


Abstract
Individual cells are basic units of life. Despite extensive efforts to characterize the cellular heterogeneity of different organisms, cross-species comparisons of landscape dynamics have not been achieved. Here, we applied single-cell RNA sequencing (scRNA-seq) to map organism-level cell landscapes at multiple life stages for mice, zebrafish and Drosophila. By integrating the comprehensive dataset of > 2.6 million single cells, we constructed a cross-species cell landscape and identified signatures and common pathways that changed throughout the life span. We identified structural inflammation and mitochondrial dysfunction as the most common hallmarks of organism aging, and found that pharmacological activation of mitochondrial metabolism alleviated aging phenotypes in mice. The cross-species cell landscape with other published datasets were stored in an integrated online portal—Cell Landscape. Our work provides a valuable resource for studying lineage development, maturation and aging.
Graphical Abstract
Graphical Abstract.
Construction of a cross-species cell landscape at the single-cell level and revealing signatures and common pathways that changed throughout the life cycle.
INTRODUCTION
How many cell types are there in nature? How do they change during the life cycle? These are two fundamental questions that researchers have been trying to understand. The rapid development of high-throughput single-cell RNA sequencing (scRNA-seq) offers unprecedented opportunities to reshape our knowledge about cell types with a universal measurement system (1–5). As the throughput of scRNA-seq increases dramatically (6–11), it is now possible to construct single-cell transcriptomic atlases at the organism level. For example, cell atlases for vertebrate systems covering fetal and adult periods have been generated, such as Xenopus (12), zebrafish embryos (13–15), Human Cell Landscape (HCL) (16), Tabula Sapiens (17), Mouse Cell Atlas (MCA) (8), Mouse Cell Differentiation Atlas (MCDA) (18), Tabula Muris (19), Tabula Muris Senis (20) and Zebrafish Cell Landscape (ZCL) (21). Several invertebrate cell atlases are also available, including for Caenorhaditis elegans (22), Drosophila embryo (11), Nematostella (23), sea squirt (24), fruitfly (25) and earthworm (26). These studies have significantly enriched our knowledge about cellular hierarchy in different species.
In addition to static cell atlases, the dynamics of cellular states from development to aging will add another dimension to our understanding of life. Extensive efforts have been made to study the molecular events during embryogenesis at the single-cell level, exploring regulatory programs that govern the trajectories of cell fate decisions (9,11–14,22,27–31). In addition, researchers have used rodents to understand cellular and molecular mechanisms of aging (20,32–34). These studies revealed common patterns in aging tissues, including significant immune cell infiltration and activation (20), increased transcriptional heterogeneity and unrestricted identity of aging cells (34–36). However, most of these analyses are limited to specialized systems and thus lack a thorough comparison of different species and tissues. The universality of scRNA-seq enables this kind of comprehensive analysis to uncover the general rules behind progression of life at both the cellular and organismal levels.
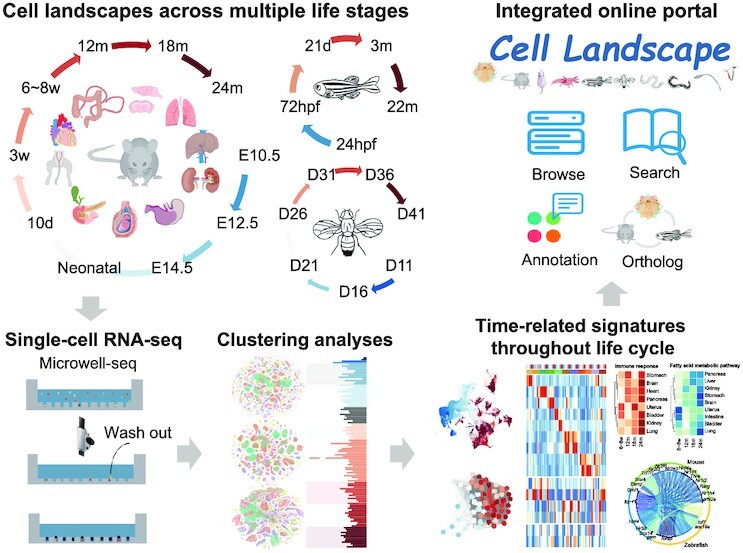
In this study, we used Microwell-seq technology (8) to profile over 2.6 million single cells from mice, zebrafish and Drosophila at different life stages (Figure 1A and B; Supplementary Table S1). Using these comprehensive datasets, we constructed a cross-species cell landscape and investigated common pathways that change throughout the life span. We found the tendency of cell types and genes to change with development and age, especially immune cells, and identified dynamics of gene expression and time-dependent trends in transcription factor (TF) regulation. Moreover, we revealed structural inflammation and mitochondrial dysfunction as the most common hallmarks of organism aging, and discovered that structural cells exhibit obvious inflammatory responses during aging. We also found that pharmacological activation of mitochondrial metabolism alleviated aging phenotypes in mice. Combined with other published datasets, we collected over 2.6 million single cells from 15 species and constructed an integrated online portal, Cell Landscape (http://bis.zju.edu.cn/cellatlas/), providing visualization, browsing, searching, annotation and cross-species analysis functions.
Figure 1.

The cross-species cell landscape was constructed using Microwell-seq. (A) Overview of the experimental and bioinformatics workflow. (B) Bar plot showing the number of analyzed cells per stage by Microwell-seq from each species. (C) t-SNE visualization of 1 130 794 single cells from all tissues across all stages of mice, colored by cluster identity. (D) t-SNE visualization of the MCDAA, colored by stage. (E) t-SNE visualization of 1 082 680 single cells from all tissues across all stages of zebrafish, colored by cluster identity. (F) t-SNE visualization of 439 201 single cells from all tissues across all stages of Drosophila, colored by cluster identity. (G) Heatmap showing the correspondence between cell types in DCL gut (this study, row) and FCA gut (tissue-wide study from Li et al., 2022, column). Blue refers to a mean area under the receiver operating characterstic curve > 0.9.
MATERIALS AND METHODS
Ethics statement
All experiments performed in this study were approved by the Animal Ethics Committee of Zhejiang University (approval number: ZJU20210067). All experiments conformed to the relevant regulatory standards at Zhejiang University Laboratory Animal Center.
Biological samples
Wild-type C57BL/6J mice were ordered from Shanghai SLAC Laboratory Animal and GemPharmatech Co., Ltd. All mice were housed at Zhejiang University Laboratory Animal Center in a specific pathogen-free (SPF) facility with individually ventilated cages. The room has controlled temperature (20–22°C), humidity (30–70%) and light (12 h light–dark cycle). Mice were provided ad libitum access to a regular rodent chow diet. All the tissues of newly collected mice samples were collected from female mice, while testis and prostate tissues were collected from male mice.
Wild-type Danio rerio (zebrafish) strain AB were raised and maintained in standard zebrafish units at Core Facilities, Zhejiang University School of Medicine.
Wild-type Drosophila melanogaster (Drosophila) were cultured under laboratory conditions at 25 ± 1°C with 70 ± 5% relative humidity under a 14:10 h light/dark photoperiod.
scRNA-seq cell preparation and sequencing
We used the Microwell-seq process to obtain single-cell RNA data for each species, while two sizes of beads (20 and 28 μm) and microwells (25 and 32 μm) were used to adapt to the cell sizes of different species. Two bead sequences were used, beads 2.0 for mice experiments and beads 3.0 for zebrafish and Drosophila experiments. The Microwell-seq process included cell and bead collection, reverse transcription, exonuclease I treatment, second-strand synthesis and cDNA amplification to generate barcoded single-cell libraries. Second-strand syntheses were introduced to zebrafish and Drosophila cells. Indexed libraries were constructed according to the modified manufacturer's protocol (Vazyme). Concentrations were measured by a Qubit 3.0 fluorometer with an Equalbit dsDNA HS Assay Kit (Vazyme). Libraries were sequenced on an Illumina HiSeq or MGI DNBSEQ-T7 sequencer with the following sequencing strategy: 150 bp read length for read 1 and 150 bp read length for read 2. Detailed methods are described in Supplementary Methods.
Immunofluorescence staining
Mouse lung and kidney tissues were fixed in 4% paraformaldehyde at 4°C overnight. Then, the tissues were cut into 15 μm thick frozen sections and mounted on pre-cleaned slides. The sections were washed three times with phosphate-buffered saline (PBS) and blocked with 5% fetal bovine serum (FBS) in PBS for 1 h at room temperature. Primary antibodies diluted in blocking solution (5% FBS in PBS) were added to cover the sections. The slides were placed in a wet box and incubated at 4°C overnight. Relevant AlexaFluor488/594-conjugated secondary antibodies (1:1,000, Invitrogen) were used for labelling. The slides were then washed three times with blocking solution and stained with 4′,6-diamidino-2-phenylindole. Glass coverslips were then attached to the slides using mounting medium. Immunofluorescence images were obtained using confocal microscopy.
Pioglitazone treatment
Seven 24-month-old female mice received pioglitazone (PGZ) intraperitoneal (i.p.) injection, and five 24-month-old female mice received vehicle control i.p. injection. For treatment, mice were injected i.p. with PGZ at a dose of 15 mg/kg of mouse weight/day for 3 weeks. Control and treated mice (n = 5) were tested for grip strength and blood lipids. Insulin sensitivity was measured by glucose tolerance test (GTT) and insulin tolerance test (ITT). For the GTT, overnight-fasted mice were given i.p. glucose (2 mg/g body weight). For the ITT, 5 h fasted mice were given 0.75 U insulin/kg body weight by i.p. injection (Humulin). Blood glucose was determined with a OneTouch glucometer (Lifescan). Forelimb grip strength was determined using a Bioseb grip strength meter (Harvard Apparatus). Lean mass composition was measured using dual-energy X-ray absorptiometry (DEXA) on a Lunar PIXImus (GE Lunar Corp). Plasma triglycerides and cholesterol were measured using a Cobas Clinical Chemistry Analyzer (Roche).
Data processing
Microwell-seq datasets were processed using the protocols described in our previously published works (8,16). Reads from three species were aligned to the Mus musculus GRCm38 genome, D.rerio GRCz11 genome and D.melanogaster BDGP6.28 genome using STAR (37). The digital gene expression (DGE) data matrices were obtained using the modified Dropseq tools (https://github.com/ggjlab/mca_data_analysis/tree/master/preprocessing/Drop-seq_tools-1.12/), and the corresponding protocol is available at http://mccarrolllab.org/dropseq/. The DGE data containing the top 10 000 cells sorted by the total number of transcriptions were obtained after this pre-process. For quality control, we filtered out cells with the detection of < 500 transcripts and 200 genes. Cells with a high proportion of transcript counts derived from mitochondria-encoded genes were also excluded. We corrected the RNA contamination using the same methods described previously (16). Then we used the R package DoubletFinder (38) to detect the potential doublets from each individual library. Approximately 5% of cells were labeled doublets and were removed. After obtaining the processed DGE matrix, we used Seurat (39) and Scanpy (40) for dimension reduction, clustering and differential gene expression analysis. Detailed methods are described in the Supplementary Methods.
Inferring the gene expression trajectory and gene regulatory network in the life cycle
We used the LOESS regression model to fit the gene expression trajectory for the three species. Hierarchical clustering was used to identify different gene expression trajectory clusters in the life cycle. For each cluster, we identified the most significant gene function by g:profiler (41) (https://biit.cs.ut.ee/gprofiler/gost). To infer the gene regulatory network in the three species, pySCENIC (42) was applied to infer the gene regulatory network. Fuzzy c-means clustering was performed on the TF-by-lineage matrix to identify lineage-specific and common TFs. Then we ran ‘map_genes.sh’ in SAMap (43) to find the homologous TFs of the same lineage across species. Detailed methods are described in the Supplementary Methods.
Identifying aging-associated genes, pathways and TFs
For mice, we collected single cells from stages of adult (6–8 weeks), 12, 18 and 24 month stages in most tissues, so this provides a good opportunity to study the dynamics of gene expression during aging. We used the MAST package (44) to identify aging-associated genes in each tissue. For each gene, the ‘zlm’ framework was applied to fit a Hurdle model for stages. Only genes with a false discovery rate (FDR) < 1e-10 were identified as aging-associated genes. We used similar methods to identify aging-associated genes in Drosophila, only genes with an FDR < 1e-3 were selected as aging-associated genes. Since the datasets for rats and zebrafish have fewer stage points, the ‘FindMarkers’ function in Seurat was applied (young versus old in rat; 3 months versus 22 months in zebrafish), only genes with a adjusted P-value < 0.05 and absolute (LogFC) > 0.1 were identified as aging-associated genes. For C.elegans, we used DNA microarray data (45) to identify genes showing significant change during aging with analysis of variance (ANOVA; P-value < 0.001). To identify aging-associated pathways, up-regulated and down-regulated genes for five species (changed in at least around half of the tissues or cell lineages), gene functional enrichment analysis was performed by the online web service g:Profiler. Then we calculated the Spearman correlation coefficient between the TF activity scores from SCENIC and the aging-associated gene set activity scores from AUCell (42) in each tissue or cell lineage, TFs with coefficient > 0.3 were identified as aging-associated TFs. Detailed methods are described in the Supplementary Methods.
Cell Landscape construction
Cell Landscape (http://bis.zju.edu.cn/cellatlas/) uses the bootstrap framework to improve overall adaptability and interactivity, with PHP, R language and MySQL for back-end development. The index of Cell Landscape is the collection of subsites for each species, each providing a global view and retrieval of the species cell landscape. Interactive t-SNE maps are provided for distribution of different clusters, specific markers for each cluster are listed in a data table, and search describes the expression of a given gene in different clusters from any selected tissue. The main functions of Cell Landscape are divided into three parts: browse, search and annotation. The Browse part presents the statistics and lists of single-cell studies used on the website. The Search part contains a direct search and ortholog search; direct search describes the expression profiles of a given gene in different clusters, and ortholog search describes the expression profiles of the given gene and its orthologs in human, mice and zebrafish. The Annotation part, including scMCA, scZCL, scDCL and their cross-species analysis function, provides the single-cell correlation analysis between the given DGE data and the reference file from the species database. The result is returned in JSON format and displayed as an interactive heatmap.
RESULTS
Constructing single-cell transcriptomic atlases of three species
Previous studies have shown that Microwell-seq is a cost-effective scRNA-seq method that is compatible with a wide range of tissue types (8,16,21,46,47). Using Microwell-seq, we aimed to construct a multispecies scRNA-seq cell landscape across multiple life stages in mice, zebrafish and Drosophila (Figure 1A). For mice, the data were obtained from > 10 major tissues with 2–3 biological replicates at 10 stages throughout the life span, including three newly collected stages (12, 18 and 24 months) (Figure 1B; Supplementary Table S1). The newly collected datasets were integrated with our previous works, MCA (8) and MCDA (18), to generate a time-series Mouse Cell Development and Ageing Atlas (MCDAA), also known as MCA v3.0, available at http://bis.zju.edu.cn/MCA/. In total, MCDAA collected more than 1.1 million single cells from embryonic day 10.5 (E10.5, 26 551 cells), embryonic day 12.5 (E12.5, 73 685 cells), embryonic day 14.5 (E14.5, 88 357 cells), neonatal (97 153 cells), 10 days (10d, 116 349 cells), 3 weeks (3w, 68 785 cells), 6–8 weeks (6–8w, 201 507 cells), 12 months (12m, 93 593 cells), 18 months (18m, 121 257 cells) and 24 months (24m, 206 758 cells), constituting the largest multi-period mouse single-cell landscape covering a wide range of life stages. The cells of the three newly collected stages accounted for 37.3% of the total cells in MCDAA. For zebrafish and Drosophila, we attempted to perform whole-body single-cell sequencing of the organism without tissue dissection. The unbiased cell landscape data with minimal batch effects from different tissues have been successfully generated by such experiments (16,26). Our analyses contained nearly 1.08 million single cells from five life stages of isolated zebrafish, i.e. pharyngula stage [24 h post-fertilization (24hpf), 44 932 cells], larval stage (72hpf, 159 128 cells; and 21 days, 121 954 cells) and adult (3m, 431 168 cells; and 22m, 330 924 cells) (Figure 1B; Supplementary Table S1), as well as from seven life stages of Drosophila (439 201 cells), from young [11 days (D11)] to senescence (D41) (Figure 1B; Supplementary Table S1). Among them, the two newly collected stages (21 d and 22 m) accounted for 41.6% of the cell numbers in zebrafish, while the five newly collected stages of Drosophila (D21 to D41) accounted for 37.0% of the Drosophila cell numbers. The newly collected datasets of zebrafish and Drosophila were integrated with our previous works (21,26) to construct the Zebrafish Cell Development Landscape (ZCDL) and Drosophila Cell Landscape (DCL), available at http://bis.zju.edu.cn/ZCL/ and http://bis.zju.edu.cn/DCL/, respectively. The scRNA-seq experiments and data analyses were performed following our previously published procedures (8,16). All information about data quality, average UMI and gene number is summarized in Supplementary Table S1.
Globally, the mouse cell landscape was categorized into 177 cell types (Figure 1C and D; Supplementary Figure S1A–C; Table S1). Each cell type was annotated according to the expression level of canonical cell type-specific markers (marked in Supplementary Table S1). In total, the 177 cell types were divided into 11 major cell lineages, namely endothelial, epithelial, erythroid, germ, immune, muscle, neuron, proliferating, secretory, stromal and other cells (Supplementary Figure S1C). Specifically, with high expression of P2ry12 and P2ry13, cluster 30 (C30) was identified as microglia. C49 was defined as astrocyte according to the remarkable marker slc1a2, whereas C70 and C150 were identified as oligodendrocyte using Plp1. Epithelial cells were composed of multiple clusters, including Alveolar Epithelial Type 1 (AT1) cell (Ager), Alveolar Epithelial Type 2 (AT2) (Sftpc), club cell (Scgb3a2), chief cell (Pga5 and Pgc), pit cell (Tff1 and Gkn2), enterocyte (Reg3b and Reg3g), proximal tubule (Aldob and Ass1), ascending loop of Henle (Umod and Slc12a1) and distal convoluted tubule (Calb1 and Pgam2). Immune cells were composed of B cell (Cd79a and Cd19), T cell (Cd3d), macrophage (Csf1r and Cd68), mast cell (Cpa3), neutrophil (Cd177) and plasma cell (Jchain and Ig genes). We also annotated fibroblast, smooth muscle cell, endothelial cell (C3 and C26), erythroid cell (C38 and C56) and secretory cell (C118) according to Col3a1, Acta2, Pecam1, HBB family and Chga expression, respectively. To validate the data, we compared our single-cell landscape with tissue-wide mouse projects (19,20,48,49) using MetaNeighbor, including Tabula Muris and Tabula Muris Senis. Among the 177 mouse cell types, > 90% cell types were consistent with tissue-specific annotations, with a mean area under the receiver operating characteristic curve (AUROC) > 0.95 (Supplementary Table S1). Moreover, we performed subclustering analysis for single tissues collected from each stage and predicted a total of 2314 cell type subclusters (Supplementary Figure S1D; Table S2). For example, kidneys collected from 24m mice could be further annotated into proximal tubule cell, ascending loop of Henle, descending loop of Henle, collecting duct intercalated cell, collecting duct principal cell and distal convoluted tubule according to the gene expression of Miox, Cldn10, Cxcl10, Atp6v1g3, Fxyd4 and Pgam2 (Supplementary Figure S1E and F). These subclusters of single tissues from each stage would help to perform cross-tissue analyses in mice. Unsurprisingly, besides the consistency between cell types in MCDAA and other tissue-wide mouse projects, we found that gene expression patterns were consistent over the same time period (Supplementary Figure S2A–D).
For zebrafish, 143 cell types were annotated with cell type-specific markers (Figure 1E; Supplementary Figure S3A and B); markers were highlighted in Supplementary Table S1. A total of 143 zebrafish cell types were divided into 10 major cell lineages, i.e. endothelial, epithelial, erythroid, germ, immune, muscle, neuron, secretory, stromal and other cells (Supplementary Figure S3B). C10, C27, C42 and C59 were defined as neurons according to elavl3 and neurod1. C61 and C92 were further identified as radial glia with high expression of fabp7a, and C91, C92 and C142 were identified as oligodendrocyte with mbpa and mpz. Epithelial cells were composed of multiple clusters, such as enterocyte (fabp6), goblet cell (arg2 and krt18), hepatocyte (apoa1b and apoa2), ionocyte (atp1a1a.4) and keratinocyte (krtt1c19e). For muscle cells, we annotated cardiomyocyte (myl7 and actc1b), mucosal muscle cell (myh11b) and smooth muscle cell (acta2 and myl9a). We also identified endothelial cell (C18), erythrocyte (C79), fibroblast (C129), pancreatic cell (C64, C78 and C82) and neurosecretory cell (C54) according to kdrl, hbba1, col1a2, prss59.1 and scg3 expression, respectively. Comparing the landscapes profiled by whole-body sampling and tissue-specific sampling (21), we found a high degree of consistency in annotations, with an average AUROC > 0.96 (Supplementary Table S1). For example, the epithelial cell types were consistent with corresponding cell types in single tissues (Supplementary Figure S3C). Then, we performed subclustering analysis for each major cell type and predicted a total of 1762 cell type subclusters in the hierarchy (Supplementary Figure S3D). For example, ionocytes (C74) could be further annotated into distinct subclusters according to atp1a1a.4, atp1b1a, slc12a10.3, ndrg1a and ca4c gene expression in a higher resolution subclustering (Supplementary Figure S3E and F).
In the Drosophila cell landscape, we annotated 85 cell types with cell type-specific markers, which grouped into 12 major cell lineages (Figure 1F; Supplementary Figure S4A and B; Table S1), comprising epithelial, gut, hemocyte, muscle, neuron, follicle, germ, male accessory gland (MAG), Malpighian tubule (MT), proliferating, fat body and other cells. C62 and C70 were defined as hemocyte according to the remarkable markers Hml, Pxn, Nplp2, PPO1 and PPO2. C37 and C43 were identified as MT with Irk2 and Irk3. C8, C10, C14, C25, C50 and C69 were defined as follicles according to known markers such as psd, Vml, peb, Fcp3C, yellow-g and Femcoat. We annotated multiple neuron-related cell types such as T1 neurons (marked by Eaat1), GABAergic neurons (marked by Gad1), glutamatergic neurons (marked by VGlut), photoreceptor cell (marked by trpl) and glial cell (marked by nrv2 and Gat). As for gut-related cell types, we identified anterior enterocytes (marked by alphaTry and Amy-p), middle enterocytes (marked by Vha100-4), posterior enterocytes (marked by lambdaTry), enteroendocrine cells (EEs, marked by AstC and Tk), cardias (marked by pgant4), and enteroblasts (marked by Sox100B). Meanwhile, we compared our single-cell landscape with a parallel fly cell atlas project (25). Of 85 Drosophila cell types, 94% were consistent with fly tissue-wide annotations (fly cell atlas, FCA), with a mean AUROC > 0.93 (Supplementary Table S1). For instance, cardia, anterior enterocyte, middle enterocyte, posterior enterocyte and EE were consistent with the corresponding cell types in FCA gut (Figure 1G). This suggest that each cell type in DCL was well annotated even when sequenced by whole-body single-cell sequencing. Furthermore, the subclustering analysis was performed for each of the 85 major cell types and predicted a total of 967 subclusters in the hierarchy, and these subclusters made sense functionally (Supplementary Figure S4C). As an example, in C58, we identify a rare class of EEs with sNPF and CCHa2 expression, but no Tk and AstC expression (Cluster 4, Supplementary Figure S4D and E). The class III EE was different from the two major classes of EEs, class I expressing AstC and class II expressing Tk, and has been experimentally validated in a previous study (50).
In our previous work (16,26), significant immune gene activity was observed in structural cells (non-immune cells), such as epithelial cells in zebrafish and Malpighian tubules in Drosophila. In the cell landscape, similar patterns were observed in all three species (Supplementary Figure S5A–E), particularly in mice. We found that mouse alveolar type II cells (AT2 cells, C42, C76, C125 and C171) expressed the AT2 cell-restricted marker gene Sftpc, as well as immune-related genes, such as Cxcl15, Lyz2, Cbr2, Cd74, Lamp3 and Chil1 (Supplementary Figure S5A and B). Some of these immune-related genes were expressed in other mouse epithelial or basal cells (C9, C11, C81, C83 and C149) (Supplementary Figure S5C). These findings gave more evidence for the term ‘structural immunity’ (51), and further demonstrated that the activity of immune genes in structural cells was evolutionarily conserved across species. Furthermore, in addition to the uncommon cell populations with interesting gene expression patterns found in our previous studies, we also found several cell types with co-expressed markers of two cell types associated with mouse development. We identified cells that co-expressed markers of EEs (Chgb, Cck and Cpe) and neurons (Nnat) in neonatal intestine (Supplementary Figure S5F). These cells might be neuroendocrine cells (52), which exhibit a number of distinct neural phenotypic features, but function like endocrine cells. In neonatal and 6–8w heart, we verified a special endothelial cell (Esam) expression muscle cell marker (Myl9) (Supplementary Figure S5G and H), which might be a progenitor or intermediate cell type that differentiates into distinct cell types of arterial vessel wall (53).
Time-related signatures throughout life cycle in mice, zebrafish and Drosophila
Our cell landscape contained multiple life stages, providing a rich resource for investigating the changes in dynamics throughout the life cycle across species. We found that the proportion of cell types changed with development and age in three species (Figure 2A–C; Supplementary Figure S6A). For example, the percentage of immune cells gradually increased with age in mice, zebrafish and Drosophila. Detailed classification of mice immune cells at different stages showed that, with age, the proportion of T cells gradually decreased, while the proportion of B cells gradually increased (Figure 2B), which was in accord with previous findings in Tabula Muris Senis (20).
Figure 2.

Time-related signatures across mice, zebrafish and Drosophila. (A) Bar plot showing the fraction of cell lineages for five life stages of mouse. (B) Bar plot showing the fraction of immune cell types for five life stages of mouse. (C) Bar plot showing the fraction of cell lineages for five life stages of zebrafish. (D) UMAP visualization of the Mouse Kidney Cell Atlas across all stages of mouse kidney, colored by stage and cell type (PT, proximal tubule cell; NPC, nephronic progenitor cell; DLOH, descending loop of Henle; ALOH, ascending loop of Henle; PC, distal collecting duct principal cell; IC, collecting duct intercalated cell; DCT, distal convoluted tubule cell; EC, endothelial cell.) (E) Heatmap showing normalized GO AUCell Scores of the Mouse Kidney Cell Atlas across all stages of mouse kidney. (F) Relatedness network for the main cell types of mouse kidney, brain, heart and testis based on similarity of regulon activities. Each dot represents the aggregated cell type within each stage (see Supplementary Methods) and colored by stage. Only nodes with > 50 cells are selected. The edges between nodes represent the Spearman correlation coefficient calculated based on the aggregated regulon activity scores and filtered with Spearman correlation coefficient > 0.9. (G) Heatmap showing the aggregated module activities of TFs clustered by fuzzy c-means from mice. (H) Sankey plot showing the homologous relationships among vertebrate developmental-related TFs.
To gain a more detailed understanding of life cycle dynamics, we analyzed how cell types change with age in representative mouse tissues (Figure 2D; Supplementary Figure S6B, D and F). In kidney, our datasets captured erythrocytes, fibroblasts, endothelial cells, immune cells and types of kidney epithelial cells, such as proximal tubule cell, distal collecting duct principal cell, collecting duct intercalated cell, distal convoluted tubule cell and loop of Henle (Figure 2D). It was observed that the progenitor cells gradually differentiated with age, developed into tissue-specific cell types and performed corresponding functions (Figure 2E). This whole developmental process continued into adulthood. In brain, heart and testis, a similar phenomenon was also observed in main cell types (Supplementary Figure S6C, E and G). Notably, we found that the number of immune cells in most tissues gradually increased with age (Figure 2D; Supplementary Figure S6B and D), while the main cell types in most tissue non-immune cells also exhibited increased immune pathway activity, such as antigen processing and presentation, cell killing and lymphocyte-mediated immunity, indicating that the inflammatory response of tissues increases with aging.
To evaluate the dynamics of gene expression throughout the life cycle, we observed gene expression trajectories in three species and found some common tendencies (Supplementary Figure S7A–C). Average trajectories for each gene were calculated across 10 major tissues in mice, 9 major cell lineages in zebrafish and 11 major lineages in Drosophila. Each trajectory represented a category of gene sets and showed distinct expression dynamics during life cycle progression. It was obvious that clusters associated with mitochondrial-related function, including ATP and oxidative phosphorylation, continued to decline over time, and immune responses were consistently activated throughout the life span, which was observed in all three species. The cluster related to cell proliferation declined rapidly during development and leveled off after reaching adulthood, the developmental process cluster increased markedly at a young age and declined immediately in adults; for other metabolic levels, the clusters had few obvious tendencies. These findings suggest that the up-regulation of the immune response and the down-regulation of mitochondrial-related metabolism might be the common signatures throughout life cycle in mice, zebrafish and Drosophila, and shared similar patterns with age across species.
Furthermore, we observed the changes of TFs throughout the life cycle in the three species. TF regulation analysis showed a time-dependent trend in TF regulation across major cell types in representative mouse tissues (Figure 2F), such as epithelial cells in kidney, neuronal cells in brain, muscle and stromal cells in heart and germ cells in testis. The same trend was also found in cell type regulatory networks in zebrafish and Drosophila (Supplementary Figure S8A and B), suggesting that the gene regulatory networks have obvious changes during the life cycle across species. By analyzing specific TFs at each stage of every lineage in mice, zebrafish and Drosophila, we found that some cell lineages were regulated by similar gene families in all three species (Figure 2G; Supplementary Figure S8C and D; Table S3). For example, the neuronal cell lineage in the three species was regulated by Sox and NeuroD gene families and corresponding orthologous genes. The muscle cell lineage in the three species was regulated by Msx1, Pitx1, Prrx1, Tbx and corresponding orthologous genes. Interestingly, we identified lineage-shared modules among mice and zebrafish, which could regulate early development (module 10–13 in mice; module 13–14 in zebrafish) and maturation and senescence (module 14–15 in mice; module 15 in zebrafish). Meanwhile, we observed a series of homologous TFs in lineage-shared modules (Figure 2H; Supplementary Figure S8E). In the early developmental modules, the Hox gene family, which play key roles in regulating morphogenesis, were mainly present (54). In the mature and senescence modules, Fos/Jun gene families playing important roles in cell type differentiation (55,56) were present.
Characterization of molecular events associated with aging in multiple species
Considering the characterization associated with aging, we found that the proportion of cells expressing the senescence marker Cdkn2a increased with age at the overall or tissue level in mice (57) (Figure 3A; Supplementary Figure S9A). Two key immunoglobulin genes, Jchain and Xbp1, were highly expressed in senescent pancreas B cells (Supplementary Figure S9B and C), consistent with previous findings (20). Moreover, we observed that intrinsic gene expression also changed during aging. For example, in both mice and zebrafish, the expression level of senescence-associated secretory phenotype (SASP) genes increased with age (Supplementary Figure S9D).
Figure 3.

Aging-related signatures across multiple species. (A) Bar plot showing the fractions of cells expressing Cdkn2a at different mouse life stages. (B) Heatmap showing consensus up-regulated genes in 14 tissues of mice. The red cell represents up-regulated genes in the aging process. (C) Heatmap showing consensus down-regulated genes in 14 tissues of mice. The blue cell represents down-regulated genes in the aging process. (D) UpSet plot showing GO terms for up-regulated genes in multiple species. Representative GO terms for up-regulated genes in mice, rats and zebrafish are shown in the red box. (E) UpSet plot showing GO terms for down-regulated genes in multiple species. Representative GO terms for down-regulated genes in five species are shown in the blue box. (F) Heatmap showing normalized AUCell score of Immune response in eight mice tissues during four aging stages. (G) A circle plot showing the up-regulated homologous TFs between mice and zebrafish, colored by TF family. (H) Ligand and receptor analysis of 3w, 6–8w, 12m and 18m kidneys using CellPhoneDB. Line thickness indicates the degree of association between cell types. (I) Heatmap showing normalized AUCell score of the Fatty acid metabolic pathway in eight mice tissues during four aging stages. (J) Circular plot showing the down-regulated homologous TFs among the three species, colored by TF family. (K) Bar plot showing that PGZ reverses aging-induced frailty and sarcopenia in mice by increasing grip strength and lean mass in aging mice (n = 5). (L) Bar plot showing that PGZ reverses aging-induced hyperlipidemia in mice by decreasing triglycerides and total cholesterol in aging mice (n = 5). (M) Line chart showing that PGZ reverses aging-induced insulin resistance (n = 5).
To further investigate the cross-species mechanisms of cellular and molecular senescence, we focus only on those genes which changed with aging in five species, i.e. mice, zebrafish, rats, Drosophila and C.elegans (45) (Supplementary Table S4). In mice, those up-regulated genes were associated with immune responses (Figure 3B), and down-regulated genes were related to mitochondrial dysfunction (Figure 3C). For systematically evaluating the general trends across species, we used Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis to profile genes that changed with aging in these five species, and integrated the functional enrichment results (Supplementary Table S5; Figure 3D and E). The GO and KEGG enrichment results indicated that those genes were involved in the same pathways and might have similar functions across species. For example, most commonly up-regulated genes in all three vertebrates were associated with immune responses, regulation of response to stress and antigen processing and presentation (Figure 3D). On the other hand, genes down-regulated with age in the five species were related to ATP metabolic process, cellular respiration, oxidative phosphorylation and fatty acid metabolic process (Figure 3E). These results indicated two important signaling pathways during aging, immune responses and mitochondrial function. Furthermore, the variation of pathway activity in different cell types in mice showed that the most obvious decrease in lipid metabolism was found in hepatocytes (Supplementary Figure S9E), and energy metabolism was down-regulated significantly in epithelial cells and muscle cells of multiple tissues (Supplementary Figure S9F and G). Notably, almost all structural cells showed up-regulation of immune activity.
For up-regulated genes, the immune response pathway (GO:0006955) was observed to be up-regulated with aging in all three vertebrates (Figure 3F; Supplementary Figure S10A and B). Calculating the correlation between TF activity and up-regulated genes, we found some TF genes that might be involved in the immune process, such as Irf3, Irf8 and Cebpb (Supplementary Table S6; Figure S10C). The ortholog TF was also found in zebrafish (Figure 3G), suggesting that these TFs may play important regulatory roles in the aging process. Then, we constructed ligand–receptor maps using CellPhoneDB to reveal cell–cell interactions at different stages (30,58). As revealed, the interaction between T cells and renal cells was enhanced with aging (Figure 3H). In the 18-month kidney, T cells showed strong interactions with cell types, such as the loop of Henle and collection duct principal cells. In addition, immunofluorescence staining experiments showed that T cells infiltrated renal cells around endothelial cells in renal tissues at 12 months instead of 6 weeks (Supplementary Figure S10D). The enhancement of the immune response was also demonstrated at the epigenetic level. Using two histone modification markers from previously published epigenetic data (59), we found that the peak intensities of H3K4me3 and H3K27ac in immune-related genes showed an increasing trend with aging in mouse liver (Supplementary Figure S10E).
Further observations revealed that vertebrate structural cells increasingly exhibited characteristics of immune activation during aging. In mice, up-regulated genes in epithelial, endothelial and stromal cells showed immune-related features (Supplementary Table S7; Figure S11A and B), such as antigen processing and presentation, regulation of T-cell-mediated immunity, regulation of leukocyte activation and inflammatory response. In particular, antigen presentation-related genes in structural cells were up-regulated with age in multiple tissues (Supplementary Figure S11C). For example, the major histocompatibility complex (MHC) genes were up-regulated in AT2 cells during mouse aging (Supplementary Figure S11D). A similar phenomenon was also found in epithelial cells of zebrafish (Supplementary Figure S11E) and a series of published scRNA-seq cell atlases, such as for mouse and human lung epithelial cells (16,35) (Supplementary Figure S11F and G).
For down-regulated genes, we have identified that mitochondrial metabolism was the most significantly down-regulated signature with aging across different cell types and species, including ATP metabolic process, oxidative phosphorylation and fatty acid metabolic process (Figure 3I; Supplementary Figure S12A–H). These pathways were down-regulated in most mouse tissues, such as liver, heart, brain, lung and uterus, and similar appearances were observed in rats, zebrafish, Drosophila and C.elegans. These observations strongly support the mitochondrial theory of aging, which explains aging in a variety of biological systems (60–63). Calculating the correlation between TF activity and down-regulated genes, we identified a series of nuclear receptor TF genes involved in lipid metabolism and energy metabolism (64), including Pparg, Nr1h4 and Rarg belonging to the THR-like family, Hnf4a, Nr2f2 and Nr2fe3 belonging to the RXR-like family and Esrrg belonging to the ESR-like family (Supplementary Table S6; Figure S12I). The corresponding TF homologs were found in zebrafish and Drosophila (Figure 3J), indicating that these TFs might play crucial regulatory roles in the aging process across multiple species.
Peroxisome proliferator activated receptor-γ (PPARγ) is a member of the nuclear hormone receptor superfamily of ligand-activated TFs, whose associated signaling might possibly be involved in the prevention of aging and age-related diseases. The FDA-approved drug PGZ, as a high-affinity PPARγ agonist, had been found to improve glucose metabolism and mitochondrial dysfunction (65–67), and to ameliorate aging-related injury, and the blunting of aging markers in aging animals reversed age-related disturbances (68). Therefore, to better understand the important hallmark of organism aging and investigate promising anti-aging agents, we tried to relieve the aging phenotype by treating 2-year-old mice with PGZ for 3 weeks. In aging organisms, some known physiological aging phenotypes corresponding to cell type-specific functions, such as muscle contraction, cholesterol metabolism and insulin resistance, decline with aging (Supplementary Figure S13A–C). Compared with the control group, the lean mass of the PGZ-treated mice increased, and the grip strength was enhanced (Figure 3K). In addition, PGZ-treated mice had lower triglycerides and total cholesterol (Figure 3L), indicating that their lipid metabolism capacity was improved. The GTT and ITT both showed that PGZ treatment reversed aging-induced insulin resistance (Figure 3M). Then we performed single-cell sequencing of multiple tissues from PGZ-treated mice to better understand the role of PGZ at single-cell resolution (Supplementary Table S8; Figure S13D). We found that immune response and inflammatory response pathways were significantly decreased, especially in PGZ-treated brain, heart, lung and uterus (Supplementary Figure S13E). In contrast, the mitochondrial metabolism-related pathways were significantly activated in a variety of structural cells in PGZ-treated mice (Supplementary Figure S13F and G). These results suggested that the inverse correlation of mitochondrial metabolism and inflammation during aging might be interconnected through intrinsic regulatory networks, and PGZ treatment is effective to modulate the oxidative and inflammatory status, cell senescence and lipid metabolism, and thereby may be a promising protective therapy of aging and age-related diseases.
Constructing the cross-species cell landscape
Besides MCDAA, ZCDL and DCL, there were several cell landscapes also performed by Microwell-seq, such as human (16), axolotl (69), Xenopus (70) and earthworm (26). Combining these cell landscapes with other single cell atlases from published works, including monkey (71), rat (32), sea squirt (24), C.elegans (22), planarian (72), Nematostella (23), hydra (73) and sponge (74), we constructed an integrated online portal, Cell Landscape (Figure 4A), which provided a foundation for a large-scale single-cell atlas and served as a powerful tool to investigate their biological importance. This cross-species cell landscape containing more than 6 million single cells from 15 species, covering multiple representative branches in the evolution of multicellular animals, is publicly available at http://bis.zju.edu.cn/cellatlas/. Users can visualize and browse the landscape for each species, and search and retrieve cell clusters or genes of interest. For customized use, users can also annotate unknown clusters and search for orthologs. Searching the stromal-associated gene Col1a2 as an example, it was easily found that Col1a2 was highly expressed in mice fibroblasts (Supplementary Figure S14A). As for its orthologs, COL1A2 in human and col1a2 in zebrafish, we could observe that these genes were highly expressed in fibroblasts and mesenchymal cells (Supplementary Figure S14B). Searching for the well-known neuronal marker (Supplementary Figure S14C and D), Elavl3, we found that this gene and its orthologs were highly expressed in mice, human and zebrafish neurons, respectively. These search functions help to observe the expression of genes and their orthologs, and find potential marker genes.
Figure 4.

The main function of Cell Landscape. (A) Overview of the Cell Landscape website construction. (B) Diagram showing the pipeline for scMCA, scZCL, scDCL and their cross-species analysis. (C) scMCA results for isolated tumors in young (6–8w) and old (20–22m) mice (n = 9966 cells). Each row represents one cell type in our reference. Each column represents data from a single cell. Pearson correlation coefficient was used to evaluate cell type gene expression similarity. Red indicates a high correlation; gray indicates a low correlation. (D) The Sankey plot showing the highest correlation coefficient pairs in each cell type between HCL and scMCA, merged by cell lineage. (E) The Sankey plot showing the highest correlation coefficient pairs in each cell type between muscle cells in HCL and corresponding cell types in scMCA. (F) t-SNE visualization of Adult-Kidney-3 in HCL, colored by cell lineage. (G) The Sankey plot showing the highest correlation coefficient pairs in each cell type between Adult-Kidney-3 in HCL and scMCA, merged by cell lineage. (H) The Sankey plot showing the highest correlation coefficient pairs in each cell type between EEs in FCA gut and corresponding cell types in scMCA.
Using the Cell Landscape database, we updated and rebuilt the single-cell mapping pipelines (scMCA, scZCL and scDCL) for the classification of cell types (Figure 4B). In mice, the newly collected single-cell datasets in MCDAA were integrated into our older version scMCA (8). In zebrafish and Drosophila, we combined the cell landscapes ZCL and DCL with published single-cell datasets focusing on single tissues and constructed the mapping pipelines (scZCL and scDCL, see Supplementary Methods). Therefore, all available cell type clusters in mapping pipelines were generated from single-cell studies, and the cell landscapes with a wide range of life stages as well as single tissue datasets would allow for more precise annotation of cell types with temporal correlation, providing tissue and age information. By using our updated scMCA, we could easy classify conventional dendritic cells (cDCs), plasmacytoid dendritic cells (pDCs), natural killer (NK) cells, neutrophils, T cells, macrophages and monocytes in isolated tumors of young (6–8 weeks) and old (20–22 months) mice (75) (Figure 4C). These cells from tumors were highly correlated with adult (6–8 weeks) and aging (18 and 24 months) mice, rather than newborn and juvenile mice. Mapping the input DGE data to our scZCL reference, we could define the main cell types in zebrafish exocrine tissues (76) (Supplementary Figure S14E), such as sst1.1 cells, pancreatic exocrine cells and endothelial cells. Then we processed the single-cell data from adult Drosophila midgut (50), and confirmed two major classes of EEs (Supplementary Figure S14F). In DCL C58, AstC was specifically co-expressed with CCHa1 and AstA, and in C81, Tk was co-localized specifically with NPF and DH31. These suggest that C0, C2, C5, C6 and C8 were class I EEs expressing AstC, while C1, C3, C4 and C7 were class II EEs expressing Tk, findings which were completely consistent with the annotations in the raw study (50).
For cross-species comparison and annotation, our database provided cross-species analysis capabilities through orthologs in single-cell mapping pipelines (Figure 4B). Through one to one orthologous gene transformation, we mapped the HCL to our scMCA and found that the cell types in human and mice showed the highest similarity (Figure 4D). For example, C33, C42 and C79 were annotated as smooth muscle cells in HCL, and they were highly associated with smooth muscle cell types in MCDAA (Figure 4E). Notably, C74 was similar to ventricular cardiomyocyte from fetal and neonatal heart in MCDAA (Figure 4E). This cell type was annotated as ventricular cardiomyocyte in HCL, 99.6% cells of which were generated from fetal heart. Then we performed cross-species comparison in single tissues (Figure 4F), and found that the cell types in Adult-Kidney-3 of HCL were well annotated (Figure 4G). Intercalated cells and proximal tubule cells were highly correlated with collecting duct intercalated cells and proximal tubule cells in MCDAA (Supplementary Figure S14G). Different loop of Henle cell types in HCL were similar with ascending loop of Henle, descending loop of Henle and distal convoluted tubules in MCDAA, respectively, suggesting that these cell types in HCL could be further annotated (Supplementary Figure S14G). We also tested the cross-species mapping pipelines in zebrafish and Drosophila. In zebrafish intestinal epithelial cells (77) and Drosophila gut EEs (25), most cell types showed the highest correlations with similar cell types in MCDAA (Figure 4H; Supplementary Figure S14H). Besides EEs from the intestine and endocrine cells from the stomach, the EEs in Drosophila gut were similar to alpha, beta, delta and gamma cells from MCDAA pancreatic islet, suggesting that regulation of gut motility and metabolism required the cross-talk among mammalian intestine, stomach and pancreas.
DISCUSSION
In this study, we generated over 2.6 million scRNA-seq cells for mice, zebrafish and Drosophila at different life stages, and constructed the Cell Landscape resource for dissecting the cellular heterogeneity of different species. Our datasets covered a wide range of stages of development and aging, providing insights into cellular development, maturation and aging. All data points were generated from fresh and unsorted single cells using the same scRNA-seq technology, and thus there was high consistency and less batch effect, enabling robust cross-species, cross-tissue and cross-stage comparison. Moreover, scRNA-seq provides a universal measurement system for the characterization of nearly all cell types, including cells from non-model species. Currently, it is not easy to correlate all single-cell signatures with the classical functional cell typing system. The highly developed spatial transcriptomic and reporter functional experiments in mice will eventually unify these two systems, and provide enormous insights into known and unknown cellular hierarchy.
Development and aging in multicellular organisms are highly intertwined processes, and developmental expression changes tend to be evolutionarily conserved. We identified the tendency of cell types and genes that changed with development and age in mice, zebrafish and Drosophila, and found that the changes in dynamics of immune cells in most tissues and cell lineages gradually increased with age in the three species. Gene expression also showed similar trends throughout life cycle across the three species: ATP metabolic process and oxidative phosphorylation continued to decline, and immune responses were consistently activated throughout the life span. Moreover, TF regulation analysis revealed that some cell lineages were regulated by similar gene families across species.
Altered intercellular communication and mitochondrial dysfunction are hallmarks of aging in different organisms, especially mammalian aging. Inflammation is the prominent aging-associated alteration in intercellular communication. In this study, cross-species trajectories and enrichment analyses revealed that genes up-regulated with age were associated with immune responses, especially in vertebrates, whereas down-regulated genes were high correlated with mitochondrial metabolic and oxidative phosphorylation processes. Our work provided more evidence for the relevance of inflammatory pathways to aging in single-cell transcriptional landscape studies. Previously, studies, including our work, have shown that immune gene regulation was widespread in mammalian structural cells, such as epithelial, endothelial and stromal cells (16,51). By analyzing different types of structural cells in aging vertebrates, we observed that structural cells exhibit increasing characteristics of immune activation during aging, as the interaction between structural cells and immune cells is gradually enhanced. In aged structural cells, immune-related features, especially antigen presentation-related genes, were significantly up-regulated. Our findings were further validated at both gene expression and epigenetic levels through the analyses of various published datasets.
According to the mitochondrial theory of aging, oxidative damage reduces mitochondrial function with age. We have demonstrated that genes involved in oxidative phosphorylation decline rapidly with aging in both invertebrates and vertebrates. Also mitochondrial damage may be closely associated with the inflammatory response during aging. Mitochondrial dysfunction is known to be a cause of cellular apoptosis, autophagy and inflammation (60,78). In this study, we used PGZ, a drug that has been shown to improve mitochondrial function (67), to treat 2-year-old aging mice. The results showed that PGZ-treated mice had improved exercise capacity, lipid metabolism and insulin response. The results of scRNA-seq showed that PGZ treatment could repair mitochondrial function and reduce structural inflammation in parallel in the aged mice. To achieve a pharmacological effect in 3 weeks, we used a relatively high dosage (15 mg/kg/day) for PGZ i.p. injection. Long-term and low-dose treatment should be considered for future clinical trials to reduce the potential side effects of PGZ.
Currently, there are several kinds of accessible single-cell RNA databases, but most of them were developed for a single study, single tissue or single organ, and only provided basic visualization and searching, and some were even difficult to access. Although some integrated databases already exist, they are more like data hubs for collection of single-cell studies, providing cell cluster visualization and downloadable function, rather than interactive, precise search functions and prediction capabilities. Therefore, we developed the atlas-level database Cell Landscape, which is not only a single-cell resource for representative animals’ atlases, but also provides rich functions and useful tools for single-cell research. The visualization function provides the global view for the cell atlas, and quick access for single cluster and marker genes. The search and the search ortholog function can easily retrieve the expression of genes of interest and their orthologs, helping to annotate unknown clusters and find potential marker genes. The single-cell mapping pipelines and cross-species mapping pipelines are easy to use to classify and annotate cell clusters, even in different species.
In summary, our study provides new insights and valuable resources for studying organism-level development, maturation and aging. To facilitate usage of our data for the wider research community, we constructed the Cell Landscape website for browsing single-cell landscapes of different species.
DATA AVAILABILITY
The accession number for the raw data files for the RNA sequencing analysis reported in this paper is GEO: GSE198832. Scripts reproducing the analysis are available at https://github.com/ggjlab/cell_landscape. Cell Landscape is publicly available at http://bis.zju.edu.cn/cellatlas/. MCDAA, ZCDL and DCL are available at http://bis.zju.edu.cn/MCA/, http://bis.zju.edu.cn/ZCL/ and http://bis.zju.edu.cn/DCL/, respectively. The DGEs and annotation files are available at https://figshare.com/s/1ab3c6d7648d12247eb2.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Annoroad for supporting this project; Vazyme for supplying the customized enzymes in the study; the core facility platform of Zhejiang University School of Medicine, Zhejiang University Medical Center and Liangzhu Laboratory for their technical support; the Center of Cryo-Electron Microscopy (CCEM) at Zhejiang University for computational support; Huanju Liu and Jun Ma for help with sample collection; and Lanfang Luo, Yan-Jiang Chua, Xiaoman Wang, Mingyong Zhou, Yuan Liao, Junqing Wu, Guodong Zhang, Xueyi Wang, Lei Yang, Haofu Niu, Tingyue Zhang, Danmei Jia and Hangjun Wu for support on the project.
Contributor Information
Renying Wang, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Peijing Zhang, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China; Liangzhu Laboratory, Zhejiang University Medical Center, Hangzhou, 311121, China.
Jingjing Wang, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China; Liangzhu Laboratory, Zhejiang University Medical Center, Hangzhou, 311121, China.
Lifeng Ma, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Weigao E, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Shengbao Suo, Guangzhou Laboratory, Guangzhou 510005, China.
Mengmeng Jiang, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China; Liangzhu Laboratory, Zhejiang University Medical Center, Hangzhou, 311121, China.
Jiaqi Li, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Haide Chen, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China; Liangzhu Laboratory, Zhejiang University Medical Center, Hangzhou, 311121, China.
Huiyu Sun, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Lijiang Fei, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Ziming Zhou, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Yincong Zhou, College of Life Sciences, Zhejiang University, Hangzhou 310058, China.
Yao Chen, Key Laboratory of Reproductive Genetics (Ministry of Education), Department of Reproductive Endocrinology, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310006, PR China.
Weiqi Zhang, CAS Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, China National Center for Bioinformation, Beijing 100101, China.
Xinru Wang, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Yuqing Mei, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Zhongyi Sun, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Chengxuan Yu, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Jikai Shao, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Yuting Fu, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Yanyu Xiao, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Fang Ye, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Xing Fang, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Hanyu Wu, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Qile Guo, Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine, Zhejiang University, Hangzhou 314400, China.
Xiunan Fang, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Xia Li, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Xianzhi Gao, Institute of Immunology and Bone Marrow Transplantation Center, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310058, China.
Dan Wang, Women's Hospital, and Institute of Genetics, Zhejiang University School of Medicine, Hangzhou 310058, China.
Peng-Fei Xu, Women's Hospital, and Institute of Genetics, Zhejiang University School of Medicine, Hangzhou 310058, China.
Rui Zeng, Division of Nephrology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
Gang Xu, Division of Nephrology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
Lijun Zhu, Zhejiang Provincial Key Laboratory for Diagnosis and Treatment of Aging and Physic-chemical Injury Diseases, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310058, China.
Lie Wang, Institute of Immunology and Bone Marrow Transplantation Center, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310058, China.
Jing Qu, Institute for Stem Cell and Regeneration, Chinese Academy of Sciences, Beijing 100101, China.
Dan Zhang, Key Laboratory of Reproductive Genetics (Ministry of Education), Department of Reproductive Endocrinology, Women's Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310006, PR China.
Hongwei Ouyang, Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine, Zhejiang University, Hangzhou 314400, China; Zhejiang Provincial Key Lab for Tissue Engineering and Regenerative Medicine, Dr. Li Dak Sum & Yip Yio Chin Center for Stem Cell and Regenerative Medicine, Hangzhou, Zhejiang 310058, China.
He Huang, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China.
Ming Chen, College of Life Sciences, Zhejiang University, Hangzhou 310058, China.
Shyh-Chang NG, Institute for Stem Cell and Regeneration, Chinese Academy of Sciences, Beijing 100101, China.
Guang-Hui Liu, Institute for Stem Cell and Regeneration, Chinese Academy of Sciences, Beijing 100101, China.
Guo-Cheng Yuan, Department of Genetics and Genomic Sciences, Charles Bronfman Institute for Personalized Medicine, Icahn School of Medicine at Mount Sinai, NY, NY 10029, USA.
Guoji Guo, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China; Liangzhu Laboratory, Zhejiang University Medical Center, Hangzhou, 311121, China; Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine, Zhejiang University, Hangzhou 314400, China; Zhejiang Provincial Key Lab for Tissue Engineering and Regenerative Medicine, Dr. Li Dak Sum & Yip Yio Chin Center for Stem Cell and Regenerative Medicine, Hangzhou, Zhejiang 310058, China.
Xiaoping Han, Center for Stem Cell and Regenerative Medicine, and Bone Marrow Transplantation Center of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang 310000, China; Zhejiang Provincial Key Lab for Tissue Engineering and Regenerative Medicine, Dr. Li Dak Sum & Yip Yio Chin Center for Stem Cell and Regenerative Medicine, Hangzhou, Zhejiang 310058, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the National Natural Science Foundation of China [grants 31922049, 31930028 and 32000461] and the National Key Research and Development Program [grant 2018YFA0800503].
Conflict of interest statement. None declared.
REFERENCES
- 1. Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B.B., Siddiqui A.et al.. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods. 2009; 6:377–382. [DOI] [PubMed] [Google Scholar]
- 2. Hashimshony T., Wagner F., Sher N., Yanai I.. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012; 2:666–673. [DOI] [PubMed] [Google Scholar]
- 3. Ramskold D., Luo S., Wang Y.C., Li R., Deng Q., Faridani O.R., Daniels G.A., Khrebtukova I., Loring J.F., Laurent L.C.et al.. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012; 30:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shalek A.K., Satija R., Adiconis X., Gertner R.S., Gaublomme J.T., Raychowdhury R., Schwartz S., Yosef N., Malboeuf C., Lu D.et al.. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013; 498:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Treutlein B., Brownfield D.G., Wu A.R., Neff N.F., Mantalas G.L., Espinoza F.H., Desai T.J., Krasnow M.A., Quake S.R.. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014; 509:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fan H.C., Fu G.K., Fodor S.P.. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science. 2015; 347:1258367. [DOI] [PubMed] [Google Scholar]
- 7. Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M.et al.. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015; 161:1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F.et al.. Mapping the mouse cell atlas by microwell-seq. Cell. 2018; 172:1091–1107. [DOI] [PubMed] [Google Scholar]
- 9. Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J.et al.. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019; 566:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W.. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015; 161:1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karaiskos N., Wahle P., Alles J., Boltengagen A., Ayoub S., Kipar C., Kocks C., Rajewsky N., Zinzen R.P.. The Drosophila embryo at single-cell transcriptome resolution. Science. 2017; 358:194. [DOI] [PubMed] [Google Scholar]
- 12. Briggs J.A., Weinreb C., Wagner D.E., Megason S., Peshkin L., Kirschner M.W., Klein A.M.. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 2018; 360:eaar5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farrell J.A., Wang Y., Riesenfeld S.J., Shekhar K., Regev A., Schier A.F.. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science. 2018; 360:eaar3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wagner D.E., Weinreb C., Collins Z.M., Briggs J.A., Megason S.G., Klein A.M.. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science. 2018; 360:981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Farnsworth D.R., Saunders L.M., Miller A.C.. A single-cell transcriptome atlas for zebrafish development. Dev. Biol. 2020; 459:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han X., Zhou Z., Fei L., Sun H., Wang R., Chen Y., Chen H., Wang J., Tang H., Ge W.. Construction of a human cell landscape at single-cell level. Nature. 2020; 581:303–309. [DOI] [PubMed] [Google Scholar]
- 17. Tabula Sapiens Consortium Jones R.C., Karkanias J., Krasnow M.A., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., Brown P.et al.. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022; 376:eabl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fei L., Chen H., Ma L.E, W. E, W. Wang R., Fang X., Zhou Z., Sun H., Wang J., Jiang M.et al.. Systematic identification of cell fate regulatory programs using a single-cell atlas of mouse development. Nat Genet. 2022; 54:1051–1061. [DOI] [PubMed] [Google Scholar]
- 19. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018; 562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tabula Muris Consortium A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. 2020; 583:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang M., Xiao Y., E W., Ma L., Wang J., Chen H., Gao C., Liao Y., Guo Q., Peng J.et al.. Characterization of the zebrafish cell landscape at single-cell resolution. Front. Cell Dev. Biol. 2021; 9:743421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cao J., Packer J.S., Ramani V., Cusanovich D.A., Huynh C., Daza R., Qiu X., Lee C., Furlan S.N., Steemers F.J.et al.. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 2017; 357:661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sebé-Pedrós A., Saudemont B., Chomsky E., Plessier F., Mailhé M.-P., Renno J., Loe-Mie Y., Lifshitz A., Mukamel Z., Schmutz S.et al.. Cnidarian cell type diversity and regulation revealed by whole-organism single-cell RNA-Seq. Cell. 2018; 173:1520–1534. [DOI] [PubMed] [Google Scholar]
- 24. Cao C., Lemaire L.A., Wang W., Yoon P.H., Choi Y.A., Parsons L.R., Matese J.C., Wang W., Levine M., Chen K.. Comprehensive single-cell transcriptome lineages of a proto-vertebrate. Nature. 2019; 571:349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H., Janssens J., De Waegeneer M., Kolluru S.S., Davie K., Gardeux V., Saelens W., David F.P.A., Brbic M., Spanier K.et al.. Fly cell atlas: a single-nucleus transcriptomic atlas of the adult fruit fly. Science. 2022; 375:eabk2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo G Inferring predictive genetic models and regulatory elements by deep learning of cross-species single-cell gene expression landscapes. 2022; Research Square doi:11 April 2022, preprint: not peer reviewed 10.21203/rs.3.rs-1544073/v1. [DOI]
- 27. Packer J.S., Zhu Q., Huynh C., Sivaramakrishnan P., Preston E., Dueck H., Stefanik D., Tan K., Trapnell C., Kim J.et al.. A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolution. Science. 2019; 365:eaax1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pijuan-Sala B., Griffiths J.A., Guibentif C., Hiscock T.W., Jawaid W., Calero-Nieto F.J., Mulas C., Ibarra-Soria X., Tyser R.C.V., Ho D.L.L.et al.. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature. 2019; 566:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park J.E., Botting R.A., Dominguez Conde C., Popescu D.M., Lavaert M., Kunz D.J., Goh I., Stephenson E., Ragazzini R., Tuck E.et al.. A cell atlas of human thymic development defines T cell repertoire formation. Science. 2020; 367:eaay3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vento-Tormo R., Efremova M., Botting R.A., Turco M.Y., Vento-Tormo M., Meyer K.B., Park J.-E., Stephenson E., Polański K., Goncalves A.et al.. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature. 2018; 563:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reynolds G., Vegh P., Fletcher J., Poyner E.F.M., Stephenson E., Goh I., Botting R.A., Huang N., Olabi B., Dubois A.et al.. Developmental cell programs are co-opted in inflammatory skin disease. Science. 2021; 371:eaba6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma S., Sun S., Geng L., Song M., Wang W., Ye Y., Ji Q., Zou Z., Wang S., He X.et al.. Caloric restriction reprograms the single-cell transcriptional landscape of Rattus norvegicus aging. Cell. 2020; 180:984–1001. [DOI] [PubMed] [Google Scholar]
- 33. Schaum N., Lehallier B., Hahn O., Pálovics R., Hosseinzadeh S., Lee S.E., Sit R., Lee D.P., Losada P.M., Zardeneta M.E.et al.. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. 2020; 583:596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang W., Zhang S., Yan P., Ren J., Song M., Li J., Lei J., Pan H., Wang S., Ma X.et al.. A single-cell transcriptomic landscape of primate arterial aging. Nat. Commun. 2020; 11:2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Angelidis I., Simon L.M., Fernandez I.E., Strunz M., Mayr C.H., Greiffo F.R., Tsitsiridis G., Ansari M., Graf E., Strom T.-M.et al.. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019; 10:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martinez-Jimenez C., Eling N., Chen H.-C., Vallejos C., Kolodziejczyk A.A., Connor F., Stojic L., Rayner T., Stubbington M., Teichmann S.et al.. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science. 2017; 355:1433–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McGinnis C.S., Murrow L.M., Gartner Z.J.. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019; 8:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M. 3rd, Hao Y., Stoeckius M., Smibert P., Satija R.. Comprehensive integration of single-cell data. Cell. 2019; 177:1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wolf F.A., Angerer P., Theis F.J.. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018; 19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J.. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019; 47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aibar S., Gonzalez-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J.et al.. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017; 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tarashansky A.J., Musser J.M., Khariton M., Li P., Arendt D., Quake S.R., Wang B.. Mapping single-cell atlases throughout metazoa unravels cell type evolution. eLife. 2021; 10:e66747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M.et al.. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015; 16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Budovskaya Y.V., Wu K., Southworth L.K., Jiang M., Tedesco P., Johnson T.E., Kim S.K.. An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell. 2008; 134:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jiang M., Chen H., Lai S., Wang R., Qiu Y., Ye F., Fei L., Sun H., Xu Y., Jiang X.et al.. Maintenance of human haematopoietic stem and progenitor cells in vitro using a chemical cocktail. Cell Discovery. 2018; 4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lai S., Huang W., Xu Y., Jiang M., Chen H., Cheng C., Lu Y., Huang H., Guo G., Han X.. Comparative transcriptomic analysis of hematopoietic system between human and mouse by Microwell-seq. Cell Discovery. 2018; 4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lukassen S., Bosch E., Ekici A.B., Winterpacht A.. Single-cell RNA sequencing of adult mouse testes. Sci Data. 2018; 5:180192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ximerakis M., Lipnick S.L., Innes B.T., Simmons S.K., Adiconis X., Dionne D., Mayweather B.A., Nguyen L., Niziolek Z., Ozek C.et al.. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 2019; 22:1696–1708. [DOI] [PubMed] [Google Scholar]
- 50. Guo X., Yin C., Yang F., Zhang Y., Huang H., Wang J., Deng B., Cai T., Rao Y., Xi R.. The cellular diversity and transcription factor code of Drosophila enteroendocrine cells. Cell Rep. 2019; 29:4172–4185. [DOI] [PubMed] [Google Scholar]
- 51. Krausgruber T., Fortelny N., Fife-Gernedl V., Senekowitsch M., Schuster L.C., Lercher A., Nemc A., Schmidl C., Rendeiro A.F., Bergthaler A.et al.. Structural cells are key regulators of organ-specific immune responses. Nature. 2020; 583:296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ihler F., Vetter E.V., Pan J., Kammerer R., Debey-Pascher S., Schultze J.L., Zimmermann W., Enders G.. Expression of a neuroendocrine gene signature in gastric tumor cells from CEA 424-SV40 large T antigen-transgenic mice depends on SV40 large T antigen. PLoS One. 2012; 7:e29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rensen S.S.M., Doevendans P.A.F.M., van Eys G.J.J.M.. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007; 15:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parker H.J. Mammalian embryo: Hox genes. eLS. 2022; 10.1002/9780470015902.a0000740.pub4. [DOI] [Google Scholar]
- 55. Liu J., Han Q., Peng T., Peng M., Wei B., Li D., Wang X., Yu S., Yang J., Cao S.et al.. The oncogene c-Jun impedes somatic cell reprogramming. Nat. Cell Biol. 2015; 17:856–867. [DOI] [PubMed] [Google Scholar]
- 56. Madrigal P., Alasoo K.. AP-1 takes centre stage in enhancer chromatin dynamics. Trends Cell Biol. 2018; 28:509–511. [DOI] [PubMed] [Google Scholar]
- 57. Liu Y., Sanoff H.K., Cho H., Burd C.E., Torrice C., Ibrahim J.G., Thomas N.E., Sharpless N.E.. Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009; 8:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R.. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020; 15:1484–1506. [DOI] [PubMed] [Google Scholar]
- 59. Benayoun B.A., Pollina E.A., Singh P.P., Mahmoudi S., Harel I., Casey K.M., Dulken B.W., Kundaje A., Brunet A.. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 2019; 29:697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Green D.R., Galluzzi L., Kroemer G.. Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science. 2011; 333:1109–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Davie K., Janssens J., Koldere D., De Waegeneer M., Pech U., Kreft L., Aibar S., Makhzami S., Christiaens V., Bravo Gonzalez-Blas C.et al.. A single-cell transcriptome atlas of the aging Drosophila brain. Cell. 2018; 174:982–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cui H., Kong Y., Zhang H.. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal. Transduct. 2012; 2012:646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lopez-Otin C., Blasco M.A., Partridge L., Serrano M., Kroemer G.. The hallmarks of aging. Cell. 2013; 153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alaynick W.A. Nuclear receptors, mitochondria and lipid metabolism. Mitochondrion. 2008; 8:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bogacka I., Xie H., Bray G.A., Smith S.R.. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005; 54:1392–1399. [DOI] [PubMed] [Google Scholar]
- 66. Sauerbeck A., Gao J., Readnower R., Liu M., Pauly J.R., Bing G., Sullivan P.G.. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp. Neurol. 2011; 227:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Takada S., Hirabayashi K., Kinugawa S., Yokota T., Matsushima S., Suga T., Kadoguchi T., Fukushima A., Homma T., Mizushima W.et al.. Pioglitazone ameliorates the lowered exercise capacity and impaired mitochondrial function of the skeletal muscle in type 2 diabetic mice. Eur. J. Pharmacol. 2014; 740:690–696. [DOI] [PubMed] [Google Scholar]
- 68. Shen D., Li H., Zhou R., Liu M.J., Yu H., Wu D.F.. Pioglitazone attenuates aging-related disorders in aged apolipoprotein e deficient mice. Exp. Gerontol. 2018; 102:101–108. [DOI] [PubMed] [Google Scholar]
- 69. Ye F., Zhang G., E W., Chen H., Yu C., Yang L., Fu Y., Li J., Fu S., Sun Z.et al.. Construction of the axolotl cell landscape using combinatorial hybridization sequencing at single-cell resolution. Nat. Commun. 2022; 13:4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liao Y., Ma L., E W., Yang L., Fang X., Guo Q., Ruan F., Wang J., Zhang P., Sun Z.et al.. Cell landscape of larval and adult Xenopus laevis at single-cell resolution. Nat. Commun. 2022; 13:4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Han L., Wei X., Liu C., Volpe G., Zhuang Z., Zou X., Wang Z., Pan T., Yuan Y., Zhang X.et al.. Cell transcriptomic atlas of the non-human primate macaca fascicularis. Nature. 2022; 604:723–731. [DOI] [PubMed] [Google Scholar]
- 72. Fincher C.T., Wurtzel O., de Hoog T., Kravarik K.M., Reddien P.W.. Cell type transcriptome atlas for the planarian Schmidtea mediterranea. Science. 2018; 360:eaaq1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Siebert S., Farrell J.A., Cazet J.F., Abeykoon Y., Primack A.S., Schnitzler C.E., Juliano C.E.. Stem cell differentiation trajectories in hydra resolved at single-cell resolution. Science. 2019; 365:eaav9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Musser J.M., Schippers K.J., Nickel M., Mizzon G., Kohn A.B., Pape C., Ronchi P., Papadopoulos N., Tarashansky A.J., Hammel J.U.et al.. Profiling cellular diversity in sponges informs animal cell type and nervous system evolution. Science. 2021; 374:717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang C., Lei L., Yang X., Ma K., Zheng H., Su Y., Jiao A., Wang X., Liu H., Zou Y.et al.. Single-cell sequencing reveals antitumor characteristics of intratumoral immune cells in old mice. J. Immunother. Cancer. 2021; 9:e002809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Singh S.P., Chawla P., Hnatiuk A., Kamel M., Silva L.D., Spanjaard B., Eski S.E., Janjuha S., Olivares-Chauvet P., Kayisoglu O.et al.. A single-cell atlas of de novo beta-cell regeneration reveals the contribution of hybrid beta/delta-cells to diabetes recovery in zebrafish. Development. 2022; 149:dev199853. [DOI] [PubMed] [Google Scholar]
- 77. Wen J., Mercado G.P., Volland A., Doden H.L., Lickwar C.R., Crooks T., Kakiyama G., Kelly C., Cocchiaro J.L., Ridlon J.M.et al.. Fxr signaling and microbial metabolism of bile salts in the zebrafish intestine. Sci. Adv. 2021; 7:eabg1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhou R., Yazdi A.S., Menu P., Tschopp J.. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011; 469:221–225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the raw data files for the RNA sequencing analysis reported in this paper is GEO: GSE198832. Scripts reproducing the analysis are available at https://github.com/ggjlab/cell_landscape. Cell Landscape is publicly available at http://bis.zju.edu.cn/cellatlas/. MCDAA, ZCDL and DCL are available at http://bis.zju.edu.cn/MCA/, http://bis.zju.edu.cn/ZCL/ and http://bis.zju.edu.cn/DCL/, respectively. The DGEs and annotation files are available at https://figshare.com/s/1ab3c6d7648d12247eb2.