

Abstract

The conjugated bis-guanidinate-stabilized zinc hydride complex (I)-precatalyzed chemoselective dehydroborylation of a wide array of terminal alkynes with excellent yields is reported. Further, precatalyst I is compared with a newly synthesized DiethylNacNac zinc hydride precatalyst (III) for selective dehydroborylation of terminal alkynes, and it is discovered that precatalyst I is more active than III. We have studied intra- and intermolecular chemoselective dehydroborylation of terminal alkynes over other reducible functionalities such as alkene, ester, isocyanide, nitro, and heterocycles. The highly efficient precatalyst I shows a turnover number of 48.5 and turnover frequency of up to 60.5 h–1 in the dehydroborylation of 1-ethynyl-4-fluorobenzene (1i). A plausible mechanism for selective dehydrogenative borylation of alkynes has been proposed based on active catalyst isolation and a series of stoichiometric reactions.
Introduction
A selective dehydrogenative borylation of terminal alkyne produces ubiquitous organoborane products, which play an essential role in many chemical transformations.1 It also provides valuable intermediates in organic and medicinal chemistry.2 Because of its wide application, many methods have been developed to synthesize organoboranes.3 In conventional methods, organoboranes have been synthesized using stoichiometric amounts of organolithium and organomagnesium reagents.3,4 The abovementioned methods suffer from the following drawbacks: poor functional group tolerance, safety issues, and large quantities of waste material. Therefore, hydroboration reactions using pinacolborane (HBpin)5 or bis(pinacolato)diboron (B2pin2)6 are efficient routes for synthesizing precious organoborane compounds. As far as zinc-mediated hydroboration of alkynes to vinyl boronates is concerned, Uchiyama and co-workers reported hydroboration of alkyne by using diethylzinc and B2pin2.7 Besides, Geetharani and co-workers described Zn(OTf)2- and [Na][HBEt3]-catalyzed hydroboration of alkynes.5d Compared to well-established hydroboration of alkynes using transition metals,5b,6,8 main group metal,5a,5c,5d,9 and metal-free catalysts,10 there are fewer reports on dehydrogenative borylation of terminal alkynes to alkynyl boronates (Scheme 1, A).
Scheme 1. (A–C) Metal-Catalyzed Dehydroborylation of Terminal Alkynes.
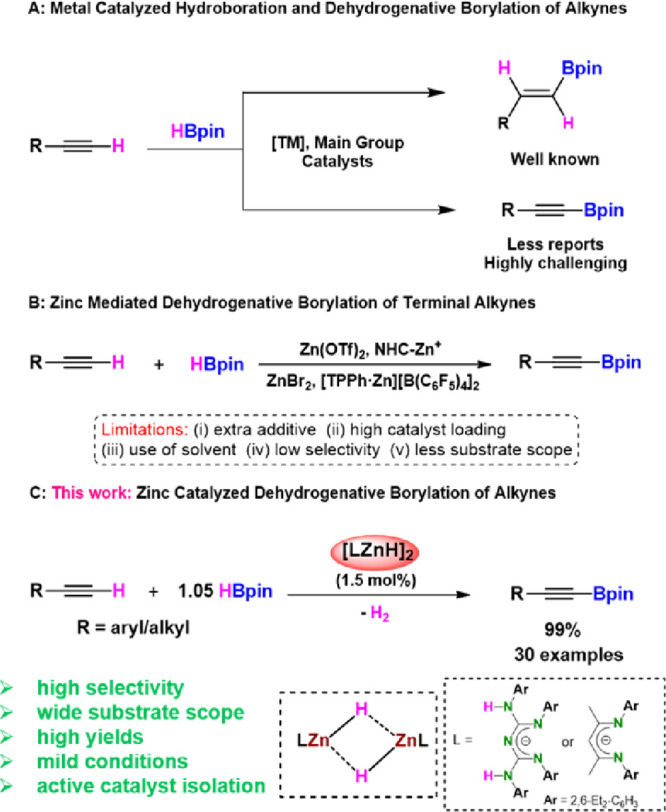
Ozerov and co-workers revealed the first catalytic dehydrogenative borylation of terminal alkynes using the iridium-based catalyst.11 Subsequently, transition (Cu,12 Fe13) and main group (Al)14 metal-based catalysts have been utilized for the dehydrogenative borylations. Moreover, a few reports on zinc-mediated dehydrogenative borylation of terminal alkynes are also known in the literature. Tsuchimoto and co-workers described Zn(OTf)2-catalyzed dehydrogenative borylation of terminal alkynes.15 Ingleson and co-workers showed the dehydrogenative borylation of terminal alkyne and hydroboration of alkynyl borate products using NHC zinc cation adducts.16
After that, Ma and co-workers reported similar reactions using ZnBr2 as a catalyst.17 During the preparation of our article, Dobrovetsky and co-workers demonstrated the zinc cation [TPPhZn2+][B(C6F5)4]2 as a catalyst for dehydrogenative borylation and hydroboration of terminal alkynes.18 All those methods suffer from drawbacks such as high catalyst loading, additive usage, low selectivity, and limited substrate scope. Thus, synthesizing selective dehydrogenative borylation of terminal alkynes by applying an efficient catalyst under mild conditions is highly attractive.
Enhancing the catalytic activities of the molecular metal complex depends upon ligand design, including steric and electronic properties. The main advantage of choosing molecular catalysts is understanding the reaction mechanism by isolating the key intermediates and influencing the selectivity. In this context, molecular main group hydrides, including zinc hydrides, are used as catalysts for various organic transformations (Scheme 1, B).19 Very recently, our group reported conjugated bis-guanidinate (CBG)-supported low oxidation state Zn–Zn bonded compounds, i.e., Cp*ZnZnL and LZnZnL {Cp* = 1,2,3,4,5-pentamethyl cyclopentadienide, L = [(ArNH)(ArN)–C=N–C=(NAr)(NHAr)], and Ar = 2,6-Et2-C6H3} as precatalysts for the dehydroborylation of terminal alkynes under mild conditions.20 However, both compounds were prepared by the previously reported zinc (I) dimer, Cp*ZnZnCp*, by Carmona and co-workers.21 Considering the zinc hydride catalysis, we presumed that easily accessible molecular Zn hydrides would be a better option for dehydroborylation of terminal alkynes. Surprisingly, there have been no reports of molecular zinc hydrides as precatalysts for selective dehydroborylation of terminal alkynes, except Ingleson’s sole example.16a
Thus, herein, we report the CBG zinc hydride (I) as a precatalyst for selective dehydroborylation of a broad range of terminal alkynes under mild conditions to afford the respective alkynyl boronate products. Further, we describe the isolation and structural characterization of the active catalysts CBG zinc alkynyl II and NacNac zinc alkynyl IV (Scheme 1, C) compounds. Moreover, the zinc hydride (I) is compared with NacNac zinc hydride (III) to understand the spectator ligand’s role and catalytic efficiency and selectivity for the dehydroborylation of terminal alkynes for the first time.
Results and Discussion
Previously, we synthesized the CBG zinc hydride compound I, [{L1ZnH}2; (L1 = {(ArNH)(ArN)–C=N–C=(NAr)(NHAr)}; Ar = 2,6-Et2-C6H3)], which can be accessed by the reaction of L1ZnI with KNH(iPr)BH3.22,23 In addition, we published that compound I catalyzed the hydroboration of heteroallenes and carbonyl compounds.22,23 As we mentioned, only a handful of examples documented the zinc-catalyzed dehydroborylation of terminal alkynes. Thus, herein we aimed to explore CBG zinc hydride(I)-catalyzed dehydroborylation of terminal alkynes. Based on the NacNac-supported zinc hydrides established by Roesky and co-workers,24 Harder and co-workers,19a and other research groups,19 the corresponding diethyl NacNacZn hydride (III) can be prepared in two steps. The reaction of [L2ZnEt]225 (DiethylNacNac (L2) = CH{(CMe)(2,6-Et2C6H3N)}2) with 2.0 equiv. of I2 in toluene produced [L2ZnI]2. A reaction between [L2ZnI]2 and 2.0 equiv. KH in toluene afforded [L2ZnH]2.
We began our investigation by taking 1:1.05 stoichiometric amounts of phenylacetylene and HBpin with 10 mol % of precatalyst I in a neat condition and gave quantitative conversion of borylated alkyne after 6 h at 60 °C (Table S1, entry 2). After that, lowering the precatalyst loadings to 5.0, 3.0, 2.0, and 1.5 mol % and decreasing the temperature from 60 °C to room temperature after 6 h gave quantitative conversion in neat conditions. Notably, 1 mol % precatalyst loading after 1 h gave a 98% yield, which shows the turnover number (TON) of 49 and turnover frequency (TOF) of 49 h–1 (Table S1, entry 7/Table 1, entry 1). However, further reduction of the catalyst loading to 0.5 mol % yielded low conversion (75%) (Table S1, entry 8). Moreover, the same reaction in benzene, toluene, and THF using 1.5 mol % of precatalyst I produced the desired product 2a quantitatively. A negligible conversion was noticed in the absence of a precatalyst, I, which indicates that a zinc hydride complex is necessary for the dehydroborylation of terminal alkynes. A wide range of aryl, cyclic, and acyclic (long-chain) terminal alkyne substrates were investigated using optimized conditions. The various alkynes, including electron-donating (1a–1g) and withdrawing substituents (1h–1n), bis(ethynyl)benzene (1o), heterocycle systems (1p and 1q), aliphatic alkynes including long-chain (1r–1z), and cyclic groups (1aa–1ad), afforded selective alkynyl boronate products with high yields. Moreover, a substrate bearing an electron-donating group, 4-ethynyltoluene (1c), exhibits a TON of 48.5 and TOF of 32.5 h–1, while a substrate bearing an electron-withdrawing group, 1-ethynyl-4-fluorobenzene (1i), gives a TON of 48.5 and TOF of 60.5 h–1. All alkynyl borates (2a–2ad) were confirmed by multinuclear magnetic resonance (1H, 13C{1H}, and 11B) and HRMS analyses (Scheme 2).
Table 1. Comparison of TON and TOF for Dehydroborylation of Terminal Alkynes of Selected Substrates with Precatalysts I and III and Catalyst A.

| entry | substrate | cat. | time (h) | conv. (%) | a/a′ | TON | TOF (h–1) |
|---|---|---|---|---|---|---|---|
| 1 | 1a | I | 1 | 98 | 99:<1 | 49 | 49 |
| 2 | 1a | III | 1 | 70 | 98:2 | 35 | 35 |
| 3 | 1a | A | 5 | 84 | 16.8 | 3.4 | |
| 4 | 1c | I | 1.5 | 97 | 99:<1 | 48.5 | 32.5 |
| 5 | 1c | III | 1.5 | 70 | 98:2 | 35 | 23.5 |
| 6 | 1c | A | 5 | 90 | 18 | 3.6 | |
| 7 | 1i | I | 0.8 | 97 | 99:<1 | 48.5 | 60.5 |
| 8 | 1i | III | 0.8 | 76 | 97:3 | 38 | 47.5 |
| 9 | 1i | A | 5 | 81 | 16.2 | 3.2 |
Reaction conditions: alkyne (0.2 mmol, 1.0 equiv.), pinacolborane (0.21 mmol, 1.05 equiv.), precatalyst I or III (1 mol %), at rt under N2. The NMR yield was determined by 1H NMR spectroscopy using mesitylene as the internal standard. TON and TOF were calculated for the conversion of product a. TON was calculated by dividing the number of moles of the product by the number of moles of catalyst used. TOF was determined by dividing TON by the reaction time. LZnH(I) and L′ZnH(III), calculating TON and TOF based on monomeric structures.
Scheme 2. Dehydrogenative Borylation of Terminal Alkynes Catalyzed by [L1ZnH]2 Precatalyst(I)a.

Reaction conditions: terminal alkynes (0.3 mmol, 1.0 equiv.), pinacolborane (0.315 mmol, 1.05 equiv.), precatalyst I (1.5 mol %), at rt under N2.
Preparative-scale reaction: 1 mmol of terminal alkynes, precatalyst I (1.5 mol %), 1.05 mmol of HBpin, 6 h, rt.
For 2a, 2c, and 2i, 1 mol % precatalyst was used and stirred for 1, 1.5, and 0.8 h, respectively, and the NMR yield was determined by 1H NMR spectroscopy using mesitylene as the internal standard.
For 2n, NMR yield was determined by 1H NMR spectroscopy using mesitylene as the internal standard.
For 2o, pinacolborane (0.63 mmol, 2.1 equiv.) was used and stirred for 12 h.
For 2x, stirred for 2 h. Based on alkyne consumption, the NMR yield was determined by 1H and 11B NMR spectroscopy and identified the Bpin peak to confirm the product.
Next, we decided to conduct some intermolecular chemoselective dehydroborylation of terminal alkynes. Accordingly, equimolar quantities of phenylacetylene, styrene, and HBpin were mixed with precatalyst I (1.5 mol %) under neat conditions for 2 h, which afforded the dehydroborylated product 2a in a quantitative yield in preference to styrene.
Likewise, phenylacetylene provided corresponding dehydrogenative borylated product 2a under the same reaction conditions in a quantitative yield over benzyl benzoate or pyridine. Similarly, equimolar amounts of 5-chloro-1-pentyne (1x), 1-pentyl isocyanide, and HBpin with 1.5 mol % precatalyst I for 2 h produced a corresponding dehydroborylated product (2x), in which isocyanide is untouched. We have noticed the excellent tolerance of alkene, ester, pyridine, and isocyanide functionalities in intermolecular chemoselective reactions (Scheme 3).
Scheme 3. Intermolecular Chemoselective Reactions.

Phenylacetylene (1a), 4-ethynyltoluene (1c), and 1-ethynyl-4-fluorobenzene (1i) substrates were selected to screen the catalytic activity of zinc precatalysts I and III and catalyst A.16b The catalytic dehydroborylation of phenylacetylene (1a) was performed under mild conditions to produce high TON, 49, and TOF, 49 h–1, compared to precatalyst III (TON, 35, and TOF, 35 h–1) and catalyst A (TON, 16.8, and TOF, 3.4 h–1) (Table 1, entries 1–3). Similarly, for substrate 4-ethynyltoluene (1c), precatalyst I displayed a better TON, 48.5, and TOF, 32.5 h–1, compared to precatalyst III (TON, 35, and TOF, 23.5 h–1) and catalyst A (TON, 18, and TOF, 3.6 h–1)16b (Table 1, entries 4–6). Table 1 (entries 7–9) indicates that compound I shows better catalytic performance (TON, 48.5, and TOF, 60.5 h–1) than compound III (TON, 38, and TOF, 47.5 h–1) and compound A (TON, 16.2, and TOF, 3.2 h–1). As shown in Table 1 (entries 1–9), it is evident that precatalyst I exhibits better catalytic activity and selectivity than precatalyst III and the Ingleson catalyst A.
Moreover, we observed a mixture of dehydroborylation and 1,1-borylated products in the presence of precatalyst III. It should be noted that Ingleson’s catalyst A also gave a mixture of products.16b The 1,1-borylated products evidenced by 11B NMR spectroscopy exhibit a characteristic signal in the range of 30–34 ppm. However, exclusively dehydroborylated products are formed in the case of compound I as a precatalyst.
We conducted a series of stoichiometric experiments to establish the mechanism of zinc-catalyzed dehydroborylation of terminal alkynes to a selective alkynyl borate product. A reaction between 1:2 stoichiometric amounts of precatalyst I and phenylacetylene afforded CBG zinc alkynyl complex(II) in d8-toluene at rt after 20 min. Compound II was characterized by NMR, HRMS, IR, and X-ray studies.
The 1H NMR spectrum shows the production of compound II by complete disappearances of alkynyl “C≡C–H” and Zn–H resonances (Scheme 4a). Next, catalyst II reacted with 2 equiv. of HBpin undergoes Zn–C/B–H bond metathesis to give the desired dehydroboration product (2a) and precatalyst I with a 30% yield, which was observed by multinuclear NMR spectroscopy. The 1H and 11B NMR spectra indicate the formation of product 2a by the appearance of new peaks at 1.03 and 24.5 ppm, corresponding to the Bpin moiety of product 2a.
Scheme 4. (a–e) Stoichiometric Experiments for Dehydroborylation of Terminal Alkynes.

Prolonged heating up to 24 h at 80 °C showed no change in the relative ratio of 2a and II (does not complete the reaction). This suggests that the equilibrium position has been reached before heating. The reaction mixture contains metathesis products I and 2a and unreacted compound II, HBpin, at room temperature, confirming the equilibrium position.
Further, a reaction between compound I and 2a has been performed in d8-toluene to confirm the reversible reaction. We noticed the formation of compound II with HBpin in 70% conversion, as shown by the characteristic peak of HBpin at 0.98 ppm in 1H and 27.7 and 29.1 ppm in the 11B spectrum (for more details, see Figures S8 and S10). However, Zn–H/C–B metathesis reaction did not complete even after heating at 80 °C for 24 h; the same ratio of compound II: 2a was observed (Scheme 4c). Finally, 2 equiv. of phenylacetylene was added to a J. Young valve NMR tube containing a mixture of compounds I and 2a (30% conversion) in d8-toluene. After 15 min, the quantitative formation of compounds 2a and II was observed. The 1H NMR spectra confirm the formation of compound II and product 2a (Scheme 4d). Next, equimolar quantities of compounds I, 1a, and 2a were mixed in d8-toluene at room temperature for 20 min. We notice the exclusive formation of compound II (Scheme 4e). This study supports the abovementioned reactions.
To further assess the reactivity of precatalysts I and III, the same stoichiometric experiments were performed with the NacNacZn system. Similarly, {L′ZnH}2(III) reacts with phenylacetylene to produce compound IV via deprotonation. However, the metathesis of zinc–alkynyl(IV) species with HBpin yielded a 25% conversion (less conversion compared to CBG systems) of the dehydroboration product (2a).
Likewise, a reaction between complex III and compound 2a in d8-toluene resulted in compound IV and HBpin with a 75% conversion. Thus, the subsequent reaction of a mixture of compounds III and 2a with a 25% conversion with additional phenylacetylene (2.0 equiv) produces a quantitative conversion of compounds 2a and IV along with 1,1-diborylated alkenes after 20 min (a slightly longer reaction time compared to CBG systems) (for more details, see Figures S16 and S33).
Further, we conducted additional stoichiometric experiments to confirm the relative ratio of alkynyl-Bpin and zinc alkynyl complexes (which are in equilibrium) via 19F{1H} NMR spectroscopy. The zinc hydride complexes (I or III) reacted with 4-(trifluoromethyl)phenylacetylene (1m) afforded corresponding CBG zinc alkynyl complex (II′) or NacNac zinc alkynyl complex (IV′) in C6D6 at rt after 15 min. Further, catalyst II′ reacted with 2 equiv of HBpin yielded the desired dehydroboration product (2m) and precatalyst I with a 20% conversion, and by reacting with 2 equiv. of 1m, it afforded the quantitative amounts of compounds 2m and II′ (for more details, see Scheme S1 and Figures S37–S47). However, catalyst IV′ reacted with 2 equiv. of HBpin yielded the corresponding dehydroboration product (2m), precatalyst III, with a 14% yield and 1,1-diborylated alkenes product. When prolonging the heating, the alkynylborates convert to the 1,1-diborylated alkenes (for more details, see Scheme S2 and Figures S48–S54). Moreover, the relative ratio between alkynyl-Bpin and zinc alkynyl complexes is confirmed by 19F{1H} NMR spectroscopy (for more details, see Figures S55). Hence, precatalyst I is more active and selective than III.
The zinc alkynyl compounds II and IV were crystallized from a benzene solution and structurally characterized. The solid-state structure of compound II or IV revealed that the distorted tetrahedral zinc atom is coordinated by two N atoms from the CBG or NacNac ligand and the other two sites from the C-atoms of bridged alkynyl moieties (Figure 1). The bond distance of Zn1-C1, 2.026(2) Å, in compound II is significantly longer than the zinc–alkynyl bond distance of compound IV, 1.979(5) Å, and NHC zinc-bis alkynyl complex [Zn–C = 1.969(4) and 1.959(4) Å]. It suggests that compound II is more active than compound IV and the zinc-bis alkynyl complex established by Ingleson. In contrast, the C1–C2 bond length, 1.167(3) Å, of compound II is shorter compared to compound IV, 1.213(7) Å, and NHC zinc-bis alkynyl complex, 1.197(4) and 1.179(4) Å.16b It revealed that compound II is more selective than compound IV and the zinc-bis alkynyl complex to produce alkynyl boronates (Figure 1). Based on the abovementioned change in structural parameters (Figure 1), stoichiometric experiment data (Scheme 4) and comparison of TON and TOF in Table 1 suggest that precatalyst I exhibits better catalytic activity and selectivity than precatalyst III and the NHC–zinc hydride complex (vide supra).16b
Figure 1.

Molecular structures of compounds II (left) and IV (right). All the ethyl groups and hydrogen atoms (except for H(4) and H(5) from structure II) are removed for clarity. Comparison of crystal parameters of compound II vs IV [selected bond lengths (Å) and angles (degrees)].
Based on the isolation of active catalysts and a series of stoichiometric experiments, a plausible catalytic cycle has been proposed for zinc hydride-precatalyzed selective dehydrogenative borylation of terminal alkynes to alkynyl borates. The catalytic cycle involves two steps: (i) metathesis and (ii) protonolysis. Initially, the zinc hydride complex reacts with the terminal alkyne formation of zinc alkynyl II or IV.
Next, either II or IV reacts with HBpin, affording the alkynyl borate and precatalyst LZnH (I or III) in an equilibrium position via Zn–C/B–H bond metathesis. Next, LZnH reacts with another molecule of terminal alkyne, gives the quantitative amount of corresponding alkynylborate product (2a) and regenerates the active catalyst (II or IV), and closes the catalytic cycle (Scheme 5).
Scheme 5. Proposed Mechanism for the Dehydroborylation of Terminal Alkynes.

Conclusions
The N,N′-chelated and six-membered dimeric zinc hydride complexes I and III are used as precatalysts for dehydrogenative borylation of a wide array of terminal alkynes. A CBG zinc hydride I precatalyst is highly efficient and exceptionally selective compared to NacNac zinc hydride III.
The precatalyst I shows excellent tolerance to several functional groups such as halide, alkene, ester, heterocycle, and isocyanide in intra- and intermolecular chemoselective reactions. Most importantly, we isolated and structurally characterized the active catalysts, zinc acetylides II and IV. Such examples are rare in the literature. The most plausible reaction mechanism has been established based on stoichiometric experiments. We are undertaking further reactivity studies of zinc acetylides and their catalytic applications in our laboratory.
Experimental Section
Unless stated, manipulations were performed under a dinitrogen atmosphere using standard glovebox and Schlenk techniques. NMR spectra were recorded on a Jeol-400 MHz spectrometer and Bruker NMR spectrometers at 400 MHz (1H), 101 MHz (13C{1H}), 128.3 MHz (11B), and 377 MHz (19F{1H}). 1H NMR and 13C{1H} NMR chemical shifts are referenced to residual protons or carbons in the deuterated solvent. 11B were calibrated using an external reference of BF3·Et2O. Multiplicities are reported as singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). Chemical shifts are reported in ppm. Coupling constants are reported in Hz. The crystal data of compounds II and IV were collected on a Rigaku Oxford diffractometer with graphite-monochromated Cu-Kα radiation (λ = 1.54184 Å) and Mo-Kα radiation (λ = 0.71073 Å), respectively, at 100 K. Selected data collection parameters and other crystallographic results are summarized in Table S2. Mass spectrometry analyses were carried out on Bruker micrOTOF-Q II and Waters XevoG2 XS Q-TOF mass spectrometers. Infrared (IR) spectra were recorded in Thermo-Nicolet FT-IR spectrophotometers. The melting points of [L2ZnI]2, [L2ZnH]2, II, III, and IV were measured from the Stuart SMP 10 instrument.
Solvents were purified by distillation over Na/benzophenone. Deuterated chloroform (CDCl3) was dried on molecular sieves, and benzene-d6 (C6D6) was dried over Na/K alloy and distilled. The ligand L1(H) (L1 = {(ArNH)(ArN)–C
=N–C=(NAr)(NHAr)}; Ar = 2,6-Et2-C6H3)] and complex {L1ZnH}2(I) are prepared according to reported literature procedures.19,22−24 Complex {L2ZnEt}2 (L2 = DiethylNacNac) was prepared according to reported literature procedures.25 For catalysis reactions, J. Young valve-sealed NMR tubes or sealed reaction vials, as per the requirement, were properly oven-dried before being used. Chemicals and reagents were purchased from Sigma-Aldrich Co. Ltd., Merck India Pvt. Ltd., and TCI chemicals were used without purification.
Synthesis of [L1ZnCC(C6H5)]2, (II) {Reaction-Scale}
A solution of [L1ZnH]2(I) 0.250 g (0.179 mmol) in 10 mL of toluene was added to 39.47 μL (0.359 mmol) of phenylacetylene in ∼5 mL of toluene at room temperature through a cannula transfer and stirred for 2 h. The solvent was then removed in a vacuum, forming a white solid compound, and dried. The resultant solid was dissolved in benzene (∼10 mL) and filtered. The filtered solution was stored at room temperature and gave block-shaped colorless crystals within 2 days. (0.240 g, 82%); m.p. 190–195 °C. 1H NMR (400 MHz, C6D6) δ 7.29–7.27 (d, J = 7.8 Hz, 4H), 7.16–7.07 (m, 12H), 6.90 (t, J = 7.7 Hz,4H), 6.85–6.81 (d, 3JHH = 13.1 Hz, 6H), 6.65–6.63 (d, 3JHH = 7.6 Hz, 8H), 5.07 (s, 4H), 3.18–3.09 (m, 8H), 2.77–2.67 (m, 8H), 2.40–2.31 (m, 8H), 2.21–2.11 (m, 8H), 1.38 (t, 3JHH = 7.5 Hz, 24H), 0.96 (t, 3JHH = 7.5 Hz, 24H). 13C{1H} NMR (101 MHz, C6D6) δ 157.5, 141.9, 141.1, 139.0, 135.0, 131.9, 128.1, 126.7, 126.6, 126.4, 126.0, 125.3, 101.7, 77.4, 24.9, 24.3, 14.5, 14.2. IR (Nujol mull) ν (cm–1): 3024, 2883, 2811, 1996, 1495, 1416, 1327, 1260, 1203, 1091, 1018. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C50H60N5Zn 794.4140, found: 794.4146.
Synthesis of [L1ZnCC(C6H5)]2, (II) {NMR-Scale}
A solution of complex I (0.020 g, 25 °C, 0.014 mmol) in a J. Young valve NMR tube was treated with phenylacetylene (3.07 μL, 0.028 mmol) at room temperature; after 20 min, it resulted in the formation of compound II with the liberation of H2 and was observed by 1H NMR spectroscopy. NMR yield: (>99%). 1H NMR (400 MHz, d8-toluene) δ 7.24–7.15 (m, 5H), 7.09–7.01 (m, 8H), 6.97–6.90 (m, 5H), 6.87–6.79 (m, 8H), 6.61–6.59 (d, 3JHH = 9.5 Hz, 8H), 5.04 (s, 4H), 3.18–3.01 (m, 8H), 2.78–2.63 (m, 8H), 2.37–2.26 (m, 8H), 2.22–2.11 (m, 8H), 1.38 (t, 3JHH = 7.6 Hz, 24H), 0.95 (t, 3JHH = 7.4 Hz, 24H). 13C{1H} NMR (101 MHz, d8-toluene) δ 157.5, 141.1, 139.0, 131.9, 131.8, 128.3, 128.0, 126.6, 126.3, 126.0, 125.3, 125.2, 77.2, 24.9, 24.3, 14.4, 14.2.
Synthesis of [L1ZnCC(4-CF3C6H4)]2, (II′) {NMR-Scale}
A solution of complex I (0.020 g, 25 °C, 0.014 mmol) in a J. Young valve NMR tube was treated with 4-(trifluoromethyl)phenylacetylene (1m) (4.5 μL, 0.028 mmol) at room temperature; after 15 min, it resulted in the formation of compound II′ with the liberation of H2 and was observed by 1H NMR spectroscopy. NMR yield: (>99%). 1H NMR (400 MHz, C6D6) δ 7.13–7.05 (m, 12H), 7.00–6.98 (d, 3JHH = 8.2 Hz, 4H), 6.94–6.89 (m, 8H), 6.66–6.64 (d, 3JHH = 7.6 Hz, 8H), 5.07 (s, 4H), 3.17–3.08 (m, 8H), 2.76–2.67 (m, 8H), 2.40–2.30 (m, 8H), 2.21–2.11 (m, 8H), 1.39 (t, 3JHH = 7.5 Hz, 24H), 0.96 (t, 3JHH = 7.5 Hz, 24H). 13C{1H} NMR (101 MHz, C6D6) δ 157.6, 141.7, 141.1, 139.0, 134.9, 131.9, 126.9, 126.5, 126.2, 125.3, 124.5, 124.5, 124.5, 124.4, 107.7, 105.6, 24.9, 24.4, 14.6, 14.2. 19F{1H} NMR (377 MHz, C6D6) δ −62.41. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C51H59F3N5Zn 862.4014, found: 862.4054.
Synthesis of [L2ZnI]2
First, 1 g (1.10 mmol) of [L2ZnEt]2 was dissolved in ∼20 mL of dry toluene. Also, 0.56 g (2.20 mmol) of iodine was dissolved in ∼10 mL of dry toluene in another Schlenk tube. Then, the iodine solution was added dropwise to the [L2ZnEt]2 solutions. The reaction mixture was stirred for 12 h; a pale yellow solution was observed during this period. The solution was filtered and evaporated in a high vacuum to get compound [L2ZnI]2. (0.855 g, 71%); m.p. 220–225 °C. 1H NMR (400 MHz, CDCl3) δ 7.05–6.99 (m, 6H), 4.98 (s, 1H), 2.52–2.35 (m, 8H), 1.70 (s, 6H), 1.12 (t, J = 7.5 Hz, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 169.2, 144.2, 136.8, 126.2, 125.6, 95.5, 24.7, 23.5, 14.3. HRMS (ASAP/Q-TOF): m/z calcd for C25H34IN2Zn: 553.1058 [M + H]+; found: 553.1049.
Synthesis of [L2ZnH]2, (III)
[L2ZnI]2 (1.5 g, 1.36 mmol) and KH (0.06 g, 1.43 mmol) were weighed individually, and both were taken in a Schlenk tube in a glovebox under nitrogen conditions. The Schlenk tube was placed in an ice bath, and 20 mL of dry THF was added. Then, the reaction mixture was allowed to achieve ambient temperature, followed by stirring at room temperature for 18 h. After evaporation of the solvent under vacuum, the residue was dissolved in toluene (20 mL). The solution was filtered off from precipitated KI and evaporated in a high vacuum to get compound III. (890 g, 77%); m.p. 210–215 °C. 1H NMR (700 MHz, d8-toluene) δ 7.02–6.99 (m, 12H), 4.93 (s, 2H), 4.26 (s, 2H), 2.60–2.55 (m, 8H), 2.45–2.39 (m, 8H), 1.59 (s, 12H), 1.16 (t, J = 7.6 Hz, 24H). 13C{1H} NMR (176 MHz, d8-toluene) δ 167.2, 146.1, 137.0, 126.4, 125.2, 95.5, 24.7, 22.7, 14.3. IR (Nujol mull) ν (cm–1): 3206, 3018, 3005, 2988, 2833, 2807, 1491, 1470, 1466, 1465, 1401, 1395, 1373. HRMS (ASAP/Q-TOF): m/z calcd for C25H35N2Zn: 427.2092 [M + H]+; found: 427.2066.
Synthesis of [L2ZnCC(C6H5)]2, (IV) {Reaction-Scale}
A solution of [L2ZnH]2 (III) 0.250 g (0.293 mmol) in 10 mL of toluene was added to 64.35 μL (0.586 mmol) of phenylacetylene in ∼5 mL of toluene at room temperature through a cannula transfer and stirred for 2 h. The solvent was then removed in a vacuum, forming a white solid compound and drying thoroughly. The resultant solid was dissolved in benzene (∼10 mL) and filtered. The filtered solution was stored at room temperature and gave block-shaped colorless crystals within 3 days. (0.245 g, 80%); m.p. 185–190 °C. 1H NMR (400 MHz, C6D6) δ 7.09–7.06 (m, 4H), 6.96–6.93 (m, 12H), 6.69–6.63 (m, 6H), 4.80 (s, 2H), 2.57–2.48 (m, 8H), 2.40–2.30 (m, 8H), 1.46 (s, 12H), 1.09 (t, J = 7.6 Hz, 24H). 13C{1H} NMR (101 MHz, C6D6) δ 168.1, 145.5, 136.8, 132.0, 131.9, 128.1, 127.5, 126.5, 125.4, 105.8, 95.5, 77.4, 24.7, 22.8, 14.3. IR (Nujol mull) ν (cm–1): 3410, 2962, 2724, 2669, 2322, 1802, 1580, 1467, 1467, 1369, 1259, 1228, 1162, 1020. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C33H39N2Zn 527.2405, found: 527.2444.
Synthesis of [L2ZnCC(C6H5)]2, (IV) {NMR-Scale}
A solution of complex III (0.020 g, 25 °C, 0.023 mmol) in a J. Young valve NMR tube was treated with phenylacetylene (5.15 μL, 0.047 mmol) at room temperature; after 20 min, it resulted in the formation of compound IV with the liberation of H2 and was observed by 1H NMR spectroscopy. NMR yield: (>99%). 1H NMR (700 MHz, d8-toluene) δ 7.15–7.12 (m, 4H), 7.03–7.01 (m, 12H), 6.82–6.78 (m, 6H), 4.91 (s, 2H), 2.64–2.58 (m, 8H), 2.49–2.44 (m, 8H), 1.58 (s, 12H), 1.21 (t, J = 7.6 Hz, 24H). 13C{1H} NMR (176 MHz, d8-toluene) δ 167.2, 146.1, 136.6, 127.4, 126.4, 125.2, 95.5, 24.7, 22.7, 14.3.
Synthesis of [L2ZnCC(4-CF3C6H4)]2, (IV′) {NMR-Scale}
A solution of complex III (0.020 g, 25 °C, 0.023 mmol) in a J. Young valve NMR tube was treated with 4-(trifluoromethyl)phenylacetylene (7.6 μL, 0.047 mmol) at room temperature; after 15 min, it resulted in the formation of compound IV′ with the liberation of H2 and was observed by 1H NMR spectroscopy. NMR yield: (>99%). 1H NMR (400 MHz, C6D6) δ 7.06 (m, 14H), 6.92–6.91 (d, 3JHH = 3.0 Hz, 6H), 4.94 (s, 2H), 2.68–2.59 (m, 8H), 2.49–2.40 (m, 8H), 1.59 (s, 12H), 1.21 (t, 3JHH = 7.5 Hz, 24H). 13C{1H} NMR (101 MHz, C6D6) δ 168.3, 145.4, 136.8, 131.9, 126.6, 125.6, 124.5, 124.4, 124.4, 124.4, 107.4, 95.7, 24.8, 22.8, 14.4. 19F{1H} NMR (377 MHz, C6D6) δ −62.41. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C34H38F3N2Zn 595.2278, found: 595.2230.
General Procedure for Catalytic Dehydrogenative Borylation of Alkynes with HBpin
In a 10 mL catalytic oven-dried airtight sealed vial, 6.3 mg (0.0045 mmol, 1.5 mol %) of precatalyst I (based on dimeric structure) and 0.3 mmol (1.0 equiv) terminal alkyne were added, followed by 45 μL (0.31 mmol, 1.05 equiv) of pinacolborane in a neat condition. This reaction mixture was stirred at room temperature for 6 h. The reactions were monitored by 1H and 13C{1H} NMR spectroscopy until maximum conversion was observed. (Note: The 11B NMR spectra of some alkynyl borates show a peak in the range of 20–22 ppm due to a small amount of B(OR)3 as a side product).
Procedure for the Preparative Scale Synthesis of Organoboranes
In a 10 mL catalytic oven-dried vial containing a stir bar, 21 mg (0.015 mmol, 1.5 mol %) of precatalyst I (based on dimeric structure) and 1 mmol (1.0 equiv) terminal alkyne were added, followed by 152.4 μL (1.05 mmol, 1.05 equiv) of pinacolborane in a neat condition under a nitrogen atmosphere. This reaction mixture was stirred at room temperature for 6 h. After completion, 8 mL of pentane was added to the reaction mixture and filtered through Celite. The resulting solution was evaporated under vacuum to obtain the corresponding organoboranes. This procedure applies to compounds 2a, 2c, 2i, 2l, and 2m.
4,4,5,5-Tetramethyl-2-(phenylethynyl)-1,3,2-dioxaborolane (2a)
White solid, isolated yield 0.200 g (88%).26 NMR yield: (99%). 1H NMR (400 MHz, CDCl3) δ 7.47–7.45 (d, J = 8.3 Hz, 2H), 7.30–7.22 (m, 3H), 1.26 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 132.4, 129.3, 128.2, 121.8, 101.7, 84.3, 24.6. 11B NMR (128 MHz, CDCl3) δ 24.25.
4,4,5,5-Tetramethyl-2-(m-tolylethynyl)-1,3,2-dioxaborolane (2b)
NMR yield: (99%, light yellow oil).141H NMR (400 MHz, CDCl3) δ 7.37–7.30 (m, 2H), 7.23–7.16 (m, 2H), 2.32 (s, 3H), 1.33 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 137.9, 133.0, 130.3, 129.6, 128.2, 121.7, 102.0, 84.3, 24.7, 21.1. 11B NMR (128 MHz, CDCl3) δ 24.26.
4,4,5,5-Tetramethyl-2-(p-tolylethynyl)-1,3,2-dioxaborolane (2c)
White solid, isolated yield 0.222 g (92%).26 NMR yield: (99%). 1H NMR (400 MHz, CDCl3) δ 7.45–7.43 (d, J = 8.0 Hz, 2H), 7.15–7.13 (d, J = 8.0 Hz, 2H), 2.36 (s, 3H), 1.34 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 139.7, 132.5, 129.0, 118.8, 102.1, 84.3, 24.7, 21.5. 11B NMR (128 MHz, CDCl3) δ 24.28.
2-((4-(tert-Butyl)phenyl)ethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2d)
NMR yield: (99%, white solid).131H NMR (400 MHz, CDCl3) δ 7.39–7.37 (d, J = 8.2 Hz, 2H), 7.26–7.24 (d, J = 8.3 Hz, 2H), 1.23 (s, 12H), 1.21 (s, 9H). 13C{1H} NMR (101 MHz, CDCl3) δ 152.7, 132.3, 125.3, 118.8, 102.1, 84.3, 34.8, 31.1, 24.7. 11B NMR (128 MHz, CDCl3) δ 23.39.
2-(Mesitylethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2e)
NMR yield: (99%, orange oil).16b1H NMR (400 MHz, CDCl3) δ 6.74 (s, 2H), 2.34 (s, 6H), 2.17 (s, 3H), 1.23 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 141.5, 138.8, 127.6, 118.8, 99.5, 84.1, 24.7, 21.4, 21.0. 11B NMR (128 MHz, CDCl3) δ 23.78.
2-((3-Methoxyphenyl)ethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2f)
NMR yield: (99%, pale yellow oil). 1H NMR (400 MHz, CDCl3) δ 7.13 (t, J = 7.9 Hz, 1H), 7.05–7.03 (d, J = 7.5 Hz, 1H), 6.99–6.97 (m, 1H), 6.84 (dd, J = 8.3, 2.4 Hz, 1H), 3.68 (s, 3H), 1.23 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 159.1, 129.4, 125.0, 122.7, 117.0, 116.3, 101.7, 84.4, 55.2, 24.7. 11B NMR (128 MHz, CDCl3) δ 23.16. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C15H20BO3 259.1542, found: 259.1531.
2-((4-Methoxyphenyl)ethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2g)
NMR yield: (99%, pale yellow solid).261H NMR (400 MHz, CDCl3) δ 7.39–7.37 (d, J = 7.9 Hz, 2H), 6.74–6.73 (d, J = 7.6 Hz, 2H), 3.69 (s, 3H), 1.22 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 160.5, 134.1, 133.5, 113.9, 102.1, 84.2, 55.2, 24.6.
2-((2-Fluorophenyl)ethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2h)
NMR yield: (99%, colorless solid). 1H NMR (400 MHz, CDCl3) δ 7.34–7.27 (m, 1H), 7.19–7.14 (m, 1H), 6.93–6.86 (m, 2H), 1.14 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 164.6, 162.0, 134.3, 131.2 (d, J = 8.0 Hz), 123.9 (d, J = 3.9 Hz), 115.6, 115.4, 110.5 (d, J = 15.4 Hz), 94.6, 84.5, 24.6. 11B NMR (128 MHz, CDCl3) δ 23.29. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C14H17BFO2 247.1342, found: 247.1326.
2-((4-Fluorophenyl)ethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2i)
White solid, isolated yield 0.211 g (86%).16b NMR yield: (99%) 1H NMR (400 MHz, CDCl3) δ 7.55–7.51 (m, 2H), 7.03 (t, J = 8.4 Hz, 2H), 1.34 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 164.4, 161.9, 134.6 (d, J = 8.4 Hz), 118.0 (d, J = 8.3 Hz), 115.8, 115.5, 100.7, 84.5, 24.6. 11B NMR (128 MHz, CDCl3) δ 24.06.
2-((2-Chlorophenyl)ethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2j)
NMR yield: (99%, dark red oil).16b1H NMR (400 MHz, CDCl3) δ 7.47–7.45 (d, J = 7.7 Hz, 1H), 7.30–7.28 (d, J = 8.0 Hz, 1H), 7.21 (dd, J = 11.0, 4.4 Hz, 1H), 7.11 (t, J = 7.6 Hz, 1H), 1.23 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 136.5, 134.3, 130.4, 129.3, 126.4, 121.9, 107.7, 84.5, 24.7. 11B NMR (128 MHz, CDCl3) δ 24.17.
2-((4-Bromophenyl)ethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2k)
NMR yield: (99%, white solid).271H NMR (400 MHz, CDCl3) δ 7.45–7.43 (d, J = 8.2 Hz, 2H), 7.38–7.36 (d, J = 8.1 Hz, 2H), 1.31 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 133.8, 131.6, 123.9, 120.8, 100.4, 84.5, 24.6. 11B{1H} NMR (128 MHz, CDCl3) δ 23.39.
4,4,5,5-Tetramethyl-2-((2-(trifluoromethyl)phenyl)ethynyl)-1,3,2-dioxaborolane (2l)
Pale yellow solid, isolated yield 0.251 g (85%). NMR yield: (99%). 1H NMR (400 MHz, CDCl3) δ 7.69 (dd, J = 12.1, 7.8 Hz, 2H), 7.50–7.42 (m, 2H), 1.32 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 135.1, 131.3, 129.0, 125.8, 125.8, 121.9, 120.1, 110.9, 84.5, 24.6. 11B{1H} NMR (128 MHz, CDCl3) δ 23.85. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C15H17BF3O2 297.1310, found: 297.1363.
4,4,5,5-Tetramethyl-2-((4-(trifluoromethyl)phenyl)ethynyl)-1,3,2-dioxaborolane (2m)
White solid, isolated yield 0.254 g (86%).13 NMR yield: (99%). 1H NMR (400 MHz, CDCl3) δ 7.57–7.55 (d, J = 8.2 Hz, 2H), 7.53–7.51 (d, J = 8.0 Hz, 2H), 1.26 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 132.6, 131.4, 131.1, 130.7, 130.4, 125.6 (d, J = 0.9 Hz), 125.2 (q, J = 3.8 Hz), 99.7, 84.6, 24.6. 11B NMR (128 MHz, CDCl3) δ 23.33.
4,4,5,5-Tetramethyl-2-((3-nitrophenyl)ethynyl)-1,3,2-dioxaborolane (2n)
NMR yield: (93%, colorless oil). 1H NMR (400 MHz, CDCl3) δ 7.72–7.67 (m, 2H), 7.43–7.39 (m, 2H), 1.24 (s, 12H). HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C14H17BNO4 274.1287, found: 274.1289.
1,3-Bis((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethynyl)benzene (2o)
NMR yield: (99%, white solid). 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 1H), 7.43–7.42 (d, J = 7.8 Hz, 2H), 7.20 (t, J = 7.8 Hz, 1H), 1.24 (s, 24H). 13C{1H} NMR (101 MHz, CDCl3) δ 135.8, 133.3, 128.4, 122.3, 100.2, 84.5, 24.6. 11B NMR (128 MHz, CDCl3) δ 23.21. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C22H29B2O4 379.2246, found: 379.2266.
4,4,5,5-Tetramethyl-2-(thiophen-3-ylethynyl)-1,3,2-dioxaborolane (2p)
NMR yield: (99%, pale yellow solid).141H NMR (400 MHz, CDCl3) δ 7.51–7.50 (d, J = 2.8 Hz, 1H), 7.18–7.15 (d, J = 1.8 Hz, 1H), 7.08–7.07 (d, J = 1.8 Hz, 1H), 1.22 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 131.6, 130.2, 125.4, 121.1, 96.8, 84.4, 24.6. 11B NMR (128 MHz, CDCl3) δ 23.24.
2-((4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)ethynyl)pyridine (2q)
NMR yield: (97%, dark red oil). 1H NMR (400 MHz, CDCl3) δ 8.51–8.50 (d, J = 4.9 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.42–7.40 (d, J = 7.7 Hz, 1H), 7.20–7.16 (d, J = 15.5 Hz, 1H), 1.22 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 150.1, 142.1, 136.0, 127.6, 123.6, 100.0, 84.5, 24.6. 11B{1H} NMR (128 MHz, CDCl3) δ 23.24. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C13H17BNO2 230.1389, found: 230.1401.
2-(3,3-Dimethylbut-1-yn-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2r)
NMR yield: (99%, colorless solid).131H NMR (400 MHz, CDCl3) δ 1.20 (s, 12H), 1.17 (s, 9H). 13C{1H} NMR (101 MHz, CDCl3) δ 83.2, 66.3, 30.8, 28.7, 24.8.
Trimethyl((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethynyl)silane (2s)
NMR yield: (99%, yellow solid).141H NMR (400 MHz, CDCl3) δ 1.09 (s, 12H), −0.00 (s, 9H). 13C{1H} NMR (101 MHz, CDCl3) δ 93.5, 84.9, 25.1, −0.0.
2-(Hex-1-yn-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2t)
NMR yield: (99%, colorless oil).111H NMR (400 MHz, CDCl3) δ 2.19 (t, J = 13.9 Hz, 2H), 1.48–1.39 (m, 2H), 1.37–1.30 (m, 2H), 1.19 (s, 12H), 0.82 (t, J = 14.4 Hz, 3H). 13C{1H} NMR (101 MHz, CDCl3) δ 104.9, 83.9, 67.9, 30.0, 24.6, 21.8, 19.1, 13.4.
2-(Hept-1-yn-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2u)
NMR yield: (99%, colorless oil).281H NMR (400 MHz, CDCl3) δ 2.25 (t, J = 7.2 Hz, 2H), 1.57–1.50 (m, 2H), 1.42–1.30 (m, 4H), 1.26 (s, 12H), 0.88 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (101 MHz, CDCl3) δ 105.1, 83.9, 68.0, 30.9, 27.7, 24.6, 22.1, 19.4, 13.8. 11B NMR (128 MHz, CDCl3) δ 23.63.
4,4,5,5-Tetramethyl-2-(oct-1-yn-1-yl)-1,3,2-dioxaborolane (2v)
NMR yield: (99%, colorless oil).271H NMR (400 MHz, CDCl3) δ 2.25 (t, J = 7.1 Hz, 2H), 1.57–1.49 (m, 2H), 1.42–1.30 (m, 6H), 1.27 (s, 12H), 0.88 (t, J = 6.6 Hz, 3H). 13C{1H} NMR (101 MHz, CDCl3) δ 105.2, 83.9, 68.0, 31.2, 28.5, 28.0, 24.6, 22.4, 19.5, 14.0. 11B NMR (128 MHz, CDCl3) δ 23.62.
4,4,5,5-Tetramethyl-2-(5-methylhex-1-yn-1-yl)-1,3,2-dioxaborolane (2w)
NMR yield: (99%, pale yellow oil). 1H NMR (400 MHz, CDCl3) δ 2.27 (t, J = 7.5 Hz, 2H), 1.76–1.65 (m, 1H), 1.47–1.41 (m, 2H), 1.27 (s, 12H), 0.89–0.87 (d, J = 6.6 Hz, 6H). 13C{1H} NMR (101 MHz, CDCl3) δ 102.5, 83.9, 36.9, 27.0, 24.6, 22.0, 17.4. 11B NMR (128 MHz, CDCl3) δ 23.54. HRMS (ASAP/Q-TOF) m/z: [M + H]+ calcd for C13H24BO2 223.1906, found: 223.1898.
2-(5-Chloropent-1-yn-1-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2x)
NMR yield: (99%, colorless oil).111H NMR (400 MHz, CDCl3) δ 3.63 (t, J = 6.4 Hz, 2H), 2.44 (t, J = 6.8 Hz, 2H), 2.00–1.93 (m, 2H), 1.25 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 102.5, 84.1, 43.4, 30.8, 24.6, 16.9. 11B NMR (128 MHz, CDCl3) δ 23.49.
N,N-Dimethyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)prop-2-yn-1-amine (2y)
NMR yield: (99%, colorless oil).291H NMR (400 MHz, CDCl3) δ 3.33 (s, 2H), 2.30 (s, 6H), 1.26 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 84.1, 83.0, 48.3, 44.0, 24.5. 11B NMR (128 MHz, CDCl3) δ 23.58.29
4,4,5,5-Tetramethyl-2-(4-phenylbut-1-yn-1-yl)-1,3,2-dioxaborolane (2z)
NMR yield: (99%, white solid).111H NMR (400 MHz, CDCl3) δ 7.17–7.15 (d, J = 6.9 Hz, 2H), 7.11–7.07 (d, J = 7.8 Hz, 3H), 2.75 (t, J = 7.8 Hz, 2H), 2.42 (t, J = 7.9 Hz, 2H), 1.16 (s, 12H).1113C{1H} NMR (101 MHz, CDCl3) δ 140.3, 128.5, 128.3, 126.4, 104.0, 84.0, 34.6, 24.7, 21.7. 11B{1H} NMR (128 MHz, CDCl3) δ 22.74.
2-(Cyclopropylethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2aa)
NMR yield: (99%, colorless solid).131H NMR (400 MHz, CDCl3) δ 1.19 (s, 12H), 0.75–0.73 (m, 5H). 13C{1H} NMR (101 MHz, CDCl3) δ 108.0, 83.8, 24.5, 8.6, −0.0. 11B NMR (128 MHz, CDCl3) δ 23.41.
2-(Cyclopentylethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2ab)
NMR yield: >99%, colorless oil).261H NMR (400 MHz, CDCl3) δ 2.63–2.54 (m, 1H), 1.92–1.77 (m, 2H), 1.66–1.55 (m, 4H), 1.50–1.46 (m, 2H), 1.20 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 109.1, 83.9, 67.4, 41.6, 33.3, 30.5, 24.6. 11B NMR (128 MHz, CDCl3) δ 22.66.
2-(Cyclohexylethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2ac)
NMR yield: (99%, white solid).12a1H NMR (400 MHz, CDCl3) δ 2.46–2.39 (m, 1H), 1.86–1.77 (m, 2H), 1.74–1.67 (m, 2H), 1.55–1.43 (m, 4H), 1.34–1.29 (m, 2H), 1.28 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 108.9, 83.9, 32.0, 29.7, 25.7, 24.8, 24.6. 11B NMR (128 MHz, CDCl3) δ 23.75.
2-(Cyclohex-1-en-1-ylethynyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2ad)
NMR yield: (99%, colorless solid).141H NMR (400 MHz, CDCl3) δ 6.30–6.27 (d, J = 11.3 Hz, 1H), 2.14–2.06 (m, 4H), 1.62–1.53 (m, 4H), 1.27 (s, 12H). 13C{1H} NMR (101 MHz, CDCl3) δ 138.8, 120.0, 104.0, 84.0, 28.5, 25.7, 24.6, 22.0, 21.2. 11B NMR (128 MHz, CDCl3) δ 24.04.
General Procedure for Intermolecular Chemoselective Reactions
In an oven-dried airtight vial, 6.3 mg (0.0045 mmol, 1.5 mol %) of precatalyst I, 0.31 mmol of pinacolborane, 0.3 mmol of alkyne, and 0.3 mmol of competitor were added in a neat condition. The reactions were regularly monitored by 1H NMR analyses, which indicated the complete formation of the C–H borylated product over unreacted alkene, ester, pyridine, and isonitrile.
Acknowledgments
The authors thank the National Institute of Science Education and Research (NISER), HBNI, Bhubaneswar, Department of Atomic Energy (DAE), Govt. of India. The Science and Engineering Research Board (SERB), India (CRG/2021/007000) is acknowledged for providing financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c07381.
The authors declare no competing financial interest.
Supplementary Material
References
- a Suzuki A. Cross-Coupling Reactions of Organoboranes: An Easy Way To Construct C-C Bonds (Nobel Lecture). Angew. Chem., Int. Ed. 2011, 50, 6722–6737. 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]; b Lennox A. J. J.; Lloyd-Jones G. C. Selection of boron reagents for Suzuki–Miyaura coupling. Chem. Soc. Rev. 2014, 43, 412–443. 10.1039/C3CS60197H. [DOI] [PubMed] [Google Scholar]; c Hartwig J. F. Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc. Chem. Res. 2012, 45, 864–873. 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- Pécharman A.-F.; Colebatch A. L.; Hill M. S.; McMullin C. L.; Mahon M. F.; Weetman C. Easy access to nucleophilic boron through diborane to magnesium boryl metathesis. Nat. Commun. 2017, 8, 15022. 10.1038/ncomms15022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. C–H Activation for the Construction of C–B Bonds. Chem. Rev. 2010, 110, 890–931. 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]; b Bose S. K.; Mao L.; Kuehn L.; Radius U.; Nekvinda J.; Santos W. L.; Westcott S. A.; Steel P. G.; Marder T. B. First-Row d-Block Element-Catalyzed Carbon–Boron Bond Formation and Related Processes. Chem. Rev. 2021, 121, 13238–13341. 10.1021/acs.chemrev.1c00255. [DOI] [PubMed] [Google Scholar]; c Tian Y.-M.; Guo X.-N.; Braunschweig H.; Radius U.; Marder T. B. Photoinduced Borylation for the Synthesis of Organoboron Compounds. Chem. Rev. 2021, 121, 3561–3597. 10.1021/acs.chemrev.0c01236. [DOI] [PubMed] [Google Scholar]
- Brown H. C.; Bhat N. G.; Srebnik M. A simple, general synthesis of 1-alkynyldiisopropoxyboranes. Tetrahedron Lett. 1988, 29, 2631–2634. 10.1016/0040-4039(88)85245-6. [DOI] [Google Scholar]
- a Magre M.; Maity B.; Falconnet A.; Cavallo L.; Rueping M. Magnesium-Catalyzed Hydroboration of Terminal and Internal Alkynes. Angew. Chem., Int. Ed. 2019, 58, 7025–7029. 10.1002/anie.201902188. [DOI] [PubMed] [Google Scholar]; b Chen J.; Shen X.; Lu Z. Cobalt-Catalyzed Markovnikov-Type Selective Hydroboration of Terminal Alkynes. Angew. Chem., Int. Ed. 2021, 60, 690–694. 10.1002/anie.202012164. [DOI] [PubMed] [Google Scholar]; c Li J.; Luo M.; Sheng X.; Hua H.; Yao W.; Pullarkat S. A.; Xu L.; Ma M. Unsymmetrical β-diketiminate magnesium(I) complexes: syntheses and application in catalytic hydroboration of alkyne, nitrile and carbonyl compounds. Org. Chem. Front. 2018, 5, 3538–3547. 10.1039/C8QO00720A. [DOI] [Google Scholar]; d Mandal S.; Mandal S.; Geetharani K. Zinc-Catalysed Hydroboration of Terminal and Internal Alkynes. Chem. – Asian J. 2019, 14, 4553–4556. 10.1002/asia.201900839. [DOI] [PubMed] [Google Scholar]; e Aelterman M.; Sayes M.; Jubault P.; Poisson T. Electrochemical Hydroboration of Alkynes. Chem. – Eur. J. 2021, 27, 8277–8282. 10.1002/chem.202101132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhong M.; Gagné Y.; Hope T. O.; Pannecoucke X.; Frenette M.; Jubault P.; Poisson T. Copper-Photocatalyzed Hydroboration of Alkynes and Alkenes. Angew. Chem., Int. Ed. 2021, 60, 14498–14503. 10.1002/anie.202101874. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bose S. K.; Fucke K.; Liu L.; Steel P. G.; Marder T. B. Zinc-Catalyzed Borylation of Primary, Secondary and Tertiary Alkyl Halides with Alkoxy Diboron Reagents at Room Temperature. Angew. Chem., Int. Ed. 2014, 53, 1799–1803. 10.1002/anie.201308855. [DOI] [PubMed] [Google Scholar]; c Neeve E. C.; Geier S. J.; Mkhalid I. A. I.; Westcott S. A.; Marder T. B. Diboron(4) Compounds: From Structural Curiosity to Synthetic Workhorse. Chem. Rev. 2016, 116, 9091–9161. 10.1021/acs.chemrev.6b00193. [DOI] [PubMed] [Google Scholar]; d Hemming D.; Fritzemeier R.; Westcott S. A.; Santos W. L.; Steel P. G. Copper-boryl mediated organic synthesis. Chem. Soc. Rev. 2018, 47, 7477–7494. 10.1039/C7CS00816C. [DOI] [PubMed] [Google Scholar]
- Nagashima Y.; Takita R.; Yoshida K.; Hirano K.; Uchiyama M. Design, Generation, and Synthetic Application of Borylzincate: Borylation of Aryl Halides and Borylzincation of Benzynes/Terminal Alkyne. J. Am. Chem. Soc. 2013, 135, 18730–18733. 10.1021/ja409748m. [DOI] [PubMed] [Google Scholar]
- Guo J.; Cheng Z.; Chen J.; Chen X.; Lu Z. Iron- and Cobalt-Catalyzed Asymmetric Hydrofunctionalization of Alkenes and Alkynes. Acc. Chem. Res. 2021, 54, 2701–2716. 10.1021/acs.accounts.1c00212. [DOI] [PubMed] [Google Scholar]
- Sarkar N.; Bera S.; Nembenna S. Aluminum-Catalyzed Selective Hydroboration of Nitriles and Alkynes: A Multifunctional Catalyst. J. Org. Chem. 2020, 85, 4999–5009. 10.1021/acs.joc.0c00234. [DOI] [PubMed] [Google Scholar]
- Fontaine F.-G.; Rochette É. Ambiphilic Molecules: From Organometallic Curiosity to Metal-Free Catalysts. Acc. Chem. Res. 2018, 51, 454–464. 10.1021/acs.accounts.7b00514. [DOI] [PubMed] [Google Scholar]
- Lee C.-I.; Zhou J.; Ozerov O. V. Catalytic Dehydrogenative Borylation of Terminal Alkynes by a SiNN Pincer Complex of Iridium. J. Am. Chem. Soc. 2013, 135, 3560–3566. 10.1021/ja311682c. [DOI] [PubMed] [Google Scholar]
- a Romero E. A.; Jazzar R.; Bertrand G. Copper-catalyzed dehydrogenative borylation of terminal alkynes with pinacolborane. Chem. Sci. 2017, 8, 165–168. 10.1039/C6SC02668K. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu X.; Ming W.; Zhang Y.; Friedrich A.; Marder T. B. Copper-Catalyzed Triboration: Straightforward, Atom-Economical Synthesis of 1,1,1-Triborylalkanes from Terminal Alkynes and HBpin. Angew. Chem., Int. Ed. 2019, 58, 18923–18927. 10.1002/anie.201909376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D.; Carboni B.; Sortais J.-B.; Darcel C. Iron-Catalyzed Dehydrogenative Borylation of Terminal Alkynes. Adv. Synth. Catal. 2018, 360, 3649–3654. 10.1002/adsc.201800588. [DOI] [Google Scholar]
- Willcox D. R.; De Rosa D. M.; Howley J.; Levy A.; Steven A.; Nichol G. S.; Morrison C. A.; Cowley M. J.; Thomas S. P. Aluminium-Catalyzed C(sp)–H Borylation of Alkynes. Angew. Chem., Int. Ed. 2021, 60, 20672–20677. 10.1002/anie.202106216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchimoto T.; Utsugi H.; Sugiura T.; Horio S. Alkynylboranes: A Practical Approach by Zinc-Catalyzed Dehydrogenative Coupling of Terminal Alkynes with 1,8-Naphthalenediaminatoborane. Adv. Synth. Catal. 2015, 357, 77–82. 10.1002/adsc.201400767. [DOI] [Google Scholar]
- a Uzelac M.; Yuan K.; Ingleson M. J. A Comparison of Two Zinc Hydride Catalysts for Terminal Alkyne C–H Borylation/Hydroboration and the Formation of 1,1,1-Triborylalkanes by Tandem Catalysis Using Zn–H and B–H Compounds. Organometallics 2020, 39, 1332–1338. 10.1021/acs.organomet.0c00086. [DOI] [Google Scholar]; b Procter R. J.; Uzelac M.; Cid J.; Rushworth P. J.; Ingleson M. J. Low-Coordinate NHC–Zinc Hydride Complexes Catalyze Alkyne C–H Borylation and Hydroboration Using Pinacolborane. ACS Catal. 2019, 9, 5760–5771. 10.1021/acscatal.9b01370. [DOI] [Google Scholar]
- Luo M.; Qin Y.; Chen X.; Xiao Q.; Zhao B.; Yao W.; Ma M. ZnBr2-Catalyzed Dehydrogenative Borylation of Terminal Alkynes. J. Org. Chem. 2021, 86, 16666–16674. 10.1021/acs.joc.1c01936. [DOI] [PubMed] [Google Scholar]
- Jaiswal K.; Groutchik K.; Bawari D.; Dobrovetsky R. An “On-Demand,” Selective Dehydrogenative Borylation or Hydroboration of Terminal Alkynes Using Zn2+-based Catalyst. ChemCatChem 2022, 14, e202200004 10.1002/cctc.202200004. [DOI] [Google Scholar]
- a Spielmann J.; Piesik D.; Wittkamp B.; Jansen G.; Harder S. Convenient synthesis and crystal structure of a monomeric zinc hydride complex with a three-coordinate metal center. Chem. Commun. 2009, 23, 3455–3456. 10.1039/B906319F. [DOI] [PubMed] [Google Scholar]; b Bose S. K.; Deißenberger A.; Eichhorn A.; Steel P. G.; Lin Z.; Marder T. B. Zinc-Catalyzed Dual C–X and C–H Borylation of Aryl Halides. Angew. Chem., Int. Ed. 2015, 54, 11843–11847. 10.1002/anie.201505603. [DOI] [PubMed] [Google Scholar]; c Roy M. M. D.; Omaña A. A.; Wilson A. S. S.; Hill M. S.; Aldridge S.; Rivard E. Molecular Main Group Metal Hydrides. Chem. Rev. 2021, 121, 12784–12965. 10.1021/acs.chemrev.1c00278. [DOI] [PubMed] [Google Scholar]; d Wiegand A.-K.; Rit A.; Okuda J. Molecular zinc hydrides. Coord. Chem. Rev. 2016, 314, 71–82. 10.1016/j.ccr.2015.08.010. [DOI] [Google Scholar]; e Bayram M.; Gondzik S.; Bläser D.; Wölper C.; Schulz S. Syntheses and Structures of Zinc Bis(phosphinimino)methanide Complexes. Z. Anorg. Allg. Chem. 2016, 642, 847–852. 10.1002/zaac.201600196. [DOI] [Google Scholar]; f Coles M. P.; El-Hamruni S. M.; Smith J. D.; Hitchcock P. B. An Organozinc Hydride Cluster: An Encapsulated Tetrahydrozincate?. Angew. Chem., Int. Ed. 2008, 47, 10147–10150. 10.1002/anie.200804224. [DOI] [PubMed] [Google Scholar]; g Bose S. K.; Marder T. B. Efficient Synthesis of Aryl Boronates via Zinc-Catalyzed Cross-Coupling of Alkoxy Diboron Reagents with Aryl Halides at Room Temperature. Org. Lett. 2014, 16, 4562–4565. 10.1021/ol502120q. [DOI] [PubMed] [Google Scholar]
- Sahoo R. K.; Rajput S.; Patro A. G.; Nembenna S. Synthesis of low oxidation state zinc(I) complexes and their catalytic studies in the dehydroborylation of terminal alkynes. Dalton Trans. 2022, 51, 16009–16016. 10.1039/D2DT02846H. [DOI] [PubMed] [Google Scholar]
- Resa I.; Carmona E.; Gutierrez-Puebla E.; Monge A. Decamethyldizincocene, a Stable Compound of Zn(I) with a Zn-Zn Bond. Science 2004, 305, 1136–1138. 10.1126/science.1101356. [DOI] [PubMed] [Google Scholar]
- a Sahoo R. K.; Sarkar N.; Nembenna S. Zinc Hydride Catalyzed Chemoselective Hydroboration of Isocyanates: Amide Bond Formation and C=O Bond Cleavage. Angew. Chem., Int. Ed. 2021, 60, 11991–12000. 10.1002/anie.202100375. [DOI] [PubMed] [Google Scholar]; b Sahoo R. K.; Mahato M.; Jana A.; Nembenna S. Zinc Hydride-Catalyzed Hydrofuntionalization of Ketones. J. Org. Chem. 2020, 85, 11200–11210. 10.1021/acs.joc.0c01285. [DOI] [PubMed] [Google Scholar]
- Peddarao T.; Baishya A.; Sarkar N.; Acharya R.; Nembenna S. Conjugated Bis-Guanidines (CBGs) as β-Diketimine Analogues: Synthesis, Characterization of CBGs/Their Lithium Salts and CBG Li Catalyzed Addition of B–H and TMSCN to Carbonyls. Eur. J. Inorg. Chem. 2021, 2034–2046. 10.1002/ejic.202100141. [DOI] [Google Scholar]
- Hao H.; Cui C.; Roesky H. W.; Bai G.; Schmidt H.-G.; Noltemeyer M. Syntheses, and structures of the first examples of zinc compounds with bridging fluorine and hydrogen atoms. Chem. Commun. 2001, 1118–1119. 10.1039/b102275j. [DOI] [Google Scholar]
- Cheng M.; Moore D. R.; Reczek J. J.; Chamberlain B. M.; Lobkovsky E. B.; Coates G. W. Single-Site β-Diiminate Zinc Catalysts for the Alternating Copolymerization of CO2 and Epoxides: Catalyst Synthesis and Unprecedented Polymerization Activity. J. Am. Chem. Soc. 2001, 123, 8738–8749. 10.1021/ja003850n. [DOI] [PubMed] [Google Scholar]
- Hu J.-R.; Liu L.-H.; Hu X.; Ye H.-D. Ag(I)-catalyzed C–H borylation of terminal alkynes. Tetrahedron 2014, 70, 5815–5819. 10.1016/j.tet.2014.06.033. [DOI] [Google Scholar]
- Desrosiers V.; Garcia C. Z.; Fontaine F.-G. Boron Recycling in the Metal-Free Transfer C–H Borylation of Terminal Alkynes and Heteroarenes. ACS Catal. 2020, 10, 11046–11056. 10.1021/acscatal.0c02682. [DOI] [Google Scholar]
- Ho H. E.; Asao N.; Yamamoto Y.; Jin T. Carboxylic Acid-Catalyzed Highly Efficient and Selective Hydroboration of Alkynes with Pinacolborane. Org. Lett. 2014, 16, 4670–4673. 10.1021/ol502285s. [DOI] [PubMed] [Google Scholar]
- Foley B. J.; Bhuvanesh N.; Zhou J.; Ozerov O. V. Combined Experimental and Computational Studies of the Mechanism of Dehydrogenative Borylation of Terminal Alkynes Catalyzed by PNP Complexes of Iridium. ACS Catal. 2020, 10, 9824–9836. 10.1021/acscatal.0c02455. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.