


Abstract
Alternative splicing (AS) is an important mechanism in the development of many cancers, as novel or aberrant AS patterns play an important role as an independent onco-driver. In addition, cancer-specific AS is potentially an effective target of personalized cancer therapeutics. However, detecting AS events remains a challenging task, especially if these AS events are novel. This is exacerbated by the fact that existing transcriptome annotation databases are far from being comprehensive, especially with regard to cancer-specific AS. Additionally, traditional sequencing technologies are severely limited by the short length of the generated reads, which rarely spans more than a single splice junction site. Given these challenges, transcriptomic long-read (LR) sequencing presents a promising potential for the detection and discovery of AS. We present Freddie, a computational annotation-independent isoform discovery and detection tool. Freddie takes as input transcriptomic LR sequencing of a sample alongside its genomic split alignment and computes a set of isoforms for the given sample. It then partitions the input reads into sets that can be processed independently and in parallel. For each partition, Freddie segments the genomic alignment of the reads into canonical exon segments. The goal of this segmentation is to be able to represent any potential isoform as a subset of these canonical exons. This segmentation is formulated as an optimization problem and is solved with a dynamic programming algorithm. Then, Freddie reconstructs the isoforms by jointly clustering and error-correcting the reads using the canonical segmentation as a succinct representation. The clustering and error-correcting step is formulated as an optimization problem—the Minimum Error Clustering into Isoforms (MErCi) problem—and is solved using integer linear programming (ILP). We compare the performance of Freddie on simulated datasets with other isoform detection tools with varying dependence on annotation databases. We show that Freddie outperforms the other tools in its accuracy, including those given the complete ground truth annotation. We also run Freddie on a transcriptomic LR dataset generated in-house from a prostate cancer cell line with a matched short-read RNA-seq dataset. Freddie results in isoforms with a higher short-read cross-validation rate than the other tested tools. Freddie is open source and available at https://github.com/vpc-ccg/freddie/.
INTRODUCTION
Alternative splicing (AS) is a cellular process that enables a single gene to code for different proteins (1), contributing to protein diversity (2,3). Recent findings show that AS plays a critical role in regulating gene expression (4) and tissue specialization (5). Abnormalities in the AS of genes are also linked to the pathogenesis of many diseases, including cancer. For example, AS aberrations contribute to a tumor’s ability to proliferate and to evade programmed cell death (6). Additionally, cancer-specific AS aberrations present potential targets for cancer therapeutics (7,8). The efficacy of these AS-centred treatments relies on the personalized, accurate and rapid detection of AS isoforms at the level of individual patients.
From the mid-2000s and until recently, short-read (SR) sequencing has been the dominant high-throughput sequencing technology for genomics and transcriptomics analysis. While SR sequencing has a very low sequencing error rate (of the order of 1 error per 1000 sequenced nucleotides), it generates reads that are too short to fully sequence an isoform molecule in a single read: 75–250 nt per read versus a median of 909 nt per isoform (9,10). SR AS detection methods use SRs to identify splicing junctions between pairs of exons (11–14). However, due to the limited span of SRs, such methods face major challenges in ‘chaining’ these splicing junctions to reconstruct complete isoforms.
More recently, long-read (LR) sequencing became a commercially viable option to study the transcriptome and genome (15). In theory, LR sequencing machines can sequence the full length of RNA molecules, generating reads that range from thousands to tens of thousands of nucleotides (16). Thus, ideally, aligning LRs to the reference genome should be enough to perfectly define the exons of the underlying isoforms. However, in practice, LRs suffer from a high sequencing error rate (10–20 errors per 100 sequenced nucleotides) that is dominated by indels (erroneous insertions and deletions) (17). This high-error profile is especially characteristic of Oxford Nanopore Sequencing, one of the leading LR sequencing platforms. When comparing aligned LRs with a reference genome, indels cause erroneous shifts in the observed boundaries of the exons and occasionally result in missing smaller exons altogether. Additionally, LR sequencing of an RNA molecule, more often than not, terminates before reaching the full length of the molecule, resulting in missing exons at the tail of the isoform (18). These main types of LR sequencing errors are summarized in Supplementary Figure S1. Current isoform detection methods based on LRs overcome these challenges by relying on existing isoform annotation databases. The current methods can be classified into three categories depending on how they use available annotation data: (i) methods that detect only isoforms that are described in the annotation which is typically done by aligning the LRs to the sequences of annotated isoforms; (ii) methods that detect isoforms with potentially novel exon chains if those exons boundaries are present in some annotated isoforms [e.g FLAIR (19)]; and (iii) de novo methods that do not rely on any annotation but can potentially benefit from using existing annotation data [e.g. StringTie2 (20)]. Approaches that rely on annotation data are limited by the fact that annotation databases are incomplete: millions of splicing genomic locations and thousands of isoforms are present in different individuals but not in major annotation databases (21,22). Figure 1 provides further detail of the hierarchy of dependence on annotations for isoform detection. Note that this whole spectrum of methods relies on the presence of a genome reference sequence against which LRs are aligned. In the absence of such genome reference sequences, some methods have been developed that attempt to reconstruct transcripts from LR sequences without genomic alignment [e.g. RATTLE (23)].
Figure 1.
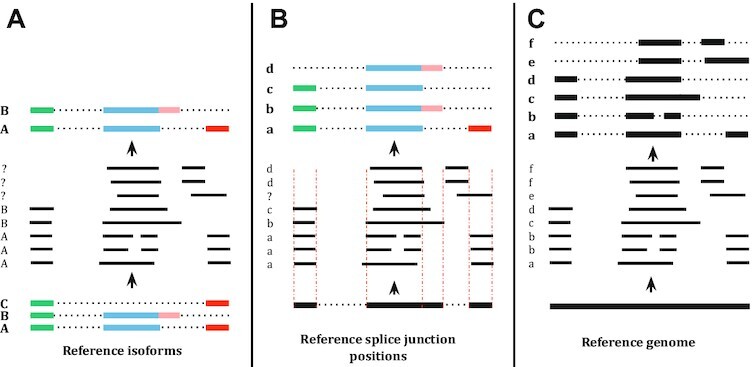
AS isoform detection tools can be put on a spectrum in terms of their reliance on reference annotations. (A) On the left of the spectrum, the tool is fully dependent on the known isoform annotations and is thus unable to discover any novel isoforms. Each read is annotated by the isoform that it best matches or is discarded if it does not match any isoform. (B) In the middle, the tool is partially reliant on the isoform annotation; novel isoforms can be detected as long as they are composed of known splice junctions (i.e. boundaries of known isoforms). The split-alignment boundary of each read is corrected to best matching splice junction position. If a read has a novel splice junction position, the tool will have difficulty identifying its isoform structure. (C) On the right, the tool does not rely on the reference annotation. Instead, it relies solely on the split alignment of the reads to the reference genome.
Contributions
In this paper, we introduce Freddie, a novel multistage computational method aimed at detecting isoforms using Oxford Nanopore LR sequencing without relying on isoform annotation data. The design of each stage in Freddie is motivated by the specific challenges of annotation-free isoform detection from noisy LRs. We compare the performance of Freddie against two alternative state-of-the-art isoform detection tools, FLAIR (19) and StringTie2 (20). We show using simulated data that Freddie achieves accuracy (as measured by the harmonized F1 score) on a par with FLAIR despite not using any annotations and outperforms StringTie2 in accuracy. Furthermore, Freddie’s F1 score is better than FLAIR’s when FLAIR is provided with only partial annotations. Finally, we demonstrate Freddie’s ability to detect novel isoforms on a real cancer cell line dataset and use reverse transcription–polymerase chain reaction (RT–PCR) to biologically validate a select set of these detected novel isoforms.
MATERIALS AND METHODS
Freddie takes as input the mapping of transcriptomic LRs to a reference genome, and outputs, in GTF format, a list of detected isoforms each described as a set of genomic intervals. Freddie assumes that the mapping is performed by a splice-aware mapper which attempts to solve the problem of finding the best read to genome alignment that accounts for introns by allowing for large deletions to not be penalized. Freddie is made up of three stages: partitioning, segmentation and clustering/error correction, as depicted in Figure 2. The design of each of these stages is motivated by the challenges of isoform detection using noisy LR sequencing that does not rely on annotation data.
Figure 2.

Overview of the Freddie stages, from genomic split alignments of LRs to detected isoforms.
Read partitioning
In this stage, we partition the aligned reads into sets with the aim that no isoform from the sequenced transcriptome has reads present in different sets. As a result, each set of the partition can be assumed to contain all the reads from a set of isoforms and can be processed independently from the other sets, which allows for highly parallel processing. We define the partition sets as follows: if two reads have split-alignment intervals that overlap, then they must be in the same set or, equivalently, if two reads are in different sets, then their split-alignment intervals cannot overlap.
Given a sorted list of the split-alignment intervals of the reads, we can compute the partition sets in linear time. This is achieved by first identifying genomic intervals with contiguous coverage, and then running a classic breadth-first search using the read membership in these contiguous genomic intervals. At the end of this stage, we have independent sets of reads that we can process independently and in parallel in the next stages. Therefore, when we refer to the reads in the next stages, we mean only the reads in a given set. Figure 2 illustrates the parallel processing of multiple partition read sets in the next stages.
Canonical segmentation of the genome
The segmentation stage addresses the challenge of detecting exon (and intron) boundaries from the alignments of LRs to a reference genome. Annotation-dependent isoform detection tools bypass this challenge by matching the LR alignments to the closest exon boundary in the annotation, assuming that any deviation from the annotation is a result of sequencing noise and not of a potentially novel AS event. To overcome this limitation, we propose a data-driven segmentation approach aiming at identifying exon boundaries by finding a set of segmentation breakpoints that are best supported by the input LR split-alignments. To find this segmentation, we devise a two-step process (illustrated in Figure 3).
Figure 3.

Illustrating the two main steps of Freddie’s segmentation stage. (A) The interval boundaries of the genomic split-alignments of the reads are represented as a discrete signal (grey). This signal is smoothed using a Gaussian filter. The peaks (red) of this filtered signal are used as candidate breakpoints for the segmentation. (B) A subset of the elements in the list of candidate breakpoints, 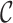 , is selected and scored. Score increases due to high coverage contrast are shown in green and score decreases due to partial coverage are shown in red. This scoring scheme is used to select the optimal subset of
, is selected and scored. Score increases due to high coverage contrast are shown in green and score decreases due to partial coverage are shown in red. This scoring scheme is used to select the optimal subset of 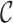 to be used as canonical segmentation in this stage.
to be used as canonical segmentation in this stage.
Identifying candidate breakpoints
To generate the set of candidate breakpoints, 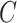 , we treat the LR split alignments as a discrete signal: for a genomic position
, we treat the LR split alignments as a discrete signal: for a genomic position 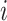 , we define
, we define 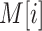 as the number of reads that have a split-alignment interval starting or ending on
as the number of reads that have a split-alignment interval starting or ending on 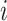 . We then apply a Gaussian filter on the signal encoded by the array
. We then apply a Gaussian filter on the signal encoded by the array 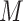 to smooth over the signal’s noise. The Gaussian filter smooths out the raw signal and makes it more robust to the noise due to indel sequencing errors on the potential splicing positions. We denote the smoothed signal by
to smooth over the signal’s noise. The Gaussian filter smooths out the raw signal and makes it more robust to the noise due to indel sequencing errors on the potential splicing positions. We denote the smoothed signal by 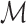 . Finally, we denote by
. Finally, we denote by 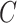 the set of genomic positions of the local peaks (i.e. local maxima) of
the set of genomic positions of the local peaks (i.e. local maxima) of 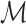 (see Figure 3A). Our implementation uses scikit-learn (24) Gaussian filter and peak-finding functions.
(see Figure 3A). Our implementation uses scikit-learn (24) Gaussian filter and peak-finding functions.
Pruning the set of the candidate breakpoints
Freddie selects a subset of breakpoints, 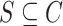 , to represent the finalized set of canonical exon boundaries inferred from the input LR alignments. Note that the breakpoint sets,
, to represent the finalized set of canonical exon boundaries inferred from the input LR alignments. Note that the breakpoint sets, 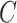 and
and 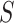 , divide the genome, respectively, into
, divide the genome, respectively, into 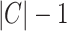 and
and 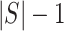 non-overlapping genomic segments. We define a scoring function,
non-overlapping genomic segments. We define a scoring function, 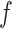 , which given a set of breakpoints simultaneously rewards individual reads for sharp changes in coverage between consecutive segments and penalizes them for having partial alignment on individual segments (see Figure 3B). Freddie selects the subset
, which given a set of breakpoints simultaneously rewards individual reads for sharp changes in coverage between consecutive segments and penalizes them for having partial alignment on individual segments (see Figure 3B). Freddie selects the subset 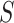 which maximizes
which maximizes 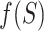 over all the subsets of
over all the subsets of 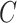 , using a dynamic programming algorithm. The details of this optimization process and its dynamic programming solution are in Section S1.1 of the Supplementary Data. Additionally, the details of the segmentation hyperparameter selection process are presented in Section S1.2 of the Supplementary Data.
, using a dynamic programming algorithm. The details of this optimization process and its dynamic programming solution are in Section S1.1 of the Supplementary Data. Additionally, the details of the segmentation hyperparameter selection process are presented in Section S1.2 of the Supplementary Data.
Vectorization of LRs
For the next stage, we use the selected breakpoints, 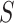 , of this stage to succinctly represent the reads: each LR is encoded as a binary vector of length
, of this stage to succinctly represent the reads: each LR is encoded as a binary vector of length 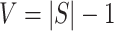 in which each bit indicates the presence (1) or absence (0) of a segment on the LR using the 90% coverage threshold used above, as illustrated in Figure 4.
in which each bit indicates the presence (1) or absence (0) of a segment on the LR using the 90% coverage threshold used above, as illustrated in Figure 4.
Figure 4.

The segmentation is projected on the reads. Each read is represented as a binary vector of size equal to the number of canonical segments. A segment has a value of 1 (filled grey) if a read has at least 90% coverage over the segment, and 0 otherwise.
Clustering and error correction
The goal of this stage is to compute a set of potential isoforms using the succinct binary vector representation of the LRs generated by the segmentation stage. Our approach is to cluster the LRs such that each cluster represents a potential isoform. A crucial point is to consider the possibility for some reads to have erroneously missed segments (1 to 0 errors) due to sequencing errors and to correct such errors by using evidence from other LRs from the same cluster. Thus, each cluster is composed of similar reads that potentially originate from the same isoform. We can then reconstruct each isoform by generating the consensus structure from the vector representation of the LRs in the cluster. We recognize two main challenges for this task that are not faced by annotation-dependent tools: we do not know a priori (i) the number of clusters (i.e. isoforms) and (ii) the structure of the isoforms in terms of segments. To overcome these challenges, we devise an iterative process inspired by the minimum error correction (MEC) problem (25) that is commonly used in haplotype assembly.
In each round of this iterative process, we assign the input reads into one of two bins: an isoform bin and a recycling bin. The isoform bin should contain similar reads that are assumed to originate from the same isoform, while the recycling bin should contain everything else. At the end of each round, the isoform bin reads are set aside and their consensus is used as the structure of a detected isoform, while the recycling bin reads are used as input to the next round (see Figure 5). The assignment of the reads in each round to one of the two bins is done according to a scoring function: each read in the isoform bin incurs a penalty proportional to the number of error corrections it requires in order to match the consensus structure of the isoform bin reads (similar to the MEC problem) while each read in the recycling bin incurs a constant penalty. The interesting feature of this approach lies in its ability to cluster the reads without specifying the number of expected clusters or their structure. We call this clustering problem the Minimum Error Clustering into Isoforms (MErCI) problem. The mathematical formulation of MErCI is presented in Section S.1.3 of the Supplementary Data.
Figure 5.
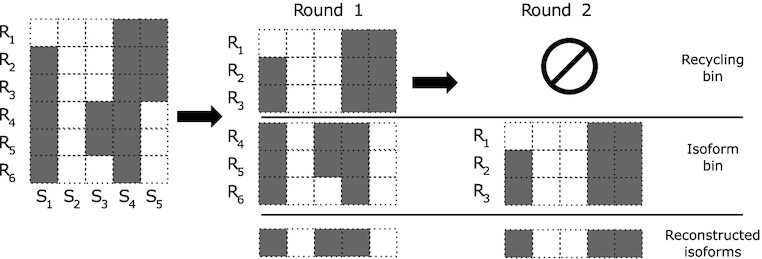
The input to the clustering stage is the vectorized reads. In each round of MErCI, the reads are assigned to either the isoform bin or the recycling bin. The isoform bin reads are set aside and the recycling bin reads are used as input to the next stage. The read vectors of each isoform cluster are used to reconstruct the isoforms employing a simple column-wise consensus strategy.
Additional considerations for transcriptomic LRs
In Freddie, we extend and modify the MErCI problem as described in the previous section and in the Supplementary Data in order to include considerations that are specific to transcriptomic LR sequencing data.
Poly(A) tail
During the biological process of transcription, the spliced RNA molecule is extended with a small sequence of A nucleotides, known as the poly(A) tail. The presence of the poly(A) sequence is used as a target in the preparation step of sequencing to extract and isolate the mRNA molecules from the sample RNA material. Note that the poly(A) tail is not part of the genomic sequence of the gene. Therefore, if an LR has successfully sequenced the poly(A) tail of its isoform, we expect to observe an A-enriched sequence (or a T-enriched sequence for cDNA sequencing) in the LR in the parts of its sequence that did not align to the genome. If we observe the A-enriched sequence after the last covered canonical segment of the LR, then we can infer that the gene of the LR isoform is on the forward strand of the genome and we describe this LR as a forward read. Similarly, if we observe the A-enriched sequence before the first covered canonical segment of the LR, we describe the LR as a reverse read. Note that the order of the segments is defined by their increasing genomic coordinates. In Freddie, we constrain the MErCI assignment to the isoform cluster to forbid assigning a forward read and a reverse read to the isoform cluster in a given round.
Truncated reads
As mentioned earlier, LRs do not always cover the full length of an RNA/cDNA molecule. Therefore, for a given LR, it makes sense not to penalize the corrected segments at the start or end of the isoform if the sequencing has terminated before they were sequenced. We, therefore, modify the correction cost function in Freddie to penalize only the internal segments of a given LR. We define the internal segments of an LR to be the segments between the first and last covered segments of the LR (i.e. segments between the first and last 1 entries in the binary representation of the read). For forward reads, we extend the definition of internal segments to include segments after its last covered segment. Similarly, for reverse reads, we include segments before its first covered segment.
Length of corrected segments
When correcting internal segments in an LR, we also take into account the lengths of the corrected segments. This is because missing shorter segments are more likely to be the result of sequencing and mapping errors. More specifically, we want to account for the possibility that the missing segments were sequenced by the LR, but, due to substitution and indel errors, the LR alignment to the reference genome missed some segments. Therefore, for each read, we extract the set of its internal contiguous missing segments and their flanking covered segments (i.e. maximal stretches of 0s surrounded by 1 on both sides) which we call ‘gaps’. The formal definition of how this constraint is incorporated into MErCI is detailed in Section S.1.3 of the Supplementary Data.
Solving MErCI using integer linear programming
The search space for assigning reads to the isoform bin or the recycling bin is exponentially large in the size of the input (i.e. number reads multiplied by the number of segments). To overcome this challenge, we formulate the MErCI problem with the extra constraints described as an integer linear program (ILP). We use the ILP solver Gurobi to obtain an optimal assignment of the reads according to the MErCI formulation.
Isoform reconstruction
Finally, to reconstruct the isoforms, we build a consensus for each cluster of reads. Here, we apply a simple rule of column-wise consensus of the reads with a plurality threshold of 30% of the reads that span a given exon segment.
RESULTS
To evaluate the accuracy and predictive power of Freddie, we assessed Freddie against two well-established methods, FLAIR (19) and StringTie2 (20), on simulated and real datasets. As the ground truth about the real datasets is not known a priori, it is essential to use controlled simulated data to perform accuracy measurements. However, the simulation can only reflect the aspects of LR sequencing that we are aware of and that we introduce in the simulation’s design. Therefore, it is also equally important to test our method on a real dataset and attempt to validate it using orthogonal technologies to provide some assurance of the usability of our method. Thus, we also tested the three tools on a prostate cancer cell line dataset that we transcriptomically sequenced using both ONT PromethION’s LR platform and Illumina’s SR platform. Note that the computational benchmarking pipeline can be fully reproduced using our Snakemake (26) scripts at https://github.com/vpc-ccg/freddie/tree/benchmarking.
Simulated data experiment
Data generation
We generated an LR transcriptomic sequencing dataset for the human genome. For this dataset, we wanted it to reflect the isoform expression distribution of a typical real dataset. Therefore, we used a publicly available prostate cancer cell line LR dataset that we previously sequenced (SRA: PRJNA726724) to estimate isoform expression levels. We mapped the LRs of this real dataset using Minimap2 (27) to the set of annotated human isoform sequences from the ENSEMBL 97 database (28) and used the number of primary alignments of the reads to each isoform as an expression profile of this real dataset. We used this expression profile to simulate LRs from the isoform sequences employing Badread (29), a simulator specifically designed for Oxford Nanopore LRs. The simulator pipeline, which wraps around the Badread simulator, is available on our GitHub repository at https://github.com/vpc-ccg/LTR-sim. Finally, we discarded any isoform (and its simulated reads) with less than three simulated reads. In total, the simulated dataset includes 1 341 487 reads from 28 025 isoforms with a median length of 1446 nt. The median number of reads simulated per isoform is eight. Additionally, using the same simulation pipeline, we generated a second simulated dataset using the publicly accessible ONT fruit fly RNA dataset (accession number ERR3588905) (30). All the results of this second simulated dataset are available in Supplementary Figures S3, S4 and S5, and Tables S3 and S4.
Tool configuration
We used Minimap2 (27) to generate input genomic split alignments and ran it without using any annotation files. We tested all tools using their default settings. We ran StringTie2 (with -L flag) and Freddie without any annotation files. FLAIR requires the use of an annotation file so we supplied it with the complete GTF file for the human transcriptome, including the annotations of the 28 025 considered isoforms. Note that this provides the best-case scenario for FLAIR in terms of the comprehensiveness of the annotation. To further investigate FLAIR’s reliance on the annotation, we tested it with subsets of the annotation with varying sampling rates of 75, 50, 25 and 1% of the annotation isoforms. We present here the accuracy results of FLAIR using 100% and 50% annotation sampling rates. The complete results for FLAIR are available in Supplementary Figure S2.
Accuracy measurement
In assessing the accuracy of AS detection by different tools, we use two approaches. The first approach is based on computing for each tool how many of its predicted isoforms are present in the set of simulated isoforms using the structure of the isoforms to test for isoform equivalency. In this approach, we consider two isoforms as equivalent if they have the same set of exon (or intron) intervals with the possibility for each coordinate to be shifted by ±10 bp. This approach provides a simple yet strict means of quantifying the accuracy metrics of different tools.
However, since the isoforms of the same gene can greatly overlap, it is justifiable to take into account highly similar isoforms, whether predicted by a tool or present in the ground truth, as related data points. The second approach is based on computing clusters of highly similar isoforms. In this approach, we construct a graph 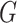 whose vertices are
whose vertices are 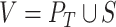 where
where 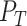 and
and 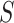 are, respectively, the sets of isoforms predicted by tool
are, respectively, the sets of isoforms predicted by tool 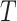 and the set of true isoforms. We add an edge between any pair of isoforms,
and the set of true isoforms. We add an edge between any pair of isoforms, 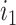 and
and 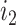 , if their pairwise sequence split-alignment score is higher than a given threshold
, if their pairwise sequence split-alignment score is higher than a given threshold 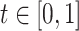 . We define the pairwise sequence split-alignment score to be:
. We define the pairwise sequence split-alignment score to be:
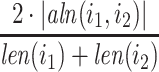 |
where 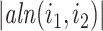 is the total lengths of the split-alignment intervals of aligning the sequences of
is the total lengths of the split-alignment intervals of aligning the sequences of 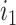 and
and 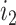 isoforms and
isoforms and 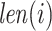 is the length of the sequence of isoform
is the length of the sequence of isoform 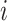 . We then extract all the (connected) components of the graph
. We then extract all the (connected) components of the graph 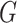 and classify them into three categories: (i) a mixed clique contains at least one predicted isoform (PI) and at least one ground truth isoform (GTI) and an edge between all pairs of its vertices; (ii) a non-mixed component contains only PIs or only GTIs and may or may not be a clique; and (iii) a mixed ambiguous component contains at least one PI and at least one GTI, with some pairs of vertices having no edge connecting them. For a given alignment score threshold, mixed cliques (category i) represent sets of pairwise similar PIs and GTIs that we interpret as true positives, while PIs and GTIs in non-mixed components (category ii) can be interpreted as false positive and false negative, respectively. The isoforms in mixed ambiguous components (category iii) cannot be unambiguously labeled as true positive, false positive or false negative. Thus we investigate the structure of each component of category (iii) as follows: We label (a) each PI connected to all GTIs as probably true positive; (b) each GTI connected to all PIs as probably true positive; (c) each PI connected to some GTIs as probably false positive; and (d) each GTI connected to some PIs as probably false negative. Note that to parallelize the computation of the split-alignment score. we use GNU Parallel (31).
and classify them into three categories: (i) a mixed clique contains at least one predicted isoform (PI) and at least one ground truth isoform (GTI) and an edge between all pairs of its vertices; (ii) a non-mixed component contains only PIs or only GTIs and may or may not be a clique; and (iii) a mixed ambiguous component contains at least one PI and at least one GTI, with some pairs of vertices having no edge connecting them. For a given alignment score threshold, mixed cliques (category i) represent sets of pairwise similar PIs and GTIs that we interpret as true positives, while PIs and GTIs in non-mixed components (category ii) can be interpreted as false positive and false negative, respectively. The isoforms in mixed ambiguous components (category iii) cannot be unambiguously labeled as true positive, false positive or false negative. Thus we investigate the structure of each component of category (iii) as follows: We label (a) each PI connected to all GTIs as probably true positive; (b) each GTI connected to all PIs as probably true positive; (c) each PI connected to some GTIs as probably false positive; and (d) each GTI connected to some PIs as probably false negative. Note that to parallelize the computation of the split-alignment score. we use GNU Parallel (31).
In order to assess the impact of sequencing errors, we also built an additional graph for the ideal case of read mapping and clustering. In this graph, we aligned the reads simulated from each isoform to that isoform’s sequence using Minimap2 splice alignment mode. We then generated a position by position consensus sequence of these alignments. Inaccuracies in this graph reflect only the sequencing error model and local alignment mistakes, but not mapping and clustering problems. Figure 6 plots various statistics on the structure of the resulting graphs for various threshold 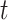 values defining edges of the graphs.
values defining edges of the graphs.
Figure 6.

Graph-based accuracy results of the simulated dataset experiment. The first and second rows plot the count/percentages of isoforms belonging to mixed clique components (category i) and non-mixed components (category ii). The last two rows describe mixed ambiguous components (category ii). The left column presents the absolute counts and percentages for GTIs in each type of component for all graphs. The middle and right columns present the absolute and percentage for PIs in each type of component for all graphs.
Simulated dataset results
The results of using the first approach for assessing the accuracy of the isoform detection tools are summarized in Table 1 and Figure 7. We observe that FLAIR (which is supplied with the full set of annotation isoforms) and Freddie achieve close F1 scores (64% and 61%, respectively), with Freddie having slightly better recall and FLAIR having better precision. Both Freddie and FLAIR achieve a much higher F1 accuracy score than StringTie2 which has an F1 score of 36%. Figure 7 shows the UpSet (32) plot of the intersections of the isoforms of the different tools and the ground truth.
Table 1.
Accuracy statistics for the simulated human dataset using the exon intervals to identify equivalent isoforms
| Tool | F1 score | Precision | Recall | True iso. | False iso. |
|---|---|---|---|---|---|
| Freddie | 61.03% | 61.89% | 60.19% | 16 778 | 10 332 |
| StringTie2 | 35.83% | 34.57% | 37.18% | 10 366 | 19 618 |
| FLAIR (100%) | 64.42% | 69.79% | 59.79% | 16 669 | 7215 |
Figure 7.
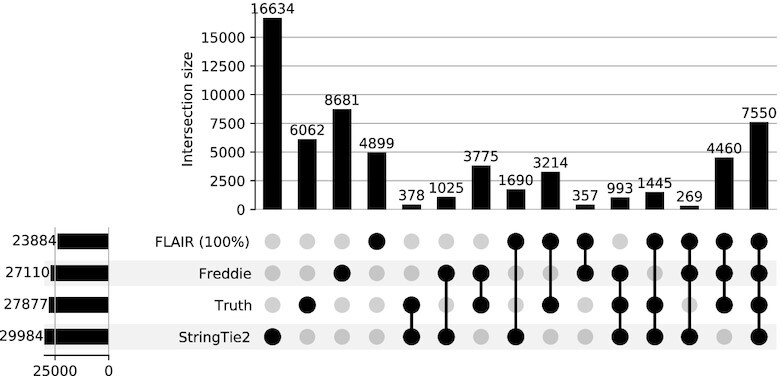
Simulated human dataset. UpSet plot showing the intersection sizes between the sets of isoforms predicted by different tools or present in the simulated ground truth using their exon intervals as a condition of equivalence. Note that UpSet plots present the same information as Venn diagrams but have the advantage of representing each intersection as a bar with its height corresponding to the intersection’s size. The members of each intersection are indicated by the solid black circles under each bar. The total size of each tool’s predictions (i.e. the number of isoforms predicted by each tool) are shown on the bar graph on the left.
While the assessment of the graphs (one for each tool output and one for isoform alignment baseline) is fairly consistent across all threshold 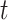 values, we use
values, we use 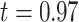 to quantify our findings since this value presents a clear inflection point in the plots. Freddie, FLAIR and StringTie2 predicted 27 243, 24 605 and 30 306 isoforms, respectively, versus a total of 28 025 GTIs. The analysis of Figure 6 shows that, overall, Freddie and FLAIR perform similarly, in terms of accuracy of the PIs compared with the GTIs; both tools outperform StringTie2 in terms of true positive and false negative, in terms of both absolute count and proportion. We note that FLAIR’s results are obtained with the exact annotation for all the GTIs in the dataset, while StringTie2 and Freddie are run without any annotations. If we run FLAIR with a partial annotation dataset, we notice a precipitous fall in true positive GTIs (see FLAIR 50% in Figure 6 and Supplementary Figure S4).
to quantify our findings since this value presents a clear inflection point in the plots. Freddie, FLAIR and StringTie2 predicted 27 243, 24 605 and 30 306 isoforms, respectively, versus a total of 28 025 GTIs. The analysis of Figure 6 shows that, overall, Freddie and FLAIR perform similarly, in terms of accuracy of the PIs compared with the GTIs; both tools outperform StringTie2 in terms of true positive and false negative, in terms of both absolute count and proportion. We note that FLAIR’s results are obtained with the exact annotation for all the GTIs in the dataset, while StringTie2 and Freddie are run without any annotations. If we run FLAIR with a partial annotation dataset, we notice a precipitous fall in true positive GTIs (see FLAIR 50% in Figure 6 and Supplementary Figure S4).
In terms of computational resources, StringTie2 had by far the smallest computational footprint, finishing in under 3 min and with <30 MB of RAM. Freddie had a memory footprint of ∼1.4GB while FLAIR memory use maxed at 9.1 GB. FLAIR outperformed Freddie in CPU time use, occupying the CPU for 77 min compared with Freddie’s 125 min. Note that roughly two-thirds of Freddie’s CPU time is spent in the Gurobi ILP solver. For the full details of the computational resource use of the different tools, including the different stages of Freddie, refer to Supplementary Tables S1 and S2.
Assessing the resolution of exon boundaries
The alignment-based similarity provides us with a good assessment of the structure of the predicted isoforms and their ground truth counterparts. However, it is also important to understand the accuracy of the exact locations of exon boundaries generated by the different tools. Therefore, we extracted the set genomic locations of the exon boundaries of the GTIs and of the predicted isoforms of each tool. For each tool, we counted for each exon boundary locus the number of ground truth exon boundaries it has as a neighbor in a neighborhood of ±10 bp. Figure 8 illustrates a histogram of these counts for the tested tools. As expected, we observe that FLAIR has the highest exon boundary resolution, with its histogram tightly concentrated at position zero. Freddie and StringTie2 have similar distributions. Generally speaking, all tools have histograms tightly concentrated around the zero position, indicating high base-level resolution across all tools.
Figure 8.
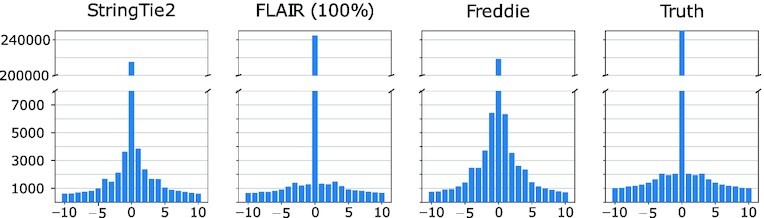
Histogram of the number of ground truth exon boundaries in the neighborhood of the predicted exon boundaries for different tools. As a baseline, we also show the histogram for the ground truth plotted against itself. For the baseline, bars not on the zero position are explained by the fact that some different annotation exons have very close starting/ending positions.
Real data experiments
Data generation
We generated a real mRNA transcriptomic dataset using the Oxford Nanopore Technologies PromethION cDNA sequencing platform (chemistry kit SQK-PBK004 and R9.4.1 flow cell). We also generated a matched RNA-seq paired SR dataset using Illumnia’s HiSeq 2x150 sequencing platform. The mRNA material for both datasets was extracted from the same LNCaP prostate cancer cell line This cell line is widely used as a first step model for stages of prostate cancer. In these experiments, we focused on a randomly selected set of 294 genes with various degrees of expected isoform complexity (according to the ENSEMBL database). The selection process is detailed on a Jupyter notebook in Section S3 of the Supplementary Data.
Short-read validation of detected isoform splice junctions
Since this is a real dataset, it is not appropriate to use the annotations as an absolute ground truth. However, it is still informative to compare the isoform calls by different tools against each other and against the annotation database. To do that, we used our approach of matching isoforms using their structures. Note that here, we use the isoform intron intervals instead of their exons to compare the different isoforms since we expect a greater degree of early sequencing termination in the real dataset compared with the simulated dataset. We also used the matching SR RNA-seq data as a means of validating the detected isoforms. Our assumption here is that if a splice junction is detected by one of the three tools using the LR dataset, it should also be detectable using the SR dataset. Thus, this SR validation offers a means to detect false-positive isoforms if they include invalidated splice junctions. We used STAR RNA-seq aligner to detect SR-supported splice junctions (33). In the following results, we assume that any LR-detected splice junction is valid if it is detected by STAR using the matched SR dataset, allowing for up to ±10 bp shifts. Then, we also assume that an LR-detected isoform is valid if all of its splice junctions are valid. Figure 9 shows, using an UpSet plot, the intersections of the detected isoforms of the three tools and the ENSEMBL database on the real LNCaP dataset and the number of validated isoforms of each intersection. Supplementary Figure S6 shows the same intersections but at the level of introns instead of isoforms.
Figure 9.
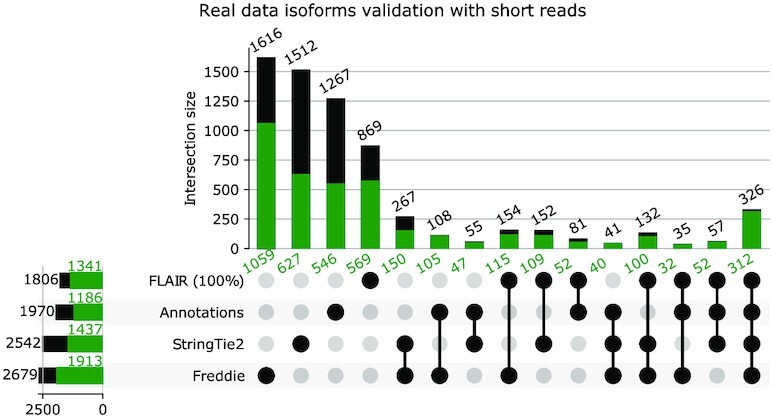
LNCaP real dataset. UpSet plot showing the intersection sizes between the sets of isoforms predicted by different tools or present in the ENSEMBL 97 annotation database using their intron intervals as a condition of equivalence. For each intersection column, the number of isoforms that have all their splice junctions validated using the STAR and the SR dataset is indicated in green.
Freddie results in the second highest validation rate among all the tested tools at 71.41% of its detected isoforms having all their splice junctions validated by the SRs. FLAIR and StringTie2 had validation rates of 74.25% and 56.53%, respectively. These results demonstrate the limitations of fully relying on the annotation dataset and Freddie’s ability to detect novel isoforms that are supported by SR alignment evidence since both Freddie and StringTie2 are able to detect isoforms not present in the ENSEBML database.
Biological validation of novel splice junctions by RT–PCR
In addition to the SR in silico validation, we have also performed reverse transcription–polymerase chain reaction (RT–PCR) to biologically validate novel splice variants (i.e. not present in the annotation) detected by Freddie and/or StringTie2 using the LNCaP prostate cancer cell line. To select candidates for RT–PCR validation, we focused on novel splice junctions for which the computational methods show strong support. Specifically, we identified novel junctions that are both supported by the SRs and that have the highest expression within the genes of their respective isoforms. We aim to design PCR primers that can cover both known and novel exon junctions when possible. In cases where the target sequences have high GC or AT ratios, dinucleotide repeats or intrasequence homology, we had to compromise by designing primers to detect novel exon junctions only. As shown in Figure 10, we validated both known and novel exons simultaneously in the ING3, KRT19, GSPT2 and PPFIA4 transcripts, and validated novel exon junctions only in the ORM2, FRG1, LYAR, TDRD3 and RABL2A transcripts. Additionally, we attempted to validate a novel exon detected by StringTie2 but not by Freddie in the EOGT gene. As shown in Figure 10, these 10 RT–PCR experiments confirmed the splice junctions detected by Freddie only or detected by Freddie and StringTie2 and did not confirm the splice junction detected only by StringTie2. These results confirmed that the novel splice variants identified by Freddie are expressed in LNCaP cells and that Freddie detects novel splice junctions that are missed by StringTie2.
Figure 10.
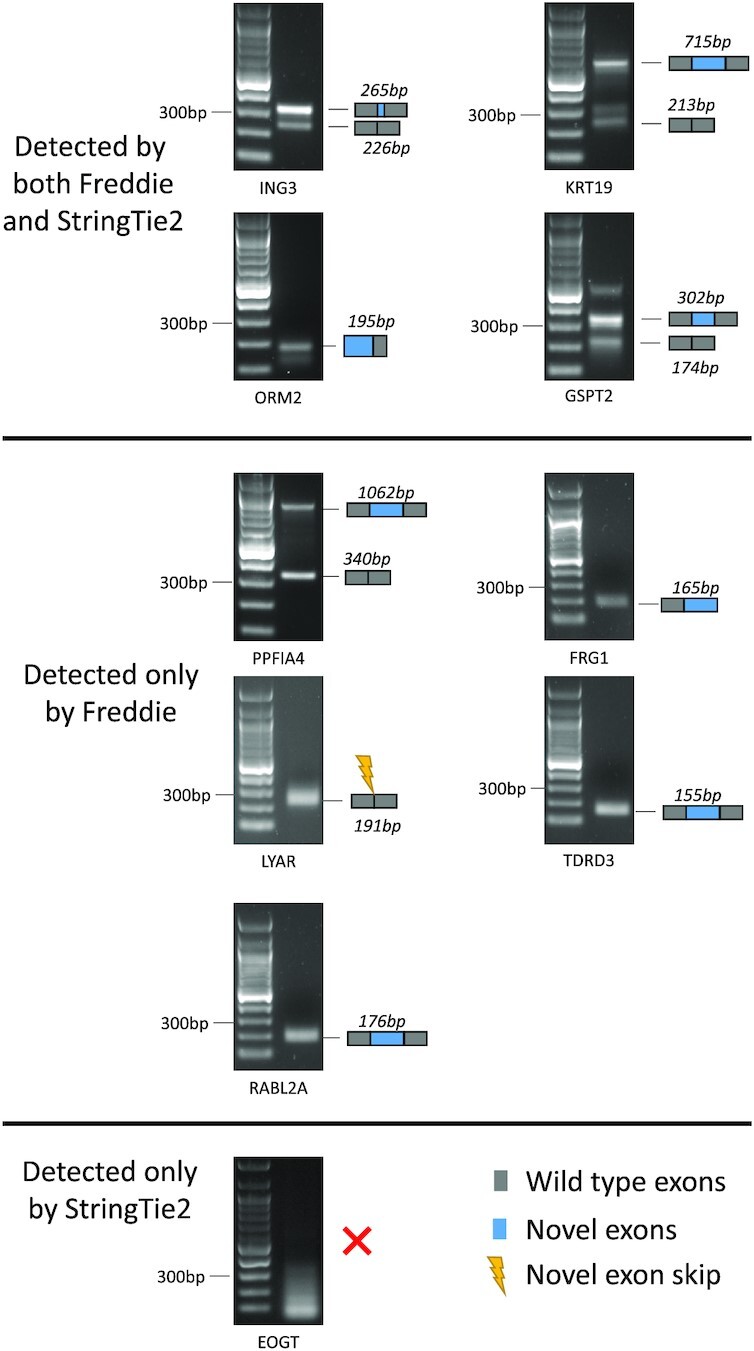
RT–PCR validation confirmed the selected novel isoforms with novel splice junctions as detected by StringTie2 or Freddie from the real LNCaP dataset. PCR products were separated by electrophoresis of DNA agarose gels along with DNA ladders. Cartoons of the novel exons and their predicted PCR product sizes are shown on the right side of each gel image.
DISCUSSION
Freddie is a novel AS isoform detection tool that does not rely on isoform annotation databases. Freddie is designed to address the characteristic sequencing errors of LRs, especially in the absence of isoform annotations. Using simulated data, we demonstrate that Freddie achieves accuracy higher than or on par with existing detection tools even when they are supplied with isoform annotations. On an LR real dataset with a matched SR dataset, we used the SRs to demonstrate Freddie’s ability to detect isoforms in a noisy real experimental set-up. Freddie shows higher rates of validated isoforms than the other annotation-free tool, StringTie2, and it performs on a par with FLAIR when FLAIR is supplied with the full annotation dataset. More importantly, these Freddie-detected isoforms with novel splice junctions can be biologically validated by RT–PCR analysis using total RNA samples extracted from the LNCaP cell line.
For future directions, we plan to further analyze the computational complexity of the MErCI problem used in Freddie. We hope to identify a tight bound on the complexity of solving MErCI and shed light on possible ways to speed it up. This includes exploring ways to intelligently subsample its input or approximating its solution. We also aim to create variants of MErCI to address the problem of detection of other transcriptomic targets, besides AS isoforms, such as circular RNA and gene fusions.
DATA AVAILABILITY
Freddie is open source and is available at https://github.com/vpc-ccg/freddie/. The benchmarking branch includes the scripts used to generate the simulation and real data results. The simulation pipeline is also open source and is available at https://github.com/vpc-ccg/LTR-sim. Both repositories are deposited to Figshare at https://doi.org/10.6084/m9.figshare.21441027. The short- and long-read real datasets are deposited in the SRA database under the accession number PRJNA763233.
Supplementary Material
Contributor Information
Baraa Orabi, Department of Computer Science, the University of British Columbia, Vancouver, BC, Canada.
Ning Xie, Vancouver Prostate Centre, Vancouver, BC, Canada.
Brian McConeghy, Vancouver Prostate Centre, Vancouver, BC, Canada.
Xuesen Dong, Vancouver Prostate Centre, Vancouver, BC, Canada; Department of Urologic Sciences, the University of British Columbia, Vancouver, BC, Canada.
Cedric Chauve, Department of Mathematics, Simon Fraser University, Burnaby, BC, Canada.
Faraz Hach, Department of Computer Science, the University of British Columbia, Vancouver, BC, Canada; Vancouver Prostate Centre, Vancouver, BC, Canada; Department of Urologic Sciences, the University of British Columbia, Vancouver, BC, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This research is funded in part by the National Science and Engineering Council of Canada (NSERC) Discovery Grants [RGPIN-05952 to F.H. and RGPIN-03986 to C.C.]; the Michael Smith Foundation for Health Research (MSFHR) Scholar Award [SCH-2020-0370 to F.H.]; the Canadian Institutes of Health Research (CIHR) grant [PJT-156150 and PJT-178063 to X.D.]; the Department of Defense (DoD) Prostate Cancer Research Program Idea Development Award [PC190327 to X.D.]; and the NSERC Alexander Graham Bell Canada Graduate Scholarship-Doctoral (CGS D) to B.O. Funding for open access charge: NSERC.
Conflict of interest statement. None declared.
REFERENCES
- 1. Deorowicz S., Gudys A., Dlugosz M., Kokot M., Danek A.. Kmer-db: instant evolutionary distance estimation. Bioinformatics. 2019; 35:133–136. [DOI] [PubMed] [Google Scholar]
- 2. International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 3. Nilsen T.W., Graveley B.R.. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010; 463:457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hughes T.A. Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006; 22:119–122. [DOI] [PubMed] [Google Scholar]
- 5. Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B.. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oltean S., Bates D.O.. Hallmarks of alternative splicing in cancer. Oncogene. 2014; 33:5311–5318. [DOI] [PubMed] [Google Scholar]
- 7. Lee S. C.-W., Abdel-Wahab O.. Therapeutic targeting of splicing in cancer. Nat. Med. 2016; 22:976–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Escobar-Hoyos L., Knorr K., Abdel-Wahab O.. Aberrant RNA splicing in cancer. Annu. Rev. Cancer Biol. 2019; 3:167–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goodwin S., McPherson J.D., McCombie W.R.. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016; 17:333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G.et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., Van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin Y.Y., Dao P., Hach F., Bakhshi M., Mo F., Lapuk A., Collins C., Sahinalp S.C.. CLIIQ: accurate comparative detection and quantification of expressed isoforms in a population. Lect. Notes Comput. Sci. 2012; 7534:178–189. [Google Scholar]
- 14. Li W., Feng J., Jiang T.. IsoLasso: a LASSO regression approach to RNA-Seq based transcriptome assembly. J. Comput. Biol. 2011; 18:1693–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amarasinghe S.L., Su S., Dong X., Zappia L., Ritchie M.E., Gouil Q.. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020; 21:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Dijk E.L., Jaszczyszyn Y., Naquin D., Thermes C.. The third revolution in sequencing technology. Trends Genet. 2018; 34:666–681. [DOI] [PubMed] [Google Scholar]
- 17. Kono N., Arakawa K.. Nanopore sequencing: review of potential applications in functional genomics. Dev. Growth Differ. 2019; 61:316–326. [DOI] [PubMed] [Google Scholar]
- 18. Sessegolo C., Cruaud C., Da Silva C., Cologne A., Dubarry M., Derrien T., Lacroix V., Aury J.-M.. Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci. Rep. 2019; 9:14908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang A.D., Soulette C.M., van Baren M.J., Hart K., Hrabeta-Robinson E., Wu C.J., Brooks A.N.. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020; 11:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kovaka S., Zimin A.V., Pertea G.M., Razaghi R., Salzberg S.L., Pertea M.. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019; 20:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morillon A., Gautheret D.. Bridging the gap between reference and real transcriptomes. Genome Biol. 2019; 20:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Workman R.E., Tang A.D., Tang P.S., Jain M., Tyson J.R., Razaghi R., Zuzarte P.C., Gilpatrick T., Payne A., Quick J.et al.. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods. 2019; 16:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de la Rubia I., Srivastava A., Xue W., Indi J.A., Carbonell-Sala S., Lagarde J., Albà M., Eyras E.. RATTLE: reference-free reconstruction and quantification of transcriptomes from Nanopore sequencing. Genome Biol. 2022; 23:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V.et al.. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 25. Lippert R., Schwartz R., Lancia G., Istrail S.. Algorithmic strategies for the single nucleotide polymorphism haplotype assembly problem. Brief. Bioinform. 2002; 3:23–31. [DOI] [PubMed] [Google Scholar]
- 26. Mölder F., Jablonski K.P., Letcher B., Hall M.B., Tomkins-Tinch C.H., Sochat V., Forster J., Lee S., Twardziok S.O., Kanitz A.et al.. Sustainable data analysis with Snakemake. F1000Research. 2021; 10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M., Bennett R., Bhai J., Billis K., Boddu S.et al.. Ensembl 2019. Nucleic Acids Res. 2019; 47:D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wick R. Badread: simulation of error-prone long reads. J. Open Source Software. 2019; 4:1316. [Google Scholar]
- 30. Sahlin K., Medvedev P.. Author Correction: Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis. Nat. Commun. 2021; 12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tange O. GNU Parallel - The Command-Line Power Tool. The USENIX Magazine. 2018; 36:42–47. [Google Scholar]
- 32. Lex A., Gehlenborg N., Strobelt H., Vuillemot R., Pfister H.. UpSet: visualization of intersecting sets. IEEE Trans. Visual. Comput. Graph. 2014; 20:1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Freddie is open source and is available at https://github.com/vpc-ccg/freddie/. The benchmarking branch includes the scripts used to generate the simulation and real data results. The simulation pipeline is also open source and is available at https://github.com/vpc-ccg/LTR-sim. Both repositories are deposited to Figshare at https://doi.org/10.6084/m9.figshare.21441027. The short- and long-read real datasets are deposited in the SRA database under the accession number PRJNA763233.