

Summary
Immune checkpoint blockade (ICB) therapy has revolutionized cancer treatment. However, only a fraction of patients respond to ICB therapy. Accurate prediction of patients to likely respond to ICB would maximize the efficacy of ICB therapy. The tumor microenvironment (TME) dictates tumor progression and therapy outcome. Here, we classify the TME by analyzing the transcriptome from 11,069 cancer patients based on angiogenesis and T cell activity. We find three distinct angio-immune TME subtypes conserved across 30 non-hematological cancers. There is a clear inverse relationship between angiogenesis and anti-tumor immunity in TME. Remarkably, patients displaying TME with low angiogenesis with strong anti-tumor immunity show the most significant responses to ICB therapy in four cancer types. Re-evaluation of the renal cell carcinoma clinical trials provides compelling evidence that the baseline angio-immune state is robustly predictive of ICB responses. This study offers a rationale for incorporating baseline angio-immune scores for future ICB treatment strategies.
Keywords: tumor angiogenesis, tumor immunity, immune checkpoint blockade
Graphical abstract
Highlights
-
•
Angiogenesis and T cell immunity are inversely correlated in the tumor microenvironment
-
•
Angiogenesis and T cell activity are used to stratify the tumor microenvironment
-
•
Angio-immune subtypes are prognostic of survival post-anti-PD1/L1 treatment
-
•
Patients with low angiogenic and high T cell subtypes most likely to benefit from ICB treatment
There is a large proportion of patients who do not mount responses to immune checkpoint blockade. Subramanian et al. show through a computational analysis that, across solid tumor types, baseline characteristics of tumor angiogenesis and T cell immunity can predict therapeutic outcomes to immune checkpoint blockade treatment.
Introduction
Heterogeneity in tumor cells and the surrounding microenvironment drive treatment resistance among patients.1 As such, there have been efforts to identify hierarchical classifications of tumor subtypes on a pan-cancer level.2,3,4,5,6 Unlike the genetically unstable tumor cells, microenvironmental features are more stable and can be conserved across tumor types. As such, delineating tumor microenvironment (TME) subtypes may enable the identification of common resistance mechanisms across tumor types and guide treatment decision-making, particularly for treatments targeting microenvironmental features such as immune checkpoint inhibitors and anti-angiogenic agents.
Abnormal tumor vasculature contributes to tumor growth and escape by remodeling the local TME. Recent evidence from pre-clinical studies suggests that vascular dysfunction in tumors provides physical and chemical barriers to the infiltration of immune cells. Hyperpermeable, immature blood vessels result in improper circulation and hypoxia in the TME and cannot adequately provide a conduit for cytotoxic T cell trafficking to the TME.7 Under inadequate perfusion, glycolytic co-option produces a highly acidic environment that suppresses T cell effector functions.8 In addition, angiogenic tumor endothelial cells express death signals like FAS ligand (FASLG) that induce apoptosis in infiltrating cytotoxic T cells.9 Importantly, recent pre-clinical evidence suggests that normalization of vasculature can reverse the immunosuppressive phenotypes, promote anti-tumor T cell immunity, and synergize with immune checkpoint blockade (ICB), suggesting an intimate interaction between angiogenesis and tumor immunity that is beyond correlative.10,11,12
While ICB has revolutionized the treatment of metastatic and solid tumors by providing lasting regression to subsets of patients, a sizable majority of patients fail to mount responses to ICB.13 High interdependence of endothelial and T cell infiltration and activation in the TME raises the question of whether baseline angiogenic state can be a determinant of tumor immune microenvironment characteristics and can be prognostic of ICB response in patients. Elucidating heterogeneity in baseline angiogenic state and corresponding immune activation might help inform treatment decision-making, allowing us to reduce toxicities faced by patients and design more efficient therapeutic regimens.
Here, we show that baseline angiogenic state is a determinant of cytotoxic T cell function and infiltration in the TME. We developed a transcriptional profile analysis pipeline to stratify patients based on baseline angiogenic and immune activity using endothelial cell and T cell functional gene sets. This highly interpretable tool provides insight into the intimate interaction between angiogenic and immune processes and enables us to identify pan-cancer molecular subtypes of tumor TME. Importantly, pan-cancer angio-immune TME subtypes are prognostic of response and survival of patients treated with ICB and outperform other reported prognostic features. Retrospective analysis of pretreatment datasets reveals that patients showing low angiogenic TME and high T cell activity significantly benefit from Food and Drug Administration-approved first-line ICB strategies. We present a reformed understanding of how angiogenesis and T cell immunity are linked across tumor types, laying the foundation for more efficient treatment decision-making approaches.
Results
Angiogenesis and T cell-mediated immunity are inversely correlated across cancer types
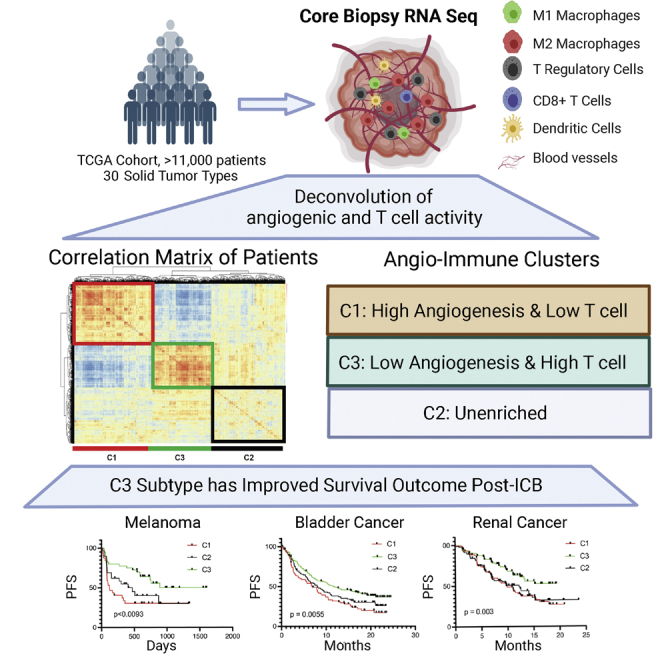
To characterize the interplay of angiogenesis and T cell-mediated immunity in tumors, 91 functional gene sets corresponding to endothelial cell and T cell activity were compiled from the Molecular Signatures Database (MSigDB, Table S1). Transcriptomic data from all solid tumor samples from The Cancer Genome Atlas (TCGA) representing 30 non-hematological tumor types were scored for the 91 functional gene sets using gene set variation analysis (GSVA, Figure S1A) to deconvolute the TME’s angiogenic and T cell functional landscape. We first investigated the distributions of gene sets across patients harboring non-hematological tumor types. A correlation matrix generated using Pearson correlation coefficients was built and revealed two functional gene set modules bound by positive correlation (Figure S1B). We found that T cell gene set scores inversely correlated with angiogenic gene set scores (Figure S1B). Gene sets corresponding to angiogenic function, including endothelial cell migration, proliferation, sprouting, and angiogenesis, were characteristic of one module. The second module was characterized by gene sets corresponding to T cell effector function, antigen processing and presentation, and trans-endothelial migration of leukocytes. The inverse relationship between angiogenesis and immunity has also been observed in mice in previous studies.11
Identification of angio-immune subtypes
A distinct inverse relationship between angiogenic and immune gene sets suggests that we may be able to stratify patients based on baseline angiogenic and T cell activity. To identify angio-immune subtypes of TME based on the distinct distribution of signatures associated with angiogenic and T cell function, we generated a correlation matrix of patients using the distinct distribution of gene signature enrichment across samples. Hierarchical clustering of the correlation matrix, depicting individual patients’ similarity to others in the cohort, was conducted using Euclidian distance. As a result, distinct subtypes of the TME based on distinct distribution of gene sets characterized by the angio-immune scores were uncovered and termed C1, C2, and C3 (Figure 1A). On one hand, C1 contained strong positive enrichment for angiogenic gene sets, including endothelial cell (EC) migration, EC proliferation, and EC sprouting. In contrast, C3 was characterized by the downregulation of functional angiogenesis signatures. C2 had no significant enrichment for functional angiogenesis signatures (Figure 1B). On the other hand, C1 had marked downregulation of T cell functional signatures, including T cell chemotaxis, T cell-mediated cytotoxicity, T cell extravasation, and antigen presentation to T cells. Conversely, C3 presented with strong positive enrichment for T cell functions. Again, C2 had no significant enrichment for T cell-related gene sets (Figure 1C). These TME subtypes were conserved across several tumor types; however, the proportion of the microenvironment subtypes varied across tumor types (Figure 1D).
Figure 1.

Three pan-cancer angio-immune subtype identification
(A) Heatmap of Pearson correlation of patients with non-hematological tumor types across 91 gene sets corresponding T cell and angiogenesis activity. Distance based clustering revealed three distinct clusters across patients. The clusters were labeled C1 (red outline), C2 (black outline), and C3 (green outline).
(B) Bar graphs depicting the average enrichment of angiogenesis signatures in the three angio-immune subtypes. Enrichment of pathways was conducted using gene set variation analysis (GSVA). The enrichment of representative gene sets is plotted. One-way ANOVA was used to determine statistical significance.
(C) Bar graphs depicting the average enrichment of T cell signatures in the three angio-immune subtypes. Enrichment of pathways was conducted using GSVA. The enrichment of representative gene sets is plotted. One-way ANOVA was used to determine statistical significance.
(D) Stacked barplots depicting relative proportion of each angio-immune subtype in all cancers queried. The proportion of all tumors of a particular cancer type belonging to individual angio-immune subtypes was calculated.
(E) Overall survival of patients belonging to three angio-immune subtypes among skin cutaneous melanoma (SKCM) patients. Survival data were derived from publicly available clinical records of TCGA patients. Log ranked test was used for survival analysis.
To ensure pan-cancer clustering is fair to individual tumor types, we also have compared the membership in angio-immune clusters and previously established RNA-based subtypes of different cancer types. We focused on cancers that contained previously established RNA-based subtypes that directly relate to immune function. mRNA-based characterization of skin cutaneous melanoma (SKCM) previously yielded three subtypes: MITF low, immune, and keratin high.14 We noticed that C3 SKCM tumors were enriched in the immune subtype. Conversely C1 SKCM tumors had the smallest proportion of the immune subtype and were enriched in the keratin-high and MITF-low subtypes (Figure S2A). Similarly, mRNA-based characterization of ovarian cancer (OV) previously yielded four subtypes: differentiated, immunoreactive, mesenchymal, and proliferative.15 Tumors classified as immunoreactive were abundant among C3 OV tumors. C1 OV tumors were enriched for mesenchymal and proliferative tumor subtypes (Figure S2B). Moreover, previous mRNA-based characterization of adrenocortical carcinoma (ACC) previously yielded four subtypes: steroid-phenotype-high, steroid-phenotype-high + proliferation, steroid-phenotype-low, and steroid-phenotype-low + proliferation.16 Interestingly, C1 ACC tumors contained an abundance of the steroid-high tumors that contain dampened immune response (Figure S2C). The observed agreement between previously identified molecular clusters in individual cancer types and the angio-immune clusters suggest that the pan-cancer clustering is fair to individual tumor types. Analysis of survival revealed that C3 conferred a marked survival benefit among SKCM patients who routinely received ICB (Figure 1E). Under challenge with standard-of-care therapies, survival differences between clusters were observed in bladder adenocarcinoma (BLCA), lung adenocarcinoma (LUAD), head and neck squamous cell carcinoma (HNSC), clear cell renal carcinoma (KIRC), low-grade glioma (LGG), pancreatic adenocarcinoma (PAAD), stomach adenocarcinoma (STAD), and renal adenocarcinoma (READ) (Figure S3). Together, this analysis provides an easily interpretable framework to classify TMEs independent of the anatomic location of the tumor that may confer survival difference when challenged with treatments like ICB.
Immune and somatic mutation characteristics of angio-immune subtypes
A signature-dependent method to estimate enrichment of cell types in the TME, xCell,17 was first used to identify the enrichment of 30 different immune and stromal cell types across TME subtypes. Notably, there was a more significant enrichment of dendritic cells, CD8+ T cells, B cells, Th1 and Th2 cells, and M1 macrophages in C3 (Figure 2A). Again, to ensure a pan-cancer analysis is fair to individual tumor types, we confirmed that pan-cancer trends in immune cell infiltration were consistent in representative tumors like SKCM (Figure S4A), bladder cancer (Figure S4B), gastric cancer (Figure S4C), and renal cancer (Figure S4D). Infiltrating CD8+ T cells in C3 have more significant effector function characteristics as evidenced by increased expression of co-stimulatory molecules in C3 compared with C1 and C2 (Figure 2B). Infiltrating CD8+ T cells also display activation as evidenced by the upregulation of exhaustion markers (Figure 2C). Conversely, C1 was characterized by a distinct increase in enrichment of endothelial cells (Figure 2D). Fibroblast score was also higher in C1 compared with C2 and C3. C1 may contain a denser stroma, evidenced by a higher stromal score (Figure 2D). To identify if C3 tumors contained more normalized vasculature, we normalized and transformed endothelial and pericyte enrichment from xCell and calculated the ratio of pericytes to endothelial cells. Since normalized vasculature contains a greater coverage of pericytes, we termed the ratio as “vessel normalization score” and assessed it across angio-immune clusters. Interestingly, C3 tumors displayed a higher ratio of pericyte to endothelial cells, suggesting a more normalized vasculature phenotype (Figure 2E).
Figure 2.

Immune characteristics of angio-immune subtypes
(A) Bar plots showing xCell enrichment results for major tumor hematopoietic cells across angio-immune subtypes. The enrichment of classical dendritic cells (cDC), plasmacytoid dendritic cells (pDC), activated dendritic cells (aDC), B cells, plasma cells, CD4+ and CD8+ T cells, Tregs, Th1 cells, Th2 cells, and M1 and M2 macrophages as derived from xCell were compared across angio-immune subtypes. One-way ANOVA was used to determine statistical significance.
(B) Heatmap of the Z-scored expression co-stimulatory molecules across angio-immune subtypes.
(C) Heatmap of the Z-scored expression T cell inhibitory molecules across angio-immune subtypes.
(D) Bar plots showing xCell enrichment results for endothelial cells, fibroblasts, and pericytes across angio-immune subtypes. One-way ANOVA was used to determine statistical significance.
(E) Vessel normalization scores across angio-immune subtypes. xCell enrichment for pericytes and endothelial cells was normalized, and the ratio of pericytes to endothelial cells was evaluated and termed the vessel normalization score and is plotted in a bar graph. One-way ANOVA was used to determine statistical significance.
(F) Violin plots depicting silent and non-silent mutational burden across three angio-immune clusters. Data were obtained from the GDC pan-cancer atlas. One-way ANOVA was used to determine statistical significance.
(G) Violin plot of neoantigen counts across three angio-immune clusters. Neoantigen counts were log transformed for visualization purposes. One-way ANOVA was used to determine statistical significance.
(H) Violin plot of TCR richness across three angio-immune clusters. TCR richness data were obtained from the GDC pan-cancer atlas. One-way ANOVA was used to determine statistical significance.
∗ = p < 0.05, ∗∗ = p < 0.01, ∗∗∗ = p < 0.001, ∗∗∗∗ = p < 0.0001.
Somatic variation in tumor cells can dictate the robustness of immune responses in the TME. A high tumor mutational burden in tumors is suspected of promoting the formation of antigenic peptides that can trigger immune responses. We analyzed a publicly available MC3 repository for somatic variants derived from whole-exome sequencing data to characterize somatic variations of tumors. The silent and non-silent mutational burden was higher in C3 than in C2 and C1 (Figure 2F). In line with this finding, the mean neoantigen load was greater in C3 than C2 and C1 (Figure 2G). Infiltrating T cells also contained greater clonality in TCR in C3 when compared with C2 and C1 (Figure 2H). Distinct actionable mutations also characterized different angio-immune subtypes in specific tumor types where ICB is considered for first-line treatment. Among patients with SKCM, MGAM and CSMD2 mutations were enriched among C2 and C3 patients (Figure S5A). Among bladder adenocarcinoma patients, MUC16 and RB1 mutations were enriched among C3 patients, whereas PCLO mutations were enriched among C1 patients (Figure S5B). In patients with STAD, FAT3 and FAT4 mutations were enriched in C2 and C3 patients. C3 patients also had higher rates of TTN mutations (Figure S5C). Last, among clear cell renal carcinoma patients, VHL and BAP1 mutations were enriched in C1 and C3 patients, respectively (Figure S5D). The distinct immune environment of C3 showing higher CD8 T cell infiltration, increased expression of exhaustion markers, and increased mutational burden suggests that this group of patients may benefit from ICB.
Response and survival post ICB
Several clinical and pre-clinical studies suggest that angiogenic control heightens response to ICB.10,11,12 In addition, there have been numerous studies suggesting that the immune profile observed in C3 (high TMB, enriched neoantigen count, improved infiltration of CD8+ T cells, higher PD-L1 expression) correspond to improved response to ICB.18,19 As such, to evaluate if the angio-immune molecular subtypes are prognostic of survival and response upon treatment with ICB, RNA sequencing data from pretreatment biopsies were queried. Four cohorts of patients were assembled consisting of three different tumor types challenged with ICB: anti-PD1-treated metastatic melanoma, metastatic gastric cancer, and metastatic bladder cancer and anti-CTLA4 treated metastatic melanoma.20,21,22,23,24
Angio-immune molecular TME subtypes were largely conserved in the anti-PD1-treated metastatic melanoma (Figure 3A), metastatic gastric cancer (Figure 3B), metastatic bladder cancer (Figure 3C), and the anti-CTLA-4-treated metastatic melanoma cohort (Figure S6A). The distribution of enrichment scores was largely consistent with pan-cancer analysis for anti-PD1-treated metastatic melanoma (Figure 3D), metastatic gastric cancer (Figure 3E), metastatic bladder cancer (Figure 3F), and anti-CTLA-4-treated metastatic melanoma (Figure S6B). C3 displayed a marked increase in response rate to anti-PD1 therapy among metastatic melanoma tumors. Improved response rate translated to improved progression-free survival (PFS; p < 0.0093) and overall survival (OS; p < 0.0027) for patients in C3 (Figure 3G). Melanoma patients in C3 had not reached median PFS. In comparison, the median PFS for C1 was 4.1 months, and the median PFS for C2 was 15.7 months. Similarly, metastatic gastric cancer patients treated with anti-PD1 belonging to C3 displayed a drastic improvement in response rate in comparison with C1 and C2 (Figure 3H). Likewise, bladder cancer patients belonging to C3 displayed more improved PFS than C2 and C1 (p = 0.0055; Figure 3I). Next, to evaluate the relative contribution of each process to the ability of the angio-immune clusters to predict survival of patients treated with ICB, we split patients based on enrichment of an angiogenic signature (Hallmark angiogenesis) and T cell cytotoxicity signature. Among patients with melanoma and bladder cancer, the two signatures individually failed to be prognostic of survival upon treatment with anti-PD1/PDL1 (Figure 3G versus 3J; Figure 3I versus 3K). This suggests that robust T cell activity or poor angiogenic activity alone does not predict response to anti-PD1/PDL1 efficiently, and the synergistic impact of the two is required for a robust prediction. Intriguingly, T cell scores, rather than angiogenic scores, appear to better predict anti-CTLA response. As such, melanoma patients receiving anti-CTLA4 therapy displayed improved survival in C2 and C3 in comparison with C1 (Figure S6C). The results above demonstrate that angio-immune molecular subtypes can be prognostic of response and survival for patients treated with ICB.
Figure 3.

Angio-immune subtypes are prognostic of response to anti-PD1/PDL1
(A) Heatmap of Pearson Correlation of 145 patients with melanoma across 91 gene sets corresponding T cell and angiogenesis activity. Angio-immune subtypes are preserved in the melanoma cohort. Response status is depicted for each patient on top of the heatmap. Green depicts responders to treatment and red depicts non-responders.
(B) Heatmap of Pearson correlation of 45 patients with gastric cancer across 91 gene sets corresponding T cell and angiogenesis activity. Response status is depicted for each patient on the top of the heatmap. Angio-immune subtypes are preserved in the gastric cancer cohort. Response status is depicted for each patient on top of the heatmap. Green depicts responders to treatment and red depicts non-responders.
(C) Heatmap of Pearson correlation of 348 patients with bladder cancer across 91 gene sets corresponding T cell and angiogenesis activity. Angio-immune subtypes are preserved in the gastric cancer cohort.
(D) Bar graphs depicting the average enrichment of angiogenesis signature and T cell signature in the three angio-immune subtypes in the melanoma cohort. Enrichment of pathways was conducted using gene set variation analysis (GSVA). The enrichment of representative gene sets is plotted. One-way ANOVA was used to determine statistical significance.
(E) Bar graphs depicting the average enrichment of angiogenesis signature and T cell signature in the three angio-immune subtypes in the gastric cancer cohort. Enrichment of pathways was conducted using GSVA. The enrichment of representative gene sets is plotted. One-way ANOVA was used to determine statistical significance.
(F) Bar graphs depicting the average enrichment of angiogenesis signature and T cell signature in the three angio-immune subtypes in the bladder cancer cohort. Enrichment of pathways was conducted using GSVA. The enrichment of representative gene sets is plotted. One-way ANOVA was used to determine statistical significance.
(G) Overall survival and progression-free survival for patients in different angio-immune clusters upon treatment with anti-PD1 in melanoma patients.
(H) Response rate for patients in different angio-immune clusters upon treatment with anti-PD1 in gastric cancer patients.
(I) Progression-free survival for patients in different angio-immune clusters upon treatment with anti-PDL1 in bladder cancer patients.
(J) Progression-free survival of patients with melanoma treated with anti-PD1 split by high (>50th percentile) and low (<50th percentile) angiogenesis and T cell cytotoxicity.
(K) Progression-free survival of patients with bladder cancer treated with anti-PDL1 split by high (>50th percentile) and low (<50th percentile) angiogenesis and T cell cytotoxicity. Log-ranked tests were used for survival analysis.
∗ = p < 0.05, ∗∗ = p < 0.01, ∗∗∗ = p < 0.001, ∗∗∗∗ = p < 0.0001.
Re-evaluation of Javelin Renal 101, Checkmate 010, and Checkmate 025 clinical trials
The approval of the combination of avelumab, an anti-PDL1, and axitinib, a small molecule tyrosine kinase inhibitor, for the first-line treatment of metastatic renal cancer was provided in 2020. The approval was granted on the back of median PFS improvement from 8.4 months with the previous standard of care, sunitinib, a tyrosine kinase inhibitor, to 13.8 months for the combination.25 Checkmate 010, phase II study, and Checkmate 025, a randomized phase III study, demonstrated the benefit of nivolumab, an anti-PD1, over everolimus, an mTOR inhibitor, among clear cell renal carcinoma patients previously treated with anti-angiogenic agents.26 We sought to identify if stratification by angio-immune subtypes can confer more significant differences in responses and survival.
The enrichment of the 91 angio-immune signatures was scored using RNA sequencing data from pretreatment biopsies. Three angio-immune molecular subtypes were conserved in the Javelin Renal 101 dataset (Figure 4A) and the Braun dataset (Checkmate 025 and Checkmate 010) (Figure S7A). Angiogenic and immune gene sets’ enrichment distribution was consistent with TCGA data in the Javelin Renal 101 dataset (Figure 4B) and the Braun dataset (Figure S7B). As such, C1 presented with an upregulation of angiogenesis functions and a downregulation of T cell functions. C3 presented with an upregulation of T cell functions and a downregulation of angiogenesis functions. C2 displayed no significant enrichment in either functional module.
Figure 4.

Re-evaluation of Javelin 101 reveals differing propensity for response by angio-immune subtypes
(A) Heatmap of Pearson correlation of 726 patients with renal cell carcinoma across 91 gene sets corresponding T cell and angiogenesis activity. Angio-immune subtypes are preserved in the renal cancer cohort.
(B) Bar graphs depicting the average enrichment of angiogenesis signatures and T cell signatures in the three angio-immune subtypes in renal cell carcinoma cohort. Enrichment of pathways was conducted using gene set variation analysis (GSVA). The enrichment of representative gene sets is plotted. One-way ANOVA was used to determine statistical significance.
(C) Progression-free survival of patients treated with sunitinib versus the combination of axitinib + avelumab.
(D) Progression-free survival of patients treated with sunitinib belonging to different angio-immune clusters.
(E) Progression-free survival of patients treated with combination of axitinib + avelumab belonging to different angio-immune clusters.
(F) Progression-free survival of patients belonging in C1 treated with sunitinib versus the combination of axitinib + avelumab.
(G) Progression-free survival of patients belonging in C2 treated with sunitinib versus the combination of axitinib + avelumab.
(H) Progression-free survival of patients belonging in C3 treated with sunitinib versus the combination of axitinib + avelumab.
∗ = p < 0.05, ∗∗ = p < 0.01, ∗∗∗ = p < 0.001, ∗∗∗∗ = p < 0.0001.
Median PFS improvement for patients with pretreatment RNA sequencing data in the Javelin Renal 101 dataset was from 8.4 months in the sunitinib arm to 12.5 months in the combination arm, consistent with the previous report (Figure 4C).25 We tracked survival across clusters to identify if angio-immune molecular subtypes can inform treatment choice in this setting. When treated with sunitinib, a tyrosine kinase inhibitor, angio-immune molecular subtypes presented no significant differences in median PFS (Figure 4D). However, when treated with the combination of avelumab, an anti-PDL1 therapy, and axitinib, a tyrosine kinase inhibitor, significant improvement in survival was observed: the median PFS of C3 had not matured, C2 had a median PFS of 12.2 months, and C1 had a median PFS of 9.7 months (Figure 4E). To determine if patients belonging to C1, C2, and C3 derive clinical benefit from the combination of avelumab and axitinib, PFS was tracked among angio-immune molecular subtypes across treatment arms. Remarkably, patients belonging to C1 derived no clinical benefit from the combination of avelumab and axitinib compared with sunitinib (median PFS = 9.7 versus 9.7; p = 0.668; Figure 4F). Patients belonging in C2 also derived no clinical benefit from the combination (median PFS = 12.2 versus 8.3 months; p = 0.1469; Figure 4G). However, among C3, patients treated with avelumab and axitinib displayed significantly improved median PFS than the sunitinib arm (not matured versus 8.2 months; p < 0.001; Figure 4H).
Median OS improvement for patients with pretreatment RNA sequencing data in the Braun dataset was from 19.7454 months in the everolimus, an mTOR inhibitor, arm to 25.9877 months in the nivolumab, an anti-PD1 therapy (Figure S7C). We tracked survival across clusters to identify if angio-immune molecular subtypes can inform treatment choice in this setting. When treated with everolimus, angio-immune molecular subtypes presented no significant differences in median OS (Figure S7D). However, when treated with the nivolumab, significant improvement in median OS was observed (p = 0.0033): the median OS of C3 was 29.8645 months, C2 had a median OS of 36.961 months, and C1 had a median OS of 16.9528 months (Figure S7E). C3 displayed a better survival than C2 in the long run (Figure S7E). To determine if patients belonging to C1, C2, and C3 derive clinical benefit from nivolumab over everolimus, OS was tracked among angio-immune molecular subtypes across treatment arms. Remarkably, patients belonging to C1 derived no clinical benefit from nivolumab compared with everolimus (median OS = 24.6735 months in the everolimus arm versus 16.9528 months in the nivolumab arm; p = 0.6347; Figure S7F). Patients belonging in C2 also derived no statistically significant clinical benefit from the combination (median OS = 36.96 months in the nivolumab arm versus 21.13 months in the everolimus arm; p = 0.0581; Figure S3G). However, among C3, patients treated with nivolumab displayed significantly improved median OS than the everolimus arm (median OS = 29.86 months in the nivolumab arm versus 13.27 months in the everolimus arm; p < 0.0043; Figure S7H). Collectively, this analysis suggests that only patients with low angiogenic status and high T cell activity (cluster C3) mount responses to ICB. As such, angio-immune molecular subtypes can be used to exclude patients from treatment that would not derive clinical benefit but are faced with adverse toxicities from therapy.
Angio-immune clusters better predict ICB survival in comparison with previous methods
Poor response rate upon ICB treatment has sparked a range of studies seeking to identify predictors of response among treated patients. The angio-immune subtypes are based on the interaction of the vascular and immune biological systems that is independent of tumor type. As such, to display the universality of the predictive power of the angio-immune subtypes, we comparatively evaluated the predictive power of CXCL9 expression,27 interferon-gamma (IFNg) signature,28 IMPRES (an immune-predictive score),29 PD-L1 expression,30 MHCI expression,31 and MHCII expression31 across anti-PD1-treated melanoma patients, anti-PD1/PD-L1-treated renal cancer patients, and anti-PD-L1-treated bladder cancer patients. Remarkably, none of these tools displayed a universal ability to predict survival upon ICB treatment (Figure 5). High CXCL9 expression was predictive of improved survival in melanoma and bladder cancer patients (IMVIGOR210), but failed to predict improved survival in renal cancer (Javelin and Braun, Figure 5A). High IFNg signature enrichment was predictive of improved survival in melanoma and bladder cancer patients, but failed to predict improved survival in renal cancer (Figure 5B). Intriguingly, high IMPRES scores were predictive of survival only in one cohort of renal cancer patients and failed to predict responses in the melanoma and bladder cancer patient cohort (Figure 5C). High PD-L1 expression predicts response in anti-PD1 treated melanoma and anti-PD-L1-treated bladder cancer patients but fails to do so among renal cancer patients (Figure 5D). Similarly, baseline MHCI expression was predictive of survival only in one cohort of renal cancer patients and melanoma but failed to predict responses in the bladder cancer cohort (Figure 5E). Baseline MHCII expression was predictive of improved survival only in the melanoma cohort (Figure 5F).
Figure 5.

Ability of previous techniques to predict ICB response
(A) Overall survival (OS) tracked for anti-PD1-treated melanoma, anti-PDL1-treated bladder cancer, and anti-PD1-treated renal cancer (Braun) and progression-free survival (PFS) tracked for anti-PDL1-treated renal cancer (Javelin) based on CXCL9 expression. Median expression was used to separate “High” and “Low” expressers.
(B) OS tracked for anti-PD1-treated melanoma, anti-PDL1-treated bladder cancer, and anti-PD1-treated renal cancer (Braun) and PFS tracked for anti-PDL1-treated renal cancer (Javelin) based on IFNG signature enrichment. Median enrichment was used to separate “High” and “Low” expressers.
(C) OS tracked for anti-PD1-treated melanoma, anti-PDL1-treated bladder cancer, and anti-PD1-treated renal cancer (Braun) and PFS tracked for anti-PDL1-treated renal cancer (Javelin) based on IMPRES scores. Median scores were used to separate “High” and “Low” scores.
(D) OS tracked for anti-PD1-treated melanoma, anti-PDL1-treated bladder cancer, and anti-PD1-treated renal cancer (Braun) and PFS tracked for anti-PDL1-treated renal cancer (Javelin) based on PD-L1 expression. Median expression was used to separate “High” and “Low” expressers.
(E) OS tracked for anti-PD1-treated melanoma, anti-PDL1-treated bladder cancer, and anti-PD1-treated renal cancer (Braun) and PFS tracked for anti-PDL1-treated renal cancer (Javelin) based on MHCI expression. Median expression was used to separate “High” and “Low” expressers.
(F) OS tracked for anti-PD1-treated melanoma, anti-PDL1-treated bladder cancer, and anti-PD1-treated renal cancer (Braun) and PFS tracked for anti-PDL1-treated renal cancer (Javelin) based on MHCII expression. Median expression was used to separate “High” and “Low” expressers.
The IMVIGOR210 bladder cancer clinical trial reported the tumor mutational burden (TMB) was predictive of improved survival in bladder cancer patients treated with anti-PDL1.22 Indeed, patients with high TMB displayed drastically improved survival in comparison with patients with low TMB (Figure S8A). Intriguingly, the survival benefit conferred by high TMB dissipates in patients belonging to clusters C1 and C2 (Figure S8A). Only C3 patients with low angiogenic activity and high T cell activity derive clinical benefit when stratified by TMB. This dependence is absent in tumors where TMB does not predict response like renal cell carcinoma (Figure S8B). This observation suggests that the biology underpinning angio-immune clusters is indispensable for the use of TMB as a predictive biomarker for ICB response. Together, we show that angio-immune subtypes display a remarkable universality and can effectively predict responses to ICB across different tumor types superior to other prognostic features.
Discussion
ICB has revolutionized cancer treatment outcomes for a subset of patients. However, a large subset of patients fails to show responses. It is imperative to (1) identify strategies to improve response rates for patients upon ICB treatment and (2) identify pretreatment characteristics of patients that can provide an a priori prediction to patients’ response to treatment. Such efforts can minimize toxicity profiles faced by patients while maximizing clinical benefits derived from ICB. Recent pre-clinical and clinical studies suggest that the status of tumor blood vessels can dictate immune responses in the TME. The vessel normalization hypothesis provides a framework for the interaction of blood vessels and infiltrating immune cells in the TME.32 Tumor vessel networks exhibiting low angiogenesis improve the infiltration of immune cells as they promote high endothelial venule (HEV) formation and as they upregulate expression of critical selectins and adhesion molecules imperative for the trafficking of leukocytes to the tumor parenchyma.11,33 Abnormal tumor blood vessels can also inhibit T cell effector activity by expressing FAS ligand and various inhibitory ligands.34
Herein, we examined whether baseline angiogenic state and corresponding T cell immune activity can provide us with tools to better inform treatment decision-making processes and delineate resistance mechanisms to ICB. We demonstrate that angiogenic activity and T cell-mediated immunity are inversely correlated across patients with 30 non-hematological solid tumor types. Distinct distribution of angiogenic and T cell activity enrichment across tumor types enabled the stratification of patients into three conserved tumor angio-immune subtypes, high angiogenesis, and low anti-tumor immunity (C1), low angiogenesis and high anti-tumor immunity (C3), and the one in-between (C2). The vasculature in C3 tumors is more normalized as evidenced by the higher pericyte to EC ratio. While tumor heterogeneity plagues efforts to develop overarching rules to define tumors independent of tissue of origin, the remarkable conservation of the distinct relationship between angiogenesis and T cell-mediated immunity across solid tumors allowed us to develop highly interpretable rules to predict ICB response across all tumor types.
The importance of baseline angiogenic state provides a strong foundation for a temporal treatment strategy characterized by vascular normalization followed by ICB. As such, patients with low angiogenesis levels and corresponding high T cell activity belonging to C3 consistently respond better to local-acting ICB in melanoma, gastric cancer, bladder cancer, and renal cell cancer. Notably, the immune features alone of the C3 fail to effectively predict responses in ICB patients with renal, gastric, and bladder cancer,35 suggesting the essential role angiogenesis may play in dictating the kind of immune response required to confer responses.
Re-evaluation of the Javelin 101, Checkmate 025, and Checkmate 010 renal cancer clinical trials provide therapeutic relevance of the identified TME subtypes. Patients categorized in the C3 angio-immune subtype demonstrated remarkable responses to the combination of axitinib, a small molecule tyrosine kinase inhibitor, and avelumab, an anti-PDL1, and received clinically significant improvements in survival compared with the previous standard of care sunitinib, a tyrosine kinase inhibitor, among the patients in the Javelin Renal 101 clinical trial. Conversely, patients in C2 and C1 did not benefit from axitinib combined with avelumab compared with the previous standard of care sunitinib. Patients harboring C3 angio-immune subtype were the only population to display the benefit of nivolumab, an anti-PD1 therapy, over the previous standard of care everolimus, an mTOR inhibitor, in the Checkmate 025 and Checkmate 010 clinical trials. This analysis suggests that the angio-immune subtypes can reliably identify a subset of patients that would derive clinical benefit from the ICB treatment in renal cell carcinoma.
ICB treatment allows the silencing of inhibitory signals on T cells to enable reactivity against tumor cells. The current clinical landscape of treatment with ICB targets two key checkpoint molecules: CTLA4 and PD1. CTLA4 is an inhibitory receptor that competes with co-stimulatory receptor CD28 for B7 (CD80/CD86) binding.36 B7 expression is primarily restricted to antigen-presenting cells in the tumor-draining lymph nodes.37 As such, the anti-CTLA4 is thought to affect the priming phase of CD8+ T cell activation primarily acting in lymph nodes. Our data that anti-CTLA4 response was better predicted predominantly by the immune scores alone probably reflects the mode of anti-CTLA4 action. In comparison, anti-PD1/PDL1 treatments affect during the effector phase in the tumor microenvironment. Tumor or tumor-associated macrophage upregulation of PD-L1 engages with inhibitory PD1 receptors on T cells.38 TME characteristics may have a more direct impact on anti-PD1/PDL1 efficacy. Immune features in C3 microenvironment include improved infiltration of anti-tumor immune cells, higher cytotoxicity activity of CD8+ T cells, higher expression of co-stimulatory molecules, and higher expression of markers of T cell exhaustion. The highly inflamed profile of C3 is partly due to the high mutational burden and neoantigen load in this category of patients. Importantly, we identified distinct differences in mutational profiles of patients belonging to different angio-immune subtypes. The distinct mutations are actionable and may present distinct treatment modalities for patients harboring differing TME subtypes. Local immune characteristics of patients belonging to C3 provide a compelling rationale for exploring treatment with locally acting anti-PD1/PDL1 ICB.
In summary, leveraging the biological relationship between angiogenesis and T cell immunity in the tumor microenvironment has allowed us to develop a framework with universal predictive power for identifying patients who will derive benefit from ICB treatment. Importantly, angio-immune subtypes outperform previous predictive methods and are remarkably conserved across tumor types. Future investigations are required to explore accessible surrogates for local angiogenic and immune activity. Serum levels of vascular endothelial growth factor (VEGF) have been shown to predict the angiogenic capacity of non-small cell lung cancer.39 Whether this finding is consistent with other tumor types remains to be seen. Similarly, circulating CD8+ T cells can be probed for their cytolytic activity against tumors. Using surrogate markers for local angiogenesis and T cell activity would allow us to classify patients into angio-immune subtypes. In addition, the newly evolved vascular normalization hypothesis calls for a revisiting of dosage and combination schemes to optimize the use of anti-angiogenics in the clinic. High toxicity profile of the current anti-angiogenics mainly targeting the VEGF pathway remains an obstacle. Recently uncovered anti-angiogenic targets that are dispensable for normal vessel maintenance like MYCT1 provide early pre-clinical promise toward a tolerable anti-angiogenic therapeutic.11 Such efforts will help to make more informed treatment decisions and enable us to maximize clinical benefit.
Limitations of the study
While this study provides insights into the importance of the association between tumor vessels and anti-tumor immune responses in the microenvironment, the nature of the study is correlative and retrospective. As such, we require robust validation with an adequately powered sample collection study to further validate the phenotypes studied. In addition, we primarily deconvoluted features of the microenvironment from heterogeneous bulk RNA sequencing data. Further validation using techniques with higher resolution, such as single-cell RNA sequencing, is required to avoid confounding variables from a heterogeneous tumor microenvironment. For the ultimate translation of the findings, appropriate surrogates for intratumoral angiogenesis and T cell activity that can be easily measured by clinicians are required.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Javelin Renal 101: Pretreatment RNA-seq | Motzer et al., 202046 | https://doi.org/10.1038/s41591-020-1044-8 |
| Hugo: Melanoma Pretreatment RNA-seq | Hugo et al., 201621 | GSE78220 |
| Gide: Melanoma Pretreatment RNA-seq | Gide et al., 201920 | ERP105482 |
| Liu: Melanoma Pretreatment RNA-seq | Liu et al., 201923 | phs000452 |
| Mariathasan: Bladder Cancer Pretreatment RNA-seq | Mariathasan et al., 201822 | EGAS00001002556 |
| Kim: Gastric cancer Pretreatment RNA-seq | Kim et al., 201824 | ERP107734 |
| TCGA Pancancer RNA Sequencing, Mutations, and clinical Data | Grossman et al., 201640 | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Braun: Renal Cancer Pretreatment RNA sequencing | Braun et al., 202026 | 10.1038/s41591-020-0839-year |
| Van Allen: Anti-CTLA4 Melanoma pretreatment RNA sequencing | Van Allen et al., 201544 | phs000452.v2.p1 |
| Nathonson: Anti-CTLA4 Melanoma pretreatment RNA sequencing | Nathanson et al., 201745 | https://doi.org/10.1158/2326-6066.CIR-16-0019 |
| Software and algorithms | ||
| GSVA | Hanzelmann et al., 2013 | https://www.bioconductor.org/packages/release/bioc/html/GSVA.html |
| pheatmap | Kolde, 201242 | https://cran.r-project.org/web/packages/pheatmap/index.html |
| msigdbr | N/A | https://cran.r-project.org/web/packages/msigdbr/ |
| maftools | Mayakonda et al., 2018 | https://bioconductor.org/packages/release/bioc/html/maftools.html |
| xCell | Aran et al., 201717 | https://github.com/dviraran/xCell |
| CIBERSORT | Chen et al., 2018 | https://cibersort.stanford.edu |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kyunghee Choi (kchoi@wustl.edu).
Materials availability
This study did not generate any new reagents.
Experimental model and subject details
Subject details
Data from all subjects analyzed has been previously published. Accession IDs and source for data can be found in the key resources table.
Method details
TCGA data
We used RNA, mutations, and clinical profiles for thirty non-hematological TCGA tumor types. Cancer types profiled include: Adrenocortical carcinoma (ACC), Bladder Urothelial Carcinoma (BLCA), Brain Lower Grade Glioma (LGG), Breast invasive carcinoma (BRCA), Colon adenocarcinoma (COAD), Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), Cholangiocarcinoma (CHOL), Esophageal carcinoma (ESCA), Glioblastoma multiforme (GBM), Head and Neck squamous cell carcinoma (HNSC), Kidney Chromophobe (KICH), Kidney renal clear cell carcinoma (KIRC), Kidney renal papillary cell carcinoma (KIRP), Liver hepatocellular carcinoma (LIHC), Lung adenocarcinoma (LUAD), Lung squamous cell carcinoma (LUSC), Mesothelioma (MESO), Ovarian serous cystadenocarcinoma (OV), Pancreatic adenocarcinoma (PAAD), Pheochromocytoma and Paraganglioma (PCPG), Prostate adenocarcinoma (PRAD), Rectum adenocarcinoma (READ), Sarcoma (SARC), Skin Cutaneous Melanoma (SKCM), Stomach adenocarcinoma (STAD), Testicular Germ Cell Tumors (TGCT), Thyroid carcinoma (THCA), Uterine Carcinosarcoma (UCS), Uterine Corpus Endometrial Carcinoma (UCEC), and Uveal Melanoma (UVM).
RNA sequencing data
RNA sequencing data for 11,069 patients was downloaded from the GDC pan cancer portal (https://gdc.cancer.gov/about-data/publications/pancanatlas).40 Data was processed using the Firehose pipeline with upper quantile normalization. For patients with more than one RNA-seq sample, primary tumor sample was favored. RNA sequencing samples from patients with DLBC and LAML were excluded.
Mutations
Version 2.8 of the mutations annotation file (MAF) generated by the MC3 group was downloaded from the GDC pan cancer portal (https://gdc.cancer.gov/about-data/publications/pancanatlas).40 Samples from patients with DLBC and LAML were excluded. Cluster annotations were added to the MAF files. The maftools R package was used for visualization purposes.
Clinical data
Survival information was derived from TCGA-Clinical Data Resource (CDR) Outcome file provided in the GDC pan cancer portal (https://gdc.cancer.gov/about-data/publications/pancanatlas).40
Patient stratification
Building of angio-immune score matrix
An unbiased selection of all gene sets relating to endothelial cell activity and T cell activity from the molecular signatures database was conducted.41 A total of 91 gene signatures were identified and compiled to curate the angio-immune gene set collection. Gene set variation analysis (GSVA) was implemented to score the enrichment of 91 gene sets among patients of 30 TCGA cohorts to generate matrix of enrichment scores.
Clustering gene sets
Correlation matrix using Pearson coefficients were generated across enrichment scores for individual gene sets. Pheatmap package in R was used to generate a hierarchically clustered heatmap.42 Two modules of gene sets were identified and characterized based on the gene set membership.
Clustering patients
Correlation matrix using Pearson coefficients were generated across enrichment scores for patients. Pheatmap package in R was used to generate a hierarchically clustered heatmap. Three angio-immune subsets were identified and characterized based on distribution of enrichment of different gene sets.
Immune characteristics of tumors
Immune and stromal cell enrichment
xCell, a gene signatures-based enrichment approach, was used to delineate enrichment of 64 immune and stromal cell types as previously described.17 Briefly, the xCell R package was used generate raw enrichment scores, transform into linear scale, and apply a spillover compensation to derive corrected enrichment scores. Distribution of enrichment scores for patients belonging to different angio-immune clusters were compared.
Immune cell deconvolution
CIBERSORT,43 which uses a nu-support vector regression algorithm to estimate cell fractions in inputted mixture files, was used to depict immune cell fractions from bulk RNA-Seq data as previously described. Briefly, the leukocyte signature matrix, LM22, containing the expression of 547 genes across 22 immune cell subtypes, was used to deconvolute immune cell fractions for each patient in all cohorts. CIBERSORT was run on the R environment (version 4.04) using 100 permutations to generate leukocyte fractions for each patient. An alpha value of 0.05 was used to filter nonsignificant deconvolution results. Patients were grouped by angio-immune cluster and distribution of cell type fraction was compared. Neoantigen load and TCR richness were downloaded from the GDC pan cancer portal (https://gdc.cancer.gov/about-data/publications/pancanatlas).
Immunotherapy datasets
Pretreatment TPM normalized RNA sequencing data from anti-PD1/L1 and anti-CTLA4 treated cohorts were downloaded for the following studies: Gide,20 Hugo,21 Liu,23 Van Allen,44 Nathanson,45 Mariathasan,22 Kim,24 Braun,26 and Motzer.46 Studies were selected based on the following criteria: >20 samples for enrichment calculation, RNA sequencing of pretreatment biopsies, and only anti-PD1/PDL1 treated patients. Gene signature enrichment and molecular subtypes were derived as described elsewhere in the manuscript. Survival and response rates to treatment when available were compared among angio-immune clusters.
Quantification and statistical analysis
Unless indicated elsewhere, all visualizations were completed in GraphPad Prism 9.
GraphPad Prism 9 was used for all statistical analysis. Data is presented as mean ± SE of mean. One-way ANOVA was used for comparison across more than two groups. Non-parametric tests were used when data was not normally distributed. Log-rank tests were used for all survival analysis. Alpha value of 0.05 was used throughout the study.
Acknowledgments
This work was supported by Siteman Investment Program Research Development Awards and NIH grants R01HL149954 and R01HL55337 (to K.C.).
Author contributions
M.S., A.U.K., and K.C. conceived the manuscript idea and wrote the paper. M.S. designed all procedures and conducted all analyses. A.U.K. and D.A.G.B. provided analysis suggestions. K.K., A.U.K., and D.A.G.B. provided editorial assistance. K.C. provided overall supervision.
Declaration of interests
The authors declare no competing interests.
Published: January 10, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100896.
Supplemental information
Data and code availability
All datasets used can be accessed using the references listed. All analysis data, gene sets, and code has been deposited in Dryad (https://doi.org/10.5061/dryad.v41ns1s11). All datasets used for analysis are indicated in the key resources table. Any further requests can be made to the lead contact.
References
- 1.Dagogo-Jack I., Shaw A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018;15:81–94. doi: 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 2.Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A., et al. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H., Li C., Peng X., Zhou Z., Weinstein J.N., Cancer Genome Atlas Research Network. Liang H. A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell. 2018;173:386–399.e12. doi: 10.1016/j.cell.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng X., Chen Z., Farshidfar F., Xu X., Lorenzi P.L., Wang Y., Cheng F., Tan L., Mojumdar K., Du D., et al. Molecular characterization and clinical relevance of metabolic expression subtypes in human cancers. Cell Rep. 2018;23:255–269.e4. doi: 10.1016/j.celrep.2018.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearson J.D., Huang K., Pacal M., McCurdy S.R., Lu S., Aubry A., Yu T., Wadosky K.M., Zhang L., Wang T., et al. Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity. Cancer Cell. 2021;39:1115–1134.e12. doi: 10.1016/j.ccell.2021.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagaev A., Kotlov N., Nomie K., Svekolkin V., Gafurov A., Isaeva O., Osokin N., Kozlov I., Frenkel F., Gancharova O., et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. 2021;39:845–865.e7. doi: 10.1016/j.ccell.2021.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Abou Khouzam R., Brodaczewska K., Filipiak A., Zeinelabdin N.A., Buart S., Szczylik C., Kieda C., Chouaib S. Tumor hypoxia regulates immune escape/invasion: influence on angiogenesis and potential impact of hypoxic biomarkers on cancer therapies. Front. Immunol. 2020;11:613114. doi: 10.3389/fimmu.2020.613114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischbeck A.J., Ruehland S., Ettinger A., Paetzold K., Masouris I., Noessner E., Mendler A.N. Tumor lactic acidosis: protecting tumor by inhibiting cytotoxic activity through motility arrest and bioenergetic silencing. Front. Oncol. 2020;10:589434. doi: 10.3389/fonc.2020.589434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Motz G.T., Santoro S.P., Wang L.P., Garrabrant T., Lastra R.R., Hagemann I.S., Lal P., Feldman M.D., Benencia F., Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014;20:607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmittnaegel M., Rigamonti N., Kadioglu E., Cassará A., Wyser Rmili C., Kiialainen A., Kienast Y., Mueller H.J., Ooi C.H., Laoui D., De Palma M. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017;9:eaak9670. doi: 10.1126/scitranslmed.aak9670. [DOI] [PubMed] [Google Scholar]
- 11.Kabir A.U., Subramanian M., Lee D.H., Wang X., Krchma K., Wu J., Naismith T., Halabi C.M., Kim J.Y., Pulous F.E., et al. Dual role of endothelial Myct1 in tumor angiogenesis and tumor immunity. Sci. Transl. Med. 2021;13:eabb6731. doi: 10.1126/scitranslmed.abb6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen E., Jabouille A., Rivera L.B., Lodewijckx I., Missiaen R., Steri V., Feyen K., Tawney J., Hanahan D., Michael I.P., Bergers G. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017;9:eaak9679. doi: 10.1126/scitranslmed.aak9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dammeijer F., Lau S.P., van Eijck C.H.J., van der Burg S.H., Aerts J.G.J.V. Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev. 2017;36:5–15. doi: 10.1016/j.cytogfr.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Assié G., Letouzé E., Fassnacht M., Jouinot A., Luscap W., Barreau O., Omeiri H., Rodriguez S., Perlemoine K., René-Corail F., et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014;46:607–612. doi: 10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- 17.Aran D., Hu Z., Butte A.J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan T.A., Yarchoan M., Jaffee E., Swanton C., Quezada S.A., Stenzinger A., Peters S. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 2019;30:44–56. doi: 10.1093/annonc/mdy495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruni D., Angell H.K., Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer. 2020;20:662–680. doi: 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- 20.Gide T.N., Quek C., Menzies A.M., Tasker A.T., Shang P., Holst J., Madore J., Lim S.Y., Velickovic R., Wongchenko M., et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell. 2019;35:238–255.e6. doi: 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., III, Koeppen H., Astarita J.L., Cubas R., et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu D., Schilling B., Liu D., Sucker A., Livingstone E., Jerby-Arnon L., Zimmer L., Gutzmer R., Satzger I., Loquai C., et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019;25:1916–1927. doi: 10.1038/s41591-019-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S.T., Cristescu R., Bass A.J., Kim K.M., Odegaard J.I., Kim K., Liu X.Q., Sher X., Jung H., Lee M., et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018;24:1449–1458. doi: 10.1038/s41591-018-0101-z. [DOI] [PubMed] [Google Scholar]
- 25.Motzer R.J., Penkov K., Haanen J., Rini B., Albiges L., Campbell M.T., Venugopal B., Kollmannsberger C., Negrier S., Uemura M., et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019;380:1103–1115. doi: 10.1056/NEJMoa1816047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braun D.A., Hou Y., Bakouny Z., Ficial M., Sant' Angelo M., Forman J., Ross-Macdonald P., Berger A.C., Jegede O.A., Elagina L., et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020;26:909–918. doi: 10.1038/s41591-020-0839-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu Y., Wen J., Thomas G., Yang W., Prior W., He W., Sundar P., Wang X., Potluri S., Salek-Ardakani S. Baseline frequency of inflammatory Cxcl9-expressing tumor-associated macrophages predicts response to avelumab treatment. Cell Rep. 2020;32:107873. doi: 10.1016/j.celrep.2020.107873. [DOI] [PubMed] [Google Scholar]
- 28.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V., et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Auslander N., Zhang G., Lee J.S., Frederick D.T., Miao B., Moll T., Tian T., Wei Z., Madan S., Sullivan R.J., et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018;24:1545–1549. doi: 10.1038/s41591-018-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doroshow D.B., Bhalla S., Beasley M.B., Sholl L.M., Kerr K.M., Gnjatic S., Wistuba I.I., Rimm D.L., Tsao M.S., Hirsch F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021;18:345–362. doi: 10.1038/s41571-021-00473-5. [DOI] [PubMed] [Google Scholar]
- 31.Rodig S.J., Gusenleitner D., Jackson D.G., Gjini E., Giobbie-Hurder A., Jin C., Chang H., Lovitch S.B., Horak C., Weber J.S., et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018;10:eaar3342. doi: 10.1126/scitranslmed.aar3342. [DOI] [PubMed] [Google Scholar]
- 32.Viallard C., Larrivée B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis. 2017;20:409–426. doi: 10.1007/s10456-017-9562-9. [DOI] [PubMed] [Google Scholar]
- 33.Asrir A., Tardiveau C., Coudert J., Laffont R., Blanchard L., Bellard E., Veerman K., Bettini S., Lafouresse F., Vina E., et al. Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to PD-1 plus CTLA-4 combination immunotherapy. Cancer Cell. 2022;40:318–334.e9. doi: 10.1016/j.ccell.2022.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Schaaf M.B., Garg A.D., Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018;9:115. doi: 10.1038/s41419-017-0061-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGrail D.J., Pilié P.G., Rashid N.U., Voorwerk L., Slagter M., Kok M., Jonasch E., Khasraw M., Heimberger A.B., Lim B., et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021;32:661–672. doi: 10.1016/j.annonc.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leach D.R., Krummel M.F., Allison J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 37.Brzostek J., Gascoigne N.R.J., Rybakin V. Cell type-specific regulation of immunological synapse dynamics by B7 ligand recognition. Front. Immunol. 2016;7:24. doi: 10.3389/fimmu.2016.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei S.C., Duffy C.R., Allison J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 39.Tamura M., Ohta Y., Kajita T., Kimura K., Go T., Oda M., Nakamura H., Watanabe G. Plasma VEGF concentration can predict the tumor angiogenic capacity in non-small cell lung cancer. Oncol. Rep. 2001;8:1097–1102. doi: 10.3892/or.8.5.1097. [DOI] [PubMed] [Google Scholar]
- 40.Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A., Staudt L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016;375:1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolde R. Pheatmap: pretty heatmaps. R package version. 2012;1:726. [Google Scholar]
- 43.Chen B., Khodadoust M.S., Liu C.L., Newman A.M., Alizadeh A.A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 2018;1711:243–259. doi: 10.1007/978-1-4939-7493-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Allen E.M., Miao D., Schilling B., Shukla S.A., Blank C., Zimmer L., Sucker A., Hillen U., Foppen M.H.G., Goldinger S.M., et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nathanson T., Ahuja A., Rubinsteyn A., Aksoy B.A., Hellmann M.D., Miao D., Van Allen E., Merghoub T., Wolchok J.D., Snyder A., Hammerbacher J. Somatic mutations and neoepitope homology in melanomas treated with CTLA-4 blockade. Cancer Immunol. Res. 2017;5:84–91. doi: 10.1158/2326-6066.CIR-16-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Motzer R.J., Robbins P.B., Powles T., Albiges L., Haanen J.B., Larkin J., Mu X.J., Ching K.A., Uemura M., Pal S.K., et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020;26:1733–1741. doi: 10.1038/s41591-020-1044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets used can be accessed using the references listed. All analysis data, gene sets, and code has been deposited in Dryad (https://doi.org/10.5061/dryad.v41ns1s11). All datasets used for analysis are indicated in the key resources table. Any further requests can be made to the lead contact.