

Abstract
The uptake transporter OATP1B1 (SLC01B1) is largely localized to the sinusoidal membrane of hepatocytes and is a known victim of unwanted drug-drug interactions (DDIs). Computational models are useful for identifying potential substrates and/or inhibitors of clinically relevant transporters. Our goal was to generate OATP1B1 in vitro inhibition data for [3H] estrone-3-sulfate (E3S) transport in CHO cells and use it to build machine learning models to facilitate a comparison of seven different classification models (DNN, Adaboosted decision trees, Bernoulli naïve bayes, k-nearest neighbors (knn), random forest, Support Vector Classifier (SVC), Logistic Regression (lreg) and XGBoost (xgb)) using ECFP6 fingerprints to perform 5-fold, nested cross validation. In addition, we compared models using 3D pharmacophores, simple chemical descriptors alone or plus ECFP6, as well as ECFP4 and ECFP8 fingerprints. Several machine learning algorithms (SVC, lreg, xgb, knn) had excellent nested cross validation statistics, particularly for accuracy, AUC and specificity. An external test set containing 207 unique compounds not in the training set demonstrated that at every threshold SVC outperformed the other algorithms based on a rank normalized score. A prospective validation test set was chosen using prediction scores from the SVC models with ECFP fingerprints and were tested in vitro with 15 of 19 compounds (84% accuracy) predicted as active (≥20% inhibition). Of these compounds 6 (abamectin, asiaticoside, berbamine, doramectin, mobocertinib and umbralisib) appear to be novel inhibitors of OATP1B1 not previously reported. These validated machine learning models can now be used to make predictions for drug-drug interactions for human OATP1B1 alongside other machine learning models for other important drug transporters in our MegaTrans software.
Keywords: Deep Learning, Drug Discovery, Machine learning, MegaTrans, OATP1B1, Support Vector Machine, Transporters
Graphical Abstract
Introduction
Membrane transporters that belong to the solute carrier and ATP-binding cassette superfamilies work to control the uptake, efflux and homeostasis of many nutrients that are physiologically relevant, as well as xenobiotics to which we are constantly exposed. These transporters can also influence the pharmacokinetics of many drugs and, in addition, may be used as potential targets for prodrug approaches. These transporters are often quite promiscuous in their selectivity, which increases the probability for potential interactions. Unfortunately, unlike enzymes involved in xenobiotic metabolism, transporters have not been as well characterized, either experimentally or computationally, which then impacts our ability to predict drug-drug interactions (DDIs) 1. In addition, several transporters have been associated with toxicity or diseases 2 supporting the need for additional characterization. Transporter genotypes can also impact biomarker levels, which may in turn affect our ability to predict these interactions 3. Recent regulatory guidance recommendations 4 have also suggested additional membrane transporters with emerging clinical relevance for assessment 5.
The membrane transporter, OATP1B1 (SLCO1B1), is widely regarded as an ‘uptake’ transporter largely restricted to the sinusoidal aspect of hepatocytes. It transports a wide variety of structurally unrelated compounds, including members of several clinically important drug families: hydroxymethylglutaryl-CoA reductase inhibitors (statins), angiotensin II receptor blockers (sartans) and angiotensin converting enzyme (ACE) inhibitors 6. OATP1B1 is a known victim of unwanted DDIs 7 as therapeutically important drugs such as cyclosporine, gemfibrozil, some statins, antibiotics, and antiretroviral drugs are clinically relevant inhibitors of OATP1B1 8, 9. Several examples highlight the importance of this: gemfibrozil caused an eightfold increase in the AUC of repaglinide, and approximately 2-3-fold increase in the AUC of drugs that are not, or only partly, metabolized by CYP2C8, including pravastatin and rosuvastatin 10. Co-administration of statins with fusidic acid in patients treated for methicillin-resistant Staphylococcus aureus infection is linked to myopathy and rhabdomyolysis due to potent inhibition of OATP1B1 by the antibiotic 11. The potentially lethal interaction between cerivastatin and gemfibrozil led to the withdrawal of cerivastatin from the market and was attributed to concomitant inhibition of OATP1B1 and CYP2C8 by gemfibrozil glucuronide 12. The known influence of OATP1B1 on hepatic uptake and disposition of drugs led to its inclusion in the FDA 13, 14 and EMA 15 guidances for the evaluation of all drug candidates as inhibitors of drug elimination via initial in vitro assessments of DDI.
Computational models of drug-transporter interactions are useful for identifying potential substrates and/or inhibitors of clinically relevant transporters and can provide insight on transporter substrate selectivity 16-32. There have been many efforts to use computational approaches to predict drug interactions with transporters, including pharmacophores, quantitative structure activity relationships (QSARs) using various descriptors, machine learning models and docking in crystal structures or homology models 17, 22, 24, 26-33. In addition, drug metabolite preferrences of OAT1 and OAT3, based on knockout mice plasma analysis, was shown to be useful in predicting OAT1/3 compound preference using machine learning 23. For example, several recent examples used in vitro data to generate Bayesian machine learning models that can, in turn, be used to score libraries of compounds and predict additional compounds as substrates and/or inhibitors of selected transporters 34-36. These models were shown to be useful in identifying important molecular features in the training sets and can be applied to the drug discovery and development process to identify valuable information on favorable and unfavorable drug-transporter interactions before additional in vitro and in vivo studies are conducted. DDIs that involve transporters can increase exposure and the risk of toxicity and or facilitate drug disposition 37. Computational modeling that is implemented early in the drug discovery and development process can refine the process of screening compounds and reduce the amount of time and effort required for lead identification and optimization. Our experience of using machine learning 38-55 has previously been focused on Bayesian approaches, which classify compounds as either active or inactive based on user-defined thresholds, by means of a simple probabilistic classification model based on Bayes' theorem. More recently we have also explored large-scale comparisons with many other state-of-the-art machine learning methods 56-58 which have been part of our software development efforts 56, 57. There has been limited comparison of different machine learning algorithms for human drug transporter datasets, unlike for other relevant drug discovery datasets 56, 59, 60. As yet there have been few larger-scale machine learning approaches that have been applied for OATP1B1 and this study uses the most recent machine learning approaches 61 and tests the models using prospective testing, which was the focus of our current research. In the process we have identified new OATP1B1 inhibitors and provide our training and test sets for others to utilize (Supplemental data).
Methods
Chemicals.
[3H]Estrone-3-sulfate (E3S) (specific activity (S.A.), 49 Ci/mmol) was purchased from Perkin-Elmer (Waltham, MA). Unlabeled E3S was purchased from Sigma-Aldrich (St. Louis, MO). The Spectrum Collection (MicroSource Discovery Systems, Inc., Gaylordsville, CT; consisting of the compounds in their US and International Drug Collections, plus their Natural Product and Discover libraries) was provided by Arizona Cancer Center Drug Discovery Program of the University of Arizona. Ham’s F12 Kaighn’s modified medium was obtained from Sigma-Aldrich Co. Other reagents were of analytical grade and obtained commercially.
Cell Culture.
Chinese Hamster Ovary that stably express OATP1B1 were generously provided by Prof. Bruno Steiger 62. Cells were passaged every 3-4 days and maintained at 37°C in a humidified environment with 5% CO2. Expression of OATP1B1 in cells was maintained through G418 (purchased from Invitrogen, Carlsbad, CA, 100 μg/mL) selective pressure. When seeded into 96-well plates (Greiner; VWR Intl., Arlington Heights, IL) for transport assays, these cells were grown to confluence in antibiotic-free media.
Transport Experiments.
Cells were seeded in 96 well plates with 200 μL of cell media containing 550,000 cells/mL or 275,000 cells/mL, and 5mM sodium butyrate was added after 24 hours for optimal protein expression. Experiments were typically performed after 24 hours of incubation in sodium butyrate media. To begin an experiment, media was aspirated, and the wells were washed for three cycles with 300 μL of room temperature Waymouth Buffer (WB; 135 mM NaCl, 13 mM HEPES, 2.5 mM CaCl2 ·2H2O, 1.2 mM MgCl2, 0.8 mM MgSO4·7H2O, 5 mM KCl, and 28 mM D-glucose; pH 7.4) using an automatic fluid aspirator/dispenser (Model 406, BioTek, Winooski, VT). Transport was then initiated by the addition into each well of 60 μL of WB containing a 20 μM concentration of test agent plus ~0.018 μM [3H]E3S. Kinetic parameters for OATP1B1-mediated E3S transport were based on estimates of initial rates of [3H]E3S uptake from six substrate concentrations, as determined by analysis of the 90 sec time courses of net E3S accumulation at each concentration (see 63).
For the purpose of determining the kinetics of E3S transport, the initial rate at time zero was estimated by fitting a hyperbolic function to each time course (refer to Sandoval et al., 2019 64): Ut = (Umax [S])/(Ku + [S]), where Ut is net substrate accumulation at time t; Umax is the extrapolated maximum accumulation (including carrier mediated uptake and any additional accumulation arising from diffusion, non-specific binding and/or incomplete rinsing of the test solution); and KU is a fitted constant. The ratio Umax/KU provided an empirical estimate of the rate at time zero, i.e., ‘the initial rate’ of total substrate accumulation. The relationship between these estimated initial rates and E3S concentration were adequately described by the Michaelis-Menten equation:
where J is the initial rate of mediated transport from a substrate concentration of [S], Jmax is the maximum rate of mediated substrate transport, and Kt is the Michaelis constant.
In all protocols, after selected time intervals, transport was terminated by rinsing with three cycles of cold WB (300 μL). Accumulated radioactivity was quantified by adding 200 μL of scintillation cocktail per well and sealing the plates (Topseal-A, Perkin Elmer). After allowing the plates to sit for at least two hours, radioactivity was determined in a twelve channel multi-well scintillation counter (Wallac Trilux 1450 Microbeta, Perkin-Elmer).
Drug Screening.
Compounds from the Spectrum Collection, distributed in 80 wells of 96 well plates (100 nmol per well in 10 μl of DMSO), were screened for their inhibitory effectiveness against transport of E3S into CHO cells expressing OATP1B1; a total of 560 compounds were used. Each compound was diluted to a concentration of 20 μM, pH 7.4, to a final concentration of 2% dimethylsulfoxide (DMSO) using a VIAFLO multichannel electronic pipet (Integra Biosciences, Hudson, NH) 65. The following compounds for prospective testing were purchased from MedChemExpress (Monmouth Junction, NJ): abamectin, asiaticoside, baloxavir, berbamine, bremelanotide, bromocriptine, cabazitaxel, doramectin, etoposide, lapatinib, mobocertinib, novobiocin, posaconazole, rifaximin, teniposide, umbralisib, vancomycin, vinblastine and vincristine. Pyronaridine tetraphosphate was purchased from BOC Sciences (Shirley, NY) and tilorone hydrochloride from Caymen Chemical Company (Ann Arbor, MI). These compounds were solubilized in DMSO at 20 mM prior to tested.
Curation for the external validation test set
Data for the external test set was a subset of the dataset downloaded from ChEMBL for target “Solute carrier organic anion transporter family member 1B1” (CHEMBL1697668). This dataset was initially filtered to only include experimental parameters that were the most comparable to those used in this study (concentration of experimental compound: 20μM, % inhibition readout). Following this, only unique compounds were retained (i.e., not identical to those in the training set). These filters left 207 unique molecules to be used as an external test set. It is noted that while some experimental parameters aligned, the cell lines and tracer used for these experiments did not, but since a strong correlation was identified with the activity found in the overlapping compounds (Figure S1) this was considered to be acceptable.
Molecular property analysis
We analyzed the active and inactive molecules from the dataset as separate groups using simple molecular descriptors generated from the ChemAxon software (Budapest, Hungary) to determine if there are any significant differences between the active/inactive groups. The molecular descriptors used to describe each dataset were molecular weight, logP, molecular fraction polar surface area, logD (pH 7.4), as well as the number of aromatic rings, hydrogen bond acceptor and donor, rings and rotatable bonds. To test for statistically significant differences, comparison tests assumed non-parametric data distributions (Mann Whitney and Kolmogorov-Smirnov tests). All graphing and statistical analysis was done using Graphpad Prism 9.3.1 for macOS. 4 compounds were removed from the analysis dataset for the following reasons: 1) after removing salts the compounds were duplicates or 2) a minimum carbon count was not met (≤1 carbon atom).
t-SNE visualization
t-SNE 66 embeds data into a lower-dimensional space. 1024 ECFP6 fingerprints were generated for all compounds. The 1024-bit fingerprints were then embedded into a 2-dimensional vector using t-SNE. All t-SNE values were generated using the scikit-learn library in python with default hyperparameters (n_components = 2, perplexity = 30, early exaggeration = 12.0, learning rate = 200, n_iter = 1000). In addition, simple chemical descriptors generated by ChemAxon (molecular weight, AlogP, molecular fraction polar surface area (PSA), logD (pH 7.4), numbers of aromatic rings, hydrogen bond acceptor and donor, rings and rotatable bonds) were also used in conjunction with, or independently of, ECFP6 fingerprints in the same manner as with ECFP6 alone to visualize molecular properties. Chemical descriptors were z-normalized prior to dimensional compression. In short, each descriptor value was transformed by subtracting the sample mean and dividing by the sample standard deviation. When merging with ECFP6, this was a necessary step as it prevented the descriptors from subduing the impact of the sparse bit-vector fingerprints. In addition, some descriptors, such as molecular weight, could massively bias the t-SNE plots irrespective of ECFP6 due to disparities in the magnitude prior to normalization.
Machine learning
Our proprietary software, Assay Central, uses multiple machine learning algorithms that are integrated in our web-based software to build classification models, as described previously 67, with the recent addition of the algorithm XGboost. These methods are described in more details in the Supplemental Materials. Machine learning model validation was performed using a nested 5-fold cross validation and with external test sets (prospective and retrospective). Nested 5-fold cross validation initially selects a random, stratified 20% hold out set that is removed from the training set prior to model building. The model is then built with the other 80% of the training data and the hyperparameters (if applicable) are optimized using a grid search using 5-fold dataset splits (20% validation sets). This optimized model is then used to predict the initial 20% hold out set. This is repeated until all compounds have been in a hold-out set (total 20 models trained). The final nested 5-fold cross validation scores are an average of each of the hold-out set metrics. In contrast, deep learning (DL) uses a 20% leave out set only due to its high computational requirement. External test sets are independent of the training data and represent true external evaluations of the models. For these external test sets we also generated consensus predictions (Ave), with a “majority rule” decision boundary based on each algorithm’s predicted classification (a tied score = 1). Models were built using various molecular descriptors including 3D-Pharmacophore (described below), ECFP4, ECFP6, ECFP8 and or simple chemical descriptors generated by ChemAxon. A detailed definition of each metric is defined in the Supplemental Methods. The applicability domain is calculated based on the reliability-density neighborhood (RDN) method, which considers not only the model overlap but also the individual bias and precision of the overlapping fingerprints 68.
3D-Pharmacophore descriptors:
We first randomly enumerated 50 3D-conformers generated using RDkit and the ETKDG method 69. Pharmacophore fingerprints were then generated for each conformer in the training set as outlined previously 70, 71. Random combinations of conformers were used for training and model validation using the SVC model and the best performing model was selected for evaluating compounds.
Prospective Model Validation Selection
Two compound libraries were scored using the OATP1B1 inhibition models generated during this study. First, a dataset was compiled for all small molecule drugs (261) that were approved by the US FDA between 2017-2021 for the purpose of finding drugs that have not previously been characterized as OATP1B1 inhibitors, as older FDA-approved drugs are often in many commercial high throughput screens used for repurposing. Salts were removed from all structures prior to predictions with our OATP1B1 models. These compounds were scored using models at the thresholds of 20 and 25%, as these models had the best statistics for the ChEMBL external test set as well as good cross validation performance. In addition, the Clinical Compound Library Plus #HY-L026P from MedChemExpress (Monmouth Junction, NJ), which contains 1747 compounds, was also scored in the same manner as the recent FDA-approved drugs dataset. From this library 15 compounds were chosen in total to be tested. Selection was not only based on prediction score but was also filtered to remove compounds with limited diversity, including steroids and flavonoids. In total, 19 compounds were selected for prospective OATP1B1 machine learning model validation.
OATP1B1 Transport Inhibition Experiments (Prospective Model Validation)
Prospective test set validation assays were performed by Eurofins (St. Charles, MO, USA) and were based on a previously published protocol 72. In short, the uptake of the substrate fluorescein methotrexate (FMTX) at 5μM was assessed in Chinese Hamster Ovary (CHO-K1) cells stably expressing OATP1B1 with or without 20μM of the experimental compound. Following a 20 min incubation at 37°C, uptake was stopped by removing the uptake solution and washing the cells four times with buffer. The cells were then solubilized in 1% Triton X-100 in PBS. Fluorescence was then measured in a microplate reader at an excitation wavelength of 485 nm and an emission wavelength of 528 nm. The percent of negative control was calculated using the following equation:
The percent of inhibition was calculated by subtracting the percent of control from 100. The positive control included 10μM of rifampicin. As we have seen substrate-dependent differences in Organic Cation Transporter inhibition previously 73, we also tested two compounds, pyronaridine and tilorone, in both assays to assess the compatibility between the fluorescent (Eurofins) and radioactive OATP1B1 uptake assays (Wright lab) with the resulting comparisons shown in Figure S2A.
Results
Characterization of OATP1B1 transport activity
OATP1B1 activity was assessed using estrone-3-sulfate as a representative transported substrate. Figure 1A shows time courses of net uptake of six concentrations of E3S into CHO cells that stably expressed OATP1B1. These time courses were not linear, owing to the backflux of accumulated E3S. In three separate experiments, the Jmax for E3S was 5.4 ± 0.47 pmol cm−2 min−1, and the Kt was 0.31 ± 0.12 μM, which was similar to previously reported results 74.
Figure 1.

(A). Forty-five second time courses of OATP1B1-dependent net uptake for six concentrations of [3H] labeled E3S. Each point is the mean (±SD) of uptakes measured in three separate experiments (each in triplicate). Individual data points from each time course were corrected for time zero background, based on first-order extrapolation to time zero of that time course. Lines describe hyperbolic rises to steady state (see text). (B) Kinetics of OATP1B1-mediated E3S transport. Symbols represent mean rates of E3S transport calculated from the data shown in Figure 1A. The solid line was fit to these data using the Michaelis-Menten equation.
Inhibition of transport activity
We determined the effect of 480 structurally distinct compounds from the Spectrum Collection on OATP1B1-mediated E3S transport. Figure 2 shows the effect of a 20 μM concentration of each compound on mediated transport of 10-15 nM [3H]E3S (total uptake corrected for uptake in wild type CHO cells). Data is presented in rank order of increasing inhibitory potency; 32 of these compounds (6.7%) inhibited E3S transport by at least 50% and 77 compounds (16.0%) inhibited transport by at least 20%. The OATP1B1 inhibition novelty was evaluated for compounds that showed the highest inhibition (≥80%) with details elaborated on in the discussion. The inhibition of each compound is shown in detail in Table S1.
Figure 2.

The inhibitory effect of 480 test compounds from the Spectrum Collection on the OATP1B1-mediated transport of ~12 μM [3H]E3S. The 60 sec accumulation of substrate was measured in the presence of a 20 μM concentration of each test agent. The height of the shaded grey region indicates the average (±SD; black lines) accumulation (expressed relative to uptake measured in the absence of inhibitor, i.e., ‘control’) determined in two-three separate experiments, each measured in triplicate and corrected for uptake measured in wild-type CHO cells. The histograms are arranged from no inhibition (left side) to complete inhibition (right side). The horizontal red dashed line indicates 20% inhibition, while the vertical red dashed line divides the ‘active’ inhibitors (≤20 % of control uptake; to the right) from the ‘inactive’ inhibitors (>20 % control, to the left).
Analysis of the OATP1B1 dataset
Using a 20% threshold cutoff, we found that there are multiple statistically significant differences in the chemical properties between the active and inactive compounds (Figure 3). Of the properties examined, we found that only the median of hydrogen bond donors was not statistically significantly different between these groups. The most substantial relative median differences were with molecular weight and lipophilicity (AlogP, logD), a trend that has been seen previously 75, 76.
Figure 3.

Comparison of the distribution of molecular properties for 476 unique compounds in the OATP1B1 dataset split into active (77 compounds) and inactive (399 compounds) groups. Median values are highlighted along with statistical analyses. The gaussians are meant to visually highlight each group and are not meant to imply a normal distribution.
Machine learning model comparison
We used the previously described experimental approach 64, 77 to first fully characterize OATP1B1-mediated transport of [3H]E3S in CHO cells. The data used to generate the OATP1B1 transporter machine learning models assessed the inhibition produced at 20 μM of experimental compounds. The average percent inhibition, which was normalized to an experimental control, was used to set a threshold, or cut-off, to distinguish active from inactive compounds prior to building classification models. Several different thresholds were chosen to assess which had the best predictive ability based on cross validation statistics. We initially compared all the models generated at five cutoffs (20%, 25%, 30%, 35% and 40%) for the OATP1B1 dataset (Table S2). Each threshold created different ratios of active to inactive compounds, affecting the “balance” of the training set. We then compared the cross-validation statistics of different algorithms, including DL, Adaboosted decision trees, Bernoulli naïve bayes, k-nearest neighbors, random forest, SVC, Logistic Regression and XGBoost using ECFP6 fingerprints (Table S2). The 20% cutoff had generally good statistics for multiple algorithms for cross validations, without any clear winner (Table 1). Interestingly, while several algorithms have good statistics with cross validation, this was not true with the prediction of the external validation dataset, which was a subset of unique molecules tested for OATP1B1 inhibition downloaded from ChEMBL (see methods) (Table S3). As a consensus model may often have improved predictive ability than a single model, we also generated statistics for this as well (Table S3; Ave). In this case, SVC outperformed all the other algorithms, including the consensus, and was therefore selected for compound prioritization for prospective testing (Table 2).
Table 1.
OATP1B1 Inhibition (20μM); 20% Threshold had 77 ‘active’ and 399 ‘inactive’ compounds. Values represent 5-fold nested cross validation averages (*DL = 20% hold-out set). The rank normalized score is a normalized average of each of these metrics.
| Method Name |
AUC | F1- Score |
ACC | Recall | Specificity | Precision | Cohen's Kappa |
MCC | Rank Normalized Score |
|
|---|---|---|---|---|---|---|---|---|---|---|
| ada | 0.71 | 0.07 | 0.84 | 0.04 | 1.00 | 0.40 | 0.05 | 0.10 | 0.52 | |
| bnb | 0.81 | 0.34 | 0.85 | 0.26 | 0.97 | 0.67 | 0.28 | 0.33 | 0.65 | |
| DL* | 0.74 | 0.42 | 0.85 | 0.31 | 0.96 | 0.63 | 0.34 | 0.37 | 0.66 | |
| knn | 0.73 | 0.40 | 0.86 | 0.30 | 0.97 | 0.64 | 0.33 | 0.37 | 0.65 | |
| lreg | 0.77 | 0.36 | 0.85 | 0.27 | 0.96 | 0.65 | 0.29 | 0.34 | 0.65 | |
| rf | 0.79 | 0.41 | 0.86 | 0.30 | 0.97 | 0.69 | 0.34 | 0.38 | 0.67 | |
| svc | 0.80 | 0.41 | 0.63 | 0.81 | 0.59 | 0.28 | 0.23 | 0.30 | 0.60 | |
| xgb | 0.78 | 0.37 | 0.85 | 0.29 | 0.95 | 0.55 | 0.29 | 0.31 | 0.64 |
Table 2.
Comparison of SVC models at different thresholds using metrics from nested, 5-fold cross validation. The rank normalized score is a normalized average of each of these metrics.
| Threshold | Method | Validation type |
AUC | F1- Score |
Accuracy | Recall | Specificity | Precision | Cohen's Kappa |
MCC | Rank Normalized Score |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 20% | svc | Cross validation | 0.80 | 0.41 | 0.63 | 0.81 | 0.59 | 0.28 | 0.23 | 0.30 | 0.60 | |
| 25% | 0.83 | 0.44 | 0.84 | 0.51 | 0.89 | 0.40 | 0.35 | 0.36 | 0.66 | |||
| 30% | 0.82 | 0.44 | 0.84 | 0.56 | 0.87 | 0.37 | 0.35 | 0.36 | 0.66 | |||
| 35% | 0.80 | 0.37 | 0.83 | 0.53 | 0.86 | 0.29 | 0.28 | 0.30 | 0.62 | |||
| 40% | 0.84 | 0.35 | 0.84 | 0.55 | 0.87 | 0.26 | 0.27 | 0.30 | 0.62 | |||
| 20% | svc | External test set | 0.75 | 0.80 | 0.72 | 0.78 | 0.59 | 0.83 | 0.35 | 0.36 | 0.73 | |
| 25% | 0.71 | 0.50 | 0.55 | 0.36 | 0.89 | 0.85 | 0.20 | 0.27 | 0.64 | |||
| 30% | 0.77 | 0.52 | 0.61 | 0.39 | 0.88 | 0.78 | 0.25 | 0.30 | 0.65 | |||
| 35% | 0.78 | 0.53 | 0.67 | 0.4 | 0.89 | 0.76 | 0.31 | 0.35 | 0.67 | |||
| 40% | 0.78 | 0.55 | 0.69 | 0.45 | 0.86 | 0.7 | 0.32 | 0.34 | 0.67 |
In addition to comparing algorithms based on an activity threshold, we also expanded our criteria to include additional descriptors. Since the 20% and 25% thresholds seemed to produce good validation statistics, these were used for a comparison of molecular descriptors. We compared the nested 5-fold cross validation scores of EFCP4, EFCP6, EFCP8, 3D pharmacophores, as well as simple chemical descriptors alone or with ECFP6 (Tables S4-S5). While the fingerprints of different radii performed similarly, the 3D pharmacophore descriptor approach generally gave poorer model statistics for cross validation. This same trend was also seen using the prediction scores for the external test sets (Tables S4-S5). These comparisons suggest that 2D molecular fingerprints are more appropriate for these data to optimize their predictive ability. Interestingly, the simple descriptors plus ECFP6 outperformed all the others with the cross-validation statistics, but the scores were generally poorer for the external test sets.
Prospective Model Validation
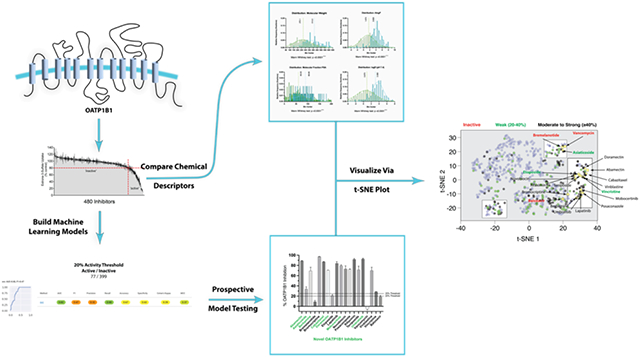
As SVC outperformed the other algorithms with the ChEMBL external test set, only the prediction scores from these models were considered for compound selection for prospective model validation. This test set was comprised of compounds predicted to be active from a recently approved FDA drug library (2017-2021) and from Clinical Compound Library Plus from MedChemExpress (Monmouth Junction, NJ) (see Methods for more details). Of those compounds from the recently approved FDA drug library, four were selected for in vitro testing after being filtered down by prediction score, drug cost and or availability and two of four were shown to be inhibitors at 20μM. While baloxavir (18.75% ± 2.47) and bremelanotide (10.8% ± 0) showed inhibition below the 20% threshold, both umbralisib (92.6% ± 1.69) and mobocertinib (79.4% ± 1.69) far exceeded it. From the 15 compounds selected from the compound library “Clinical Compound Library Plus #HY-L026P” from MedChemExpress (Monmouth Junction, NJ) 14 of 15 showed inhibition of ≥20% at 20μM, with the majority (11/15) of these compounds showing ≥70% inhibition. Of these active compounds, six were novel as we could not find any previous mention of their inhibition of OATP1B1 in the literature. We were unaware that four of the compounds chosen for prospective testing had previously been tested in a related assay, so their inhibition was used to assess assay compatibility (Figure S2B). These compounds were not in our training set and therefore still represent a valid external test set for model evaluation. A synopsis of these results, as well as the validation statistics for the prospective test set, are shown in Figure 4. A more comprehensive list of validation statistics using SVC, including models built with different molecular descriptors and either a 20% or 25% activity threshold, is shown in Tables S4-S5. The structures, prediction scores, model applicability domain, chemical descriptors, inhibition values, as well as ChemAxon descriptors for the experimental validation set are also shown in Table S6.
Figure 4.

Prospective test set inhibition. (A) in vitro inhibition (CHO-K1 cells stably expressing OATP1B1) of the uptake of 5μM fluorescein-methotrexate (FMTX) by OATP1B1 via test compounds (20μM, n=2, error bars represent SD) predicted to be inhibitors by SVC model(s). Compounds with names in green are previously unknown OATP1B1 inhibitors. (B) Truth tables and various statistics showing the performance of models at thresholds 20% and 25% inhibition. It should be noted that there were either none or only a single compound predicted to be inactive by the 20% and 25% models, respectively, some of these statistics are not very descriptive of the model’s performance.
As the experimental validation assay varied from that used to generate the inhibition data and build the machine learning models, we also assessed assay compatibility using the arbitrarily chosen molecules pyronaridine and tilorone (Figure S2A). While tilorone showed no inhibition in both assays, the inhibition of pyronaridine showed a statistically significant difference between these assays (unpaired t-test). It should be noted that the standard deviation for pyronaridine in the fluorescent assay using FMTX was the highest of all the compounds tested (±10%), suggesting the possibility of assay interference. Regardless, this does suggest a partial incompatibility between the assays, though the accuracy of the prospective test set suggests this incompatibility to be minor.
t-SNE Plot for OATP1B1 Model Property Space Analysis
Using the OATP1B1 inhibition data generated in this study we were able to observe some separation between active and inactive compounds using simple chemical descriptors visualized in a 2D t-SNE plot, which compresses multiple dimensions to two (Figure 5A). ECFP6 descriptors alone do not show any obvious enrichment using a t-SNE plot (Figure 5B) and a combination of both EFCP6 and the chosen chemical descriptors also did not enhance the separation between groups (Figure 5C). To assess if this separation could be enhanced, the external test set from ChEMBL (148/59; active/Inactive) was added to our dataset and these plots were regenerated (Figure 5D-F). In addition, compounds chosen for prospective testing based on model predictions were also plotted in these figures. The compounds chosen for the prospective test set were not based on the t-SNE plots, but by model predictions built with ECFP6 alone. Interestingly, the same separation appears as when using simple chemical descriptors (Figure 5D), but this became more pronounced with the addition of predominantly new actives. The combination of these data does not enhance the separation when using ECFP6 fingerprints (Figure 4E) or with ECFP6 plus simple chemical descriptors (Figure 5F). A more comprehensive assessment of this data shows that the separation created by the t-SNE plot seems to be able to predict not only activity, but the degree as well (Figure S3). Categorizing actives from the training and ChEMBL external test sets as either weakly (≥20 - <40%) or moderately/highly (≥40) active shows additional separation. All of the compounds chosen for the prospective test set that fell within the region highly enriched with moderate to strong inhibitors, with the exception of vincristine, were all shown to have moderate to high inhibition. In summary, this suggests that both classification models and t-SNE plots used in conjunction may lead to an enhanced predictive ability. Interestingly, while the t-SNE plot based on simple chemical descriptors appears to be able to distinguish between inhibitors and non-inhibitors in both the prospective and external test sets, ML models built with these same descriptors were unable to predict this (Tables S4-S5).
Figure 5.

t-SNE plots of compounds tested against OATP1B1 classified by activity (Active ≥ 20% inhibition at 20μM) using various descriptors. The descriptors used were either simple chemical descriptors alone (A,D), fingerprints (ECFP6) (B,E) or simple chemical descriptors plus ECFP6 (C,F). The compounds in the training set (tested in this study alone) (A-C) show the most separation using chemical descriptors alone as highlighted by a white box (A). In addition to those compounds used in the training set, D-F also have the compounds from the external test as well as those chosen for a prospective test set. The most visible separation was also seen with the simple descriptors alone as highlighted by the white box. The highlighted separation has been manually selected and is meant as a visual aid and not a separation metric.
To ensure that there was sufficient diversity within the prospective test set, we also compared the Tanimoto similarity of each molecule (Figure S4), which shows that few molecules have greater than 0.15 and 0.7 when using the ECFP6 and MACCS fingerprints, respectively, suggesting a low overlap in the chemical space of the prospective test set.
DISCUSSION
To our knowledge the first computational model for human OATP1B1 substrates was a series of pharmacophores generated with small datasets from different cell lines that suggested multiple hydrophobic and hydrogen bond features were key 78. Following this (in roughly chronological order) several other studies were published, for example a set of 146 molecules were assessed as inhibitors of OATP1B1 and used to build an orthogonal partial least squares projection to latent squares which showed inhibitors were more lipophilic, larger, and have a larger polar surface area 79. Another group tested 2000 molecules for OATP1B1 and OATP1B3 inhibition, which was then used to train a proteochemometrics-based random forest model with FCFP6 and simple chemical descriptors. These models achieved excellent prospective accuracy, with an 80% accuracy with a 54-compound test set 80. A further study used a genetic algorithm-SVM approach for modeling 284 compounds from a proprietary database and then performed an external validation with 1738 compounds from ChEMBL resulting in a classification accuracy of 77.72% 81. The logD descriptor was also indicated as important 81. We are not aware of any efforts to ensure the training and test sets were performed under similar assay conditions. A more recent further study used a small set of 80 FDA approved drugs to understand the features related to liver transporters like OATPs versus organic anion transporters (OATs) and suggested the former prefer more hydrophobic molecules, larger structure with greater complexity76. As a final example, a very recent study used a training set of 1377 compounds with known OATP1B1 activity and focused on utilizing multiple types of models, including proteochemometric, conformal prediction, and XGBoost models, in conjunction with molecular docking to predict OATP1B1 inhibitors. This approach yielded promising results, with strong cross validation statistics as well as the correct prediction of 36% of the 44 compounds tested prospectively 82.
In this current study we generated OATP1B1 inhibitor models using the % inhibition data that we determined for 476 compounds. The OATP1B1 inhibition novelty was evaluated for compounds in this set that showed the highest inhibition (≥80%). Many of these are drugs sold in the US market and include the compounds docusate, atorvastatin, mesalamine, tiratricol, aceclofenac, leoidin and epalrestat. While several of these compounds have previously been identified as OATP1B1 inhibitors or substrates, tiratricol, aceclofenac and epalrestat have not. Each compound represents different drug classes where tiratricol, aceclofenac and epalrestat are a thyroid hormone analog, a nonsteroidal anti-inflammatory drug (NSAID) and an aldose reductase inhibitor, respectively. While all these compounds are available in the US, only aceclofenac is an FDA-approved drug. Interestingly, the Cmax for the typical oral dosing of aceclofenac leads to plasma levels of between 25-30 μM 83, 84 suggesting that the high inhibition identified at 20μM may be clinically relevant at the approved dosing concentrations, though as this compound is known to be highly protein-bound (>99%) (based on information in the FDA package insert) the relevance is unknown. Aceclofenac is also available as a gel, and various formulations have been shown to have significant effects on the unbound plasma levels in mice 85.
We have also curated 207 compounds with OATP1B1 inhibition data from ChEMBL to act as an external test set. Our machine learning models demonstrated that cross validation generated poorer statistics than using external validation with the 207 molecules from ChEMBL and that a 20% cutoff was optimal for SVC (Tables S2-S3). This SVC model was then used to predict the activity of compounds from several libraries, including a recent FDA approved drugs list, and ultimately 19 compounds were chosen for experimental validation of this model. Of these 19 compounds tested, 15 were shown to be inhibitors of OATP1B1 with ≥ 20% inhibition.
After further analysis of the literature and package inserts for the FDA approved drugs we had selected for our prospective test set, we found additional information relating to OATPs for several of these molecules. For example, the kinase inhibitor lapatinib and the antifungal posaconazole were both shown previously to be OATP1B1 inhibitors with IC50s of ~4μM 86 and <3μM 87, respectively. Rifaximin at 3μM was also shown to inhibit the uptake of multiple OATPs, with OATP1B1, OATP1B3 and OATP1A2 substrate uptake being inhibited by 64%, 70% and 40%, respectively. The inhibitory potential of rifaximin on these transporters at the clinically relevant concentrations is unknown. In addition, rifaximin has been shown in vitro to be a substrate of P-glycoprotein, OATP1A2, OATP1B1, and OATP1B3, but not a substrate of OATP2B1. The insecticide and anthelmintic abamectin has not been shown to inhibit OATP1B1 directly, but does inhibit the related transporter OATP1B3 88 and is structurally similar to ivermectin, which is a known inhibitor of OATP1B1 inhibition 75. The anticancer drug cabazitaxel was originally found not to be a substrate of mouse Oatp1b2 89, but has been suggested to inhibit OATP1B1 and OATP1B3 at high concentrations 90. Based on the FDA package insert, the kinase inhibitor mobocertinib was found not to be a OATP1B1 substrate or inhibitor, which is contrary to our findings. Mobocertinib was also not shown to inhibit OATP1B3, OAT1, OAT3, OCT1, or OCT2. Finally, umbralisib was identified as not being a substrate P-gp, BCRP, OAT1, OAT3, OCT2, OATP1B1, OATP1B3, MATE1, or MATE2-K, although we showed inhibition of 92.6 ± 1.7% of OATP1B1 substrate uptake at 20μM.
In addition to building and testing our ML models with various molecular descriptors, we have also performed an extensive analysis of simple molecular descriptors independently. Comparison of individual molecular properties did suggest significant differences between actives and inactives at the 20% inhibition cutoff for both molecular weight and lipophilicity (AlogP, logD), which is in line with the earlier studies 76. Once compressed to two dimensions, the visualization of all the chemical descriptors generated led to some surprising results. We found reasonable separation between actives and inactives using a t-SNE plot that not only appears to help to classify OATP1B1 inhibitors but also offers a semi-quantitative element to the inhibition prediction. Surprisingly, machine learning models built with the same descriptors used to generate these t-SNE plots showed essentially no predictive ability for the prospective or external test sets. This suggests that the multidimensional compression utilized to generate these t-SNE plots can aid in distinguishing differences between inhibitors and non-inhibitors of OATP1B1 in a manner that is not recapitulated in SVC ML models even with the same descriptors. Such predictions can perhaps be enhanced by utilizing both methodologies in conjunction with one another in future.
These combined in vitro and in silico efforts demonstrate the value of comparing multiple classification model cut-offs for activity, different algorithms, and molecular descriptors, to find the optimal predictive model. In the process of developing these machine learning models for OATP1B1 we identified several previously unknown inhibitors from the initial screen and, additionally, from the machine learning model prospective validation.
During our compiling of the FDA approved data set we discovered a trend of recently for approved FDA approved drugs which shows an increase in Lipinski’s rule of 5 violations (Figure S5). Following this, we also carried out an analysis to assess if the chemical characteristics we confirmed are important to predict OATP1B1 inhibition also showed a similar trend. This analysis of drugs approved by the FDA between 2013-2021 showed that while the percent of rule of 5 violations has increased in recent years, the mean molecular weight, number of hydrogen bond donors, hydrogen bond acceptors and LogP have remained fairly constant (Figure S5). These findings would suggest, based on the size of the molecules and lipophilicity alone, that we are not likely to see an increase in the number of potential inhibitors of OATP1B1 if these trends continue.
Conclusion
The enrichment from the machine learning approach undertaken suggests that it may represent a reliable approach to select or avoid compounds that are potential inhibitors of OATP1B1 for use in early drug discovery to avoid potential drug-transporter interactions. The approach we have used can also be applied to other drug transporters of relevance for predicting DDIs and is being used to develop our MegaTrans software (Supplemental Methods) which contains additional uptake and efflux transporters. Providing a suite of computational models for clinically relevant transporters like OATP1B1 and others 16-32 will go some way towards enabling the computational prediction of potential DDIs as highlighted in the regulatory guidances from the FDA 13, 14 and EMA 15.
Supplementary Material
ACKNOWLEDGMENTS
Ms. Kimberley Zorn is kindly acknowledged for her assistance with preliminary models and input for this project.
Grant information
This work was supported by National Institutes of Health National Institute of General Medical Sciences grants: 1R41GM131433 to SE and SW and R44GM122196 to SE.
ABBREVIATIONS USED
- ABDT
AdaBoost
- ADME
absorption, distribution, metabolism, and excretion
- ANN
artificial neural networks
- AUC
area under the curve
- BNB
Bernoulli Naive Bayes
- DT
Decision Tree
- DNN
Deep Neural Networks
- ECFP6
extended connectivity fingerprints of maximum diameter 6
- HTS
high throughput screening
- kNN
k-Nearest Neighbors
- RF
Random forest
- ROC
receiver operating characteristic
- RP
Recursive partitioning
- SVM
support vector machines
- XV ROC AUC
cross-validated receiver operator characteristic curve’s area under the curve
Footnotes
Competing interests:
SE is owner of Collaborations Pharmaceuticals Inc., and TRL, FU, JG are employees. All other authors have no conflicts of interest.
Supporting Information
Supporting further details on the machine learning methods and models including molecule structures, are available. This material is available free of charge via the Internet at http://pubs.acs.org. Models generated with our software can also be made available to academic researchers upon written request.
REFERENCES
- 1.Schlessinger A; Welch MA; van Vlijmen H; Korzekwa K; Swaan PW; Matsson P Molecular Modeling of Drug-Transporter Interactions-an International Transporter Consortium Perspective. Clin Pharmacol Ther 2018, 104, 818–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuetz JD; Swaan PW; Tweedie DJ The Role of Transporters in Toxicity and Disease. Drug Metab Dispos 2014, 42, 541–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yee SW; Giacomini MM; Shen H; Humphreys WG; Horng H; Brian W; Lai Y; Kroetz DL; Giacomini KM Organic Anion Transporter Polypeptide 1b1 Polymorphism Modulates the Extent of Drug-Drug Interaction and Associated Biomarker Levels in Healthy Volunteers. Clin Transl Sci 2019, 12, 388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Transporter C; Giacomini KM; Huang SM; Tweedie DJ; Benet LZ; Brouwer KL; Chu X; Dahlin A; Evers R; Fischer V; Hillgren KM; Hoffmaster KA; Ishikawa T; Keppler D; Kim RB; Lee CA; Niemi M; Polli JW; Sugiyama Y; Swaan PW; Ware JA; Wright SH; Yee SW; Zamek-Gliszczynski MJ; Zhang L Membrane Transporters in Drug Development. Nat Rev Drug Discov 2010, 9, 215–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zamek-Gliszczynski MJ; Taub ME; Chothe PP; Chu X; Giacomini KM; Kim RB; Ray AS; Stocker SL; Unadkat JD; Wittwer MB; Xia C; Yee SW; Zhang L; Zhang Y; International Transporter C Transporters in Drug Development: 2018 Itc Recommendations for Transporters of Emerging Clinical Importance. Clin Pharmacol Ther 2018, 104, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu L; Cui Y; Chung AY; Shitara Y; Sugiyama Y; Keppler D; Pang KS Vectorial Transport of Enalapril by Oatp1a1/Mrp2 and Oatp1b1 and Oatp1b3/Mrp2 in Rat and Human Livers. J Pharmacol Exp Ther 2006, 318, 395–402. [DOI] [PubMed] [Google Scholar]
- 7.Hinton LK; Galetin A; Houston JB Multiple Inhibition Mechanisms and Prediction of Drug-Drug Interactions: Status of Metabolism and Transporter Models as Exemplified by Gemfibrozil-Drug Interactions. Pharm Res 2008, 25, 1063–74. [DOI] [PubMed] [Google Scholar]
- 8.Smith NF; Figg WD; Sparreboom A Role of the Liver-Specific Transporters Oatp1b1 and Oatp1b3 in Governing Drug Elimination. Expert Opin Drug Metab Toxicol 2005, 1, 429–45. [DOI] [PubMed] [Google Scholar]
- 9.Garrison DA; Talebi Z; Eisenmann ED; Sparreboom A; Baker SD Role of Oatp1b1 and Oatp1b3 in Drug-Drug Interactions Mediated by Tyrosine Kinase Inhibitors. Pharmaceutics 2020, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niemi M; Backman JT; Neuvonen M; Neuvonen PJ Effects of Gemfibrozil, Itraconazole, and Their Combination on the Pharmacokinetics and Pharmacodynamics of Repaglinide: Potentially Hazardous Interaction between Gemfibrozil and Repaglinide. Diabetologia 2003, 46, 347–51. [DOI] [PubMed] [Google Scholar]
- 11.Eng H; Scialis RJ; Rotter CJ; Lin J; Lazzaro S; Varma MV; Di L; Feng B; West M; Kalgutkar AS The Antimicrobial Agent Fusidic Acid Inhibits Organic Anion Transporting Polypeptide-Mediated Hepatic Clearance and May Potentiate Statin-Induced Myopathy. Drug Metab Dispos 2016, 44, 692–9. [DOI] [PubMed] [Google Scholar]
- 12.Shitara Y; Hirano M; Sato H; Sugiyama Y Gemfibrozil and Its Glucuronide Inhibit the Organic Anion Transporting Polypeptide 2 (Oatp2/Oatp1b1:Slc21a6)-Mediated Hepatic Uptake and Cyp2c8-Mediated Metabolism of Cerivastatin: Analysis of the Mechanism of the Clinically Relevant Drug-Drug Interaction between Cerivastatin and Gemfibrozil. J Pharmacol Exp Ther 2004, 311, 228–36. [DOI] [PubMed] [Google Scholar]
- 13.Food US; Drug A Guidance for Industry: Drug Interaction Studies - Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations; 2012; pp 1–79. [Google Scholar]
- 14.FDA, In Vitro Metabolism- and Transporter- Mediated Drug-Drug Interaction Studies Guidance for Industry Draft Guidance. 2017. [Google Scholar]
- 15.EuropeanMedicinesAgency , Guideline on the Qualification and Reporting of Physiologically Based Pharmacokinetic (Pbpk) Modelling and Simulation Agency EM, Ed.http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/clinical_pharmacology_pharmacokinetics/general_content_001729.jsp&mid=WC0b01ac0580032ec5, 2016; pp 1–18. [Google Scholar]
- 16.Astorga B; Ekins S; Morales M; Wright SH Molecular Determinants of Ligand Selectivity for the Human Multidrug and Toxin Extruder Proteins Mate1 and Mate2-K. 2012, 341, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ekins S; Polli JE; Swaan PW; Wright SH Computational Modeling to Accelerate the Identification of Substrates and Inhibitors for Transporters That Affect Drug Disposition. Clin Pharmacol Ther 2012, 92, 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain S; Ecker GF, In Silico Approaches to Predict Drug-Transporter Interaction Profiles: Data Mining, Model Generation, and Link to Cholestasis. Springer New York: 2019; pp 383–396. [DOI] [PubMed] [Google Scholar]
- 19.Zdrazil B; Hellsberg E; Viereck M; Ecker GF From Linked Open Data to Molecular Interaction: Studying Selectivity Trends for Ligands of the Human Serotonin and Dopamine Transporter. MedChemComm 2016, 7, 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin L; Yee SW; Kim RB; Giacomini KM Slc Transporters as Therapeutic Targets: Emerging Opportunities. Nature Reviews Drug Discovery 2015, 14, 543–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang C; Swaan PW Computational Approaches to Modeling Drug Transporters. European Journal of Pharmaceutical Sciences 2006, 27, 411–424. [DOI] [PubMed] [Google Scholar]
- 22.Liu HC; Goldenberg A; Chen Y; Lun C; Wu W; Bush KT; Balac N; Rodriguez P; Abagyan R; Nigam SK Molecular Properties of Drugs Interacting with Slc22 Transporters Oat1, Oat3, Oct1, and Oct2: A Machine-Learning Approach. J Pharmacol Exp Ther 2016, 359, 215–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nigam AK; Li JG; Lall K; Shi D; Bush KT; Bhatnagar V; Abagyan R; Nigam SK Unique Metabolite Preferences of the Drug Transporters Oat1 and Oat3 Analyzed by Machine Learning. J Biol Chem 2020, 295, 1829–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsson P; Englund G; Ahlin G; Bergstrom CA; Norinder U; Artursson P A Global Drug Inhibition Pattern for the Human Atp-Binding Cassette Transporter Breast Cancer Resistance Protein (Abcg2). J Pharmacol Exp Ther 2007, 323, 19–30. [DOI] [PubMed] [Google Scholar]
- 25.Matsson P; Pedersen JM; Norinder U; Bergstrom CA; Artursson P Identification of Novel Specific and General Inhibitors of the Three Major Human Atp-Binding Cassette Transporters P-Gp, Bcrp and Mrp2 among Registered Drugs. Pharm Res 2009, 26, 1816–31. [DOI] [PubMed] [Google Scholar]
- 26.Pan Y; Chothe PP; Swaan PW Identification of Novel Breast Cancer Resistance Protein (Bcrp) Inhibitors by Virtual Screening. Mol Pharm 2013, 10, 1236–48. [DOI] [PubMed] [Google Scholar]
- 27.Sedykh A; Fourches D; Duan J; Hucke O; Garneau M; Zhu H; Bonneau P; Tropsha A Human Intestinal Transporter Database: Qsar Modeling and Virtual Profiling of Drug Uptake, Efflux and Interactions. Pharm Res 2013, 30, 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eric S; Kalinic M; Ilic K; Zloh M Computational Classification Models for Predicting the Interaction of Drugs with P-Glycoprotein and Breast Cancer Resistance Protein. SAR QSAR Environ Res 2014, 25, 939–66. [DOI] [PubMed] [Google Scholar]
- 29.Montanari F; Ecker GF Bcrp Inhibition: From Data Collection to Ligand-Based Modeling. Mol Inform 2014, 33, 322–31. [DOI] [PubMed] [Google Scholar]
- 30.Belekar V; Lingineni K; Garg P Classification of Breast Cancer Resistant Protein (Bcrp) Inhibitors and Non-Inhibitors Using Machine Learning Approaches. Comb Chem High Throughput Screen 2015, 18, 476–85. [DOI] [PubMed] [Google Scholar]
- 31.Montanari F; Cseke A; Wlcek K; Ecker GF Virtual Screening of Drugbank Reveals Two Drugs as New Bcrp Inhibitors. SLAS Discov 2017, 22, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang D; Lei T; Wang Z; Shen C; Cao D; Hou T Admet Evaluation in Drug Discovery. 20. Prediction of Breast Cancer Resistance Protein Inhibition through Machine Learning. J Cheminform 2020, 12, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ekins S; Clark AM; Wright SH Making Transporter Models for Drug–Drug Interaction Prediction Mobile. Drug Metabolism and Disposition 2015, 43, 1642–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller SR; Zhang X; Jilek JL; Hau RK; Jennings EQ; Galligan JJ; Foil DH; Zorn KM; Ekins S; Wright SH; Cherrington NJ Predicting Drug Interactions with Human Equilibrative Nuceloside Transporters 1 and 2 Using Functional Knockout Cell Lines and Bayesian Modeling. Mol. Pharmacol 2021, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandoval PJ; Zorn KM; Clark AM; Ekins S; Wright SH Assessment of Substrate-Dependent Ligand Interactions at the Organic Cation Transporter Oct2 Using Six Model Substrates. Molecular Pharmacology 2018, 94, 1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Guerrero LJ; Morales M; Ekins S; Wright SH Lack of Influence of Substrate on Ligand Interaction with the Human Multidrug and Toxin Extruder, Mate1. 2016, 90, 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng Y; El-Kattan AF; Zhang Y; Ray AS; Lai Y Involvement of Drug Transporters in Organ Toxicity: The Fundamental Basis to Drug Discovery and Development. 2016. [DOI] [PubMed] [Google Scholar]
- 38.Clark AM; Dole K; Coulon-Spector A; McNutt A; Grass G; Freundlich JS; Reynolds RC; Ekins S Open Source Bayesian Models: 1. Application to Adme/Tox and Drug Discovery Datasets. J Chem Inf Model 2015, 55, 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kortagere S; Ekins S Troubleshooting Computational Methods in Drug Discovery. J Pharmacol Toxicol Methods 2010, 61, 67–75. [DOI] [PubMed] [Google Scholar]
- 40.Gupta RR; Gifford EM; Liston T; Waller CL; Bunin B; Ekins S Using Open Source Computational Tools for Predicting Human Metabolic Stability and Additional Adme/Tox Properties. Drug Metab Dispos 2010, 38, 2083–2090. [DOI] [PubMed] [Google Scholar]
- 41.Ekins S; Williams AJ Precompetitive Preclinical Adme/Tox Data: Set It Free on the Web to Facilitate Computational Model Building to Assist Drug Development. Lab on a Chip 2010, 10, 13–22. [DOI] [PubMed] [Google Scholar]
- 42.Ekins S; Honeycutt JD; Metz JT Evolving Molecules Using Multi-Objective Optimization: Applying to Adme. Drug Discov Today 2010, 15, 451–460. [DOI] [PubMed] [Google Scholar]
- 43.Bahadduri PM; Polli JE; Swaan PW; Ekins S Targeting Drug Transporters - Combining in Silico and in Vitro Approaches to Predict in Vivo. Methods Mol Biol 2010, 637, 65–103. [DOI] [PubMed] [Google Scholar]
- 44.Ekins S; Bugrim A; Brovold L; Kirillov E; Nikolsky Y; Rakhmatulin E; Sorokina S; Ryabov A; Serebryiskaya T; Melnikov A; Metz J; Nikolskaya T Algorithms for Network Analysis in Systems-Adme/Tox Using the Metacore and Metadrug Platforms. Xenobiotica 2006, 36, 877–901. [DOI] [PubMed] [Google Scholar]
- 45.Ekins S; Andreyev S; Ryabov A; Kirillov E; Rakhmatulin EA; Sorokina S; Bugrim A; Nikolskaya T A Combined Approach to Drug Metabolism and Toxicity Assessment. Drug Metab Dispos 2006, 34, 495–503. [DOI] [PubMed] [Google Scholar]
- 46.Ekins S Systems-Adme/Tox: Resources and Network Approaches. J Pharmacol Toxicol Methods 2006, 53, 38–66. [DOI] [PubMed] [Google Scholar]
- 47.Chang C; Ekins S, Pharmacophores for Human Adme/Tox-Related Proteins. In Pharmacophores and Pharmacophore Searches, Langer T; Hoffman RD, Eds. Wiley-VCH: Weinheim, 2006; pp 299–324. [Google Scholar]
- 48.Ekins S; Nikolsky Y; Nikolskaya T Techniques: Application of Systems Biology to Absorption, Distribution, Metabolism, Excretion and Toxicity. Trends Pharmacol Sci 2005, 26, 202–9. [DOI] [PubMed] [Google Scholar]
- 49.Ekins S; Andreyev S; Ryabov A; Kirillov E; Rakhmatulin EA; Bugrim A; Nikolskaya T Computational Prediction of Human Drug Metabolism. Expert Opin Drug Metab Toxicol 2005, 1, 303–24. [DOI] [PubMed] [Google Scholar]
- 50.Balakin KV; Ivanenkov YA; Savchuk NP; Ivaschenko AA; Ekins S Comprehensive Computational Assessment of Adme Properties Using Mapping Techniques. Curr Drug Disc Tech 2005, 2, 99–113. [DOI] [PubMed] [Google Scholar]
- 51.Ekins S; Swaan PW Computational Models for Enzymes, Transporters, Channels and Receptors Relevant to Adme/Tox. Rev Comp Chem 2004, 20, 333–415. [Google Scholar]
- 52.Ekins S; Boulanger B; Swaan PW; Hupcey MA Towards a New Age of Virtual Adme/Tox and Multidimensional Drug Discovery. Mol Divers 2002, 5, 255–75. [DOI] [PubMed] [Google Scholar]
- 53.Ekins S; Wrighton SA Application of in Silico Approaches to Predicting Drug--Drug Interactions. J Pharmacol Toxicol Methods 2001, 45, 65–9. [DOI] [PubMed] [Google Scholar]
- 54.Ekins S; Waller CL; Swaan PW; Cruciani G; Wrighton SA; Wikel JH Progress in Predicting Human Adme Parameters in Silico. J Pharmacol Toxicol Methods 2000, 44, 251–72. [DOI] [PubMed] [Google Scholar]
- 55.Ekins S; Ring BJ; Grace J; McRobie-Belle DJ; Wrighton SA Present and Future in Vitro Approaches for Drug Metabolism. J Pharmacol Toxicol Methods 2000, 44, 313–24. [DOI] [PubMed] [Google Scholar]
- 56.Korotcov A; Tkachenko V; Russo DP; Ekins S Comparison of Deep Learning with Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Datasets. Mol Pharm 2018, 14, 4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lane T; Russo DP; Zorn KM; Clark AM; Korotcov A; Tkachenko V; Reynolds RC; Perryman AL; Freundlich JS; Ekins S Comparing and Validating Machine Learning Models for Mycobacterium Tuberculosis Drug Discovery. Mol Pharm 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Russo DP; Zorn KM; Clark AM; Zhu H; Ekins S Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol Pharmaceutics 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baskin II; Winkler D; Tetko IV A Renaissance of Neural Networks in Drug Discovery. Expert Opin Drug Discov 2016, 11, 785–795. [DOI] [PubMed] [Google Scholar]
- 60.Ekins S The Next Era: Deep Learning in Pharmaceutical Research. Pharm Res 2016, 33, 2594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lane TR; Foil DH; Minerali E; Urbina F; Zorn KM; Ekins S Bioactivity Comparison across Multiple Machine Learning Algorithms Using over 5000 Datasets for Drug Discovery. Mol Pharm 2021, 18, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gui C; Miao Y; Thompson L; Wahlgren B; Mock M; Stieger B; Hagenbuch B Effect of Pregnane X Receptor Ligands on Transport Mediated by Human Oatp1b1 and Oatp1b3. Eur J Pharmacol 2008, 584, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sandoval PJ; Morales MN; Secomb TW; Wright SH Kinetic Basis of Metformin-Mpp Interactions with the Organic Cation Transporter Oct2. Am J Physiol Renal Physiol 2019, 317, F720–F734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sandoval PJ; Morales M; Secomb TW; Wright SH Kinetic Basis of Metformin-Mpp Interactions with Organic Cation Transporter Oct2. Am J Physiol Renal Physiol 2019, 317, F720–F734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martinez-Guerrero LJ; Morales MH; Ekins S; Wright SH Lack of Influence of Substrate on Ligand Interaction with Human Mate1. Mol Pharmacol 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van der Maaten L; Hinton G Visualizing Data Using T-Sne. J Machine Learning Research 2008, 9, 2579–2605. [Google Scholar]
- 67.Lane T; Russo DP; Zorn KM; Clark AM; Korotcov A; Tkachenko V; Reynolds RC; Perryman AL; Freundlich JS; Ekins S Comparing and Validating Machine Learning Models for Mycobacterium Tuberculosis Drug Discovery. Mol Pharm 2018, 15, 4346–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aniceto N; Freitas AA; Bender A; Ghafourian T A Novel Applicability Domain Technique for Mapping Predictive Reliability across the Chemical Space of a Qsar: Reliability-Density Neighbourhood. Journal of Cheminformatics 2016, 8, 69. [Google Scholar]
- 69.Riniker S; Landrum GA Better Informed Distance Geometry: Using What We Know to Improve Conformation Generation. Journal of Chemical Information and Modeling 2015, 55, 2562–2574. [DOI] [PubMed] [Google Scholar]
- 70.Kutlushina A; Khakimova A; Madzhidov T; Polishchuk P Correction: Kutlushina, A., Et Al. Ligand-Based Pharmacophore Modeling Using Novel 3d Pharmacophore Signatures. Molecules, 2018, 23, 3094. Molecules 2019, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kutlushina A; Khakimova A; Madzhidov T; Polishchuk P Ligand-Based Pharmacophore Modeling Using Novel 3d Pharmacophore Signatures. Molecules 2018, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gui C; Obaidat A; Chaguturu R; Hagenbuch B Development of a Cell-Based High-Throughput Assay to Screen for Inhibitors of Organic Anion Transporting Polypeptides 1b1 and 1b3. Curr Chem Genomics 2010, 4, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martinez Guerrero LJ; Zhang X; Zorn KM; Ekins S; Wright SH Cationic Compounds with Sars-Cov-2 Antiviral Activity and Their Interaction with Oct/Mate Secretory Transporters. J Pharmacol Exp Ther 2021, 379, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miyagawa M; Maeda K; Aoyama A; Sugiyama Y The Eighth and Ninth Transmembrane Domains in Organic Anion Transporting Polypeptide 1b1 Affect the Transport Kinetics of Estrone-3-Sulfate and Estradiol-17beta-D-Glucuronide. J Pharmacol Exp Ther 2009, 329, 551–7. [DOI] [PubMed] [Google Scholar]
- 75.Karlgren M; Vildhede A; Norinder U; Wisniewski JR; Kimoto E; Lai Y; Haglund U; Artursson P Classification of Inhibitors of Hepatic Organic Anion Transporting Polypeptides (Oatps): Influence of Protein Expression on Drug-Drug Interactions. J Med Chem 2012, 55, 4740–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nigam AK; Ojha AA; Li JG; Shi D; Bhatnagar V; Nigam KB; Abagyan R; Nigam SK Molecular Properties of Drugs Handled by Kidney Oats and Liver Oatps Revealed by Chemoinformatics and Machine Learning: Implications for Kidney and Liver Disease. Pharmaceutics 2021, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sandoval PJ; Zorn KM; Clark AM; Ekins S; Wright SH Assessment of Substrate-Dependent Ligand Interactions at the Organic Cation Transporter Oct2 Using Six Model Substrates. Mol Pharmacol 2018, 94, 1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chang C; Pang KS; Swaan PW; Ekins S Comparative Pharmacophore Modeling of Organic Anion Transporting Polypeptides: A Meta-Analysis of Rat Oatp1a1 and Human Oatp1b1. J Pharmacol Exp Ther 2005, 314, 533–41. [DOI] [PubMed] [Google Scholar]
- 79.Karlgren M; Ahlin G; Bergstrom CA; Svensson R; Palm J; Artursson P In Vitro and in Silico Strategies to Identify Oatp1b1 Inhibitors and Predict Clinical Drug-Drug Interactions. Pharm Res 2012, 29, 411–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Bruyn T; van Westen GJ; Ijzerman AP; Stieger B; de Witte P; Augustijns PF; Annaert PP Structure-Based Identification of Oatp1b1/3 Inhibitors. Mol Pharmacol 2013, 83, 1257–67. [DOI] [PubMed] [Google Scholar]
- 81.You H; Lee K; Lee S; Hwang SB; Kim KY; Cho KH; No KT Computational Classification Models for Predicting the Interaction of Compounds with Hepatic Organic Ion Importers. Drug Metab Pharmacokinet 2015, 30, 347–51. [DOI] [PubMed] [Google Scholar]
- 82.Tuerkova A; Bongers BJ; Norinder U; Ungvari O; Szekely V; Tarnovskiy A; Szakacs G; Ozvegy-Laczka C; van Westen GJP; Zdrazil B Identifying Novel Inhibitors for Hepatic Organic Anion Transporting Polypeptides by Machine Learning-Based Virtual Screening. J Chem Inf Model 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rhim SY; Park JH; Park YS; Lee MH; Shaw LM; Kang JS Bioequivalence and Pharmacokinetic Evaluation of Two Branded Formulations of Aceclofenac 100 Mg: A Single-Dose, Randomized, Open-Label, Two-Period Crossover Comparison in Healthy Korean Adult Volunteers. Clin Ther 2008, 30, 633–40. [DOI] [PubMed] [Google Scholar]
- 84.Gowda KV; Rajan DS; Mandal U; Selvan PS; Sam Solomon WD; Bose A; Sarkar AK; Pal TK; Chattaraj TK Evaluation of Bioequivalence of Two Formulations Containing 100 Milligrams of Aceclofenac. Drug Dev Ind Pharm 2006, 32, 1219–25. [DOI] [PubMed] [Google Scholar]
- 85.Somagoni J; Boakye CH; Godugu C; Patel AR; Mendonca Faria HA; Zucolotto V; Singh M Nanomiemgel--a Novel Drug Delivery System for Topical Application--in Vitro and in Vivo Evaluation. PLoS One 2014, 9, e115952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Midgley RS; Kerr DJ; Flaherty KT; Stevenson JP; Pratap SE; Koch KM; Smith DA; Versola M; Fleming RA; Ward C; O'Dwyer PJ; Middleton MR A Phase I and Pharmacokinetic Study of Lapatinib in Combination with Infusional 5-Fluorouracil, Leucovorin and Irinotecan. Ann Oncol 2007, 18, 2025–9. [DOI] [PubMed] [Google Scholar]
- 87.Eisenmann ED; Garrison DA; Talebi Z; Jin Y; Silvaroli JA; Kim JG; Sparreboom A; Savona MR; Mims AS; Baker SD Interaction of Antifungal Drugs with Cyp3a- and Oatp1b-Mediated Venetoclax Elimination. Pharmaceutics 2022, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van de Steeg E; Venhorst J; Jansen HT; Nooijen IH; DeGroot J; Wortelboer HM; Vlaming ML Generation of Bayesian Prediction Models for Oatp-Mediated Drug-Drug Interactions Based on Inhibition Screen of Oatp1b1, Oatp1b1 *15 and Oatp1b3. Eur J Pharm Sci 2015, 70, 29–36. [DOI] [PubMed] [Google Scholar]
- 89.Nieuweboer AJ; Hu S; Gui C; Hagenbuch B; Ghobadi Moghaddam-Helmantel IM; Gibson AA; de Bruijn P; Mathijssen RH; Sparreboom A Influence of Drug Formulation on Oatp1b-Mediated Transport of Paclitaxel. Cancer Res 2014, 74, 3137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anon Jevtana Package Insert. https://products.sanofi.us/jevtana/jevtana.pdf [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.