

SUMMARY
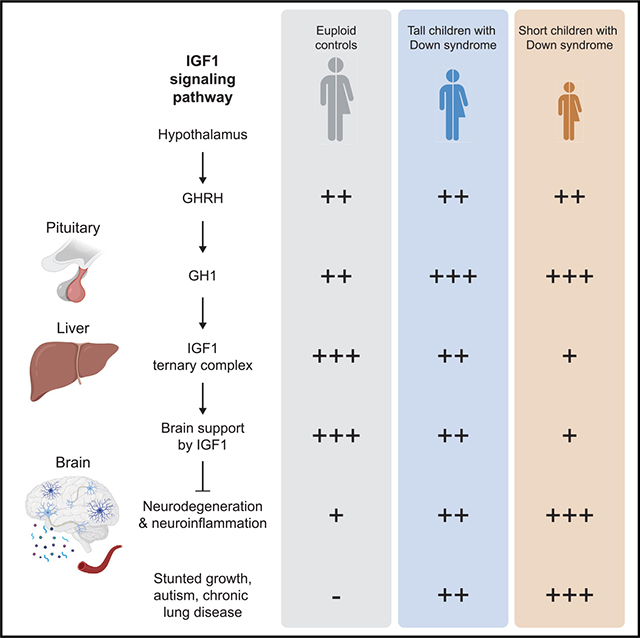
Down syndrome (DS), the genetic condition caused by trisomy 21 (T21), is characterized by stunted growth, cognitive impairment, and increased risk of diverse neurological conditions. Although signs of lifelong neurodegeneration are well documented in DS, the mechanisms underlying this phenotype await elucidation. Here we report a multi-omics analysis of neurodegeneration and neuroinflammation biomarkers, plasma proteomics, and immune profiling in a diverse cohort of more than 400 research participants. We identified depletion of insulin growth factor 1 (IGF1), a master regulator of growth and brain development, as the top biosignature associated with neurodegeneration in DS. Individuals with T21 display chronic IGF1 deficiency downstream of growth hormone production, associated with a specific inflammatory profile involving elevated tumor necrosis factor alpha (TNF-α). Shorter children with DS show stronger IGF1 deficiency, elevated biomarkers of neurodegeneration, and increased prevalence of autism and other conditions. These results point to disruption of IGF1 signaling as a potential contributor to stunted growth and neurodegeneration in DS.
In brief
Individuals with Down syndrome display stunted growth, chronic inflammation, and elevated signs of neurodegeneration across their lifespan. Araya et al. show that chronic deficiency in IGF1, a master regulator of human development, is associated with stunted growth, a specific inflammatory profile, and elevated signs of neurodegeneration in Down syndrome.
Graphical Abstract
INTRODUCTION
Down syndrome (DS) is caused by trisomy 21 (T21), the most common chromosomal abnormality, affecting ~1 in 700 newborns.1 T21 affects the development of multiple organ systems, leading to stunted growth, craniofacial abnormalities, dysregulated brain morphogenesis, and high frequency of congenital heart defects.2 Individuals with T21 experience a differential disease spectrum relative to the typical population, where they are protected from developing certain conditions, such as most solid malignances,3 while also being predisposed to multiple autoimmune disorders, leukemias, specific cardiovascular and cardiopulmonary conditions, Alzheimer’s disease (AD), autism spectrum disorders (ASDs), and seizure disorders.4,5 Therefore, elucidation of the mechanisms underlying these developmental and clinical hallmarks will serve people with DS as well as the general population affected by these conditions.
Individuals with DS present a distinct neurological profile across their lifespan. The cognitive phenotype of DS involves impairments in morphosyntax, verbal short-term memory, and explicit long-term memory.6 In addition to the likely deleterious effects of decreased brain volume, reduced neuronal density, and dysregulation of diverse neurotransmitter systems, cognitive function can also be affected by vision and hearing issues prevalent in DS.6,7 Co-occurring neurological disorders more common in DS, such as ASD and seizure disorders, can exacerbate cognitive impairment.6,8 Later in life, the early onset of AD pathology leads to cognitive decline and dementia, with high inter-individual variability.9 With a few exceptions, the mechanisms by which T21 causes these phenotypes are unclear. The high prevalence of AD can be attributed to triplication of the amyloid precursor protein (APP) gene encoded on chromosome 21 (chr21); however, other triplicated genes may also contribute to AD pathology.9 A clear definition of gene-phenotype associations has likely been impeded by the fact that multiple triplicated genes may contribute to a given phenotype. In terms of neurodevelopment and cognitive function, dysregulation of highly intertwined molecular and cellular processes are likely to make important contributions to these hallmarks.6,8 Therefore, identification of core mechanisms underlying the neurodevelopmental profile of DS could illuminate therapeutic strategies to ameliorate the ill effects of the trisomy.
Recently, validated biomarkers have been used to assess neurodegeneration in multiple neurological conditions.10 Neurofilament light chain (NfL), a structural component of the neural cytoskeleton, and the microtubule-associated protein TAU (TAU) are used to assess axonal degeneration or injury; ubiquitin carboxyl-terminal hydroxyl L1 (UCHL1) is employed as a biomarker of neuronal cell death; and glial fibrillary acid protein (GFAP) serves as a proxy of reactive astrocytes and neuroinflammation. These biomarkers are elevated in diverse dementias, multiple sclerosis, movement disorders, and traumatic brain injury.10,11 Numerous studies have reported that individuals with DS present elevated levels of these biomarkers in plasma and cerebrospinal fluid (CSF).12–18 Plasma levels of NfL have prognostic value for AD progression in DS.13,14,16,19–21 Despite these advances, the mechanisms driving neurodegeneration in DS are poorly understood. Individuals with DS present signs of chronic inflammation and immune dysregulation, including heightened neuroinflammation, across their lifespan.22–27 Activation of astrocytes and microglia and increased production of inflammatory cytokines are observed in the brains of those with DS from early childhood,28,29 suggesting a potential contribution of inflammatory processes to neurodegeneration in DS. In mouse models of DS, multiple anti-inflammatory agents have been shown to improve cognition.30–33
In this context, we investigated the interplay between neurodegeneration, inflammation, and other physiological processes dysregulated in DS by completing a multi-omics analysis using matched measurements of NfL, TAU, UCHL1, and GFAP, along with plasma proteomics and inflammatory marker profiling, in a cohort of more than 400 participants, more than 300 of them with T21, from early childhood to late adult life. These efforts revealed that the top biosignature associated with neurodegeneration in DS is depletion of the insulin growth factor 1 (IGF1) complex, a master regulator of human growth and brain development. Immune profiling revealed that neurodegeneration and IGF1 deficiency are associated with a distinct inflammatory profile in DS, marked by elevated tumor necrosis factor alpha (TNF-α) and other inflammatory factors. We found that children with DS of shorter stature display stronger depletion of the IGF1 complex and increased markers of neurodegeneration and neuroinflammation. Last, analysis of clinical data from more than 1,000 children and young adults with DS revealed that shorter stature is associated with increased risk of specific co-occurring conditions, most prominently ASD, autoimmune diagnoses, and chronic lung disease. These results indicate that inflammation-associated disruption of IGF1 signaling could contribute to stunted growth, neurodegeneration, and the appearance of key co-occurring conditions in DS.
RESULTS
Neurodegeneration in DS is associated with decreased IGF1 signaling
To investigate signaling pathways associated with neurodegeneration and neuroinflammation in DS, we measured biomarkers associated with these processes in plasma samples obtained from a large cohort of individuals with and without T21 for whom matching multi-omics datasets are available (Figure S1A). We measured NfL, total TAU, UCHL1, and GFAP using single molecular array assays (SIMOA) in a total of 419 research participants, 316 of them from individuals with DS (T21), and 103 age- and sex-matched euploid controls (D21) (STAR Methods; Figure S1A; Tables S1 and S2). Overall, the cohort with DS shows significantly higher levels of NfL, TAU, and UCHL1 but not GFAP relative to euploid controls (Figure S1B). Age trajectory plots revealed differential behaviors of each marker across the lifespan (Figure S1C). For example, although NfL increases with age throughout adulthood, GFAP levels decrease in adolescents with or without T21 (Figure S1C). We then completed a correlation analysis between the four neurodegeneration/neuroinflammation biomarkers in individuals with and without T21, which revealed significant positive correlations between some of these biomarkers (Figure S1D). In the T21 cohort, NfL levels are significantly positively correlated with levels of UCHL1 and GFAP but not with TAU. GFAP is positively correlated with the other three neurodegeneration biomarkers. TAU is positively associated with UCHL1 and GFAP levels in individuals with DS (Figure S1D). When these correlations were defined in the smaller euploid control cohort, we observed that NfL was again significantly positively associated with GFAP, but GFAP, TAU, and UCHL1 did not show any other significant associations (Figure S1D). When results are analyzed by different age groups encompassing children (0–18 years old), young adults (18+ through 30), and older adults (30+), NfL is significantly higher in DS in all age brackets with age-associated increases in both karyotypes, TAU is significantly elevated only in children and older adults with DS, UCHL1 is significantly increased in all age groups with DS but without a strong age effect, and GFAP is significantly higher only in older adults with DS (Figure 1A). These results indicate that, although neurodegeneration and neuroinflammation may be more pronounced in older adults with DS and potentially linked to early onset of AD, biochemical evidence suggestive of neurodegeneration is clear even among young children with T21.
Figure 1. Proteomic biosignatures of neurodegeneration in DS associated with IGF1 deficiency.
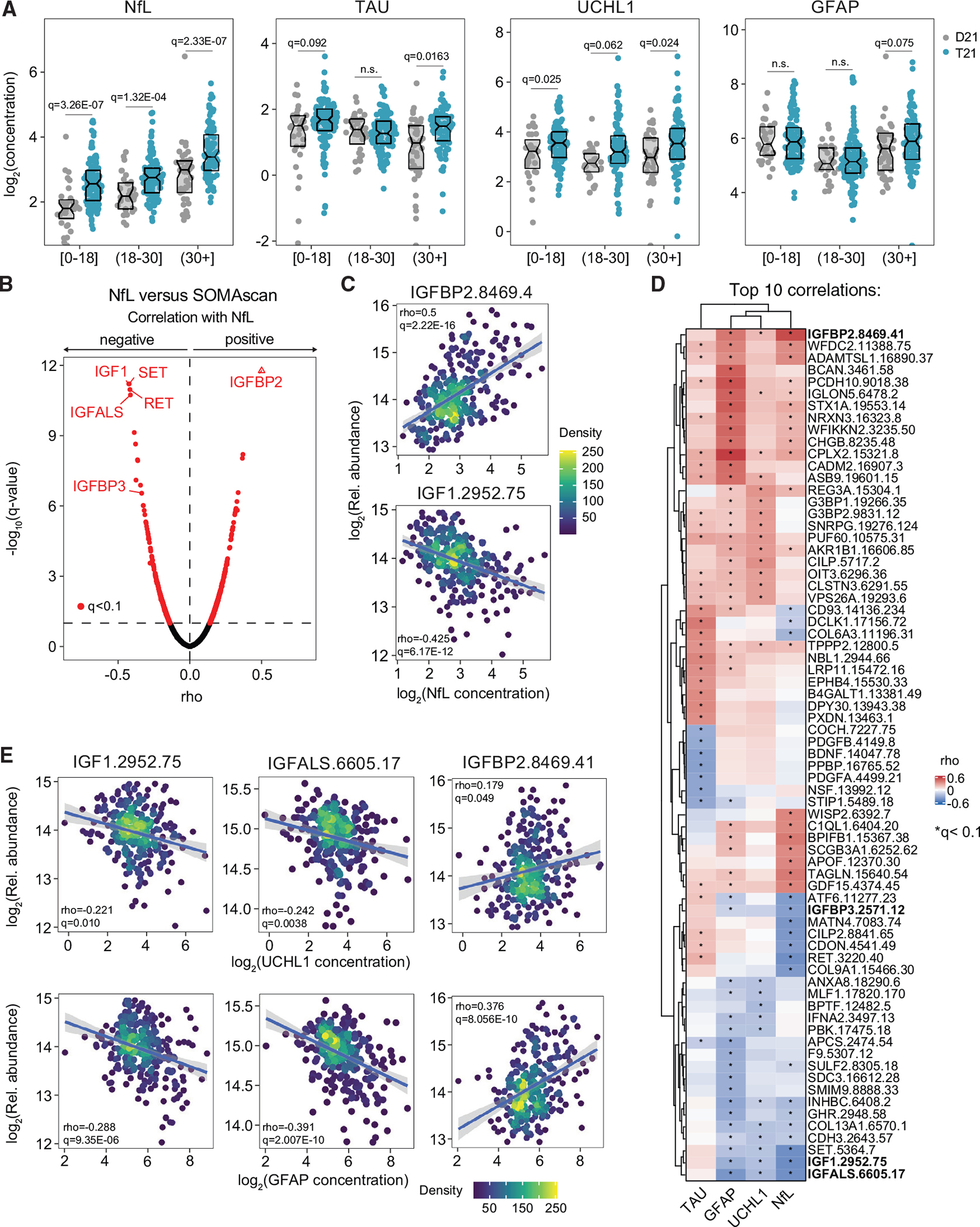
Levels of neurodegeneration/neuroinflammation biomarkers were measured in plasma samples from 419 research participants, 316 of them with trisomy 21 (T21), versus 103 age- and sex-matched euploid controls (D21).
(A) Sina plots displaying levels of the indicated biomarkers at different age brackets. Data are presented as sina plots, with boxes indicating median and interquartile range. Differences between groups were determined with a multivariable linear regression with age, sex, and source as covariables with Benjamini-Hochberg (BH) correction of p values. Significance is defined by q < 0.1.
(B) Volcano plot for Spearman correlations between circulating NfL levels and plasma proteins.
(C) Scatterplots for levels of IGFBP2 (top) and IGF1 (bottom) correlated with NfL levels among individuals with T21. Values shown represent Spearman rho values. Points are colored by density. Lines represent a simple linear regression with 95% confidence interval.
(D) Heatmap representing the top 10 proteins that are significantly correlated, positively or negatively, with the indicated biomarkers. Values displayed represent Spearman rho values.
(E) Scatterplots for levels of IGF1, IGFALS, and IGFBP2 correlated with levels of UCHL1 (top) and GFAP (bottom) among individuals with T21. Other details are as described in (C).
Next, to identify biosignatures associated with each of these biomarkers in DS, we defined Spearman correlations for each of them versus mor than 4,500 epitopes measured by SOMAscan proteomics in the same plasma samples. This exercise revealed hundreds of significant correlations for each biomarker (Figures 1B and S1E–S1G; Table S2). For example, the most positively correlated protein with NfL is IGF binding protein 2 (IGFBP2), a negative regulator of IGF1 signaling.34 The most negatively correlated proteins with NfL are IGF1, SET, RET, and IGFBP acid labile subunit (IGFALS) (Figure 1B; Table S2). IGF1 is a master regulator of human development necessary for brain growth, maturation, and regeneration.35,36 SET is a multifunctional protein for which loss-of-function mutations have been associated with neurodevelopmental disorders.37 RET is a receptor for the glial cell line-derived neurotrophic factor (GDNF), which is critical for development of the nervous system.38 IGFALS is a subunit of the IGF1 signaling ternary complex, which stabilizes and increases bioavailability of IGF1 in circulation39 and for which loss-of-function mutations have been associated with diverse growth disorders.40 The third subunit of the IGF1 ternary complex, IGFBP3, which also increases the half-life of IGF1,41 is the 10th most negatively correlated protein with NfL (Figure 1B; Table S2). Therefore, circulating levels of NfL are negatively correlated with IGF1 signaling, as denoted by a positive correlation with the IGF1 repressor IGFBP2 and negative correlations with subunits of the IGF1/IGFALS/IGFBP3 ternary complex (Figure 1C).
To compare the patterns of correlations for the four biomarkers in individuals with T21, we visualized the top 10 positively and negatively correlated proteins for each of them, along with unsupervised hierarchical clustering (Figure 1D). This exercise produced several interesting observations. Most prominently, IGF1 signaling is also negatively associated with circulating levels of UCHL1 and GFAP because both markers are significantly anti-correlated with IGF1 and IGFALS and significantly positively correlated with IGFBP2 (Figure 1E). IGFALS is the most negatively correlated protein with UCHL1 and GFAP (Figures S1F and S1G). Another shared feature is a positive association between all four markers and Complexin-2 (CPLX2), a factor involved in synaptic vesicle exocytosis dysregulated in diverse neurological disorders.42 IGLON5, a member of the IgLON (immunoglobulin-like domain) family of neural cell adhesion molecules known to regulate neurite outgrowth and synapse formation,43 is positively correlated with NfL, UCHL1, and GFAP (Figure 1D). Among the four markers, TAU displays the most dissimilar pattern of associations (Figure 1D; Table S2). The most positively associated protein with TAU is doublecortin-like kinase 1 (DCLK1) (Figure S1E), a protein kinase involved in neuronal migration, retrograde transport, and dendritic remodeling.44 In contrast, TAU levels are negatively associated with circulating levels of the brain-derived neurotrophic factor (BDNF), a key neurotrophic factor45 (Figure S1E). When we analyzed the pattern of correlations in euploid controls, we observed similarities and differences (Figure S1D; Table S2). Although some differences in the results can be accounted for by the lesser statistical power in the euploid cohort, other differences are clearly indicative of different physiologies between the two groups. For example, NfL levels are significantly associated with levels of IGFBP2 and negatively correlated with levels of IGF1 and IGFALS in euploid controls as well (Figure S1D). However, TAU was significantly positively associated with IGF1 among euploid controls, which is not observed in DS (Figure S1D).
These results indicate that neurodegeneration in DS is highly variable among individuals and that it associates with specific signaling pathways reflected in plasma protein signatures, with a negative correlation with IGF1 signaling being the most prominent example. Thus, hereafter, we focused our analyses on the IGF1 pathway, which has well-recognized neuroprotective effects in humans and mice, with critical roles in early brain development, neurogenesis, and synaptogenesis.35,36
IGF1 levels are decreased downstream of growth hormone production in DS
IGF1 is a master regulator of human development, acting downstream of the pituitary growth hormone (GH1)46 (Figure S2A). Upon release of GH-releasing hormone (GHRH) from the hypothalamus, the pituitary gland releases GH1 into the bloodstream. In the liver, GH1 stimulates production of IGF1 and several IGFBPs.46 Although IGF1 is expressed in multiple tissues, more than 75% of it is produced by the liver.47 In the bloodstream, IGF1 exists mostly as a ternary complex with IGFBP3 and IGFALS, which stabilize IGF1 and increase its availability throughout the human body.48 IGF1 crosses the blood-brain barrier (BBB) to reach the central nervous system (CNS), where it promotes brain development and function.35 We therefore investigated the proteomics dataset for potential differences in the levels of multiple factors involved in GH1/IGF1 signaling in DS.
Individuals with DS show significantly decreased levels of all three subunits of the IGF1 complex (Figure 2A). Age trajectory plots show that levels of the ternary complex increase in the first two decades of life and decrease later in adult life, with all three subunits being depleted in DS compared with euploid individuals, particularly at younger ages (Figure 2B). GH1 is mildly but significantly elevated in people with T21 compared with euploid controls (Figure 2A), but the levels of GHRH are not significantly different (Figure S2B). Elevated GH1 without a concomitant increase in GHRH could be explained by decreased negative feedback by IGF1 on the pituitary/hypothalamus axis46 (Figure S2A). On average, levels of IGFBP2, the major IGF1 antagonist, are not obviously different in DS (Figure S2C). However, its age trajectory is reversed relative to IGF1, decreasing in the first two decades of life but increasing with age in adults (Figure S2C).
Figure 2. Individuals with DS display chronic IGF1 deficiency downstream of GH1 production.
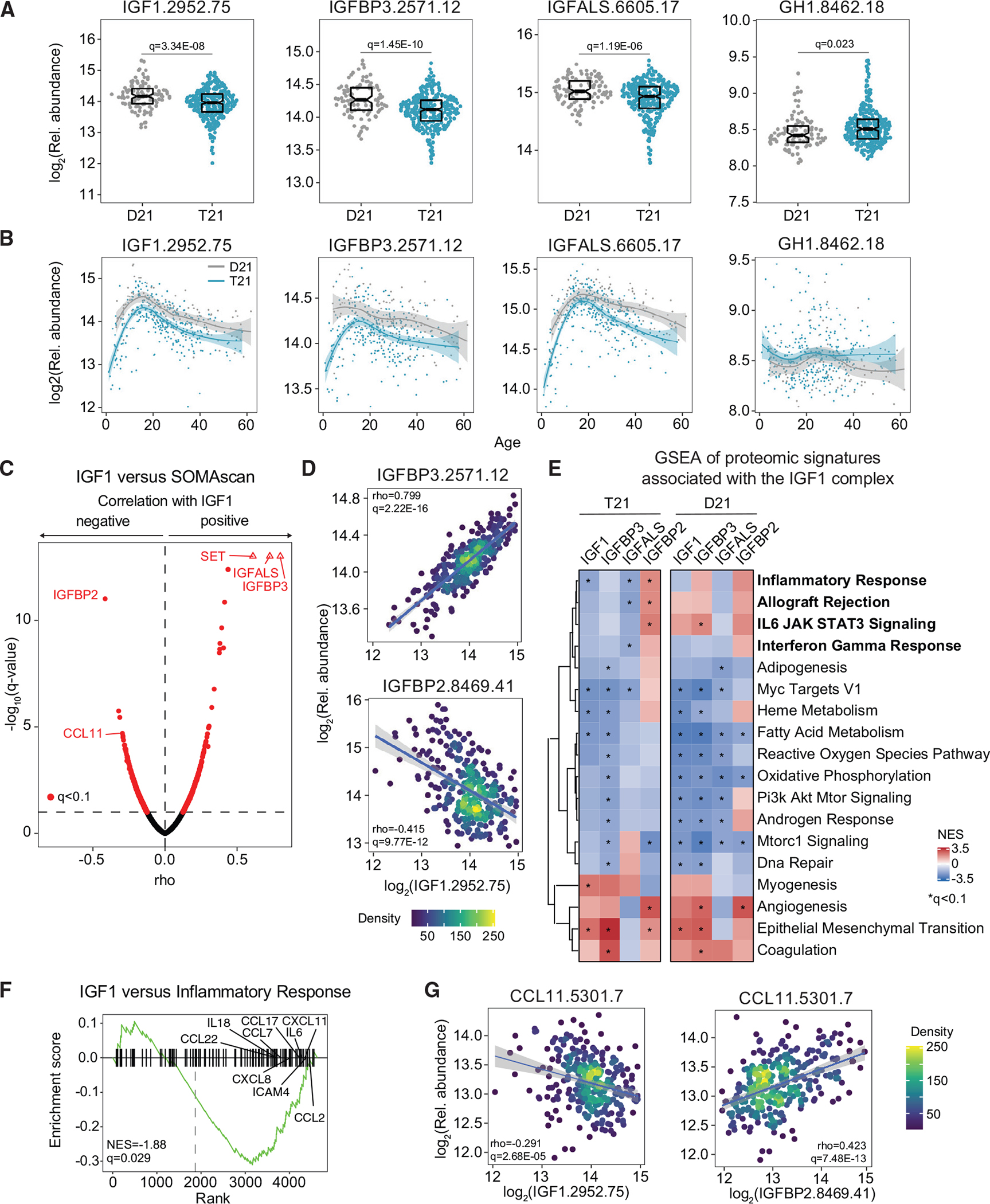
Factors in the GH1/IGF1 signaling pathway were measured in plasma samples from 419 research participants, 316 of them with T21, versus 103 age- and sex-matched euploid controls (D21).
(A) Sina plots displaying levels of IGF1, IGFBP3, IGFALS, and GH1 in individuals with and without T21. Data are presented as sina plots, with boxes indicating median and interquartile range. Differences between groups were determined with a multivariable linear regression with age, sex, and source as covariables with BH correction of p values.
(B) LOESS age trajectory plots for IGF1, IGFBP3, IGFALS, and GH1 in individuals with and without T21.
(C) Volcano plot of Spearman correlations between circulating levels of IGF1 and plasma proteins in individuals with T21.
(D) Scatterplots for levels of IGFBP3 (top) and IGFBP2 (bottom) versus IGF1 among individuals with T21. Values shown represent Spearman rho. The q values were calculated with the BH method. Points are colored by density. Lines represent a simple linear regression with 95% confidence interval.
(E) Pathways significantly enriched by normalized enrichment score (NES) from weighted gene set enrichment analysis (GSEA) of proteins that are significantly positively and negatively correlated with IGF1 in individuals with and without T21.
(F) Enrichment plot of the hallmark inflammatory response gene set negatively correlated with IGF1 levels in people with DS.
(G) Scatterplots for levels of CCL11 (eotaxin) protein correlated with levels of IGF1 (left) and IGFBP2 (right) among individuals with T21. Details are as in (D).
To investigate pathways associated with IGF1 deficiency, we defined biosignatures associated with varying levels of IGF1, IGFBP3, IGFALS, and IGFBP2 in DS by calculating Spearman correlations between these factors and the rest of the proteomics dataset (Figures 2C and S2D; Table S3). The top positive correlations with IGF1 are IGFBP3 and IGFALS, whereas the most negative correlated protein is IGFBP2 (Figures 2C and 2D). This pattern of correlations is conserved for IGFBP3 and IGFALS and reversed for IGFBP2 (Figure S2D), consistent with the notion that IGFBP3 and IGFALS stabilize IGF1 in circulation, whereas IGFBP2 destabilizes the ternary complex.41 Gene set enrichment analysis (GSEA) of proteome-wide correlations identified numerous pathways associated with varying levels of the IGF1 complex. Multiple inflammatory pathways are consistently associated with depletion of the IGF1 complex with an inverse pattern for IGFBP2, including inflammatory response, allograft rejection, interleukin-6 (IL-6)/JAK (Janus kinase)/STAT3 (Signal Transducer and Activator of Transcription 3) signaling, and interferon gamma response (Figures 2E and 2F; Table S3). Proteins in the inflammatory response signature have statistically significant negative associations with IGF1 and IGFALS and significant positive associations with IGFBP2. An inflammatory protein illustrating this behavior is CCL11 (eotaxin), a chemokine with chemotactic activity for eosinophils and T cells,49 which is the fourth most negatively correlated protein with IGF1 (Figures 2C and 2G) and also negatively associated with levels of IGFBP3 and IGFALS but positively associated with IGFBP2 (Figure S2D). Next we analyzed proteomics signatures associated with IGF1 signaling factors among euploid controls (Table S3). IGF1 levels also correlate positively with levels of IGFBP3 and IGFALS and negatively with levels of IGFBP2 among controls (Figures S2E and S2F). Pathway analysis revealed similarities and differences in the signatures associated with IGF1 signaling in DS versus controls (Figure 2E). Although a few inflammatory markers where also associated with lower IGF1 levels among euploids (e.g., CCL11, Figure S2F; Table S3), the overall relationship between elevated signatures of inflammation and IGF1 deficiency observed in DS is not obviously conserved among euploid controls (Figure 2E).
These results reveal lifelong IGF1 deficiency downstream of GH1 production in DS, with clear ties to proteomic changes indicative of inflammation.
IGF1 deficiency is associated with a specific immune profile in DS involving TNF-α
Given the known role of inflammation in attenuating liver production of IGF1,50 and the fact that individuals with DS present signs of chronic immune dysregulation,23–27 we investigated the interplay between inflammation, IGF1 deficiency, and increased neurodegeneration in people with DS. Toward this end, we defined correlations between levels of the three subunits of the IGF1 complex, IGFBP2, and the four neurodegeneration/neuroinflammation biomarkers versus 54 circulating inflammatory factors measured in the same plasma samples (STAR Methods). Using a multivariable linear model with age and sex and random effect for sample source as covariables, we identified 20 inflammatory markers significantly elevated and 12 significantly decreased in DS (Figure S3A; Table S4). The correlation analysis revealed that IGF1 depletion and neurodegeneration are associate with a specific inflammatory profile in DS (Figures 3A and 3B; Table S4). Most prominently, the pro-inflammatory cytokine TNF-a, which is significantly elevated in DS (Figure 3C), is significantly negatively associated with all three subunits of the IGF1 complex and significantly positively associated with IGFBP2 and the four neurodegeneration biomarkers (Figures 3A–3C and S3B). Other relevant examples are IL-2, IL-17A, and IL-12/IL-23p40, which also correlate with decreased IGF1 levels and elevated neurodegeneration and neuroinflammation biomarkers (Figures 3A and 3B). These results are consistent with reports showing that TNF-α can shut down GH1-induced IGF1 production in the liver.51,52 Remarkably, key markers of liver inflammation elevated in DS are not associated with IGF1 depletion. For example, levels of the acute-phase protein CRP (C-reactive protein), which is strongly elevated in DS (Figure S3C), are instead negatively associated with levels IGFBP2, NfL, and GFAP (Figures 3A, 3B, and S3D).
Figure 3. IGF1 deficiency is associated with a specific inflammatory profile in DS.
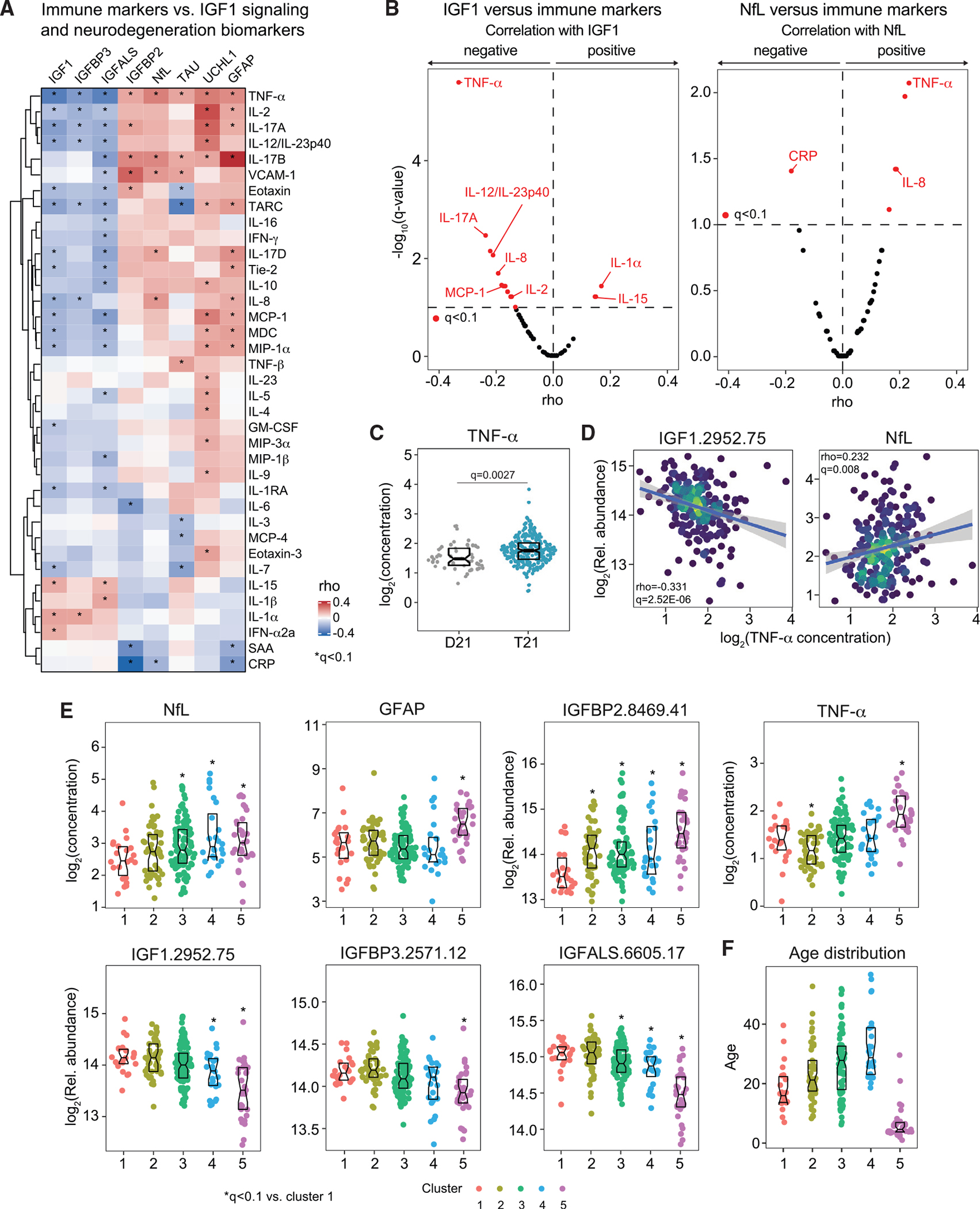
54 inflammatory factors were measured in plasma samples from 309 research participants, 259 of them with T21, versus 50 age- and sex-matched euploid controls (D21) for whom matched measurements of IGF1 signaling factors and neurodegeneration markers were available.
(A) Heatmap displaying correlations in circulating levels of inflammatory markers adjusted by age, sex, and source that are significantly associated, positively or negatively, with IGF1, IGFBP3, IGFALS, IGFBP2, and four neurodegeneration/neuroinflammation biomarkers. Values displayed represent Spearman rho values, and asterisks indicate a significant correlation after multiple hypothesis correction with the method (q < 0.1).
(B) Volcano plots for rho values from Spearman correlations between circulating IGF1 (left) and NfL (right) levels versus circulating levels of inflammatory factors in individuals with T21 after adjustment for age, sex, and source.
(C) Sina plots displaying levels of TNF-α in individuals with and without T21. Boxes indicate median and interquartile range. Differences between groups were determined with a multivariable linear regression with age, sex, and source as covariables, with BH correction of p values.
(D) Scatterplots displaying adjusted values for levels of IGF1 and NfL correlated with levels of TNF-α among individuals with DS. Values shown represent rho values from Spearman correlations and q values calculated with the BH method. Points are colored by density. Lines represent a simple linear regression with 95% confidence interval.
(E and F) Sina plots displaying distributions of the indicated proteins (E) and ages (F) across the five molecular clusters identified among 259 participants with T21. Asterisks indicate q < 0.1 for Mann-Whitney tests of each cluster against cluster 1. Boxes represent medians and interquartile ranges.
To investigate this phenomenon, we performed a consensus clustering analysis in all individuals with DS, integrating the multiple datasets into the clustering algorithm (STAR Methods). This exercise identified five major clusters among individuals with DS, which we ranked from lowest (cluster 1) to highest (cluster 5) NfL levels (Figure S3E). In agreement with the correlation analyses, the three clusters with the highest NfL median levels (3, 4, and 5) also show lower levels of the IGF1 complex subunits and higher levels of IGFBP2 and TNF-α, albeit with different degrees of statistical significance relative to cluster 1 (Figure 3E). The clustering exercise revealed interesting differences not obvious from the correlation analysis. For example, cluster 3 has the highest TAU levels, and UCHL1 levels are similar among clusters (Figure S3F). Each cluster has a unique inflammatory marker profile, with multiple factors being significantly different in each individual cluster relative to all others (Figure S3E). Mean age increases steadily from cluster 1 to cluster 4 (Figure 3F), but cluster 5 is primarily composed of children, an observation that can be linked to results described later in this study, demonstrating that the anti-correlation between NfL levels and levels of the IGF1 complex subunits is strongest among children.
These results indicate that dysregulation of IGF1 signaling is associated with a specific inflammatory subtype in DS involving elevation of TNF-α and other cytokines, which likely leads to disruption of IGF1 production in the liver and a GH resistance phenomenon in DS.
Interplay between IGF1 deficiency and overexpression of proteins encoded on chr21
Next we investigated the relationship between overexpression of proteins encoded on chr21, IGF1 deficiency, and elevation of neurodegeneration and neuroinflammation biomarkers in DS. We measured 46 proteins encoded on chr21 using the SOMAscan platform, 24 of which are significantly more abundant in the plasma of individuals with DS (Figure 4A; Table S5). We then defined correlations between these chr21-encoded proteins and the IGF1 complex subunits, IGFBP2, the four neurodegeneration/neuroinflammation biomarkers, and TNF-α and visualized the correlation matrix via unsupervised clustering analysis (Figure 4B; Table S5). This exercise identified many chr21-encoded proteins whose expression is associated with decreased levels of the IGF1 complex, elevated IGFBP2, and increased signs of neurodegeneration, including the interferon (IFN) receptors IFNAR1 and IFNGR2, carbonyl reductase (CBR3), transcription trefoil factor 2 (TFF2), the ADAM (a disintegrin and metaloproteinase) metallopeptidases with thrombospondin motifs 1 and 5 (ADAMTS1 and ADAMTS5), and APP, among others (Figures 4B–4D and S4). A few of these proteins also show significant positive associations with TNF-α levels, such as the pyridoxal kinase PDXK, often mutated in patients with primary axonal polyneuropathy,53 the interferon-inducible protein MX1, the junctional adhesion molecule 2 (JAM2), and small ubiquitin-like modifier 3 (SUMO3) (Figures 4B–4D). A subset of proteins preferentially associates with IGF1 deficiency, such as the superoxidase dismutase SOD1, the chromatin protein HMGN1, and the phosphodiesterase PDE9A (Figures 4B and 4C). In contrast, a different subset is associated only with neurodegeneration biomarkers and IGFBP2, such as the neuronal cell adhesion molecule NCAM2 and transcription factor 3 (TFF3) (Figures 4B and 4C). Many proteins encoded on chr21 and overexpressed in DS show the oppositive pattern in relation to IGF1 depletion and neurodegeneration/neuroinflammation markers, such as ITGB2, IL10RB, and BACH1 (Figure 4B).
Figure 4. Interplay between overexpression of chr21 proteins, IGF1 deficiency, and elevated markers of neurodegeneration in DS.
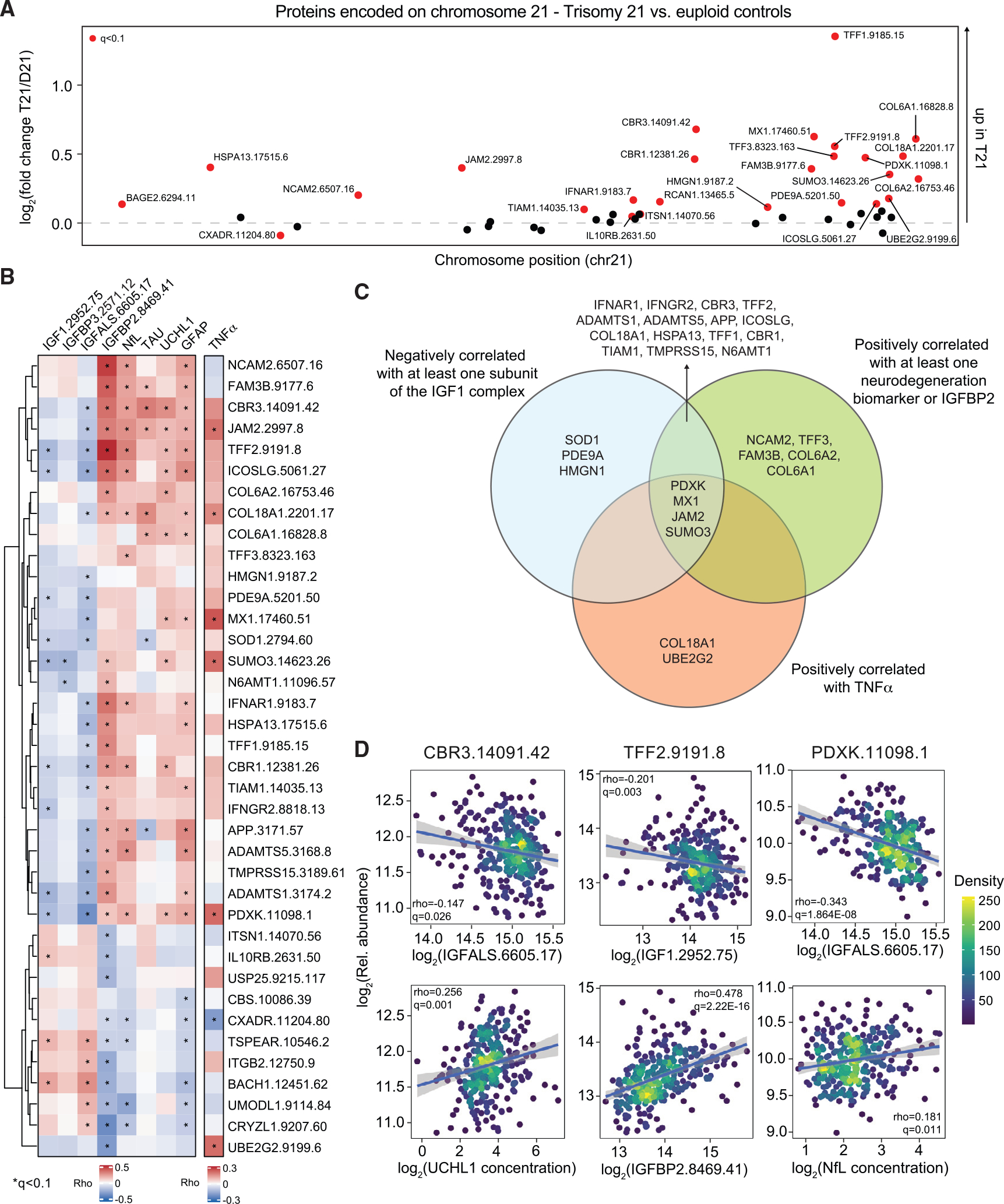
Levels of proteins encoded on chromosome (chr21) were measured in plasma samples from 419 research participants, 316 of them with T21, versus 103 age- and sex-matched euploid controls (D21).
(A) Manhattan plot of chr21, displaying the results of proteomics analysis by karyotype as estimated by multivariable linear model for log2(fold change T21/D21) with adjustment for age, sex, and source as covariables. Proteins passing the statistical cutoff (q < 0.1) are highlighted in red.
(B) Heatmap indicating the proteins encoded on chr21 that are significantly correlated, positively or negatively, with the IGF1 ternary complex and IGFBP2 proteins, the four neurodegeneration/neuroinflammation biomarkers, and TNF-α, adjusted by age, sex, and source as covariables. Values displayed represent Spearman rho values.
(C) Venn diagram displaying the overlaps in proteins encoded on chr21 that are negatively or positively correlated with various factors as described in (B).
(D) Scatterplots displaying adjusted values for levels of select proteins encoded on chr21 (CBR3, TFF2, and PDXK) versus factors involved in IGF1 signaling and neurodegeneration/neuroinflammation biomarkers among individuals with T21. Values shown represent Spearman rho values and q values. Points are colored by density. Lines represent a simple linear regression with 95% confidence interval.
See also Figure S4 and Table S5.
Although the proteomics dataset does not include measurements of all chr21-encoded proteins, it is nonetheless possible that IGF1 deficiency and elevated markers of neurodegeneration may be linked to one or more specific gene products encoded on chr21, a line of future investigation that could illuminate much needed cause-effect relationships between gene overexpression and phenotypes of DS.
Shorter children with DS display stronger IGF1 deficiency and neurodegeneration
Next we investigated the relationship between stature and GH1/IGF1 signaling in DS. Stunted growth is a hallmark of DS, with decreased growth velocity being more pronounced during infancy and adolescence.54 Having observed that levels of the IGF1 complex increase throughout the first two decades of life (Figure 2B), we reasoned that the stronger the IGF1 deficiency during childhood, the more pronounced the stunted growth would be in children with DS. To test this, we generated a growth curve for participants with T21 in our cohort based on height and age LOESS (locally estimated scatterplot smoothing) curves for females and males separately (STAR Methods; Figures 5A and S5A; Table S6). We then assigned each participant to a Short or Tall group based on their relative height within each sex and evaluated for differences in the circulating levels of the IGF1 complex subunits, IGFBP2, GH1, TNF-α, and the neurodegeneration/neuroinflammation markers (Figures 5A–5D). This analysis revealed that, when assessing all ages combined, individuals in the Short group tend to present decreased levels of the IGF1 complex, concurrent with elevated levels of NfL and GFAP (Figure 5B; Table S6). When analyzing the different age groups separately, we observed that children (ages 0–18) in the Short group have significantly lower circulating levels of all three subunits of the IGF1 complex, accompanied by significantly elevated levels of IGFBP2, NfL, and GFAP (Figures 5B–5D, S5B, and S5C), but these differences are attenuated with age. In young adults (ages 18–30), only GFAP is significantly elevated in the Short group, whereas no significant differences were observed in older adults (ages 30+) (Figure 5B). Levels of GH1 and TNF-α were no different by stature (Figure 5B). The trend for total TAU diverges from the overall trend seen for NfL, being actually elevated in the Tall group (Figure 5B).
Figure 5. Shorter children with DS display stronger IGF1 deficiency and elevated markers of neurodegeneration.
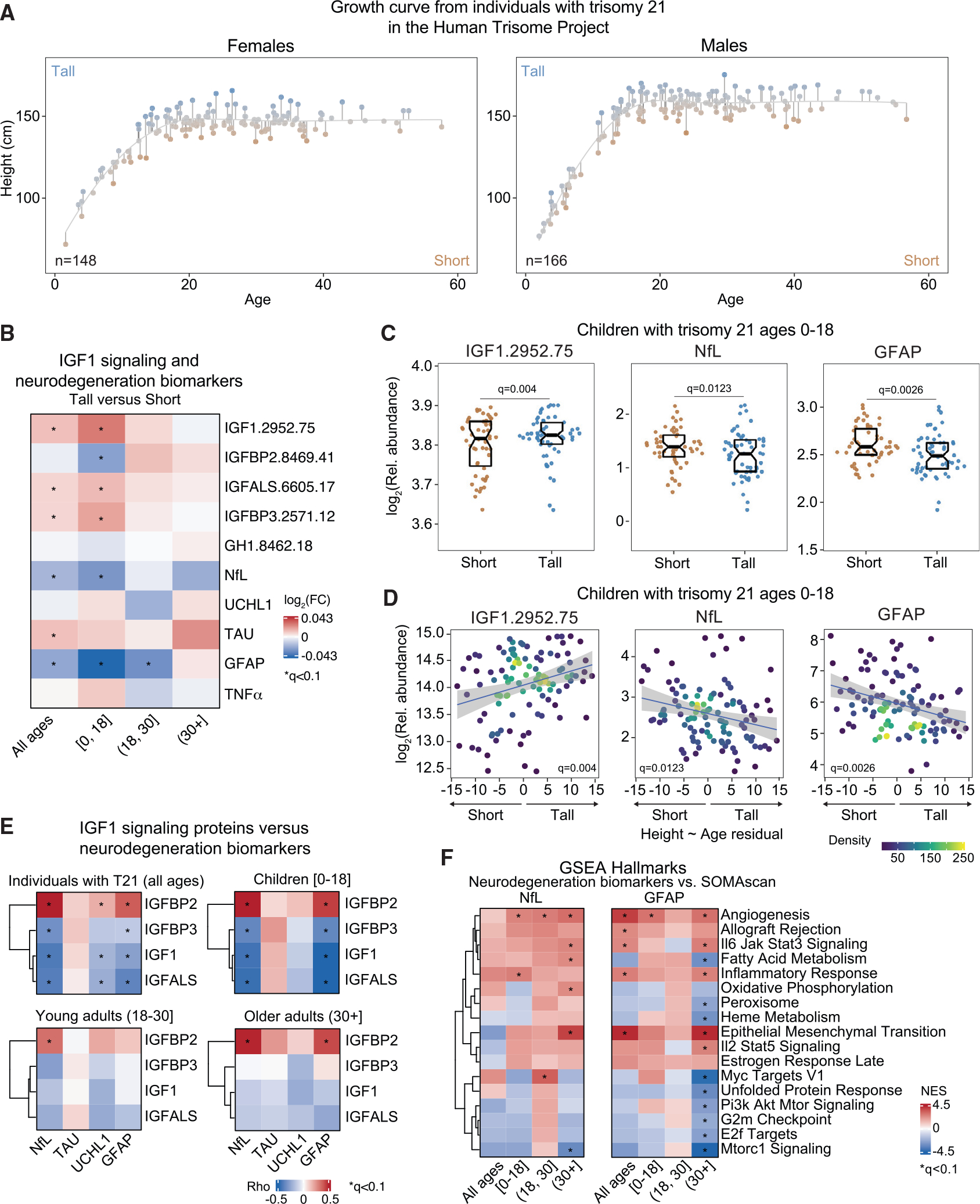
Relationship between stature with GH1/IGF1 signaling and neurodegeneration was explored in 314 individuals with Down syndrome (DS).
(A) Growth curve showing the height versus age LOESS curve from females (n = 148) and males (n = 166) with DS in the Human Trisome Project cohort. Short or tall stature was assigned to each participant based on whether the distance between the participant’s height and the line from the height ~ age LOESS regression (residual) was positive or negative.
(B) Heatmap representing differences in levels of GH1/IGF1 signaling proteins, the indicated neurodegeneration/neuroinflammation biomarkers, and TNF-α per unit of residual value from the height ~ age LOESS regression in individuals with T21. Values displayed are the log2 fold change of concentration levels per unit of residual, and asterisks indicate a significant correlation after multiple hypothesis correction with the BH method (q < 0.1).
(C) Sina plots showing the levels of IGF1, NfL, and GFAP in short versus tall children (0–18) with T21 (short, n = 50–51; tall, n = 55–56). Data are presented as sina plots, with boxes indicating median and interquartile range. Differences between groups were determined as in (B).
(D) Scatterplots for levels of IGF1, NfL, and GFAP versus the residuals from the height ~ age LOESS regression in children with DS. Points are colored by density. Lines represent a simple linear regression with 95% confidence interval.
(E) Heatmap representing the correlation between four neurodegeneration/neuroinflammation biomarkers and IGF1 signaling proteins. Values displayed represent Spearman rho values.
(F) Pathways significantly enriched by NES from weighted GSEA of proteins that are significantly positively and negatively correlated with NfL and GFAP at specific age brackets in individuals with T21.
Then we completed the same analysis for the euploid cohort, which revealed similarities and differences (Figures S5D–S5H). Euploid controls in the Short group show a trend toward lower IGF1 levels and higher NfL and GFAP levels, as seen in DS, but without reaching statistical significance (Figures S5E and S5F). In contrast, euploid controls in the Tall group display significantly higher levels of TAU among all ages and among children and young adults (Figures S5E and S5F). Last, in the older adult euploid population, we observed significantly decreased UCHL1 levels in the Tall group, which is not seen in DS (Figures S5E and S5H).
We then investigated potential differences in biosignatures associated with neurodegeneration and IGF1 signaling during early development versus later in life. The interplay between depressed IGF1 signaling and neurodegeneration and neuroinflammation is stronger among children with DS, as demonstrated by the multiple significant associations with NfL and GFAP (Figure 5E). Pathway analysis of the global proteomics correlations for NfL and GFAP at different age brackets in DS revealed that, although some pathways are consistently associated with levels of these biomarkers at all ages, some are preferentially associated at a specific age range. For example, the inflammatory response signature is more strongly associated with NfL levels in the 0–18 age bracket and with GFAP levels at all ages combined and at the 30+ age bracket (Figure 5F).
These results indicate a somewhat expected association between IGF1 levels and stature, along with a notable association between the degree of stunted growth and levels of NfL and GFAP, two key biomarkers of neurodegeneration and neuroinflammation in DS.
Stature is associated with a differential burden of co-occurring conditions in DS
Next we hypothesized that the degree of stunted growth among children with DS might be linked to the prevalence of co-occurring conditions in this population. To test this, we evaluated associations between stature and various diagnoses in a separate cohort of 1,147 individuals with T21, 1.5–25.1 years old, with rich annotation of clinical information (Figure 6A; Table S1; STAR Methods). Individuals were assigned to the Short or Tall group according to their height in reference to a LOESS fit curve generated separately for males and females (Figure S6A). Then we investigated potential differences in the prevalence of co-occurring conditions in the Short versus Tall groups. This analysis revealed eight diagnoses significantly associated with stature in DS, including ASD, other autoimmune diagnoses (i.e., type 1 diabetes, juvenile rheumatoid arthritis, and vitiligo), chronic lung disease, neonatal acute respiratory distress syndrome (ARDS), hypothyroidism (including Hashimoto’s disease), use of an audiology pressure-equalizing (PE) tube, and neonatal intensive care unit (NICU) stay after birth, all of which are significantly more prevalent in shorter individuals, and hyperthyroidism (including Graves’ disease), which is more prevalent in taller individuals (Figure 6B; Table S1). The increased prevalence of ASD in the Short group may be related to reports of IGF1 deficiency in ASD, leading to testing of IGF1-based therapies for this condition.55–57 Dysregulation of IGF1 signaling has been implicated in the etiology of various autoimmune diseases,58–61 which may contribute to the increased proportion of autoimmune disorders in the Short group. We also observed a significant increase in neonatal ARDS and chronic lung disease in the Short group, conditions that have been associated with IGF1 deficiency because IGF1 is essential for normal lung development.62 Increased use of audiology PE tubes in the Short group, a proxy for recurrent ear infections, could be tied to the immune-regulatory effects of IGF1.58–61 We also found that NICU stay after birth was more common in the Short group, which may be linked to the known role of IGF1 in early fetal development.63 Conversely, hyperthyroidism, including Graves’ disease, was more prevalent in the Tall group, which is consistent with the fact that typical individuals with hyperthyroidism present elevated levels of IGF1 in circulation.64
Figure 6. Shorter children with DS exhibit increased incidence of specific co-occurring conditions.
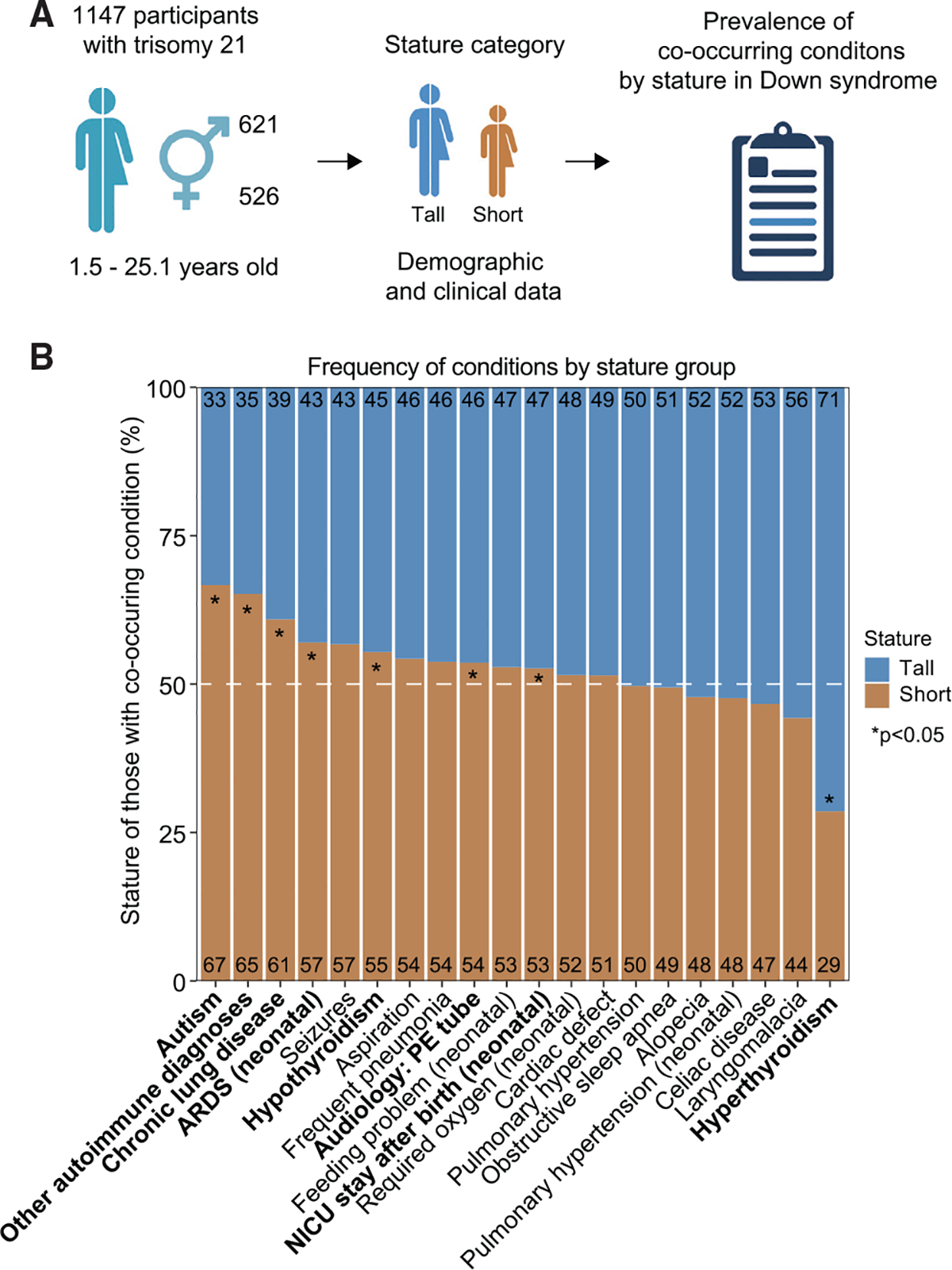
(A) Associations between stature and co-occurring conditions were defined in a cohort of 1,147 research participants with DS aged 1.5–25.1 years. Demographics and clinical data were analyzed by stature group as shown in Figure S6.
(B) Bar plots showing the distribution of the indicated co-occurring conditions in individuals with T21 by stature category. History of autism, other autoimmune diagnoses, chronic lung disease, neonatal acute respiratory distress syndrome (ARDS), hypothyroidism (including Hashimoto’s disease), audiology pressure-equalizing (PE) tubes, and neonatal intensive care unit (NICU) stay after birth were more common in shorter children. Hyperthyroidism, including Graves’ disease, was more common in taller children. Asterisks indicate a significant difference using two-sided Fisher’s exact tests; *p < 0.05.
We then tested whether the conditions associated with short stature co-segregated in DS (Table S1). This exercise revealed that, although some of the diagnoses associated with stature display significant co-segregation (e.g., neonatal ARDS with chronic lung disease), others do not (e.g., autism with “other autoimmune diagnoses”). Therefore, these results indicate that, although multiple diagnoses are significantly more common in shorter children with DS, they do not necessarily always “cluster” together, suggesting that stunted growth may affect different individuals with DS in diverse ways.
Our analysis revealed a relationship between stunted growth and increased prevalence of specific diagnoses in DS, suggesting that IGF1 deficiency may contribute to the etiology of certain co-occurring conditions more common in DS.
DISCUSSION
Despite substantial research efforts, with a few notable exceptions, the mechanisms by which T21 causes the multiple phenotypes of DS remain unclear. Elucidation of these mechanisms could illuminate strategies to attenuate the ill effects of the trisomy through personalized medicine approaches. For example, the established link between APP triplication and the high prevalence of AD in this population justifies pursuit of strategies to ameliorate the burden of chronic amyloidosis.9 Likewise, the demonstration that triplication of IFN receptors contributes to multiple hallmarks of DS in mice65,66 and immune hypersensitivity in human cell types with T2126,27,67,68 justifies ongoing testing of JAK inhibitors to decrease the burden of autoimmune diseases in DS.69,70 In this context, we report here that individuals with DS display lifelong IGF1 deficiency downstream of GH1 production and that IGF1 deficiency is the top biosignature associated with neurodegeneration and neuroinflammation in DS. Disruption of IGF1 signaling may contribute to key hallmarks of DS, such as stunted growth, including microcephaly, and dysregulated neurodevelopment. The fact that IGF1 depletion is significantly associated with a specific inflammatory profile may link the chronic immune dysregulation observed in DS with the stunted growth and neurodevelopmental delays characteristic of this condition.
Our results demonstrate that individuals with T21 present signs of neurodegeneration across their lifespan, with unique age trajectories for different biomarkers. Although some of these changes could be potentially attributed to accelerated aging in DS (e.g., elevated NfL across the lifespan), others cannot (e.g., elevated TAU in children and other adults). These findings extend previous reports demonstrating elevated levels of NfL, GFAP, and TAU in DS.12,19 Increased levels of plasma NfL and TAU were substantially higher in individuals with DS with AD dementia.13,14,16,19–21 However, these studies did not define the broader biosignatures associated with neurodegeneration in DS. Our findings linking IGF1 deficiency to neurodegeneration in DS, particularly early in life, align with a vast body of work documenting key roles for IGF1 signaling in brain development and homeostasis.35 IGF1 is produced by multiple cell types in the CNS, mainly microglia cells, with important actions on neurons, oligodendrocytes, and astrocytes, driving cell proliferation and promoting maturation and myelination.71 Single-nucleus transcriptome analyses revealed that microglia with T21 display decreased expression of IGF1, concomitant with increased expression of IGFBP2 in pericytes, indicating decreased IGF1 signaling in the CNS of individuals with DS.72 Circulating IGF1 can cross the BBB, and some studies have observed a direct relationship between peripheral levels of IGF1 versus hippocampal IGF1 protein levels.73 Liver-specific IGF1-deficient mice, which have an ~70% reduction in systemic IGF1 levels, manifest early hippocampus-dependent cognitive deficits despite maintained autocrine IGF1 production.74,75 Upon binding to the IGFR, IGF1 induces canonical signaling pathways, such as the phosphatidylinositol 3-kinase (PI3K)-AKT and Ras-Raf-mitogen-activated protein kinase (MAPK) pathways, with potent effects on neuroplasticity. The GPE (glycine-proline-glutamate) tripeptide produced by cleavage of IGF1 and its derivative cGP (cyclic glycine-proline) harbor additional neuroprotective properties independent of IGFR76. Accordingly, multiple clinical trials are currently testing recombinant human IGF1 (rhIGF1/mecasermin/Increlex), GPE analogs (e.g., trofinetide/NNZ-2566), and cGP analogs (e.g., NNZ-2591) for a range of neurological conditions. Mecasermin is approved for treatment of severe primary IGF1 deficiency (SPIGFD), a condition defined by normal GH1 but low IGF1 and short stature, and is currently being tested for ASD, Rett syndrome, amyotrophic lateral sclerosis, and multiple sclerosis.57,76
Our work points to chronic IGF1 deficiency in DS despite mildly elevated levels of GH1 in circulation, a hallmark of GH resistance disorders. Loss-of-function mutations in IGF1, IGFBP3, IGFALS, and IGF1R cause diverse GH resistance disorders, characterized by stunted growth with varying degrees of neurological manifestations.77 Although GH levels have consistently been found to be normal in DS,78–80 dysregulation of downstream steps in GH signaling has been proposed in the past. In 1983, Sara et al.81 measured levels of IGF1 and IGF2 in children with DS, documenting for the first time differential depletion of IGF1 in DS. These results led to a few small clinical trials of GH therapy in DS, with diverse beneficial effects being reported. In a pioneer study of five children of ages 3–6 treated with GH for 6 months, growth velocity was clearly accelerated during the treatment period but decelerated again when the treatment stopped.78 Serum IGF1 levels were increased during GH treatment, indicating that IGF1 deficiency could be mitigated by elevating GH to supra-physiological levels.78 In an independent study, 13 children with DS were treated with GH for a year, leading to accelerated growth and increased head circumference.82 In a study of 15 children with DS treated with daily injections of GH for 3 years,83 long-term follow-up determined that adolescents with DS treated previously with GH had a larger head circumference, and their mean raw scores were consistently higher in diverse subsets of cognitive assessments.84 Therefore, although children with T21 produce normal levels of GH, downstream IGF1 deficiency could have profound effects not only on overall growth but also on brain development and function.
Our analyses demonstrate that IGF1 deficiency in DS is associated with a specific immune profile characterized by elevation of TNF-α and other pro-inflammatory markers. There is substantial evidence demonstrating that TNF-α and other cytokines disrupt the GH/IGF1 axis through diverse mechanisms. For example, TNF-α can impair IGF1 production via SOCS (Suppressor Of Cytokine Signaling)-mediated attenuation of JAK2/STAT5 signaling downstream of GH-GHR engagement.85–87 TNF-α can also downregulate GHR expression in hepatocytes52,88 and alter IGF1 bioavailability through reductions in IGFBP3 levels.89 We observed that many circulating immune factors elevated in DS were not associated with IGF1 deficiency or elevated NfL, such as CRP, a broad marker of inflammation. These observations suggest the existence of distinct immune subtypes in DS, with potential for different inflammatory pathways to contribute to different phenotypes of DS.
Although our analysis of the interplay between overexpression of specific proteins encoded on chr21, IGF1 deficiency, and elevated biomarkers of neurodegeneration was not comprehensive, it lends support to the notion that IGF1 depletion may be linked to overexpression of a distinct subset of chr21 genes. Assuming that IGF1 deficiency is a consequence of chronic inflammation, special attention should be devoted to key immune regulators encoded on chr21, such as the four IFN receptors. However, other genes may also contribute, including APP and other factors whose abundance in plasma was associated with IGF1 depletion. Elucidation of these cause-effect relationships will require additional investigations, including mechanistic studies in animal models.
Last, our findings illuminate an interplay between IGF1 deficiency, neurodegeneration, stunted growth, and burden of co-occurring conditions in DS. Shorter children with DS display stronger IGF1 deficiency. However, they also show elevated markers of neurodegeneration and neuroinflammation as well as increased risk of key co-occurring conditions, such as ASD and chronic lung disease. These findings indicate that the stunted growth phenotype of DS could be associated with an increased risk of several co-occurring conditions in this population. For example, microcephaly, a hallmark of DS and IGF1 deficiency disorders and likely tied to the effects of IGF1 on bone morphogenesis,90 could impair brain development and induce neurodegeneration and neuroinflammation in DS, with consequent increased risk of specific neurological conditions. Likewise, stunted growth associated with IGF1 deficiency could impair lung and heart development and function, increasing the risk of some cardiopulmonary conditions in DS. IGF1 signaling has prominent roles in lung and heart development, and IGF1 deficiency has been associated with multiple cardiopulmonary conditions.62,91 Therefore, a comprehensive study of the effects of IGF1 deficiency in DS would not only advance our understanding of the developmental and clinical effects of T21 but also of other genetic disorders displaying hyperinflammation and stunted growth, such as monogenic interferonopathies.92 Although our study does not propose that stature on its own is a viable biomarker in DS, we hypothesize that the degree of stunted growth will correlate with the risk of some co-occurring conditions, particularly those involving organ systems regulated by IGF1.
These findings pave the road for mechanistic investigations to elucidate the multidimensional effects of IGF1 deficiency in DS while justifying the pursuit of clinical investigations to define the value of IGF1-based therapies in this population.
Limitations of the study
Although this study shows co-regulation of IGF1 signaling factors, neurodegeneration biomarkers, and a subset of inflammatory markers among individuals with DS, caution should be exercised when interpreting cause-effect relationships. Showing that IGF1 deficiency causes stunted growth and neurodegeneration in DS will require additional investigations because it is formally possible that IGF1 deficiency is a late response to neurodegeneration. Likewise, although TNF-α has been shown to impair IGF1 signaling in diverse experimental settings, whether TNF-α contributes to IGF1 deficiency in DS awaits elucidation. Another limitation is the difficulty of interpreting the variable circulating levels of proteins in plasma. Although some proteins highlighted in this study are secreted into plasma (e.g., IGF1 and TNF-α), others are found in circulation upon cellular damage (e.g., NfL and GFAP). Another limitation is the fact that our study involved roughly three times more individuals with DS than controls. Therefore, some differences observed in the control group could be explained by decreased statistical power. Our study did not identify important differences between males and females, but larger sample sizes may reveal sex-specific differences. Last, our study did not measure levels of the various biomarkers in the CNS, only in the bloodstream.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Joaquin Espinosa (joaquin.espinosa@cuanschutz.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Deidentified plasma proteomics data, plasma neurodegeneration biomarker data, and plasma inflammatory marker data have been deposited in the Synapse platform and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Critical commercial assays | ||
|
| ||
| Neurology 4-plex B kit | Single Molecular Array Technology (SIMOA® SR-XTM Analyzer, Quanterix Corporation, MA) | Cat# 103345 |
| U-PLEX Biomarker Group 1 (hu) 71-Plex | Meso Scale Discovery (MSD) | Cat# K15081K |
| V-PLEX Vascular Injury Panel 2 Human Kit | Meso Scale Discovery (MSD) | Cat# K15198D |
| V-PLEX Angiogenesis Panel 1 Human Kit | Meso Scale Discovery (MSD) | Cat# K15190D |
|
| ||
| Deposited data | ||
|
| ||
| Combined multi-omics dataset | Human Trisome Project | Synapse data: https://www.synapse.org/#!Synapse:syn35991637.1/datasets/ |
| Plasma proteomics data | Human Trisome Project | Synapse data: https://doi.org/10.7303/syn31488781 |
| Neurodegeneration biomarker data | Human Trisome Project | Synapse data: https://doi.org/10.7303/syn35993837 |
| Inflammatory marker data | Human Trisome Project | Synapse data: https://doi.org/10.7303/syn31475487 |
|
| ||
| Software and algorithms | ||
|
| ||
| R | R Foundation for Statistical Computing | v3.6.1 RRID:SCR_001905, https://www.R-project.org/ |
| RStudio | RStudio, Inc. | v1.2.5025 RRID:SCR_000432, http://www.rstudio.com/ |
| Bioconductor | N/A | v3.11 RRID:SCR_006442, https://bioconductor.org/ |
| limma package for R | N/A | v3.44.3 RRID:SCR_010943, https://bioconductor.org/packages/release/bioc/html/limma.html |
| SomaDataIO R package | SomaLogic | v3.1.0 RRID: SCR_022198, https://github.com/SomaLogic/SomaDataIO |
| ggplot2 package for R | N/A | v3.3.1 RRID:SCR_014601, https://ggplot2.tidyverse.org/ |
| fgsea package for R | N/A | v 1.14.0 RRID:SCR_020938, https://github.com/ctlab/fgsea |
| ConsensusClusterPlus package for R | Bioconductor | v1.52.0 RRID:SCR_016954, https://git.bioconductor.org/packages/ConsensusClusterPlus |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study design
Research participants were enrolled to the Crnic Institute Human Trisome Project (HTP) under a study protocol approved by the Colorado Multiple Institutional Review Board (COMIRB #15-2170, www.trisome.org, NCT02864108). Written informed consent was obtained from parents or guardians of each participant, and assent was obtained from participants over the age of seven years who were cognitively able to assent. Biological data was generated from deidentified biospecimens and linked to demographics and clinical metadata procured via participant/family surveys and abstraction of electronic medical records (EMRs). The HTP cohort used for this study consisted of 419 individuals, 316 of them with DS. Cohort information can be found in the Table S1. All samples were processed directly ex vivo. The cohort used for analysis of co-occurring conditions in Figure 6 consists of children and young adults with DS who received care from the Anna and John J. Sie Center for Down Syndrome (SCDS) at Children’s Hospital Colorado (CHCO). Study data was approved by the Institutional Review Board of the University of Colorado School of Medicine, which determined that the need for informed consent was exempted. EMR data on 1,147 children and young adults with DS from 1.5 to 25.1 years of age was reviewed. All participants met inclusion criteria of having a diagnosis of DS and attended at least one clinic appointment. Patients were identified from the SCDS Clinical Research Electronic Data Capture (REDCap) Database hosted at CHCO.
METHOD DETAILS
Blood processing
Blood samples were collected into BD Vacutainer K2 EDTA tubes (BD, Cat# 366643). After centrifugation, EDTA plasma was separated and used for SIMOA measurements of neurodegeneration and neuroinflammation biomarkers, SOMAscan proteomics, as well as multiplex immunoassays using MSD technology for cytokine profiles assays.
Neurodegeneration biomarker analysis by SIMOA
Plasma biomarker assays for Neuro-Filament light (NfL), Total TAU (TAU), Ubiquitin Carboxy-Terminal Hydrolase L1 (UCHL1), and Glial fibrillary acidic protein (GFAP), were conducted at the University of Colorado Alzheimer’s and Cognition Center (CUACC) using the SR-X Single Molecular Array Technology (SIMOA SR-XTM Analyzer, Quanterix Corporation, MA) and the Neurology 4-plex B kit (Cat# 103345). Following manufacturer’s recommendations for handling and analyzing plasma samples, each SIMOA run included an 8-point calibration curve for each marker and two internal controls. To avoid batching effects, experiments were predesigned to include a similar proportion of individuals from each karyotype matched by sex and age. Investigators running the experiments were blinded to study group assignments. The samples were thawed once on ice before each run, centrifuged for 10 min at 10,000xg, diluted 1:4 in sample diluent and centrifuged again. Each diluted sample was run in duplicate or triplicate, based on sample availability, and the average value of the runs was used for analyses.
Plasma proteomics by SOMAscan assays
125 μL of EDTA plasma was analyzed by SOMAscan assays using previously established protocols.93 Briefly, each of the 4500 + SOMAmer reagents binds a target peptide and is quantified on a custom Agilent hybridization chip. Normalization and calibration were performed according to SOMAscan Data Standardization and File Specification Technical Note (SSM-020).93 The output of the SOMAscan assay is reported in relative fluorescent units (RFU).
Multiplex immunoassays for measurements of cytokines, chemokines, and immune markers
Multiplex immunoassays were performed on EDTA plasma aliquots using a Meso QuickPlex SQ120 instrument following manufacturer’s instructions (Meso Scale Discovery, MSD). A list of immune factors measured by MSD can be found in Table S4. Values were extrapolated against a standard curve using provided calibrators and are reported in pg/mL.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data preprocessing, statistical analysis, and plot generation for all datasets were carried out using R (R 3.6.1/RStudio 1.2.5025/Bioconductor v 3.11),94,95 as detailed below. Extreme outliers were classified per-karyotype and per-analyte if the measurement was above Q3 + 3xIQR or below Q1 − 3xIQR and removed for analyses. Note: Q1: first quartile, Q3: third quartile, IQR: interquartile range (IQR = Q3 − Q1). Method schematics were generated using Biorender.com. All figures were finalized in Adobe Illustrator v24.1.
Neurodegeneration biomarkers data
Plasma concentration values (pg/mL) for each of the neurodegeneration/neuroinflammation biomarkers measured across multiple SIMOA assay plates was imported into R. Data were adjusted for age, sex, and source where indicated using the removeBatchEffect function from the limma package (v 3.44.3).96
SOMAscan proteomics data
Normalized data (RFU) was imported and converted from a SOMAscan.adat file using the SomaDataIO R package (v3.1.0, https://github.com/SomaLogic/SomaDataIO). Data were adjusted for age, sex and source where indicated using the removeBatchEffect function from the limma package (v 3.44.3).96
MSD cytokine profiling data
Plasma concentration values (pg/mL) for each of the immune factors measured across multiple MSD assay plates were imported to R, combined, and analytes with >10% of values outside of detection or fit curve range flagged. For each analyte, missing values were replaced with either the minimum (if below fit curve range) or maximum (if above fit curve range) calculated concentration and means of duplicate wells used in all further analysis. Data were adjusted for age, sex, and source where indicated using the removeBatchEffect function from the limma package (v 3.44.3).96
Correlation analysis
To identify features in each dataset that correlate with the four neurodegeneration/neuroinflammation biomarkers, the IGF1/IGFBP3/IGFALS ternary complex and IGFBP2 proteins in samples from individuals with T21, Spearman rho values and p values were calculated between each feature and the respective values for each dataset using the rcorr function from the Hmisc package (v 4.4–0), with Benjamini-Hochberg (BH) correction of p values and an estimated false discovery rate threshold (FDR) of 10%, q < 0.1. Data were adjusted for age and sex where indicated using the removeBatchEffect function from the limma package (v 3.44.3).96 For visualization, XY scatterplots with points colored by local density were generated using a custom density function and the ggplot2 (v3.3.1) package.
Gene set enrichment analysis (GSEA)
GSEA97 was carried out using the fgsea package (v 1.14.0)98 in R, using Hallmark gene sets and Spearman rho values (for the four neurodegeneration/neuroinflammation biomarkers, IGF1 ternary complex, and IGFBP2 proteins correlations) as the ranking metric.
Consensus clustering
For the consensus cluster described in Figures 3 and S3, Z-scores were calculated from the circulating levels of 54 cytokines/chemokines measured by MSD, the four neurodegeneration/neuroinflammation biomarkers measured by SIMOA, and the relative abundance of four SOMAscan analytes for IGF1, IGFBP2, IGFBP3, and IGFALS in samples from individuals with T21 and used as input to the ConsensusClusterPlus() function from the ConsensusClusterPlus package99 with 100-fold subsampling, Pearson as the distance measure, and agglomerative hierarchical clustering. Examination of the delta area plot indicated 5 clusters gave a reasonable compromise between gains in cluster stability and number of clusters. The Mann-Whitney U test was employed to test for differences in the levels of relative abundance or concentration of the analytes for each cluster in comparison to cluster 1, using the wilcox_test() function from the rstatix package. Unadjusted values were used for the analysis, and correction for multiple comparisons was performed using the Benjamini-Hochberg (FDR) approach.
Growth curve definition and stature classification in individuals with T21
To investigate the relationship between stature and various biomarkers, a LOESS curve was fit to all the individuals with DS (or euploid controls) with the formula Height ~ Age, using a span value of 0.75 for smoothing the LOESS curve. Since females and males have a different growth velocity, a separate growth curve was generated for females and males. Stature classification for each participant was based on distance (i.e., residuals), from the original data to the LOESS fit curve for each sex, with positive values defined as Tall and negative values defined as Short.
Analysis of proteins levels by stature
To determine whether neurodegeneration/neuroinflammation biomarkers, IGF1/GFBP3/IGFALS, IGFBP2, GH1, and TNFα levels had a relationship with height in individuals with T21, we fit a linear model to all the individuals with DS in our research cohort with the form Value ~ Residual. Benjamini-Hochberg (BH) correction of p values was performed and an estimated false discovery rate threshold (FDR) of 10%, q < 0.1 was employed. For Figures 5D, S5C, S5G, and S5H, Age ~ Height residuals were plotted against raw values for individual proteins.
Analysis of distribution of co-occurring conditions by stature in the SCDS cohort
To annotate individual co-occurring conditions, a query of ICD-9 and ICD-10 codes was used from the EMR data of 1,147 children and young adults with DS from 1.5 to 25.1 years of age. Additional criteria were used to establish pulmonary hypertension, cardiac defect, and autoimmune diagnoses. For this study, pulmonary hypertension was defined as a ratio of estimated pulmonary arterial pressure (PAP)/SAP >1/3, a mean PAP greater than 25 mm Hg, interventricular septal flattening, right ventricular (RV) dilation, or presence of RV hypertrophy. Cardiac defects consisted of atrial septal defect, ventricular septal defect, atrioventricular canal, patent ductus arteriosus requiring surgical repair, valve defect (bicuspid, dysplastic, stenosis, etc.), aberrant subclavian artery, coarctation, Tetralogy of Fallot, and vascular ring. The ‘other autoimmune diagnoses’ group included type 1 diabetes, juvenile rheumatoid arthritis, and vitiligo. The chronic lung disease category included agenesis, hypoplasia, and dysplasia of lung; bronchopulmonary dysplasia; alveolar capillary dysplasia; bronchogenic cyst; congenital bronchiectasis; congenital cystic lung; congenital diaphragmatic hernia; congenital lobar emphysema; cystic fibrosis; Scimitar syndrome; congenital lung disorder; and other congenital lung disorders. Except for six cases which had telehealth appointments, all patient comorbidity data was queried at the time of height collection. In cases with telehealth appointments, height was measured within two months of the comorbidity data pull. Separate height for age LOESS curves were generated for males (n = 621) and females (n = 526), using a span value of 0.75 for smoothing the LOESS curve. Patients were then assigned to the Short or Tall group based upon their stature in reference to these curves. Number and percent of cases by stature were reported for each comorbidity or medical complication. Two-sided Fisher’s exact tests were conducted at an α-level 0.05 significance. To account for age at identification in standard clinical care, children under the age of five years were excluded in the autism analysis (reducing the total to n = 909) and under three years for the celiac disease analysis (reducing the total to n = 1,075).
Supplementary Material
Highlights.
Biomarkers of neurodegeneration are associated with IGF1 deficiency in Down syndrome
IGF1 deficiency is associated with a specific inflammatory profile in Down syndrome
Short stature is associated with lower IGF1 and higher neurodegeneration markers
Stunted growth is associated with elevated rates of key co-occurring conditions
ACKNOWLEDGMENTS
This work was supported primarily by the NIH Office of the Director through NIAID grant R01AI150305 (to J.M.E). Additional funding was provided by NIH grants R01AI145988 (to K.D.S.), 5UL1TR002535-02, and P30CA046934; the Linda Crnic Institute for Down Syndrome; the Global Down Syndrome Foundation; the GI & Liver Innate Immune Program; the Human Immunology and Immunotherapy Initiative; the University of Colorado School of Medicine; and the Boettcher Foundation.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
J.M.E. has provided consulting services for Elli Lily and Co. and Gilead Sciences Inc. and serves on the advisory board of Perha Pharmaceuticals and on the editorial board of Cell Reports.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111883.
REFERENCES
- 1.de Graaf G, Buckley F, and Skotko BG (2017). Estimation of the number of people with Down syndrome in the United States. Genet. Med. 19, 439–447. 10.1038/gim.2016.127. [DOI] [PubMed] [Google Scholar]
- 2.Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, Sherman SL, and Reeves RH (2020). Down syndrome. Nat. Rev. Dis. Primers 6, 9. 10.1038/s41572-019-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasle H, Friedman JM, Olsen JH, and Rasmussen SA (2016). Low risk of solid tumors in persons with Down syndrome. Genet. Med. 18, 1151–1157. 10.1038/gim.2016.23. [DOI] [PubMed] [Google Scholar]
- 4.Alexander M, Petri H, Ding Y, Wandel C, Khwaja O, and Foskett N (2016). Morbidity and medication in a large population of individuals with Down syndrome compared to the general population. Dev. Med. Child Neurol. 58, 246–254. 10.1111/dmcn.12868. [DOI] [PubMed] [Google Scholar]
- 5.Chicoine B, Rivelli A, Fitzpatrick V, Chicoine L, Jia G, and Rzhetsky A (2021). Prevalence of common disease conditions in a large cohort of individuals with down syndrome in the United States. J. Patient. Cent. Res. Rev. 8, 86–97. 10.17294/2330-0698.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lott IT, and Dierssen M (2010). Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 9, 623–633. 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- 7.Gardiner K, Herault Y, Lott IT, Antonarakis SE, Reeves RH, and Dierssen M (2010). Down syndrome: from understanding the neurobiology to therapy. J. Neurosci. 30, 14943–14945. 10.1523/JNEUROSCI.3728-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lott IT (2012). Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res. 197, 101–121. 10.1016/B978-0-444-54299-1.00006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snyder HM, Bain LJ, Brickman AM, Carrillo MC, Esbensen AJ, Espinosa JM, Fernandez F, Fortea J, Hartley SL, Head E, et al. (2020). Further understanding the connection between Alzheimer’s disease and Down syndrome. Alzheimers Dement. 16, 1065–1077. 10.1002/alz.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehrenberg AJ, Khatun A, Coomans E, Betts MJ, Capraro F, Thijssen EH, Senkevich K, Bharucha T, Jafarpour M, Young PNE, et al. (2020). Relevance of biomarkers across different neurodegenerative diseases. Alzheimer’s Res. Ther. 12, 56. 10.1186/s13195-020-00601-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashton NJ, Hye A, Rajkumar AP, Leuzy A, Snowden S, Suárez-Calvet M, Karikari TK, Schöll M, La Joie R, Rabinovici GD, et al. (2020). An update on blood-based biomarkers for non-Alzheimer neurodegenerative disorders. Nat. Rev. Neurol. 16, 265–284. 10.1038/s41582-020-0348-0. [DOI] [PubMed] [Google Scholar]
- 12.Mengel D, Liu W, Glynn RJ, Selkoe DJ, Strydom A, Lai F, Rosas HD, Torres A, Patsiogiannis V, Skotko B, and Walsh DM (2020). Dynamics of plasma biomarkers in Down syndrome: the relative levels of Aβ42 decrease with age, whereas NT1 tau and NfL increase. Alzheimer’s Res. Ther. 12, 27. 10.1186/s13195-020-00593-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortea J, Carmona-Iragui M, Benejam B, Fernández S, Videla L, Barroeta I, Alcolea D, Pegueroles J, Muñoz L, Belbin O, et al. (2018). Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study. Lancet Neurol. 17, 860–869. 10.1016/s1474-4422(18)30285-0. [DOI] [PubMed] [Google Scholar]
- 14.Fortea J, Vilaplana E, Carmona-Iragui M, Benejam B, Videla L, Barroeta I, Fernández S, Altuna M, Pegueroles J, Montal V, et al. (2020). Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study. Lancet 395, 1988–1997. 10.1016/s0140-6736(20)30689-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carmona-Iragui M, Alcolea D, Barroeta I, Videla L, Muñoz L, Van Pelt KL, Schmitt FA, Lightner DD, Koehl LM, Jicha G, et al. (2021). Diagnostic and prognostic performance and longitudinal changes in plasma neurofilament light chain concentrations in adults with Down syndrome: a cohort study. Lancet Neurol. 20, 605–614. 10.1016/s1474-4422(21)00129-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendrix JA, Airey DC, Britton A, Burke AD, Capone GT, Chavez R, Chen J, Chicoine B, Costa ACS, Dage JL, et al. (2021). Cross-Sectional exploration of plasma biomarkers of Alzheimer’s disease in down syndrome: early data from the longitudinal investigation for enhancing down syndrome research (LIFE-DSR) study. J. Clin. Med. 10, 1907. 10.3390/jcm10091907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strydom A, Heslegrave A, Startin CM, Mok KY, Hardy J, Groet J, Nizetic D, and Zetterberg H; LonDownS Consortium (2018). Neurofilament light as a blood biomarker for neurodegeneration in Down syndrome. Alzheimer’s Res. Ther. 10, 39. 10.1186/s13195-018-0367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinomoto M, Kasai T, Tatebe H, Kondo M, Ohmichi T, Morimoto M, Chiyonobu T, Terada N, Allsop D, Yokota I, et al. (2019). Plasma neurofilament light chain: a potential prognostic biomarker of dementia in adult Down syndrome patients. PLoS One 14, e0211575. 10.1371/journal.pone.0211575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henson RL, Doran E, Christian BT, Handen BL, Klunk WE, Lai F, Lee JH, Rosas HD, Schupf N, Zaman SH, et al. (2020). Cerebrospinal fluid biomarkers of Alzheimer’s disease in a cohort of adults with Down syndrome. Alzheimers Dement. 12, e12057. 10.1002/dad2.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashton NJ, Janelidze S, Al Khleifat A, Leuzy A, van der Ende EL, Karikari TK, Benedet AL, Pascoal TA, Lleó A, Parnetti L, et al. (2021). A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat. Commun. 12, 3400. 10.1038/s41467-021-23620-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petersen ME, Rafii MS, Zhang F, Hall J, Julovich D, Ances BM, Schupf N, Krinsky-McHale SJ, Mapstone M, Silverman W, et al. (2021). Plasma total-tau and neurofilament light chain as diagnostic biomarkers of Alzheimer’s disease dementia and mild cognitive impairment in adults with down syndrome. J. Alzheimers Dis. 79, 671–681. 10.3233/JAD-201167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Che M, Yuan J, Yu Y, Cao C, Qin XY, and Cheng Y (2017). Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: a meta-analysis. Oncotarget 8, 84489–84496. 10.18632/oncotarget.21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, Rachubinski AL, Wolter-Warmerdam K, Hickey F, Espinosa JM, and Blumenthal T (2017). Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep. 7, 14818. 10.1038/s41598-017-13858-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huggard D, Doherty DG, and Molloy EJ (2020). Immune dysregulation in children with down syndrome. Front. Pediatr. 8, 73. 10.3389/fped.2020.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huggard D, Kelly L, Ryan E, McGrane F, Lagan N, Roche E, Balfe J, Leahy TR, Franklin O, Doherty DG, and Molloy EJ (2020). Increased systemic inflammation in children with Down syndrome. Cytokine 127, 154938. 10.1016/j.cyto.2019.154938. [DOI] [PubMed] [Google Scholar]
- 26.Araya P, Waugh KA, Sullivan KD, Núñez NG, Roselli E, Smith KP, Granrath RE, Rachubinski AL, Enriquez Estrada B, Butcher ET, et al. (2019). Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc. Natl. Acad. Sci. USA 116, 24231–24241. 10.1073/pnas.1908129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waugh KA, Araya P, Pandey A, Jordan KR, Smith KP, Granrath RE, Khanal S, Butcher ET, Estrada BE, Rachubinski AL, et al. (2019). Mass cytometry reveals global immune remodeling with multi-line-age hypersensitivity to type I interferon in down syndrome. Cell Rep. 29, 1893–1908.e4. 10.1016/j.celrep.2019.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ponroy Bally B, and Murai KK (2021). Astrocytes in down syndrome across the lifespan. Front. Cell. Neurosci. 15, 702685. 10.3389/fncel.2021.702685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilcock DM (2012). Neuroinflammation in the aging down syndrome brain; lessons from Alzheimer’s disease. Curr. Gerontol. Geriatr. Res. 2012, 170276. 10.1155/2012/170276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guedj F, Siegel AE, Pennings JLA, Alsebaa F, Massingham LJ, Tantravahi U, and Bianchi DW (2020). Apigenin as a candidate prenatal treatment for trisomy 21: effects in human amniocytes and the Ts1Cje mouse model. Am. J. Hum. Genet. 107, 911–931. 10.1016/j.ajhg.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pinto B, Morelli G, Rastogi M, Savardi A, Fumagalli A, Petretto A, Bartolucci M, Varea E, Catelani T, Contestabile A, et al. (2020). Rescuing over-activated microglia restores cognitive performance in juvenile animals of the Dp(16) mouse model of down syndrome. Neuron 108, 887–904.e12. 10.1016/j.neuron.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hunter CL, Quintero EM, Gilstrap L, Bhat NR, and Granholm AC (2004). Minocycline protects basal forebrain cholinergic neurons from mu p75-saporin immunotoxic lesioning. Eur. J. Neurosci. 19, 3305–3316. 10.1111/j.0953-816X.2004.03439.x. [DOI] [PubMed] [Google Scholar]
- 33.Lockrow J, Prakasam A, Huang P, Bimonte-Nelson H, Sambamurti K, and Granholm AC (2009). Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp. Neurol. 216, 278–289. 10.1016/j.expneurol.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan S (2019). IGFBP-2 signaling in the brain: from brain development to higher order brain functions. Front. Endocrinol. 10, 822. 10.3389/fendo.2019.00822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dyer AH, Vahdatpour C, Sanfeliu A, and Tropea D (2016). The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 325, 89–99. 10.1016/j.neuroscience.2016.03.056. [DOI] [PubMed] [Google Scholar]
- 36.Wrigley S, Arafa D, and Tropea D (2017). Insulin-like growth factor 1: at the crossroads of brain development and aging. Front. Cell. Neurosci. 11, 14. 10.3389/fncel.2017.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deliu E, Arecco N, Morandell J, Dotter CP, Contreras X, Girardot C, Käsper EL, Kozlova A, Kishi K, Chiaradia I, et al. (2018). Haploin-sufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci. 21, 1717–1727. 10.1038/s41593-018-0266-2. [DOI] [PubMed] [Google Scholar]
- 38.Mahato AK, and Sidorova YA (2020). RET receptor tyrosine kinase: role in neurodegeneration, obesity, and cancer. Int. J. Mol. Sci. 21, 7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baxter RC, Martin JL, and Beniac VA (1989). High molecular weight insulin-like growth factor binding protein complex. Purification and properties of the acid-labile subunit from human serum. J. Biol. Chem. 264, 11843–11848. 10.1016/S0021-9258(18)80143-0. [DOI] [PubMed] [Google Scholar]
- 40.Savage MO, Hwa V, David A, Rosenfeld RG, and Metherell LA (2011). Genetic defects in the growth hormone-IGF-I Axis causing growth hormone insensitivity and impaired linear growth. Front. Endocrinol. 2, 95. 10.3389/fendo.2011.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allard JB, and Duan C (2018). IGF-binding proteins: why do they exist and why are there so many? Front. Endocrinol. 9, 117. 10.3389/fendo.2018.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hass J, Walton E, Kirsten H, Turner J, Wolthusen R, Roessner V, Sponheim SR, Holt D, Gollub R, Calhoun VD, and Ehrlich S (2015). Complexin2 modulates working memory-related neural activity in patients with schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 265, 137–145. 10.1007/s00406-014-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubick N, Brösamle D, and Mickael M-E (2018). Molecular evolution and functional divergence of the IgLON family. Evol. Bioinform. Online 14, 1176934318775081. 10.1177/1176934318775081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin E, Kashiwagi Y, Kuriu T, Iwasaki H, Tanaka T, Koizumi H, Gleeson JG, and Okabe S (2013). Doublecortin-like kinase enhances dendritic remodelling and negatively regulates synapse maturation. Nat. Commun. 4, 1440. 10.1038/ncomms2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miranda M, Morici JF, Zanoni MB, and Bekinschtein P (2019). Brain-derived neurotrophic factor: a key molecule for memory in the healthy and the pathological brain. Front. Cell. Neurosci. 13, 363. 10.3389/fncel.2019.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Junnila RK, List EO, Berryman DE, Murrey JW, and Kopchick JJ (2013). The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 9, 366–376. 10.1038/nrendo.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sjögren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Törnell J, Isaksson OG, Jansson JO, and Ohlsson C (1999). Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc. Natl. Acad. Sci. USA 96, 7088–7092. 10.1073/pnas.96.12.7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Renes JS, van Doorn J, and Hokken-Koelega ACS (2014). Ternary complex formation and IGFBP-3 proteolytic activity during childhood: age-dependent changes. J. Clin. Endocrinol. Metab. 99, E1988–E1996. 10.1210/jc.2013-3814. [DOI] [PubMed] [Google Scholar]
- 49.Jinquan T, Quan S, Feili G, Larsen CG, and Thestrup-Pedersen K (1999). Eotaxin activates T cells to chemotaxis and adhesion only if induced to express CCR3 by IL-2 together with IL-4. J. Immunol. 162, 4285–4292. [PubMed] [Google Scholar]
- 50.Witkowska-Sędek E, and Pyrzak B (2020). Chronic inflammation and the growth hormone/insulin-like growth factor-1 axis. Cent. Eur. J. Immunol. 45, 469–475. 10.5114/ceji.2020.103422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Picardi A, Gentilucci UV, Zardi EM, Caccavo D, Petitti T, Manfrini S, Pozzilli P, and Afeltra A (2003). TNF-alpha and growth hormone resistance in patients with chronic liver disease. J. Interferon Cytokine Res. 23, 229–235. 10.1089/107999003321829944. [DOI] [PubMed] [Google Scholar]
- 52.DiFedele LM, He J, Bonkowski EL, Han X, Held MA, Bohan A, Menon RK, and Denson LA (2005). Tumor necrosis factor alpha blockade restores growth hormone signaling in murine colitis. Gastroenterology 128, 1278–1291. 10.1053/j.gastro.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 53.Chelban V, Wilson MP, Warman Chardon J, Vandrovcova J, Zanetti MN, Zamba-Papanicolaou E, Efthymiou S, Pope S, Conte MR, Abis G, et al. (2019). PDXK mutations cause polyneuropathy responsive to pyridoxal 5’-phosphate supplementation. Ann. Neurol. 86, 225–240. 10.1002/ana.25524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cronk C, Crocker AC, Pueschel SM, Shea AM, Zackai E, Pickens G, and Reed RB (1988). Growth charts for children with Down syndrome: 1 month to 18 years of age. Pediatrics 81, 102–110. [PubMed] [Google Scholar]
- 55.Riikonen R (2016). Treatment of autistic spectrum disorder with insulin-like growth factors. Eur. J. Paediatr. Neurol. 20, 816–823. 10.1016/j.ejpn.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Vahdatpour C, Dyer AH, and Tropea D (2016). Insulin-like growth factor 1 and related compounds in the treatment of childhood-onset neurodevelopmental disorders. Front. Neurosci. 10, 450. 10.3389/fnins.2016.00450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Costales J, and Kolevzon A (2016). The therapeutic potential of insulin-like growth factor-1 in central nervous system disorders. Neurosci. Biobehav. Rev. 63, 207–222. 10.1016/j.neubiorev.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raisingani M, Preneet B, Kohn B, and Yakar S (2017). Skeletal growth and bone mineral acquisition in type 1 diabetic children; abnormalities of the GH/IGF-1 axis. Growth Horm. IGF Res. 34, 13–21. 10.1016/j.ghir.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nambam B, and Schatz D (2018). Growth hormone and insulin-like growth factor-I axis in type 1 diabetes. Growth Horm. IGF Res. 38, 49–52. 10.1016/j.ghir.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki S, Morimoto S, Fujishiro M, Kawasaki M, Hayakawa K, Miyashita T, Ikeda K, Miyazawa K, Yanagida M, Takamori K, et al. (2015). Inhibition of the insulin-like growth factor system is a potential therapy for rheumatoid arthritis. Autoimmunity 48, 251–258. 10.3109/08916934.2014.976631. [DOI] [PubMed] [Google Scholar]
- 61.Skarlis C, Nezos A, Mavragani CP, and Koutsilieris M (2019). The role of insulin growth factors in autoimmune diseases. Ann. Res. Hosp. 3, 10. 10.21037/arh.2019.03.02. [DOI] [Google Scholar]
- 62.Wang Z, Li W, Guo Q, Wang Y, Ma L, and Zhang X (2018). Insulin-like growth factor-1 signaling in lung development and inflammatory lung diseases. BioMed Res. Int. 2018, 6057589. 10.1155/2018/6057589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hellström A, Ley D, Hansen-Pupp I, Hallberg B, Löfqvist C, van Marter L, van Weissenbruch M, Ramenghi LA, Beardsall K, Dunger D, et al. (2016). Insulin-like growth factor 1 has multisystem effects on foetal and preterm infant development. Acta Paediatr. 105, 576–586. 10.1111/apa.13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tseng FY, Chen YT, Chi YC, Chen PL, and Yang WS (2019). Serum levels of insulin-like growth factor 1 are negatively associated with log transformation of thyroid-stimulating hormone in Graves’ disease patients with hyperthyroidism or subjects with euthyroidism: a prospective observational study. Medicine (Baltim.) 98, e14862. 10.1097/md.0000000000014862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Waugh KA, Minter R, Baxter J, Chi C, Tuttle KD, Eduthan NP, Galbraith MD, Kinning KT, Andrysik Z, Araya P, et al. (2022). Interferon receptor gene dosage determines diverse hallmarks of Down syndrome. bioRxiv. 10.1101/2022.02.03.478982. [DOI] [Google Scholar]
- 66.Tuttle KD, Waugh KA, Araya P, Minter R, Orlicky DJ, Ludwig M, Andrysik Z, Burchill MA, Tamburini BAJ, Sempeck C, et al. (2020). JAK1 inhibition blocks lethal immune hypersensitivity in a mouse model of down syndrome. Cell Rep. 33, 108407. 10.1016/j.celrep.2020.108407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, et al. (2016). Trisomy 21 consistently activates the interferon response. Elife 5, e16220. 10.7554/eLife.16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Powers RK, Culp-Hill R, Ludwig MP, Smith KP, Waugh KA, Minter R, Tuttle KD, Lewis HC, Rachubinski AL, Granrath RE, et al. (2019). Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat. Commun. 10, 4766. 10.1038/s41467-019-12739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rachubinski AL, Estrada BE, Norris D, Dunnick CA, Boldrick JC, and Espinosa JM (2019). Janus kinase inhibition in Down syndrome: 2 cases of therapeutic benefit for alopecia areata. JAAD Case Rep. 5, 365–367. 10.1016/j.jdcr.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pham AT, Rachubinski AL, Enriquez-Estrada B, Worek K, Griffith M, and Espinosa JM (2021). JAK inhibition for treatment of psoriatic arthritis in Down syndrome. Rheumatology 60, e309–e311. 10.1093/rheumatology/keab203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Joseph D’Ercole A, and Ye P (2008). Expanding the mind: insulin-like growth factor I and brain development. Endocrinology 149, 5958–5962. 10.1210/en.2008-0920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palmer CR, Liu CS, Romanow WJ, Lee M-H, and Chun J (2021). Altered cell and RNA isoform diversity in aging Down syndrome brains. Proc. Natl. Acad. Sci. USA 118, e2114326118. 10.1073/pnas.2114326118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan H, Mitschelen M, Bixler GV, Brucklacher RM, Farley JA, Han S, Freeman WM, and Sonntag WE (2011). Circulating IGF1 regulates hippocampal IGF1 levels and brain gene expression during adolescence. J. Endocrinol. 211, 27–37. 10.1530/joe-11-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trejo JL, Piriz J, Llorens-Martin MV, Fernandez AM, Bolós M, LeRoith D, Nuñez A, and Torres-Aleman I (2007). Central actions of liver-derived insulin-like growth factor I underlying its pro-cognitive effects. Mol. Psychiatry 12, 1118–1128. 10.1038/sj.mp.4002076. [DOI] [PubMed] [Google Scholar]
- 75.Svensson J, Diez M, Engel J, Wass C, Tivesten A, Jansson JO, Isaksson O, Archer T, Hökfelt T, and Ohlsson C (2006). Endocrine, liver-derived IGF-I is of importance for spatial learning and memory in old mice. J. Endocrinol. 189, 617–627. 10.1677/joe.1.06631. [DOI] [PubMed] [Google Scholar]
- 76.Cacciatore I, Cornacchia C, Baldassarre L, Fornasari E, Mollica A, Stefanucci A, and Pinnen F (2012). GPE and GPE analogues as promising neuroprotective agents. Mini Rev. Med. Chem. 12, 13–23. 10.2174/138955712798868995. [DOI] [PubMed] [Google Scholar]
- 77.Puche JE, and Castilla-Cortázar I (2012). Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J. Transl. Med. 10, 224. 10.1186/1479-5876-10-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Annerén G, Sara VR, Hall K, and Tuvemo T (1986). Growth and somatomedin responses to growth hormone in Down’s syndrome. Arch. Dis. Child. 61, 48–52. 10.1136/adc.61.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Myrelid A, Frisk P, Stridsberg M, Annerén G, and Gustafsson J (2010). Normal growth hormone secretion in overweight young adults with Down syndrome. Growth Horm. IGF Res. 20, 174–178. 10.1016/j.ghir.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 80.Ruvalcaba RH, Thuline HC, and Kelley VC (1972). Plasma growth hormone in patients with chromosomal anomalies. Arch. Dis. Child. 47, 307–309. 10.1136/adc.47.252.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sara VR, Gustavson KH, Annerén G, Hall K, and Wetterberg L (1983). Somatomedins in Down’s syndrome. Biol. Psychiatry 18, 803–811. [PubMed] [Google Scholar]
- 82.Torrado C, Bastian W, Wisniewski KE, and Castells S (1991). Treatment of children with Down syndrome and growth retardation with recombinant human growth hormone. J. Pediatr. 119, 478–483. 10.1016/s0022-3476(05)82068-2. [DOI] [PubMed] [Google Scholar]
- 83.Annerén G, Tuvemo T, Carlsson-Skwirut C, Lönnerholm T, Bang P, Sara VR, and Gustafsson J (1999). Growth hormone treatment in young children with Down’s syndrome: effects on growth and psychomotor development. Arch. Dis. Child. 80, 334–338. 10.1136/adc.80.4.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Myrelid Å, Bergman S, Elfvik Strömberg M, Jonsson B, Nyberg F, Gustafsson J, and Annerén G (2010). Late effects of early growth hormone treatment in Down syndrome. Acta Paediatr. 99, 763–769. 10.1111/j.1651-2227.2009.01679.x. [DOI] [PubMed] [Google Scholar]
- 85.Yumet G, Shumate ML, Bryant DP, Lang CH, and Cooney RN (2006). Hepatic growth hormone resistance during sepsis is associated with increased suppressors of cytokine signaling expression and impaired growth hormone signaling. Crit. Care Med. 34, 1420–1427. 10.1097/01.Ccm.0000215113.66070.E0. [DOI] [PubMed] [Google Scholar]
- 86.Chen Y, Sun D, Krishnamurthy VMR, and Rabkin R (2007). Endotoxin attenuates growth hormone-induced hepatic insulin-like growth factor I expression by inhibiting JAK2/STAT5 signal transduction and STAT5b DNA binding. Am. J. Physiol. Endocrinol. Metab. 292, E1856–E1862. 10.1152/ajpendo.00581.2006. [DOI] [PubMed] [Google Scholar]
- 87.Zhao Y, Xiao X, Frank SJ, Lin HY, and Xia Y (2014). Distinct mechanisms of induction of hepatic growth hormone resistance by endogenous IL-6, TNF-α, and IL-1β. Am. J. Physiol. Endocrinol. Metab. 307, E186–E198. 10.1152/ajpendo.00652.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Denson LA, Menon RK, Shaufl A, Bajwa HS, Williams CR, and Karpen SJ (2001). TNF-alpha downregulates murine hepatic growth hormone receptor expression by inhibiting Sp1 and Sp3 binding. J. Clin. Invest. 107, 1451–1458. 10.1172/jci10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.De Benedetti F, Meazza C, Oliveri M, Pignatti P, Vivarelli M, Alonzi T, Fattori E, Garrone S, Barreca A, and Martini A (2001). Effect of IL-6 on IGF binding protein-3: a study in IL-6 transgenic mice and in patients with systemic juvenile idiopathic arthritis. Endocrinology 142, 4818–4826. 10.1210/endo.142.11.8511. [DOI] [PubMed] [Google Scholar]
- 90.Tahimic CGT, Wang Y, and Bikle DD (2013). Anabolic effects of IGF-1 signaling on the skeleton. Front. Endocrinol. 4, 6. 10.3389/fendo.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ren J, Samson WK, and Sowers JR (1999). Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J. Mol. Cell. Cardiol. 31, 2049–2061. 10.1006/jmcc.1999.1036. [DOI] [PubMed] [Google Scholar]
- 92.Rodero MP, and Crow YJ (2016). Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J. Exp. Med. 213, 2527–2538. 10.1084/jem.20161596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gold L, Walker JJ, Wilcox SK, and Williams S (2012). Advances in human proteomics at high scale with the SOMAscan proteomics platform. N. Biotechnol. 29, 543–549. 10.1016/j.nbt.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 94.RStudio Team (2020). RStudio: integrated development for R. (RStudio, PBC; ). http://www.rstudio.com. [Google Scholar]
- 95.R Core Team (2020). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). http://www.rstudio.com. [Google Scholar]
- 96.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sergushichev AA (2016). An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. 10.1101/060012. [DOI] [Google Scholar]
- 99.Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Deidentified plasma proteomics data, plasma neurodegeneration biomarker data, and plasma inflammatory marker data have been deposited in the Synapse platform and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Critical commercial assays | ||
|
| ||
| Neurology 4-plex B kit | Single Molecular Array Technology (SIMOA® SR-XTM Analyzer, Quanterix Corporation, MA) | Cat# 103345 |
| U-PLEX Biomarker Group 1 (hu) 71-Plex | Meso Scale Discovery (MSD) | Cat# K15081K |
| V-PLEX Vascular Injury Panel 2 Human Kit | Meso Scale Discovery (MSD) | Cat# K15198D |
| V-PLEX Angiogenesis Panel 1 Human Kit | Meso Scale Discovery (MSD) | Cat# K15190D |
|
| ||
| Deposited data | ||
|
| ||
| Combined multi-omics dataset | Human Trisome Project | Synapse data: https://www.synapse.org/#!Synapse:syn35991637.1/datasets/ |
| Plasma proteomics data | Human Trisome Project | Synapse data: https://doi.org/10.7303/syn31488781 |
| Neurodegeneration biomarker data | Human Trisome Project | Synapse data: https://doi.org/10.7303/syn35993837 |
| Inflammatory marker data | Human Trisome Project | Synapse data: https://doi.org/10.7303/syn31475487 |
|
| ||
| Software and algorithms | ||
|
| ||
| R | R Foundation for Statistical Computing | v3.6.1 RRID:SCR_001905, https://www.R-project.org/ |
| RStudio | RStudio, Inc. | v1.2.5025 RRID:SCR_000432, http://www.rstudio.com/ |
| Bioconductor | N/A | v3.11 RRID:SCR_006442, https://bioconductor.org/ |
| limma package for R | N/A | v3.44.3 RRID:SCR_010943, https://bioconductor.org/packages/release/bioc/html/limma.html |
| SomaDataIO R package | SomaLogic | v3.1.0 RRID: SCR_022198, https://github.com/SomaLogic/SomaDataIO |
| ggplot2 package for R | N/A | v3.3.1 RRID:SCR_014601, https://ggplot2.tidyverse.org/ |
| fgsea package for R | N/A | v 1.14.0 RRID:SCR_020938, https://github.com/ctlab/fgsea |
| ConsensusClusterPlus package for R | Bioconductor | v1.52.0 RRID:SCR_016954, https://git.bioconductor.org/packages/ConsensusClusterPlus |