

ABSTRACT
A phase 1b, randomized, placebo-controlled, double-blind, multiple ascending dose study (NCT02858973) was conducted to assess the safety, tolerability, and pharmacokinetics of the new antituberculosis agent telacebec (Q203). A total of 47 healthy adult subjects entered the study; 36 received telacebec, and 11 received placebo. Telacebec at doses of 20, 50, 100, 160, 250, and 320 mg was orally administered once daily with a standard meal for 14 days. Multiple oral doses of telacebec up to 320 mg daily for 14 days appeared to be safe and well tolerated by healthy adult subjects in this study. There were no deaths, serious adverse events, or subject discontinuations due to adverse events. Following oral doses of telacebec, the overall extent (AUCτ) and peak (Cmax) exposures of telacebec increased from 538.94 to 10,098.47 ng·h/mL and from 76.43 to 1502.33 ng/mL, respectively, with increasing telacebec doses from 20 mg to 320 mg. A steady state was achieved for plasma telacebec by day 12, and there was 1.9- to 3.1-fold accumulation in the extent of telacebec exposure after daily doses for 14 days. Analysis of plasma samples from the participants indicated that telacebec was the primary circulating entity with no significant metabolites. Three potential metabolites of telacebec have been identified, which may be relatively minimal compared to the parent drug. Consistent with findings from preclinical and previous single-dose clinical studies, these results also support the potential of telacebec for further development as a safe and effective agent for the treatment of tuberculosis.
KEYWORDS: telacebec, multiple ascending doses, antituberculosis, safety, pharmacokinetics, metabolism
INTRODUCTION
Tuberculosis (TB) caused by Mycobacterium tuberculosis persists as a major cause of morbidity and mortality worldwide, particularly in developing countries. TB has been posed as one of the top 10 causes of death and the leading cause of death attributed to a single infection before the outburst of coronavirus disease 2019 (COVID-19) (1, 2). Unfortunately, recent COVID-19 pandemic has even reversed years of global progress in struggle against TB (3–5). While the estimated number of TB deaths has gradually decreased every year since 2005, the elaborating trend of decline was set back in the face of COVID-19 pandemic (4, 6). Reduced access to diagnosis and treatment of TB led to an increase in mortality as well as the number of people dying from TB for the first time in the last 15 years. In 2020, the number of deaths from TB had increased to more than 1.5 million, including deaths from human immunodeficiency virus (HIV) coinfection, returning to the level in 2017 (6). Moreover, the devastating impact of the pandemic on TB is anticipated to continue and worsen in 2021 and 2022 (4, 6).
Besides immediate actions to rebuild the TB care and services disrupted by COVID-19, unceasing long-term efforts are required to restore progress toward the goal “End TB” set by World Health Organization (WHO) (4). Endeavors to develop effective therapeutic strategies to overcome TB should be continued. In fact, breakthroughs in therapeutics are needed more than ever to rapidly reduce the mortality rate of TB worldwide and ultimately terminate TB. The most urgent clinical need is to develop a highly effective agent capable of treating multi and extensively drug-resistant TB (7, 8). While effective therapies have been available to cure drug-susceptible TB, treatment of drug-resistant TB requires long courses of treatment, with lower success rates and high rates of drug-associated toxicity (9, 10).
Two drugs with new mechanisms of action, i.e., bedaquiline and delamanid, have been approved for the treatment of multidrug-resistant TB by the US Food and Drug Administration (FDA) and by the European Medicines Agency, respectively (11, 12). Both bedaquiline and delamanid, however, have been associated with potential risk of cardiotoxicity due to QT prolongation (13, 14), which also limits their combination with other antituberculosis agents. In 2019, the US FDA conditionally approved another drug, pretomanid, in combination with bedaquiline and linezolid in patients with highly resistant TB (10). Despite the high efficacy of the combination of three drugs, the extra benefits of adding pretomanid to the bedaquiline-linezolid combination has yet to be further evaluated (15). Thus, there is a need for new therapies with a novel mechanism of action that offer efficacy against drug-sensitive and drug-resistant TB with an acceptable tolerability profile to provide more options for combination regimens in the fight against TB.
As of 2021, 25 drugs, including 16 new compounds and 6 repurposed drugs, are in Phase I, II, or III trials for the treatment of drug-susceptible TB, multidrug-resistant TB, or TB infection (6). Among others, telacebec (Q203) is a new antibiotic that inhibits the growth of Mycobacterium tuberculosis by targeting the energy metabolism, in particular, the oxidative phosphorylation pathway (15). Bedaquiline is an inhibitor of mycobacterial ATP synthase, thereby validating oxidative phosphorylation. On the other hand, telacebec is a first-in-class imidazopyridine amide (IPA) that interferes with a different element of the oxidative phosphorylation pathway, i.e., cytochrome bc1 complex, which is essential for the electron transport required for ATP synthesis (16–19). Thus, telacebec causes rapid depletion of intracellular ATP in Mycobacterium tuberculosis, leading to bacterial cell death. The efficacy of telacebec has been well demonstrated in vitro as well as in vivo animal models (17). Telacebec was active against the reference strain Mycobacterium tuberculosis H37Rv at a MIC required for 50% growth (MIC50) of the low nanomolar range in cultured broth medium and inside macrophages (17, 20). In particular, telacebec has shown its potency across multidrug-resistant, and extensively drug-resistant Mycobacterium tuberculosis isolates in vitro, indicating its potential as part of drug combinations for the treatment of multidrug-resistant TB. The MIC90 of telacebec was conserved against clinical isolates of multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis strains that mostly belonged to the Beijing family (17). It was also shown that the MIC of telacebec was 3.0 to 7.4 nM against isoniazid, rifampicin, and fluoroquinolone drug-resistant Mycobacterium tuberculosis strains (21).
A good safety profile of telacebec with a wide range of doses was also indicated by preclinical studies in mice, rats, and dogs (17). Preclinical studies have shown the high metabolic stability of telacebec in microsomes and cryopreserved hepatocytes from human, monkey, rat, and dog origin (17). These results suggest that telacebec may achieve good systemic exposure in humans. Since telacebec was neither an inhibitor nor an inducer of cytochrome P450 isozymes, the drug-drug interaction potential may be low. In addition, favorable safety profiles and adequate pharmacokinetic characteristics of telacebec in healthy volunteers in the first-in-human study have recently been reported (22). In the Phase 1a trial, telacebec doses from 10 to 800 mg demonstrated tolerability and safety without any significant or severe adverse events in healthy subjects (22).
Given the promising outcomes of preclinical and clinical studies, this report presents the results of a Phase 1b, randomized, placebo-controlled, double-blind, multiple ascending dose study of telacebec (NCT02858973). The present study was designed to evaluate the safety, tolerability, and pharmacokinetics of telacebec in healthy subjects for 14 days, prior to a 14-day early bactericidal activity (EBA) study as an early phase 2 clinical trial (23). The dose levels of the present multiple ascending dose study were selected based on the safety and pharmacokinetic results from the previously completed single ascending dose study (22) and the in vitro and in vivo activity of telacebec (17, 20). A total of 47 healthy adult subjects were randomly allocated into 6 cohorts and received daily doses of either placebo or telacebec at 20 to 320 mg for 14 days with a standard meal for safety and pharmacokinetic evaluation. In addition, the presence of potential metabolites of telacebec has been explored in the plasma samples from the participants. This report would be helpful for the accurate assessment of telacebec as an antituberculosis drug for further development.
RESULTS
Study subjects.
A total of 47 healthy adult subjects sequentially entered the study to assess the safety, tolerability, and pharmacokinetics of multiple oral doses of telacebec administered once daily for 14 days. Subjects were randomized to receive either telacebec or matching placebo (36 active telacebec and 11 placebo) in 1 of 6 cohorts, with each subject participating in only 1 cohort. The decision to study higher dose levels was based upon a comprehensive review of the available safety, tolerability, and pharmacokinetic data from the prior cohorts treated with the lower doses. One subject who received a placebo was discontinued early on day 3 due to an electrocardiogram (ECG)-related abnormality that was considered to be a preexisting condition. Finally, a total of 46 subjects completed the study, including Cohort 1: 20 mg telacebec (6 active, 2 placebo), Cohort 2: 50 mg (6 active, 1 placebo), Cohort 3: 100 mg (6 active, 2 placebo), Cohort 4: 160 mg (6 active, 2 placebo), Cohort 5: 250 mg (6 active, 2 placebo), and Cohort 6: 320 mg (6 active, 2 placebo). Table 1 shows the demographic descriptions of the subjects by overall characteristics. Although every attempt was made to include at least 2 female subjects in each cohort, there was only 1 female subject in this study. Among 36 subjects in the treatment cohorts, most subjects were white (21 of 36; 58%) or black/African American (13 of 36; 36%) with an average age of 34.6 ± 10.8 years and body mass index (BMI) of 27.2 ± 3.4 kg/m2. All 36 subjects who received telacebec treatments were included in the pharmacokinetic analysis, and all 47 were included in the safety analysis.
TABLE 1.
Demographic summary of study subjectsa
| Statistic | Placebo | Telacebec |
||||||
|---|---|---|---|---|---|---|---|---|
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | Cohort 6 | Total | ||
| Dose, mg | 0 | 20 | 50 | 100 | 160 | 250 | 320 | |
| n | 11 | 6 | 6 | 6 | 6 | 6 | 6 | 36 |
| Sex | ||||||||
| Female | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (3%) |
| Male | 11 (100%) | 5 (83%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 35 (97%) |
| Race | ||||||||
| American Indian or Alaska Native | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Black or African American | 5 (45%) | 4 (67%) | 2 (33%) | 1 (17%) | 2 (33%) | 3 (50%) | 1 (17%) | 13 (36%) |
| White | 6 (55%) | 2 (33%) | 4 (67%) | 5 (83%) | 4 (67%) | 3 (50%) | 3 (50%) | 21 (58%) |
| White, Black or African American | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Ethnicity | ||||||||
| Hispanic or Latino | 1 (9%) | 0 (0%) | 0 (0%) | 2 (33%) | 0 (0%) | 1 (17%) | 1 (17%) | 4 (11%) |
| Not Hispanic or Latino | 10 (91%) | 6 (100%) | 6 (100%) | 4 (67%) | 6 (100%) | 5 (83%) | 5 (83%) | 32 (89%) |
| Age, yrs | 33.1 ± 8.9 | 35.7 ± 11.2 | 31.3 ± 9.0 | 31.0 ± 11.7 | 32.0 ± 10.6 | 44.5 ± 9.1 | 32.8 ± 10.7 | 34.6 ± 10.8 |
| ht, cm | 178.4 ± 6.2 | 173.5 ± 8.2 | 177.5 ± 4.4 | 174.5 ± 4.3 | 177.5 ± 2.6 | 175.5 ± 8.9 | 174.2 ± 8.2 | 175.4 ± 6.3 |
| wt, kg | 83.7 ± 9.1 | 87.7 ± 12.9 | 85.2 ± 10.4 | 78.4 ± 14.6 | 82.5 ± 5.5 | 90.4 ± 13.0 | 78.4 ± 14.2 | 83.7 ± 12.1 |
| Body Mass Index, kg/m² | 26.225 ± 1.877 | 29.042 ± 3.401 | 27.062 ± 3.276 | 25.797 ± 4.693 | 26.182 ± 1.753 | 29.213 ± 2.848 | 25.632 ± 3.201 | 27.154 ± 3.407 |
Data were presented as the mean ± SD.
Safety and tolerability.
Multiple oral doses of telacebec, up to 320 mg (or matching placebo) daily for 14 days, appeared to be safe and well tolerated by the healthy adult subjects. There were no deaths, serious adverse events, or subject discontinuations due to adverse events throughout the study. Overall, a total of 66 mild treatment-emergent adverse events were reported by 23 (64%) subjects taking telacebec and 14 (91%) subjects taking placebo. The treatment-emergent adverse event frequency by treatment groups is summarized in Table 2. The incidence of adverse events did not increase with increasing telacebec dose levels. The most common adverse event was mild contact dermatitis (due to ECG lead adhesive), which was reported by 13 (36%) subjects in the telacebec cohorts and 4 (36%) subjects in the placebo group. All contact dermatitis events were considered to be unrelated to the study treatment. Mild headache was the next most common adverse event, reported by 7 (19%) subjects taking telacebec, with 2 events probably related, 9 possibly related, and 4 unlikely related to the study drug. In subjects taking placebo, nasal congestion was the second most common, which was experienced by 2 (18%) subjects.
TABLE 2.
Treatment-emergent adverse event frequency by treatment
| Adverse events | Placebo | Telacebec |
||||||
|---|---|---|---|---|---|---|---|---|
| Cohort 1 (20 mg) |
Cohort 2 (50 mg) |
Cohort 3 (100 mg) |
Cohort 4 (160 mg) |
Cohort 5 (250 mg) |
Cohort 6 (320 mg) |
Total | ||
| No. of subjects dosed | 11 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 36 (100%) |
| No. of subjects with adverse events | 10 (91%) | 5 (83%) | 3 (50%) | 4 (67%) | 3 (50%) | 5 (83%) | 3 (50%) | 23 (64%) |
| Gastrointestinal disorders | 1 (9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 2 (33%) | 0 (0%) | 2 (6%) |
| Constipation | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (3%) |
| Nausea | 1 (9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (3%) |
| General disorders and administration site conditions | 1 (9%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (17%) | 1 (17%) | 3 (8%) |
| Fatigue | 1 (9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Feeling hot | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (17%) | 2 (6%) |
| Pain | 1 (9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (3%) |
| Vessel puncture site erythema | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (3%) |
| Injury, poisoning, and procedural complications | 2 (18%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (3%) |
| Excoriation | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (3%) |
| Foreign body | 1 (9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Laceration | 1 (9%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Investigations | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (17%) | 3 (8%) |
| Blood creatinine increased | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (3%) |
| Electrocardiogram ST-T segment abnormal | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (3%) |
| Urine output increased | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Musculoskeletal and connective tissue disorders | 1 (9%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (17%) | 2 (6%) |
| Arthralgia | 1 (9%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 1 (3%) |
| Myalgia | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Nervous system disorders | 1 (9%) | 2 (33%) | 1 (17%) | 1 (17%) | 2 (33%) | 2 (33%) | 0 (0%) | 8 (22%) |
| Headache | 1 (9%) | 2 (33%) | 1 (17%) | 1 (17%) | 2 (33%) | 1 (17%) | 0 (0%) | 7 (19%) |
| Somnolence | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (3%) |
| Respiratory, thoracic, and mediastinal disorders | 3 (27%) | 2 (33%) | 2 (33%) | 0 (0%) | 0 (0%) | 3 (50%) | 2 (33%) | 9 (25%) |
| Cough | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Dry throat | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Dysphonia | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Nasal congestion | 2 (18%) | 2 (33%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 3 (8%) |
| Oropharyngeal pain | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 2 (6%) |
| Productive cough | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (3%) |
| Rhinorrhoea | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (17%) | 2 (6%) |
| Sinus congestion | 0 (0%) | 0 (0%) | 2 (33%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 2 (6%) |
| Sneezing | 1 (9%) | 1 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 2 (6%) |
| Sputum discolored | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Throat irritation | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 1 (3%) |
| Skin and subcutaneous tissue disorders | 4 (36%) | 5 (83%) | 3 (50%) | 2 (33%) | 0 (0%) | 3 (50%) | 2 (33%) | 15 (42%) |
| Dermatitis contact | 4 (36%) | 5 (83%) | 3 (50%) | 1 (17%) | 0 (0%) | 3 (50%) | 1 (17%) | 13 (36%) |
| Dry skin | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 1 (3%) |
| Pruritus | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 2 (6%) |
| Skin hyperpigmentation | 0 (0%) | 0 (0%) | 0 (0%) | 1 (17%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (3%) |
No clear dose-related changes in serum chemistry, hematology, coagulation, and urinalysis were observed after taking telacebec compared to placebo. One subject in Cohort 5 showed the laboratory-related adverse event of increased serum creatinine of 1.49 – 1.64 mg/dL (versus reference level of 0.69 – 1.2 mg/dL) from day 8 to day 16, which was resolved 20 days after onset. Of note, the subject also had elevated creatinine levels at Screening, on day −1 with values ranging from 1.23 to 1.41 mg/dL. All vital sign parameters, i.e., systolic and diastolic blood pressure, heart rate, respiration rate, and body temperature, were within normal limits. There were no vital sign-related adverse events in this study, and all individual vital sign abnormalities were considered clinically insignificant.
There were no clinically significant ECG-related adverse events or abnormalities in this study. Mean safety 12-lead ECG parameters for heart rate, PR, QRS, QT, and QTcF remained within normal limits. There was one subject in Cohort 1 who experienced the ECG-related adverse event of abnormal ST-T segment that was resolved spontaneously and considered unlikely related to the study drug. One subject receiving placebo was discontinued from the study on day 3 due to an ECG abnormality, which was determined as a preexisting condition. There were no other remarkable findings in the adverse events, vital signs, ECGs, clinical laboratory, or physical examination assessments concerning subject safety.
Pharmacokinetics of telacebec after multiple oral doses.
Average plasma concentrations of telacebec versus time profiles following multiple oral administrations of telacebec at 20 to 320 mg once daily for 14 days in healthy adult subjects are shown in Fig. 1. The pharmacokinetic profiles after the first dose on day 1 and the last dose on day 14 were highlighted in Fig. 1A and 1B, respectively. The pharmacokinetic parameters of telacebec are presented in Table 3. On day 1, following oral administration, plasma concentrations of telacebec increased, achieved the peak plasma concentrations (Cmax) at approximately 2.75 to 4.00 h, and declined in a multiexponential manner across all treatment cohorts. The overall plasma concentrations of telacebec increased with increasing telacebec doses from 20 mg to 320 mg. Correspondingly, Cmax and the area under the plasma concentration-time curve (AUCτ) of telacebec increased with increasing telacebec doses. Other parameters, including half-life (t1/2) and time to reach Cmax (Tmax) were comparable across all cohorts with no dose-related trends.
FIG 1.

Average plasma concentrations of telacebec versus time profiles after (A) the first dose on day 1, (B) the last dose on day 14, and (C) trough concentrations (Ctrough) following oral doses of telacebec at 20 – 320 mg once daily for 14 days in healthy adult subjects (mean ± SD, n = 6).
TABLE 3.
Pharmacokinetic parameters of telacebec (Q203) following multiple oral administrations in healthy subjects (Q203-TB-PI-US002)a
| Parameter | 20 mg | 50 mg | 100 mg | 160 mg | 250 mg | 320 mg |
|---|---|---|---|---|---|---|
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 6) | |
| Day 1 | ||||||
| Tmax (h) | 3.25 (1.50–5.00) | 3.25 (2.50–5.00) | 2.75 (2.00–3.50) | 2.75 (2.50–5.00) | 4.00 (2.00–5.00) | 3.75 (2.50–6.00) |
| Cmax (ng/mL) | 76.43 ± 11.40 | 185.50 ± 33.93 | 496.83 ± 279.99 | 733.17 ± 321.67 | 834.50 ± 226.93 | 1502.33 ± 509.28 |
| AUCτ (ng·h/mL) | 538.94 ± 147.83 | 1512.38 ± 323.53 | 3275.32 ± 1377.62 | 5683.77 ± 2190.29 | 6199.16 ± 2191.32 | 10098.47 ± 3781.43 |
| Day 14 | ||||||
| t1/2,z (h) | 162.03 ± 58.39 | 332.64 ± 75.70 | 173.00 ± 51.17 | 472.79 ± 134.25 | 391.77 ± 104.34 | 419.92 ± 276.11 |
| t1/2,eff (h) | 25.09 ± 7.94 | 33.67 ± 16.93 | 35.87 ± 22.51 | 21.87 ± 13.84 | 41.89 ± 22.80 | 29.17 ± 9.73 |
| Tmax (h) | 3.25 (2.00–5.00) | 5.00 (2.00–6.00) | 4.00 (1.50–6.00) | 4.25 (2.00–5.00) | 5.00 (3.00–6.00) | 5.00 (3.50–5.00) |
| Cmax (ng/mL) | 114.48 ± 19.06 | 408.00 ± 131.43 | 819.00 ± 375.69 | 1088.67 ± 395.80 | 1781.67 ± 559.23 | 2510.00 ± 670.70 |
| AR_Cmax | 1.52 ± 0.32 | 2.27 ± 0.92 | 2.03 ± 1.11 | 1.64 ± 0.77 | 2.28 ± 1.03 | 1.77 ± 0.53 |
| AUCτ (ng·h/mL) | 1083.26 ± 289.13 | 3837.70 ± 1487.62 | 8014.28 ± 3596.45 | 9805.71 ± 2754.53 | 17465.71 ± 5940.17 | 22977.17 ± 10148.91 |
| AR_AUCτ | 2.07 ± 0.45 | 2.57 ± 1.00 | 2.72 ± 1.31 | 1.89 ± 0.81 | 3.06 ± 1.34 | 2.31 ± 0.57 |
Data were presented as the mean ± SD, expect for Tmax, which was presented as the median (minimum-maximum); t1/2,z, terminal half-life; t1/2,eff, effective half-life; AR, accumulation ratio.
Following daily doses of telacebec, the predose concentrations (Ctrough) gradually increased compared to that after the 1st dose on day 1 (Fig. 1C). Based on the steady-state assessment using the Helmert contrast method, steady-state was achieved for plasma concentrations of telacebec by day 12, i.e., Ctrough day 11, after oral administration of telacebec at 20 to 320 mg daily doses (P values > 0.05). Since the steady-state analysis was based on the limited number of Ctrough comparisons, the true steady-state profile may not be reflected, which might have been reached earlier.
Similar to day 1, the average Cmax and AUCτ values also increased with increasing telacebec doses from 20 mg to 320 mg on day 14. On the other hand, telacebec plasma concentrations after the last dose on day 14 were higher than those obtained after 1st dose on day 1 in all dose cohorts (Fig. 1A and 1B). The average accumulation ratio (AR) was calculated as 1.89 to 3.06 for AUCτ and 1.52 to 2.28 for Cmax after multiple daily doses for 14 days across the dose levels studied (Table 3). The terminal half-life (t1/2,z) calculated based on the data obtained after the last dose was variable and ranged from 162.03 to 472.79 h, whereas the average effective half-life (t1/2,eff) calculated based on the accumulation at steady state ranged from 21.87 to 41.89 h. The apparent clearance (CL/F) was variable across the dose cohorts with an average of 14.3 to 19.9 L/h with no dose-related trends.
Dose proportionality was assessed with respect to the systemic exposure parameters of Cmax and AUCτ obtained on day 1 and day 14. Individual and mean ln-transformed pharmacokinetic parameters versus ln-transformed telacebec doses are presented in Fig. 2. Dose proportional increases of Cmax and AUCτ on day 1 were observed in telacebec dose from 20 mg to 320 mg with the slope estimate of 1.020 (95% confidence interval [CI]; 0.888 to 1.151) and 1.012 (95% CI; 0.882 to 1.143), respectively. Cmax and AUCτ on day 14 also increased with the dose of telacebec with the slope of 1.054 (95% CI; 0.931 to 1.178) and 1.049 (95% CI; 0.917 to 1.182), respectively. As the 95% CIs for the slopes of tested pharmacokinetic parameters included the value of 1, dose proportionality was statistically significant, suggesting the overall extent and peak exposures of telacebec increased proportionately over the entire telacebec doses of 20 to 320 mg daily for 14 days.
FIG 2.

Dose proportionality of telacebec with respect to (A) Cmax on day 1, (B) Cmax on day 14, (C) AUCτ on day 1, and (D) AUCτ on day 14. Closed symbols represent the mean and open symbols represent the individual data (n = 6).
Metabolism of telacebec by LC/MS/TOF analysis.
Human plasma samples collected during the study were pooled from 6 subjects across time points (1.5 to 24 h after drug administration) under steady-state dosing conditions and analyzed for the presence of telacebec and metabolites by liquid chromatography with quadrupole time-of-flight mass spectrometry (LC/MS/TOF). Possible metabolite identities were assigned based on the accurate mass data. A chromatographic peak profile of the parent and metabolites were generated.
Table 4 shows the potential metabolites of telacebec found in the plasma from the study subjects. Three potential metabolites of telacebec were observed by LC/MS/TOF. The tentatively characterized metabolite, M1 appeared to be loss of C19H19F3NO from the parent, while M2 was unknown, and M3 might result from hydroxylation. The structures of the parent and proposed metabolites based on the masses and chemical precedent are also shown (Table 4). The relative abundances were calculated assuming that ionization efficiency and matrix effects were uniform for all analytes at all elution times in the profile. The parent molecule telacebec was the major circulating analyte in the plasma of all subjects, representing 90.6% of the total peak height. Among the identified metabolites, M1 was the highest at 4.1%, followed by M3 and M2 at 3.6% and 1.7%.
TABLE 4.
Potential metabolites of telacebec in human plasmaa
| Name | TR, min | Found m/z | Theoretical m/z | Proposed structure |
|---|---|---|---|---|
| Telacebec | 7.95 | 557.1931 | 557.1931 |
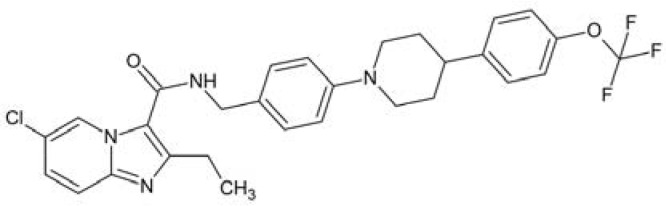
|
| M1 | 6.90 | 224.0602 | 224.0591 |
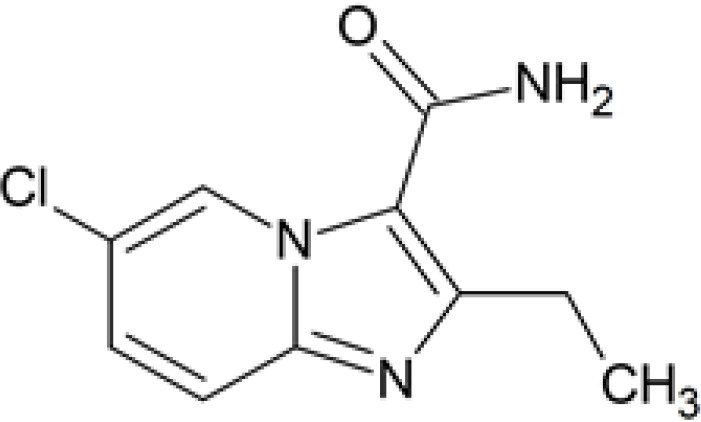
|
| M2 | 6.25 | 551.1435 | –b | Unknown |
| M3 | 6.60 | 573.1906 | 573.1880 | Hydroxylation |
TR, retention time; m/z, mass to charge ratio.
Dash indicates the theoretical m/z of M2 (unknown metabolite).
DISCUSSION
Following the first-in-human trial (22), this study aimed to further evaluate the safety, tolerability, and pharmacokinetics of multiple ascending doses of the new antituberculosis drug telacebec in healthy adult subjects. As a 14-day early bactericidal activity (EBA) study was planned as an early phase 2 clinical trial, this study was designed to support the EBA study following daily oral doses of telacebec for 14 days. In all subjects given telacebec doses at 20 to 320 mg once daily for 14 days, telacebec was well tolerated, and no serious or life-threatening adverse events were observed. Pharmacokinetic analysis showed that dose-proportional increase in the systemic exposure of telacebec and approximately 2- to 3-fold accumulation in the extent of exposure following multiple oral administrations of telacebec for 14 days, by which a steady state has been achieved.
Major shortcomings of recently approved drugs for the treatment of drug-resistant TB include limited utility in combinatory regimens associated with potential safety concerns. Bedaquiline (Sirturo) and delamanid (Deltyba) exhibited potential cardiotoxicity due to prolonging the QT interval (13, 14, 24). Since bedaquiline and delamanid can be part of complex multidrug regimens, combination with other drugs with potential QT prolonging effect such as clofazimine and the fluoroquinolones (25, 26) may increase the safety concern. In this regard, the safety profile of telacebec is promising. There were no clinically significant findings associated with the drug compared to placebo during the entire study period. Particularly, in terms of electrocardiograms, there was one subject in Cohort 1 (20 mg) experienced mild, temporary ST-T segment abnormality on day 6, and one subject in the placebo group discontinued due to an ECG abnormality on day 3. There were no other ECG-related adverse events observed in this study.
The safety profile of telacebec is in good agreement with preclinical and clinical findings. In vitro studies showed that telacebec was not cytotoxic or genotoxic and did not inhibit hERG in hERG potassium channel patch-clamp assay, indicating a low risk of cardiotoxicity (17). Telacebec was well tolerated in mice up to 1,000 mg/kg in an acute toxicity study as well as in rats after administration of 10 mg/kg for 20 days in long-term study (17). No drug-associated serious adverse events were reported in the first-in-human Phase 1a, randomized, placebo-controlled, double-blind, dose-escalation study in healthy male and female subjects (22). In any clinical studies conducted so far, telacebec was not associated with the potential risk of cardiotoxicity, suggesting the compatibility of telacebec in combination with other agents with QT-prolongation effects without further cardiovascular complications.
Phase 1a study allowed to determine the systemic exposure (AUC) at 10 – 800 mg, which was significantly enhanced in the fed state (22). It was also observed that plasma concentrations after 24 h of telacebec administration at all dose levels except the lowest, were well above the MIC50, i.e., 2.7 nM (17, 22). Thus, it was indicated that a daily dose over 10 mg might lead to sufficient systemic exposure to telacebec for its activity. On the other hand, in a chronic mouse model of tuberculosis, telacebec treatment at 0.4 to 10 mg/kg for 4 weeks resulted in 90 to 99.9% reductions in Mycobacterium tuberculosis H37Rv bacterial load (17). Based on these results, the maximum effective dose level was predicted by calculating the dose that would achieve a comparable systemic exposure to that in the animal model at 10 mg/kg, which was approximately 300 mg in the fed condition once daily. Therefore, the present study evaluated daily dosing of telacebec for 14 days, starting from 20 mg and escalating up to 320 mg once daily. In a recently completed phase 2 clinical study (19), telacebec 100, 200, and 300 mg once a day for 14 days indeed demonstrated the dose-dependent early bactericidal activity in patients with tuberculosis, suggesting daily dosing of 300 mg for a more extended treatment period as a potential target regimen.
Consistent with the Phase 1a study (22), this study further demonstrated that the overall extent and peak exposures, represented by AUCτ and Cmax, respectively, increased proportionately with increasing telacebec doses from 20 to 320 mg (Fig. 2). The increased systemic exposures with increasing telacebec doses are likely associated with its dose-dependent bactericidal activity (19). In the phase 2 study, greater reductions in viable mycobacterial sputum load were observed with increasing doses of telacebec from 100 mg to 300 mg once daily for 14 days in tuberculosis patients (19). On the other hand, the systemic exposure after 1st dose in the present study was significantly higher than those obtained in the single-dose study, which is attributed to the effect of food. The enhanced exposure of telacebec by the presence of food has already been described by the first-in-human trial (22). Correspondingly, the Cmax and AUC values obtained after telacebec dose with a standard meal in the present study are well comparable with those obtained previously at 100 mg at the fed state. Following multiple oral administrations, plasma concentrations of telacebec achieved a steady-state by day 12 (Ctrough day 11) with 1.9- to 3.1-fold accumulation in the extent of drug exposure and an approximate 1.5- to 2.3-fold increase in telacebec peak levels with no dose-related trends noted on day 14. Despite the accumulation, multiple oral doses of telacebec up to 320 mg for 14 days appeared to be safe and well tolerated. Since Helmert contrast, which is a standard method for evaluating steady state, indicated that a steady state was achieved for plasma concentrations of telacebec by day 12, a longer duration of telacebec treatment may not lead to further accumulation. However, the steady state should be confirmed by a more quantitative modeling approach. The potential risk associated with drug accumulation for a more extended treatment duration needs to be evaluated by further studies.
Although the terminal half-life (t1/2,z) estimates based on the terminal phase following the last telacebec dose were variable with an average of 162.03 – 472.79 h, the average effective half-life ranged from 21.87 – 41.89 h (Table 3). As the plasma concentration versus time profiles of telacebec show multiexponential declines, there are more than one half-lives to describe the pharmacokinetics of the drug. Terminal half-life describing the terminal phase of the concentration versus time profile may only represent a small fraction of the total systemic exposure and, thus, is relatively unimportant in defining the accumulation upon multiple dosing (27). On the other hand, the effective half-life (t1/2,eff) of telacebec based on the accumulation at steady-state has been estimated to be significantly shorter.
The variability in the pharmacokinetics has already been indicated by the previous study, particularly in the estimates of the apparent terminal half-life (22). As the pharmacokinetics of telacebec showed a multiexponential decline, determining the terminal phase is likely variable leading to the variability in parameter estimates related to the terminal slope. Further studies are required to identify the potential factors associated with the pharmacokinetic variability of telacebec.
Analysis of the plasma samples from the subjects via LC/MS/TOF also indicated that the major circulating entity after oral administration was telacebec with no significant metabolites. There were three potential metabolites of telacebec identified, but the relative abundances of these metabolites were marginal compared to the parent. However, it should be noted that the relative abundances of the parent and metabolites were determined by the integrated peak heights of ion chromatograms. Due to potential differences in ionization efficiencies among analytes, the peak heights may not accurately represent actual abundances. Nevertheless, the high metabolic stability of telacebec has also been indicated in vitro microsomes and hepatocytes as well (17). In addition, it has been suggested that telacebec is neither an inhibitor nor an inducer of cytochrome P450 (CYP450) isozymes (17). These characteristics of metabolism of telacebec suggest that metabolism-related drug interaction may be insignificant, which makes telacebec more favorable as part of a combination with other agents for multidrug resistant TB.
In summary, telacebec was well tolerated when administered orally at daily doses 20 to 320 mg for 14 days with no safety concerns. The dose-proportional increase in the systemic exposure with increasing telacebec dose was also demonstrated. Telacebec plasma concentrations achieved steady-state by day 12 following multiple doses with an accumulation index of 1.89 to 3.06, suggesting a shorter effective half-life than the terminal half-life. Taken together with the efficacy outcomes (19), these results further support the potential of telacebec as a safe and efficient agent for the treatment of TB. In particular, the advantages of telacebec in terms of its novel mechanism, potency, safety profile, and pharmacokinetics suggest its capability as part of drug combinations for treating multidrug resistant TB. This study may be helpful for further evaluation of telacebec as a new antituberculosis drug in the future development process.
MATERIALS AND METHODS
Study design.
A randomized, double-blind, placebo-controlled, multiple ascending dose (MAD) study (Q203-TB-PI-US002) was conducted. The study protocol was reviewed and approved by the Chesapeake Institutional Review Board (IRB) prior to study initiation. Six cohorts of 8 subjects (6 active and 2 placebo) were sequentially enrolled for evaluation. Prior to initiation of any study-specific procedures, all subjects provided written informed consent. In each cohort, subjects received multiple oral doses of telacebec or placebo once daily for 14 days with a standard meal. Subjects were housed from day −1 to day 16 and returned approximately 7 days (days 21 ± 2) and 14 days (day 28 ± 3) after the last dose for follow-up procedures and monitoring potential adverse events (AEs). Additional follow-up procedures for pharmacokinetics and coagulation sample collection and monitoring AEs were conducted on day 44 ± 2 for Cohorts 2, 4, 5, and 6. Safety, including physical examinations, vital signs, electrocardiograms (ECGs), clinical laboratory tests, and AEs, was assessed throughout the study. Serial blood and urine samples were collected for the safety assessments. Blood samples were also collected for the PK assessment of telacebec.
Treatments.
Each cohort was planned to include 8 subjects, with 2 subjects randomized to receive placebo and 6 subjects randomized to receive the active drug. The doses of telacebec were 20 mg (2 × 10 mg tablets, cohort 1), 50 mg (5 × 10 mg tablets, cohort 2), 100 mg (1 × 100 mg tablet, cohort 3), 160 mg (6 × 10 mg tablets and 1 × 100 mg tablet, cohort 4), 250 mg (5 × 10 mg tablets and 2 × 100 mg tablets, cohort 5), and 320 mg (2 × 10 mg tablets and 3 × 100 mg tablets, cohort 6). Two subjects in each cohort received matching placebo tablets. Doses were administered following a standard breakfast on days 1 to 14 once daily for 14 days.
Safety evaluations.
Full physical examinations were performed within 28 days prior to the first drug administration (screening), on day −1 (check-in), and predose at days 2, 8, 14, and 16 (end of study or early termination). Symptom-driven physical examination has been performed at other times. Vital signs, including systolic and diastolic blood pressure, pulse rate, respiratory rate, and body temperature, were measured at screening, check-in, and each day on days 1 to 14: predose, 1, 2, 4, 6, 8, 12, and 16 h after drug administration, and on days 15 and 16. Twelve-lead ECGs were taken in triplicate, and the average of each parameter from triplicate ECGs was used for summarization. Triplicate 12-lead ECGs readings were collected at screening, check-in, and each day on days 1 to14: predose, 2, 4, 6, and 8 h after drug administration, and day 16. In addition, continuous 12-lead ECG data were collected from the Holter monitor on days 1, 7, and 14 for approximately 2 h before dosing until up to 8 h after drug administration. Hematology, serum chemistry, and urinalysis were assessed at screening, check-in, and on days 2, 8, 14, and 16 for all cohorts, and additionally on days 5 and 11 only for Cohorts 3, 4, 5, and 6. Samples for serum chemistry were obtained following a fast of at least 12 h, except for dropouts or rechecks. Coagulation test was performed at screening, check-in, and on days 1 to 14: approximately 5 h after drug administration, and on days 15, 16, 21, and 28 for all cohorts, and additionally on day 44 only for Cohorts 2, 4, 5, and 6. For female subjects, a serum pregnancy test was performed at screening, check-in, and on day 16. All AEs occurring during the clinical trial and concomitant medication were monitored and recorded throughout the study.
Pharmacokinetic analysis.
For all subjects, blood samples were collected on days 1 and 14 at predose (0 h) and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 12, 16, and 24 h after drug administration. Additional predose (0 h) samples were collected prior to dosing on days 4, 7, 10, 12, and 13, and after day 14 dosing on days 16, 21 ± 2, and 28 ± 2. From subjects in Cohorts 2, 4, 5, and 6, blood samples were also collected on day 44 ± 2. Plasma concentrations of telacebec were determined by a validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) method as described previously (22). The analytical range was 5.00 to 5,000 ng/mL in the human plasma.
Pharmacokinetic parameters of telacebec were obtained by analyzing the individual plasma telacebec concentration versus time data via the noncompartmental method. The accumulation ratio (AR) for the area under the concentration versus time curve (AUC) was calculated by the ratio of AUC during a dosing interval (AUCτ) on day 14 and AUC0 to 24 on day 1, i.e., AUCτ,day14/AUCτ,day1. AR for the maximum plasma concentration (Cmax) was calculated by the ratio of Cmax on day 14 and Cmax on day 1, i.e., Cmax,day14/Cmax,day1. Effective half-life (t1/2,eff) was calculated by ln2·τ/ln(AR/[AR-1]) by using the AR for AUC (27).
Metabolite analysis.
The collected plasma samples were analyzed to identify potential metabolites of telacebec. Plasma samples were pooled per time point at equal volumes and extracted using three volumes of acetonitrile. After centrifugation at 16,000 × g for 5 min, the supernatants were transferred to autosampler vials for analysis. The LC/MS/TOF system consisted of a Waters Acquity UPLC system with an in-line photodiode array detector coupled to a Micromass Q-Tof Premier mass spectrometer (Waters, Milford, MA). Initial chromatographic separation was conducted on the ACE Excel 2 C18 PFP column (100 × 2.1 mm, 2.0 μm) with the mobile phases composed of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Following nebulization using heated nitrogen in a Z-spray source/interface, the ionized compounds were detected using time-of-flight spectrometry with a lock mass of leucine enkephalin (m/z 556.2771 for ESI positive mode). The full scan data (50 to 950 amu) were analyzed for the presence of potential metabolites using MetaboLynx XS 2.0 software (Waters). Possible metabolite identities were assigned based on the accurate mass data. The product ion spectra of telacebec and potential metabolites were also collected. The relative abundance of metabolites compared to the parent was estimated based using the peak heights. Based on an assumption that the ionization potentials for the metabolites are similar to telacebec, the AUC of each peak for parent and the metabolites was calculated and compared.
Statistical analysis.
Dose proportionality was evaluated for AUCτ and Cmax on day 1 and AUCτ and Cmax on day 14. Several considerations were to be taken into account for assessing dose proportionality such as: results derived from the power model analysis (22, 28), qualitative assessments, and clinical relevance.
A steady-state analysis was performed on the log-transformed Ctrough values on days 11, 12, and 13, corresponding to the predose concentrations on days 12, 13, and 14, respectively, using Helmert contrasts (29). A steady state was concluded if the comparison did not show a statistically significant difference at a 5% level of significance (α = 0.05).
ACKNOWLEDGMENTS
Jeongjun Kim, Jinho Choi, Hwankyu Kang, Jiye Ahn, Jane Hutchings, Christo van Niekerk, Jaeseung Kim, Yeejin Jeon, and Kiyean Nam are employees of Qurient Co., Ltd.
Contributor Information
Beom Soo Shin, Email: bsshin@skku.edu.
Soyoung Shin, Email: shins@wku.ac.kr.
REFERENCES
- 1.World Health Organization. 2020. Global tuberculosis report.
- 2.Ahmed S, Nandi S, Saxena AK. 2022. An updated patent review on drugs for the treatment of tuberculosis (2018-present). Expert Opin Ther Pat 32:243–260. 10.1080/13543776.2022.2012151. [DOI] [PubMed] [Google Scholar]
- 3.Coronel Teixeira R, Aguirre S, Perez Bejarano D. 2021. Thinking about tuberculosis in times of COVID-19. J Intern Med 289:589–590. 10.1111/joim.13192. [DOI] [PubMed] [Google Scholar]
- 4.Pai M, Kasaeva T, Swaminathan S. 2022. COVID-19's Devastating effect on tuberculosis care —a path to recovery. N Engl J Med 386:1490–1493. 10.1056/NEJMp2118145. [DOI] [PubMed] [Google Scholar]
- 5.Zimmer AJ, Klinton JS, Oga-Omenka C, Heitkamp P, Nawina Nyirenda C, Furin J, Pai M. 2022. Tuberculosis in times of COVID-19. J Epidemiol Community Health 76:310–316. 10.1136/jech-2021-217529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization. 2021. Global tuberculosis report.
- 7.Abubakar I, Zignol M, Falzon D, Raviglione M, Ditiu L, Masham S, Adetifa I, Ford N, Cox H, Lawn SD, Marais BJ, McHugh TD, Mwaba P, Bates M, Lipman M, Zijenah L, Logan S, McNerney R, Zumla A, Sarda K, Nahid P, Hoelscher M, Pletschette M, Memish ZA, Kim P, Hafner R, Cole S, Migliori GB, Maeurer M, Schito M, Zumla A. 2013. Drug-resistant tuberculosis: time for visionary political leadership. Lancet Infect Dis 13:529–539. 10.1016/S1473-3099(13)70030-6. [DOI] [PubMed] [Google Scholar]
- 8.Gunther G. 2014. Multidrug-resistant and extensively drug-resistant tuberculosis: a review of current concepts and future challenges. Clin Med (Lond) 14:279–285. 10.7861/clinmedicine.14-3-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dheda K, Gumbo T, Maartens G, Dooley KE, McNerney R, Murray M, Furin J, Nardell EA, London L, Lessem E, Theron G, van Helden P, Niemann S, Merker M, Dowdy D, Van Rie A, Siu GKH, Pasipanodya JG, Rodrigues C, Clark TG, Sirgel FA, Esmail A, Lin H-H, Atre SR, Schaaf HS, Chang KC, Lange C, Nahid P, Udwadia ZF, Horsburgh CR, Churchyard GJ, Menzies D, Hesseling AC, Nuermberger E, McIlleron H, Fennelly KP, Goemaere E, Jaramillo E, Low M, Jara CM, Padayatchi N, Warren RM. 2017. The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respiratory Medicine 5:291–360. 10.1016/S2213-2600(17)30079-6. [DOI] [PubMed] [Google Scholar]
- 10.Ignatius EH, Dooley KE. 2019. New drugs for the treatment of tuberculosis. Clin Chest Med 40:811–827. 10.1016/j.ccm.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahajan R. 2013. Bedaquiline: first FDA-approved tuberculosis drug in 40 years. Int J Appl Basic Med Res 3:1–2. 10.4103/2229-516X.112228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan NJ, Lo JH. 2014. Delamanid: first global approval. Drugs 74:1041–1045. 10.1007/s40265-014-0241-5. [DOI] [PubMed] [Google Scholar]
- 13.Gler MT, Skripconoka V, Sanchez-Garavito E, Xiao H, Cabrera-Rivero JL, Vargas-Vasquez DE, Gao M, Awad M, Park S-K, Shim TS, Suh GY, Danilovits M, Ogata H, Kurve A, Chang J, Suzuki K, Tupasi T, Koh W-J, Seaworth B, Geiter LJ, Wells CD. 2012. Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med 366:2151–2160. 10.1056/NEJMoa1112433. [DOI] [PubMed] [Google Scholar]
- 14.Diacon AH, Pym A, Grobusch MP, de los Rios JM, Gotuzzo E, Vasilyeva I, Leimane V, Andries K, Bakare N, De Marez T, Haxaire-Theeuwes M, Lounis N, Meyvisch P, De Paepe E, van Heeswijk RPG, Dannemann B. TMC207-C208 Study Group. 2014. Multidrug-resistant tuberculosis and culture conversion with bedaquiline. N Engl J Med 371:723–732. 10.1056/NEJMoa1313865. [DOI] [PubMed] [Google Scholar]
- 15.Lee BS, Pethe K. 2022. Telacebec: an investigational antibacterial for the treatment of tuberculosis (TB). Expert Opin Invest Drugs 31:139–144. 10.1080/13543784.2022.2030309. [DOI] [PubMed] [Google Scholar]
- 16.Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, Dawes SS, Rubin H, Coppel RL, Mizrahi V. 2005. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J Bacteriol 187:6300–6308. 10.1128/JB.187.18.6300-6308.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim SA, Nam J, Kang H, Kwon H, Oh CT, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SP, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han SJ, No Z, et al. 2013. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 19:1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 18.Bald D, Villellas C, Lu P, Koul A. 2017. Targeting energy metabolism in mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio 8. 10.1128/mBio.00272-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Jager VR, Dawson R, van Niekerk C, Hutchings J, Kim J, Vanker N, van der Merwe L, Choi J, Nam K, Diacon AH. 2020. Telacebec (Q203), a new antituberculosis agent. N Engl J Med 382:1280–1281. 10.1056/NEJMc1913327. [DOI] [PubMed] [Google Scholar]
- 20.Kang S, Kim RY, Seo MJ, Lee S, Kim YM, Seo M, Seo JJ, Ko Y, Choi I, Jang J, Nam J, Park S, Kang H, Kim HJ, Kim J, Ahn S, Pethe K, Nam K, No Z, Kim J. 2014. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J Med Chem 57:5293–5305. 10.1021/jm5003606. [DOI] [PubMed] [Google Scholar]
- 21.Moraski GC, Deboosere N, Marshall KL, Weaver HA, Vandeputte A, Hastings C, Woolhiser L, Lenaerts AJ, Brodin P, Miller MJ. 2020. Intracellular and in vivo evaluation of imidazo[2,1-b]thiazole-5-carboxamide anti-tuberculosis compounds. PLoS One 15:e0227224. 10.1371/journal.pone.0227224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Choi J, Kang H, Ahn J, Hutchings J, van Niekerk C, Park D, Kim J, Jeon Y, Nam K, Shin S, Shin BS. 2022. Safety, tolerability, and pharmacokinetics of telacebec (Q203), a new antituberculosis agent, in healthy subjects. Antimicrob Agents Chemother 66:e0143621. 10.1128/AAC.01436-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.US FDA. 2013. Guidance for industry-pulmonary tuberculosis: developing drugs for treatment.
- 24.Pontali E, Sotgiu G, Tiberi S, D'Ambrosio L, Centis R, Migliori GB. 2017. Cardiac safety of bedaquiline: a systematic and critical analysis of the evidence. Eur Respir J 50:1701462. 10.1183/13993003.01462-2017. [DOI] [PubMed] [Google Scholar]
- 25.Amankwa K, Krishnan SC, Tisdale JE. 2004. Torsades de pointes associated with fluoroquinolones: importance of concomitant risk factors. Clin Pharmacol Ther 75:242–247. 10.1016/j.clpt.2003.11.376. [DOI] [PubMed] [Google Scholar]
- 26.Choudhri SH, Harris L, Butany JW, Keystone JS. 1995. Clofazimine induced cardiotoxicity–a case report. Lepr Rev 66:63–68. [DOI] [PubMed] [Google Scholar]
- 27.Sahin S, Benet LZ. 2008. The operational multiple dosing half-life: a key to defining drug accumulation in patients and to designing extended release dosage forms. Pharm Res 25:2869–2877. 10.1007/s11095-008-9787-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith BP, Vandenhende FR, DeSante KA, Farid NA, Welch PA, Callaghan JT, Forgue ST. 2000. Confidence interval criteria for assessment of dose proportionality. Pharm Res 17:1278–1283. 10.1023/A:1026451721686. [DOI] [PubMed] [Google Scholar]
- 29.Maganti L, Panebianco DL, Maes AL. 2008. Evaluation of methods for estimating time to steady state with examples from phase 1 studies. AAPS J 10:141–147. 10.1208/s12248-008-9014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]