integrating prior biological knowledge via regulatory sequence analysis to infer TF-target gene interactions. We designed CellOracle to apply inferred GRNs to simulate gene expression changes in response to TF perturbation. This unique feature enables inferred GRN configurations to be interrogated in silico, facilitating their interpretation. We have benchmarked CellOracle against ground-truth TF-gene interactions, demonstrating its efficacy to recapitulate known regulatory changes across hematopoiesis (Kamimoto et al., 2020). Further, we have applied CellOracle to predict TFs regulating medium spiny neuron maturation in human fetal striatum development (Bocchi et al., 2021). Other groups have successfully used the method to investigate mouse and human T cell differentiation (Chopp et al., 2020; Nie et al., 2022), T cell dysfunction in glioblastoma (Ravi et al., 2022), and pharyngeal organ development (Magaletta et al., 2022).
Here, we apply CellOracle to interrogate GRN reconfiguration during direct lineage reprogramming of fibroblasts to induced endoderm progenitors (iEPs), a prototypical TF-mediated fate conversion. Via single-cell lineage tracing, we previously demonstrated that this protocol comprises two distinct trajectories leading to reprogrammed and dead-end fates (Biddy et al., 2018). We expand on this lineage tracing strategy to experimentally define state-fate relationships, supporting the inference of early network states associated with defined reprogramming outcomes. These analyses reveal the early GRN configurations associated with the successful conversion of cell identity. Using principles of graph theory to identify critical regulatory nodes in conjunction with in silico simulation predicts several novel regulators of reprogramming, which we experimentally validate. We also demonstrate that one of these TFs, Fos, plays roles in both iEP reprogramming and maintenance, where interrogation of inferred Fos targets reveals a role for AP1-Yap1. We validate these findings to demonstrate that Fos and Yap1 overexpression significantly enhances reprogramming efficiency. Together, these results demonstrate the efficacy of CellOracle to infer and interpret cell-type-specific GRN configurations at high resolution, enabling new mechanistic insights into reprogramming. CellOracle code and documentation are available at https://github.com/morris-lab/CellOracle.
Results
CellOracle GRN inference applied to direct lineage reprogramming
CellOracle is designed to infer GRN configurations, revealing how networks are rewired during the establishment of defined cellular identities and states, highlighting known and putative regulatory factors of fate commitment (Kamimoto et al., 2020). In the first step of the CellOracle pipeline, single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq) is used to assemble a “base” GRN structure, representing a list of all potential regulatory genes associated with each defined DNA sequence (Figures 1A and 1B). The second step in the CellOracle pipeline uses single-cell RNA sequencing (scRNA-seq) data to convert the base GRN into context-dependent GRN configurations for each defined cell cluster. Removal of inactive connections refines the base GRN structure, selecting the active edges that represent regulatory connections associated with a specific cell type or state (Figures 1C, 1D, and S1A). Here, we apply CellOracle to infer GRN reconfiguration during TF-mediated direct lineage reprogramming.
Figure 1.

Application of CellOracle to assess reprogramming GRN dynamics
(A and B) Overview of CellOracle. (A) First, CellOracle uses scATAC-seq data to identify accessible regulatory elements, which are scanned for TF binding motifs, generating a Base GRN—a list of potential regulatory connections between a TF and its target genes (B).
(C) Using single-cell expression data, active connections are identified from all potential connections in the base GRN.
(D) Cell type- and state-specific GRN configurations are constructed by pruning insignificant or weak connections.
(E) Hnf4α and Foxa1-mediated fibroblast to iEP reprogramming.
(F) (Left) Force-directed graph: 15 clusters of cells are grouped into five cell types; fibroblasts (Fib), early transition (Early), transition (Tran), dead-end, and reprogrammed iEPs (iEP). (Right) Projection of Apoa1 (iEP marker) and Col1a2 (fibroblast marker) expression.
(G) CellOracle analysis. Heatmap (left) and boxplot (right) of network edge strength between Hnf4α-Foxa1 and its target genes. ∗∗∗p < 0.001.
(H) Degree and eigenvector centrality scores for Hnf4α-Foxa1.
(I) Hnf4α-Foxa1 network cartography terms for each cluster.
(J and K) Scatterplots of degree centrality scores between specific clusters.
(J) Degree centrality score comparison between Fib_1 cluster GRN and other early and transition reprogramming cluster GRNs.
(K) Degree centrality score comparison between iEP_1 and Dead-end_0 cluster GRNs.
The generation of induced endoderm progenitors (iEPs) from mouse embryonic fibroblasts (MEFs) represents a prototypical lineage reprogramming protocol, which, like most conversion strategies, is inefficient and lacks fidelity. Initially reported as hepatocyte-like cells that functionally engraft the liver (Sekiya and Suzuki, 2011), we demonstrated that these cells also harbor intestinal identity and can functionally engraft the colon, prompting their re-designation as iEPs (Guo et al., 2019; Morris et al., 2014). More recently, we have shown that iEPs transcriptionally resemble injured biliary epithelial cells (BECs) and exhibit BEC-like behavior in 3D-culture models (Kong et al., 2022). Building on these findings, our single-cell lineage tracing revealed two distinct trajectories: one to a successfully reprogrammed iEP state, and one to a dead-end, mesenchymal-like state (Figure 1E; Biddy et al., 2018).
Our previously published MEF to iEP reprogramming scRNA-seq dataset consists of eight time points collected over 28 days (n = 27,663 cells) (Biddy et al., 2018). We reprocessed this dataset using partition-based graph abstraction (PAGA; Wolf et al., 2019), manually annotating 15 clusters based on marker gene expression, identifying the expected trajectories (Figures 1F and S1B–S1D). Relative to reprogrammed cells, dead-end cells only weakly express iEP markers, Cdh1 and Apoa1, accompanied by higher expression levels of fibroblast markers, such as Col1a2 (Figures 1F, S1B, and S1C). Using CellOracle with a base GRN generated using a mouse scATAC-seq atlas (Cusanovich et al., 2018), we inferred GRN configurations for each cluster, calculating network connectivity scores to analyze GRN dynamics during reprogramming.
Analysis of network reconfiguration during reprogramming
We initially assess the network configuration associated with the exogenous reprogramming TFs, Hnf4α and Foxa1, focusing on the strength of their connections to target genes. Hnf4α and Foxa1 receive a combined score in these analyses since they are expressed as a single transcript that produces two independent factors via 2A-peptide-mediated cleavage. Network strength scores show significantly stronger connectivity of Hnf4α-Foxa1 to its inferred target genes in early reprogramming, followed by decreasing connection strength (Early_2 versus iEP_2: p < 0.001, Wilcoxon test; Figure 1G). We next evaluated the inferred GRN structures using traditional graph theory methods. We examined (1) degree centrality of each gene, a straightforward measure reporting how many edges are directly connected to a node; and (2) eigenvector centrality, a measure of influence via connectivity to other well-connected genes (Klein et al., 2012). Hnf4α-Foxa1 receives high degree and eigenvector centrality scores in the early conversion stages, gradually decreasing as reprogramming progress (Figure 1H). In agreement with a central role for the transgenes early in reprogramming, network cartography analysis (Guimerà and Amaral, 2005) classified Hnf4α-Foxa1 as a prominent "connector hub" in the early_2 cluster network configuration (Figures 1I and S1E). Together, these analyses show that Hnf4α-Foxa1 network configuration connectivity and strength peak in early reprogramming phases.
Next, we analyzed the Hnf4α-Foxa1 network configuration in later conversion, following bifurcation into reprogrammed and dead-end trajectories (Figures 1F and S1B–S1D). The reprogrammed clusters (iEP_0, iEP_1, iEP_2) exhibit stronger network connectivity scores relative to the dead-end clusters 1 and 2 (Figure 1G; iEP versus dead-end; p < 0.001, Wilcoxon test). We also identify a smaller dead-end cluster (Dead-end_0); cells within this cluster only weakly initiate reprogramming, retaining robust fibroblast gene expression signatures and expressing significantly lower levels of reprogramming initiation markers such as Apoa1 (Figure S1C; p < 0.001, permutation test). This cluster also exhibits significantly lower Hnf4α-Foxa1 connectivity scores relative to Dead-end_1 and 2 (Figure 1G; p < 0.001, Wilcoxon test), accompanied by lower degree centrality and eigenvector centrality scores (Figure 1H). However, CellTag lineage data reveal that most cells (93% of tracked cells) on this unique path derive from a single clone, representing a rare reprogramming event captured due to clonal expansion (Figure S1F).
We next turned to global GRN reconfiguration to identify candidate TFs initiating reprogramming. Comparing degree centrality scores between fibroblast and early reprogramming clusters reveals differential connectivity of a handful of key TFs. For example, Hes1, Eno1, Fos, Foxq1, and Zfp57 receive relatively high degree centrality scores in the early reprogramming clusters, whereas Klf2 and Egr1 degree centrality increases in later transition stages (Figure 1J). These factors remain highly connected on the reprogramming trajectory relative to the dead-end (Figure 1K), suggesting that the GRN configurations controlling reprogramming outcome are remodeled at the initiation of fate conversion.
Altogether, reprogramming network analysis suggests that Hnf4α-Foxa1 function peaks at conversion initiation. These early, critical changes in GRN configuration determine reprogramming outcome, with dysregulation or loss of this program leading to dead-ends, where cells either do not successfully initiate or complete reprogramming. This hypothesis is consistent with our previous CellTag lineage tracing, showing the establishment of reprogramming outcomes from early stages of the conversion process (Biddy et al., 2018). We next performed new experimental lineage tracing targeting cells at reprogramming initiation to further investigate how early GRN configuration relates to the successful generation of iEPs.
Clonal tracing links early network state to reprogramming fate
Barcoding and tracking cells via scRNA-seq represents a powerful method to investigate how the early molecular state of a cell relates to its eventual fate (Biddy et al., 2018; Jindal et al., 2022; Weinreb et al., 2020). Cells are labeled with combinations of heritable random barcodes, CellTags, delivered using lentivirus, enabling cells to be uniquely labeled and tracked over time; cells sharing identical barcodes are identified as clonal relatives; thus, early cell state can be directly linked to reprogramming outcome (Biddy et al., 2018; Kong et al., 2020; Figure 2A). However, our previous lineage tracing study was not designed to maximize the capture of clones early in reprogramming; thus, we did not meet the minimum cell number required for accurate GRN inference (50 cells; Kamimoto et al., 2020). Here, we performed new lineage tracing experiments to associate early-stage cells with reprogramming outcome.
Figure 2.

Lineage tracing links early network state to reprogramming outcome
(A) Overview of CellTag-based clonal tracking. Cells are transduced with the random CellTag lentiviral library so that each cell expresses three to four CellTags, resulting in a unique, heritable barcode signature. CellTags are transcribed and captured during single-cell profiling, enabling clonally related cells to be tracked throughout an experiment.
(B) Experimental strategy to capture state-fate relationships. MEFs are transduced with Hnf4α-Foxa1 for 48 h, then transduced with CellTags. The end of this period is considered reprogramming day 0. Cells are expanded, and 25% of the population is profiled at day 4; this is termed the state population. The remaining cells are reseeded and profiled again on days 10 and 28 to capture reprogramming fate.
(C) Captured state-fate cells. Time point information projected onto the Uniform Manifold Approximation and Projection (UMAP) embedding. A total of 24,799 cells were sequenced: 8,440 on day 4, 4,836 on day 10, and 11,523 on day 28.
(D) Projection of fibroblast, iEP, and dead-end identity scores and (E) fate annotations onto the UMAP embedding.
(F) A randomized test identified day 4 state clones whose day 10 and 28 fate sisters were iEP-enriched or iEP-depleted. (Top) Kernel density estimation of iEP-enriched day 4 state clones and their day 10 and 28 fates, outlining the reprogramming trajectory (n = 1,347 cells). (Bottom) iEP-depleted state-fate cells outlining the dead-end trajectory (n = 4,802 cells).
(G) Projection of iEP-enriched and iEP-depleted clones onto the UMAP embedding.
(H) Comparison of degree centrality scores between native fibroblasts and day 4 reprogrammed-destined cells (left) and day 4 reprogrammed- and dead-end-destined cells (right).
Cells were reprogrammed with Hnf4α-Foxa1, as above, and CellTagged at the end of the reprogramming TF transduction period. After 4 days of expansion (reprogramming day 4), we collected 25% of the cell population for scRNA-seq, reseeding the remaining cells. A total of 24,799 cells were sequenced: 8,440 on day 4, 4,836 on day 10, and 11,523 on day 28 (Figures 2B and 2C). Using our previous method to score cell identity along with established marker gene expression (Biddy et al., 2018), we identify reprogrammed and dead-end fates (reprogrammed n = 1,895; dead-end n = 6,324; Figures 2D, S2A, and S2B). Next, using clonal information, we identify the day 4 clones whose day 10 and day 28 descendants are significantly enriched or depleted of successfully reprogrammed cells. From CellTag processing (supplemental experimental procedures), we recovered 1,158 clones, containing a total of 10,927 cells across all time points. Using randomized testing, we identified two groups of day 4 cells: iEP-enriched (55 cells in nine clones) and iEP-depleted (50 cells in 43 clones), from which reprogramming and dead-end trajectories stem (Figures 2F and 2G), reproducing our earlier observations (Biddy et al., 2018).
Pooling the day 4 clones by outcome, we meet the minimum number of cells required for GRN inference (Figure S2C). We first compared the global GRN configurations for each of these states relative to MEFs to assess early GRN reconfiguration on each trajectory. For example, comparing degree centrality between day 4 cells destined to reprogram and native fibroblasts agrees with our above analysis comparing early transition to fibroblast states (Figure 1J), showing high connectivity of similar factors, such as Klf9, and Mef2a, in fibroblasts and Fos and Foxq1 in day 4 reprogrammed-destined clones (Figure 2H, left). Additional highly connected TFs also emerge in this reprogramming group, including the known induced pluripotency factor, Klf4 (Takahashi and Yamanaka, 2006), and Klf5, Mybl2, and Foxk2. The appearance of several additional factors here is likely due to assessing the early cells with known reprogramming descendants rather than the early reprogramming cluster as a whole, in which many cells will not successfully reprogram, highlighting how these state-fate experiments can further dissect population heterogeneity.
Indeed, the state-fate experimental design allows us to compare those early cells destined to reprogram versus early cells that fail to reprogram, for which clonal information is essential. A comparison of these two groups reveals subtle differences in GRN configuration, with Klf6, Tbx5, Tfapb2, and Foxs1 demonstrating higher connectivity in cells failing to reprogram, in contrast to Fos, Klf5, and Junb in cells destined to attain full iEP identity (Figure 2H, right). Differential expression analysis between day 4 reprogramming and dead-end groups did not identify these TFs (Table S3). CoSpar, a computational tool designed to identify lineage-specific gene markers based on single-cell lineage tracing data (Wang et al., 2022b), identified only Foxs1 and Junb. Overall, this new experimental state-fate analysis reveals the highly connected fibroblast TFs decoupled upon reprogramming initiation, representing potential targets to extinguish fibroblast identity. Further, we identify many TFs that are highly connected early on the successful reprogramming trajectory, representing potential candidates to improve iEP yield. We next use CellOracle’s in silico perturbation function to identify putative regulators of reprogramming in a systematic, unbiased manner.
Systematic in silico simulation of TF knockout to identify novel regulators of iEP reprogramming
While network structure can point to how gene regulation changes during reprogramming, it offers a static picture that does not necessarily provide functional insight. CellOracle bridges this gap by using its unique GRN inference model to interrogate networks to gain mechanistic insight into how specific TFs regulate cell identity (Kamimoto et al., 2020). CellOracle simulates the transition of cell identity following candidate TF perturbation (knockout [KO] or overexpression), using cluster-specific GRNs to model subsequent expression changes in regulated genes. The simulated values are then converted into a transition vector map and visualized in the dimensional reduction space, enabling an intuitive interpretation of how a candidate TF regulates cell identity (Kamimoto et al., 2020); Figures 3A–3C and S3A–S3C; supplemental experimental procedures).
Figure 3.

Systematic in silico simulation of TF KO to identify novel regulators of iEP reprogramming
(A) Monocle-based pseudotemporal ordering of 48,515 cells from Biddy et al. (2018), two independent biological replicates.
(B) Schematic for perturbation score calculations. CellOracle calculates a perturbation score by comparing the direction of the simulated cell state transition with the direction of cell differentiation. First, the pseudotime data is summarized by grid points and converted into a 2D gradient vector field. The results of the perturbation simulation are converted into the same vector field format, and the inner product of these vectors is calculated to produce a perturbation score.
(C) A positive perturbation score (green) suggests that the perturbation is predicted to promote reprogramming. In contrast, the negative perturbation score (magenta) represents impaired reprogramming.
(D) Ranked list of TFs based on the sum of the negative perturbation score.
(E) Representative examples of TF KO simulation (top row). Expression of respective genes (bottom row).
(F) Experimental validation of candidate TFs: colony-formation assay.
(G) Colony quantification. n = 5 independent biological replicates for non-targeting scramble shRNA control, Fosb, Id1; n = 4 independent biological replicates for Eno1, Klf4; n = 3 independent biological replicates for Fos; unpaired t test with Welch's correction, two-tailed; ∗p < 0.05, ∗∗p < 0.01.
In silico TF perturbation comprises four steps: (1) GRN configurations are constructed (as in Figure 1A). (2) Using these GRN models, shifts in target gene expression in response to TF perturbation are calculated. This step applies the GRN model as a function to propagate the shift in gene expression rather than the absolute gene expression value, representing TF-to-target gene signal flow. This signal is propagated iteratively to calculate the broad, downstream effects of TF perturbation, allowing the global transcriptional shift to be estimated (Figures S3A and S3B). (3) The probability of a cell identity transition is estimated by comparing this gene expression shift with the gene expression of local neighbors (Figure S3C). (4) The transition probability is converted into a weighted local average vector to represent the simulated directionality of cell state transition for each cell upon candidate TF perturbation. This final step converts the simulation results into a 2D vector map, enabling robust predictions by mitigating the effect of errors or noise derived from scRNA-seq data and the preceding simulation (Figures 3B middle; S3C). The resulting small-length vectors allow the directionality of cell identity transitions to be feasibly predicted rather than interpreting long-ranging terminal effects from initial states.
To enable the simulation results to be assessed systematically and unbiasedly, we consider the changes in cell identity induced by reprogramming, together with the predicted effects from the perturbation. Taking the relatively densely sampled time course from Biddy et al. (2018), we use semi-supervised Monocle analysis (Trapnell et al., 2014) to order cells in pseudotime based on the expression of the fibroblast marker Col1a2 and the iEP marker Apoa1, capturing the distinctive reprogramming and dead-end trajectories as distinguished by their respective lineage-restricted clones (n = 48,515 cells, two independent biological replicates; Figures 3A and S3D). We use the pseudotime information to calculate a vector gradient, representing the direction of reprogramming as a vector field (Figures 3B, left; S3E; supplemental experimental procedures). We then quantify the similarity between the reprogramming and perturbation simulation vector fields by calculating their inner-product value, which we term perturbation score (Figure 3B). A negative perturbation score implies that the TF perturbation blocks reprogramming (Figure 3C, magenta). Conversely, a positive perturbation score indicates that reprogramming is promoted following TF perturbation (Figure 3C, green). By calculating the sum of the negative perturbation scores, we rank TFs by their potential to regulate the reprogramming process, where a greater negative score indicates that reprogramming is impaired upon KO of the candidate TF. Using these metrics, we can interpret perturbation effects on cell fate quantitatively and objectively.
We used this approach to perform a systematic in silico simulation of TF KOs during iEP generation to identify novel reprogramming regulators (Figure S3F). Following GRN inference for each of the seven Monocle states identified (Figure S3D), we performed KO simulations for all TFs with inferred connections to at least one other gene (“active” TFs, n = 180), calculating the sum of the negative perturbation scores to rank TFs by the predicted inhibition of reprogramming following their KO. This in silico screen prioritizes factors for experimental validation. In the top-ranked TFs, many factors are shared between independent biological replicates ((Figure 3D; Pearson’s, r = 0.72). The Hnf4α-Foxa1 transgene is ranked top, as expected since these factors are driving the reprogramming process. Only half of the remaining top-ranked factors are differentially expressed in reprogrammed cells (Table S1). Further, only three of these prioritized TFs (Jun, Junb, Hes1) were identified by orthogonal analysis using CoSpar (Wang et al., 2022b) (Table S3), highlighting the utility of CellOracle to recover novel candidate regulators.
For experimental validation, we further prioritized candidate genes based on GRN degree centrality, enrichment of gene expression along the entire reprogramming trajectory, and ranking agreement across biological replicates, yielding eight candidates: Eno1, Fos, Fosb, Foxd2, Id1, Klf2, Klf4, and Klf15 (Figure 3E). For all TFs, CellOracle predicts impaired reprogramming following their KO. We performed an initial screen for all eight TFs, using a short hairpin RNA (shRNA)-based strategy to knock down each TF during reprogramming (confirmed by qRT-PCR; Figure S3G), followed by colony-formation assay to quantify clusters of successfully reprogrammed cells based on E-cadherin expression. From this initial screen, reprogramming was impaired following the knockdown of six of the eight TFs (Eno1, Fos, Fosb, Id1, Klf4, and Klf15), with 20%–50% fewer colonies formed (Figures S3H and S3I). We selected Eno1, Fos, Fosb, Id1, and Klf4 for additional colony-formation assays, confirming that their knockdown significantly reduces reprogramming efficiency (n = 5 independent biological replicates for non-targeting scramble shRNA control, Fosb, Id1; n = 4 for Eno1, Klf4; n = 3 for Fos; unpaired t test with Welch’s correction, two-tailed; ∗p < 0.05, ∗∗p < 0.01; Figures 3F and 3G).
Overall, our systematic perturbation simulation and experimental validation revealed several novel regulators of MEF to iEP reprogramming. Of these TFs, we identified Fos as a positive regulator of reprogramming. Further, our above state-fate analysis identified Fos as a highly connected factor in day 4 reprogrammed-destined clones, suggesting a role for this TF from the early stages of cell fate conversion. Indeed, we noted an enrichment of genes associated with the activator protein-1 TF (AP-1), a dimeric complex primarily containing members of the FOS and JUN factor families (Eferl and Wagner, 2003). AP-1 establishes cell-type-specific enhancers and gene expression programs (Vierbuchen et al., 2017) and reconfigures enhancers during reprogramming to pluripotency (Knaupp et al., 2017). As part of the AP-1 complex, Fos plays broad roles in proliferation, differentiation, and apoptosis, both in development and tumorigenesis (Eferl and Wagner, 2003; Jochum et al., 2001). We next focused on further in silico simulation and experimental validation of Fos, a core component of AP-1.
The AP-1 TF subunit Fos is central to reprogramming initiation and maintenance of iEP identity
Comparing degree centrality scores between fibroblast and early reprogramming clusters, Fos receives relatively high degree and eigenvector centrality scores, along with connector hub classification (Figures 1J, 4A, 4B, and S4A). Clonal analysis of early ancestors destined to reprogram successfully agrees with a central role for Fos (Figure 2H). Indeed, perturbation simulation and reduced reprogramming efficiency following experimental knockdown (Figures 3 and S3) led us to select Fos for deeper mechanistic investigation as a candidate gene playing a critical role in initiating iEP conversion.
Figure 4.

CellOracle analysis and experimental validation of Fos in establishing and maintaining iEP identity
(A) Degree centrality, betweenness centrality, and eigenvector centrality of Fos for each cluster.
(B) Network cartography terms of Fos for each cluster.
(C) Fos expression projected onto the force-directed graph.
(D) Violin plot of Fos expression across reprogramming stages. ∗∗∗p < 0.001.
(E and F) (E) Fos gene overexpression simulation with reprogramming GRN configurations. (Left) The projection of simulated cell transitions onto the force-directed graph. The Sankey diagram summarizes the simulation of cell transitions between cell clusters. For overexpression simulation, Fos expression was set to 1.476, representing its maximum value in the imputed gene expression matrix (F) Fos gene KO simulation.
(G) Colony-formation assay with addition of Fos to Hnf4α-Foxa1. (Left) E-cadherin immunohistochemistry. (Right) Boxplot of colony numbers (n = 6 technical replicates, two independent biological replicates; ∗∗∗p < 0.001, t test, one sided).
(H) qPCR for Fos and iEP marker expression (Apoa1 and Chd1) following addition of Fos to Hnf4α-Foxa1 (n = 3 independent biological replicates; ∗∗∗p < 0.001, ∗∗p < 0.01, t test, one sided).
(I) Fos gene KO simulation in expanded, long-term cultured iEPs.
(J) CRISPR-Cas9 Fos KO in expanded iEP cells. (Left) Kernel density estimation was applied with the t-SNE (t-distributed stochastic neighbor embedding) to compare cell density between control guide RNAs and guide RNAs targeting Fos. (Right) Quantification of changes in cell ratio following Fos KO.
During MEF to iEP reprogramming, Fos is gradually and significantly upregulated (Figures 4C and 4D; p < 0.001, permutation test, one sided). Several Jun AP-1 subunits are also expressed in iEPs, classifying as connectors and connector hubs across various reprogramming stages (Figures S4C–S4E). Fos and Jun are among a battery of genes reported to be upregulated in a cell-subpopulation-specific manner in response to cell dissociation-induced stress, potentially leading to experimental artifacts (van den Brink et al., 2017). Considering this report, we performed qRT-PCR for Fos on dissociated and undissociated cells. This orthogonal validation confirms an 8-fold upregulation (p < 0.01, t test, one sided) of Fos in iEPs, relative to MEFs, revealing no significant changes in gene expression in cells that are dissociated and lysed versus cells lysed directly on the plate (Figure S4F). Further, analysis of unspliced and spliced Fos mRNA levels reveals an accumulation of spliced Fos transcripts in reprogrammed cells (la Manno et al., 2018). This observation suggests that these transcripts accumulated over time rather than by rapid induction of expression by cell dissociation (Figure S4G).
To further investigate the role of Fos, we simulated its overexpression. In these analyses, to assess the in silico perturbation of a specific candidate, we use a Markov simulation to predict how cell identity shifts within the overall cell population, visualizing the results as a Sankey diagram. Overexpression simulation for Fos predicts a major cell state shift from the early transition to transition clusters, in addition to predicting shifts in identity from dead-end to reprogrammed clusters (Figure 4E). In contrast, the simulation of Fos KO produces the opposite results. (Figure 4F). We experimentally validated this simulation by adding Fos to the iEP reprogramming cocktail. As expected, we see a significant increase in the number of iEP colonies formed (n = 10, p < 0.001, t test, one sided; Figure 4G), increasing reprogramming efficiency more than 2-fold, accompanied by significant increases in iEP marker expression as measured by qRT-PCR (n = 3, p < 0.001, t test, one sided; Figure 4H).
Turning our attention to the later stages of reprogramming, Fos continues to receive relatively high network scores in the iEP GRN configurations (Figure 4A). Fos also classifies as a connector hub (Figure 4B) in iEPs, suggesting a role for Fos in the stabilization and maintenance of the reprogrammed fate. To test this hypothesis, we use CellOracle to perform KO simulation, followed by experimental KO validation in an established iEP cell line. Here, we leverage the ability to culture iEPs, long term, where they retain a range of phenotypes (from fibroblast-like to iEP states; Figure S4H) and functional engraftment potential (Guo et al., 2019; Morris et al., 2014). Simulation of Fos KO using these long-term cultured iEP GRN configurations predicts the loss of iEP identity upon Fos KO (Figure 4I). To test this prediction, we used CRISPR-Cas9 to knock out Fos in established iEPs. Quantitative comparison of the cell proportions between control and KO groups confirms that fully reprogrammed iEPs regress toward an intermediate state upon Fos KO, confirming a role for this factor in maintaining iEP identity (Figure 4J), in addition to the establishment of iEPs, as we demonstrate in our systematic simulation and experimental validation in Figure 3.
Fos target inference uncovers a role for the Hippo signaling effector Yap1 in reprogramming
To gain further insight into Fos regulation of reprogramming, we interrogated a list of the top 50 inferred Fos targets (Figure 5A; Table S2). We also assembled a list of genes predicted to be downregulated following Fos KO simulation (Figure S5A). From this analysis, we noted the presence of direct targets of YAP1, a central downstream transducer of the Hippo signaling pathway (Ramos and Camargo, 2012). These targets include Cyr61, Amotl2, Gadd45g, and Ctgf. Previous associations between Yap1 and Fos support these observations; YAP1 is recruited to the same genomic regions as FOS via complex formation with AP-1 (Zanconato et al., 2015). Moreover, AP-1 is required for YAP1-regulated gene expression and liver overgrowth caused by Yap overexpression, where FOS induction contributes to the expression of YAP/TAZ downstream target genes (Koo et al., 2020).
Figure 5.

Inferred Fos targets reveal a role for the Hippo signaling effector, Yap1, in reprogramming
(A) Heatmap of expression of the top 50 inferred Fos targets across reprogramming. Established YAP1 targets are highlighted in red.
(B) Colony-formation assay with the addition of Yap1 and Fos to Hnf4α-Foxa1. (Left) E-cadherin immunohistochemistry. (Right) Boxplot of colony numbers (n = 6 independent biological replicates; ∗∗∗p < 0.001, t-test, one sided).
(C) Brightfield and epifluorescence images of cells reprogrammed with Hnf4α-Foxa1 or Hnf4α-Foxa1-Fos-Yap1. Scale bar, 500 μm.
(D) scRNA-seq of cells reprogrammed with Hnf4α-Foxa1 (n = 7,414 cells), Hnf4α-Foxa1-Fos (n = 8,771 cells), Hnf4α-Foxa1-Yap1 (n = 8,549 cells), and Hnf4α-Foxa1-Fos-Yap1 (n = 10,507 cells), profiled at day 20. Projection of fibroblast and iEP identity scores onto the UMAP embedding.
(E) Kernel density estimation of cell density for each reprogramming cocktail from (D).
(F) Violin plot of iEP identity scores for each reprogramming cocktail. ∗∗∗∗p < 0.0001, Wilcoxon test.
(G) Unsupervised cell type classification for each reprogramming cocktail, using normal and injured mouse liver as a reference. BEC, biliary epithelial cells. ∗p < 0.0001, randomized test.
Together, this evidence suggests that Fos may play a role in reprogramming via an AP-1-Yap1-mediated mechanism. Since Yap1 does not directly bind to DNA, we cannot deploy CellOracle to perform perturbation simulations, highlighting a limitation of our approach. However, in lieu of this analysis, we again turn to our previous reprogramming data (Biddy et al., 2018). Using an established active signature of Yap1 (Dong et al., 2007), we find significant enrichment of this signature as reprogramming progresses (Figures S5B and S5C; p < 0.001, permutation test, one-sided). Together, these results suggest a role for the Hippo signaling component Yap1 in reprogramming, potentially affected via its interactions with Fos/AP-1. Indeed, the Hippo signaling axis plays a role in liver regeneration (Pepe-Mooney et al., 2019) and regeneration of the colonic epithelium (Yui et al., 2018), in line with the known potential of iEPs to functionally engraft the liver and intestine (Guo et al., 2019; Morris et al., 2014; Sekiya and Suzuki, 2011). Further, we have recently demonstrated that iEPs transcriptionally resemble injured BECs (Kong et al., 2022), the target of YAP signaling in the context of liver regeneration (Pepe-Mooney et al., 2019).
To test the role of Yap1 in iEP reprogramming, we first performed colony-formation assays. We find that the addition of Yap1 to the Hnf4α-Foxa1 cocktail significantly enhances reprogramming efficiency, where the addition of Fos and Yap1 together increase colony formation almost 3-fold, accompanied by significant increases in iEP marker expression (Figures 5B, S5D, and S5E, p < 0.001, t test, one sided). Further, we note the formation of extremely dense colonies (Figure 5C). To further characterize this distinctive phenotype, we performed scRNA-seq on cells reprogrammed with Hnf4α-Foxa1 (n = 7,414 cells), Hnf4α-Foxa1-Yap1 (n = 8,549 cells), Hnf4α-Foxa1-Fos (n = 8,771 cells), and Hnf4α-Foxa1-Yap1-Fos (n = 10,507 cells), profiled at day 20 (Figure S5F).
We scored cells using established markers of MEFs and iEPs (Biddy et al., 2018), revealing a significant increase in reprogramming efficiency, particularly following the addition of Yap1 (p < 0.0001, Wilcoxon test; Figures 5F and S5F), which is also accompanied by a reduction in fibroblast marker expression (Figure S5G). We further classify cell identity using our unsupervised method for cell-type classification, Capybara (Kong et al., 2022). In agreement with our previous reports, using a healthy and regenerating liver atlas, iEPs generated with Hnf4α-Foxa1 alone classify mainly as stromal cells (Figure 5G). However, following the addition of Fos and Yap1, a significant population (p < 0.0001, randomized test) of injured BECs emerges, in similar proportions to those observed in long-term cultured iEPs (Kong et al., 2022). We also observe a significant expansion of a normal BEC population, from ∼4% to ∼12%–35%, upon the addition of Yap1 to the reprogramming cocktail (p < 0.0001, randomized test), where endogenous Fos expression is also upregulated (Figure S5G). We observed a similar expansion of the normal BEC population when long-term iEPs were cultured in a 3D Matrigel sandwich culture (Kong et al., 2022). Here, our results are consistent with these previous observations and point to the molecular regulation driving changes in cell identity. In summary, CellOracle analysis and in silico prediction, combined with experimental validation, have revealed several new factors and putative regulatory mechanisms to enhance the efficiency and fidelity of reprogramming.
Discussion
Our application of CellOracle to iEP reprogramming has revealed new insight into this lineage conversion paradigm. Using CellTag-based lineage tracing, we had previously demonstrated the existence of distinct conversion trajectories: one path leading to successfully reprogrammed cells and a route to a dead-end state, accompanied by fibroblast gene re-expression (Biddy et al., 2018). From lineage analysis, we found that sister cells follow the same reprogramming trajectories, suggesting that conversion outcome is established shortly after overexpression of the reprogramming TFs. The network analysis we present in this study, powered by CellOracle, supports these earlier observations, revealing GRN reconfiguration within the first few days of reprogramming.
From our analysis of early GRN rewiring, we find that Mef2a and Klf6 are highly connected in fibroblasts and that these connections are largely decommissioned in successfully converting cells. Although better known as a cardiac factor (Filomena and Bang, 2018), Mef2a expression is enriched in the dead-end population, whereas Klf6 is enriched in early transition states, followed by its downregulation as reprogramming progresses. In this study, we have mainly focused on the TFs associated with installing new cell identities. From our clonal analysis of GRN reconfiguration in reprogrammed-destined cells, we find many previously unreported regulators of iEP reprogramming, such as Klf5, Mybl2, Foxq1, Fos, and Junb. The recovery of these factors is likely due to the clonal analysis, which further breaks down population heterogeneity to target those rare cells that successfully reprogram.
To explore the role of these factors in reprogramming, we leverage the unique feature of CellOracle: simulation of cell identity transition following candidate TF perturbation (KO or overexpression). From systematic in silico KO simulation and experimental validation, we identified five new regulators of iEP reprogramming: Id1, Fosb, Fos, Eno1, and Klf4. Klf4 is one of the previously described core pluripotency reprogramming factors (Takahashi and Yamanaka, 2006). The reduction of iEP reprogramming efficiency following its knockdown also suggests that Klf4 plays a role in this direct lineage conversion paradigm. Similarly, Id1 has also been shown to play a positive role in reprogramming to pluripotency (Hayashi et al., 2016), suggesting parallels with direct lineage conversion. We also noted the involvement of several AP-1 factors, both from our network analyses and in silico simulations, including Fos, Fosb, Fosl2, and Junb. The FOS-JUN-AP1 complex has been reported to regulate reprogramming to pluripotency (Xing et al., 2020) and direct reprogramming to cardiomyocytes (Wang et al., 2022a); thus, we selected Fos for further investigation.
The CellOracle analyses presented here provide new mechanistic insight into the reprogramming process, revealing a role for the Fos-Yap1 axis, which we experimentally validated. In a parallel study, we found that iEPs resemble post-injury BECs (Kong et al., 2022). Considering that Yap1 plays a central role in liver regeneration (Pepe-Mooney et al., 2019), these results raise the possibility that iEPs represent a regenerative cell type, explaining their Yap1 activity, self-renewal in vitro, and capacity to functionally engraft the liver (Sekiya and Suzuki, 2011) and intestine (Guo et al., 2019; Morris et al., 2014). Indeed, our unsupervised cell type classification of iEPs reprogrammed with the addition of Fos and Yap to the Hnf4α-Foxa1 reprogramming cocktail suggests that these factors can directly expand the injured and normal BEC populations, supporting the notion that iEPs may resemble a regenerative population. Altogether, these new mechanistic insights have been enabled by CellOracle analysis, placing it as a powerful tool for the dissection of cell identity, aiding improvements in reprogramming efficiency and fidelity.
Experimental procedures
Detailed experimental procedures can be found in the supplemental information.
Resource availability
Corresponding author
Samantha A. Morris, s.morris@wustl.edu.
Materials availability
Pooled CellTag libraries have been deposited at Addgene: https://www.addgene.org/pooled-library/morris-lab-celltag/
Computational methods
CellOracle code is open source and available on GitHub: (https://github.com/morris-lab/CellOracle). For alignment, digital gene expression matrix generation, the Cell Ranger v6.0.1 pipeline (https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest) was used to process data generated using the 10x Chromium platform. For clone calling, we used our CellTag analysis pipeline: https://github.com/morris-lab/newCloneCalling. Cell type classification was performed using Capybara: https://github.com/morris-lab/Capybara.
Experimental methods
MEFs were derived from E13.5 C57BL/6J embryos (the Jackson laboratory: 000664). Retroviral particles were produced by transfecting 293T-17 cells (ATCC: CRL-11268) with the pGCDN-Sam construct containing Hnf4α-t2a-Foxa1/Fos/Yap1, along with packaging construct pCL-Eco (Imgenex). Lentiviral particles were produced with the envelope construct pCMV-VSV-G (Addgene plasmid 8454), the packaging construct pCMV-dR8.2 dvpr (Addgene plasmid 8455), and the shRNA expression vector for the respective candidate TF to be knocked down. For generation of the complex CellTag library, lentiviral particles were produced by transfecting 293T-17 cells (ATCC: CRL-11268) with the pSMAL-CellTag construct, along with packaging constructs pCMV-dR8.2 dvpr (Addgene plasmid 8455) and pCMV-VSVG (Addgene plasmid 8454). For iEP reprogramming, MEFs (< passage 6) were converted to iEPs as in Biddy et al. (2018), modified from (Sekiya and Suzuki, 2011). Colony-formation assays were performed as in Biddy et al. (2018). Perturb-seq was performed as previously described (Adamson et al., 2016). Single-cell libraries were prepared using the 10x Genomics Chromium platform.
Author contributions
Conceptualization and methodology, K.K. and S.A.M.; software, K.K.; formal analysis, K.K., M.T.A., K.J., C.M.H., and S.A.M; investigation, K.K., M.T.A., K.J., C.M.H., X.Y., and S.A.M.; data curation, K.K., M.T.A., and K.J.; writing – original draft, K.K. and S.A.M.; writing – review & editing, K.K., M.T.A., K.J., C.M.H., and S.A.M.; visualization, K.K. and S.A.M.; funding acquisition and resources, Supervision, S.A.M.
Acknowledgments
We thank members of the Morris laboratory for critical feedback. This work was funded by the National Institute of General Medical Sciences (R01 GM126112) and Silicon Valley Community Foundation, Chan Zuckerberg Initiative grant HCA2-A-1708-02799, both to S.A.M. S.A.M. is supported by an Allen Distinguished Investigator Award (through the Paul G. Allen Frontiers Group), a Vallee Scholar Award, a Sloan Research Fellowship, and a New York Stem Cell Foundation Robertson Investigator Award. K.K. is supported by a Japan Society for the Promotion of Science Postdoctoral Fellowship. C.M.H. is supported by a National Science Foundation Graduate Research Fellowship (DGE-1745038).
Conflict of interests
S.A.M. is a co-founder of CapyBio LLC.
Published: December 29, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2022.11.010.
Supplemental information
Top-ranked genes from CellOracle in silico perturbation are marked in red.
Confirmed YAP1 targets are highlighted in red.
Genes in bold are also identified by CoSpar analysis. The right column shows TFs prioritized by CellOracle analysis. Genes in bold are also identified by CoSpar analysis.
Data and code availability
All source data, including sequencing reads and single-cell expression matrices, are available from the Gene Expression Omnibus (GEO) under accession codes GSE99915 (Biddy et al., 2018) and GSE217675 for the new scRNA-seq data presented in this manuscript. CellOracle code, documentation, and tutorials are available on GitHub: (https://github.com/morris-lab/CellOracle).
References
- Adamson B., Norman T.M., Jost M., Cho M.Y., Nuñez J.K., Chen Y., Villalta J.E., Gilbert L.A., Horlbeck M.A., Hein M.Y., et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell. 2016;167:1867–1882.e21. doi: 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddy B.A., Kong W., Kamimoto K., Guo C., Waye S.E., Sun T., Morris S.A. Single-cell mapping of lineage and identity in direct reprogramming. Nature. 2018;564:219–224. doi: 10.1038/s41586-018-0744-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchi V.D., Conforti P., Vezzoli E., Besusso D., Cappadona C., Lischetti T., Galimberti M., Ranzani V., Bonnal R.J.P., De Simone M., et al. The coding and long noncoding single-cell atlas of the developing human fetal striatum. Science. 2021;372:eabf5759. doi: 10.1126/science.abf5759. [DOI] [PubMed] [Google Scholar]
- van den Brink S.C., Sage F., Vértesy Á., Spanjaard B., Peterson-Maduro J., Baron C.S., Robin C., van Oudenaarden A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods. 2017;14:935–936. doi: 10.1038/nmeth.4437. [DOI] [PubMed] [Google Scholar]
- Chopp L.B., Gopalan V., Ciucci T., Ruchinskas A., Rae Z., Lagarde M., Gao Y., Li C., Bosticardo M., Pala F., et al. An integrated epigenomic and transcriptomic map of mouse and human αβ T cell development. Immunity. 2020;53:1182–1201.e8. doi: 10.1016/J.IMMUNI.2020.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D.E., Melton D. Turning straw into gold: directing cell fate for regenerative medicine. Nat. Rev. Genet. 2011;12:243–252. doi: 10.1038/nrg2938. [DOI] [PubMed] [Google Scholar]
- Cusanovich D.A., Hill A.J., Aghamirzaie D., Daza R.M., Pliner H.A., Berletch J.B., Filippova G.N., Huang X., Christiansen L., DeWitt W.S., et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018;174:1309–1324.e18. doi: 10.1016/J.CELL.2018.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson E.H., Erwin D.H. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311:796–800. doi: 10.1126/science.1113832. [DOI] [PubMed] [Google Scholar]
- Dong J., Feldmann G., Huang J., Wu S., Zhang N., Comerford S.A., Gayyed M.F., Anders R.A., Maitra A., Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/J.CELL.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R., Wagner E.F. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- Filomena M.C., Bang M.L. In the heart of the MEF2 transcription network: novel downstream effectors as potential targets for the treatment of cardiovascular disease. Cardiovasc. Res. 2018;114:1425–1427. doi: 10.1093/CVR/CVY123. [DOI] [PubMed] [Google Scholar]
- Guimerà R., Amaral L.A.N. Cartography of complex networks: modules and universal roles. J. Stat. Mech. 2005;2005:P02001-1–P02001-13. doi: 10.1088/1742-5468/2005/02/P02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Kong W., Kamimoto K., Rivera-Gonzalez G.C., Yang X., Kirita Y., Morris S.A. CellTag Indexing: genetic barcode-based sample multiplexing for single-cell genomics. Genome Biol. 2019;20:90. doi: 10.1186/s13059-019-1699-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y., Hsiao E.C., Sami S., Lancero M., Schlieve C.R., Nguyen T., Yano K., Nagahashi A., Ikeya M., Matsumoto Y., et al. BMP-SMAD-ID promotes reprogramming to pluripotency by inhibiting p16/INK4A-dependent senescence. Proc. Natl. Acad. Sci. USA. 2016;113:13057–13062. doi: 10.1073/PNAS.1603668113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochum W., Passegué E., Wagner E.F. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–2412. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- Jindal K., Adil M.T., Yamaguchi N., Wang H.C., Yang X., Kamimoto K., Rivera-Gonzalez G.C., Morris S.A. Multiomic single-cell lineage tracing to dissect fate-specific gene regulatory programs. Preprint at bioRxiv 2022.10.23.512790. 2022;20 doi: 10.1101/2022.10.23.512790. [DOI] [Google Scholar]
- Kamimoto K., Hoffmann C.M., Morris S.A. CellOracle: dissecting cell identity via network inference and in silico gene perturbation. bioRxiv. 2020 doi: 10.1101/2020.02.17.947416. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C., Marino A., Sagot M.-F., Vieira Milreu P., Brilli M. Structural and dynamical analysis of biological networks. Brief. Funct. Genomics. 2012;11:420–433. doi: 10.1093/bfgp/els030. [DOI] [PubMed] [Google Scholar]
- Knaupp A.S., Buckberry S., Pflueger J., Lim S.M., Ford E., Larcombe M.R., Rossello F.J., de Mendoza A., Alaei S., Firas J., et al. Transient and permanent reconfiguration of chromatin and transcription factor occupancy drive reprogramming. Cell Stem Cell. 2017;21:834–845.e6. doi: 10.1016/J.STEM.2017.11.007. [DOI] [PubMed] [Google Scholar]
- Kong W., Biddy B.A., Kamimoto K., Amrute J.M., Butka E.G., Morris S.A. CellTagging: combinatorial indexing to simultaneously map lineage and identity at single-cell resolution. Nat. Protoc. 2020;15:750–772. doi: 10.1038/s41596-019-0247-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W., Fu Y.C., Holloway E.M., Garipler G., Yang X., Mazzoni E.O., Morris S.A. Capybara: a computational tool to measure cell identity and fate transitions. Cell Stem Cell. 2022;29:635–649.e11. doi: 10.1016/J.STEM.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo J.H., Plouffe S.W., Meng Z., Lee D.-H., Yang D., Lim D.-S., Wang C.-Y., Guan K.-L. Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev. 2020;34:72–86. doi: 10.1101/gad.331546.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magaletta M.E., Lobo M., Kernfeld E.M., Aliee H., Huey J.D., Parsons T.J., Theis F.J., Maehr R. Integration of single-cell transcriptomes and chromatin landscapes reveals regulatory programs driving pharyngeal organ development. Nat. Commun. 2022;13:457. doi: 10.1038/s41467-022-28067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S.A., Daley G.Q. A blueprint for engineering cell fate: current technologies to reprogram cell identity. Cell Res. 2013;23:33–48. doi: 10.1038/cr.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S.A., Cahan P., Li H., Zhao A.M., San Roman A.K., Shivdasani R.A., Collins J.J., Daley G.Q. Dissecting engineered cell types and enhancing cell fate conversion via CellNet. Cell. 2014;158:889–902. doi: 10.1016/j.cell.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie J., Carpenter A.C., Chopp L.B., Chen T., Balmaceno-Criss M., Ciucci T., Xiao Q., Kelly M.C., McGavern D.B., Belkaid Y., et al. The transcription factor LRF promotes integrin β7 expression by and gut homing of CD8αα+ intraepithelial lymphocyte precursors. Nat. Immunol. 2022;23:594–604. doi: 10.1038/s41590-022-01161-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe-Mooney B.J., Dill M.T., Alemany A., Ordovas-Montanes J., Matsushita Y., Rao A., Sen A., Miyazaki M., Anakk S., Dawson P.A., et al. Single-cell analysis of the liver epithelium reveals dynamic heterogeneity and an essential role for YAP in homeostasis and regeneration. Cell Stem Cell. 2019;25:23–38.e8. doi: 10.1016/J.STEM.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A., Camargo F.D. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012;22:339–346. doi: 10.1016/J.TCB.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi V.M., Neidert N., Will P., Joseph K., Maier J.P., Kückelhaus J., Vollmer L., Goeldner J.M., Behringer S.P., Scherer F., et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022;13:925. doi: 10.1038/s41467-022-28523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya S., Suzuki A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature. 2011;475:390–393. doi: 10.1038/nature10263. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T., Ling E., Cowley C.J., Couch C.H., Wang X., Harmin D.A., Roberts C.W.M., Greenberg M.E. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell. 2017;68:1067–1082.e12. doi: 10.1016/J.MOLCEL.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Yang Y., Qian Y., Liu J., Qian L. Delineating chromatin accessibility re-patterning at single cell level during early stage of direct cardiac reprogramming. J. Mol. Cell. Cardiol. 2022;162:62–71. doi: 10.1016/J.YJMCC.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.W., Herriges M.J., Hurley K., Kotton D.N., Klein A.M. CoSpar identifies early cell fate biases from single-cell transcriptomic and lineage information. Nat. Biotechnol. 2022;40:1066–1074. doi: 10.1038/s41587-022-01209-1. [DOI] [PubMed] [Google Scholar]
- Weinreb C., Rodriguez-Fraticelli A., Camargo F.D., Klein A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science. 2020;367:eaaw3381. doi: 10.1126/SCIENCE.AAW3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Hamey F.K., Plass M., Solana J., Dahlin J.S., Göttgens B., Rajewsky N., Simon L., Theis F.J. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20:59. doi: 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Q.R., el Farran C.A., Gautam P., Chuah Y.S., Warrier T., Toh C.X.D., Kang N.Y., Sugii S., Chang Y.T., Xu J., et al. Diversification of reprogramming trajectories revealed by parallel single-cell transcriptome and chromatin accessibility sequencing. Sci. Adv. 2020;6:18. doi: 10.1126/SCIADV.ABA1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui S., Azzolin L., Maimets M., Pedersen M.T., Fordham R.P., Hansen S.L., Larsen H.L., Guiu J., Alves M.R.P., Rundsten C.F., et al. YAP/TAZ-Dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell. 2018;22:35–49.e7. doi: 10.1016/j.stem.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F., Forcato M., Battilana G., Azzolin L., Quaranta E., Bodega B., Rosato A., Bicciato S., Cordenonsi M., Piccolo S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015;17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Top-ranked genes from CellOracle in silico perturbation are marked in red.
Confirmed YAP1 targets are highlighted in red.
Genes in bold are also identified by CoSpar analysis. The right column shows TFs prioritized by CellOracle analysis. Genes in bold are also identified by CoSpar analysis.
Data Availability Statement
All source data, including sequencing reads and single-cell expression matrices, are available from the Gene Expression Omnibus (GEO) under accession codes GSE99915 (Biddy et al., 2018) and GSE217675 for the new scRNA-seq data presented in this manuscript. CellOracle code, documentation, and tutorials are available on GitHub: (https://github.com/morris-lab/CellOracle).