

ABSTRACT
Background
Mitochondrial functions are controlled by genes of both mitochondrial and nuclear DNA. Pathogenic variants affecting any of these are responsible for primary mitochondrial disorders (MIDs), which can be diagnosed during adulthood. Kidney functions are highly dependent on mitochondrial respiration. However, the prevalence of MID-associated nephropathies (MIDANs) is unknown in the adult population. We aimed to address this point and to provide a full characterization of MIDANs in this population.
Methods
We retrospectively included for observational study adults (≥16 years of age) with genetically diagnosed MID between 2000 and 2020 in our tertiary care academic centre when they had a chronic kidney disease (CKD) evaluation. MIDANs were ascertained by CKD occurring in MIDs. The phenotypic, biological, histopathological and genotypic characteristics were recorded from the medical charts.
Results
We included 80 MID-affected adults and ascertained MIDANs in 28/80 (35%). Kidney diseases under the care of a nephrologist occurred in only 14/28 (50%) of the adults with MIDAN. MIDANs were tubulointerstitial nephropathy in 14/28 patients (50%) and glomerular diseases in 9/28 (32.1%). In adults with MID, MIDAN was negatively associated with higher albumin levels {odds ratio [OR] 0.79 [95% confidence interval (CI) 0.67–0.95]} and vision abnormalities [OR 0.17 (95% CI 0.03–0.94)] and positively associated with hypertension [OR 4.23 (95% CI 1.04–17.17)].
Conclusion
MIDANs are frequent among adult MIDs. They are mostly represented by tubulointerstitial nephropathy or glomerular disease. Vision abnormalities, hypertension and albumin levels were independently associated with MIDANs. Our results pave the way for prospective studies investigating the prevalence of MIDANs among undetermined kidney disease populations.
Keywords: adult, glomerular disease, kidney disease, mitochondrial disorders, tubulointerstitial nephropathy
Graphical Abstract
Graphical Abstract.

INTRODUCTION
Mitochondria integrate essential cell functions, including the regulation of reactive oxygen species, apoptosis signalling and energy production through oxidative phosphorylation (OXPHOS) by the electron transport chain [1]. These functions are dependent on both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) [2, 3]. As a consequence, defects in either nDNA or mtDNA can lead to primary mitochondrial disorders (MIDs) [4]. Mitochondrial genetics rules include maternal inheritance, but de novo mtDNA pathogenic variants are common [5, 6]. It is now clear that MIDs are also considered an adulthood disease, with an estimated prevalence of 1 in 4300 [7]. Mitochondria are present in all nucleated cells. The coexistence of two or more different mtDNA populations (e.g. wild-type and pathogenic variants) in an individual defines heteroplasmy. There is a heteroplasmic threshold above which a mitochondrial defect becomes clinically relevant, depending on the extent to which the affected tissue relies on OXPHOS [8]. In line with this, all organs can be affected by MIDs, with susceptibility depending in part on energy consumption [9].
The kidneys are second only to the heart in terms of energy consumption, to ensure podocyte functions in the glomerular filtration barrier and to guarantee tubular epithelial cell reabsorption [10–12]. A considerable number of case reports or small series describe MID-associated nephropathies (MIDANs) in an adult population. Most of them are tubulointerstitial nephropathy and glomerular diseases with focal segmental glomerulosclerosis (FSGS) [13–20]. Clinical manifestations are heterogeneous, often referring to systemic manifestations such as mitochondrial encephalopathy, lactic acidosis, stroke-like episode syndrome (MELAS) [15, 17] and maternal inherited diabetes and deafness syndrome [21], deafness, developmental disabilities and cardiomyopathies [16, 22]. Nevertheless, a significant number of different types of MIDANs have been isolated at diagnosis [16, 18, 23]. The incidence and prevalence of MIDANs, as well as their clinicopathological characteristics among the adult population, need to be clarified. Indeed, existing adult cohorts were limited to specific syndromes and mutations [e.g. maternally inherited deafness and diabetes (MIDD)] or did not use physician bedside definitions applicable in practice (e.g. proteomic characterization of MIDANs) [24, 25].
This study aimed to establish the prevalence of MIDANs among an adult population with genetically diagnosed MID in different departments of our academic tertiary care hospital; to characterize MIDANs with clinical, biological and histopathological descriptions; and to investigate the association between MIDAN outcome and MID characteristics.
MATERIALS AND METHODS
Study population
Adult patients (≥16 years of age) with a genetically confirmed MID diagnosis made at the University Hospital of Bordeaux between January 2000 and January 2020 were screened. We included patients with a chronic kidney disease (CKD) evaluation defined by two or more measurements ≥3 months apart of serum creatinine, blood electrolytes, urinalysis (quantification and composition of proteinuria, leukocyturia and haematuria) and at least one kidney imaging [26]. Exclusion criteria were evidence for a kidney disease with another aetiology, unavailability of the medical records and the patient's refusal to participate. The follow-up period included the period from the genetic confirmation of the MID to the last kidney disease assessment. Patients were censored if they died or were lost to follow-up. Our study population included unrelated patients. This study was approved by the local institutional review board (CE-GP-2020-32) and followed the Declaration of Helsinki and the Declaration of Istanbul.
MID ascertainment
Our criteria included adults exhibiting compatible MID symptoms and genetic confirmation of a mutation known to cause MID. Molecular genetic testing of mtDNA was performed on muscle biopsy when available, and on urine and/or blood samples using long-range mtDNA polymerase chain reaction covering the whole mtDNA, followed by next-generation sequencing (NGS) on Ion Torrent Personal Genome Machine (Thermo Fisher Scientific, Waltham, MA, USA) (see Supplemental Methods) or by a conventional targeted method (Sanger sequencing or restriction fragment length polymorphism). Individuals with mtDNA pathogenic variants (point mutations, duplications or single deletions) were included. Pathogenicity was defined according to the American College of Medical Genetics/Association of Molecular Pathology standards and guidelines for mtDNA pathogenic variant interpretation [27]. We included those with multiple mtDNA deletions if muscle clinical features were consistent with MID and if they met the following criteria: >3% of cyclooxygenase (COX)-deficient fibres in biochemical analysis and the absence of evidence of other muscle pathology. Molecular genetic testing of nDNA pathogenic variants in genes coding for mitochondrial functions was based on target genes analysis in other French reference centres for mitochondrial disorders, using Sanger sequencing or NGS at the behest of the physician.
MIDAN definition
We broadly defined MIDAN in our study as a CKD, i.e. abnormalities of kidney structure or function >3 months, according to the Kidney Disease: Improving Global Outcomes (KDIGO) guidelines [26], occurring in a MID-affected patient, without evidence for another cause. The only way to assess a pathological connection between MID and kidney disease is to confirm a genetically defective OXPHOS function in nephron segment cells. We then assumed that kidney diseases could also be the consequence of an indirect consequence of MID (e.g. diabetes mellitus, cardiorenal syndrome).
Glomerular filtration rate (GFR) was estimated from creatinine serum dosages and Chronic Kidney Disease Epidemiology Collaboration formula [28]. We defined as ‘underappreciated kidney disease’, any kidney disease that was not under the care of a nephrologist before the study. To this purpose, we telephoned each included living patient, or his former attending clinician if the patient was dead, and we asked if the patient was under the care of a nephrologist.
We distinguished different CKDs based on analyses of blood (creatinine, urea and blood electrolytes), urinalyses (proteinuria, urine sediment abnormalities, renal excretion of electrolytes), imaging (echography or abdominal scanner) and histopathological findings, including proximal convoluted tubulopathy (Fanconi syndrome); other tubulopathy (evidence for Bartter-like or Gitelman-like syndromes); tubulointerstitial nephropathy, defined as evidence of tubulointerstitial damage (tubular proteinuria, aseptic leukocyturia) in the absence of evidence for Fanconi, Bartter-like or Gitelman-like syndrome [29]; glomerular disease, then classified as FSGS, or another diagnosis regarding histological lesions in kidney biopsies and congenital anomalies of the kidney and urinary tract (CAKUT).
Renal biopsy tissues were checked by two independent nephropathologists: the first analysis was performed at the time of diagnosis (S.L.) and was reviewed for the study (A.V.). Tubulointerstitial changes and arteriolar hyaline thickening were scored following the Banff lesion score 2017 [30].
Clinical, biological and histopathological assessments
Non-renal manifestations were hypertension, defined as systolic blood pressure ≥140 mmHg and/or the use of antihypertensive drugs; muscular affliction, defined as subjective exercise intolerance and/or an objective muscular change and/or an electromyogram myogenic abnormality and/or histopathological abnormalities based on open biopsies performed and interpreted by the same pathologist (M.L.M.N); hypertrophic and dilated cardiomyopathy assessed by echocardiography; and retinal abnormalities, Leber's optic neuropathy and macular dystrophy assessed by fundus, optical coherence tomography and fluorescein angiography. We checked for age, sex, height, weight, seizures, ataxia, stroke-like episodes, encephalopathy, vision abnormalities, ophthalmoparesis, ptosis, audiogram hearing impairment, liver and gastrointestinal afflictions, diabetes mellitus and biological data in the medical records.
Phenotypes were assessed in PhenoTips (Gene42, Toronto, ON, Canada) using Human Phenotype Ontology (HPO) terms. HPO terms relative to clinical data appear in different tables. A ‘>’ following the HPO term accounts for all phenotypes in the subtree of the corresponding HPO term. A phenotypic similarity matrix was performed in Python (https://www.python.org/) using the HPO terms [31]. Each sample was defined by a vector of n HPO terms. The Resnik similarity between two samples has been computed as a semantic distance between their vectors using the library ssmpy [32]. The pairwise matrix was then computed to produce a clustered heatmap where each cell represents the distance between two samples.
Statistical analyses
Quantitative values are presented as median [1st–3rd quartile (IQR)]. Qualitative data are presented as numbers (percentages). Quantitative variables between groups were compared with the Wilcoxon test and qualitative variables were compared with the chi-squared test. P-values <.05 were considered significant. Univariate and multivariable logistic regression analyses were used to estimate odds ratios (ORs) for independent variables associated with the occurrence of MIDAN. Variables with P < .25 in the univariate analysis were considered in the multivariable analysis using a backward selection procedure. The best-fitting model was defined according to the Akaike information criterion (AIC). The significance of the parameters was evaluated with the Wald test and the likelihood test. Factors displaying a P-value <.05 in the multivariable analysis were considered to be independently associated with the tested outcome. Missing data were addressed with complete case analyses. Results are presented with their 95% confidence intervals (CIs). Statistical analyses were performed with R version 4.0.1 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
Characteristics of MID-affected adults
Among 134 genetically confirmed MIDs, 116 were adults and 80 were included in our study (Fig. 1). The median follow-up was 26 months (IQR 12–36); 25/80 (31.2%) were lost to follow-up. Twelve deaths (15% of patients) not related to kidney disease occurred during the follow-up (Supplemental Table S1).
Figure 1:
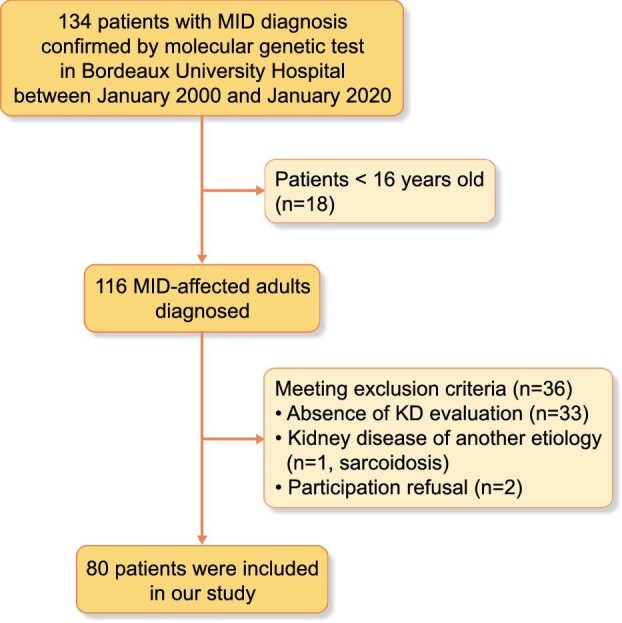
Flow chart of the studied MID adult population.
The baseline clinical characteristics of the study population at the MID diagnosis are presented in Supplemental Table S2. The median age at diagnosis was 52 years (IQR 34–64). Abnormalities of the nervous system (e.g. morphological abnormalities, seizures) were described in most cases [61/80 (76.2%)], followed by abnormalities of the eyes (e.g. abnormalities of vision, Leber's optic neuropathy) [57/80 (71.2%)] and abnormalities of the cardiovascular system (e.g. hypertension, hypertrophic cardiomyopathy) [44/80 (55%)]. MIDANs, defined as CKD according to KDIGO guidelines associated with genetically confirmed MID, accounted for 28/80 (35%) of MID-affected adults with kidney disease assessment, ranking fourth among MID manifestations.
MID genotypic features are presented in Supplemental Table S3. A large proportion of MID was due to mtDNA pathogenic variants [56/80 (70%)], mostly a consequence of point mutations [43/80 (53.8%)].
MIDANs are frequent and underappreciated in MID-affected adults
MIDANs were ascertained in 28 MID-affected adults and their characteristics are listed in Table 1. We noted that 14/28 (50%) patients with MIDANs were under the care of a nephrologist (e.g. kidney disease that had been detected before the study and under the care of a nephrologist; see corresponding methods section). We also observed that 9/28 (32.1%) were in end-stage renal disease (ESRD): 5/28 (17.9%) received dialysis before a MID diagnosis and 3/28 (10.7%) during the year after a MID diagnosis (Table 1).
Table 1:
Description of patients presenting with MIDANs.
| Characteristics | MIDANs (N = 28) |
|---|---|
| Demographics | |
| Age at diagnosis (years), median (IQR) | 52 (34–64) |
| Sex (male/female), n/n | 19/9 |
| BMI (kg/cm2), median (IQR) | 24 (20–27) |
| Kidney disease, n (%) | |
| Tubulointerstitial nephropathy | 14 (50) |
| Documented by kidney biopsy | 6 (21.4) |
| Glomerular disease | 9 (32.1) |
| FSGS | 3 (10.7) |
| Nephrotic syndrome | 3 (10.7) |
| Documented by kidney biopsy | 3 (10.7) |
| CAKUT | 4 (14.3) |
| Kidney cysts | 2 (7.1) |
| Urinary tract abnormalities | 2 (7.1) |
| Fanconi syndrome | 2 (7.1) |
| Other tubulopathy | 2 (7.1) |
| Kidney disease under the care of a nephrologist | 14 (50) |
| ESRD | 9 (32.1) |
| Transplantation | 4 (14.3) |
| Biological characteristicsa | |
| Urinalysis | |
| Protein/creatinine (g/g), median (IQR) | 0.6 (0.36–2.07) |
| Albumin/creatinine (g/g), median (IQR) | 0.37 (0–1.82) |
| Haematuria, n (%) | 1 (3.6) |
| Leukocyturia, n (%) | 12 (42.9) |
| Glycosuria, n (%) | 1 (3.6) |
| Blood ionogram, median (IQR) | |
| Calcium (mg/dl) | 9.3 (8.2–9.7) |
| Phosphorus (mg/dl) | 3.7 (3.4–4.3) |
| Sodium (mmol/l) | 139 (137–141) |
| Chloride (mmol/l) | 103.5 (101–106) |
| Glycemia (mg/dl) | 98.5 (88.3–108.1) |
| Potassium (mmol/l) | 4.41 (3.9–4.7) |
| Proteins (g/l) | 70.5 (64.75–74) |
| Albumin (g/l) | 38.1 (34.7–42.5) |
| Increased uric acid (female >5 mg/dl; male >6 mg/dl), n (%) | 11 (39.3) |
| GFR at diagnosis (ml/min/1.73 m2), median (IQR) | 60 (27–84) |
| GFR slopeb (ml/min/1.73 m2/year), median (IQR) | −3 (−6–0) |
BMI: body mass index; CPK: creatine phosphor-kinase.
aBiological characteristics at the time of MIDAN diagnosis.
bGFR slope during a median observation period of 26 months.
Conversion factors for units: serum calcium in mg/dl to mmol/l, × 0.2495; serum phosphorus in mg/dl to mmol/l, × 0.3229; serum glucose in mg/dl to mmol/l, × 0.05 551; uric acid in mg/dl to µmol/l, × 59.58.
Among the patients with MIDANs, 14 (50%) experienced tubulointerstitial nephropathy, 9 (32.1%) glomerular disease, 4 (14.3) CAKUT, 2 (7.1%) Fanconi syndrome and 2 (7.1%) other tubulopathies. We observed an overlap, as three patients with CAKUT also had tubulopathies. We subsequently focused on describing tubulointerstitial nephropathy and glomerular diseases, which were overrepresented.
Tubulointerstitial nephropathy was documented by kidney biopsy in 6/28 (21.4%) patients. Four of these biopsies were reviewed and none exhibited granular swollen epithelial cells or hypertrophied podocytes (Fig. 2A and Table 2). An electron microscopy (EM) analysis was performed on one specimen and showed an increase in the number of swollen mitochondria with loss of the cristae structure inside the kidney tubular epithelial cells (Fig. 2B and 100).
Figure 2:

Histopathological manifestations related to MIDAN. (A) Pathology study by optical microscopy. Magnification ×10, Masson's trichrome staining. Global aspect of severe interstitial fibrosis and tubular atrophy. (B, C) Pathological electron microscopic studies detected abnormal tubular mitochondria that were swollen, with loss of cristae structures. (D) Pathology study by optical microscopy. Magnification ×20, Masson's trichrome staining. Glomerulus presented segmental lesions (e.g. affecting only a portion of the glomerular tuft) that associated proliferation and hypertrophy of podocytes and synechiae between the Bowman's capsule and the tuft. These lesions are compatible with FSGS. (E) Pathology study by optical microscopy. Magnification ×20, Masson's trichrome staining. Glomerulus presented with hypertrophied podocyte lesions (black arrows), but without FSGS lesion.
Table 2:
Histopathological characteristics of reviewed kidney biopsies.
| MIDAN | Glomeruli, n | Global sclerosis, n (%) | Segmental sclerosis | Tubulointerstitial fibrosis, n | AH, n | GSEC | PH | Swollen mitochondria |
|---|---|---|---|---|---|---|---|---|
| TIN | 12 | 0 (0) | − | 3 | 1 | − | − | + |
| TIN | 11 | 5 (37) | − | 3 | 0 | − | − | N/A |
| TIN | 5 | 3 (60) | − | 3 | 0 | − | − | N/A |
| TIN | 15 | 9 (60) | − | 3 | 2 | − | − | N/A |
| GD | 10 | 10 (1) | + | 0 | 0 | − | + | N/A |
| GD | 14 | 5 (36) | + | 2 | 3 | − | − | N/A |
| GD | 2 | 1 (50) | + | 3 | 0 | − | − | N/A |
AH: arteriolar hyaline thickening; GD: glomerular disease; GSEC: granular swollen epithelial cells; N/A/: not available; PH: podocyte hypertrophy; TIN: tubulointerstitial nephropathy; −: negative; + positive. Grading of severity for tubulointerstitial fibrosis and arteriolar hyaline thickening followed the 2017 Banff Lesion Score [30].
Glomerular disease was documented by kidney biopsy in 3/28 (10.7%) patients. The histopathological findings were FSGS for all three patients (Fig. 2D and E, Table 2). One kidney specimen exhibited hypertrophied podocytes, with no possibility to perform EM. Of note, none of the patients who experienced glomerular disease had haematuria and 3/28 (10.7%) suffered from nephrotic syndrome (Table 1). The MID-affected adults with glomerular disease were more likely to suffer from abnormalities of the endocrine system, such as diabetes mellitus, and to carry the MT-TL1 m.3243A>G pathogenic variants than patients presenting with tubulointerstitial nephropathy (Supplemental Table S4; P = .038 and P = .010, respectively).
Clinical, biological and genotypic features associated with MIDANs
The baseline phenotypic, biological and genotypic characteristics of the study population according to the occurrence of MIDANs are presented in Supplemental Table S5, Supplemental Table S6 and Supplemental Table S7, respectively. In our cohort, MID-affected patients who suffered from abnormality of the myocardium, such as hypertrophic and dilated cardiomyopathy, and hypertension were more likely to have MIDANs (P = .008, P = .024, P = .018 and P = .001, respectively). The same association was observed for patients with diabetes mellitus (P = .038). Conversely, patients with MIDANs seemed less affected by abnormalities of vision (P = .037). Biological analyses highlighted lower calcium and albumin concentrations in patients with MIDANs (P = .040 and P = .004). Genetic analyses showed that the MIDAN population experienced more frequent mtDNA point mutations (P = .020).
The multivariable model with the best AIC for MIDANs among MID-affected patients integrated the following variables: gender, hypertension, abnormality of vision and albuminemia (Table 3). After adjustment to the other variables, multivariable analysis with logistic regression identified abnormality of vision [OR 0.17 (95% CI 0.03–0.94), P = .042] and arterial hypertension [OR 4.23 (95% CI 1.04–17.17), P = .044] as independently associated with MIDAN. Among the biological characteristics, higher serum albumin level was negatively associated with MIDANs [OR 0.79 (95% CI 0.67–0.95), P = .010].
Table 3:
Factors associated with MIDAN.
| Univariate analysis | Multivariable analysis | |||||
|---|---|---|---|---|---|---|
| Factors | OR | 95% CI | P-valuea | OR | 95% CI | P-valueb |
| Phenotypic characteristics | ||||||
| Age (years) | 1 | 0.98–1.03 | .836 | |||
| Sex (women as reference) | 1.43 | 0.54–3.76 | .469 | 0.47 | 0.1–2.15 | .332 |
| Abnormality of the myocardium (0 001 637>) | 3.73 | 1.38–10.1 | .010 | |||
| Hypertrophic cardiomyopathy (0 001 639> and 0 001 714>) | 3.63 | 1.13–11.62 | .030 | |||
| Dilated cardiomyopathy (001 644) | 11.09 | 1.23–100.34 | .032 | |||
| Hypertension (0 000 822>) | 4.97 | 1.83–13.53 | .002 | 4.23 | 1.04–17.17 | .044 |
| Abnormality of vision (0 000 504>) | 0.31 | 0.11–0.85 | .023 | 0.17 | 0.03–0.94 | .042 |
| Ophthalmoparesis (0 000 597>) | 0.09 | 0.01–0.73 | .024 | |||
| Ptosis (0 000 508>) | 0.12 | 0.02–1.01 | .051 | |||
| Diabetes mellitus (0 000 819>) | 3.06 | 1.04–8.99 | .043 | |||
| Biological characteristics | ||||||
| Calcium (mg/dl) | 0.02 | 0–0.73 | .033 | |||
| Albuminemia (g/l) | 0.82 | 0.71–0.93 | .003 | 0.79 | 0.67–0.95 | .01 |
| Increased uric acid (female >5 mg/dl; male >6 mg/dl) | 14.67 | 3.29–65.29 | <0.001 | |||
| Genotypic characteristics | ||||||
| Point mutations | 3.15 | 1.18–8.45 | .022 | |||
| Single deletions | 0.16 | 0.02–1.28 | .084 | |||
HPO terms are in parentheses. The ‘>’ refers to the preceding HPO term associated with the subtree HPO terms.
aLogistic regression and
bbackward multivariable analysis including factors with P < .2 in the univariate analysis.
We performed a phenotypic similarity matrix according to HPO terms for every MID-affected adult associated with his genetic diagnosis and observed three phenotypic clusters, as shown in Fig. 3. We then studied patients’ phenotypic characteristics and compared every cluster (Supplemental Table S8). The majority of patients with MIDANs were part of cluster 3, with 11/20 (55%). These patients were more likely to have hypertension [14/20 (70%)], no abnormality of vision [1/20 (5%)] and lower albumin levels [37.5 g/l (IQR 35.4–42.3)].
Figure 3:

Phenotypic similarity matrix of MID-affected adults related to their genetic diagnosis. The matrix was colour-coded according to the phenotypic similarity score from 0 (black, absence of similarity) to 5 (light red-orange, very similar). Ontological subtrees and clusters were obtained according to HPO term semantics. Adults with MIDAN are coloured in green, whereas non-renal MID patients are in red. We were able to identify three clusters of patients with great phenotypic similarities based on HPO term semantics. For clarity, we numbered these clusters 1–3, identified with parentheses.
DISCUSSION
Our study is among the first to evaluate the prevalence of MIDANs in a large population of MID-affected adults. We showed that patients presenting with MIDAN accounted for 35% of this population. This cohort is one of the largest MID-affected adult populations analysed, providing robustness to our findings. Furthermore, we did not restrict our study population to specific mutations or syndromic manifestations to benefit from a heterogeneous, representative adult population of MIDs based on a genetic diagnosis. The same proportions have been reported in more restricted populations, such as in MIDD syndrome (28–46%) [21, 24] and MELAS cohorts (39%) [20].
The definitions of MIDANs we used correspond to physician bedside practice, involving routinely performed analyses. Nevertheless, with routine clinical definitions of MIDAN types, we found the same proportion of kidney disease as Hall et al. [25], who screened 117 MID-affected adults using proteomic and metabolomic urinalyses and observed tubular afflictions in 39% and glomerular afflictions in 31% of patients. Considering the high frequency of low muscle mass in patients suffering from neuromuscular manifestations, estimating GFR using serum creatinine clearance equations may fail to assess kidney disease correctly [25]. Indeed, Imasawa et al. [22] recently provided evidence through a cohort of 81 patients with MIDAN showing that proteinuria is more common than decreased GFR. Thus great care was taken regarding blood analyses of electrolytes and urinalyses, both available for all included patients, to ensure that MIDANs were not misestimated. We believe that our approach based on simple clinical and biological data is the most relevant for day-to-day practice.
We showed that MIDANs in our adult population were predominantly represented by tubulointerstitial nephropathy (50%) and glomerular disease (32.1%), which is consistent with reported cases [15–18, 20, 25]. It should be noted that none of the patients presented with haematuria and a minority presented with nephrotic syndrome. These are huge differences compared with the paediatric population in which specific tubulopathies (e.g. Fanconi, Gitelman-like and Bartter-like tubulopathies) and nephrotic syndrome are predominant [22, 33, 34]. The metabolism of the renal tubular segments is highly dependent on OXPHOS to ensure ion homeostasis [12]. Because the apical membrane of epithelial cells is impermeable to ions, basolateral Na+/K+ ATPases use the energy from adenosine triphosphate (ATP) hydrolysis to create an electrochemical gradient, allowing apical reabsorption and secretion through specific channels and transporters. As such, it is not surprising that we can observe numerous mitochondria all along the tubular epithelial cells. Tubulointerstitial nephropathy could be the consequence of subclinical tubular manifestations evolving to fibrosis and CKD through various phenomena mediated by mitochondria dysfunctions (e.g. epithelial– and endothelial–mesenchymal transition) [35].
Glomerular diseases, such as FSGS, are the consequence of alteration of the glomerular filtration barrier. Podocytes have a central role in maintaining the integrity of this structure, including structural support, maintenance of glomerular capillary hydrostatic pressure, glomerular basement membrane synthesis, intercellular crosstalk [e.g. vascular endothelial growth factor (VEGF) secretion that acts on the VEGF receptors of endothelial cells], slit diaphragm protein synthesis (nephrin and podocin) and permselectivity modulation through actin cytoskeleton interactions with slit diaphragm proteins [36]. These functions require ATP synthesis by the mitochondrial respiratory chain and thus the intracellular importation and oxidation of carbohydrates, fatty acids and amino acids [11]. Glomerular diseases observed in MID-affected patients are thus the direct consequence of energy inadequacy within podocytes, which compromises the glomerular filtration barrier. Indeed, numerous studies have shown the relation between MID and glomerular affections both in paediatric and adult populations [15, 20, 22, 34]. The prevalence of glomerular disease and tubulointerstitial nephritis has to be mitigated, as the two types can merge. Tubulointerstitial damage could lead to glomerular lesions. Our clinical and biological definition helped us to divide these MIDANs for bedside practice.
We have defined MIDANs as CKDs occurring in MID-affected adults in the absence of any evidence of another cause of kidney disease. We have to keep in mind that MID is a systemic disease and that MIDAN could potentially be a consequence of another MID-affected organ (e.g. cardiorenal syndrome, diabetes mellitus nephropathy, hypertensive nephropathy) that may induce indirect renal damage. Researchers have provided evidence that kidney diseases are more likely to be MIDANs rather than diabetes mellitus complications [21, 24]. In contrast, analysis of a large population of patients with MIDANs underscored the fact that 10% of biopsied patients presented diabetic nephropathy lesions [22]. It might be challenging to ascertain the pathogenic connection between MID and kidney disease occurrence. The high proportion of kidney disease in our cohort is indirect evidence of this connection. Massin et al. [24] showed that kidney disease is more frequent and more severe in MID-affected adults with MIDD syndrome than in patients with diabetes mellitus without MID. This is reinforced by other studies that found the same proportion of MIDANs in both adult and paediatric populations [20, 21, 25, 33]. We believe that mitochondrial abnormalities observed in EM analysis of kidney biopsies provide further evidence [15, 16]. Last but not least, we highlighted mtDNA pathogenic variants at a high level of heteroplasmy using NGS on kidney biopsy specimens [16]. Extensive studies using this method might be of great interest to assess the pathogenic connection of MIDANs. Another interesting method to develop would be the use of biopsied renal tissue to measure OXPHOS enzyme activity on kidney cells to ascertain MIDANs. Pragmatically, direct or indirect renal damage in the context of MIDAN does not change the fact that kidney diseases are frequent in MID-affected adults and that nephrologist referral is necessary to initiate supportive care and follow-up.
We have emphasized that MIDANs were underappreciated, as only 50% of patients with MIDANs were under the care of a nephrologist at the time of the study. Martìn-Hernandez et al. [33] screened 42 MID paediatric patients for kidney disease and found that 13/21 (63%) with MIDAN had mild subclinical tubular impairment, thus they were undiagnosed and not under the care of a nephrologist. Kidney involvement is not in the foreground of multisystemic manifestations, so it might go unnoticed and underappreciated [16]. Nevertheless, a delayed MID diagnosis and MIDAN referral to nephrologists could have serious consequences, as we observed that 32.1% of patients with MIDAN reached ESRD and 88.9% developed ESRD before or during the first year following the MID diagnosis. Parikh et al. [37] evaluated 35 solid organ transplant recipients and showed that MID was diagnosed after transplantation for 50% of them. These data must of course be confirmed by larger prospective studies; however, we encourage clinicians to systematically screen MID-affected patients for kidney disease (analysing serum creatinine, ionic abnormalities and dipstick for proteinuria, haematuria and leukocyturia) and to refer patients with potential MIDAN to nephrologists. Indeed, an early MIDAN diagnosis is mandatory to initiate supportive care and aerobic exercise conditioning to potentially slow down the progression of kidney disease and MID [38].
Following the best AIC multivariable model after adjusting on numerous MID features and analysing an interesting phenotypic similarity matrix using HPO terms to group MID-affected patients related to their genetic diagnosis, we proved the robustness of this interesting association with MIDANs. We demonstrated that abnormality of vision was negatively associated with the occurrence of MIDANs. This could be a consequence of ophthalmological or neurological manifestations (e.g. Leber's optic neuropathy) preceding MIDAN, or of recruitment bias. Albuminemia levels were also associated with MIDAN, probably as a consequence of albumin loss during glomerular disease. More interestingly, hypertension was positively associated with MIDANs. It is well established that the kidney is both a cause and a victim of high blood pressure [39]. Hypertension has been demonstrated to be responsible for altered OXPHOS in endothelial cells [40], podocytes [41] and tubular epithelial cells [42]. But mitochondrial dysfunctions are also related to promoting and maintaining hypertension through endothelial dysfunction (e.g. impairment of vasculature relaxation) and increasing proximal tubule sodium reabsorption [43]. Thus the association observed in our study could be the consequence of a vicious circle of mitochondrial dysfunctions, hypertension and MIDANs, each worsening the other. It is interesting to note that Imasawa et al. [22] showed that 10% of biopsied MIDANs had nephrosclerosis lesions. Interpreting such intricate findings is complex and requires a specially designed prospective evaluation. However, this finding enhances the importance of the prompt initiation of supportive care, such as antihypertensive drugs (e.g. renin–angiotensin system inhibitors) in MIDAN.
Our study was an observational retrospective study based on an examination of medical files and therefore has some limitations. It might have been underpowered, as 33/116 patients (28.4%) were excluded because they lacked a kidney disease assessment. The other limitation point was the absence of longitudinal data allowing us to better characterize the clinical course of MID and MIDAN. Although genetic diagnoses were available for each patient, numerous patients had mtDNA multiple deletions without nDNA analyses. The proportion of nDNA pathogenic variants, and the association with MIDANs, has probably been underestimated. Unfortunately, we were unable to evaluate the impact of heteroplasmy level on the occurrence of MIDANs, which would have been of great interest [44]. Variations in molecular techniques during the inclusion period and in the patients’ samples that were analysed made it unmatchable.
Overall, our results pave the way for further larger prospective studies that would aim to evaluate kidney disease among a MID adult population with systematic and longitudinal clinical, biological and pathological evaluations in order to develop a predictive score for the occurrence of MIDAN, to screen undetermined kidney disease (such as tubulointerstitial nephropathy and glomerular disease) for a MIDAN diagnosis using mtDNA NGS in urine samples to assess MIDAN epidemiology and to establish diagnostic scores to mentor clinicians.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge archives and medical secretaries for their valued assistance. We are grateful to Dr Sébastien Lepreux and the technicians of the pathology and genetic laboratories for their technical expertise.
Contributor Information
Hugo Bakis, CHU de Bordeaux, Service de Néphrologie, Transplantation, Dialyse et Aphérèses, Bordeaux, France; CHU de Bordeaux, Centre de Référence pour les Maladies Mitochondriales de l’Enfant à l’Adulte (CARAMMEL), Bordeaux, France.
Aurélien Trimouille, CHU de Bordeaux, Service de Génétique Médicale, Bordeaux, France; Université de Bordeaux, INSERM U1211, Bordeaux, France.
Agathe Vermorel, CHU de Bordeaux, Service de Néphrologie, Transplantation, Dialyse et Aphérèses, Bordeaux, France; CHU de Bordeaux, Service de Pathologie, Bordeaux, France.
Cyril Goizet, CHU de Bordeaux, Service de Génétique Médicale, Bordeaux, France; CHU de Bordeaux, Centre de Référence pour les Maladies Mitochondriales de l’Enfant à l’Adulte (CARAMMEL), Bordeaux, France; Université de Bordeaux, INSERM U1211, Bordeaux, France.
Yaniss Belaroussi, Université de Bordeaux, INSERM, Bordeaux Population Health Center, ISPED, Bordeaux, France; CHU de Bordeaux, Bordeaux, France; Institut Bergonié, INSERM CIC1401, Clinical and Epidemiological Research Unit, Bordeaux, France.
Sacha Schutz, CHU de Brest, Laboratoire de Génétique Moléculaire, Brest, France; Université de Brest, INSERM, EFS, UMR1078, GGB, Brest, France.
Guilhem Solé, CHU de Bordeaux, Département de Neurologie, Unité Nerf-Muscle, Bordeaux, France; CHU de Bordeaux, AOC National Reference Center for Neuromuscular Disorders, Bordeaux, France.
Christian Combe, CHU de Bordeaux, Service de Néphrologie, Transplantation, Dialyse et Aphérèses, Bordeaux, France; Tissue Bioengineering, U1026, INSERM, Bordeaux, France.
Marie-Laure Martin-Negrier, CHU de Bordeaux, Service de Génétique Médicale, Bordeaux, France; CHU de Bordeaux, Centre de Référence pour les Maladies Mitochondriales de l’Enfant à l’Adulte (CARAMMEL), Bordeaux, France; Université de Bordeaux, Institut des Maladies Neurodégénératives, Bordeaux, France; CNRS, Institut des Maladies Neurodégénératives, Bordeaux, France.
Claire Rigothier, CHU de Bordeaux, Service de Néphrologie, Transplantation, Dialyse et Aphérèses, Bordeaux, France; CHU de Bordeaux, Centre de Référence pour les Maladies Mitochondriales de l’Enfant à l’Adulte (CARAMMEL), Bordeaux, France; Tissue Bioengineering, U1026, INSERM, Bordeaux, France.
FUNDING
The authors have no funding to declare.
AUTHORS’ CONTRIBUTIONS
H.B., A.T., M.L.M.N. and C.R. designed the study and wrote the manuscript. A.T., A.V. and M.L.M.N. carried out the experiments. H.B., A.T and M.L.M.N. collected the data. H.B., A.T, Y.B., S.S., M.L.M.N. and C.R. analysed the data. A.V., G.S., C.C. and C.G. were involved in the critical revision of the manuscript. All authors approved the final version of the manuscript.
DATA AVAILABILITY STATEMENT
Data are available upon request.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to disclose.
REFERENCES
- 1. Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–12. 10.1126/science.281.5381.1309 [DOI] [PubMed] [Google Scholar]
- 2. Anderson S, Bankier AT, Barrell BGet al. Sequence and organization of the human mitochondrial genome. Nature 1981;290:457–65. 10.1038/290457a0 [DOI] [PubMed] [Google Scholar]
- 3. Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol 1988;4:289–331. 10.1146/annurev.cb.04.110188.001445 [DOI] [PubMed] [Google Scholar]
- 4. DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci 2008;31:91–123. 10.1146/annurev.neuro.30.051606.094302 [DOI] [PubMed] [Google Scholar]
- 5. Thorburn DR. Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis 2004;27:349–62. 10.1023/B:BOLI.0000031098.41409.55 [DOI] [PubMed] [Google Scholar]
- 6. Sallevelt SCEH, de Die-Smulders CEM, Hendrickx ATMet al. De novo mtDNA point mutations are common and have a low recurrence risk. J Med Genet 2017;54:73–83. 10.1136/jmedgenet-2016-103876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gorman GS, Schaefer AM, Ng Yet al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 2015;77:753–9. 10.1002/ana.24362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lightowlers RN, Chinnery PF, Turnbull DMet al. Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet 1997;13:450–5. 10.1016/S0168-9525(97)01266-3 [DOI] [PubMed] [Google Scholar]
- 9. DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med 2003;348:2656–68. 10.1056/NEJMra022567 [DOI] [PubMed] [Google Scholar]
- 10. Wang Z, Ying Z, Bosy-Westphal Aet al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr 2010;92:1369–77. 10.3945/ajcn.2010.29885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Imasawa T, Rossignol R.. Podocyte energy metabolism and glomerular diseases. Int J Biochem Cell Biol 2013;45:2109–18. 10.1016/j.biocel.2013.06.013 [DOI] [PubMed] [Google Scholar]
- 12. Wirthensohn G, Guder WG. Renal substrate metabolism. Physiol Rev 1986;66:469–97. 10.1152/physrev.1986.66.2.469 [DOI] [PubMed] [Google Scholar]
- 13. Parikh S, Goldstein A, Koenig MKet al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med 2015;17:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emma F, Montini G, Parikh SMet al. Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol 2016;12:267–80. 10.1038/nrneph.2015.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guéry B, Choukroun G, Noël L-Het al. The spectrum of systemic involvement in adults presenting with renal lesion and mitochondrial tRNA(Leu) gene mutation. J Am Soc Nephrol 2003;14:2099–108. 10.1097/01.ASN.0000080180.51098.02 [DOI] [PubMed] [Google Scholar]
- 16. Bakis H, Trimouille A, Vermorel Aet al. Adult onset tubulo-interstitial nephropathy in MT-ND5-related phenotypes. Clin Genet 2020;97:628–33. 10.1111/cge.13670 [DOI] [PubMed] [Google Scholar]
- 17. Jansen JJ, Maassen JA, van der Woude FJet al. Mutation in mitochondrial tRNA(Leu(UUR)) gene associated with progressive kidney disease. J Am Soc Nephrol 1997;8:1118–24. 10.1681/ASN.V871118 [DOI] [PubMed] [Google Scholar]
- 18. Connor TM, Hoer S, Mallett Aet al. Mutations in mitochondrial DNA causing tubulointerstitial kidney disease. PLos Genet 2017;13:e1006620. 10.1371/journal.pgen.1006620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tabebi M, Mkaouar-Rebai E, Mnif Met al. A novel mutation MT-COIII m.9267G>C and MT-COI m.5913G>A mutation in mitochondrial genes in a Tunisian family with maternally inherited diabetes and deafness (MIDD) associated with severe nephropathy. Biochem Biophys Res Commun 2015;459:353–60. [DOI] [PubMed] [Google Scholar]
- 20. Damian MS, Hertel A, Seibel Pet al. Follow-up in carriers of the ‘MELAS’ mutation without strokes. Eur Neurol 1998;39:9–15. 10.1159/000007892 [DOI] [PubMed] [Google Scholar]
- 21. Guillausseau PJ, Massin P, Dubois-LaForgue Det al. Maternally inherited diabetes and deafness: a multicenter study. Ann Intern Med 2001;134:721–8. 10.7326/0003-4819-134-9_Part_1-200105010-00008 [DOI] [PubMed] [Google Scholar]
- 22. Imasawa T, Hirano D, Nozu Ket al. Clinicopathologic features of mitochondrial nephropathy. Kidney Int Rep 2022;7:580–90. 10.1016/j.ekir.2021.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hotta O, Inoue CN, Miyabayashi Set al. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int 2001;59:1236–43. 10.1046/j.1523-1755.2001.0590041236.x [DOI] [PubMed] [Google Scholar]
- 24. Massin P, Dubois-Laforgue D, Meas Tet al. Retinal and renal complications in patients with a mutation of mitochondrial DNA at position 3,243 (maternally inherited diabetes and deafness). A case-control study. Diabetologia 2008;51:1664–70. 10.1007/s00125-008-1073-1 [DOI] [PubMed] [Google Scholar]
- 25. Hall AM, Vilasi A, Garcia-Perez Iet al. The urinary proteome and metabonome differ from normal in adults with mitochondrial disease. Kidney Int 2015;87:610–22. 10.1038/ki.2014.297 [DOI] [PubMed] [Google Scholar]
- 26. Kidney Disease: Improving Global Outcomes . CKD Evaluation and Management. https://kdigo.org/guidelines/ckd-evaluation-and-management/ (accessed29 May 2020). [Google Scholar]
- 27. McCormick EM, Lott MT, Dulik MCet al. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum Mutat 2020;41:2028–57. 10.1002/humu.24107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levey AS, Stevens LA, Schmid CHet al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–12. 10.7326/0003-4819-150-9-200905050-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perazella MA. Clinical approach to diagnosing acute and chronic tubulointerstitial disease. Adv Chronic Kidney Dis 2017;24:57–63. 10.1053/j.ackd.2016.08.003 [DOI] [PubMed] [Google Scholar]
- 30. Roufosse C, Simmonds N, Clahsen-van Groningen Met al. A 2018 reference guide to the Banff Classification of Renal Allograft Pathology. Transplantation 2018;102:1795–814. 10.1097/TP.0000000000002366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Köhler S, Carmody L, Vasilevsky Net al. Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res 2019;47:D1018–27. 10.1093/nar/gky1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Couto FM, Lamurias A.. Semantic similarity definition. In: Ranganathan S, Gribskov M, Nakai Ket al., eds. Encyclopedia of Bioinformatics and Computational Biology. Oxford: Academic Press, 2019:870–6. [Google Scholar]
- 33. Martín-Hernández E, García-Silva MT, Vara Jet al. Renal pathology in children with mitochondrial diseases. Pediatr Nephrol 2005;20:1299–305. 10.1007/s00467-005-1948-z [DOI] [PubMed] [Google Scholar]
- 34. Govers LP, Toka HR, Hariri Aet al. Mitochondrial DNA mutations in renal disease: an overview. Pediatr Nephrol 2021;36:9–17. 10.1007/s00467-019-04404-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Che R, Yuan Y, Huang Set al. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol 2014;306:F367–78. 10.1152/ajprenal.00571.2013 [DOI] [PubMed] [Google Scholar]
- 36. Jefferson JA, Alpers CE, Shankland SJ.. Podocyte biology for the bedside. Am J Kidney Dis 2011;58:835–45. 10.1053/j.ajkd.2011.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parikh S, Karaa A, Goldstein Aet al. Solid organ transplantation in primary mitochondrial disease: proceed with caution. Mol Genet Metab 2016;118:178–84. 10.1016/j.ymgme.2016.04.009 [DOI] [PubMed] [Google Scholar]
- 38. Taivassalo T, Shoubridge EA, Chen Jet al. Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann Neurol 2001;50:133–41. 10.1002/ana.1050 [DOI] [PubMed] [Google Scholar]
- 39. Bakris GL, Ritz E, World Kidney Day Steering Committee. The message for World Kidney Day 2009: hypertension and kidney disease: a marriage that should be prevented. J Am Soc Hyperten 2009;3:80–3. 10.1016/j.jash.2009.02.001 [DOI] [PubMed] [Google Scholar]
- 40. Nargesi AA, Zhu X-Y, Saadiq IMet al. Experimental renovascular disease induces endothelial cell mitochondrial damage and impairs endothelium-dependent relaxation of renal artery segments. Am J Hypertens 2020;33:765–74. 10.1093/ajh/hpaa047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Su M, Dhoopun A-R, Yuan Yet al. Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am J Physiol Renal Physiol 2013;305:F520–31. 10.1152/ajprenal.00570.2012 [DOI] [PubMed] [Google Scholar]
- 42. Nargesi AA, Zhu X-Y, Conley SMet al. Renovascular disease induces mitochondrial damage in swine scattered tubular cells. Am J Physiol Renal Physiol 2019;317:F1142–53. 10.1152/ajprenal.00276.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li XC, Zhou X, Zhuo JL. Evidence for a physiological mitochondrial angiotensin II system in the kidney proximal tubules: novel roles of mitochondrial Ang II/AT1a/O2− and Ang II/AT2/NO signaling. Hypertension 2020;76:121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fayssoil A, Laforêt P, Bougouin Wet al. Prediction of long-term prognosis by heteroplasmy levels of the m.3243A>G mutation in patients with the mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome. Eur J Neurol 2017;24:255–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request.