

Abstract
Many risk loci for Parkinson’s disease (PD) have been identified by genome-wide association studies (GWAS), but target genes and mechanisms remain largely unknown. We linked the GWAS-derived chromosome 7 locus (sentinel SNP rs199347) to GPNMB, through colocalization analyses of expression quantitative trait locus (eQTL) and PD risk signals, confirmed by allele-specific expression studies in human brain. In cells, Glycoprotein Nonmetastatic Melanoma Protein B protein (GPNMB) co-immunoprecipitated and co-localized with alpha-synuclein (aSyn). In induced pluripotent stem cell-derived neurons, loss of GPNMB resulted in loss of ability to internalize aSyn fibrils and develop aSyn pathology. In 731 PD and 59 control biosamples, GPNMB was elevated in PD plasma, associating with disease severity. Thus, GPNMB represents a PD risk gene, with potential for biomarker development and therapeutic targeting.
One-Sentence Summary:
Computational, cell biological, and human tissue-based studies establish GPNMB as a risk gene and therapeutic target for PD.
Parkinson’s disease is a progressive neurodegenerative disease affecting an estimated 5 million people worldwide (1). While genome-wide association studies (GWAS) have nominated >80 genetic loci as PD risk factors (2), target genes and mechanisms for these loci remain largely unknown. To date, drug development in PD has focused on pathways related to alpha-synuclein (aSyn), the main component of disease-defining neuropathological lesions, or a handful of other genes causal for PD in a small minority of patients (3, 4). However, these efforts have not yet yielded successful disease-modifying therapies.
Here, we sought to widen the net for PD therapeutic targets by dissecting a common variant GWAS-derived PD risk locus. We used computational, cell biological, and human tissue-based approaches to ascertain that GPNMB is a bona fide PD risk gene. Moreover, we implicated GPNMB-aSyn interactions and GPNMB expression levels in the development of PD.
GPNMB is the target gene at the rs199347 PD risk locus
Multiple GWAS have linked a chromosome 7 locus (sentinel SNP rs199347) with PD (2, 5). Because rs199347 and linked variants are non-coding, we hypothesized that this locus confers PD risk through modulation of expression levels of one or more target genes, as has been shown for a number of common variant GWAS-derived risk factors in other diseases (6–9). To nominate candidate target genes, we used the Genotype Tissue Expression (GTEx) database (10), finding genes for which a shared causal variant could be linked to both PD risk and expression levels in normal human tissue. Target genes within a 1 Mb window of the sentinel SNP were evaluated in colocalization analyses (11, 12): GPNMB, KLHL7, and NUPL2 (Figure S1). Among these, GPNMB demonstrated the greatest overlap between PD risk and expression quantitative trait locus (eQTL) effects, with the most significant colocalization effects seen in the PD-affected caudate brain region (Figure S1B). Moreover, when we examined eQTL effects more broadly, we found that rs199347 genotypes associated significantly with GPNMB expression levels in all GTEx-characterized brain regions as well as in whole blood (Figure 1A). Thus, computational analyses of existing databases strongly implicated GPNMB as the target gene at this locus, with higher levels of brain and blood expression linked to PD risk.
Fig. 1. Chromosome 7 PD risk locus is a multi-tissue GPNMB eQTL and shows allele-specific expression (ASE).
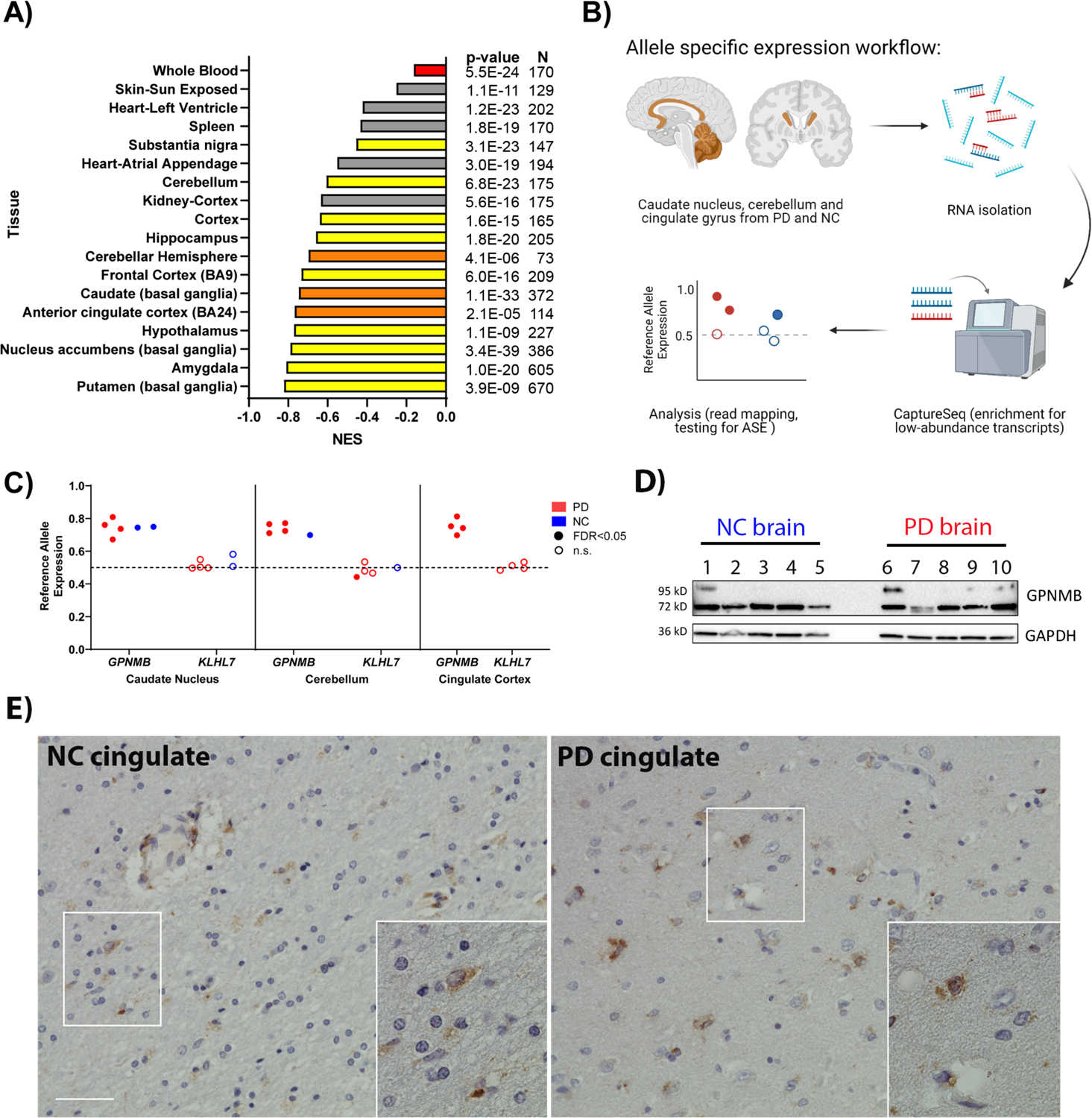
(A) GTEx normalized expression size (NES) coefficients for rs199347’s effect on GPNMB expression in various tissues (10). Red indicates whole blood, yellow and orange indicate brain regions. Orange bars correspond to brain regions analyzed for ASE. The PD risk allele at rs199347 (allele = A) is uniformly associated with higher GPNMB mRNA expression. (B) Workflow for ASE experiment in brain samples from PD patients (n = 4) and neurologically normal controls (NC, n = 2). (C) ASE analysis for rs199347 target gene candidates GPNMB and KLHL7 in caudate, cerebellum, and cingulate cortex brain samples of PD (red) and NC (blue) individuals heterozygous at this locus. Shaded dots indicate significant (BH-adjusted p-value < 0.05) ASE, whereas empty dots do not show significant ASE. In the absence of ASE, the allelic ratio would be 0.5. For GPNMB, the allelic ratio approached 0.8, with higher expression for the GPNMB allele carrying the PD risk haplotype. (D) Immunoblots showing GPNMB expression in caudate brain lysates from NC (n=5) and PD (n=5) individuals. A 72kD band was detected by the E1Y7J antibody in all cases, with variable expression of a higher-molecular weight glycosylated form. A GAPDH loading control is also shown. (E) Representative immunohistochemical staining of GPNMB in NC and PD brain demonstrates expression in multiple cell types. Scale bar = 50um.
The mechanistic basis for eQTL effects lies in allele-specific expression (ASE), whereby different alleles at a given locus show different levels of expression (13). Thus, to validate the database-predicted eQTL relationship between rs199347 and GPNMB expression, we developed a capture RNA-seq-based (14) ASE assay for human brain tissue (Figure 1B) and used it to ask whether individuals carrying two different alleles for the rs199347 haplotype demonstrated ASE for GPNMB. Moreover, in order to understand whether rs199347 haplotype effects on brain tissue expression extended to PD brain, we characterized multiple brain regions with varying degrees of pathological involvement in PD (in order of greatest to least aSyn pathological involvement: caudate, cingulate, cerebellum) from both PD cases (n=4) and neurologically normal controls (n=2, extended demographic information in Table S1).
We found a high degree of ASE for GPNMB regardless of PD status or brain region, with 15/15 brain samples showing significant ASE (Figure 1C). Moreover, all samples displayed the same direction of effect, with the PD risk haplotype associating with higher expression levels, averaging ~3-fold higher than the PD-protective haplotype. In contrast, ASE analyses for another gene at this locus, KLHL7, did not show preferential expression of either haplotype (Figure 1C). Finally, we confirmed that GPNMB is expressed at the protein level in human brain, examining multiple postmortem cases (Table S1) by immunoblotting (Figure 1D), which demonstrated a main band at the predicted molecular weight of 72kD across all cases, as well as a variably-expressed higher-molecular-weight band that collapses to 72kD with deglycosylation (Figure S2), consistent with prior reports of GPNMB’s variably glycosylated forms (15). In order to better understand what cell types might be expressing GPNMB, we also performed immunohistochemical staining of human brain tissue (Figure 1E, Table S1), finding GPNMB expression in multiple cell types, consistent with reports from spinal cord tissue (16). Thus, both database-predicted and empirically-validated eQTL studies in human PD samples support GPNMB as the target gene at the rs199347 locus, with protein expression in the brain.
Loss of GPNMB in iPSC-neurons decreases synaptic α-synuclein
Having nominated GPNMB as the mechanistic link between a GWAS locus and PD risk, we sought to define its function in a disease-relevant context. GPNMB encodes the GPNMB protein, a transmembrane glycoprotein first identified in a screen of human melanoma cells with varying potential for metastasis (17). While GPNMB has subsequently been found to enhance malignant phenotypes in a number of cancers (18), characterization of its function in the brain or in neurons is scant (15, 16, 19–21). We thus turned to a rapidly-inducible human pluripotent stem cell-derived cortical neuron (iPSC-N) model (22) in which we could directly manipulate GPNMB levels in order to investigate its neuronal function.
Using CRISPR-Cas9 genome editing, we generated iPSC-N lines with normal levels of GPNMB (wild type, WT), heterozygous loss of GPNMB (Het), or homozygous loss of GPNMB (2 lines: KO1 and KO2, Figure S3–S4) and assessed them by confocal microscopy. At a time point when morphology, expression of neuronal markers, and ability to fire trains of action potentials indicated neuronal maturity (14 days after neuronal induction (22)), loss of GPNMB was accompanied by a marked reduction in aSyn at the synapse (Figure 2A, 2B). Moreover, similar levels of decrease were observed in the Het, KO1, and KO2 lines, when we quantified microscopy results (Figure 2C). In contrast, staining of synapsin-1, another pre-synaptic terminal protein, did not differ significantly among the iPSC-N lines (Figure 2C).
Fig. 2. Heterozygous and homozygous loss of GPNMB results in altered α-synuclein expression and localization.
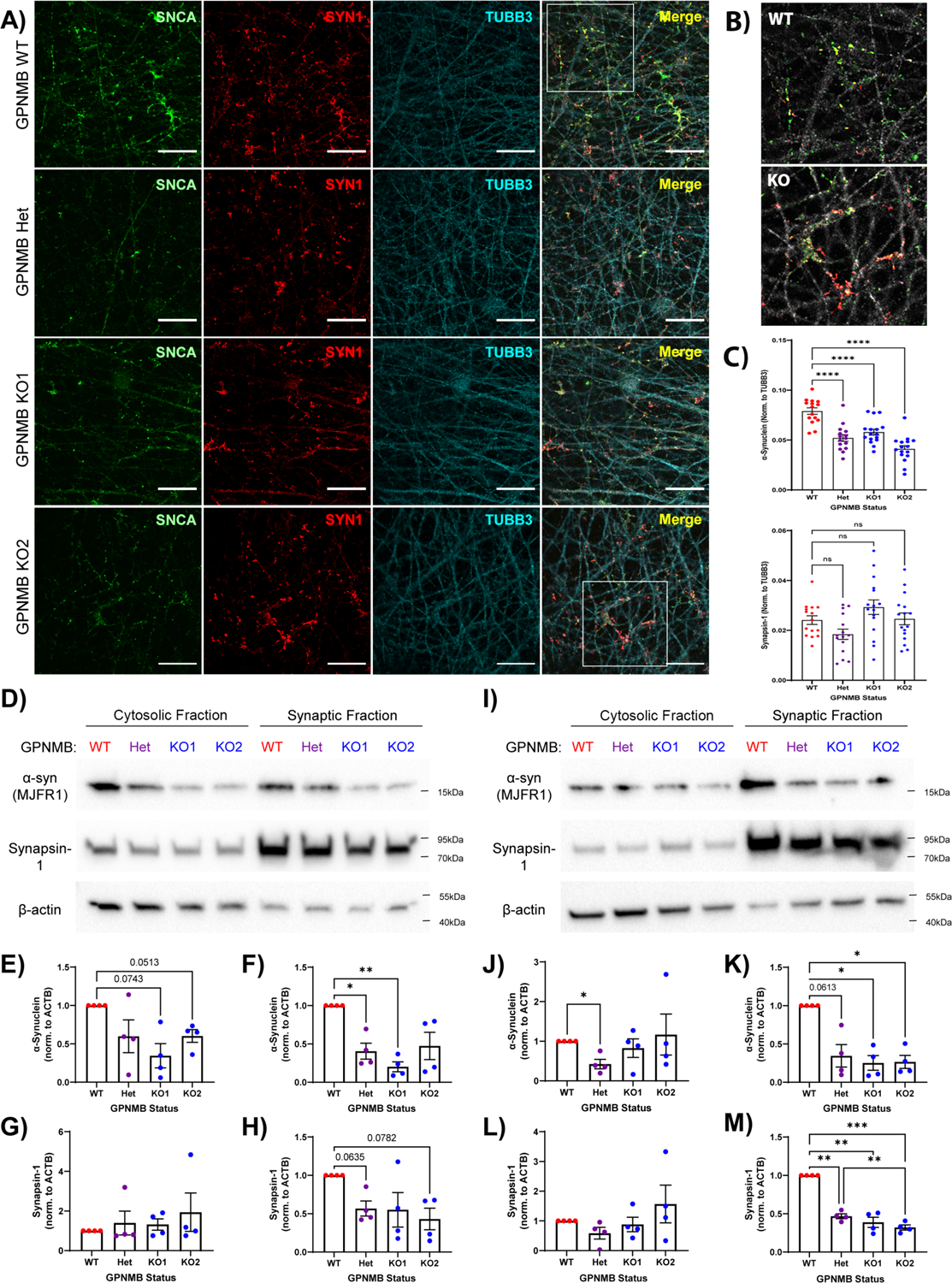
(A) Immunofluorescence imaging of α-synuclein (SNCA, green, stained with MJFR1 antibody), synapsin-1 (SYN1, red), and β-tubulin (TUBB3, cyan) in GPNMB WT, Het, KO1, and KO2 iPSC-N acquired 14 days after induction of neuronal differentiation. Scale bar = 20μm. (B) 2x magnified images of merged insets from (A) demonstrate greatly reduced localization of α-synuclein (SNCA, green) to synapses (labeled with SYN1, red) in GPNMB KO iPSC-N, compared to WT. TUBB3 converted to grey. (C) Quantification of α-synuclein (top) and synapsin-1 (bottom). N = 15 images (5 images from each of 3 wells) per condition. Mean +/− SEM, as well as individual data points, shown. Statistics = one-way ANOVA followed by post-hoc Dunnett’s test comparing all other groups to WT. ns = p > 0.05, **** p < 0.0001. (D,I) Western blots showing cytosolic and synaptosomal expression of α-synuclein, synapsin-1, and β-actin in iPSC-N lysates isolated at day 14 (D) and day 21 (I) after neuronal induction. (E-H) Quantification of D14 immunoblots for either α-synuclein (E-F) or synapsin-1 (G-H) expression in the cytosolic (E,G) or synaptosomal (F,H) fractions. (J-M) Quantification of D21 immunoblots for α-synuclein (J-K) or synapsin-1 (L-M) expression in the cytosolic (J,L) or synaptosomal (K,M) fractions. N = 4 blots per timepoint from 4 independent differentiations. Means +/− SEM, as well as individual data points, shown. Statistics = one-way ANOVA with repeated measures, followed by post-hoc Tukey test. * p < 0.05, ** p < 0.01, *** p < 0.001.
To corroborate our immunofluorescence findings, we isolated synaptosomes from the WT, Het, KO1, and KO2 iPSC-N lines and quantified aSyn by immunoblot in both the synaptosomal and cytosolic fractions (Figure 2D–2M). At the same time point as our microscopy experiments, synaptosomal aSyn was decreased in the Het and KO1 lines (Figure 2D, 2F). In contrast, neither synaptosomal nor cytosolic synapsin-1 levels differed significantly among the WT, Het, KO1, and KO2 lines (Figure 2G, 2H).
Because aSyn expression at the synapse is known to increase as neurons mature (23, 24), we asked whether the observed loss of aSyn could be due to a delay in neuronal maturation that would improve over time. However, after an additional week of neuronal differentiation, differences in aSyn only increased, with the GPNMB Het, KO1, and KO2 lines showing ~75% reduction in the synaptosomal aSyn fraction compared to WT (Figure 2I, 2K). In addition, by 21 days after induction, both aSyn and synapsin-1 were found primarily in the synaptosomal fraction, and synapsin-1 levels were also significantly decreased in the GPNMB Het, KO1, and KO2 lines compared to WT (Figure 2M).
Thus, decreased expression of GPNMB in neurons leads to loss of aSyn at the synapse, suggesting direct links between the rs199347 risk haplotype, GPNMB expression, and aSyn. Moreover, loss of GPNMB may result in a broader synaptic defect.
Transcriptomic profiling of iPSC-neurons reveals GPNMB effects on synaptic biology and α-synuclein
To obtain an unbiased view of biological changes induced by loss of GPNMB in neurons, we transcriptionally profiled five independent replicates each of the GPNMB WT, Het, and KO2 (hereafter referred to as KO) iPSC-N lines. While all three lines showed similar gene expression at baseline, upon neuronal differentiation, distinct profiles emerged for the WT vs. Het and KO cells (Figure S5). Specifically, regardless of whether we performed hierarchical clustering using the top 35 differentially-expressed genes (Figure 3A, Figure S5), or principal component analysis (PCA) using all expressed genes (Figure 3B, Figure S5), GPNMB WT separated from the Het and KO lines, while within-line replicates clustered together. As expected, across all cell lines, as neuronal differentiation occurred, neuronal genes (e.g. MAP2, TUBB3, SYN1) were upregulated, while pluripotency genes (e.g. SOX2) were downregulated (Figure S6).
Fig. 3. Transcriptomic profiling of iPSC-N with heterozygous or homozygous loss of GPNMB reveals altered synaptic biology.
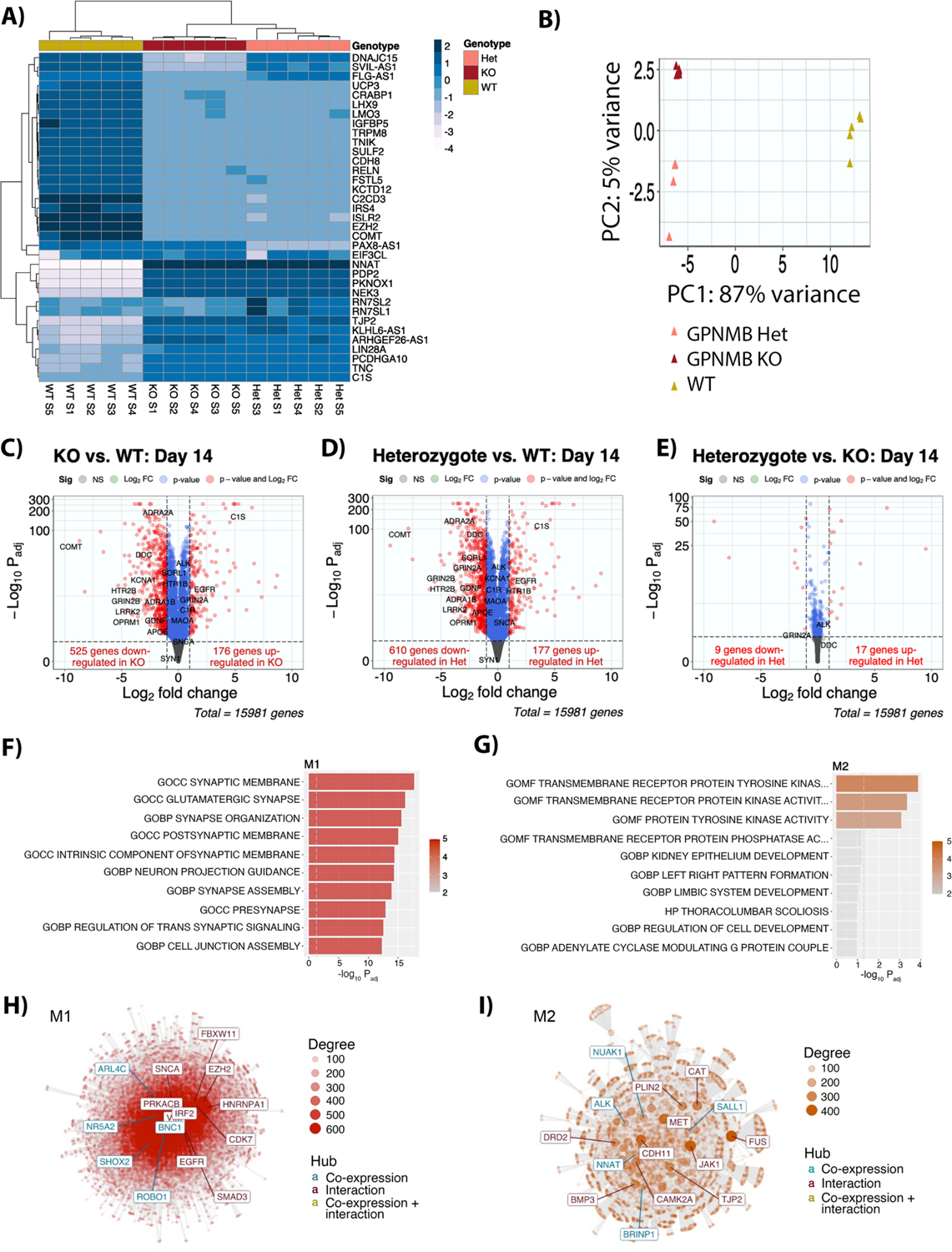
(A) Heatmap showing clustering of iPSC-N (day 14 after neuronal induction) based on the top 35 genes. N=5 replicate samples from 3 independent neuronal differentiations per GPNMB genotype. (B) Principal component analysis (PCA) using all expressed genes. (C-E) Volcano plots for pairwise comparison of GPNMB KO cells compared to WT (C); GPNMB Het cells compared to WT (D); and GPNMB KO cells compared to Het (E). The horizontal axis indicates the log2 fold-change (log2FC) in gene expression. The vertical axis indicates the −log10 of BH-adjusted p-value (Padj), with dotted line indicating a Padj = 0.01 significance threshold. (F,G) Two co-expression modules (M1 and M2) differentiated WT iPSC-N from GPNMB Het and KO iPSC-N. The top 10 enriched pathways for M1 (F), decreased in GPNMB Het and KO iPSC-N, and M2 (G), increased in GPNMB Het and KO iPSC-N, are listed. (H,I) Network analysis of genes in modules M1 (H) and M2 (I) constructed using the GeneMANIA (25) protein-protein interaction dataset. Each point represents a gene, with edges showing protein-protein interactions or co-expression. The most connected genes, or hubs, are labeled, with teal representing co-expression hubs and maroon representing protein-protein interaction hubs.
In the neuronally-differentiated lines, overlap among genes dysregulated in the GPNMB Het and GPNMB KO lines compared to WT was considerable, with 142 and 449 overlapping upregulated and downregulated genes respectively (out of 177/610 up/down-regulated genes in the Het condition, and 176/525 up/down-regulated genes in the KO condition), while relatively few genes showed differential expression comparing the GPNMB Het vs. KO lines (Figure 3C–3E).
Globally, two modules of co-expressed genes distinguished WT from GPNMB Het and KO iPSC-Ns, with one module (M1) increased in expression in WT compared to GPNMB Het and KO iPSC-Ns and one module (M2) decreased in expression in WT (Figure S5). Biological pathway analysis of M1 genes revealed a highly significant over-representation of synapse-related categories (Figure 3F, Table S2), corroborating our cell biological findings. The top three categories over-represented in M2 genes related to receptor tyrosine kinases (Figure 3G, Table S3).
Employing module co-expression network analysis using the GeneMANIA (25) database of protein-protein interactions (Figure 3H, 3I), we found that aSyn (encoded by SNCA) served as a key protein-protein interaction hub for M1 genes with dysregulated expression in GPNMB Het and KO states. These data – from unbiased “bottom-up” analyses of global gene expression – highlight the strong functional effects of GPNMB loss on aSyn.
GPNMB is necessary and sufficient for cellular internalization of α-synuclein
Because data from the edited iPSC-N lines supported a functional interaction between GPNMB and aSyn from both targeted and unbiased lines of investigation, we asked whether these two proteins might directly interact. In iPSC-N, GPNMB expression is not high, making visualization and co-immunoprecipitation experiments challenging, so we began with immortalized cell lines. Here, confocal microscopy of GPNMB and aSyn revealed colocalization between GPNMB and aSyn in LAMP1+ cytoplasmic punctae when transfected in both HeLa (Figure 4A) and HEK293 (Figure S2) cells. Moreover, when expressed in both cell types, GPNMB and aSyn co-immunoprecipitated, regardless of whether the immunoprecipitated protein was GPNMB or aSyn (Figure 4B, 4C, Figure S2).
Figure 4: GPNMB is necessary and sufficient for internalization of fibrillar alpha-synuclein.
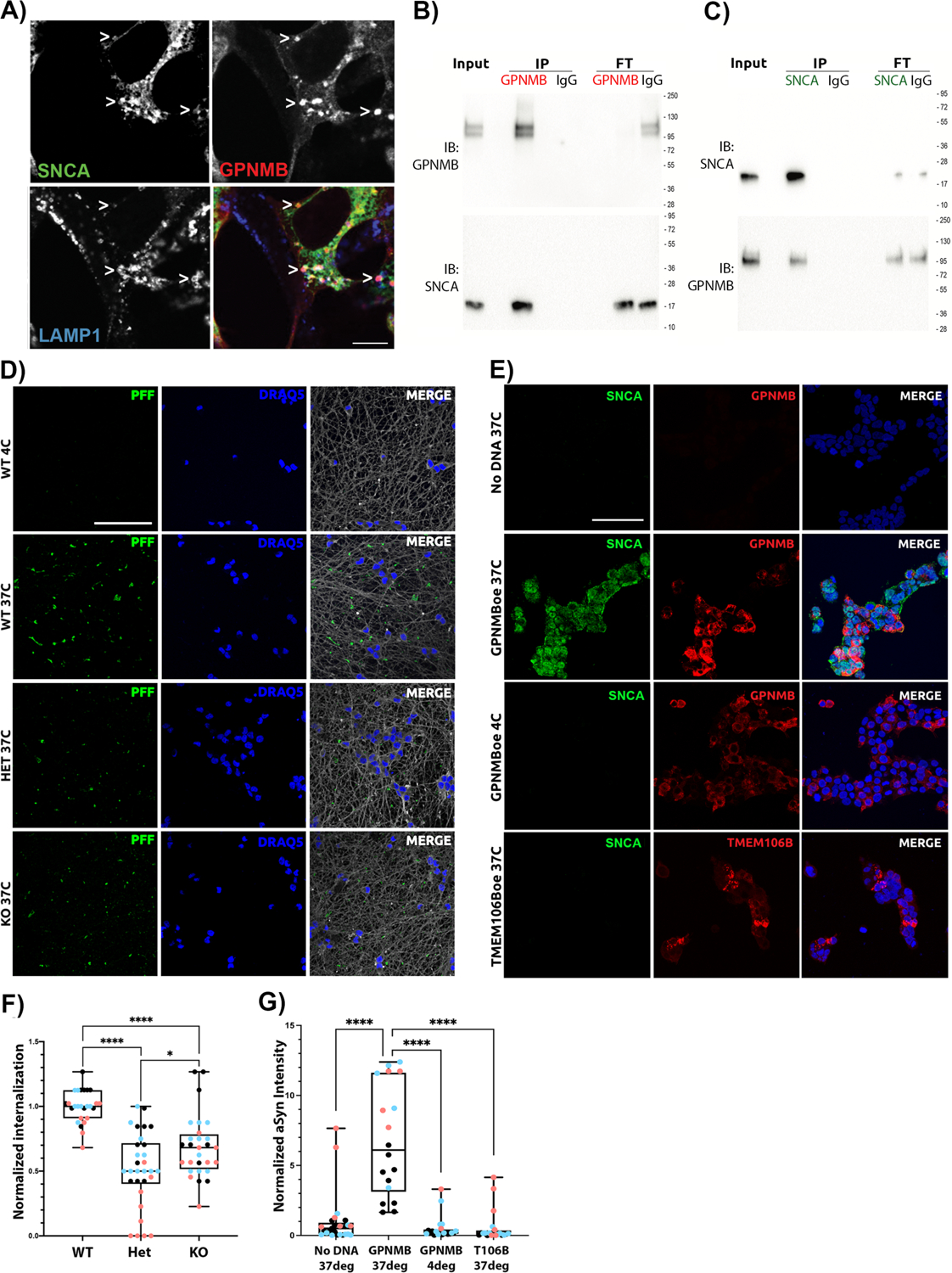
(A) Immunofluorescence images of HeLa cells transfected with GPNMB and α-synuclein (SNCA, stained with MJFR1) expression constructs shows colocalization between GPNMB (AF2550 antibody) and SNCA in LAMP1+ vesicles. Scale bar = 7.5μm. (B) HeLa cells over-expressing GPNMB and SNCA were IP’d for GPNMB, showing co-IP of SNCA (bottom, MJFR1); or (C) IP’d for SNCA, showing co-IP of GPNMB (bottom, AF2550). Bi-directional co-IP of GPNMB and SNCA was confirmed 8 times total (4 replicate IP experiments from each direction). GPNMB appears as 1–2 bands that collapse to the predicted molecular weight upon deglycosylation (see Figure S2). 1:10:1 Input:IP:FT (Flow-through) for protein loading on immunoblots. (D) Representative images of Alexa Fluor 594 (AF594)-labeled human α-synuclein pre-formed fibrils (PFFs) internalized in iPSC-N (DIV14). All experiments were started at 4°C to allow for addition of PFF in the absence of active uptake. Cells were then maintained at 4°C (top row: negative control) or warmed to 37°C for 90 min to permit uptake (WT, GPNMB Het, and GPNMB KO shown in rows 2, 3, and 4, respectively). Monochrome images were captured by confocal microscopy, then converted to color for visualization using FIJI software. In each case, PFFs are shown in green, nuclei are shown in blue, and β-tubulin (neuronal structural marker) is shown in grey. Scale bar = 50μm. Only wild-type (WT) iPSC-N demonstrate definite uptake of α-synuclein PFF. (E) Representative images of HEK293 cells, a human cell line that does not internalize α-synuclein PFF at baseline (top row: negative control). Over-expression of GPNMB (red, stained with D9 antibody) enables internalization of human WT PFFs (SNCA, green, stained with MJFR1 antibody) at 37°C (row 2). However, no internalization is observed when GPNMB-over-expressing cells are maintained at 4°C (row 3), or at 37°C when another protein (TMEM106B) is over-expressed in HEK293 cells (row 4, TMEM106B is shown in red for this row only, FLAG tag stained). Monochrome images were captured by confocal microscopy, then processed using FIJI imaging software. Nuclei are shown in blue. Scale bar = 50μm. (F) Quantification of α-synuclein PFF uptake in iPSC-N lines. N=30 images from 3 independent differentiations (blue, pink, and black dots) per iPSC-N line. Groups compared by Mann-Whitney test. * p < 0.05, **** p < 0.0001. (G) Quantification of α-synuclein PFF uptake in HEK293 cells. N=30 images from 3 independent transfections (blue, pink, and black dots) per condition. Groups compared by Mann-Whitney test. **** p < 0.0001. For F and G, boxplot depicts median, 25th, 75th quartiles, while whiskers are full range.
Encouraged by these data suggesting that a GPNMB-aSyn protein-protein interaction might form the basis for the profound effects on aSyn caused by loss of GPNMB, we next investigated the functional interaction of these two proteins in a cellular model of PD. Specifically, when aSyn is fibrillized and introduced in the culture medium to neurons, including iPSC-N, these fibrillar forms of aSyn are known to be internalized by the cells, which will subsequently develop aSyn inclusions with pathological hallmarks including phosphorylation at serine 129 and decreased solubility (26). We thus used this PD model to probe the effects of neuronal loss of GPNMB by introducing fibrillar aSyn to cultures of our GPNMB WT, Het, and KO iPSC-derived neurons.
While WT iPSC-N at a mature stage of differentiation readily internalized labeled preformed fibrils (PFF) of aSyn (Figure 4D, second row), we observed a significant reduction in aSyn internalization in the Het and KO iPSC-N lines, to levels barely above background (Figure 4D, 4F). Thus, GPNMB is necessary for the robust internalization of aSyn into iPSC-N.
To test whether GPNMB is also sufficient for aSyn internalization, we turned to a cell line that does not internalize aSyn at baseline. In HEK293 cells, GPNMB is expressed at very low levels endogenously and, in the absence of exogenous GPNMB expression, aSyn was not taken up by cells (Figure 4E, top row). However, expression of GPNMB, but not of a control transmembrane protein (TMEM106B), conferred the ability to internalize aSyn (Figure 4E, 4G). Thus, GPNMB is sufficient for internalization of aSyn.
GPNMB expression is permissive for development of α-synuclein pathology in neurons
Having demonstrated that GPNMB is needed for robust internalization of fibrillar aSyn in iPSC-N, we asked whether loss of GPNMB would rescue the development of aSyn pathology in these cells. To test this, we introduced aSyn PFF into the culture medium of GPNMB WT, Het, and KO iPSC-N lines at the same time point as the internalization experiments (Day 14 post neuronal induction), then continued to culture the neurons for 14 more days after the one-time addition of fibrillar aSyn. At the end of this period, in WT neurons, as expected, we observed abundant examples of S129-phosphorylated aSyn (Figure 5A, second row), often along neuronal processes (Figure 5C). In GPNMB Het or KO neurons, however, minimal phospho-aSyn staining was found (Figure 5A). Moreover, by extracting soluble proteins from our neuronal cultures, we found that WT GPNMB iPSC-N developed insoluble aSyn aggregates to a much greater degree than GPNMB Het or KO neurons (Figure 5B, second row). These insoluble aggregates had a dense appearance and were often found in a perinuclear location (Figure 5E). Moreover, quantifications of S129-phosphorylated aSyn, with (Figure 5F) or without (Figure 5D) first extracting soluble proteins, revealed >70% reduction in hyperphosphorylated aSyn species in the GPNMB Het and KO neurons.
Figure 5: GPNMB is necessary for development of a-Syn pathology in iPSC-N.
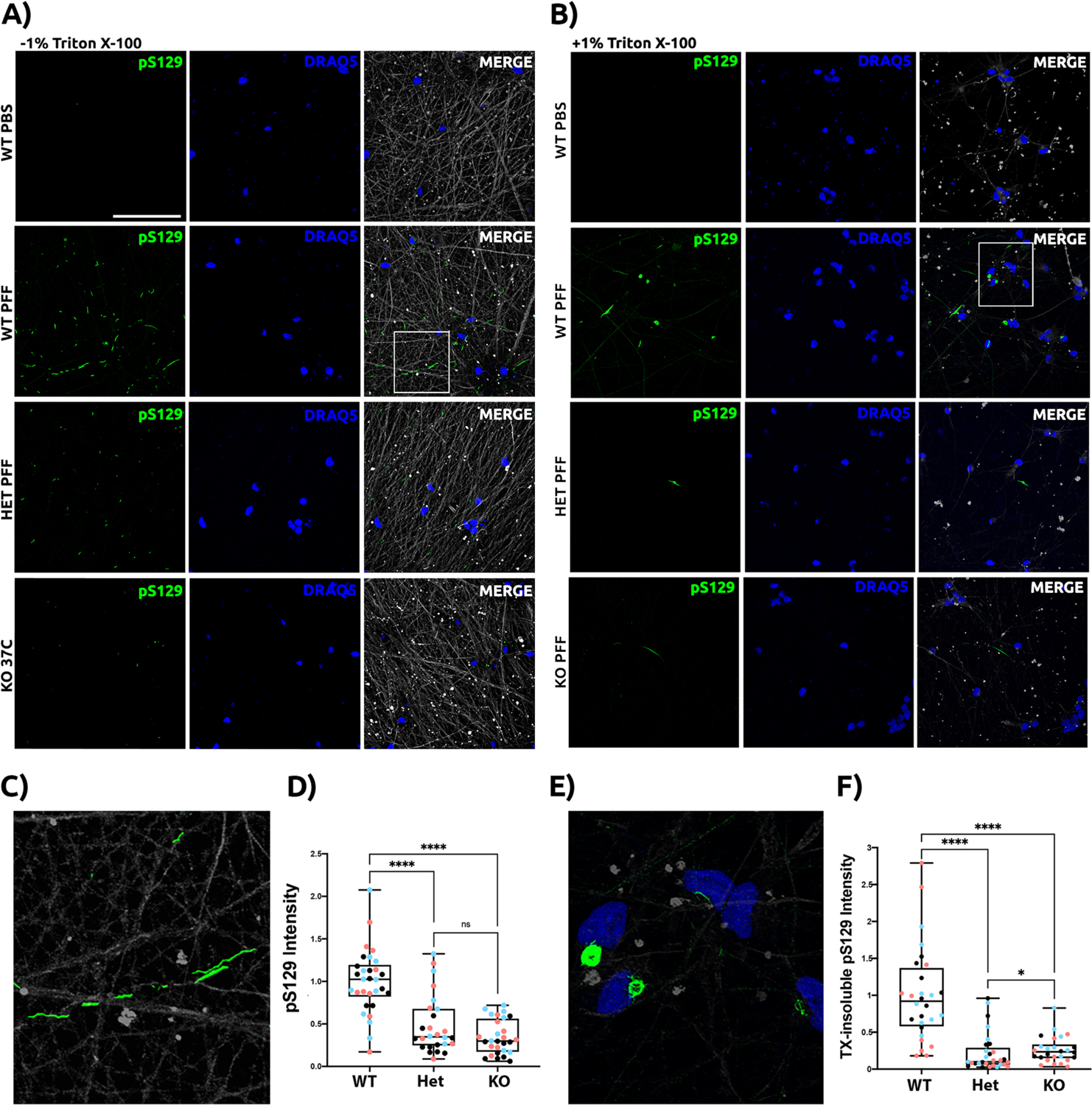
Untagged human aSyn PFF were added to iPSC-N at DIV14. Neuronal cultures were maintained for 14 additional days to allow for development of aSyn pathology before cells were washed, fixed, and stained under conditions without 1% Triton X-100 (TX) extraction (A) or after extraction of soluble proteins with 1% TX (B). 81a antibody against aSyn phospho-Ser129 (pS129) was used to stain pathology (green). Neurons were also stained for nuclear DNA (blue) and tubulin (grey), although this structural protein was largely removed under 1% TX extraction conditions. Scale bar = 50um. For each set, a negative control condition without addition of aSyn PFF is shown in the top row, followed by addition of aSyn PFF in WT (row 2), GPNMB Het (row 3), and GPNMB KO (row 4) iPSC-N. Only the WT iPSC-N demonstrate abundant pS129 staining. Moreover, the pS129 staining shows a neuritic pattern (C, inset from panel A) in the absence of 1% TX extraction and a dense perinuclear aggregate pattern (E, inset from panel B) after extraction of soluble proteins, only in the WT iPSC-N. Quantification of pS129 pathology scores is shown under conditions without 1% TX (D) and after extraction of soluble proteins with 1% TX (F). N=30 images from 3 independent differentiations (shown as blue, pink, and black dots) per line. Boxplots depict median, 25th, 75th quartiles, while whiskers are full range. Groups were compared by Mann-Whitney test. **** p < 0.0001, * p > 0.05.
Thus, GPNMB functions in the development of PD pathology through mechanisms related to the cellular internalization of fibrillar aSyn.
GPNMB measures reflect clinical status in PD
Findings from our human eQTL/ASE analyses and iPSC-N experiments support a model in which higher GPNMB expression levels confer risk for PD through interactions with aSyn that are permissive for neuronal internalization of fibrillar aSyn. Because the extracellular domain of GPNMB is known in cancer contexts to be shed and to circulate in bodily fluids, we asked whether GPNMB might be detected in plasma and cerebrospinal fluid (CSF) samples from 731 PD and 59 neurologically normal control individuals (Figure 6, also see demographic data in Tables S4–S7). Moreover, we sought to understand whether GPNMB measures in biofluids from living subjects might reflect either rs199347 genotype or disease.
Fig. 6. Biofluid GPNMB protein levels associate with rs199347 genotype, PD diagnosis, and disease severity.
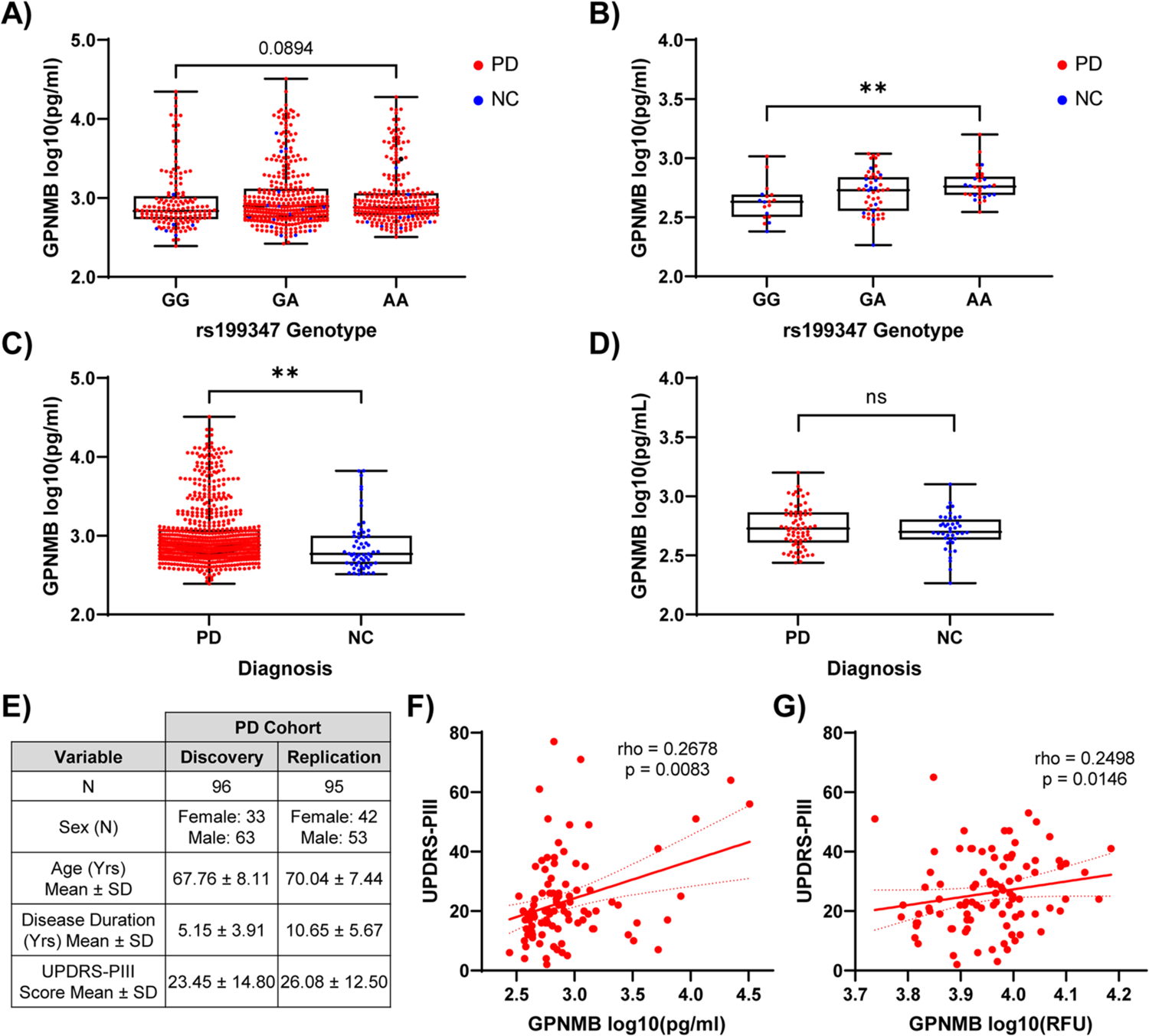
(A) Plasma and (B) CSF GPNMB levels grouped by rs199347 genotype. Genotypes compared with Kruskal-Wallis test, followed by post-hoc Dunn’s test. ** p < 0.01. (C) Plasma and (D) CSF GPNMB levels grouped by diagnosis. PD (red) vs. neurologically normal controls (NC, blue) compared by Mann-Whitney test. Boxplots depict median, 25th, 75th quartiles, while whiskers are full range. ** p < 0.01. (E) Demographics for discovery and replication cohorts used for UPDRS-PIII analysis. (F-G) Scatterplots showing positive correlation between UPDRS-PIII scores and plasma GPNMB values in discovery (F) and replication (G) cohorts. Spearman’s rho and p-value displayed in text.
Echoing our results from eQTL and ASE analyses in human brain samples (Figure 1), we found that rs199347 genotypes associated with GPNMB levels in CSF in the direction predicted by GTEx (10), while no protein quantitative trait locus (pQTL) effect was observed in the plasma (Figure 6A, 6B). Notably, however, plasma GPNMB levels were elevated in PD individuals compared to NC (Figure 6C), and this elevation persisted after adjustment for age, sex, and rs199347 genotype (βPDvs.NC = 0.141, p = 0.0397, Tables S8–S9). Moreover, in a subset of 96 PD individuals with clinical severity measures (Figure 6E), plasma GPNMB levels associated with disease severity as measured by the Unified Parkinson’s Disease Rating Scale (UPDRS) Part III score (27), with higher plasma GPNMB levels found in more severely affected PD patients (Figure 6F, Table S10).
We confirmed these results in a second cohort of PD individuals in whom we had previously obtained GPNMB plasma measures using an aptamer-based method (28). Despite differences in measurement platform, disease duration, and severity between the original cohort of 96 individuals and this cohort of 95 additional, non-overlapping PD individuals, GPNMB levels replicated their association with disease severity (Figure 6G, Table S11).
Discussion:
Using statistical genetic methods, direct measures of ASE in human brain samples, CRISPR-based manipulation of gene levels in iPSC-derived neurons, and human biomarker studies, we here establish GPNMB as a PD risk gene. Moreover, using a PD cellular model, we nominate GPNMB as a potential therapeutic target for interrupting the transmission of pathological aSyn. Finally, we demonstrate the importance of GPNMB expression levels, with even partial loss of expression exerting profound effects on aSyn and the synapse, and increased expression associating with clinical severity in PD. Our results support a model in which PD risk-associated haplotypes at the rs199347 locus result in increased expression of GPNMB at the RNA and protein levels. Increase in GPNMB expression, in turn, may confer risk for development of PD through effects on the synapse and specifically on the cellular uptake of pathological forms of aSyn.
Here, we integrated computational analyses of large databases with direct experimental verification of key predictions from these analyses. For example, statistical colocalization techniques and eQTL analyses suggested that the rs199347 haplotype impacts GPNMB expression levels, while direct measures of GPNMB ASE in human brain samples confirmed these effects. Similarly, transcriptomic profiling of GPNMB-edited iPSC-Ns revealed broad effects on the synapse, with aSyn emerging as a hub protein from network analyses of genes impacted by loss of GPNMB, while our cell biological studies confirmed a functional GPNMB-aSyn interaction with repercussions for the cellular transmission of pathological aSyn.
In addition, we integrated the use of patient-derived samples with bench-based manipulation in a model system. The failure, to date, of many cell- and mouse-model-based findings to translate to clinical utility in PD underlines the importance of verifying that biological pathways implicated in model systems show corresponding signals in human disease. Here we used ~800 human samples to ground our study in clinical relevance. Direct manipulation of GPNMB in iPSC-derived neurons then allowed us to establish a causal effect for GPNMB in cellular processes that may mediate PD pathology.
What remains to be seen is whether GPNMB interacts with both normal and pathological conformations of aSyn to a similar extent and whether our neuronal findings will translate in vivo. A detailed analysis of the interaction between these two proteins, including whether the interaction with aSyn occurs with the soluble form of GPNMB vs. the transmembrane protein prior to cleavage, would be key to downstream attempts at targeting. We note, however, that the discovery of a new protein, expressed at the cell surface, which is both necessary and sufficient for internalization of fibrillar aSyn in neurons, opens up new therapeutic avenues in PD.
Materials and Methods:
A more detailed version of the Materials and Methods is provided in the supplementary materials.
RNA isolation and library preparation from human brain samples:
Human postmortem brain samples from neurologically normal controls (NC, n = 2) and PD (n = 4) individuals were obtained from the University of Pennsylvania Center for Neurodegenerative Disease Research (CNDR) Brain Bank and dissected as previously described (29). Demographics are summarized in Table S1. The regions analyzed included the caudate nucleus, cingulate gyrus, and cerebellum. Samples comprised predominantly gray matter. Genotypes for the brain samples were obtained as previously described (29, 30).
Library preparation was performed with the KAPA RNA HyperPrep Kit (KR1350, Illumina Platforms, Roche, WI). Briefly, RNA was fragmented using heat and magnesium for 8 minutes at 94°C to obtain 100–200bp fragments. The first cDNA strand was then synthesized using a thermocycler, followed by the second strand synthesis combined with A-tailing. Unique adapters were then ligated onto the library insert fragments and amplified using high-fidelity, low-bias PCR. The products then underwent a bead-based cleanup for purification of the cDNA libraries.
SeqCap RNA ChoiceTM probe pool design:
The SeqCap RNA probe design pool was designed by Roche Sequencing Solutions Custom Design (Roche, WI). The pool was designed to contain probes about 60bp in length with no more than 20 “close matches” in the genome, as determined by the Sequence Search and Alignment by Hashing Algorithm (SSAHA), for the purposes of providing sufficient coverage of transcripts of interest while minimizing potential off-target effects. A “close match” was defined as any genomic sequence that differed from one of the probe sequences by five or fewer singlebase insertions, deletions, or substitutions. The majority of included probes had no off-target matches, with an exceedingly small percentage of probes displaying very few off-target matches. Only exonic probes covering the transcripts of interest were included and, to minimize SNP-mediated capture bias, probes that overlapped any SNPs in linkage disequilibrium (r2 > 0.2) with the sentinel PD GWAS SNP were excluded.
Target cDNA enrichment and sequencing:
Target cDNA enrichment and sequencing was performed as previously described (14) using the SeqCap EZ Accessory kit v2 (07145594001, Roche, WI). Briefly, equimolar amounts of cDNA libraries were combined for a total mass of 1ug. Each of these libraries underwent multiplexed PCR with unique index oligonucleotides. The libraries were then hybridized with capture probes using the SeqCap EZ Hybridization and Wash Kit (05634261001, Roche, WI). Libraries were dried with heat in a vacuum and resuspended with hybridization reagents. Pooled capture probes for each region were added to the resuspended libraries and incubated for 20 hrs in a thermocycler at 47°C, with the lid temperature at 57°C. The captured multiplex cDNA samples then underwent stringent washing steps and were amplified using ligation-mediated PCR (LM-PCR). These post-capture PCR amplified libraries were pooled and sequenced on two lanes of an Illumina HiSeq 2500 with 150bp paired-end reads, yielding ~150 million read pairs per lane.
Read mapping and allele specific expression analyses:
To assess RNA-seq reads quality we employed FastQC (31), while for reads quality filtering and trimming we used Trimmomatic (Version 032) (32). We ran Trimmomatic to remove low quality fragments in a 4 base wide sliding window (average window quality below PHRED 20), and low quality leading and trailing bases (below PHRED 10). We also dropped all the reads with average PHRED quality below 25, as well as reads shorter than 75 bases. Depending on the sample, 65–80% of reads passed this trimming and filtering step, resulting in 6.5–24 million read pairs per sample for mapping.
To perform unbiased allele specific read mapping to the reference human genome (hg19) we applied WASP–STAR pipeline (Figure S7). First, we mapped reads with STAR (33) applying 2 step alignment and filtering them for mapping bias using WASP (34). Before proceeding with variant calling, we removed duplicate reads using rmdup_pe.py script incorporated into WASP pipeline. To call and filter SNV we used GATK tools; HaplotypeCaller, SelectVariants and VariantFiltration. We obtained allele specific read counts by GATK – ASEReadCounter. In order to filter out intergenic variants, we functionally annotated SNVs using VariantAnnotation (35) and TxDb.Hsapiens.UCSC.hg19.knownGene (36) R packages. To test for allele specific expression (ASE) at the gene level, we first selected proxy SNP’s that were highly linked (r2 > 0.6) with rs199347 and located within a coding region for the gene of interest in order to assign allele of origin. For GPNMB, we assigned the allele of origin for each transcript read based on genotype at rs199355, and for KLHL7, we assigned the allele of origin based on genotype at rs2072368. We then tested for allelic imbalance with a beta-binomial model with overdispersion using the MBASED R package (13). P-values were adjusted for false discovery rate using the Benjamini-Hochberg method (37).
Colocalization analysis:
Colocalization analysis was performed as previously described (11) using the ‘coloc’ package in R (version 4.0–2). The prior probabilities of p1, p2, and p12 were set to 1e-4, 1e-4, and 1e-5 respectively. Significant colocalization was determined as having a PPH4 > 75%. Sensitivity analysis was performed to determine whether PPH4 is robust over plausible values of p12 (1e-5 to 1e-6) (38). Association plots were generated using LocusComparer package (version 1.0.0) (39) in R.
Immortalized cell line transfection and collection:
For co-immunoprecipitation (co-IP) experiments, HEK293 or HeLa cells were plated at 700,000 or 1 million cells per well, respectively, and 18 hours after plating, transfection was performed with 4ug of each DNA construct and Lipofectamine 2000 (Thermo Scientific) in serum-free DMEM. Cells were transfected with either GPNMB-myc-DDK pCMV6-Entry (Origene) and 5’ UTR-Syn pcDNA3.1+ (obtained from Dr. Kelvin Luk) for the GPNMB-flag IP or GPNMB untagged clone pCMV6-XL4 (Origene) and Syn-Myc pcDNA3.1+ (obtained from Dr. Kelvin Luk) for the Syn-Myc IP. 4 hours post-transfection, media was changed to DMEM with 10% FBS 1% L-Glut and 1% Pen-Strep. 20 hours post-transfection, cells were washed in dPBS and lysed in CHAPS buffer (25mM Tris, 150mM NaCl, 1mM EDTA 1% CHAPS, 5% glycerol, pH to 7.4). Lysates were collected and spun down at 4°C for 30 minutes at 21380 xg. BCA assays (Thermo Scientific) were used to determine protein concentrations.
For immunofluorescence experiments, HEK293 or HeLa cells were plated at either 100,000 or 200,000 cells per well on either PDL-coated or untreated 12mm glass coverslips in a 12-well format. 18 hours after plating, the cells were transfected with 1.6ug of each DNA construct (GPNMB untagged clone pCMV6-XL4 and 5’ UTR-Syn pcDNA3.1+) and Lipofectamine 2000 (Thermo Scientific) in serum-free DMEM.
Co-immunoprecipitation:
To immunoprecipitate (IP) GPNMB, 300 μL anti-Flag-conjugated beads (Sigma A2220) or mouse IgG-conjugated beads (Sigma A0919, used as a negative control) were used to IP from 1,000 μg of lysate from GPNMB-myc-DKK and 5’UTR-syn double-transfected cells in CHAPS buffer overnight at 4°C. To IP α-synuclein, 300 μL anti-c-Myc-conjugated beads (Sigma A7470) or rabbit IgG-conjugated beads (Sigma A2909, used as a negative control) were used to IP from 1,000 μg of lysate from GPNMB untagged clone and Syn-Myc double-transfected cells in CHAPS buffer overnight at 4°C. After 24 hours, the protein-conjugated beads were washed 3x with CHAPS buffer and the bound protein was competitively eluted from the beads using either 250 μM 3x flag peptide (Sigma) or 250 mM myc peptide (Sigma) for 1 hour at 4°C.
CRISPR-Cas9 knock-out of human iPSC’s:
Human induced pluripotent stem cells (iPSC) with Neurog1 and Neurog2 in a bicistronic doxycycline-inducible expression cassette (22) were used for generation of GPNMB knock-out (KO) iPSC and rapid induction of iPSC-derived neurons (iPSC-N). The protocol for generating CRISPR edited iPSC’s is summarized in Figure S3 and detailed in the Supplementary Methods.
Culture and differentiation of iPSC-Neurons:
Prior to differentiation, iPSC cultures were maintained on Matrigel-coated 6cm tissue culture plates with mTeSR1 media and mechanically passaged every 4 days with StemMACS passaging solution XF. The neural induction protocol is summarized in Figure S4, based on prior reports (40), and detailed in the Supplementary Methods.
Immunofluorescence:
The media was aspirated from coverslip-containing wells, and the cells were fixed in 2% paraformaldehyde in dPBS for 15 minutes at room temperature. After fixing, the coverslips were rinsed 5x with dPBS and blocked/permeabilized with blocking buffer (3% bovine serum albumin + 0.05% saponin in dPBS) for 1hr prior to incubating in primary antibody overnight. Image stacks of 1um thickness were acquired by confocal microscopy (Leica SP5) using a 40x oil immersion objective with 2x Zoom. Antibody concentrations are summarized in the Key Resources Table.
Key Resources Tables:
| Antibodies | ||
|---|---|---|
| Name | Product ID | Dilution |
| Anti-Alpha-synuclein antibody (MJFR1) | Abcam (ab138501) | IF: 1:300 – 1:1000* WB: 1:1000 |
| Anti-Synapsin-1 | SySy (106-011) | IF: 1:500 WB: 1:1000 |
| Anti-α-synuclein Antibody (syn211) | Santa Cruz (sc-12767) | WB: 1:200 |
| Anti-α-synuclein (phospho S129) Antibody (81a) | Provided by Center for Neurodegenerative Disease Research (CNDR) at University of Pennsylvania | IF: 1:5000 |
| Anti-Human Osteoactivin/GPNMB Antibody | RnD Systems (AF2550) | IF: 1:500 IHC: 1:200 WB: 1:1000 |
| Anti-GPNMB (E1Y7J) Rabbit mAb | Cell Signaling Technology (#13251) | WB: 1:1000 |
| Anti-GPNMB Antibody (D-9) | Santa Cruz (sc-271415) | IF: 1:500 WB: 1:250 |
| Anti-beta Actin antibody (AC-15) | Abcam (ab6276) | WB: 1:5000 |
| Anti-beta-III Tubulin Antibody | Novus Biologicals (NB100-1612) | IF: 1:500 |
| Anti-Flag M2 Mouse Monoclonal Antibody | Sigma Aldrich (F1804) | IF: 1:1000 |
| Anti-GAPDH Antibody | Advanced Immunochemical Anti-GAPDH | WB: 1:3000 |
| Anti-LAMP1 Antibody (H4A3) | University of Iowa Hybridoma Bank | IF: 1ug/mL |
| Goat Anti-Chicken Alexa Fluor 647 | Invitrogen (A-32933) | IF: 1:1000 |
| Goat Anti-Rabbit Alexa Fluor 488 | Invitrogen (A-21206) | IF: 1:1000 |
| Donkey Anti-Mouse Alexa Fluor 594 | Invitrogen (A-21203) | IF: 1:1000 |
| DRAQ5 | Thermo-Fischer (62251) | IF: 1:5000 |
| Goat Anti-Mouse HRP | Jackson Immuno Research (115-035-062) | WB: 1:3000 |
| Goat Anti-Rabbit HRP | Jackson Immuno Research (111-035-144) | WB: 1:3000 |
| Biotinylated Rabbit Anti-Goat | Vector Laboratories (BA-5000-1.5) | IHC: 1:5000 |
| Cell Lines | |||
|---|---|---|---|
| Name | Species | Obtained from | Citations |
| HeLa | Homo sapiens | Michael S. Marks | |
| QBI293 | Homo sapiens | Penn Center for Neurodegenerative Disease Research (CNDR) | |
| iPSC-N | Homo sapiens | George Church | (22) |
| Deglycosylation and dephosphorylation | ||
|---|---|---|
| Name | Product ID | Protocol |
| PNGase F (Deglycosylation) | New England Biolabs Inc., Catalog # P0704L | As per product instructions |
| Lambda Protein Phosphatase (Dephosphorylation) | New England Biolabs Inc., Catalog #P0753S | As per product instructions |
MJFR1 was used at 1:300 to stain endogenous α-synuclein in iPSC-N and 1:1000 to stain overexpressed α-synuclein in HEK293 and HeLa cells.
Image processing and quantification:
Image processing to quantify synaptic proteins was performed using a Cell Profiler pipeline based on Danielson et. al.’s previously published work (41). A single slice was chosen from each stack to focus our analysis on the plane with the most abundant synapsin-1 staining. TUBB3 images underwent image enhancement for neurite-like features and were used to calculate TUBB3+ area for normalization and expanded to generate a mask. Synapsin-1 and α-synuclein images underwent enhancement for speckle-like features, followed by object identification and characterization of object size/intensity. The total integrated intensity (i.e. sum for all particles) was used for analysis. The pipeline used has been made publicly available in the supplementary material.
aSyn pathology in iPSC-N was also quantified using CellProfiler. Maximum intensity projections of z-stack images were created using FIJI. Desired 81a+ objects were identified with intensity thresholding and the total area of 81a+ objects were normalized with total area of DRAQ5+ objects. To quantify MJFR1+ staining in HEK293 overexpressing GPNMB, nuclei artifact staining was removed by generating DRAQ5+ nuclei mask and excluding MJFR1+ speckles within the mask. Total area of MJFR1+ objects was normalized by total area of DRAQ5+ objects.
A researcher blinded to sample identity was provided with randomly ordered maximum intensity projection of z-stack images of iPSC-N treated with AF594-PFF. Scores ranging from 0 to 3 were given for each criterion: intensity, length and frequency of internalized PFF. The three scores were added and were normalized to the average score of WT iPSC-N.
Synaptosome extraction:
Synaptosomes were extracted using Syn-PER™ Synaptic Protein Extraction Reagent (Thermo-Fisher) per the user instructions.
Immunoblotting:
Samples were diluted in 5X concentrated sample buffer (10g Sucrose, 1.85mL 0.5M Tris, pH 6.8, 1.0mL 0.1M EDTA, 1.0mL of 0.1% Bromophenol Blue, 1.0mL of 0.05% Pyronine Yellow, 0.615g of DTT, 10mL of 10% SDS, and adjusted to a final volume of 20mL with Milli-Q Water) and boiled at either 100°C for 10 minutes or 70°C for 15 minutes for heat-sensitive proteins (GPNMB). Samples were run on 4–20% polyacrylamide TGX gels (Bio-Rad Laboratories) and transferred onto 0.2 nitrocellulose membrane (Bio-Rad Laboratories). When blotting for α-synuclein, membranes were fixed in 0.4% PFA for 30 minutes (42). Membranes were blocked in 5% milk in TBS for 1 hour and blotted overnight at 4°C with specific antibodies. The membranes were incubated for 2 hours in HRP-conjugated secondary antibodies and developed using Western Bright ECL and Sirius HRP substrates (Advansta). Antibody concentrations are summarized in the Key Resources Table. Densitometry was performed using the Bio-Rad Image Lab Software.
RNA sequencing of iPSC-N:
RNA was extracted from iPSC-N on days 0 and 14 after doxycycline induction using QIAGEN RNAeasy ® mini kit. RNA integrity was measured using RNA nano chips (Agilent) on an Agilent 2100 Bioanalyzer. All samples had RIN > 9.4. Library preparation was performed with the TruSeq Stranded mRNA Library Prep kit (Illumina cat #20020595) using 225ng total RNA and following manufacturer instructions. High sensitivity DNA chips (Agilent) were used to balance libraries prior to sequencing. In total 30 RNA samples (3 genotypes (WT, Het and KO) × 2 differentiation time (days 0 and 14) × 5 biological replicates) were sequenced. All 30 samples were sequenced together on a single S2 flow-cell generating >1.1 × 109 of 100 bp single end reads in total (on average 3.69 × 107 reads per sample, ranging from 2.9 × 107 to 5.3 × 107). A summary of the Illumina NovaSeq run is provided in Table S12.
Read quality control and filtering:
A quality check of the raw reads was assessed using FastQC (31) and summarized with MultiQC (43). Next, the adapters were removed and filtering of the low quality reads was conducted employing Trimmomatic, Version 0.39 (32) with the following parameters: ILLUMINACLIP:TruSeq3-SE.fa:2:30:10 LEADING:5 TRAILING:5 SLIDINGWINDOW:5:20 MINLEN:50 AVGQUAL:30 HEADCROP:10. The adapters that matched the sequences provided by TruSeq3-SE.fa were removed. The low-quality bases were cut off the start and off the end of reads if their PHRED score was below 5. The sliding window of 5 bp was applied to trim the bases if the window PHRED score dropped below 20. The 10 bases from the start of the reads were also cropped. Finally, the reads were discarded if the average PHRED score of the read was below 30.
Read alignment and quantification:
The sequence alignment BAM files generated by Bowtie 2 were used as the input to RSEM software tool, in order to quantify gene expression levels (44). On average, 3.61 × 107 (97.8%) of reads per sample passed the filtering steps and were mapped versus reference genome GRCh38 using Bowtie 2 the with the following parameters: bowtie2 -q --phred33 --sensitive --dpad 0 --gbar 99999999 --mp 1,1 --np 1 --score-min L,0,−0.1 --nofw -p 32 -k 200 -x, as suggested by RSEM manual. The read alignment rate was on average >89% (Table S12). To obtain the raw read counts at the gene level, an expectation maximization algorithm (RNA-seq by Expectation Maximization) was run by the following RSEM command: rsem-calculate-expression --bowtite2 --forwardprob 0.
Differential gene expression analysis:
To detect significant differences in iPSC-N gene expression data between Day 0 and Day 14 and between the genotypes (WT, Het, and KO), DESeq function form DESeq2 Bioconductor’s R package (45) was used. RNA-seq data was modeled using the negative binomial distribution that accounts for overdispersion. To conduct the pairwise comparisons (Day 14 vs. Day 0 in WT, KO vs. WT, Het vs. WT, and KO vs Het at Day 14) for each gene the nbinomWaldTest statistic was used. The Wald test P-values were calculated by scaling the coefficients by their standard errors and then compared to a standard Normal distribution. After DGE analysis, the lfcShrink function from R package apeglm (46) was applied to shrink the log2FC. We corrected for FDR using the Benjamini & Hochberg method (37). Genes with a BH corrected P-value < 0.01 and a |log2 fold-change| > 1 were considered to be significantly differentially expressed. To visualize the results of DGE analyses we used EnhancedVolcano R package version 1.10.0 (47).
Modular co-expression network analysis:
Modular co-expression network analysis of iPSC-Ns was performed using R package CEMiTool (Co-Expression Molecules identification Tool) version 1.14.1 (48). The rlog normalized expression data (15981 genes in 15 samples) was used as an input to cemitool function. As a first step, cemitool conducted an unsupervised filtering of expression data using the inverse gamma distribution to model the variance of genes. Out of 15981 genes, 1100 genes with P-value < 0.1 survived this filtering and were used for downstream analyses. The genes were then separated into modules using the dissimilarities measures and the Dynamic Tree Cut package (49). The minimum number of genes per module was set to 20, and similar modules were merged together based on correlation.
To determine biological functions associated with the co-expression modules, the Gene Ontology gene sets c5.all.v7.4, which contain GO resources (BP = biological process, CC = cellular component and MF = molecular function) and human phenotype ontology (HPO) from MSigDB database (50), were used in overrepresentation analysis (ORA). The module gene set enrichment analysis (GSEA) was performed using the fgsea R package (51) within the CEMitool pipeline. First, a z-score gene normalization on all input genes was performed following by calculation of the mean for each sample class (GPNMB WT, Het, and KO). Next, a pre-ranked GSEA was performed independently for each GPNMB genotype. The module activity was visualized by normalized enrichment score (NES).
To construct the gene interactions network in the co-expression modules, the combined human protein-protein interaction data, downloaded from GeneMANIA, were added to the CEMiTool object using the function interactions_data. The interactions between the genes were visualized using the plot_interactions function.
aSyn pre-formed fibril internalization and immunofluorescence experiments:
Human wild-type pre-formed fibrils (PFF) and fluorescent fibrils including Alexa Fluor 594 (AF594) conjugated aSyn were provided by Dr. Kelvin Luk at the Penn CNDR. All fibrils were kept at −80°C until use. After the stock of 5mg/mL was diluted to a working concentration of 0.1mg/mL in dPBS, fibrils were sonicated (Diagenode Biorupter® Plus) on high, sonicating for 30 sec followed by 30 sec rest for a total of 10 minutes. Sonicated PFFs were then added to classic neuronal media according to appropriate concentrations.
For AF594-PFF internalization experiments using iPSC-N, 10ug of PFF were added per well of a 12-well plate. iPSC-N were incubated in 4°C for 30 min, followed by an additional 30 min incubation at 4°C for the negative control or 1 hour 30 min in 37°C for experimental wells. All cells were immediately fixed and stained afterwards.
To demonstrate aSyn pathology following PFF transduction, we replaced regular media of iPSC-N at day 14 with classical neuronal media containing total of 1ug of human wild-type PFF per well. iPSC-N were fixed and stained after 14 days (at day 28 of neuronal induction). To extract soluble proteins, coverslips were fixed with 2% paraformaldehyde and 1% Triton X-100 in dPBS. 3% bovine serum albumin without saponin in dPBS was used as blocking buffer.
For wild-type PFF internalization experiments using HEK293, cells were plated at 200,000 cells per well in PDL-coated 12mm glass coverslips in a 12-well format. 18 hours after plating, the cells were transfected with 1ug of DNA (GPNMB untagged clone pCMV6-XL4 or TMEM106B-Flag) and Lipofectamine 2000 (Thermo Scientific) in serum-free DMEM. At 20 hours post-transfection, media was replaced with DMEM containing 1ug of PFF per well. The plates were incubated for 30 min at 4°C followed by an additional 30 min incubation at 4°C for negative control (to inhibit endocytosis) or 1 hr 30 min in 37°C for experimental wells. All cells were immediately fixed and stained for immunofluorescence afterwards.
Patient samples and genotypes:
Plasma and CSF samples of PD patients and neurologically normal controls were obtained as part of the Penn CNDR Neuropathology, Biomarker, and Genetics Biobank and the Parkinson’s Disease Molecular Integration in Neurological Diagnosis Initiative (MIND) studies. PD patients had a clinical diagnosis of PD made by a movement disorders specialist at the Parkinson’s Disease and Movement Disorder Clinic (PDMDC) at the University of Pennsylvania, while controls had no known neurological disorder. Data were stored in the Penn Integrated Neurodegenerative Disease Database (INDD) (30). These studies were approved by the UPenn Institutional Review Board (IORG0000029). Informed consent was obtained at study enrollment. Participant demographics are reported in Tables S4 – S7. Individuals with known GBA1 mutations (N370S, E365K, L444P) were excluded from the analysis, since GPNMB elevation has been reported in patients with Gaucher disease, a lysosomal storage disorder caused by homozygous mutations in GBA1 (52, 53). Genotyping of SNP rs199347 was performed by Infinium Global Screening Assay (Illumina), NeuroX genotyping platform, PANDoRA (Sequenom) panel (30), or MIND panel based on allele-specific PCR performed using FlexSix Dynamic Array integrated fluidic circuits (Fluidigm) and genotyping using BioMark HD system (Fluidigm) (54).
A subset of PD individuals from the previously-described University of Pennsylvania U19 Cohort (55) had Unified Parkinson’s Disease Rating Scale (UPDRS-III) scores for disease severity.
Enzyme-Linked Immunosorbent Assay (ELISA):
GPNMB protein levels within human plasma and CSF samples were measured with ELISA kits (R&D systems) according to manufacturer’s instructions. CSF and plasma samples were diluted by factors of 1 in 2 and 1 in 30 respectively to obtain optical density measurements within the standard range. All samples were run in duplicates and absorbance at 450nm was determined by a microplate reader (Berthold Technologies, Tristar LB 941). Only duplicate samples with a coefficient of variation (CV) <25% were retained for analysis, and the average CV across all samples used was 3.3%. Moreover, replicate samples assayed by ELISA on different days, by different operators, across multiple freeze-thaw cycles, demonstrated excellent reproducibility (Pearson r=0.97).
Multiple Linear Regression Analyses:
Multiple linear regression was used to determine factors that are significant predictors of GPNMB concentration in plasma and CSF. Age, sex, PD status, and rs199347 genotype were included as independent variables. A codominant genetic model, which considers each allele combination (GG, AG, AA) as a separate factor (56), was used to model the effect of rs199347. Regression coefficients were calculated with GG as the reference.
The relationship between GPNMB levels and disease severity was established in two different PD cohorts. GPNMB values for the discovery cohort were generated by ELISA (R&D systems), whereas GPNMB values for the replication cohort were generated using an aptamer-based platform (26). UPDRS-PIII values were downloaded from the Penn INDD.
Statistical analyses:
Statistical analyses were performed with either PRISM or R. Data was tested for normality with a Shapiro-Walk test. Data with more than two categorical groups was analyzed by either 1-way ANOVA (followed by post-hoc Tukey or Dunnett tests) or Kruskal-Wallis test (followed by post-hoc Dunn’s test) depending on the data’s distribution. Data with only two groups was analyzed by either a Welch’s two-sample t-test or Mann-Whitney-U test depending on the data distribution. Correlations were determined using Spearman’s rank correlation. Multiple linear regression (glm) was used to determine associations between two variables adjusting for potentially confounding covariates. Outlier determination was performed using ROUT method, Q (FDR)=1%.
Immunohistochemistry
Formalin-fixed, paraffin-embedded cingulate and temporal cortex samples were obtained from the Penn CNDR. Patient demographics are reported in Table S1. 6μm sections were cleared in xylenes and a descending EtOH series. Endogenous peroxidases were quenched in 30% H2O2 and 70% MeOH solution for 30 minutes. Slides were microwaved in citric acid Antigen Unmasking Solution (Vector Laboratories). After cooling, slides were rinsed in TBS-T (0.1 M Tris Buffer/0.05% TWEEN) and blocked (TBS-T/2%FBS/3%BSA). Sections were incubated overnight at 4°C in the primary antibody (see Key Resources table for antibody conditions). Once washed with TBS-T, sections were incubated for 1 hour at room temperature in the secondary antibody (see Key Resources table for antibody conditions). VECTASTAIN ABC Standard (Vector Laboratories) was applied for 1 hour at room temperature followed by ImmPACT DAB (Vector Laboratories). Sections were counterstained with Harris Hematoxylin (Thermo Scientific) for 40 seconds. Slides were dehydrated in an ascending EtOH series and xylenes then coverslipped with Cytoseal (Thermo Scientific).
Dephosphorylation and deglycosylation
For deglycosylation of cell and brain lysates, 5.0uL of PNGase F was added to 125ug of brain lysates or 60ug of cell lysates and incubated at 37°C for 30 minutes. For dephosphorylation experiments, 1uL of Lambda protein phosphatase was added to 125ug of brain lysates or 50ug of cell lysates and incubated at 30°C for 30 minutes.
Supplementary Material
Acknowledgments:
The authors would like to thank George Church for providing the iPSC-N cell line. We would also like to thank the Penn Institute for Regenerative Medicine (IRM) iPSC Core members (Director: Wenli Yang, Rachel Truitt, and John McCormick) for guidance on iPSC culture/experimental design as well as the Penn Next Generation Sequencing (NGS) Core members (Director: Jonathan Schug, Kiya Rutherford, and Olga Smirnova) for assistance in library preparation and RNA sequencing services. Lastly, we are grateful to all the patients who donated their brain, blood, and CSF samples for research and to John Trojanowski for his vision in creating a neurodegenerative disease biobank that has served so many.
Funding:
National Institutes of Health grant K23NS114167 (TT)
National Institutes of Health grant F31NS113481 (MDO)
National Institutes of Health grant R01NS115139 (ACP)
National Institutes of Health grant P30AG072979 (ACP, EL, VVD)
National Institutes of Health grants U19 AG062418 and P50 NS053488 (ACP, DW, VVD).
Biomarkers Across Neurodegenerative Diseases (BAND) grant from the Michael J. Fox
Foundation/Alzheimer’s Association/Weston Institute (ACP, TT)
Alice Chen-Plotkin is additionally supported by the Parker Family Chair, the Chan Zuckerberg Initiative Neurodegeneration Challenge Network, and the AHA/Allen Brain Health Initiative.
Footnotes
Data and materials availability:
All transcriptome data have been deposited in NCBI’s Gene Expression (GEO dataset GSE206327). Human data are available upon request subject to a data use agreement to ensure maintenance of personal privacy. Requests for data or materials should be addressed to chenplot@pennmedicine.upenn.edu.
References:
- 1.Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM, Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 68, 384 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simón-Sánchez J, Schulte C, Sharma M, Krohn L, Pihlstrøm L, Siitonen A, Iwaki H, Leonard H, Faghri F, Gibbs JR, Hernandez DG, Scholz SW, Botia JA, Martinez M, Corvol J-C, Lesage S, Jankovic J, Shulman LM, Sutherland M, Tienari P, Majamaa K, Toft M, Andreassen OA, Bangale T, Brice A, Yang J, Gan-Or Z, Gasser T, Heutink P, Shulman JM, Wood NW, Hinds DA, Hardy JA, Morris HR, Gratten P. M. Visscher, Graham RR, Singleton AB, 23andMe Research Team, System Genomics of Parkinson’s Disease Consortium, International Parkinson’s Disease Genomics Consortium, Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. The Lancet Neurology. 18, 1091–1102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McFarthing K, Simuni T, Clinical Trial Highlights: Targeting Alpha-Synuclein. Journal of Parkinson’s Disease. 9, 5–16 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Schneider SA, Alcalay RN, Precision medicine in Parkinson’s disease: emerging treatments for genetic Parkinson’s disease. Journal of Neurology. 267, 860–869 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR, I. P. D. G. Consortium, 23andMe Research Team, A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nature Genetics. 49, 1511–1516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallagher MD, Posavi M, Huang P, Unger TL, Berlyand Y, Gruenewald AL, Chesi A, Manduchi E, Wells AD, Grant SFA, Blobel GA, Brown CD, Chen-Plotkin AS, A Dementia-Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. The American Journal of Human Genetics (2017), doi: 10.1016/j.ajhg.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, v Sachs K, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, Racie T, Ejebe KG, Orho-Melander M, Melander O, Koteliansky V, Fitzgerald K, Krauss RM, Cowan CA, Kathiresan S, Rader DJ, From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 466, 714–719 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, Abdennur NA, Liu J, Svensson P-A, Hsu Y-H, Drucker DJ, Mellgren G, Hui C-C, Hauner H, Kellis M, FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. New England Journal of Medicine. 373, 895–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R, Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature. 533, 95–99 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.GTEx Consortium, Genetic effects on gene expression across human tissues. Nature. 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V, Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLOS Genetics. 10, e1004383– (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murthy MN, Blauwendraat C, Guelfi S, Hardy J, Lewis PA, Trabzuni D, UKBEC, IPDGC, Increased brain expression of GPNMB is associated with genome wide significant risk for Parkinson’s disease on chromosome 7p15.3. neurogenetics. 18, 121–133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayba O, Gilbert HN, Liu J, Haverty PM, Jhunjhunwala S, Jiang Z, Watanabe C, Zhang Z, MBASED: allele-specific expression detection in cancer tissues and cell lines. Genome Biology. 15, 405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mercer TR, Clark MB, Crawford J, Brunck ME, Gerhardt DJ, Taft RJ, Nielsen LK, Dinger ME, Mattick JS, Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nature Protocols. 9, 989–1009 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Tanaka H, Shimazawa M, Kimura M, Takata M, Tsuruma K, Yamada M, Takahashi H, Hozumi I, Niwa J, Iguchi Y, Nikawa T, Sobue G, Inuzuka T, Hara H, The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Scientific Reports. 2, 573 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagahara Y, Shimazawa M, Ohuchi K, Ito J, Takahashi H, Tsuruma K, Kakita A, Hara H, GPNMB ameliorates mutant TDP-43-induced motor neuron cell death. Journal of Neuroscience Research. 95, 1647–1665 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Weterman MAJ, Ajubi N, van Dinter IMR, Degen WGJ, van Muijen GNP, Ruiter DJ, Bloemers HPJ, nmb, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. International Journal of Cancer. 60, 73–81 (1995). [DOI] [PubMed] [Google Scholar]
- 18.Rose AAN, Biondini M, Curiel R, Siegel PM, Targeting GPNMB with glembatumumab vedotin: Current developments and future opportunities for the treatment of cancer. Pharmacology & Therapeutics. 179, 127–141 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Hüttenrauch M, Ogorek I, Klafki H, Otto M, Stadelmann C, Weggen S, Wiltfang J, Wirths O, Glycoprotein NMB: a novel Alzheimer’s disease associated marker expressed in a subset of activated microglia. Acta Neuropathologica Communications. 6, 108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moloney EB, Moskites A, Ferrari EJ, Isacson O, Hallett PJ, The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiology of Disease. 120, 1–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neal ML, Boyle AM, Budge KM, Safadi FF, Richardson JR, The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. Journal of Neuroinflammation. 15 (2018), doi: 10.1186/s12974-018-1100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Busskamp V, Lewis NE, Guye P, Ng AHM, Shipman SL, Byrne SM, Sanjana NE, Murn J, Li Y, Li S, Stadler M, Weiss R, Church GM, Rapid neurogenesis through transcriptional activation in human stem cells. Molecular Systems Biology. 10, 760 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galvin JE, Schuck TM, Lee VMY, Trojanowski JQ, Differential Expression and Distribution of α-, β-, and γ-Synuclein in the Developing Human Substantia Nigra. Experimental Neurology. 168, 347–355 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM-Y, Synucleins Are Developmentally Expressed, and α-Synuclein Regulates the Size of the Presynaptic Vesicular Pool in Primary Hippocampal Neurons. The Journal of Neuroscience. 20, 3214 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, Shao Q, Wright G, Bader GD, Morris Q, The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Research. 38, W214–W220 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volpicelli-Daley LA, Luk KC, Lee VM-Y, Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite–like aggregates. Nature Protocols. 9, 2135–2146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fahn S, Elton R, members U, in Recent developments in Parkinson’s disease, Fahn S, Marsden C, Goldstein M, Calne D, Eds. (Macmillan Healthcare Information, Florham Park, NJ, 1987), pp. 153–163. [Google Scholar]
- 28.Posavi M, Diaz-Ortiz M, Liu B, Swanson CR, Skrinak RT, Hernandez-Con P, Amado DA, Fullard M, Rick J, Siderowf A, Weintraub D, McCluskey L, Trojanowski JQ, Dewey RB Jr, Huang X, Chen-Plotkin AS, Characterization of Parkinson’s disease using blood-based biomarkers: A multicohort proteomic analysis. PLOS Medicine. 16, e1002931– (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplementary References for Materials and Methods:
- 29.Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, Grossman M, van Deerlin VM, Trojanowski JQ, Lee VM-Y, Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Human Molecular Genetics. 17, 1349–1362 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toledo JB, van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL, Xie SX, McBride J, Wood EM, Schuck T, Irwin DJ, Gross RG, Hurtig H, McCluskey L, Elman L, Karlawish J, Schellenberg G, Chen-Plotkin A, Wolk D, Grossman M, Arnold SE, Shaw LM, Lee VM-Y, Trojanowski JQ, A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer’s & Dementia. 10, 477–484.e1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrews S, FastQC: A Quality Control Tool for High Throughput Sequence Data [online] (2010), (available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
- 32.Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A, Gingeras TR, in Data Mining Techniques for the Life Sciences, Carugo O, Eisenhaber F, Eds. (Springer New York, New York, NY, 2016; 10.1007/978-1-4939-3572-7_13), pp. 245–262. [DOI] [Google Scholar]
- 34.van de Geijn B, McVicker G, Gilad Y, Pritchard JK, WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nature Methods. 12, 1061–1063 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, Morgan M, VariantAnnotation : a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics. 30, 2076–2078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlson M, Maintainer BP, TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation package for TxDb object(s) (2015).
- 37.Benjamini Y, Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society B. 57, 289–300 (1995). [Google Scholar]
- 38.Wallace C, Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLOS Genetics. 16, e1008720– (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB, Abundant associations with gene expression complicate GWAS follow-up. Nature Genetics. 51, 768–769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, v Prabhu A, Fernandopulle MS, Patel R, Abshari M, Ward ME, Kampmann M, CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron. 104, 239–255.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danielson E, Perez de Arce K, Cimini B, Wamhoff E-C, Singh S, Cottrell JR, Carpenter AE, Bathe M, eneuro, in press, doi: 10.1523/ENEURO.0286-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee BR, Kamitani T, Improved Immunodetection of Endogenous α-Synuclein. PLOS ONE. 6, e23939– (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ewels P, Magnusson M, Lundin S, Käller M, MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li B, Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu A, Ibrahim JG, Love MI, Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 35, 2084–2092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blighe K, Rana S, Lewis M, EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling (2021), (available at https://github.com/kevinblighe/EnhancedVolcano).
- 48.Russo PST, Ferreira GR, Cardozo LE, Bürger MC, Arias-Carrasco R, Maruyama SR, Hirata TDC, Lima DS, Passos FM, Fukutani KF, Lever M, Silva JS, Maracaja-Coutinho V, Nakaya HI, CEMiTool: a Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinformatics. 19, 56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langfelder P, Zhang B, Horvath S, Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 24, 719–720 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP, Molecular signatures database (MSigDB) 3.0. Bioinformatics. 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sergushichev AA, An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv, 060012 (2016). [Google Scholar]
- 52.Zigdon H, Savidor A, Levin Y, Meshcheriakova A, Schiffmann R, Futerman AH, Identification of a Biomarker in Cerebrospinal Fluid for Neuronopathic Forms of Gaucher Disease. PLOS ONE. 10, e0120194– (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kramer G, Wegdam W, Donker-Koopman W, Ottenhoff R, Gaspar P, Verhoek M, Nelson J, Gabriel T, Kallemeijn W, Boot RG, Laman JD, Vissers JPC, Cox T, Pavlova E, Moran MT, Aerts JM, van Eijk M, Elevation of glycoprotein nonmetastatic melanoma protein B in type 1 Gaucher disease patients and mouse models. FEBS Open Bio. 6, 902–913 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tropea TF, Amari N, Han N, Rick J, Suh E, Akhtar RS, Dahodwala N, Deik A, Gonzalez-Alegre P, Hurtig H, Siderowf A, Spindler M, Stern M, Thenganatt MA, Weintraub D, Willis AW, van Deerlin V, Chen-Plotkin A, Whole Clinic Research Enrollment in Parkinson’s Disease: The Molecular Integration in Neurological Diagnosis (MIND) Study. Journal of Parkinson’s Disease. 11, 757–765 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pigott K, Rick J, Xie SX, Hurtig H, Chen-Plotkin A, Duda JE, Morley JF, Chahine LM, Dahodwala N, Akhtar RS, Siderowf A, Trojanowski JQ, Weintraub D, Longitudinal study of normal cognition in Parkinson disease. Neurology. 85, 1276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lewis CM, Genetic association studies: Design, analysis and interpretation. Briefings in Bioinformatics. 3, 146–153 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All transcriptome data have been deposited in NCBI’s Gene Expression (GEO dataset GSE206327). Human data are available upon request subject to a data use agreement to ensure maintenance of personal privacy. Requests for data or materials should be addressed to chenplot@pennmedicine.upenn.edu.