

Abstract
Background
Valosin-containing protein (VCP) disease, caused by mutations in the VCP gene, results in myopathy, Paget’s disease of bone (PBD) and frontotemporal dementia (FTD). Natural history and genotype–phenotype correlation data are limited. This study characterises patients with mutations in VCP gene and investigates genotype–phenotype correlations.
Methods
Descriptive retrospective international study collecting clinical and genetic data of patients with mutations in the VCP gene.
Results
Two hundred and fifty-five patients (70.0% males) were included in the study. Mean age was 56.8±9.6 years and mean age of onset 45.6±9.3 years. Mean diagnostic delay was 7.7±6 years. Symmetric lower limb weakness was reported in 50% at onset progressing to generalised muscle weakness. Other common symptoms were ventilatory insufficiency 40.3%, PDB 28.2%, dysautonomia 21.4% and FTD 14.3%. Fifty-seven genetic variants were identified, 18 of these no previously reported. c.464G>A (p.Arg155His) was the most frequent variant, identified in the 28%. Full time wheelchair users accounted for 19.1% with a median time from disease onset to been wheelchair user of 8.5 years. Variant c.463C>T (p.Arg155Cys) showed an earlier onset (37.8±7.6 year) and a higher frequency of axial and upper limb weakness, scapular winging and cognitive impairment. Forced vital capacity (FVC) below 50% was as risk factor for being full-time wheelchair user, while FVC <70% and being a full-time wheelchair user were associated with death.
Conclusion
This study expands the knowledge on the phenotypic presentation, natural history, genotype–phenotype correlations and risk factors for disease progression of VCP disease and is useful to improve the care provided to patient with this complex disease.
INTRODUCTION
Valosin-containing protein (VCP) is an AAA+ (ATPases associated with diverse cellular Activities) ubiquitous protein involved in protein degradation by the ubiquitin–proteasome system and cellular homoeostasis regulation.1–3 Hereditary inclusion body myopathy with Paget’s disease of the bone and frontotemporal dementia (IBMPFD) is an autosomal dominant disorder caused by mutations in the VCP gene,4–6 with an estimated prevalence of 0.66/100 000 in the UK,7 although precise worldwide prevalence remains unknown. Myopathy, involving the pelvic and shoulder girdle muscles, is the most common clinical feature of IBMPFD8–13 presenting between the fourth and fifth decade.14 IBMPFD is a disabling condition, leading to death due to respiratory complications or end stage dementia.8 15 Other reported presentations include facioscapulohumeral muscular weakness,16 distal myopathy,17 amyotrophic lateral sclerosis (ALS),18 parkinsonism,19 hereditary spastic paraplegia,20 Charcot-Marie-Tooth disease,21 22 Huntington’s disease15 23 and cardiomyopathy.24 This broad phenotype requires a multidisciplinary team for patients’ care and makes its diagnosis challenging if there are atypical symptoms or an unclear family history.9
The IBMPFD nomenclature is deficient and has led to the use of the term multisystem proteinopathy (MSP) to encompass all phenotypes described.6 However, the different tissues affected in MSP share pathological features of ubiquitinated proteins accumulation, autophagic debris and TDP-43 inclusions.4 Mutations in VCP impairs autophagy and endocytic trafficking and increase ATPase activity due to structural changes.25 26
As a rare condition, MSP natural history information is limited, and detailed genotype–phenotype associations have not yet been established.7 9–11 13 14 21 27
This study aims to (1) describe the clinical and genetic features of a large, international cohort of patients with mutations in the VCP gene and (2) decipher genotype–phenotype associations.
MATERIALS AND METHODS
This is an international descriptive retrospective study collecting data obtained on routine clinical care visits from patients with MSP. Fifty-two centres from 24 countries participated.
The inclusion criteria were (1) patients heterozygous for a pathogenic(P)/likely pathogenic (LP) variant in the VCP gene (transcript reference NM_007216.3)28 and (2) sufficient data available in the clinical notes to answer some of the following clinical questions: age of disease onset, genetic diagnosis, signs/symptoms at onset, clinical progression, ambulatory status and ancillary test results, if performed. Patients who harboured a variant of unknown significance (VUS) were included if disease causality was supported by in silico analysis and clinical judgement based on item (2) of the inclusion criteria and/or the pedigree indicated a dominant disease. Once the clinical and family history data were collected for all the patients, the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG) criteria of each variant was recalculated (https://www.medschool.umary-land.edu/Genetic_Variant_Interpretation_Tool1.html/).
Statistical analysis
Quantitative variables were analysed using the Kolmogorov-Smirnov or Shapiro-Wilko tests to assess normal distribution. Data were expressed as number and percentage for categorical variables, and as mean±SD or as median, first and third inter-quartile and minimum and maximum for quantitative variables as appropriate. The χ2 or Fisher’s exact tests were used for the association between signs/symptoms and the four most frequent mutation types, with Bonferroni correction for multiple comparisons. Analysis of covariance, with adjustment for age at last assessment, was used to compare means of signs/symptoms onset among the four most frequent mutation types. A two-step analysis to select variables associated with being a full-time wheelchair user/confined to bed or death was performed. A p<0.05 level of significance was allowed. SPSS software V.27 from IBM was used for statistical analysis.
An extended version of materials and methods29 is available in online supplemental material 1.
RESULTS
A total of 255 patients, 194 families, from 24 countries were collected. Twenty-one patients were excluded from the analysis: 12 patients did not show signs/symptoms at last assessment, 7 patients had insufficient clinical information and 2 patients were duplicated.30
Asymptomatic individuals (n=12, 7 females), relatives of symptomatic patients, were diagnosed following genetic counselling and each belonged to an independent family. Their mean age at last assessment was 40.0±94 years (median 38.5; min 29.0; max 61.0 years) and the mean follow-up was 5.2±5.1 years (min 0.0; max 12.8) after the first appointment triggered by the family proband. Two of the asymptomatic carriers have passed the ages of symptom onset observed in their families, one has reached the age of onset in the family, eight has not passed the eldest age of symptom onset expected in their families and for one patient no relatives were included in the study.
A total of 234 symptomatic patients were included in the final analysis. Seventy per cent (163/234) were males and the mean age at last assessment was 56.8±9.6 years. Demographics and time to disease milestones are described in table 1. The number of patients, families and variants by country are described in online supplemental table 1.
Table 1.
Demographic and disease times data in the overall population
| Total population | n=234 (186 families) |
|---|---|
| Male/female (%) | 163 (70.0%)/71 (30.0%) |
| Mean age of disease onset (years), n=232 | 45.6±9.3 (18–71) |
| Mean age at last assessment (years), n=234 | 56.8±9.6 (31–82) |
| Mean time of disease progression (years), n=232 | 11.3±6.9 (1–45) |
| Mean age at genetic diagnosis (years), n=63 | 54.5±9.5 (30–81) |
| Mean time to genetic diagnosis (years), n=58 | 10.4±6.3 (1–29) |
| Mean age of death (years), n=36 | 63.9±8.1 (45–81) |
| Mean time to death (years), n=36 | 15.8±6.6 (2–31) |
Order of data shown: n number of patients in which the data was available, mean, SD, minimum and maximum.
Genetic variants
Fifty-seven variants in the VCP gene were reported. All variants were single-nucleotide changes except for one small deletion-insertion. Most of the variants were in exon 5 (what represented 63% of the patients, 147/234 and 30% of the variants, 17/57) and exon 3 (14% of the patients, 32/234 and 19% of the variants 11/57). The four most frequent variants identified were: c.464G>A, p.Arg155His (28.6%, 67/234); c.463C>T, p.Arg155Cys (11.1%, 26/234); c.476G>A, p.Arg159His (7.7%, 18/234); and c.277C>T, p.Arg93Cys (7.3%, 17/234). Eighteen previously unreported variants were identified. The list of variants, their frequency in this study, their exonic location, gnomAD frequency, ACGM score and the clinical phenotype of the VUS and unreported variants are described in online supplemental table 2.1 and 2.2.
Clinical features
Data on symptoms at onset were available for 226 patients. Muscle weakness was the first symptom in 90.7% (205/226). Disease onset was as a symmetric lower limb weakness in 50.0% (113/226), affecting the proximal muscles in 35.0% (79/226) and the distal muscles in 15.0% (34/226). Either proximal symmetric upper limb weakness (9.3%, 21/226) or a combination of upper and lower limb girdle weakness (9.3%, 21/226) were next most frequent symptoms at onset (figure 1). Eight per cent (18/234) of the patients showed an asymmetric weakness either in the upper or lower limbs at onset. Symptom presentation at onset was slowly progressive in 89.1% (196/220) of the patients and subacute in the remaining 10.9% (24/220). Meadian CK levels at onset (n=37) was 254.0 UI/L (IQ1:199.0–IQ3:410.5; minimum 48.0–maximum 1822.0).
Figure 1.
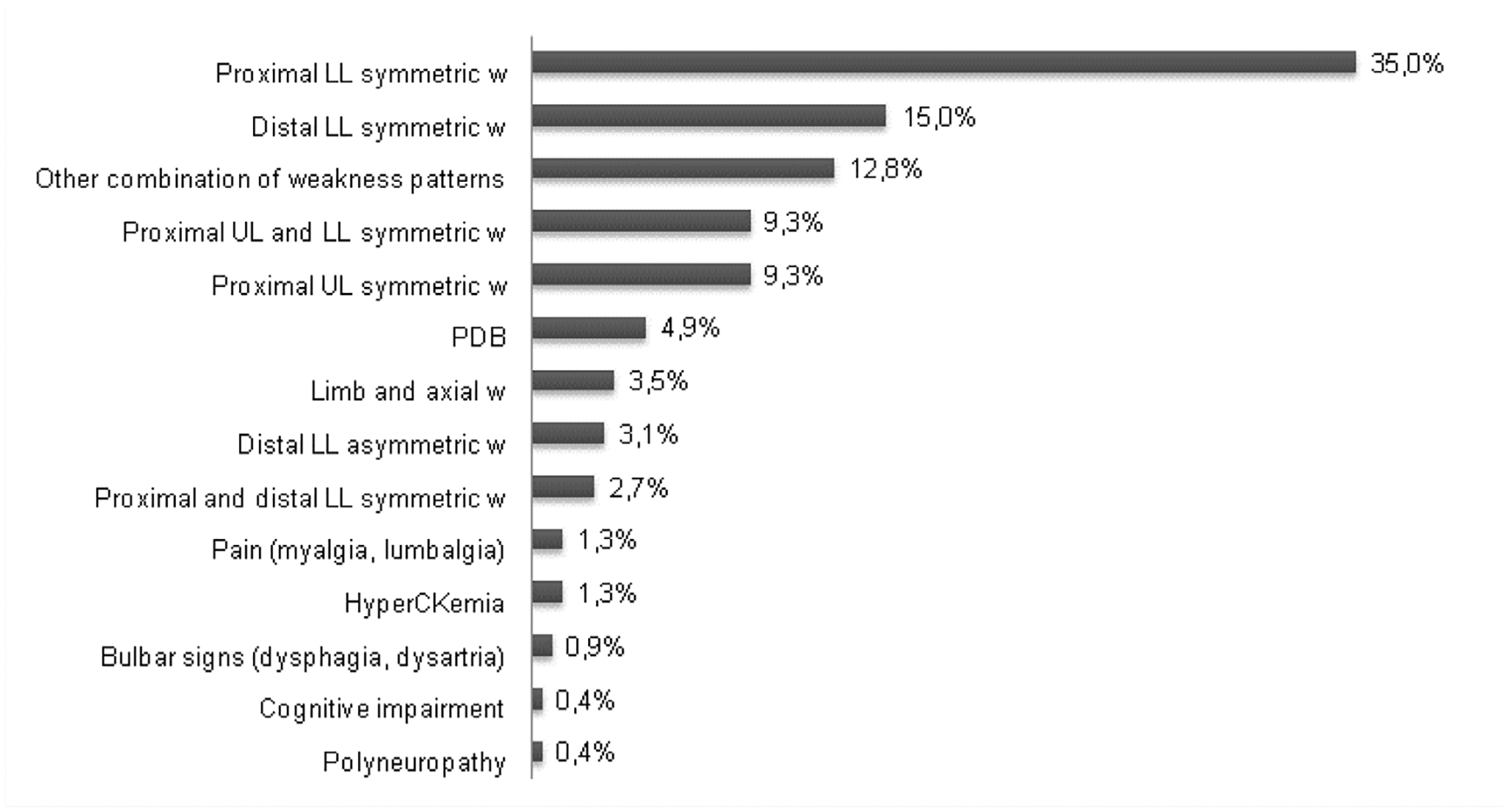
Frequency of first symptoms (n=226). PDB the category other combination of weakness patterns includes: distal UL symmetric W, five cases; proximal Ll asymmetric W, five cases; distal UL and Ll W, four cases; distal and proximal UL and Ll W, four cases; distal UL and proximal and distal Ll W, three cases; proximal and distal UL W, three cases; proximal UL asymmetric; W, two cases; distal UL asymmetric W, two cases and proximal UL and proximal and distal Ll W, one case. LL, lower limbs; PDB, Paget’s disease of the bone; UL, upper limbs; w, weakness.
Mean time of disease progression was 11.3±6.9 years (range 1–45). The frequency of signs and symptoms identified at last assessment is shown in figure 2. The median time of development of each clinical feature from disease onset is shown in figure 3. Age of patients at each clinical feature presentation is shown in online supplemental table 3.
Figure 2.
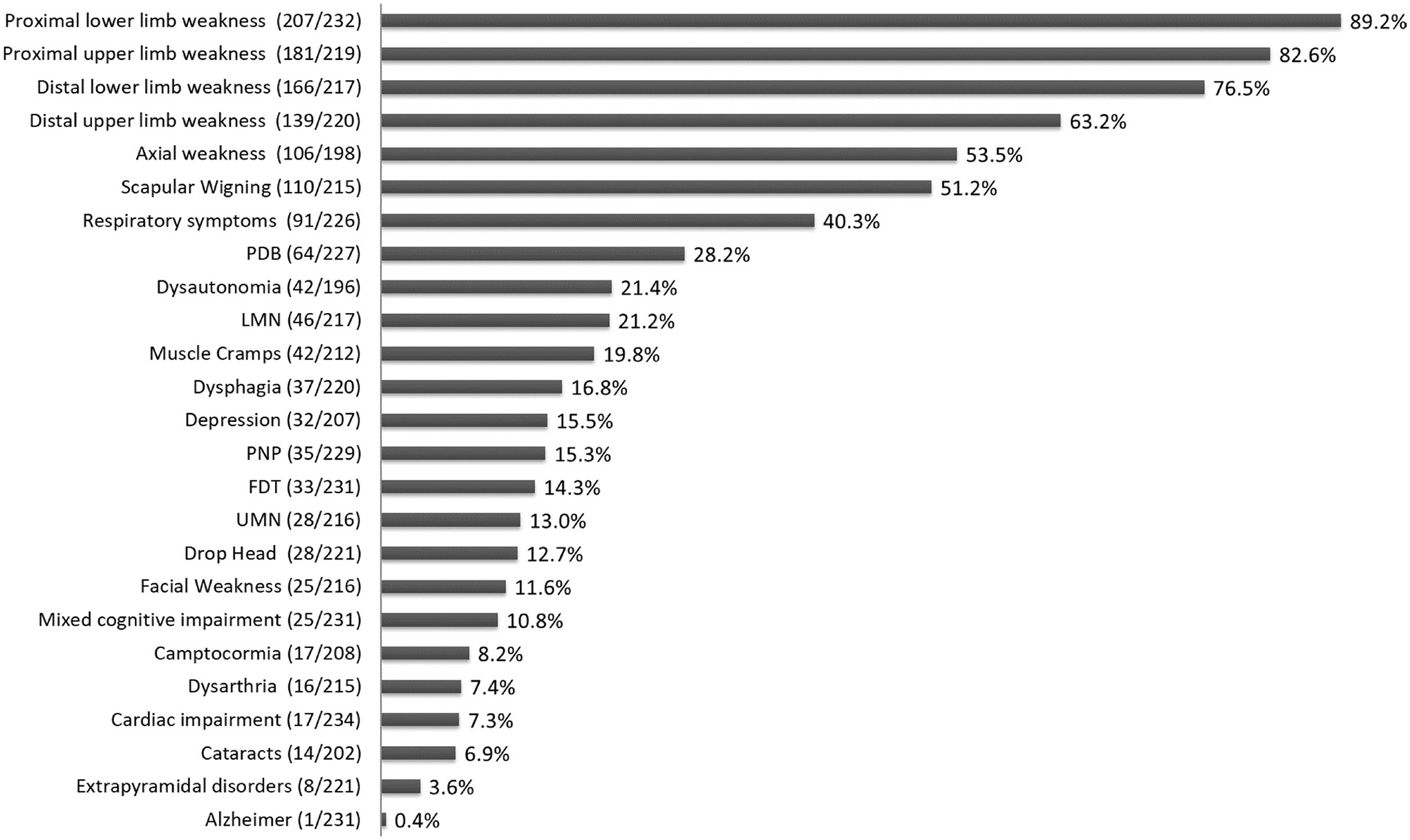
Frequency of signs/symptoms at last assessment. Data in brackets represent the number of patients that reported the sign/symptom at last assessment among those in which the data were reported. FTD, frontotemporal dementia; LMN, lower motor neuron signs; PDB, Paget’s disease of the bone; PNP, polyneuropathy; UMN, upper motor neuron signs.
Figure 3.
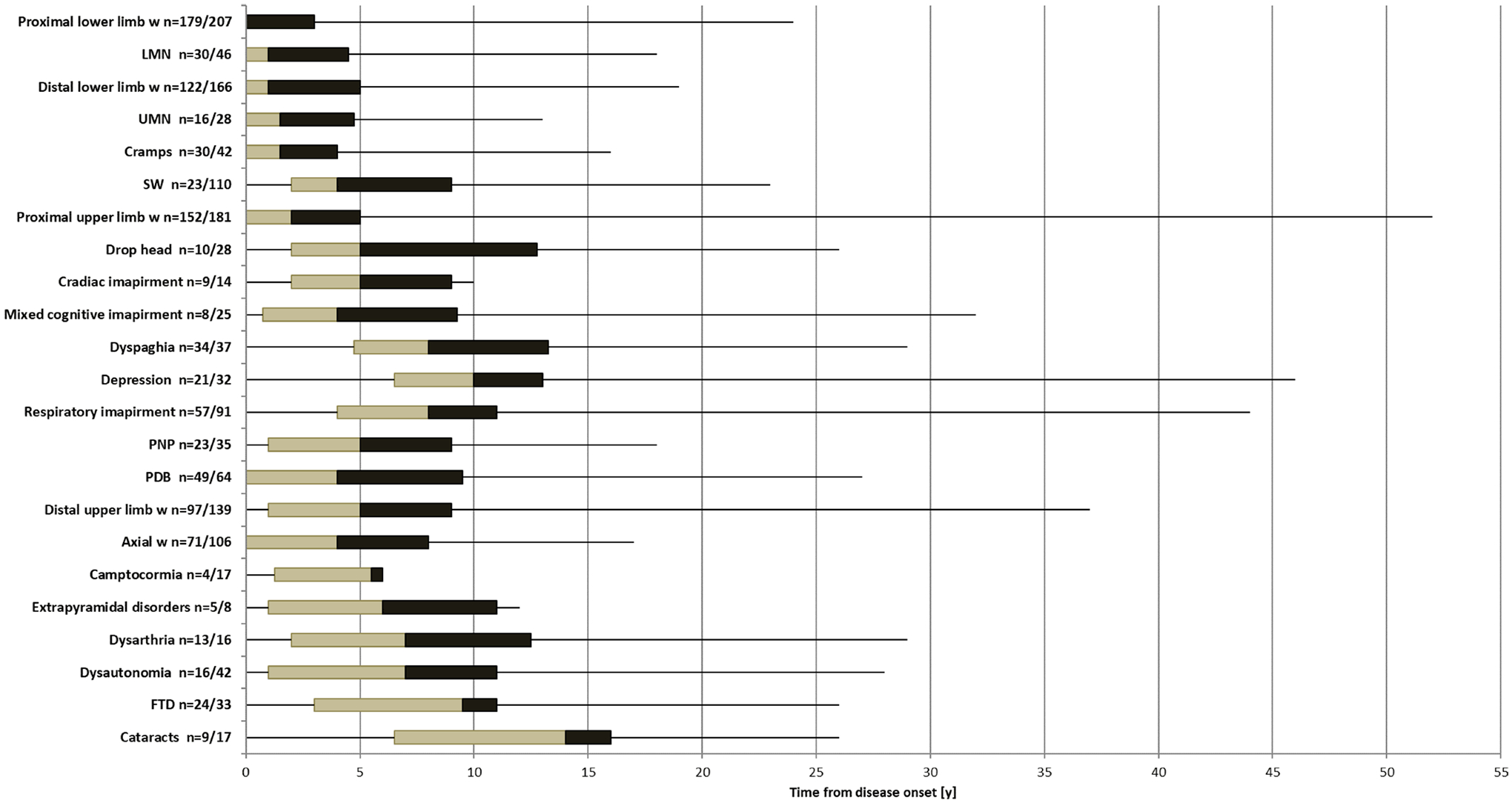
Time of signs/symptoms development from disease onset (y). The box figures show the time of signs/symptoms development from disease onset in years. The limits of the box are the 25th and 75th percentile. The middle line within the boxes represents the median. The ends of the lines extending from the boxes represent the minimum and maximum values. n=number of patients in which data about the time from disease onset was available among those patients who reported the sign/ symptom at last assessment. FTD, frontotemporal dementia; LMN, lower motor neuron signs; PDB, Paget’s disease of the bone; PNP, polyneuropathy; UMN, upper motor neuron; W, weakness.
At last assessment, all patients had developed muscle weakness except for one patient who manifested isolated Paget’s disease of bone (PDB) was the second most frequent clinical feature (28.2%, 64/227). Among patients with PDB, 76.5% (49/64) had bone lesions detected on a bone radiography. Bone lesion’s location was available in 43 cases: dorsal and lumbar spine (29/43), hip (15/43), pelvic bone (15/43), skull (11/43) and femur (6/43). An elevated serum ALP level was identified in 54.7% (35/64) of patients with PDB. Median ALP value 153.5 UI/L (IQ1:80.8–IQ3:364.3; minimum 39.0; maximum 1901.0). Bone pain was reported in 48.4% (31/64).
Cognitive impairment was identified in 25.5% (59/231) of the patients in which frontotemporal dementia (FTD) was the most frequent pattern (33/59) followed by a mixed cognitive impairment (25/59). Only one patient had a diagnoses of Alzheimer disease (see clinical details of this case in online supplemental case report 1). Thirty-two patients reported depression, with 16 cases not having associated cognitive impairment on clinical examination.
Dyspnoea on exertion was reported in 25.3% (56/221) of the patients, nocturnal hypoventilation in 15.6% (34/218) and recurrent respiratory infection in 3.2% (7/221). Percentage predicted forced vital capacity (FVC) was available for 116 patients and it was below the 80% predicted in 52.6% of the cases (61/116). The FVC percentage distribution among them was: 31.1% (19/61) between 70% and 80%, 21.3% (13/61) between 60% and 70%, 21.3% (13/61) between 60% and 50% and 26.2% (16/61) below 50% of predicted. Twelve per cent of the patients (27/224) required nocturnal non-invasive ventilation (NIV), 4.0% (9/224) full time NIV and 0.9% (2/224) used invasive ventilation.
Cardiac impairment was reported in 7.3% of the patients (17/234). Changes compatible with hypertrophic cardiomyopathy or with a dilated cardiomyopathy on echocardiogram were reported in seven and five patients, respectively. Four patients had changes on ECG including atrial fibrillation, right or left bundle branch block and persistent tachycardia. None of the patients required a pacemaker or a defibrillator. Left ventricular ejection fraction (LVEF) value, as measured by echocardiogram, was available for 28 patients. Only four patients had an LVEF of 50%–55% while no patients had an LVEF less than 50%.
Dysautonomia was reported in 21.4% (42/196). Sixty-six (7%) of the patients reported only one symptom of dysautonomia (28/42, 12 urinary incontinence, 13 combined constipation and diarrhoea, and 3 erectile dysfunction), 26.2% (11/42) 2 symptoms and 7.1% (3/42) 3 symptoms. Among the 42 patients with dysautonomia, nine had a concomitant diagnosis of polyneuropathy, six of FTD and three of extrapyramidal disorders.
Motor neuron involvement was reported in 25.0% (54/216) of which 48.1% (26/54) showed exclusively lower motor neuron signs. Thirty-seven per cent (20/54) had lower and upper motor neuron signs and 14.8% (8/54) had exclusively upper motor neuron signs. Six per cent of the patients (13/212) fulfilled clinically probable ALS as defined by El Escorial criteria.31
Among the 18.0% (39/217) of patients with signs of bulbar involvement, isolated dysphagia was reported in 59.0% (23/39), a combination of dysarthria and dysphagia in 35.9% (14/39) and only 2 patients had dysarthria exclusively. Of these 39 patients, 6 had motor neuron involvement and five fulfilled ALS criteria.
Extrapyramidal disorders were reported in eight patients. The frequency by mutation types was: 3 cases in variant c.476G>A and one case on each of the following variants c.463C>T, c.376A>G, c.410C>T, c.625T>G and c.572G>A. The clinical features were: Meige syndrome (one), asymmetric hand tremor (three), symmetric rigid-akinetic syndrome (two),and in two patients a detailed clinical description of the symptoms was not available.
The classic triad of myopathy, FTD and PDB was reported in only seven patients (2.9%, 7/234). Seventeen patients had myopathy and isolated FTD (7.2%, 17/234) and 38 patients showed myopathy associated with isolated PDB (16.2%, 38/234). The number of patients who had any type of muscle weakness, PDB and any type of cognitive impairment was 19 (8.1, 19/234%).
Of the 186 families included in the study, 42% (78/186) consisted of index cases without family history and 58% (108/186) were families in which the index case and at least one relative was included. In 48% of the families (89/186), more than one phenotype was identified being IBM, PDB, FDT and ALS the most frequent signs/symptoms combined (online supplemental figure 1).
Neurophysiology
The results of 182 nerve conduction studies (NCS) and needle EMG were available. Of the 35 patients (15.4%) with reported clinical polyneuropathy, NCS results were available for 33 patients: 14 patients had a sensorimotor neuropathy, 10 a sensory neuropathy and 9 a motor neuropathy. The NCS pattern was pure axonal in 20 patients, axonal but with intermediate conduction velocities in 12 patients and conduction velocities compatible with demyelination in 3 patients. All patients with NCS compatible with an underlying neuropathy had muscle weakness and 42.8% of them (15/35) had associated lower motor neuron signs including muscle atrophy, fasciculations and/or absence of muscle reflexes.
Needle EMG examination revealed an exclusively myopathic pattern in 46.7% (85/182), an exclusively neurogenic pattern in 20.9% (38/182), a mixed (myopathic and neurogenic) pattern in 20.3% (37/182) and it was normal in the 12.1% (22/182). The presence of spontaneous activity was reported in 44.5% (81/182).
Ambulatory status
The frequency of ambulatory status at last assessment, the mean age at each ambulatory status and the median ambulatory status time from disease onset are shown in table 2.
Table 2.
Ambulatory status al last assessment
| Ambulatory status | Frequency* | Mean age (n) | Mean age (SD, min, max) (years) | Time from disease onset (n) | Time from disease onset Median (min - IQ1 - IQ3- max) (years) |
|---|---|---|---|---|---|
| Walk independently | 35.1% (79/225) | 63 | 52.2±9.1 (30–75) | 63 | Not applicable |
| Cane/stick user | 28.0% (63/225) | 66 | 53.9±10.4 (29–77) | 66 | 8.0 (0–IQ1 5.0–IQ3 12.0–29) |
| Outdoor wheelchair user | 13.8% (31/225) | 36 | 55.8±10.1 (30–80) | 36 | 9.5 (1–IQ1 7.0–IQ3 14.0–27) |
| Outdoor and indoor wheelchair user (full-time wheelchair user) | 19.1% (43/225) | 20 | 53.6±11.2 (30–70) | 20 | 8.5 (1–IQ1 5.3–IQ3 12.5–25) |
| Confined to bed | 4.0% (9/225) | 2 | 56.5±9.2 (50–63) | 2 | 15.0 (7–IQ1 7.0–IQ3 NA–23) |
Numbers in brackets: the numerator represents the number of patients on each category and the denominator the number of patients in which the ambulatory status data was collected at last assessment.
IQ, IQR; max, maximum; min, minimum; NA, not available.
Causes of death
Thirty-seven patients i died during the follow-up at a mean age of 63.9 years (range 45–81 years) and a mean time of 15.8 years (range 2–31) from disease onset (table 1). Cause of death was available for 14 patients: 7 due to respiratory insufficiency, 5 due to rapidly progressive dementia, 1 due to myocardial infarction and 1 related to COVID-19 infection.
Genotype–phenotype associations
To study potential genotype–phenotype correlations, we compared the frequency of signs and symptoms in patients carrying the four most frequent variants (online supplemental table 4). We did not find any symptom exclusively present in patients with one particular genetic variant. However, we identified significant differences in symptoms frequency across variants. Distal upper limb weakness, scapular winging and axial weakness were significantly more frequent in variant c.463C>T (p.Arg155Cys) than in variant c.464G>A (p.Arg155His) (figure 4). There were no differences in the frequency of cognitive symptoms across these variants except for mixed cognitive impairment that was more frequent in variant c.463C>T (p.Arg155Cys) than in variant c.464G>A (p.Arg155His) (figure 4). We did not observe association between the frequency of PDB, respiratory or cardiac involvement and specific mutations. The frequency of extrapyramidal disorders between the four most frequent variants could not be assessed accurately due to the low number of patients.
Figure 4.
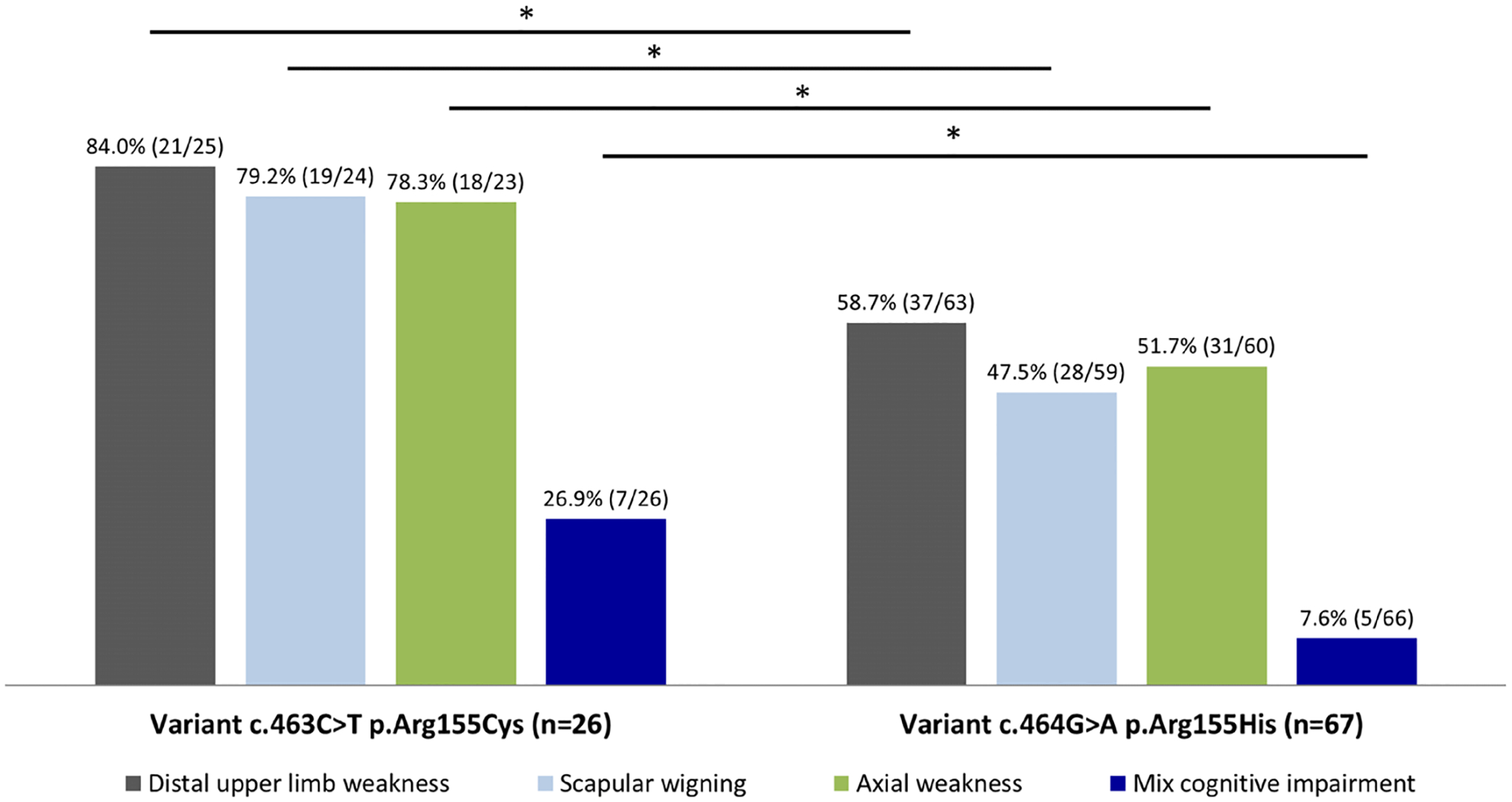
Frequency of signs/symptoms between the two most frequent variants. The bar graph shows the percentage of signs/symptoms in which significant differences were found between the two most frequent variants identified in this study. Numbers in brackets above the bars represent: the numerator the frequency of patients in whom the sign/symptom was present and the denominator the number of patients in which the data was available by variant. *P<0.05, χ2 test.
Variant c.463C>T (p.Arg155Cys) was highly frequent in females (56.7%, 15/26) compared with the frequency in the whole cohort (30.0%, 71/234).
After adjusting for age at last assessment, variant c.463C>T (p.Arg155Cys) had an earlier age of onset compared to variants c.476G>A (p.Arg159His) and c.277C>T (p.Arg93Cys) (37.8±7.6 years vs 49.6±9.0 and 52.4±5.7 years, respectively, analysis of covariance,ANCOVA, p=0.004). Similarly, variant c.464G>A (p.Arg155His) was associated with an earlier age of onset than variant c.277C>T (p.Arg93Cys) (42.6±6.7 vs 37.8±7.6 years respectively, ANCOVA, p=0.04).
No significant differences were found for the mean ages of onset of respiratory impairment, cardiac impairment, FTD, dysautonomia, ambulatory status at last assessment and age of death among the four most frequent variants after adjusting for age at the last assessment (ANCOVA test, p>0.05 in all cases).
Variables associated with loss of ambulation and death
We carried out a univariate analysis to test whether the presence of a severely reduced FVC, cardiac impairment, FTD, motor neuron signs, extrapyramidal disorders, polyneuropathy, axial weakness, dysautonomia, proximal and/or distal lower limb weakness were associated with requiring a full-time wheelchair. The following variables were significantly associated: proximal lower limb weakness, distal lower limb weakness, axial weakness, dysautonomia, FVC <50% and FTD (table 3).
Table 3.
Variables associated with loss of ambulation and deat
| Variables associated with loss of ambulation | |||
|---|---|---|---|
| Variable | Full-time wheelchair/confined to bed | Ambulant | P value |
| FTD | 23.5% (12/51) | 11.6% (20/172) | 0.03* |
| FVC <50% | 35.5% (11/31) | 6.0% (5/84) | <0.001† |
| Distal lower limb weakness | 88.5% (46/52) | 72.2% (117/162) | 0.02* |
| Proximal lower limb weakness | 98.1% (51/52) | 86.5% (148/171) | 0.02† |
| Axial weakness | 69.6% (32/46) | 49.0% (73/149) | 0.03* |
| Dysautonomia | 41.0% (16/39) | 17.4% (26/149) | 0.002* |
| Variables associated with death | |||
| Variable | Deceased | Alive | p value |
| FTD | 27.8% (10/36) | 10.8% (17/157) | 0.02† |
| FVC <50% | 35.3% (6/17) | 11.0% (9/82) | 0.02† |
| Dysphagia | 31.4% (11/35) | 16.3% (24/147) | 0.04* |
| Drop-head | 29.4% (10/34) | 9.5% (14/147) | 0.002† |
| Full-time wheelchair user/confined to bed | 47.2% (17/36) | 19.6% (30/153) | 0.001* |
χ2 test.
Fisher’s exact test.
FTD, frontotemporal dementia; FVC, forced vital capacity.
We also identified a strong positive correlation between age at which these previous symptoms started and age at disease onset with the age of full-time wheelchair use/confined to bed (online supplemental table 5). All previous variables were included in a binary logistic regression analysis after which only a FVC <50% remained associated with full-time wheelchair use (online supplemental table 6). A Cox regression analysis was performed to determine which of these variables were associated with a higher risk of being a full-time wheelchair user and only a FVC <50% remained as representing a risk for this outcome (online supplemental table 7). The time to full-time wheelchair use by FVC <50% was analysed through a Kaplan-Meier estimator and showed significant difference (figure 5).
Figure 5.
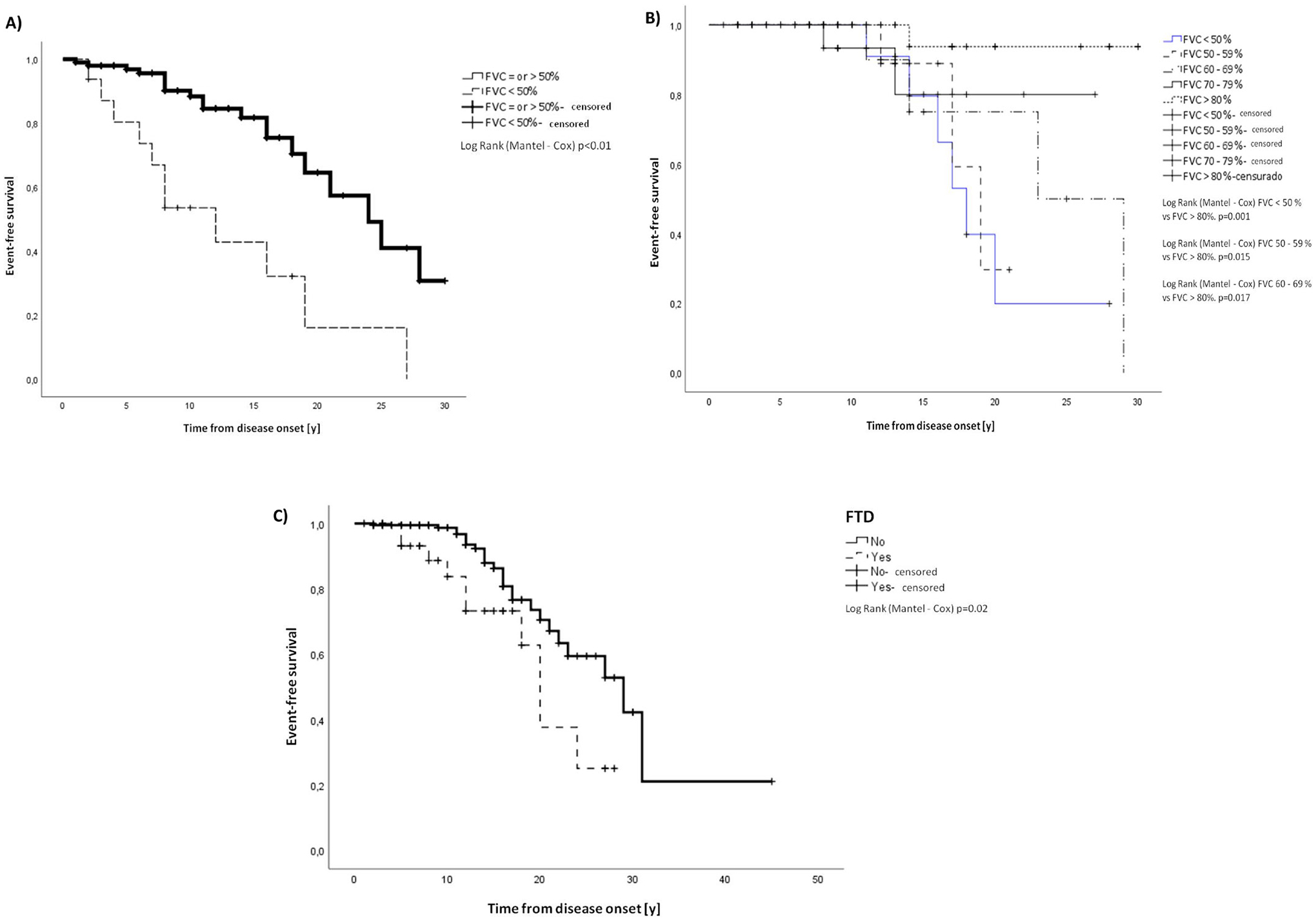
Kaplan-Meier estimator for full time wheelchair user/confined to bed and death. (A) Kaplan-Meier estimator for full time wheelchair user/confined to bed by FVC <50%. Mean time to being a full-time wheelchair user/confined to bed for FVC >50% was 22.2 years (SE 1.3, 95% CI 19.6 to 24.8 years) from disease onset and for FVC <50% 12.9 (SE 2.4, 95% CI 8.1 to 17.7). (B) Kaplan-Meier estimator for death by FVC value. Mean time to death from disease onset for FVC <50% 18.9 years (SE 2.4, 95% CI 15.2 to 22.6), FVC 50%–59% 18.2 years (SE1.1, 95% CI 16.0 to 20.4), FVC 60%–69% 23.5 years (SE 2.9, 95% CI 17.7 to 29.2), FVC 70%–79% 23.9 years (SE 2.0, 95% CI 19.8 to 27.8) and for FVC >80% 29 years (SE 0.9, 95% CI 27.1 to 30.9). (C) Kaplan-Meier estimator for death by the presence of FTD. Mean time to death from disease onset for patients with FTD 19.4 years (SE 1.8, 95% CI 15.9 to 22.9) and for patients without FTD 27.8 years (SE 2.7, 95% CI 22.6 to 33.1). All the comparisons were significant by log rank (Mantex-Cox) test as detailed in the figures. FVC, vital force capacity; FTD, frontotemporal dementia.
We tested if the presence of reduced FVC, cardiac impairment, dysphagia, dysarthria, dropped head, axial weakness, FTD, motor neuron signs and full-time wheelchair use/confinement to bed could be associated with death. The following variables were significantly associated with death: FTD, FVC 70% or less, dysphagia, drop-head and being a full-time wheelchair user (table 3). A strong positive correlation was identified between age at which these previous symptoms started, as well as the age of symptom onset, with the age of death (online supplemental table 8) These variables were included in a binary logistic regression analysis after which only the following variables remained associated with death: FVC <70% and full-time wheelchair use (online supplemental table 9). A Cox regression analysis showed that the variables FVC <70%, FTD and age at first symptom represented a risk for death (online supplemental table 10). The time to death by FVC value and FTD was analysed through a Kaplan-Meier estimator and showed significant differences (figure 5).
DISCUSSION
We present the largest serie of patients with mutations in the VCP gene reported so far. The VCP gene was found to be the cause of IBMPFD in 2004 when six missense pathogenic variants were identified in patients presenting the triad of myopathy, PDB and FTD.32 Subsequent cohorts confirmed this triad as common in patients with mutations in the VCP gene but also extended the phenotype to involvement of the CNS, motor neurones, sensory and/or motor peripheral nerves and the skeletal muscle.1 7 10 13 16–21 33–36 Our study confirms that muscle weakness, affecting both proximal or distal muscles of the lower and/or upper limbs, is the main symptom at onset, turning MSP into a challenging diagnosis whereas patients can be classified as having limb girdle muscle weakness, distal myopathy or scapuloperoneal syndrome.37
In this study, 8% of the patients showed an asymmetric muscle weakness at onset extending the diagnostic challenge to motor neuron diseases and motor neuropathies. Regardless of the pattern of weakness at onset, most of the patients showed a generalised muscle weakness and atrophy at last assessment. Remarkably, axial weakness, scapular winging and motor neuron signs presented within the first 5 years of the disease complicating the differential diagnosis process during early stages.
Twelve carriers of pathogenic variants did not develop symptoms after a mean of 5.2 years of follow-up. Penetrance variabilityin VCP is not completely understood.6 In our study, the majority of the asymptomatic carriers have not yet passed the highest age of onset expected in their families, and family members who might carry a variant in the VCP gene but were not genetically tested or were not followed by the physicians collaborating in this study, were not included in the analysis limiting the interpretation of the penetrance in this condition. Studies that include and follow-up a larger number of asymptomatic carriers could investigate weather reduce penetrance is a common feature in VCP as well as if the age of onset of the relatives could be used as an estimator of the expected age of onset in the asymptomatic carriers. More studies are required to investigate if incomplete penetrance varies based on the clinical phenotype and to examine other factors associated with an incomplete penetrance, such as genetic modifiers or environmental factors. These studies would aid in tailoring the care provided to asymptomatic carriers.38
We found a high frequency of some extra muscular symptoms in our cohort. Dysautonomia was a common feature, present in the 21.4%, as previously reported.7 We also observed a higher-than-expected peripheral neuropathy frequency, in 15.3% of the patients.13 22 Unfortunately, ancillary tests results or specific examinations to assess the autonomous nervous system, which could have provided a more accurate description, were not collected.39 Further studies evaluating the frequency and pathogenesis of these symptoms and their impact on quality of life are needed to improve the diagnosis and care guidelines.12 On the other hand, and similarly to what has been published,11 extrapyramidal disorders can be present in patients with VCP reinforcing the high phenotypic variability of this disease. Interestingly, the variants associated with extrapyramidal disorders reported here are different from variant p.Arg159Cys reported in the literature.9 However, the low number of patients by variant with extrapyramidal disorders prevented any genotype–phenotype association.
Despite the large number of variants identified in this study, just four of them accounted for 54.7% of the patients and were present in almost all countries. Exon 5 and 3 represent hotspot locations.13 All variants resulted in a missense change within the VCP gene. Fifteen of the 57 variants, in 164 patients, replaced an arginine for another aminoacid at different domains of the VCP protein: the N-domain (12/15), the linking 1 domain (2/15) or the ATPase D2 domain (1/15). Arginine has a key role in maintaining the structure and function of VCP protein as it is essential for the interaction between the N-domain and the D1-domain required for the formation of the hexameric ring where the hydrolytic activity takes place,33 and for binding of polyubiquitinated proteins.34
Similar to other cohorts, we did not identify variants that were exclusively associated with specific symptom/s.10 13 However, among the two most frequent variants, variant c.463C>T (p.Arg155Cys) showed a more severe phenotype with an earlier onset, as previously reported.9 13 A limitation for genotype–phenotype correlations studies in MSP is the large number of identified variants, but with a low number of patients in each group reducing the statistical power.9 10 13 33 Genotype–phenotype associations focusing on protein function impairment, rather than merely variant type or its location within the gene need further exploration. Finally, a number of variants previously categorised as VUS were upgraded to LP/P thanks to the identification of multiple independent families as well as their familial segregation data. This highlights the importance and usefulness of large international cohorts where clinical and genetic data is shared and analysed as a whole, rather than in independent case reports.
In our study, as in a previous cohort,19 the frequency of VCP was higher in males. Previously published cohorts have shown either a male predominance or male/female parity.9 10 14 Interestingly, variant c.463C>T (p.Arg155Cys) was more frequent in females in our study. The reasons of this gender predominance are not well understood. The male predominance in this study might not be attributed to specific population distribution as patients from 24 different countries and from independent families were included. Gender predominance was also reported in other neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease and ALS, in which sex hormones might play a role in the cell ability to cope with oxidative stress related with ageing what could be aggravated under pathological conditions.40 Moreover, male predominance has also been described in other muscular dystrophies, such as ANO5.41 Whether the hormones or other genetic modifiers related to the gender regulate VCP gene expression or influence the cell response to VCP variants needs to be explored.
Muscle weakness in MSP seems to develop more rapidly compared to other adult-onset inherited muscle conditions.10 42 In our cohort, 23.1% of the patients were no longer ambulant at a median of 8.5 years. As expected, lower limb weakness was associated with loss of ambulation but, interestingly, the presence of FTD, dysautonomia and FVC below 50% also influenced the ambulatory status. Moreover, FTD, severe respiratory and bulbar involvement were associated with death. However, after a multivariate analysis respiratory involvement, with FVC below 50%, remained as the only key risk factor associated with loss of ambulation while FVC below 70% and FTD where both associated with a higher risk of death.9 As FTD and particularly respiratory involvement speed-up disease progression, patients showing these symptoms requires a closer follow-up and improved care.
Limitations of this study include its retrospective design. Data was obtained from clinical notes implying missing data, non-homogeneous collection of data, as manifested in the variability of the number of patients in which the data was reported, and underestimation of mortality rates. Bone, cardiac and cognitive impairment might be underestimated as specific and repeated diagnostic tests were not required. In addition, the prevailing participation of neuromuscular specialist without consistently reaching out other specialists, including neurologists working primarily on dementia, and the autonomous nervous system or rheumatologists, might have led to information selection preferences. Only patients with genetic confirmation were included, excluding symptomatic relatives without a genetic test, in order to increase diagnostic accuracy but so reducing the population number and limiting the phenotypic heterogeneity.
In conclusion, this study expands the genotype and phenotype spectrum of VCP disease, better informs about disease progression and describes disease progression factors that could be used for the design of VCP natural history studies.
Supplementary Material
WHAT IS ALREADY KNOWN ON THIS TOPIC.
-
⇒
Valosin-containing protein disease is an autosomal dominant heterogeneous multisystemic condition caused by mutations in the valosin-containing protein (VCP) gene that can result in muscle weakness combined with other neurological signs, making its diagnosis challenging. Clear genotype–phenotype correlations have not yet been identified.
WHAT THIS STUDY ADDS
-
⇒
This large international cohort of patients with VCP disease expands our knowledge of the clinical phenotypes and natural history of the disease. We have identified genotype–phenotype correlations associated with variant c.463C>T (p.Arg155Cys), which is linked to an earlier age of onset and higher frequency of axial weakness. This large patient cohort allowed the identification of a reduced vital force capacity and been a full-time wheelchair use as risk factors associated with death.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
-
⇒
This study describes the phenotypic spectrum of VCP disease, which should be considered for differential diagnosis in clinical practice and to drive improvements in patient care. In addition, it identified genotype-phenotype associations, when comparing the two most frequent variants, and factors associated with unfavourable outcomes to be considered for inclusion criteria/exclusion criteria and outcomes assessment in clinical trials.
Acknowledgements
We are grateful to Dr Pol Camps-Renom (Department of Neurology, Hospital de la Santa Creu I Sant Pau, Barcelona, Spain) for statistical support on specific analysis. We are grateful to Dr Mark Baker (Department of Clinical Neurophysiology, Royal Victoria Infirmary, Newcastle Upon Tyne, UK) and Dr Mark Davis (Department of Diagnostic Genomics, Path West Laboratory Medicine Western Australia, University of Western Australia, Australia), for contacting us with clinicians attending patients with VCP. We would like to acknowledge members of the ERN-NMD (Dr Montse Olivé, Dr Marta Caballero, Dr Rodrigo Alvarez and Dr Jorge Alonso) for contributing to this project. We are grateful to Ms Cathy Turner (Duchenne Programme and TACT Coordinator at the John Walton Muscular Dystrophy Research Centre, Newcastle Upon Tyne, UK) for assistance in editing the English style of the manuscript. We are thankful to Dr David Hilton-Jones (Neurosciences Group, Nuffield Department of Clinical Neurosciences, Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, UK) for supporting clinical diagnosis of a patient in this cohort. Finally, we are grateful to all families, site investigators, clinical evaluators, research nurses, geneticist, pathologists and physiotherapists who actively collaborated in the collecting data process.
Funding
The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Collaborators
VCP International Study Group: Adolfo López de Munain, Biodonostia Neurosciences Area Group of Neuromuscular Diseases Biodonostia-Osakidetza Basque Health Service, San Sebastián, Spain; Alba Ramos-Fransi, Neuromuscular Unit, Neurology Department, Hospital Germas Trias i Pujol, Spain; Aleksandra Nadaj-Pakleza, Centre de Reference des Maldies Neuromusculaires Nord-Est-Ile de France, Department of Neurology, University Hospital of Strasbourg, France; Alicia Alonso-Jiménez, Department of Neurology, Neuromuscular Reference Centre, Antwerp University Hospital, Universiteit Antwerpen, Instituut Born Bunge, Antwerpen, Belgium; Alicia Martinez-Piñeiro, Neuromuscular Unit, Neurology Department, Hospital Germas Trias i Pujol, Spain; Ana Töpf, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Anders Oldfors, Department of Laboratory Medicine, Institute of Biomedicine, University of Gothenburg, Gothenburg Sweden; Anna Kaminska, Department of Neurology Medical University of Warsaw, European Reference Network EURO-N, Varsovia, Poland; Anna Kostera-Pruszczyk, Department of Neurology, Medical University of Warsaw, European Reference Network ERN-NMD, Warsaw, Poland; Anna Mayhew, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Anna Rydelius, Department of Clinical Sciences, Division of Neurology, Lund University, Lund Sweden; Anthony Behin, Unité clinique de pathologie neuromusculaire, Institut de Myologie Hôpital Pitié-Salpêtrière, Paris, France; Antonio Toscano, Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy; Aurelio Hernández Laín, Centro de Referencia Nacional para Enfermedades Neuromusculares Raras CIBERER, Madrid, Spain and Hospital Universitario 12 de octubre, Madrid, España; Beatrice Lannes, Department of Pathology, University Hospital of Strasbourg, France; Beatriz Velez, Neurology Department, Neuromuscular Disorders Unit, Hospital Universitario Virgen del Rocío. Instituto de Biomedicina de Sevilla and Center for Biomedical Network Research on Neurodegenerative Disorders (CIBERNED), Instituto de Salud Carlos III, Madrid Spain; Benedikt Schoser, Friedrich-Baur-Institute, Department of Neurology, LMU Clinics, Munich, Germany; Biruta Kierdaszuk, Department of Neurology, Medical University of Warsaw, European Reference Network EURO-N, Varsovia, Poland; Bjarne Udd, Tampere Neuromuscular Center, Tampere University Hospital, Finland and Folkhalsan Genetic Institute, Helsinki University, Finland; Boel De Paepe, Neurology Department and Neuromuscular Reference Centre, Gent Blegium part of the ERN-NMD; Bruno Eymard, APHP Centre de référence des maladies neuromusculaires Institut de Myologie, Sorbonne Université APHP Hôpital Pitié-Salpêtrière, Paris, France; Carla Marco Cazcarra, Neurology Department, Bellvitge University Hospital, Bellvitge, Spain; Carmelo Rodolico, Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy; Carmen Paradas, Neurology Department, Neuromuscular Disorders Unit, Hospital Universitario Virgen del Rocío; Instituto de Biomedicina de Sevilla and Center for Biomedical Network Research on Neurodegenerative Disorders (CIBERNED), Instituto de Salud Carlos III, Madrid, Spain; Carola Hedberg-Oldfors, Department of Laboratory Medicine, Institute of Biomedicine University of Gothenburg, Gothenburg, Sweden; Channa Hewamadduma, Sheffield Institute for translational neurosciences (SITRAN), Neuroscience Institute, University of Sheffield, Sheffield, UK; Cheryl Longman, Institute of Neurological Sciences, Queen Elizabeth University Hospital, Glasgow, UK; Chiara Marini Bettollo, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Chiseko Ikenaga, Johns Hopkins University School of Medicine, Baltimore, USA; Christopher Nance, Department of Neurology, Carver College of Medicine at the University of Iowa, Iowa City, Iowa, USA; Colin Quinn, Neuromuscular Division, Neurology Department, University of Pennsylvania, Philadelphia, Pennsylvania, USA; Conrad Weihl, Department of Neurology, Washington University School of Medicine, St. Louis, Missouri, USA; Constantinos Papadopoulos, First Department of Neurology, Medical School, Eginition Hospital, National and Kapodistrian University of Athens, Athens, Greece; Corinne Metay, Sorbonne Université - APHP Centre de Génétique Moléculaire et Chromosomique UF Cardiogénétique et Myogénétique Moléculaire et Cellulaire GH Pitié-Salpêtrière, Paris, France; Cristina Domínguez-González, Neurology Service, Hospital Universitario 12 de Octubre, Madrid, Spain and Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain; David Hilton-Jones, Department of Neurology, John Radcliffe Hospital, Oxford, UK; Edmar Zanotelli, Department of Neurology, School of Medicine, Universidade de São Paulo (FMUSP), São Paulo, Brazil; Edoardo Malfatti, APHP, Neuromuscular Reference Center Nord-Est-Ile-de-France, Henri Mondor Hospital, Université Paris Est, U955, INSERM, Créteil, IMRB, France.; Elena Pegoraro, Department of Neurosciences, University of Padova, Padova, Italy; Elizabeth A. Harrington, Department of Neurology, The Eleanor and Lou Gehrig ALS Center, Columbia University Medical Center, USA; Ellen Eline, Johns Hopkins University School of Medicine, Baltimore, USA; Ellen Gelpi, Division of Neuropathology and Neurochemistry, Department of Neurology, Medical University of Vienna, Vienna, Austria; Eloy Rivas, Neuropathology Department, University Hospital Virgen del Rocío, Sevilla, Spain and Center for Biomedical Network Research on Neurodegenerative Disorders (CIBERNED), Instituto de Salud Carlos III, Madrid, Spain; Endre Pál, Department of Neurology, University of Pécs, Hungary; Francesc Miralles, Unitat de Patologia Neuromuscular i Gabinet d’electrodiagnòstic, Servei de Neurologia, Hospital Universitari Son Espases, Palma de Mallorca, Spain; George K Papadimas, First Department of Neurology, Medical School, Eginition Hospital and National and Kapodistrian University of Athens, Athens, Greece; Gergious Manousakis, Department of Neurology, University of Minnesota Hospital, Minneapolis, Minnesota, USA; Gianni Sorarù, Department of Neurosciences, University of Padova, Padova, Italy; Giorgio Tasca, Unità Operativa Complessa di Neurologia, Fondazione Policlinico Universitario A Gemelli IRCCS, Rome, Italy; Giulia Bisogni, Unità Operativa Complessa di Neurologia, Fondazione Policlinico Universitario A Gemelli IRCCS, Rome, Italy; Giuseppe Lucente, Neuromuscular Unit, Neurology Department, Hospital Germas Trias i Pujol, Spain; Guillaume Bassez, APHP Centre de référence des maladies neuromusculaires Institut de Myologie, Sorbonne Université, APHP Hôpital Pitié-Salpêtrière, Paris, France; Hakan Cetin, Department of Neurology, Medical University of Vienna, Vienna, Austria; Hani Kushlaf, Department of Neurology & Rehabilitation Medicine, University of Cincinnati, Cincinnati, Ohio, USA; Ichizo Nishino, Department of Neuromuscular Research National Institute of Neuroscience National Center of Neurology and Psychiatry (NCNP), Japan; Jan L De Bleecker, Department of Neurology and Neuromuscular Reference Center, Ghent University Hospital, Ghent, Belgium, part of the ERN-NMD; Jean François, Deux APHP Centre de référence des maladies neuromusculaires, Institut de Myologie, Sorbonne Université, APHP Hôpital Pitié-Salpêtrière, Paris, France; Jean-Baptiste Chanson, Centre de Reference des Maldies Neuromusculaires Nord-Est-Ile de France, Department of Neurology, University Hospital of Strasbourg, France; Jie Lin, Department of Neurology, Huashan Hospital, Fudan University, Shanghai, China and National Center for Neurological Disorders, Shanghai, China; Jill Skeoch, Southern General Hospital, Glasgow, UK; Jin-Hong Shin, Laboratory of Molecular Neurology, Pusan National University Yangsan Hospital, Yangsan, Republic of Korea; Jodi Warman, Department of Medicine, Ottawa Neuromuscular Centre, Ottawa Hospital, Canada; Johanna Palmio, Tampere Neuromuscular Center, Tampere University Hospital and Tampere University, Finland; Jonathan Baets, Neuromuscular Reference Center, Neurology Department, University Hospital of Antwerp, Belgium; Jordi Díaz-Manera, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK and Institut de Recerca de l’Hospital de la Santa Creu i Sant Pau, Barcelona, Spain; Jorge A Bevilacqua, Unidad Neuromuscular, Departamento de Neurología y Neurocirugía, Hospital Clínico Universidad de Chile y Departamento de Neurología y Neurocirugía Clínica, Clínica Dávila, Santiago Chile, Chile; Jorge Alonso Pérez, Unidad de Enfermedades Neuromusculares, Servicio de Neurología, Hospital de la Santa Creu i Sant Pau de Barcelona, España; Jorge Díaz, Centro de Imagenología, Hospital Clínico Universidad de Chile, Chile; Jorge García-García, Neurology department, Complejo Hospitalario Universitario de Albacete, Albacete, Spain; Juan J Vilchez, Neuromuscular Unit, Department of Neurology, Hospital Universitari i Politècnic La Fe, Valencia, Spain; Neuromuscular and Ataxias Research Group, Instituto de Investigación Sanitaria La Fe, Valencia, Spain and Biomedical Network Research Centre on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Madrid, Spain; Judith Hudson, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Kate Taylor, Southern General Hospital, Glasgow, UK; Kinga Hadzsiev, Department of Pathology Neuropathology, Unit University of Pécs, Hungary; Kristl G Claeys, Department of Neurology, University Hospitals Leuven, Leuven, Belgium and KU Leuven Laboratory for Muscle Diseases and Neuropathies, Leuven, Belgium; Lindsay N Alfano, Center for Gene Therapy the Abigail Wexner Research Institute at Nationwide Children’s Hospital Columbus Ohio and Department of Pediatrics, The Ohio State University College of Medicine Columbus Ohio, USA; Luca Bello, Department of Neurosciences, University of Padova, Padova, Italy; Maria Elena Farrugia, Institute of Neurological Sciences, Queen Elizabeth University Hospital, Glasgow, UK; Marianela Schiava, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Marianne de Visser, Department of Neurology, Academic Medical Center, Amsterdam, The Netherlands; Mario Campero, Unidad Neuromuscular, Departamento de Neurología y Neurocirugía, Hospital Clínico Universidad de Chile, Santiago Chile, Chile; Mario Sabatelli, Unità Operativa Complessa di Neurologia, Fondazione Policlinico Universitario A Gemelli IRCCS, Rome, Italy; Marion Masingue, APHP Centre de référence des maladies neuromusculaires Institut de Myologie, Sorbonne Université APHP Hôpital Pitié-Salpêtrière, Paris, France; Marta Caballero-Ávila, Unidad de Enfermedades Neuromusculares Servicio de Neurología Hospital de la Santa Creu i Sant Pau de Barcelona, España; Matthew B Harms, NewYork Presbyterian Columbia University Irving Medical Centre, New York, New York, USA; Matthias Vorgerd, Heimer Institut for Muscle Research Klinikum Bergmannsheil Ruhr University Bochum, Germany; Mauro Monforte, Unità Operativa Complessa di Neurologia, Fondazione Policlinico Universitario A Gemelli IRCCS, Rome, Italy; Meredith James, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Michela Guglieri, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Michio Inoue, Department of Neuromuscular Research, National Institute of Neuroscience National Center of Neurology and Psychiatry (NCNP), Japan; Mónica Povedano, Neurology Department, Bellvitge University Hospital, Bellvitge, Spain; Monika Hofer, Department of Neuropathology, Oxford University Hospitals, NHS Foundation Trust, Oxford, UK; Montse Olivé, Neuromuscular Diseases Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain; Biomedical Research Institute Sant Pau (IIB Sant Pau), Barcelona, Spain and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Spain; Natalia Garcia-Angarita, Friedrich-Baur-Institute, Department of neurology LMU Clinics, Munich, Germany; Nicholas Earle, Departamento de Neurología y Neurocirugía, Clínica Dávila y Clínica Santa María, Santiago Chile, Chile; Noemi Vidal Sarró, Bellvitge University Hospital, Bellvitge, Spain; Nuria Muelas, Neuromuscular Unit, Department of Neurology, Hospital Universitari i Politècnic La Fe, Valencia, Spain; Neuromuscular and Ataxias Research Group, Instituto de Investigación Sanitaria La Fe, Valencia, Spain and Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain; Pascal Lafôret, Neurology department, Raymond-Poincaré hospital, APHP, UVSQ, Paris-Saclay University, Paris, France; Pascale Rihard, Sorbonne Université - APHP Centre de Génétique Moléculaire et Chromosomique UF Cardiogénétique et Myogénétique Moléculaire et Cellulaire GH Pitié-Salpêtrière, Paris, France; Paulo Victor Sgobbi Souza, Disciplina de Neurologia, Universidade Federal de São Paulo (UNIFESP), São Paulo, Brasil; Peter de Jonghe, Neuromuscular Reference Center, Neurology Department, University Hospital of Antwerp, Belgium; Phillipa J. Lamont, Department of Neurology, Royal Perth Hospital, Western Australia, Australia; Pietro Riguzzi, Department of Neurosciences, University of Padova, Padova, Italy; Pilar Camaño, Biodonostia Neurosciences, Area Group of Neuromuscular Diseases, Biodonostia-Osakidetza Basque Health Service, Molecular Diagnosis Platform, San Sebastián, Spain; Raúl Domínguez Rubio, Neurology Department, Bellvitge University Hospital, Bellvitge, Spain; Robert Carlier, Hôpital Raymond-Poincaré – APHP-Hôpitaux universitaires Paris Ile-de-France Ouest – Garches, France; Robert Muni-Lofra, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Roberto Fernández-Torrón, Neurology Department, Biodonostia Health Research Institute, Neuromuscular Area, Hospital Donostia, Basque Health Service, Doctor Begiristain, Donostia-San Sebastian, Spain; Rocío Nur Villar-Quiles, APHP Centre de référence des maladies neuromusculaires Institut de Myologie Sorbonne Université APHP Hôpital Pitié-Salpêtrière, Paris, France; Rodrigo Alvarez, Neuromuscular Diseases Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain; Biomedical Research Institute Sant Pau (IIB Sant Pau), Barcelona, Spain and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Spain; Rudi Kley, Department of Neurology and Clinical Neurophysiology, St Marien-Hospital Borken, Borken, Germany; Sabine Krause, Friedrich-Baur-Institute, Department of Neurology, LMU Clinics, Munich, Germany; Sarah Leonard-Louis, APHP Centre de référence des maladies neuromusculaires Institut de Myologie Sorbonne Université APHP Hôpital Pitié-Salpêtrière, Paris, France; Sarah Souvannanorath, Centre de référence des maladies neuromusculaires hôpital Henri-Mondor Assistance publique-Hôpitaux de Pars Créteil, France; Sigrid Klotz, Division of Neuropathology and Neurochemistry, Department of Neurology, Medical University of Vienna, Vienna, Austria; Simone Thiele, Friedrich-Baur-Institute, Department of Neurology, LMU Clinics, Munich, Germany; Sofia Xirou, First Department of Neurology, Medical School, Eginition Hospital and National and Kapodistrian University of Athens, Athens, Greece; Sruthi S Nair, Department of Neurology, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Thiruvananthapuram, Kerala, India; Stefen Brady, Department of Neurology, John Radcliffe Hospital, Oxford, UK; Stojan Peric, Neurology Clinic, Clinical Centre of Serbia, Faculty of Medicine, University of Belgrade, Belgrade, Serbia; Stuart Ralston, Centre for Genomic and Experimental Medicine, Institute of Genetics and Cancer, University of Edinburgh, Western General Hospital Edinburgh, UK; Sushan Luo, Department of Neurology, Huashan Hospital, Fudan University, Shanghai China and National Center for Neurological Disorders, Shanghai, China; Tanya Stojkovic, APHP Centre de référence des maladies neuromusculaires Institut de Myologie, Sorbonne Université, APHP Hôpital Pitié-Salpêtrière, Paris, France; Teresinha Evangelista, APHP Centre de référence des maladies neuromusculaires Institut de Myologie Sorbonne Université APHP Hôpital Pitié-Salpêtrière, Paris, France; Thomas E Lloyd, Johns Hopkins University School of Medicine, Baltimore, USA; Tiffany Grider, Department of Neurology, Carver College of Medicine at the University of Iowa, Iowa, USA; Timothy Williams, Newcastle Motor Neurone Disease Care Centre, Royal Victoria Infirmary, Newcastle, UK; Umesh Badrising, Department of Neurology, Leiden University Medical Centre, Leiden, The Netherlands; Velina Nedkova-Hristova, Neurology Department, Bellvitge University Hospital, Bellvitge, Spain; Vidosava Rakocevic-Stojanovic, Neurology Clinic, Clinical Centre of Serbia, Faculty of Medicine, University of Belgrade, Serbia; Virginia Kimonis, Department of Pediatrics, Division of Genetics and Genomic Medicine, University of California-Irvine Medical Center; Children’s Hospital of Orange County Orange, USA; Volker Straub, John Walton Muscular Dystrophy Research Centre, Newcastle University and Newcastle Hospitals NHS Foundation Trusts, Newcastle, UK; Wenhua Zhu, Department of Neurology, Huashan Hospital Fudan University, Shanghai, China and National Center for Neurological Disorders, Shanghai, China; Willem de Ridder, Neuromuscular Reference Center, Neurology Department, University Hospital of Antwerp, Belgium; William Kelly, Johns Hopkins University School of Medicine, Baltimore, USA; Yoshihiko Saito, Department of Neuromuscular Research National Institute of Neuroscience National Center of Neurology and Psychiatry (NCNP), Japan; Young-Eun Park, Laboratory of Molecular Neurology, Pusan National University Yangsan Hospital, Yangsan, Republic of Korea; Yukako Nishimori, Department of Neuromuscular Research National Institute of Neuroscience National Center of Neurology and Psychiatry (NCNP), Japan; Zarife Sahenk, Center for Gene Therapy the Abigail Research Institute at Nationwide Children’s Hospital, Columbus, Ohio and Department of Pediatrics, The Ohio State University College of Medicine, Columbus, Ohio, USA.
Footnotes
Correction notice Since this article was first published, the author Jodi Warman has been updated to Jodi Warman-Chardon.
Competing interests None declared.
Ethics approval Study approval was obtained from the Newcastle upon Tyne Hospitals Register Audit, Newcastle, UK (project number 10833, Caldicott Approval: 7918). Institutional Review Boards approvals were obtained from the LMU Klinikum at Ludwig-Maximilians University in Munich, Germany (project 21-0071), from the Washington University School of Medicine Institutional Review Board, USA (no 201103416) and the Johns Hopkins Hospital Institutional Review Board, Baltimore, USA (no 00288171). These ethics committees catalogued the present study as an audit as it was collecting deidentified retrospective data of patients with VCP. In these cases, there is no need for patients to sign a consent form.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Additional supplemental material is published online only. To view, please visit the journal online (http://dx.doi.org/10.1136/jnnp-2022-328921).
Data availability statement
Data are available from the corresponding author on reasonable request.
REFERENCES
- 1.Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010;68:857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun X, Qiu H. Valosin-containing protein, a calcium-associated ATPase protein, in endoplasmic reticulum and mitochondrial function and its implications for diseases. Int J Mol Sci 2020;21. doi: 10.3390/ijms21113842. [Epub ahead of print: 28 May 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeo BK, Yu S-W. Valosin-containing protein (VCP): structure, functions, and implications in neurodegenerative diseases. Animal Cells Syst 2016;20:303–9. [Google Scholar]
- 4.Kimonis VE, Kovach MJ, Waggoner B, et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med 2000;2:232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kovach MJ, Waggoner B, Leal SM, et al. Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, paget disease of bone, and frontotemporal dementia. Mol Genet Metab 2001;74:458–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evangelista T, Weihl CC, Kimonis V, et al. 215th ENMC International workshop VCP-related multi-system proteinopathy (IBMPFD) 13–15 November 2015, Heemskerk, the Netherlands. Neuromuscul Disord 2016;26:535–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Figueroa-Bonaparte S, Hudson J, Barresi R, Polvikoski T, et al. Mutational spectrum and phenotypic variability of VCP-related neurological disease in the UK. J Neurol Neurosurg Psychiatry 2016;87:680–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nalbandian A, Donkervoort S, Dec E, et al. The multiple faces of valosin-containing protein-associated diseases: inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J Mol Neurosci 2011;45:522–31. [DOI] [PubMed] [Google Scholar]
- 9.Mehta SG, Khare M, Ramani R, et al. Genotype-Phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet 2013;83:422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stojkovic T, Hammouda EH, Richard P, et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord 2009;19:316–23. [DOI] [PubMed] [Google Scholar]
- 11.Plewa J, Surampalli A, Wencel M, et al. A cross-sectional analysis of clinical evaluation in 35 individuals with mutations of the valosin-containing protein gene. Neuromuscul Disord 2018;28:778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller TD, Jackson AP, Barresi R, et al. Inclusion body myopathy with Paget disease and frontotemporal dementia (IBMPFD): clinical features including sphincter disturbance in a large pedigree. J Neurol Neurosurg Psychiatry 2009;80:583–4. [DOI] [PubMed] [Google Scholar]
- 13.Al-Obeidi E, Al-Tahan S, Surampalli A, et al. Genotype-Phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet 2018;93:119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikenaga C, Findlay AR, Seiffert M. Phenotypic diversity in an international cure VCP Disease registry. Orphanet J Rare Dis 2020:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul Disord 2009;19:308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sacconi S, Camaño P, de Greef JC, et al. Patients with a phenotype consistent with facioscapulohumeral muscular dystrophy display genetic and epigenetic heterogeneity. J Med Genet 2012;49:41–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmio J, Sandell S, Suominen T, et al. Distinct distal myopathy phenotype caused by VCP gene mutation in a finnish family. Neuromuscul Disord 2011;21:551–5. [DOI] [PubMed] [Google Scholar]
- 18.González-Pérez P, Cirulli ET, Drory VE, et al. Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology 2012;79:2201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spina S, Van Laar AD, Murrell JR, et al. Phenotypic variability in three families with valosin-containing protein mutation. Eur J Neurol 2013;20:251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Bot ST, Schelhaas HJ, Kamsteeg E-J, et al. Hereditary spastic paraplegia caused by a mutation in the VCP gene. Brain 2012;135:e223. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez MA, Feely SM, Speziani F, et al. A novel mutation in VCP causes charcot-marie-tooth type 2 disease. Brain 2014;137:2897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gite J, Milko E, Brady L, et al. Phenotypic convergence in charcot-marie-tooth 2Y with novel VCP mutation. Neuromuscul Disord 2020;30:232–5. [DOI] [PubMed] [Google Scholar]
- 23.Mariani L-L, Tesson C, Charles P, et al. Expanding the spectrum of genes involved in huntington disease using a combined clinical and genetic approach. JAMA Neurol 2016;73:1105–14. [DOI] [PubMed] [Google Scholar]
- 24.Wang SC, Smith CD, Lombardo DM, et al. Characteristics of VCP mutation-associated cardiomyopathy. Neuromuscul Disord 2021;31:701–5. [DOI] [PubMed] [Google Scholar]
- 25.Wani A, Weihl CC. Loss-of-function mutation in VCP mimics the characteristic pathology as in FTLD-TARDBP. Autophagy 2021;17:4502–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blythe EE, Gates SN, Deshaies RJ, et al. Multisystem proteinopathy mutations in VCP/p97 increase NPLOC4·UFD1L binding and substrate processing. Structure 2019;27:1820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shinjo SK, Oba-Shinjo SM, Lerario AM, et al. A Brazilian family with inclusion body myopathy associated with paget’s disease of bone and frontotemporal dementia linked to the VCP pGly97Glu mutation. Clin Rheumatol 2018;37:1129–36. [DOI] [PubMed] [Google Scholar]
- 28.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jankovic J, Tolosa E. Parkinson’s Disease and Movement Disorders. In: Wolters Kluwer. Sixth edit, 2015. [Google Scholar]
- 30.De Ridder W, Azmi A, Clemen CS, et al. Multisystem proteinopathy due to a homozygous p.Arg159His VCP mutation: A tale of the unexpected. Neurology 2020;94:e785–96. [DOI] [PubMed] [Google Scholar]
- 31.Ludolph A, Drory V, Hardiman O. A revision of the El Escorial criteria - 2015. Amyotroph Lateral Scler Frontotemporal Degener 2015;2015;16(5–6):291–2. [DOI] [PubMed] [Google Scholar]
- 32.Watts GDJ, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377–81. [DOI] [PubMed] [Google Scholar]
- 33.Shi Z, Hayashi YK, Mitsuhashi S, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol 2012;19:501–9. [DOI] [PubMed] [Google Scholar]
- 34.Schröder R, Watts GDJ, Mehta SG, et al. Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann Neurol 2005;57:457–61. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura T, Kawarabayashi T, Koh K, et al. Spastic paraplegia with paget’s disease of bone due to a VCP gene mutation. Intern Med 2021;60:141–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liewluck T, Milone M, Mauermann ML, et al. A novel VCP mutation underlies scapuloperoneal muscular dystrophy and dropped head syndrome featuring lobulated fibers. Muscle Nerve 2014;50:295–9. [DOI] [PubMed] [Google Scholar]
- 37.Jerath NU. Resolving a Multi-generational neuromuscular mystery in a family presenting with a variable scapuloperoneal syndrome in a c.464G>A, p.Arg155His VCP Mutation. Case Rep Genet 2019;2019:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korb M, Peck A, Alfano L. Orphanet journal of rare diseases 2022;17:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freeman R, Chapleau MW. Testing the Autonomic Nervous System. In: Elsevier BV, ed. 1St. 115, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Sumien N, Cunningham JT, Davis DL, et al. Neurodegenerative disease: roles for sex, hormones, and oxidative stress. Endocrinology 2021;162:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarkozy A, Hicks D, Hudson J, et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Hum Mutat 2013;34:1111–8. [DOI] [PubMed] [Google Scholar]
- 42.Mercuri E, Muntoni F. Muscular dystrophies. Lancet 2013;381:845–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author on reasonable request.