

Abstract
The vast amount of gene expression profiling data of bulk tumors and cell lines available in the public domain represents a tremendous resource. For any major cancer type, expression data can identify molecular subtypes, predict patient outcome, identify markers of therapeutic response, determine the functional consequences of somatic mutation, and elucidate the biology of metastatic and advanced cancers. This review provides a broad overview of gene expression profiling in cancer (which may include transcriptome and proteome levels) and the types of findings made using these data. This review also provides specific examples of accessing public cancer gene expression datasets and generating unique views of the data and the resulting genes of interest. These examples involve pan-cancer molecular subtyping, metabolism-associated expression correlates of patient survival involving multiple cancer types, and gene expression correlates of chemotherapy response in breast tumors.
Introduction
For more than 20 years, the research community has extensively profiled human cancers for gene expression, with the associated data representing thousands of studies being made available in the public domain. Of the various “-omics” levels in cancer that can be profiled, transcriptomics would have the most data generated to date, given the early adoption by academic laboratories of DNA microarrays, starting in the late 1990s1,2. With the advent of next-generation sequencing3, RNA sequencing (RNA-seq) as a transcriptomics platform has become increasingly common. Gene expression would include protein as well as mRNA, where the two may not always be strongly correlated4,5. Historically, proteomics profiling has represented additional challenges over transcriptomics, given the diverse chemistries that proteins represent, requiring experienced laboratories. Reverse phase protein arrays—typically representing 150–300 targeted proteins—have been more widely adopted as a proteomics profiling platform in recent years6. Also, recent technological advancements in mass spectrometry-based proteomics technologies, profiling thousands of proteins, have accelerated its application to study greater and greater numbers of cancer specimens7,8.
In addition to gene expression profiling data generated by individual laboratories for smaller and more independent studies, major team science efforts have generated multi-omics data on thousands of human tumors of various cancer types defined by tumor lineage or histology. The Cancer Genome Atlas (TCGA) consortium, which went from 2006 to 2018, generated multi-omics data, including RNA-seq and RPPA proteomic data, on over 10,000 human tumors9,10. Parallel to TCGA efforts focused mainly within the United States, the International Cancer Genomic Consortium (ICGC) carried out multi-omics profiling of thousands of cancers on a similar scale, with the cooperation of multiple countries11. In recent years, the Clinical Proteomic Tumor Analysis Consortium (CPTAC) and the International Cancer Proteogenome Consortium (ICPC) have generated multi-omics data on over 2,000 human cancers5, including proteomics by mass spectrometry platform.
The vast amount of gene expression profiling data made available by published studies and consortiums represents a most valuable resource for ongoing studies. As no original study can comprehensively mine an expression profile dataset for all genes of potential relevance, future studies may analyze previously published data with different questions in mind from those of the original authors. This review will provide a broad overview of gene expression profiling in cancer and the types of findings made using these data. The figures of this review showcase specific examples of accessing public cancer gene expression datasets and generating unique views of the data and the resulting genes of interest. Due partly to space constraints, this review focuses on expression profiling of bulk tumors and cell lines, where single-cell RNA sequencing (scRNA-seq) represents another expression platform profiling individual cells within a tumor12.
Molecular subtyping
Due in part to the advent of gene expression profiling technologies, it is now universally understood that multiple and distinct molecular subtypes would exist within any given cancer type as defined by tissue of origin. Early studies of breast cancer using DNA microarrays13,14 revealed five major gene expression-based subtypes: luminal A, luminal B, ERBB2+, basal-like, and normal-like. These subtypes reflected previous observations of breast cancer subtypes based on histology13, with the luminal subtypes expressing the estrogen receptor, denoting sensitivity to estrogen therapy, and the ERBB2+ subtype expressing the Her2 receptor, denoting sensitivity to therapies blocking Her2. Breast cancer might represent the most well-known example of molecular subtypes having therapeutic implications. Gene expression profiling of other tissue-based cancer types has also defined molecular subtypes existing within these diseases. For example, for most cancer types studied by TCGA consortium, expression-based subtypes could be defined15,16. These subtypes may involve histologic features of the cancer cells (e.g., basal, luminal, or squamous characteristics), cancer cell differentiation level, associated DNA-level mutations, or infiltration of non-cancer cells (including immune cells or fibroblasts).
Beyond identifying molecular subtypes within tissue-based cancer types, pan-cancer analyses can define subtypes that may either align closely with cell or tissue of origin9,17 or would transcend tumor lineage5,15,18,19. One of the advantages of team science efforts such as TCGA is that tumors from different cancer types are often profiled by the same laboratory using the same analytical platform. This aspect should allow cross-cancer type analyses defining molecular subtypes and associated pathways relevant to multiple cancer types. Figure 1 provides an example of using TCGA data to define pan-cancer molecular subtypes, reflecting the tissue of origin (Figure 1a) or transcending tissue of origin (Figure 1b), depending on the analytical approach used. In our pan-cancer study of TCGA RNA-seq data15, we classified 10224 cancers, representing 32 major types, into ten molecular-based subtypes or “classes,” whereby we first computationally removed expression patterns representing dominant tissue or histologic effects. For example, one of our pan-cancer subtypes expressed neuroendocrine markers such as CHGA. Another subtype represented basal-like breast cancer and MYC expression. Two of our subtypes expressed mesenchymal markers (e.g., VIM). Another subtype expressed immune checkpoint pathway markers (e.g., CD274) and molecular signatures of immune infiltrates. Using mass spectrometry-based proteomics data from CPTAC and ICPC, we could similarly identify pan-cancer subtypes reflected in the mRNA data, but with notable exceptions5,19. For example, a proteomic-based subtype expressed proteins in the complement pathway, distinct from the subtype expressing lymphocytic markers.
Figure 1. Pan-cancer molecular subtypes as identified using different analytical approaches.
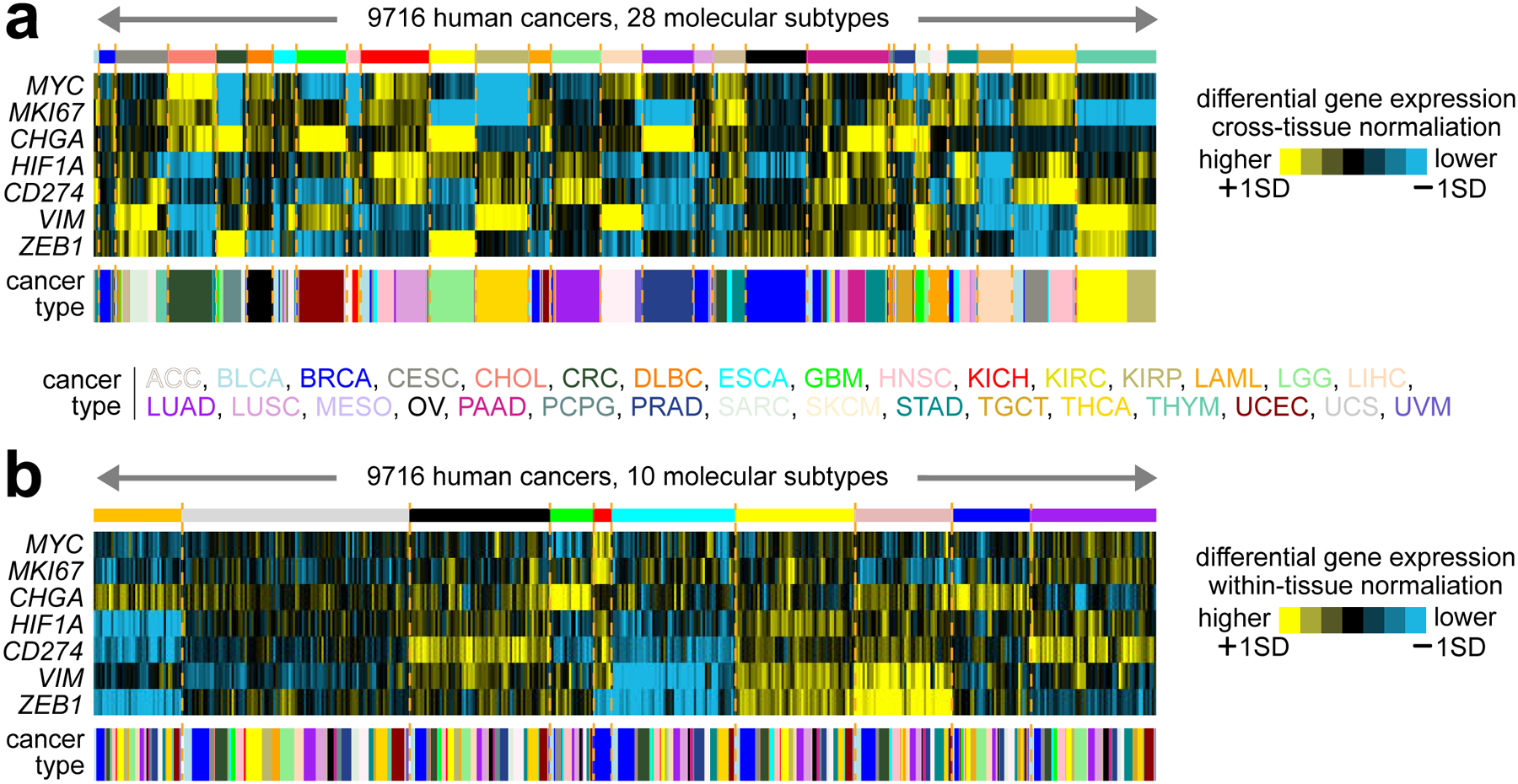
(a) Across 9716 tumors represented in TCGA datasets, TCGA Network previously defined 28 pan-cancer subtypes closely following the cancer tissue of origin9. With the tumors ordered by molecular subtype, the heat map shows differential mRNA expression patterns (values normalized across all cancers to standard deviations from the median) for a select set of genes representing pathways of particular interest: MYC, oncogene; MKI67, proliferation marker; CHGA, marker of neuroendocrine tumors; HIF1A, transcription factor inducing hypoxia; CD274, PD-L1 gene and immunotherapy target; VIM, vimentin gene and marker of mesenchymal cells; ZEB1, transcription factor activating epithelial-mesenchymal transition. (b) Using an alternate analytical approach to define molecular subtypes that would transcend tumor lineage and tissue of origin, we could classify TCGA tumors into ten major subtypes15. The heat map shows differential mRNA expression patterns (values normalized within each cancer type to standard deviations from the median) for the same set of genes from part a. While TCGA RNA-seq datasets allow for cross-cancer type comparisons, as carried out in defining the subtypes in part a9, an alternative approach to molecular classification, represented in part b, involves computationally subtracting the gene expression differences between cancer types18. As applied to TCGA RNA-seq data, this alternative approach had the effect of consolidating the individual subtypes that might be discoverable in individual cancer types into super-types or pan-cancer “classes” that transcend tissue or histology distinctions.
Prognostic gene signatures
Gene expression profiles of tumor samples taken from the initial surgery can predict the patient’s eventual outcome. Early studies first demonstrated this means of prognostication in breast cancer, establishing a 70-gene prognosis profile that could segregate patients into good versus poor prognosis20,21, consistent with patient follow-up data. Studies from other groups could establish prognostic gene signatures in most other cancer types, including lung22,23, prostate24,25, colon26, medulloblastoma27, leukemia28, lymphoma29, and so on. Gene signature information has generally represented an independent factor in predicting disease outcome, along with relevant clinical variables such as age, tumor size, histology, pathological grade, etc.20 Given the clinical application of cancer patient prognosis, commercial gene panel assays with genes selected based on gene expression profiling data, have been developed and approved for clinical use, such as the Oncotype DX assays for breast30, colon31, and prostate32 cancers. A prognostic gene signature may consist of a discrete number of genes, often a function of statistical methods and cutoffs. At the same time, many more genes not included in a given signature may also have prognostic information.
In addition to their potential for clinical application, prognostic gene signatures can provide molecular clues regarding the biological drivers and pathways underlying aggressive cancers. Genes that may inform tumor biology would not be limited to the top ~100 most significant genes but could additionally involve hundreds of genes that meet statistical significance for survival association. An example of gaining insight from gene survival correlates involves my work with TCGA consortium in clear cell renal cell carcinoma33, where we defined molecular correlates of patient survival at mRNA, microRNA, protein, and DNA methylation levels. When viewed in the context of metabolism, aggressive renal cancers demonstrated evidence of a metabolic shift, involving downregulation of TCA cycle genes, decreased AMPK and PTEN, upregulation of the pentose phosphate pathway and glutamine transporter genes, and increased acetyl-CoA carboxylase33. Along these lines, Figure 2 of this review shows a pathway diagram representing core metabolic pathways, with the genes denoting any survival associations at the mRNA level as observed in breast cancer34, clear cell renal cell carcinoma33, or across the entire TCGA pan-cancer dataset35. Other pathways would underlie prognostic gene signatures, which might be uncovered, for example, by domain knowledge or by using methods and software such as Gene Set Enrichment Analysis (GSEA)36.
Figure 2. Gene expression correlates of cancer patient survival involving metabolic pathways.
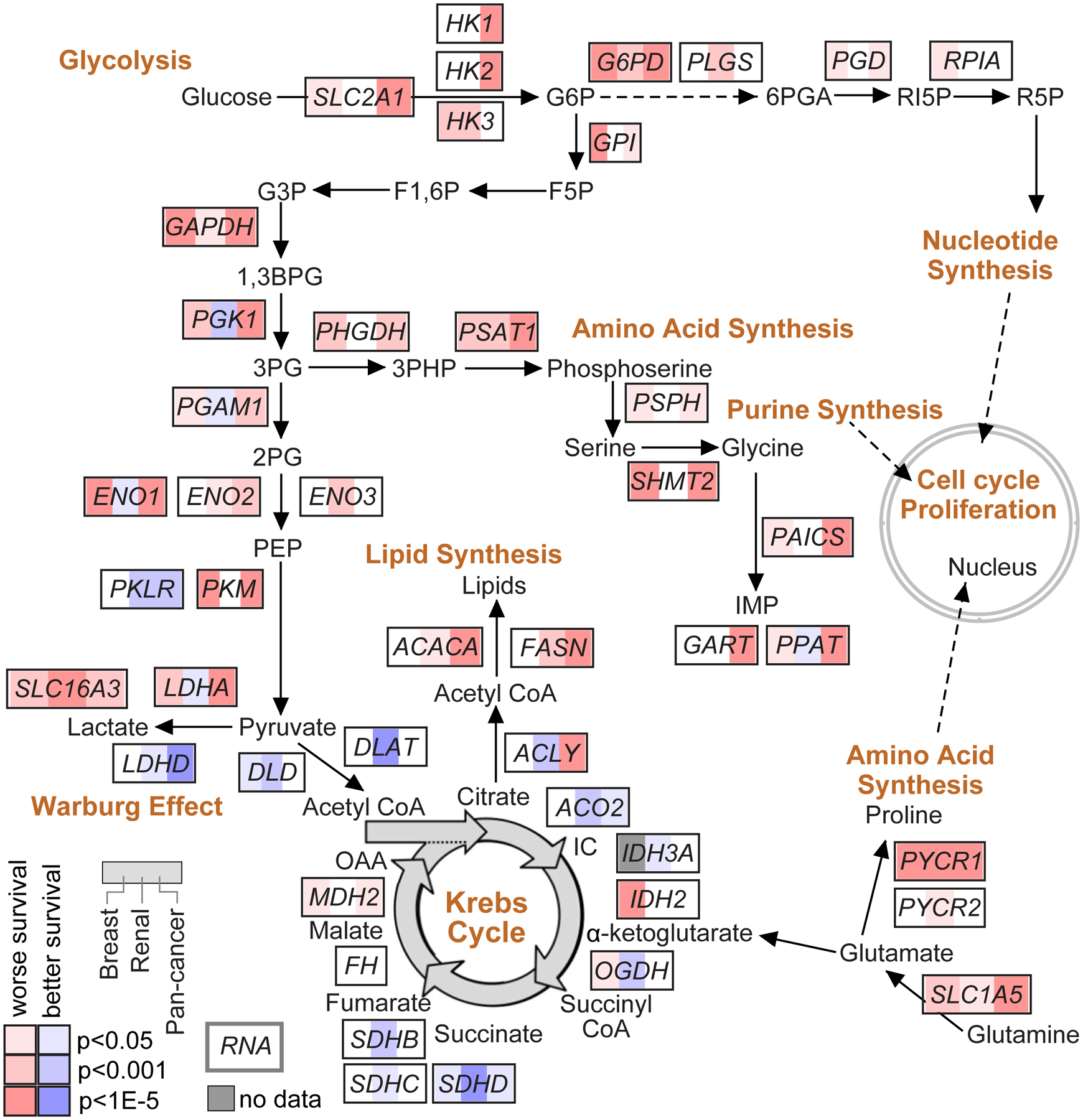
Gene expression correlates of patient survival can be examined for clues as to the molecular biology underlying the more aggressive cancers. Pathway diagram representing core metabolic pathways33,75, with corresponding mRNA correlations with patient survival. Red and blue shading respectively represent the association of increased mRNA expression with worse or better survival, by univariate Cox. For each gene, survival correlations across three cancer expression profiling datasets are represented: breast cancer dataset from Pereira et al.34 (left, n=1904 patients, overall survival endpoint), renal cell carcinoma dataset from TCGA (middle, n=417 patients, overall survival endpoint), pan-cancer dataset from TCGA (right, n=10152 patients, overall survival endpoint, p-values correcting for cancer type).
Correlation with drug response in cell lines
Cancer cell lines have historically been the most commonly used models for studying cancer biology. Using in vitro cell line models would be a typical first step in validating functional gene targets or drug responses in the laboratory, where results may be further investigated using more complicated in vivo models. Extensive molecular data (including mRNA, protein, copy number alteration, and somatic mutation), gene knockout data, and drug response data have been generated across over 1000 human cancer cell lines. These data are available via team science efforts, including the Cancer Cell Line Encyclopedia (CCLE)37,38 and the Genomics of Drug Sensitivity in Cancer (GDSC)39 projects. GDSC datasets include half maximal inhibitory concentration (IC50) data on over 400 drugs across cell lines, denoting which cell lines are most or least sensitive to a given drug in vitro. Gene expression data may be integrated with drug IC50 data to define gene correlates of drug response. CCLE data include corresponding CRISPR and RNAi data40,41, denoting which cell lines depend on a specific gene for proliferation. These resources may be combined to identify new gene targets with functional roles in a subset of cell lines for follow-up functional studies. For example, the ERBB2 gene has high expression in cell lines most sensitive to either HER2 inhibitors39 or loss of HER2 function. Candidate gene targets involving other drugs and other cell lines may be similarly identified.
Therapeutically predictive gene signatures in patient tumors
Cancer cell lines represent models that would capture some but not all aspects of cancer cells within patient tumors. Breast cancer perhaps provides the best-known examples of therapeutically predictive markers, namely estrogen receptor and HER2 (ERBB2), with high expression predicting patient response to therapies targeting these receptor pathways. Gene expression profiling datasets of human tumors, combined with treatment data, including patient response, could yield signatures of therapeutic response involving up to hundreds of genes. Patient treatment response data may include short-term as well as long-term responses. With long-term response data, there is a need to distinguish gene markers that would be therapeutically predictive versus those that are merely prognostic. In identifying markers of treatment response, numerous studies have carried out gene expression profiling of pre-treatment breast tumor biopsies from patients treated with neoadjuvant chemotherapy, with patient response recorded at the end of treatment42–48. Many of the gene expression markers from these studies are associated with basal-like breast cancer, as this subtype tends to be more responsive to chemotherapy49. For Figure 3 of this review, we assembled a compendium of eight different public breast cancer expression datasets. We used this to define a top set of genes correlated with pathologic chemotherapy response, independent of molecular subtype (Figure 3a). By enrichment analysis50, these genes represent functional gene categories of interest to cancer biology (Figure 3b). In addition, one can combine expression data from human tumors with expression data from cell lines having drug response data to identify treatment response markers that arise in both settings45.
Figure 3. Gene expression correlates of therapeutic response to chemotherapy in breast cancer patients.
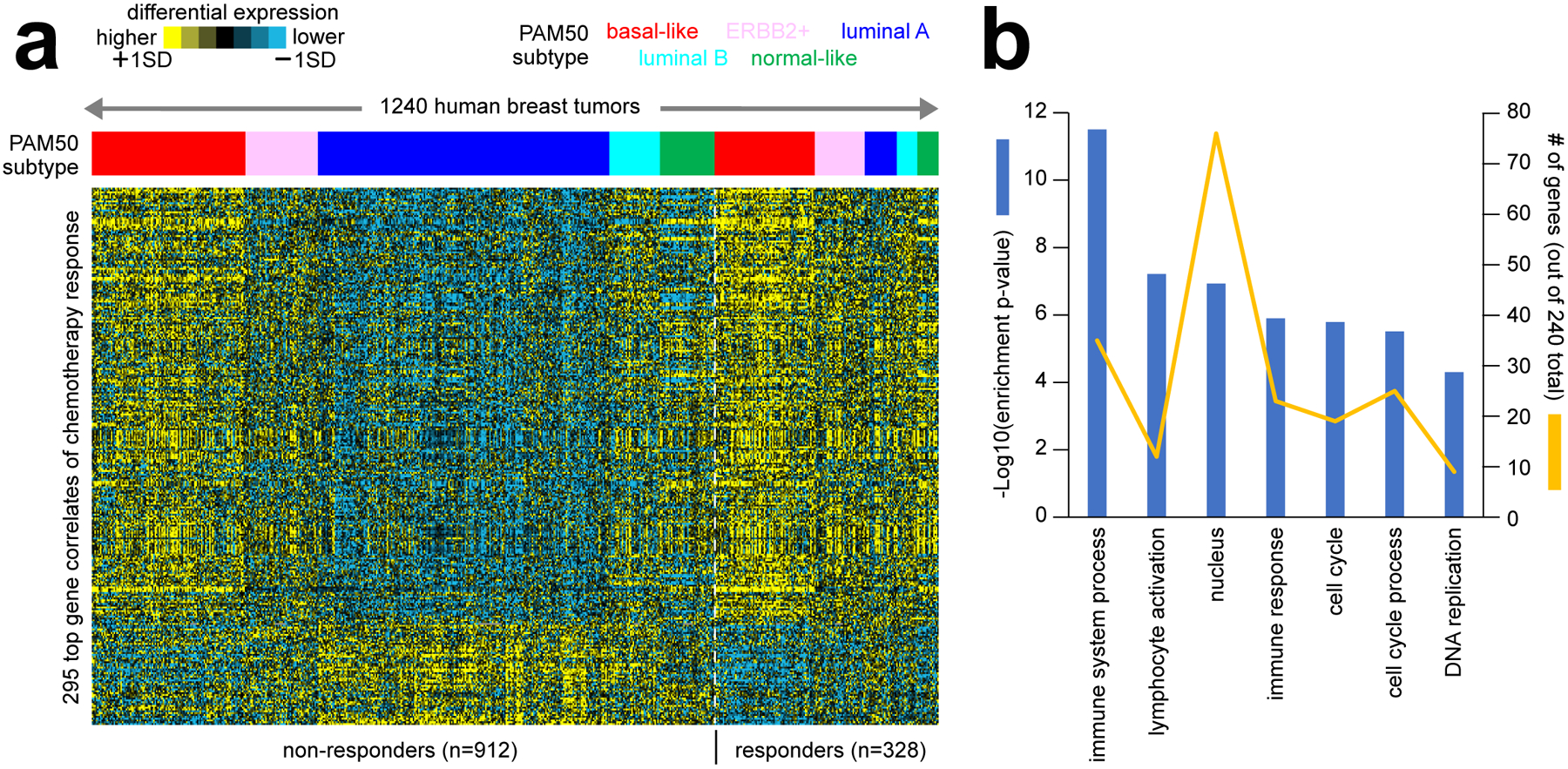
(a) Numerous studies have carried out gene expression profiling of pre-treatment breast tumor biopsies from patients treated with neoadjuvant chemotherapy, with patient response recorded at the end of treatment42–48. As part of this review, we assembled a compendium of eight separate datasets from the above studies, representing 1240 tumor expression profiles (GEO accession numbers provided in Data File S1). All datasets were generated using the same Affymetrix gene array platform. In the same manner as carried out in our previous studies5,15,76, we transformed log2 gene expression values to standard deviations from the median within each dataset, removing batch effect differences among datasets. We assessed the correlation of expression with pathologic chemotherapy response (path CR) for each gene feature after correcting for Pam50 subtype76 by linear modeling. The heat map shows expression patterns for a top set of 295 gene features (p<0.001, out of 22269 total). (b) Selected significantly enriched GO terms77 within the genes higher in breast tumors from patients with path CR (from part a). Enrichment p-values and numbers of genes in the path CR-associated gene set are indicated for each GO term. Enrichment p-values by one-sided Fisher’s exact test.
Integration of genome with transcriptome or proteome
Expression profiling data can be integrated with DNA-level somatic mutation data to examine the functional consequences of specific mutations. For example, gene copy alterations in cancer directly and widely impact gene expression, as these alterations represent a dosage effect in how much a gene can be transcribed51. Molecular pathways in cancer involve multiple genes and pathway intermediates. For a given pathway, somatic mutation—including point mutations, insertions-deletions, and copy number alterations—may impact different genes in different tumors52. The gene expression level often reflects the downstream consequences of mutation, where the diverse set of alterations at the pathway signaling level would converge upon the same set of transcriptionally regulated genes53–56. Cell line models can identify the top set of genes altered in expression when a specific pathway is experimentally perturbed. These genes can then define pathway signatures by which tumors or cell lines with expression data may be scored, with higher signature scoring indicative of higher pathway activity57. Gene signatures of pathways can also help discover unexpected connections involving genes previously unrealized or underappreciated as members of the given pathway. We demonstrated this approach in our multi-omics survey of the PI3K/AKT/mTOR pathway across TCGA cancers, whereby IDH1 and VHL mutations, previously underappreciated as impacting the pathway, were strongly associated with increased pathway activation55.
The impact of somatic alterations on gene expression is not limited to the gene coding regions. The non-coding genome provides the regulatory framework of the coding genome, and non-coding somatic alterations often impact the expression of nearby genes. One well-known example of this involves TERT, where specific point mutations or structural rearrangement breakpoints that occur directly upstream of TERT can result in up-regulation of the gene58–60. Recently, the Pan-Cancer Analysis of Whole Genomes (PCAWG) consortium comprehensively surveyed the non-coding somatic landscape of 2658 tumors from TCGA and ICGC, 1220 of these tumors having RNA-seq data61–63. Few genes with “hotspot” non-coding mutations (i.e., non-coding mutations at a specific coordinate that recurrently occur across many tumors) were found, which included TERT63. On the other hand, somatic structural variation showed a widespread impact on the transcription of hundreds of genes, where structural variant breakpoints may fall at different coordinates in relation to the gene but which can alter regulation by various mechanisms, including enhancer hijacking and TAD disruption62. In addition, non-coding point mutations that fall within a wider genomic region, as opposed to recurrent hotspot mutations targeting a specific nucleotide, can similarly impact the expression of certain genes64.
Expression profiling of advanced and metastatic cancers
To date, the vast majority of tumors with expression data in the public domain or available through large-scale efforts such as TCGA are primary tumors. Metastatic tumors, on the other hand, represent a more advanced cancer that has left its primary site to grow elsewhere in the body. By some estimates, as much as 90% of cancer deaths result from metastasis65. There is a need to understand better the genes and processes involved in metastasis. Public repositories such as the Gene Expression Omnibus (GEO)66 provide expression profiling data on tumor metastases from individual published studies. These include data allowing for paired metastasis versus primary comparisons within the same patient67,68, to help assess the changes associated with metastatic cancer cells. Pan-cancer multi-omics initiatives to profile tumor metastases from multiple cancer types include the recent MET50069 and POG57070 studies of 500 and 570 patients, respectively. The POG570 datasets include patient treatment information. As advanced and metastatic tumors involve patients who have typically been heavily treated at this stage, these data offer the opportunity to assess gene expression features associated with specific therapies70,71.
Future directions
More and more gene expression profiling data on cancers will continue to go into the public domain. Expression profiling data from different studies representing different cellular contexts may be re-analyzed, with the individual results sets brought together in interesting ways to gain insights into cancer biology and therapeutic approaches. Data from cancer cell lines or from PDX models72 could be integrated with data from human tumors, e.g., to identify gene targets for follow-up bench experiments. Bulk tumor expression profiles represent a mixture of cancer and non-cancer cells. By profiling individual cells within the tumor, the scRNA-seq platform provides insights into the tumor cell populations and how these may change over time or with treatment. At the same time, scRNA-seq studies often do not involve many samples or patients, where a study may need large numbers to establish robust associations. With all the available expression data, more sophisticated data portals could make the results available and accessible to non-computational researchers, e.g., making data for gene-level results available by a point-and-click user interface73,74.
Supplementary Material
Data File S1. Results represented in the figures. Provided as an Excel file. Results include pan-cancer molecular subtypes and associated genes (Figure 1), mRNA-level correlates of patient survival involving metabolism-related genes in breast cancer, clear cell renal cell carcinoma, and pan-cancer datasets (Figure 2), and gene expression correlates of pathologic chemotherapy response (pCR) in breast tumors (correcting for PAM50 subtype), along with GEO datasets and sample profiles analyzed.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant P30CA125123 (C. Creighton). We thank Yiqun Zhang for technical assistance regarding the gene expression correlation with therapeutic response to chemotherapy in breast cancer patients.
Acronyms:
- TCGA
The Cancer Genome Atlas
- ICPC
International Cancer Proteogenome Consortium
- CPTAC
Clinical Proteomic Tumor Analysis Consortium
- RNA-seq
RNA sequencing
- CNA
Copy Number Alteration
Footnotes
Declaration of Interests
The author discloses the following interest: Roche Diagnostics (scientific advisory board/consultancy).
References
- 1.Lockhart D, Dong H, Byrne M, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol 1996;14:1675–80. [DOI] [PubMed] [Google Scholar]
- 2.Schena M, Shalon D, Davis R, Brown P. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995;270:467–70. [DOI] [PubMed] [Google Scholar]
- 3.Fullwood M, Wei C, Liu E, Ruan Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res 2009;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang B, Wang J, Wang X, et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014;513:382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Chen F, Chandrashekar D, Varambally S, Creighton C. Proteogenomic characterization of 2002 human cancers reveals pan-cancer molecular subtypes and associated pathways. Nat Commun 2022;13:2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Creighton C, Huang S. Reverse phase protein arrays in signaling pathways: a data integration perspective. Drug Des Devel Ther 2015;9:3519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macklin A, Khan S, Kislinger T. Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clin Proteomics 2020;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mani D, Krug K, Zhang B, et al. Cancer proteogenomics: current impact and future prospects. Nat Rev Cancer 2022;22:298–313. [DOI] [PubMed] [Google Scholar]
- 9.Hoadley K, Yau C, Hinoue T, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018;173:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding L, Bailey M, Porta-Pardo E, et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018;173:305–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.International_Cancer_Genome_Consortium. International network of cancer genome projects. Nature 2010;464:993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bykov Y, Kim S, Zamarin D. Preparation of single cells from tumors for single-cell RNA sequencing. Methods Enzymol 2020;632:295–308. [DOI] [PubMed] [Google Scholar]
- 13.Perou C, Sørlie T, Eisen M, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52. [DOI] [PubMed] [Google Scholar]
- 14.Sørlie T, Perou C, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001;98:10869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen F, Zhang Y, Gibbons D, et al. Pan-cancer molecular classes transcending tumor lineage across 32 cancer types, multiple data platforms, and over 10,000 cases. Clin Cancer Res 2018;24:2182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martínez E, Yoshihara K, Kim H, Mills G, Treviño V, Verhaak R. Comparison of gene expression patterns across 12 tumor types identifies a cancer supercluster characterized by TP53 mutations and cell cycle defects. Oncogene 2015;34:2732–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoadley K, Yau C, Wolf D, et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014;158:929–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akbani R, Ng P, Werner H, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun 2014;E-pub May 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen F, Chandrashekar D, Varambally S, Creighton C. Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat Commun 2019;10:5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009. [DOI] [PubMed] [Google Scholar]
- 21.van ‘t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415:530–6. [DOI] [PubMed] [Google Scholar]
- 22.Bhattacharjee A, Richards W, Staunton J, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 2001;98:13790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 2002;8:816–24. [DOI] [PubMed] [Google Scholar]
- 24.Glinsky G, Glinskii A, Stephenson A, Hoffman R, Gerald W. Gene expression profiling predicts clinical outcome of prostate cancer. J Clin Invest 2004;113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu Y, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol 2004;22:2790–9. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Jatkoe T, Zhang Y, et al. Gene expression profiles and molecular markers to predict recurrence of Dukes’ B colon cancer. J Clin Oncol 2004;22:1564–71. [DOI] [PubMed] [Google Scholar]
- 27.Pomeroy S, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 2002;415:436–42. [DOI] [PubMed] [Google Scholar]
- 28.Chiaretti S, Li X, Gentleman R, et al. Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood 2004;103:2771–8. [DOI] [PubMed] [Google Scholar]
- 29.Rosenwald A, Wright G, Chan W, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002;346:1937–47. [DOI] [PubMed] [Google Scholar]
- 30.Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351 2817–26. [DOI] [PubMed] [Google Scholar]
- 31.Gray R, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9. [DOI] [PubMed] [Google Scholar]
- 32.Knezevic D, Goddard A, Natraj N, et al. Analytical validation of the Oncotype DX prostate cancer assay - a clinical RT-PCR assay optimized for prostate needle biopsies. BMC Genomics 2013;14:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The_Cancer_Genome_Atlas_Research_Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013;499:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pereira B, Chin S, Rueda O, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Lichtenberg T, Hoadley K, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018;173:400–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha V, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghandi M, Huang F, Jané-Valbuena J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garnett M, Edelman E, Heidorn S, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012;483:570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dempster J, Boyle I, Vazquez F, et al. Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome biology 2021;22:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsherniak A, Vazquez F, Montgomery P, et al. Defining a Cancer Dependency Map. Cell 2017;170:564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horak C, Pusztai L, Xing G, et al. Biomarker analysis of neoadjuvant doxorubicin/cyclophosphamide followed by ixabepilone or Paclitaxel in early-stage breast cancer. Clin Cancer Res 2013;19:1587–95. [DOI] [PubMed] [Google Scholar]
- 43.Iwamoto T, Bianchini G, Booser D, et al. Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J Natl Cancer Inst 2011;103:264–72. [DOI] [PubMed] [Google Scholar]
- 44.Hatzis C, Pusztai L, Valero V, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011;305:1873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen K, Qi Y, Song N, et al. Cell line derived multi-gene predictor of pathologic response to neoadjuvant chemotherapy in breast cancer: a validation study on US Oncology 02–103 clinical trial. BMC Med Genomics 2012;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korde L, Lusa L, McShane L, et al. Gene expression pathway analysis to predict response to neoadjuvant docetaxel and capecitabine for breast cancer. Breast Cancer Res Treat 2010;119:685–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prat A, Bianchini G, Thomas M, et al. Research-based PAM50 subtype predictor identifies higher responses and improved survival outcomes in HER2-positive breast cancer in the NOAH study. Clin Cancer Res 2014;20:511–21. [DOI] [PubMed] [Google Scholar]
- 48.Miyake T, Nakayama T, Naoi Y, et al. GSTP1 expression predicts poor pathological complete response to neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci 2012;103:913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nunnery S, Mayer I, Balko J. Triple-Negative Breast Cancer: Breast Tumors With an Identity Crisis. Cancer J 2021;27:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Creighton C, Nagaraja A, Hanash S, Matzuk M, Gunaratne P. A bioinformatics tool for linking gene expression profiling results with public databases of microRNA target predictions. RNA 2008;14:2290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pollack J, Sørlie T, Perou C, et al. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci U S A 2002;99:12963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018;173:321–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cancer_Genome_Atlas_Research_Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.The_Cancer_Genome_Atlas_Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, Kwok-Shing Ng P, Kucherlapati M, et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017;E-pub May 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanahan D, Weinberg R. The hallmarks of cancer. Cell 2000;100:57–70. [DOI] [PubMed] [Google Scholar]
- 57.Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006;439:353–7. [DOI] [PubMed] [Google Scholar]
- 58.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science 2013;339:957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horn S, Figl A, Rachakonda P, et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013;339:959–61. [DOI] [PubMed] [Google Scholar]
- 60.Davis C, Ricketts C, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014;26:319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.The_ICGC-TCGA_Pan-Cancer_Analysis_of_Whole_Genomes_Network. Pan-cancer analysis of whole genomes. Nature 2020;578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Chen F, Fonseca N, et al. High-coverage whole-genome analysis of 1220 cancers reveals hundreds of genes deregulated by rearrangement-mediated cis-regulatory alterations. Nat Commun 2020;11:736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rheinbay E, Nielsen MM, Abascal F, et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020;578:102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen F, Zhang Y, Creighton C. Systematic identification of non-coding somatic single nucleotide variants associated with altered transcription and DNA methylation in adult and pediatric cancers. NAR Cancer 2021;3:zcab001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chaffer C, Weinberg R. A perspective on cancer cell metastasis. Science 2011;331:1559–64. [DOI] [PubMed] [Google Scholar]
- 66.Barrett T, Wilhite S, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res 2013;41:D991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cosgrove N, Varešlija D, Keelan S, et al. Mapping molecular subtype specific alterations in breast cancer brain metastases identifies clinically relevant vulnerabilities. Nat Commun 2022;13:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Siegel M, He X, Hoadley K, et al. Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J Clin Invest 2018;128:1371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson D, Wu Y, Lonigro R, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pleasance E, Titmuss E, Williamson L, et al. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat Cancer 2020;1:452–68. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Chen F, Pleasance E, et al. Rearrangement-mediated cis-regulatory alterations in advanced patient tumors reveal interactions with therapy. Cell Rep 2021;37:110023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun H, Cao S, Mashl R, et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidatesfor targeted treatment. Nat Commun 2021;12:5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chandrashekar D, Bashel B, Balasubramanya S, et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017;19:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Monsivais D, Vasquez Y, Chen F, et al. Mass-spectrometry-based proteomic correlates of grade and stage reveal pathways and kinases associated with aggressive human cancers. Oncogene 2021;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Creighton C. The molecular profile of luminal B breast cancer. Biologics 2012;6:289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data File S1. Results represented in the figures. Provided as an Excel file. Results include pan-cancer molecular subtypes and associated genes (Figure 1), mRNA-level correlates of patient survival involving metabolism-related genes in breast cancer, clear cell renal cell carcinoma, and pan-cancer datasets (Figure 2), and gene expression correlates of pathologic chemotherapy response (pCR) in breast tumors (correcting for PAM50 subtype), along with GEO datasets and sample profiles analyzed.