

Abstract

PDE10A is an important regulator of striatal signaling that, when inhibited, can normalize dysfunctional activity. Given the involvement of dysfunctional striatal activity with schizophrenia, PDE10A inhibition represents a potentially novel means for its treatment. With the goal of developing PDE10A inhibitors, early optimization of a fragment hit through rational design led to a series of potent pyrimidine PDE10A inhibitors that required further improvements in physicochemical properties, off-target activities, and pharmacokinetics. Herein we describe the discovery of an isomeric pyrimidine series that addresses the liabilities seen with earlier compounds and resulted in the invention of compound 18 (MK-8189), which is currently in Phase 2b clinical development for the treatment of schizophrenia.
Introduction
Phosphodiesterases (PDEs) are an important class of enzymes that catalyze the hydrolysis of the second messengers cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), both of which play an important role in neuronal signaling.1 Of the 11 PDE families, PDE10A is highly expressed and localized in the mammalian striatum.2 Abnormal striatal output is strongly implicated in the pathophysiology of schizophrenia, and PDE10A inhibition provides a new mechanism to restore impaired striatal output.3,4
Schizophrenia is a chronic debilitating disorder with a lifetime prevalence estimated at 5 per 1000 in the U.S. population5 and is characterized by three symptom domains, including positive symptoms, negative symptoms, and cognitive impairment. Positive symptoms include psychosis, hallucinations, and delusions, while negative symptoms include apathy and amotivation. By increasing striatal cAMP and cGMP signaling, PDE10A inhibition has the potential to treat positive symptoms of schizophrenia, improve cognition, and address some of the limitations of current therapies. Atypical antipsychotics are the current standard of care medications used to treat schizophrenia. Although an advance over first generation antipsychotics, the atypical antipsychotics lack significant efficacy against negative and cognitive symptoms and are associated with significant side effects.6 Tolerability issues lead to poor patient compliance and include weight gain, extrapyramidal effects, increases in prolactin release, and sedation. Pre-clinical studies support the hypothesis that PDE10A inhibitors can address a number of these issues and provide an alternative to standard of care medicines.7−9 In this article, we describe the identification of an oral PDE10A lead molecule that is currently under clinical evaluation.
Results and Discussion
Our laboratories previously described the identification of 2-chloro-3-methyl pyrimidine 1, identified from a fragment screen, as a PDE10A inhibitor with high ligand binding efficiency (LBE, Figure 1).10 Rational design, aided by inhibitor-bound X-ray crystal structures and parallel library synthesis techniques, rapidly improved potency down to picomolar levels. Replacement of one of the chlorines in 1 with an aminomethyl-thiazole and modification of the cyclopropane to a quinoline-propyl-ether to engage the key Tyr683 residue in the PDE10A selectivity pocket resulted in compound 2. Despite excellent potency and LBE, compound 2 was not advanceable due to a poor pharmacokinetic (PK) profile (low oral bioavailability and high unbound clearance),11 low aqueous solubility, off-target ion channel activity against hERG, as determined by displacement of the ligand MK499 in a binding assay,12 reversible inhibition of cytochrome P450 enzymes (CYP2C9 and CYP3A4), and CYP3A4 PXR activation.
Figure 1.

Summary of early optimization in lead pyrimidine series.
The team continued efforts to improve the overall profile of the series by exploration of the western ether. Initial optimization identified trans-cyclopropyl pyridines as superior for metabolic stability, resulting in lower unbound clearance and higher oral bioavailability, while maintaining the bioactive conformation for potency.13 Scanning less lipophilic eastern heteroaryl rings to replace the dimethyl-thiazole of 2 then identified methyl-pyrazole 3. Compound 3 improved on many of the properties the team had been targeting, however further improvements in CYP inhibition, PXR activation, and unbound clearance were needed before a compound could be advanced from this series.
The team hypothesized that the chloro-methyl-pyrimidine core was likely responsible for the remaining issues within the series and needed to be modified. As a result, the team embarked on two strategies in parallel to replace the chloro-methyl-pyrimidine core in 3. The first strategy was to replace the pyrimidine with bicyclic ring systems. Evaluation of the original fragment screen data identified a pyrazolopyrimidine hit that led to the incorporation of a bicyclic pyrazolopyrimidine core in place of the chloro-methyl-pyrimidine core to give Pyp-1.13Pyp-1 displayed high passive permeability (Papp = 36 × 10–6 cm/s) and was not a substrate of the efflux transporter P-glycoprotein (P-gp, BA/AB ratio = 0.7), both of which are important features for developing CNS-active small molecules that cross the blood–brain barrier.14 Of note, replacing the core in 3 with alternate rings had a positive impact on the overall profile and further improved CYP inhibition and rat PK.
The second strategy was to modify the substitution on the monocyclic heteroaryl central core and interrogate the type of monocyclic 6-membered heteroaryl central core. To this end, several different types of central cores were prepared. Generally, these modular targets were prepared by sequential incorporation of an amine and alcohol on an activated heteroaryl core via displacement or coupling chemistry. As shown in Scheme 1, the 2-pyridyl-cyclopropyl-methanol precursors common to these targets were prepared by cyclopropanation of a vinyl pyridine or by Corey–Chaykovsky modification of an unsaturated ester, followed by ester reduction and SFC resolution, to give the (S,S) alcohols I-3, I-7, and I-11. Analogs 4 and 5 containing isomeric pyridine cores were prepared in 2 steps via sequential Mitsunobu ether formation with I-3 followed by SNAr mono-displacement of the resultant dichloro-pyridine with the aminomethyl-pyrazole (Scheme 2). The less substituted pyridine core in 6 was prepared by sequential palladium-catalyzed C–O and C–N couplings. Quinoline 7, 1,7-naphthyridine 9, and 1,5-naphthyridines 11 and 12 were prepared by incorporating the N-methyl-pyrazole- or methylthiadiazole-methylamine via SNAr displacement under basic conditions followed by palladium catalyzed C–O couplings with alcohol I-3 (Scheme 3).13,15 The opposite order of C–N and C–O bond formations was used for the 1,8-naphthyridine 8 and 1,6-naphthyridine 10 with the ether bond being formed first followed by the amine linkage. In the case of 1,8-naphthyridine 8, regioselective C–O coupling to 2,4-dichloro-1,8-naphthyridine (I-16) under palladium-catalyzed conditions was followed by SNAr displacement with N-methyl-pyrazole-methylamine. The isomeric 1,6-naphthyridine 10 was prepared via first Mitsunobu reaction between 2-hydroxy-4-chloro-1,6-naphthyridine and alcohol I-3 followed by palladium-catalyzed C–N coupling. Finally, the 2-methyl-pyrimidines 13–18 were prepared by sequential SNAr displacements first by the amine followed by the alcohol under basic conditions (Scheme 4).
Scheme 1. Synthesis of the Cyclopropyl-methanol Intermediates I-6, I-7, and I-11.

Reagents and conditions: (A) a) ethyl diazoacetate, toluene, 100 °C, 3 h, 44%; b) LAH, THF, 0 °C to rt, 20 min, then SFC, 98.7% ee. (B) c) potassium vinyl trifluoroborate, Pd(dppf)2Cl2, Cs2CO3, dioxane, water, 100 °C, 2 h; d) ethyl diazoacetate, toluene, 100 °C, 15 h, 41% over 2 steps; e) LAH, THF, 0 °C, 30 min, 85%, then SFC, >99% ee; (C) f) ethyl acrylate, Pd(OAc)2, K2CO3, Bu4NCl, DMF, 72%; g) trimethyl sulfoxonium iodide, NaH, DMSO, 50 °C, 40 min then I-9, DMSO, 5 min, 57%; h) LAH, THF, 0 °C to rt, 20 min, then SFC, >99% ee.
Scheme 2. Synthesis of Pyridine Core Analogs 4–6.

Reagents and conditions: (A) a) I-3, PS-triphenylphosphine, DIAD, THF, rt, 16 h, 77%. b) (1-methyl-1H-pyrazol-4-yl)methanamine, TEA, DMSO, 200 °C, 20 min, microwave, 21% (4), 5% (5); (B) c) I-3 (racemic), [1,1′-binaphthalen]-2-yldi-tert-butylphosphane, Pd(OAc)2, Cs2CO3, toluene, 80 °C, 15 h, 10%; d) (1-methyl-1H-pyrazol-4-yl)methanamine, DavePhos, Pd2(dba)3, NaOtBu, toluene, 80 °C, 2 h, 60%.
Scheme 3. Synthesis of Quinoline 7 and Naphthyridines 8–12.

Reagents and conditions: (A) a) (1-methyl-1H-pyrazol-4-yl)methanamine or (5-methyl-1,3,4-thiadiazol-2-yl)methanamine, DIPEA, NMP, 100–140 °C, 3–16 h, 45%–72%; b) I-3, [1,1′-binaphthalen]-2-yldi-tert-butylphosphane, Pd(OAc)2, Cs2CO3, toluene, 80 °C, 15 h, 31% (7); or I-3, Josiphos SL-J009-1, Pd2(dba)3, Cs2CO3, toluene, 140–160 °C, 0.5–15 h, 19%–51% (9, 11, 12); (B) c) I-3 (racemic), [1,1′-binaphthalen]-2-yldi-tert-butylphosphane, Pd(OAc)2, Cs2CO3, toluene, 80 °C, 1.5 h, 47% (I-17); d) I-3, triphenylphosphine, DIAD, THF, rt, 5 min, 37% (I-19); e) (1-methyl-1H-pyrazol-4-yl)methanamine, DIPEA, NMP, 100 °C, 48 h, 80% (8); f) (1-methyl-1H-pyrazol-4-yl)methanamine, DavePhos, Pd2(dba)3, NaOtBu, toluene, 120 °C, 40 min, 76% (10).
Scheme 4. Synthesis of 2-Methyl-pyrimidines 13–18.

Reagents and conditions: a) (1-methyl-1H-pyrazol-4-yl)methanamine or (5-methyl-1,3,4-thiadiazol-2-yl)methanamine, TEA, dioxane, 150 °C, 0.5 h, microwave, 70–83%; b) I-3, I-7, or I-11, NaH, THF, 100 °C, 1.5 h, microwave, 37–82%.
The importance of each nitrogen in the pyrimidine ring of 3 was evaluated first. As shown in Table 1, replacing each nitrogen of the pyrimidine in 3 demonstrated that the nitrogen in pyridine 4 was important for potency (Table 1 Entry 4 versus Entry 5). Removal of the 5-chloro substituent from 4 resulted in significant loss in potency, indicating that both substituents were important for potency (Table 1 Entry 4 versus Entry 6). Cyclization between the 5- and 6-positions of 4 gave quinoline 7, which showed excellent potency and selectivity. Not surprisingly, the quinoline ring resulted in poor pH 7 solubility. It was reasoned that incorporation of an additional nitrogen in the ring system may improve solubility, therefore the naphthyridine isomers were scanned as core replacements. Despite being more soluble, the 1,8-naphthyridine (8) did not show acceptable potency. The 1,7-naphthyridine 9 had a good balance between PDE10A potency, PDE selectivity, and pH 7 solubility, however 9 was a substrate for P-glycoprotein (P-gp) transport, which could limit CNS activity. The P-gp liability was even more pronounced in the 1,6-naphthyridine 10, precluding further investigation of this isomer. The best overall balance of PDE10A potency, selectivity, solubility, and P-gp ratio was observed with the 1,5-naphthyridine 11.
Table 1. Early Core Modifications Lead to Naphthyridines.


Each Ki value reported is an average of at least two measurements with a 10-point dose–response curve.
Given the more balanced profile of 11, improvements in potency and solubility were targeted with the 1,5-naphthyridine core. Replacing the N-methyl-pyrazole with a 5-methyl-1,3,4-thiadiazole (12) resulted in an 8-fold improvement in PDE10A potency and slightly better pH 7 solubility (Figure 2). Although MK499 binding indicated ion channel activity against hERG was slightly worse, it was encouraging that naphthyridine 12 improved on the profile of pyrimidine 3 with respect to PXR activation, reversible CYP inhibition, and rat unbound clearance.
Figure 2.

Profile of 1,5-naphthyridine improved by incorporation of eastern thiadiazole.
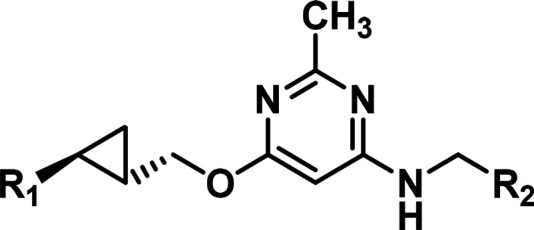
At this point on the program, the strategies to replace the 4-chloro-5-methyl-pyrimidine core in 3 had resulted in compounds with improved solubility, CYP, and PK profiles. Specifically, two lead compounds with bicyclic cores, Pyp-1 and 12, showed marked improvements over pyrimidine 3. While additional structure–activity relationships (SAR) on the eastern and western substituents were being investigated in these bicyclic series (data not shown), the team continued exploring new cores. It was hypothesized there might be an advantage to re-exploring monocyclic cores that were less lipophilic. Comparing both of the preferred bicyclic ring systems found in Pyp-1 and 12 revealed that each optimized bicyclic core had incorporated a nitrogen in proximity to the amine-linked substituent. Combining a proximal nitrogen with a monocyclic core resulted in an isomeric pyrimidine 13 (Figure 3). Although the potency of 13 against PDE10A was slightly compromised, the rest of the profile of 13 was remarkable. The selectivity over the other PDEs shifted favorably by more than an order of magnitude, the aqueous solubility was improved, no P-gp transporter activity and no binding to the hERG channel was observed, the reversible CYP inhibition and PXR activation were absent, the unbound clearance in rats was low, and the oral rat PK was excellent.
Figure 3.

Origins of isomeric 2-methyl-pyrimidine core.
In an effort to improve the potency of 13 while maintaining the excellent overall profile, the SAR of the eastern and western substituents was explored. Based on prior SAR with the 4-chloro-5-methyl-pyrimidine core, substituting the pendant pyridine ring of the western ether was expected to significantly improve potency.13 As shown in Table 2, incorporation of a methoxy (14) or a methyl (15) on the pyridine ring resulted in a 10-fold improvement in potency. Unfortunately, both 14 and 15 showed high unbound clearance in rats. Replacing the methyl-pyrazole in 13 with a methyl-1,3,4-thiadiazole (16) resulted in comparable potency and unbound clearance in rats. Combining the potency-enhancing methoxy-pyridine with the methyl-1,3,4-thiadiazole resulted in a highly potent and selective PDE10A inhibitor (17) with modest unbound clearance in rats. Incorporation of the methyl-pyridine combined with the methyl-1,3,4-thiadiazole (18) improved the PDE10A potency and increased the PDE selectivity, and the unbound clearance in rats remained in a good range. As such, compound 18 showed an optimal balance for the series and was chosen for further profiling.
Table 2. 2-Methyl-pyrimidine Core SARa.

Each Ki value reported is an average of at least two measurements with a 10-point dose–response curve. Rat CLu was calculated by dividing measured rat clearance by rat plasma free fraction.
The influence of 18 on the functional activity of PDE10A was evaluated using a fluorescence polarization assay measuring the ability of the test compound to inhibit hydrolysis of cAMP. The functional Ki of 18 on the human PDE10A enzyme is 0.029 nM. In addition, 18 is highly selective for PDE10A with greater than 500,000-fold selectivity over the other PDE enzyme families (PDE1–PDE11, Table SI-2). Compound 18 shows an IC50 of 1.6 nM in cells recombinantly expressing full-length human PDE10A. Kinetic analysis of 18 binding to full length human PDE10A2 using surface plasmon resonance indicates the on-rate of 18 is ∼8.0 × 107 M–1·s–1, the in vitro half-life on the enzyme is ∼6.7 min at 25 °C, and the binding is completely reversible.
The X-ray crystal structure of PDE10A catalytic domain (residues 439–779) in complex with 18 was determined at 2.1 Å resolution (Table SI-1). Residues are numbered as in the PDE10A1 splice variant (UniprotQ9Y233-1). Compound 18 forms several interactions with key residues in the active site of PDE10A (Figure 4). The pendant 5-methyl-pyridine reaches into the “selectivity pocket” and engages in a hydrogen-bond interaction with Tyr683.16 The pyrimidine core of 18 forms π-stacking interactions with the side chain of Phe719 and the N1 nitrogen hydrogen bonds with the side chain of Gln716. Additionally, the N3 nitrogen of the pyrimidine core and the 4-position amine linker form water-mediated hydrogen bonds with the side chains of Ser667 and Tyr514, respectively. The N3 nitrogen of the pyrimidine core is ideally positioned to pick up additional interactions with the binding site of PDE10A and is consistent with the favorable profile of 18 over previous lead series.
Figure 4.

Crystal structure of PDE10A catalytic domain in complex with 18 (PDB ID: 8DI4).
Compound 18 has low molecular weight (MW = 382) and reasonable lipophilicity (LogD = 2.1), resulting in an extremely efficient compound in terms of both ligand binding efficiency (LBE = 0.54) as well as ligand-lipophilicity efficiency (LLE = 7.8) (Figure 5). Remarkably the LBE was maintained throughout the optimization process from the initial fragment 1 (LBE = 0.57) to 18. Furthermore, compound 18 has excellent pharmaceutical properties. High-throughput solubility at pH7 is high (167 μM) for compound 18, which aligns well with the crystalline free base solubility of 0.17 mg/mL in simulated intestinal fluid (FaSSIF) and 5 mg/mL in acidic simulated gastric fluid (SGF).
Figure 5.

Profile of MK-8189 (18).
The pre-clinical PK profile of 18 in Wistar–Hannover rats and rhesus monkeys is summarized in Table 3. Compound 18 exhibited moderate plasma clearance and relatively low volume of distribution in rats and rhesus monkeys, resulting in a half-life of 4.8 h in rats and 4.2 h in rhesus monkeys. Oral bioavailability ranged from 41% in rhesus monkeys to 46% in rats. Compound 18 was significantly bound to plasma proteins in rat, monkey, and human. The mean values for the unbound fraction in plasma were 8.2% in rat, 8.7% in monkey, and 4.0% in human plasma. Compound 18 was not a potent reversible inhibitor of CYP2C9 or CYP3A4 (IC50 ≥ 50 μM), and 18 was not active in the PXR assay (EC50 > 30 μM).
Table 3. Pharmacokinetic Profile of 18 in Rats and Rhesus Monkeysa.
| Species | CLp (mL/min/kg) | Vdss (L/kg) | T1/2 (h) | F (%) |
|---|---|---|---|---|
| Rat | 21.3 | 0.93 | 4.8 | 46 |
| Rhesus | 9.3 | 1.4 | 4.2 | 41 |
Data represent mean values (n = 3). Rats were dosed 2 mg/kg IV and 10 mg/kg PO; rhesus were dosed 0.5 mg/kg IV and 5 mg/kg PO.
Compound 18 shows a promising in vitro safety and off-target profile (Figure 5). Compound 18 has an excellent profile against ion channels (Iks, Cav1.2, and Nav1.5 > 30 μM, and functional hERG Ikr IC50 = 33 μM). One off-target activity was identified in a broad Panlabs panel (somatostatin receptor type 2 (SSTR2) IC50 = 2.8 μM in a radioligand binding assay). Compound 18 is negative in a microbial mutagenesis assay (5-strain Ames test) and an assay for chromosomal aberrations in Chinese hamster ovary cells.
Transporter studies indicate high potential for CNS penetration with 18. Compound 18 has high passive permeability (35.4 to 42.6 × 10–6 cm/s) and is not a substrate of human and monkey P-gp (B-A/A-B ratio of <2). Despite being a weak substrate of rat P-gp (B-A/A-B ratio of ∼2.6), 18 achieved full enzyme occupancy in the rat striatum. In an in vivo enzyme occupancy study in rats, oral administration of 18 displaced [3H]MK-819317 in a plasma-concentration dependent manner (Figure 6). [3H]MK-8193 was previously disclosed by our labs as a high affinity PDE10A inhibitor with high-density binding sites localized primarily in the caudate-putamen and accumbens nucleus of rat brains.18 In this study, compound 18 (0.1–10 mg/kg, PO) was administered to male Wistar–Hannover rats 1 h prior to [3H]MK-8193 (20 mCi, IV injection). Striatal tissue was collected 0.5 h after [3H]MK-8193 injection, and an enzyme occupancy curve was generated. Compound 18 achieved 50% enzyme occupancy at plasma levels of 52 nM, and full enzyme occupancy was achieved at higher concentrations.
Figure 6.

Plasma concentration vs PDE10A occupancy in the rat striatum for 18, as determined by in vivo studies with [3H]MK-8193.
Consistent with the ability to achieve high levels of PDE10A target engagement, compound 18 showed the expected pharmacology in rodent models of psychosis and cognitive performance. Compound 18 was fully efficacious in the MK-801-induced psychomotor activity assay, an assay recognized to be predictive of antipsychotic potential in the clinic (Figure 7A).19 In this study, the compound was administered orally (0.25–0.75 mg/kg) 1 h prior to treatment with the non-competitive N-methyl-d-aspartate receptor antagonist MK-801, and locomotor activity was measured over 90 min.20 Compound 18 produced a robust dose-dependent and concentration-dependent decrease in the psychostimulant-induced hyperlocomotion in rats following PO dosing. Plasma concentrations at the end of the experiment ranged from 17 nM in the 0.25 mg/kg dose group to 50 nM in the 0.75 mg/kg dose group (Table 4). Striatal PDE10A occupancy derived from the plasma exposures revealed that efficacy was observed at approximately 25% enzyme occupancy and above.
Figure 7.

A) Attenuation of MK-801-induced locomotor activity in rats following oral administration of MK-8189 (18). Graph depicts mean ± standard error of mean (SEM) of total distance traveled for 90 min following administration of MK-801 for each treatment group. All doses of 18 (0.25, 0.50, and 0.75 mg/kg) significantly reduced distance traveled relative to vehicle + MK-801-treated animals. * indicates significantly greater distance than vehicle-treated animals (p-values <0.05), and α indicates significantly less distance than vehicle + MK-801-treated animals (p-values <0.05). B) Improvement of novel objection recognition in rats following oral administration of MK-8189 (18) and donepezil (Don; 0.56 mg/kg). MK-8189 (18) (0.16 and 0.25 mg/kg) and the positive control donepezil significantly improved recognition relative to vehicle-treated animals (p-values <0.05). * indicates significantly different than vehicle (Veh).
Table 4. MK-8189 (18) Dose, Plasma Exposure, and Striatal Occupancy Derived from Plasma Taken at the End of Rat Behavioral Studies.
| Rodent model | MK-8189 (18) dose (mg/kg) | Plasma at the end of the study (nM) | PDE10A Occ (%) derived from plasma |
|---|---|---|---|
| MK-801-induced locomotor activitya | 0.25 | 17 (4) | 25 (5) |
| 0.50 | 36 (10) | 41 (11) | |
| 0.75 | 50 (16) | 50 (16) | |
| Novel objection recognitionb | 0.08 | 16 (4) | 24 (6) |
| 0.16 | 18 (4) | 29 (6) | |
| 0.25 | 29 (8) | 34 (9) | |
Values reported as Mean (SEM), n = 8/group, and plasma samples taken 2.5 h post dose.
Values reported as Mean (SEM), n = 4/group, and plasma samples taken 1 h post dose.
Furthermore, compound 18 was efficacious in a test of episodic memory, the novel objection recognition assay in rats. As shown in Figure 7B, animals treated with 18 or the acetylcholinesterase inhibitor donepezil (Don) were able to differentiate an object they had been exposed to 48 h previously from a novel object.20 Improvement of novel objection recognition in rats was similar between Don dosed at 0.56 mg/kg and 18 dosed at 0.16 mg/kg and 0.25 mg/kg. Plasma concentrations at the end of the experiment ranged from 18 nM in the 0.16 mg/kg dose group to 29 nM in the 0.25 mg/kg dose group (Table 4). Striatal PDE10A occupancy derived from the plasma exposures revealed that significant effects were achieved at approximately 29% enzyme occupancy and above. The improvement of novel objection recognition in rats following oral administration of 18 indicates its potential ability to attenuate cognitive impairment in the patients with schizophrenia. Efficacy in the MK-801-induced psychomotor activity and novel object recognition tests in rats occurred at similar doses, indicating that similar extents of target engagement result in positive effects for both psychosis and cognitive performance.
In line with previous reports on PDE10A inhibitors,8 compound 18 showed a lack of effect on circulating levels of prolactin and reduced weight gain in rats (data not shown). Compared with atypical antipsychotics, compound 18 has a larger therapeutic window between antipsychotic-like effects and catalepsy in the rats (data not shown). Compound 18 was advanced into non-clinical safety studies, where it demonstrated a profile that supported progression to the clinic. Collectively, the favorable profile of 18 in terms of PDE10A potency, PDE selectivity, pharmaceutical properties, efficacy, tolerability, and toxicity, supported advancing 18 into humans, and compound 18 was approved for clinical development as MK-8189.
Conclusion
MK-8189 (18) is a potent and highly selective PDE10A inhibitor that is being developed as a novel therapeutic for the treatment of schizophrenia. Optimization of an early fragment hit led to a promising pyrimidine series. Modification to the core resulted in an isomeric pyrimidine series with improved physicochemical properties, off-target activities, and pharmacokinetics. Further refinements to the eastern and western heteroaryl rings resulted in the invention of MK-8189. MK-8189 has excellent PDE10A potency, PDE selectivity, pharmaceutical properties, ancillary profile, and oral PK in pre-clinical species. In vivo studies in rodents demonstrate PDE10A target engagement and efficacy in models of psychosis and cognition following oral administration. MK-8189 has been evaluated in several Phase 1 clinical studies and was found to be generally well tolerated at doses achieving high levels of trough PDE10A enzyme occupancy. The high level of sustained PDE10A occupancy achieved by MK-8189 enabled a more thorough test of PDE10A mechanism than compounds previously explored in the clinic,21,22 and an initial proof of concept in patients experiencing an acute episode of schizophrenia has provided evidence for antipsychotic efficacy.23 MK-8189 is currently in Phase 2b clinical development for the treatment of schizophrenia.24
Experimental Section
Chemistry: General Materials and Methods
All reagents and solvents were purchased from commercial sources and used as is without further purification. Reaction progress and synthetic intermediate analysis were assessed by liquid chromatography–mass spectrometry (LCMS) [UV detection with electrospray ionization (ESI) mass detection] when applicable using an Agilent instrument with a MeCN/water gradient with either a trifluoroacetic acid (TFA) or NH4HCO3 modifier. All reported yields are isolated yields. Silica gel and reverse-phase flash column chromatography were conducted with Teledyne ISCO CombiFlash instruments and commercially available pre-packed columns. Reverse-phase preparative high-pressure liquid chromatography (HPLC) purification of final analogs was performed on an Agilent preparative HPLC instrument with UV and MS detection using a MeCN/water gradient with either a TFA or a NH4OH modifier. All reported compounds tested in the assays were ≥95% pure as determined by LCMS or HPLC analysis. The 1H NMR spectra were collected at room temperature using Varian VXR spectrometers. Chemical shifts are reported in ppm relative to the listed deuterated solvent, and multiplicities, coupling constants (where applicable), and signal integrations are listed parenthetically. High-resolution mass spectrometry (HRMS) data were obtained using either a Waters Synapt G1 mass spectrometer under positive ESI mode or a Bruker Daltonics FTICR/MS in ESI mode.
((1S,2S)-2-(Pyridin-2-yl)cyclopropyl)methanol (I-6)
Step 1: A solution of 2-vinylpyridine (2.0 g, 19 mmol) in toluene (40 mL) was treated with ethyl diazoacetate (2.0 mL, 19 mmol) and stirred at reflux overnight. The mixture was concentrated in vacuo, and the residue was purified by gradient elution on silica gel (0 to 50% EtOAc in hexanes) to elute racemic trans ethyl 2-(pyridin-2-yl)cyclopropanecarboxylate as a yellow oil (1.6 g, 44%). ESI MS m/z = 192.1 [M+1]. 1H NMR (500 MHz, CDCl3): δ 8.44 (m, 1H), 7.56 (td, J = 7.6, 1.7 Hz, 1H), 7.22 (dd, J = 7.8, 1.0 Hz, 1H), 7.08 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H), 4.17 (q, J = 7.3 Hz, 2H), 2.58 (ddd, J = 10.0, 6.1, 3.9 Hz, 1H), 2.25 (ddd, J = 9.5, 5.6, 3.9 Hz, 1H), 1.61, (m, 2H), 1.28 (t, J = 7.1 Hz, 3H). Step 2: A solution of I-4 (0.75 g, 3.9 mmol) in THF (20 mL) was cooled to 0 °C and treated slowly with lithium aluminum hydride (3.9 mL, 3.9 mmol, 1 M solution in THF). The solution was warmed to room temperature and stirred for 20 min. The reaction mixture was then re-cooled to 0 °C and treated sequentially dropwise with 0.15 mL of water, 0.15 mL of 15% NaOH, and 0.45 mL of water. Sodium sulfate was added to the mixture. After stirring at room temperature for 10 min, the mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuo. Enantiomers of racemic trans-2-(pyridin-2-yl)cyclopropyl)methanol were resolved by chiral preparative SFC (3.0 cm i.d. × 25 cm ChiralPak AD-H, 3:7:90 MeCN/MeOH/CO2, 70 mL/min) and analyzed by chiral analytical SFC (4.6 mm i.d. × 25 cm ChiralPak AD-H, 3:7:90 MeCN/MeOH/CO2, 2.4 mL/min) ent1= 7.5 min, ent2= 8.4 min to afford ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol as a pale yellow oil in 98.7% ee. ESI MS m/z = 150.1 [M+1]. 1H NMR (500 MHz, CDCl3): δ 8.41 (d, J = 4.2 Hz, 1H), 7.52 (td, J = 7.6, 1.7 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 7.03 (ddd, J = 7.3, 4.9, 0.7 Hz, 1H), 3.72 (dd, J = 11,2, 6.4 Hz, 1H), 3.57 (dd, J = 11.2, 7.1 Hz, 1H), 2.26 (bs, 1H), 1.98 (m, 1H), 1.74 (m, 1H), 1.25 (m, 1H), 0.96 (m, 1H).
((1S,2S)-2-(5-Methylpyridin-2-yl)cyclopropyl)methanol (I-7)
Step 1: To a stirred solution of 2-bromo-5-methylpyridine (24 g, 0.14 mol) and potassium vinyl trifluoroborate (20 g, 0.15 mol) in dioxane (0.24 L) under nitrogen were added Pd(dppf)2Cl2 (11 g, 14 mmol), cesium carbonate (0.14 kg, 0.42 mol), and water (41 mL). The resulting mixture heated to 100 °C for 2 h. The reaction was partitioned between ethyl acetate and saturated sodium bicarbonate solution. The organic phase was concentrated, and flash column separation using a 0–30% ethyl acetate/hexane gradient gave 2-vinyl-5-methylpyridine as a volatile oil, which was carried on directly to the next step. Step 2: To a stirred solution of 2-vinyl-5-methylpyridine (16 g, 0.14 mol) in toluene (0.30 L) was added ethyl diazoacetate (43 mL, 0.41 mol), and the resulting mixture was heated to 100 °C for 3 h. The mixture was allowed to cool and partitioned between ethyl acetate and saturated sodium bicarbonate solution. The organic phase was concentrated, and flash column separation using a 0–20% ethyl acetate/hexane gradient gave ethyl 2-(5-methylpyridin-2-yl)cyclopropanecarboxylate as an oil (12 g, 41%). ESI MS m/z = 206.2 [M+1]. Step 3: A solution of ethyl 2-(5-methylpyridin-2-yl)cyclopropanecarboxylate (5.0 g, 24 mmol) in THF (80 mL) was cooled to 0 °C and treated slowly with lithium aluminum hydride (55 mL, 55 mmol, 1 M solution in THF). The solution was stirred for 30 min at 0 °C. The reaction mixture was then treated sequentially dropwise with 2.0 mL of water, 2.0 mL of 15% NaOH, and 6.0 mL of water. Sodium sulfate was added to the mixture. After stirring at room temperature for 4 h, the mixture was filtered, and the filtrate was concentrated in vacuo to afford racemic trans 2-(5-methylpyridin-2-yl)cyclopropyl]methanol as a pale yellow oil (3.4 g, 85%). The mixture was resolved by chiral preparative SFC (3.0 cm i.d. × 25 cm ChiralPak AS-H, 10% iPrOH/CO2 + 0.1% DEA, 80 mL/min) and analyzed by chiral analytical SFC (4.6 mm i.d. × 15 cm ChiralPak AS-H, 15% iPrOH/CO2 + 0.1% DEA, 2.4 mL/min) ent1= 3.7 min, ent2= 4.5 min to give ((1S,2S)-2-(5-methylpyridin-2-yl)cyclopropyl)methanol as the second eluting peak in >99% ee. ESI MS m/z = 164.2 [M+1]. 1H NMR (400 MHz, DMSO-d6): δ 8.21 (m, 1H), 7.41 (m, 1H), 7.12 (m, 1H), 4.57 (m, 1H) 3.44 (m, 1H), 3.33 (m, 1H), 2.21 (s, 3H), 1.88 (m, 1H), 1.44 (m, 1H), 0.98 (m, 1H), 0.81 (m, 1H).
((1S,2S)-2-(5-Methoxypyridin-2-yl)cyclopropyl)methanol (I-11)
Step 1: To a 25 mL microwave vial was added 2-bromo-5-methoxypyridine (1.9 g, 10 mmol), ethyl acrylate (5.4 mL, 50 mmol), Pd(OAc)2 (0.22 g, 1.0 mmol), K2CO3 (4.2 g, 30 mmol), and tert-butylammonium chloride hydrate (3.0 g, 10 mmol). The slurry was heated in the microwave at 160 °C for 1 h. Upon cooling to room temperature, the mixture was diluted with EtOAc (100 mL) and washed with sat. aq. NaHCO3 (100 mL). The aqueous layer was extracted with additional EtOAc (2 × 50 mL). The combined organic layers were then washed with water and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was purified by gradient elution on silica gel (0 to 50% EtOAc in hexanes) to afford ethyl 3-(5-methoxypyridin-2-yl)prop-2-enoate (1.5 g, 72%). ESI MS m/z = 208.0 [M+1]. 1H NMR (500 MHz, CDCl3): δ 8.35 (d, J = 3.0 Hz, 1H), 7.65 (d, J = 15.7 Hz, 1H), 7.38 (d, J = 8.6 Hz, 1H), 7.18 (dd, J = 8.6, 3.0 Hz, 1H), 6.76 (d, J = 15.7 Hz, 1H), 4.26 (q, J = 7.1 Hz, 2H), 3.89 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H). Step 2: To a solution of trimethyl sulfoxonium iodide (1.6 g, 7.2 mmol) in DMSO (24 mL) was added NaH (250 mg, 6.3 mmol). This mixture was stirred for 40 min at 50 °C. The reaction mixture was then cooled to room temperature, and to it was added a solution of ethyl 3-(5-methoxypyridin-2-yl)prop-2-enoate (1.0 g, 4.83 mmol) in DMSO (14 mL). The reaction mixture was stirred at room temperature for 5 min and then diluted with EtOAc (500 mL) and washed with sat. aq. NaHCO3 (4 × 100 mL). The organics were dried over MgSO4, filtered, and concentrated in vacuo. The resulting residue was purified by gradient elution on silica gel (20 to 100% EtOAc in hexanes) to afford ethyl 2-(5-methoxypyridin-2-yl)cyclopropanecarboxylate as a light yellow solid (500 mg, 47%). ESI MS m/z = 222.3 [M+1]. 1H NMR (500 MHz, CDCl3): δ 8.15 (d, J = 2.9 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 7.10 (dd, J = 8.0, 2.9 Hz, 1H), 4.16 (q, J = 7.1 Hz, 2H), 3.82 (s, 3H), 2.58–2.52 (m, 1H), 2.18–2.12 (m, 1H), 1.56 (m, 1H), 1.54 (m, 1H), 1.27 (t, J = 7.1 Hz, 3H). Step 3: Reduction was performed as described in Step 3 for compound I-7. The racemic mixture was resolved by chiral preparative SFC (3.0 cm i.d. × 25 cm ChiralPak AD-H, 6.7/13.3/80 MeCN/MeOH/CO2, + 0.1% DEA, 70 mL/min) and analyzed by chiral analytical SFC (4.6 cm i.d. × 25 cm ChiralPak AD-H, 6.7/13.3/80 MeCN/MeOH/CO2, + 0.1% DEA, mL/m) ent1= 3.7 min, ent2= 4.4 min to give ((1S,2S)-2-(5-methoxypyridin-2-yl)cyclopropyl)methanol as the second eluting peak and was isolated in >99% ee as a light yellow oil. ESI MS m/z = 180.1 [M+1]. 1H NMR (500 MHz, CDCl3): δ 8.14 (d, J = 2.9 Hz, 1H), 7.09 (dd, J = 8.6, 2.9 Hz, 1H), 7.04 (d, J = 8.6 Hz, 1H), 3.82 (s, 3H), 3.72–3.66 (m, 1H), 3.61–3.55 (m, 1H), 1.97–1.91 (m, 1H), 1.69–1.62 (m, 1H), 1.20–1.14 (m, 1H), 0.91 (dt, J = 8.71, 5.06 Hz, 1H).
2-Chloro-3-methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)-6-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyridin-4-amine (4)
Step 1: To a reaction vessel containing PS-triphenylphosphine resin (0.73 g, 2.6 mmol) was added a solution of 4,6-dichloro-5-methylpyridin-2-ol (0.21 g, 1.2 mmol) in THF (9 mL). The suspension was allowed to stand for 5 min, then a solution of DIAD (0.38 mL, 1.9 mmol) in THF (4.5 mL) was added. The mixture was agitated at room temperature for 30 min. A solution of ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (0.18 g, 1.2 mmol) in THF (4.5 mL) was added, and the reaction was stirred for 16 h. The mixture was filtered to remove resin, and the filtrate was treated with water (20 mL). The mixture was extracted with EtOAc (20 mL × 3), and combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure to give 850 mg of crude product. The crude material was purified by silica gel flash column chromatography, eluting with 0–100% EtOAc/hexanes over 20 min to give 0.28 g (77% yield) of 2,4-dichloro-3-methyl-6-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyridine as a clear oil. Step 2: To a 2 mL microwave vial were added 2,4-dichloro-3-methyl-6-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyridine (45 mg, 0.15 mmol), (1-methyl-1H-pyrazol-4-yl)methanamine (0.16 g, 1.5 mmol), triethylamine (41 μL, 0.29 mmol), and DMSO (0.45 mL). The mixture was heated under microwave irradiation at 200 °C for 20 min. After allowing the reaction to cool to room temperature, it was concentrated under a stream of N2 gas overnight. The reaction was carried out four times in a batchwise fashion. The four batches were combined to give 1.1 g of crude product as a brown oil, which was purified by silica gel flash column chromatography, eluting with 0–100% EtOAc/hexanes over 20 min to give 110 mg of a mixture of 2-chloro-3-methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)-6-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyridin-4-amine (4) and 4-chloro-3-methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)-6-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyridin-2-amine (5). Separation of 4 and 5 was carried out by reversed-phase HPLC (10–30%, 0.1% TFA in H2O/acetonitrile). The purified products were individually free-based by shaking with MP-Carbonate resin in dichloromethane. Filtration of the resin and concentration of the filtrate gave 47 mg (21% yield) of 4 and 11 mg (4.9% yield) of 5. HRMS (ESI+) m/z: [M+H]+ calculated for C20H22ClN5O + H, 384.1586; found, 384.1593. 1H NMR (500 MHz, DMSO-d6): δ 8.42 (d, J = 4.2 Hz, 1H), 7.70 (m, 1H), 7.55 (s, 1H), 7.33 (s, 1H), 7.32 (d, J = 7.0 Hz, 1H), 7.18 (m, 1H), 6.52 (m, 1H), 5.82 (s, 1H), 4.16 (m, 3H), 4.04 (dd, J = 12.0, 7.7 Hz, 1H), 3.76 (s, 3H), 2.15 (m, 1H), 2.06 (s, 3H), 1.77 (m, 1H), 1.18 (m, 1H), 1.05 (m, 1H).
4-Chloro-3-methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)-6-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyridin-2-amine (5)
Compound 5 was isolated in 5% yield from step 2 of the synthesis of 4. HRMS (ESI+) m/z: [M+H]+ calculated for C20H22ClN5O + H, 384.1586; found, 384.1590. 1H NMR (500 MHz, DMSO-d6): δ 8.39 (d, J = 4.2 Hz, 1H), 7.62 (m, 1H), 7.52 (s, 1H), 7.33 (s, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.11 (m, 1H), 6.60 (m, 1H), 5.99 (s, 1H), 4.35 (d, J = 5.6 Hz, 2H), 4.29 (dd, J = 11.2, 6.5 Hz, 1H), 4.12 (dd, J = 11.2, 7.6 Hz, 1H), 3.75 (s, 3H), 2.10 (m, 1H), 2.02 (s, 3H), 1.80 (m, 1H), 1.15 (m, 1H), 1.00 (m, 1H).
5-Methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyridin-4-amine (6)
Step 1: 2,4-Dichloro-5-methylpyridine, ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (racemic), [1,1′-binaphthalen]-2-yldi-tert-butylphosphane, Pd(OAc)2, Cs2CO3, toluene, 80 °C, 15 h, 10%. Step 2: 4-Chloro-5-methyl-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyridine, (1-methyl-1H-pyrazol-4-yl)methanamine, DavePhos, Pd2(dba)3, NaOtBu, toluene, 80 °C, 2 h, 60%. HRMS (ESI+) m/z: [M+H]+ calculated for C20H23N5O + H, 350.1975; found, 350.1978. 1H NMR (499 MHz, DMSO-d6): δ 8.38 (d, J = 4.4 Hz, 1H), 7.62 (t, J = 7.6 Hz, 1H), 7.55 (s, 1H), 7.48 (s, 1H), 7.33 (s, 1H), 7.27 (d, J = 7.8 Hz, 1H), 7.15–7.08 (m, 1H), 6.09 (s, 1H), 5.78 (s, 1H), 4.21–4.12 (m, 3H), 4.05 (dd, J = 11.2, 7.6 Hz, 1H), 3.76 (s, 2H), 2.12–2.06 (m, 1H), 1.95 (s, 3H), 1.79–1.72 (m, 1H), 1.16–1.10 (m, 1H), 0.99 (d, J = 8.7 Hz, 1H).
N-((1-Methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)quinolin-4-amine (7)
Step 1: Step 1 was conducted according to the procedure described in Step 5 for compound 12. Step 2: 2-Chloro-N-((1-methyl-1H-pyrazol-4-yl)methyl)quinolin-4-amine (18.3 mg, 0.067 mmol), ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (10 mg, 0.067 mmol), TrixiePhos (3.3 mg, 0.0084 mmol), cesium carbonate (55 mg, 0.17 mmol), and Pd(OAc)2 (1.5 mg, 0.0067 mmol) were combined in a 2–5 mL microwave vial, and toluene (0.45 mL) was added. The mixture was degassed and purged with nitrogen. The reaction mixture was heated at 80 °C for 15 h. The reaction mixture was filtered through a plug of Celite, washing with MeOH (2 × 5 mL), and then concentrated. The residue was dissolved in MeOH (4 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–65% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (50 mL). The organic layer was dried (MgSO4), filtered, and concentrated to afford N-((1-methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)quinolin-4-amine (8.2 mg, 31%) as an off-white solid. HRMS (ESI+) m/z: [M+H]+ calculated for C23H23N5O + H, 386.1975; found, 386.1964. 1H NMR (499 MHz, DMSO-d6): δ 8.39 (d, J = 4.6 Hz, 1H), 8.09 (d, J = 8.3 Hz, 1H), 7.66–7.59 (m, 2H), 7.56 (d, J = 7.9 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H), 7.43–7.36 (m, 2H), 7.30 (d, J = 7.8 Hz, 1H), 7.26 (t, J = 7.5 Hz, 1H), 7.12 (dd, J = 7.1, 5.1 Hz, 1H), 5.87 (s, 1H), 4.38 (dd, J = 11.3, 6.4 Hz, 1H), 4.30 (d, J = 5.5 Hz, 2H), 4.25 (dd, J = 11.3, 7.6 Hz, 1H), 3.77 (s, 3H), 2.18 (dt, J = 8.8, 4.6 Hz, 1H), 1.85 (m, 1H), 1.18 (dt, J = 8.6, 4.3 Hz, 1H), 1.07 (td, J = 9.1, 8.1, 3.5 Hz, 1H).
N-((1-Methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,8-naphthyridin-4-amine (8)
Step 1: 2,4-Dichloro-1,8-naphthyridine (27 mg, 0.13 mmol), ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (20 mg, 0.13 mmol), TrixiePhos (6.7 mg, 0.017 mmol), cesium carbonate (0.11 g, 0.34 mmol), and Pd(OAc)2 (3.0 mg, 0.013 mmol) were combined in a 2–5 mL microwave vial, and toluene (0.75 mL) was added. The mixture was degassed and purged with nitrogen. The reaction mixture was heated at 80 °C for 1.5 h. The reaction mixture was filtered through a plug of Celite, washing with MeOH (2 × 5 mL), and then concentrated. The residue was dissolved in MeOH (4 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–65% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (50 mL). The organic layer was dried (MgSO4), filtered, concentrated to afford 4-chloro-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,8-naphthyridine (20 mg, 47%) as an off-white solid. ESI MS m/z = 312.1 [M+1]. 1H NMR (499 MHz, DMSO-d6): δ 9.00 (d, J = 2.6 Hz, 1H), 8.53 (dd, J = 8.1, 1.5 Hz, 1H), 8.42 (d, J = 4.5 Hz, 1H), 7.70 (t, J = 7.2 Hz, 1H), 7.61 (dd, J = 8.1, 4.4 Hz, 1H), 7.48 (s, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.26–7.05 (m, 1H), 4.50 (ddd, J = 31.6, 11.4, 7.2 Hz, 2H), 2.29 (dt, J = 8.8, 4.6 Hz, 1H), 1.97 (d, J = 18.3 Hz, 1H), 1.33–1.10 (m, 2H). Step 2: To 4-chloro-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,8-naphthyridine (15 mg, 0.048 mmol) in NMP (0.50 mL) was added (1-methyl-1H-pyrazol-4-yl)methanamine (11 mg, 0.096 mmol). The resulting mixture was heated at 100 °C for 48 h. The reaction mixture was diluted with MeOH (1 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–95% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (50 mL). The organic layer was dried (MgSO4), filtered, and concentrated to afford N-((1-methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,8-naphthyridin-4-amine (15 mg, 80%) as a white solid. HRMS (ESI+) m/z: [M+H]+ calculated for C22H22N6O + H, 387.1928; found, 387.1932. 1H NMR (499 MHz, DMSO-d6): δ 8.73 (d, J = 2.7 Hz, 1H), 8.56 (d, J = 8.4 Hz, 1H), 8.39 (d, J = 4.5 Hz, 1H), 7.64 (d, J = 8.9 Hz, 2H), 7.39 (s, 1H), 7.29 (dd, J = 11.6, 6.2 Hz, 2H), 7.15–7.09 (m, 1H), 5.94 (s, 1H), 4.39 (dd, J = 11.4, 6.6 Hz, 1H), 4.31 (q, J = 7.5, 7.1 Hz, 2H), 3.77 (s, 3H), 2.22–2.17 (m, 1H), 1.88 (s, 2H), 1.21–1.16 (m, 1H), 1.12–1.04 (m, 2H).
N-((1-Methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,7-naphthyridin-4-amine (9)
Step 1: Step 1 was conducted according to the procedure described in Step 5 for compound 12. Step 2: 2-Chloro-N-((1-methyl-1H-pyrazol-4-yl)methyl)-1,7-naphthyridin-4-amine (27 mg, 0.10 mmol), ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (15 mg, 0.10 mmol), Josiphos (11 mg, 0.020 mol), cesium carbonate (98 mg, 0.30 mmol), and Pd2(dba)3 (9.2 mg, 0.010 mmol) were combined in a 0.5–2 mL microwave vial, and toluene (0.5 mL) was added. The mixture was degassed and purged with nitrogen. The reaction mixture was heated at 140 °C for 30 min in the microwave. The reaction mixture was filtered through a plug of Celite, washing with MeOH (2 × 5 mL), and then concentrated. The residue was dissolved in MeOH (4 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–75% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (100 mL). The organic layer was dried (MgSO4), filtered, and concentrated to afford N-((1-methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,7-naphthyridin-4-amine (7.6 mg, 19%) as an off-white solid. HRMS (ESI+) m/z: [M+H]+ calculated for C22H22N6O + H, 387.1928; found, 387.1933. 1H NMR (499 MHz, DMSO-d6): δ 9.15 (s, 1H), 8.54 (d, J = 5.7 Hz, 2H), 8.39 (d, J = 5.9 Hz, 1H), 8.00 (s, 1H), 7.67 (s, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.43 (s, 1H), 6.25 (s, 1H), 4.54–4.46 (m, 2H), 4.46–4.31 (m, 2H), 3.78 (s, 3H), 3.70 (s, 2H), 2.36 (s, 1H), 2.01 (s, 1H), 1.37 (d, J = 4.4 Hz, 1H), 1.32 (s, 1H).
N-((1-Methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,6-naphthyridin-4-amine (10)
Step 1: 4-Chloro-1,6-naphthyridin-2-ol (0.24 g, 1.3 mmol), ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (0.20 g, 1.3 mmol), and triphenylphosphine (0.45 g, 1.7 mmol) were combined in THF (6.6 mL) followed by DIAD (0.34 mL, 1.7 mmol). The resulting mixture was stirred at 0 °C for 5 min. A ∼3:2 mixture of regioisomers is observed by LCMS (major is the desired O-alkylated product which is the less polar (2nd eluting) peak by LCMS). The reaction mixture was concentrated. The residue was dissolved in MeOH (4 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–50% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (50 mL). The organic layer was dried (MgSO4), filtered, concentrated to afford 4-chloro-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,6-naphthyridine (Peak 2 from reverse-phase; 155 mg, 37%) as a colorless oil. HRMS (ESI+) m/z: [M+H]+ calculated for C17H14ClN3O + H, 312.0898; found, 312.0904. 1H NMR (499 MHz, DMSO-d6): δ 9.36 (s, 1H), 8.74 (d, J = 5.8 Hz, 1H), 8.40 (d, J = 4.4 Hz, 1H), 7.73 (d, J = 5.8 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.47 (s, 1H), 7.32 (d, J = 7.8 Hz, 1H), 7.22–7.02 (m, 1H), 4.50 (ddd, J = 48.2, 11.3, 7.2 Hz, 2H), 2.27 (dt, J = 8.7, 4.5 Hz, 1H), 1.92 (s, 1H), 1.19 (ddt, J = 46.9, 9.0, 4.5 Hz, 2H). Step 2: 4-Chloro-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,6-naphthyridine (30 mg, 0.096 mmol), (1-methyl-1H-pyrazol-4-yl)methanamine (16 mg, 0.14 mmol), DavePhos (5.7 mg, 0.014 mol), sodium tert-butoxide (11 mg, 0.12 mmol), and Pd2(dba)3 (4.4 mg, 0.0048 mmol) were combined in a 0.5–2 mL microwave vial, and toluene (0.5 mL) was added. The mixture was degassed and purged with nitrogen. The reaction mixture was heated at 120 °C for 40 min in the microwave. The reaction mixture was filtered through a plug of Celite, washing with MeOH (2 × 5 mL), and then concentrated. The residue was dissolved in MeOH (2 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–75% CH3CN/water w/0.1% TFA modifier over 15 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (50 mL). The organic layer was dried (MgSO4), filtered, and concentrated to afford N-((1-methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,6-naphthyridin-4-amine (28 mg, 76%) as an off-white solid. HRMS (ESI+) m/z: [M+H]+ calculated for C22H22N6O + H, 387.1928; found, 387.1934. 1H NMR (499 MHz, DMSO-d6): δ 9.38 (s, 1H), 8.47 (d, J = 5.7 Hz, 1H), 8.39 (d, J = 4.0 Hz, 1H), 7.91 (s, 1H), 7.63 (d, J = 10.9 Hz, 2H), 7.40 (s, 1H), 7.30 (d, J = 7.9 Hz, 1H), 7.16–7.10 (m, 1H), 5.95 (s, 1H), 4.41 (dd, J = 11.2, 6.6 Hz, 1H), 4.35–4.25 (m, 3H), 3.77 (s, 3H), 3.34 (m, 2H), 2.22–2.16 (m, 1H), 1.21–1.16 (m, 1H), 1.12–1.06 (m, 1H).
N-((1-Methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,5-naphthyridin-4-amine (11)
Step 1: To 2,4-dichloro-1,5-naphthyridine (0.12 g, 0.60 mmol) in NMP (1.0 mL) was added (1-methyl-1H-pyrazol-4-yl)methanamine (74 mg, 0.66 mmol). The resulting mixture was heated at 140 °C for 3 h. More (1-methyl-1H-pyrazol-4-yl)methanamine (50 mg, 0.45 mmol) was added, and the mixture was heated at 140 °C for another 3 h. The reaction mixture was diluted with MeOH (1 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–65% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were concentrated to afford 2-chloro-N-((1-methyl-1H-pyrazol-4-yl)methyl)-1,5-naphthyridin-4-amine (0.10 g, 62%) as a tan oil. ESI MS m/z = 274.2 [M+H]. 1H NMR (499 MHz, DMSO-d6): δ 8.77 (dd, J = 4.1, 1.4 Hz, 1H), 8.31 (s, 1H), 8.11 (d, J = 8.5 Hz, 1H), 7.74 (dd, J = 8.5, 4.2 Hz, 1H), 7.67 (s, 1H), 7.44 (s, 1H), 6.68 (s, 1H), 4.42 (d, J = 5.7 Hz, 2H), 3.78 (s, 3H). Step 2: 2-Chloro-N-((1-methyl-1H-pyrazol-4-yl)methyl)-1,5-naphthyridin-4-amine (0.10 g, 0.37 mmol), ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (50 mg, 0.34 mmol), Josiphos (37 mg, 0.067 mol), cesium carbonate (0.33 g, 1.0 mmol), and Pd2(dba)3 (31 mg, 0.034 mmol) were combined in a 0.5–2 mL microwave vial, and toluene (1.7 mL) was added. The mixture was degassed and purged with nitrogen. The reaction mixture was heated at 160 °C for 15 h. The reaction mixture was filtered through a plug of Celite, washing with MeOH (2 × 5 mL), and then concentrated. The residue was dissolved in MeOH (3 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–75% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (100 mL). The organic layer was dried (MgSO4), filtered, and concentrated to afford N-((1-methyl-1H-pyrazol-4-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,5-naphthyridin-4-amine (34 mg, 26%) as an off-white solid. HRMS (ESI+) m/z: [M+H]+ calculated for C22H22N6O + H, 387.1928; found, 387.1929. 1H NMR (499 MHz, DMSO-d6): δ 8.72 (s, 1H), 8.51 (d, J = 4.6 Hz, 1H), 8.01 (d, J = 8.5 Hz, 1H), 7.87 (s, 1H), 7.79 (s, 1H), 7.71 (s, 1H), 7.52–7.38 (m, 2H), 7.34 (s, 1H), 6.39 (s, 1H), 4.53 (s, 2H), 4.48–4.39 (m, 1H), 3.78 (s, 3H), 3.10 (s, 2H), 2.34 (s, 1H), 1.99 (s, 1H), 1.36 (d, J = 4.2 Hz, 1H), 1.27 (s, 1H).
N-((5-Methyl-1,3,4-thiadiazol-2-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,5-naphthyridin-4-amine (12)
Step 1: To 3-aminopicolinic acid (21 g, 0.14 mol) in EtOH (80 mL) was added sulfuric acid (25 mL, 0.47 mol). The resulting mixture was stirred at reflux for 70 h. The reaction mixture was cooled to ambient temperature and then concentrated under reduced pressure to about half the volume. The resulting mixture was cooled in an ice bath at 0 °C and basified with NH4OH to pH ∼8 with stirring to form a white precipitate. The mixture was filtered, and the solids were washed with water (2 × 15 mL) and dried in vacuo to afford ethyl 3-aminopicolinate as a white solid (18.2 g, 76%). ESI MS m/z = 167.1 [M+H]. Step 2: To diethyl malonate (73 mL, 0.48 mol) stirring at 120 °C was added ethyl 3-aminopicolinate (10 g, 60 mmol) in portions. The resulting mixture stirred at the same temperature overnight. The reaction mixture was cooled to ambient temperature and concentrated under reduced pressure. To the resulting mixture was added sodium ethoxide (21% solution in EtOH, 22 mL, 60 mmol), and the mixture was stirred at 100 °C for 1 h. The mixture was cooled to ambient temperature, Et2O (100 mL) was added to the reaction mixture, and the mixture was filtered. The solids were washed with Et2O (2 × 25 mL) and then dried in vacuo to afford ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,5-naphthyridine-3-carboxylate as an off-white solid (13.6 g, 96%). ESI MS m/z = 235.0715 [M+H]. Step 3: Ethyl 4-hydroxy-2-oxo-1,2-dihydro-1,5-naphthyridine-3-carboxylate (14 g, 60 mmol) was suspended in water (1 mL), and sodium hydroxide (10 M, 54 mL, 0.54 mol) was added. The resulting mixture stirred at 100 °C for 2 h. The reaction mixture was cooled to ambient temperature and carefully acidified with acetic acid to pH ∼5. The mixture was filtered, washed with water (2 × 25 mL), and dried in vacuo to afford 4-hydroxy-1,5-naphthyridin-2(1H)-one as a pale yellow solid (9.4 g, 96%). ESI MS m/z = 163.1 [M+H]. Step 4: To 4-hydroxy-1,5-naphthyridin-2(1H)-one (12 g, 74 mmol) was added POCl3 (0.14 L, 1.5 mol). The resulting mixture stirred at reflux for 30 min. The reaction mixture was cooled to ambient temperature and then carefully concentrated under reduced pressure. The residue was cooled to 0 °C in an ice bath and was treated with water, followed by NH4OH to pH ∼8. The mixture was filtered. The solids were washed with water (2 × 25 mL) and dried in vacuo to afford 2,4-dichloro-1,5-naphthyridine as a gray solid (11.7 g, 79%). ESI MS m/z = 199.0 [M+H]. Step 5: To a solution of 2,4-dichloro-1,5-naphthyridine (0.47 g, 2.4 mmol) in NMP (3.9 mL) was added (5-methyl-1,3,4-thiadiazol-2-yl)methanamine (0.61 g, 4.7 mmol) followed by Hunig’s base (1.2 mL, 7.1 mmol). The resulting mixture stirred at 140 °C for 3 h. The reaction mixture was purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–65% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (150 mL). The organic layer was dried (MgSO4), filtered, and concentrated to afford 2-chloro-N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)-1,5-naphthyridin-4-amine as a yellow solid (0.50 g, 72%). ESI-MS m/z = 292.1 [M+H]. Step 6: 2-Chloro-N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)-1,5-naphthyridin-4-amine (0.34 g, 1.2 mmol), ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (0.16 g, 1.1 mmol), Josiphos SL-J009-1 (59 mg, 0.11 mol), cesium carbonate (0.52 g, 1.6 mmol), and Pd2(dba)3 (49 mg, 0.054 mmol) were combined in a 2–5 mL microwave vial, and toluene (4.3 mL) was added. The mixture was degassed and purged with nitrogen. The vial was sealed, and the reaction mixture was heated at 140 °C for 30 min in the microwave. The reaction mixture was filtered through a plug of Celite, washed with MeOH (2 × 5 mL), and then concentrated. The residue was dissolved in MeOH (4 mL) and purified directly by preparative reverse-phase chromatography (Waters Sunfire 5 μm OBD C18, 5–65% CH3CN/water w/0.1% TFA modifier over 20 min). Pure fractions were combined, neutralized by the addition of aq. NaHCO3 (5 mL), and extracted with EtOAc (100 mL). The organic layer was dried (MgSO4), filtered, and concentrated to afford N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)-2-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)-1,5-naphthyridin-4-amine as an off-white solid (0.22 g, 51%). HRMS (ESI+) m/z: [M+H]+ calculated for C21H20N6OS + H, 405.1491; found, 405.1492. 1H NMR (499 MHz, DMSO-d6): δ 8.63 (dd, J = 4.2, 1.5 Hz, 1H), 8.39 (d, J = 4.0 Hz, 1H), 8.12 (t, J = 6.5 Hz, 1H), 8.00 (dd, J = 8.4, 1.5 Hz, 1H), 7.63 (ddd, J = 7.6, 4.9, 3.1 Hz, 2H), 7.30 (d, J = 7.8 Hz, 1H), 7.15–7.09 (m, 1H), 6.13 (s, 1H), 4.92 (d, J = 6.5 Hz, 2H), 4.39 (dd, J = 11.3, 6.6 Hz, 1H), 4.28 (dd, J = 11.3, 7.6 Hz, 1H), 2.64 (s, 3H), 2.19 (dt, J = 8.8, 4.7 Hz, 1H), 1.90–1.79 (m, 1H), 1.19 (dq, J = 11.1, 6.7, 5.4 Hz, 1H), 1.12–1.06 (m, 1H).
2-Methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)-6-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyrimidin-4-amine (13)
Step 1: To a stirred solution of 4,6-dichloro-2-methylpyrimidine (0.10 g, 0.61 mmol) in dioxane (1 mL) were added triethylamine (0.30 mL, 2.2 mmol) and (1-methyl-1H-pyrazol-4-yl)methanamine (68 mg, 0.61 mmol). The resulting mixture was microwave irradiated at 150 °C for 30 min. The reaction was diluted with ethyl acetate and washed with saturated sodium bicarbonate solution. The organic phase was concentrated, and flash column separation using a 10–100% ethyl acetate/hexane gradient gave 6-chloro-2-methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)pyrimidin-4-amine as a white solid (0.12 g, 83%). HRMS (ESI+) m/z: [M+H]+ calculated for C10H12ClN5 + H, 238.0854; found, 238.0857. Step 2: To a stirred solution of ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol (0.19 g, 1.3 mmol) in THF (2.5 mL) was added a sodium hydride 60% dispersion (74 mg, 1.8 mmol), and the resulting solution was stirred at room temperature for 20 min. To this was added 6-chloro-2-methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)pyrimidin-4-amine (0.20 g, 0.84 mmol), and the resulting mixture was microwave irradiated at 100 °C for 90 min. The reaction was concentrated and purified using reverse-phase chromatography (10–30%, 0.1% TFA in H2O/acetonitrile) to give 2-methyl-N-[(1-methyl-1H-pyrazol-4-yl)methyl]-6-[(2-pyridin-2-ylcyclopropyl)methoxy] pyrimidin-4-amine as a white solid (0.23 g, 78%). HRMS (ESI+) m/z: [M+H]+ calculated for C19H22N6O + H, 351.1928; found, 351.1927. 1H NMR (499 MHz, DMSO-d6): δ 8.39 (d, J = 3.4 Hz, 1H), 7.63 (dt, J = 7.6, 3.8 Hz, 1H), 7.56 (s, 1H), 7.33 (s, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.21 (m, 1H), 7.15–7.09 (m, 1H), 5.55 (s, 1H), 4.24 (m, 3H), 4.12 (m, 1H), 3.76 (s, 3H), 2.28 (s, 3H), 2.13 (m, 1H), 1.75 (m, 1H), 1.21–1.12 (m, 1H), 1.02 (m, 1H).
6-(((1S,2S)-2-(5-Methoxypyridin-2-yl)cyclopropyl)methoxy)-2-methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)pyrimidin-4-amine (14)
Compound 14 was prepared from (1-methyl-1H-pyrazol-4-yl)methanamine and ((1S,2S)-2-(5-methoxypyridin-2-yl)cyclopropyl)methanol according to the procedures described for compound 13. HRMS (ESI+) m/z: [M+H]+ calculated for C20H24N6O2 + H, 381.2034; found, 381.2039. 1H NMR (499 MHz, DMSO-d6): δ 8.12 (d, J = 2.7 Hz, 1H), 7.56 (s, 1H), 7.33 (s, 1H), 7.29–7.19 (m, 2H), 5.55 (s, 1H), 4.21 (m, 3H), 4.11 (dd, J = 11.0, 7.7 Hz, 1H), 3.77 (d, J = 3.6 Hz, 6H), 2.28 (s, 3H), 2.09 (dt, J = 8.8, 4.6 Hz, 1H), 1.65 (m, 1H), 1.12–1.04 (m, 1H), 0.96 (dd, J = 8.0, 4.8 Hz, 1H).
2-Methyl-N-((1-methyl-1H-pyrazol-4-yl)methyl)-6-(((1S,2S)-2-(5-methylpyridin-2-yl)cyclopropyl)methoxy)pyrimidin-4-amine (15)
Compound 15 was prepared from (1-methyl-1H-pyrazol-4-yl)methanamine and ((1S,2S)-2-(5-methylpyridin-2-yl)cyclopropyl)methanol according to the procedure described for compound 13. HRMS (ESI+) m/z: [M+H]+ calculated for C20H24N6O + H, 365.2084; found, 365.2091. 1H NMR (499 MHz, DMSO-d6): δ 8.22 (s, 1H), 7.56 (s, 1H), 7.45 (dd, J = 7.9, 1.8 Hz, 1H), 7.32 (s, 1H), 7.20 (m, 1H), 7.17 (d, J = 7.9 Hz, 1H), 5.55 (s, 1H), 4.22 (m, 3H), 4.11 (dd, J = 11.2, 7.6 Hz, 1H), 3.76 (s, 3H), 2.27 (s, 3H), 2.22 (s, 3H), 2.08 (dt, J = 8.7, 4.6 Hz, 1H), 1.70 (m, 1H), 1.11 (dt, J = 8.6, 4.4 Hz, 1H), 0.98 (m, 1H).
2-Methyl-N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)-6-(((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methoxy)pyrimidin-4-amine (16)
Compound 16 was prepared from (5-methyl-1,3,4-thiadiazol-2-yl)methanamine and ((1S,2S)-2-(pyridin-2-yl)cyclopropyl)methanol according to the procedure described for compound 13. HRMS (ESI+) m/z: [M+H]+ calculated for C18H20N6OS + H, 369.1492; found, 369.1493. 1H NMR (499 MHz, DMSO-d6): δ 8.39 (d, J = 4.0 Hz, 1H), 7.83 (m, 1H), 7.64 (td, J = 7.6, 1.6 Hz, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.16–7.11 (m, 1H), 5.65 (s, 1H), 4.77 (d, J = 5.7 Hz, 2H), 4.25 (dd, J = 11.2, 6.6 Hz, 1H), 4.15 (dd, J = 11.2, 7.5 Hz, 1H), 2.65 (s, 3H), 2.31 (s, 3H), 2.15 (dt, J = 8.8, 4.7 Hz, 1H), 1.76 (m, 1H), 1.17 (dt, J = 8.8, 4.5 Hz, 1H), 1.03 (dt, J = 9.1, 5.2 Hz, 1H).
6-(((1S,2S)-2-(5-Methoxypyridin-2-yl)cyclopropyl)methoxy)-2-methyl-N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)pyrimidin-4-amine (17)
Compound 17 was prepared from (5-methyl-1,3,4-thiadiazol-2-yl)methanamine and ((1S,2S)-2-(5-methoxypyridin-2-yl)cyclopropyl)methanol according to the procedure described for compound 13. HRMS (ESI+) m/z: [M+H]+ calculated for C18H20N6OS + H, 399.1598; found, 399.1586. 1H NMR (499 MHz, DMSO-d6): δ 8.12 (d, J = 2.7 Hz, 1H), 7.82 (m, 1H), 7.26 (dd, J = 8.5, 2.8 Hz, 1H), 7.22 (d, J = 8.6 Hz, 1H), 5.64 (s, 1H), 4.77 (d, J = 5.6 Hz, 2H), 4.22 (dd, J = 11.1, 6.7 Hz, 1H), 4.13 (dd, J = 11.1, 7.5 Hz, 1H), 3.77 (s, 3H), 2.65 (s, 3H), 2.31 (s, 3H), 2.09 (dt, J = 8.9, 4.8 Hz, 1H), 1.70–1.61 (m, 1H), 1.11–1.04 (m, 1H), 0.99–0.92 (m, 1H).
2-Methyl-N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)-6-(((1S,2S)-2-(5-methylpyridin-2-yl)cyclopropyl)methoxy)pyrimidin-4-amine (18)
Step 1: To a stirred solution of 4,6-dichloro-2-methylpyrimidine (0.25 g, 1.5 mmol) in dioxane (3 mL) were added triethylamine (0.70 mL, 5.0 mmol) and (5-methyl-1,3,4-thiadiazol-2-yl)methanamine hydrochloride (0.31 g, 1.9 mmol). The resulting mixture was microwave irradiated at 150 °C for 30 min. The reaction was diluted with ethyl acetate and washed with saturated sodium bicarbonate solution. The organic phase was concentrated, and flash column separation using a 10–80% ethyl acetate/hexane gradient gave 6-chloro-2-methyl-N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)pyrimidin-4-amine as a white solid (0.28 g, 72%). HRMS (ESI+) m/z: [M+H]+ calculated for C9H10ClN5S + H, 256.0418; found, 256.0411. Step 2: To a stirred solution of ((1S,2S)-2-(5-methylpyridin-2-yl)cyclopropyl)methanol (0.46 g, 2.8 mmol) in THF (7.5 mL) was added sodium hydride 60% dispersion (0.16 g, 4.1 mmol), and the resulting solution was stirred at room temperature for 20 min. To this was added 6-chloro-2-methyl-N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)pyrimidin-4-amine (0.48 g, 1.9 mmol), and the resulting mixture was microwave irradiated at 100 °C for 90 min. The reaction was concentrated and purified using reverse-phase chromatography (10–30%, 0.1% TFA in H2O/acetonitrile) gave 2-methyl-N-((5-methyl-1,3,4-thiadiazol-2-yl)methyl)-6-(((1S,2S)-2-(5-methylpyridin-2-yl)cyclopropyl)methoxy)pyrimidin-4-amine as a white solid (0.31 g, 43%). HRMS (ESI+) m/z: [M+H]+ calculated for C19H22N6OS + H, 383.1649; found, 383.1648. 1H NMR (400 MHz, DMSO-d6): δ 8.22 (s, 1H), 7.82 (br t, J = 5.0 Hz, 1H), 7.45 (d, J = 7.9 Hz, 1H), 7.17 (d, J = 7.7 Hz, 1H), 5.64 (s, 1H), 4.76 (d, J = 5.0 Hz, 2H), 4.23 (m, 1H), 4.14 (m, 1H), 2.65 (s, 3H), 2.31 (s, 3H), 2.22 (s, 3H), 2.09 (m, 1H), 1.71 (m, 1H), 1.12 (m, 1H), 0.98 (m, 1H).
In Vitro Pharmacology: General Materials and Methods
PDE Biochemical Assays
Phosphodiesterase (PDE) activity was determined using an IMAP FP kit (Molecular Devices, Sunnyvale, CA) as previously described (Huang et al., 2002). Human PDE10A2 and rhesus monkey PDE2A3 enzymes were prepared from cytosolic fractions of transiently transfected AD293, and PDE6 was purified from bovine retina. All other PDEs (PDE1A, PDE3A, PDE4A1A, PDE5A1, PDE7A, PDE8A1, PDE9A2, and PDE11A4) were GST-tagged human enzymes expressed in insect cells and were obtained from BPS Bioscience (San Diego, CA). PDE activity assays were performed in duplicate at room temperature. The apparent inhibition constant (Ki) for MK-8189 against all 11 PDEs were determined as described by Mosser25 using the following apparent Km values for each enzyme and substrate combination: human PDE1A (cGMP, 70 nM), rhesus PDE2A3 (cAMP, 10,000 nM), human PDE3A (cAMP, 50 nM), human PDE4A1 (cAMP, 1500 nM), human PDE5A1 (cGMP, 400 nM), human PDE6C (cGMP, 700 nM), human PDE7A (cAMP, 150 nM), human PDE8A1 (cAMP, 50 nM), human PDE9A2 (cGMP, 60 nM), human PDE10A2 (cAMP, 150 nM), and human PDE11A4 (cAMP, 1000 nM).
Dissociation Rate Determination by Surface Plasmon Resonance (SPR)
SPR was performed on a Biacore T-200 instrument (BIAcore, GE Lifesciences). A CM5-NTA chip activated with both nickel sulfate and EDC/NHS was used to attach His-tagged-PDE10A. In practice 500 μM nickel sulfate in ddH2O was injected over the surface for 60 s followed by a solution of 200 mM EDC/50 mM NHS for 420 s at a flow rate of 10 μL/min with running buffer (10 mM Tris·HCl, pH 7.2, 10 mM MgCl2, 0.01% P-20, 1 mM DTT, and 2.00% DMSO, filtered through a 0.2 μm PES membrane). PDE10A-His-tagged construct from Proteos (Lot 2 07-29-09. NTH-Thrombin-449-789) was diluted from 5.4 mg/mL to 5 μg/mL in running buffer and injected over flowcell 2 for 300 s at a flow rate of 10 μL/min, followed by deactivation with 1.0 M, pH 8.5 ethanolamine for 150 s to achieve approximately 7000 RU of bound enzyme. The binding assay was performed by injecting 2.00% DMSO solutions of MK-8189 in running buffer at 10 different concentrations, from 1.3 × 10–3 nM to 5.0 nM. The 2.5× serial dilution of compound was made in 100% DMSO at 50× concentration followed by subsequent dilution and mixing with running buffer. Injections were performed simultaneously over the immobilized and reference surfaces, and the net signal was obtained by subtracting the reference signal from the signal with immobilized enzyme. The association phase for the compound was followed for 100 s and the dissociation phase for 600 s. Four replicate dilution series were run. Solvent corrections were run with 8 DMSO concentrations spanning the range from 1.60% to 2.40% according to the manufacturer recommendations.
In Vivo Pharmacokinetics and Ex Vivo Occupancy of Compound 18
The protocols involving the care and use of animals were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC). The care and use of animals were in accordance with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Pharmacokinetic (PK) Animal Studies
Rats: Male Wistar–Hannover rats (n = 3) weighing approximately 300 g were used for the PK studies. Wistar–Hannover rats were obtained from Charles River Laboratories. Blood samples were collected in EDTA-containing tubes at the times specified below using an automated blood sampling machine (Instech, Plymouth Meeting, PA). For IV administration to rats, the dose solution was prepared by dissolving compound 18 in (1:1) DMSO/PEG400 at a concentration of 5 mg/mL. The rats were administered 0.4 mL/kg of the dose solution intravenously for a final dose of 2 mg/kg. For the IV leg of the study, a cannula was implanted in the jugular vein for dose administration and blood sampling. Compound 18 was administered as a bolus via the jugular vein at a dose of 2 mg/kg. Blood samples were collected at pre-dose and at 0.03, 0.13, 0.25, 0.5, 1, 2, 4, 8, 12, 18, and 24 h following administration. For PO administration to rats, compound 18 was dissolved in acidified 10% Tween 80, pH 2, to give a final concentration of 2 mg/mL. The rats were administered 5 mL/kg of the solution via oral gavage for a dose of 10 mg/kg. For the oral leg of the study, a solution of compound 18 was administered by oral gavage at 10 mg/kg. Blood samples were collected pre-dose and at 0.25, 0.5, 1, 2, 4, 8, 12, 18, and 24 h following administration. The blood samples were immediately centrifuged following collection to yield the plasma fraction, which was removed and frozen at −20 °C until analyzed by LC-MS/MS. Rhesus monkeys: Rhesus monkeys were obtained from the Mannheimer Foundation, Covance Research Product, University of Texas MD Anderson Cancer Center and University of Louisiana at Lafayette. For IV administration to rhesus monkeys, the dose solution was prepared by dissolving compound 18 in DMSO:PEG400 (1:1) at a concentration of 5 mg/mL. The monkeys were administered 0.1 mL/kg of the dose solution intravenously for a final dose of 0.5 mg/kg. Intravenous PK studies were conducted in 3 male rhesus monkeys following a 0.5 mg/kg bolus administration into the saphenous or cephalic vein. Blood samples were collected from the saphenous vein into EDTA-coated tubes pre-dose and at 0.03, 0.8, 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h. Oral dose PK studies were conducted in 3 rhesus monkeys (2 female and 1 male) following a 5 mg/kg oral gavage. For PO administration to rhesus monkeys, compound 18 was dissolved in 0.5% aqueous methylcellulose following addition of 1 mol equiv of 5 N HCl to give a concentration of 1 mg/mL. The monkeys were administered 5 mL/kg of the solution via oral gavage for a dose of 5 mg/mg. The blood samples were immediately centrifuged following collection to yield the plasma fraction, which was removed and frozen at −20 °C until analyzed by LC-MS/MS. Analytical Procedures: The concentrations of compound 18 in Wistar–Hannover rat and rhesus monkey plasma samples in the 0 to 24 h and 0 to 48 h studies were determined by LC-MS/MS analysis in the positive ion mode using the heated nebulizer (APCI) interface. Internal standard was added, and samples were extracted by protein precipitation with acetonitrile. The supernatant was transferred to a clean 96-well plate, and aliquots were injected into the LC-MS/MS system. Pharmacokinetic Analysis: PK parameters were obtained using non-compartmental methods (Watson). The area under the plasma concentration–time curve (AUC0-t) was calculated from the first time point (0 min) up to the last time point with measurable drug concentration using the linear trapezoidal or linear/log-linear trapezoidal rule. The remaining area under the plasma concentration–time curve (AUCt-inf) was estimated by dividing the observed concentration at the last time point by the terminal elimination rate constant. This value was added to AUC0-t to estimate the AUC0-inf. The percentage IV plasma clearance was calculated by dividing the dose by AUC0-inf. The terminal half-life of elimination was determined by unweighted linear regression analysis of the log-transformed data. The time points for determination of terminal half-life were selected by graphical inspection of the data. The volume of distribution at steady state (Vdss) was obtained from the product of plasma clearance and mean residence time (determined by dividing the area under the first moment curve by the area under the curve). The maximum plasma concentration (Cmax) and the time at which maximum concentration occurred (Tmax) were obtained by graphical analysis of the plasma concentration–time data. Absolute oral bioavailability was determined from dose-adjusted IV and PO (AUC0-inf) ratios.
Ex Vivo PDE10 Occupancy in Rats
Male Wistar–Hannover (Charles River Laboratories) rats weighing approximately 300 g received oral administration of either vehicle or compound 18 at doses of 0.1, 0.187, 0.3, 0.375, 1.5, 3, and 10 mg/kg (n = 5 per dose), followed by bolus intravenous injection of [3H]MK-8193 (12 μCi) 2 h later. For PO administration, compound 18 was formulated in acidified 10% Tween80, pH 2, and administered via oral gavage at a dosing volume of 5 mL/kg. For IV administration, [3H]MK-8193 (12 μCi) was formulated in saline and administered through lateral tail vein at a fixed volume of 0.1 mL per rat. Rats were anesthetized with isoflurane (1–5% isoflurane with O2 at a rate of 2 L/min) 30 min after [3H]MK-8193 administration and euthanized via decapitation. Trunk blood was collected into EDTA-containing tubes and centrifuged at 3000 rpm for 10 min to yield plasma. Plasma was frozen at −80 °C until LC-MS/MS analysis for quantification of drug concentrations. Striatum and brainstem were dissected and processed to obtain total binding and non-specific binding, respectively. Samples were homogenized using a Polytron homogenizer in 20 mL/g of tissue of ice-cold binding buffer containing 50 mM Tris-HCl, 120 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 1 mM CaCl2 (pH 7.5). Tissue homogenates were loaded on type A/E glass fiber filters pre-soaked with 0.1% PEI, placed on Hoeffer filter unit, washed with ice-cold binding buffer, and placed into scintillation vials containing Ultima Gold LSC cocktail. [3H] radioactivity for each sample was quantified using a Tri-Carb liquid scintillation counter. The specific binding was calculated by subtracting the value of non--specific binding from that of total binding for each striatal sample. PDE10A % occupancy was calculated as 100% × (1 – (specific binding in drug-treated/specific binding mean in vehicle-treated)). EC50 value was determined from % occupancy plotted as a function of plasma drug concentrations, fit to sigmoidal dose–response curve with variable slope using nonlinear regression analysis (GraphPad Prism).
Crystallographic Methods for Compound 18
Human PDE10A protein was concentrated to 10 mg/mL in buffer containing 20 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM 2-mercaptoethanol, and 1 mM EDTA. Apo crystals of human PDE10A were formed by the hanging drop vapor diffusion method at room temperature with a reservoir solution containing 100 mM HEPES pH 7.0, 18% PEG3350, 200 mM MgCl2, and 10 mM 2-mercaptoethanol. Crystals were transferred to a harvest buffer containing 100 mM HEPES pH 7.0, 30% PEG3350, 200 mM MgCl2, and 10 mM 2-mercaptoethanol and soaked with 5 mM compound 18 for 1 h. After soaking, a solution containing 80% of the harvest buffer and 20% PEG400 was used to cryo-protect the crystal for data collection. Diffraction data processing and refinement were conducted in autoPROC26 and BUSTER,27 respectively, using PDB 5C2H as a starting model.
Acknowledgments
We would like to thank John McCauley, Guillermo Fernandez, Justine Kent, Jamie McCabe Dunn, and Chris Lines for helpful commentary on the manuscript, and to Joe Salata for his contributions to the Nonclinical Drug Safety studies. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Glossary
Abbreviations Used
- CLp
plasma clearance
- CLu
unbound plasma clearance
- Cmax
maximal plasma concentration
- DIPEA
N,N-diisopropylethylamine
- EtOAc
ethyl acetate
- EtOH
ethanol
- LBE
ligand binding efficiency
- LLE
lipophilic ligand efficiency
- MeOH
methanol
- MeCN
acetonitrile
- TEA
triethylamine
- Vdss
volume of distribution at steady state
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01521.
Accession Codes
PDB code of new crystal (X-ray) structure: 8DI4 (compound 18/MK-8189). The authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
GlaxoSmithKline, plc, Philadelphia, PA 19104, United States
Author Present Address
The Janssen Pharmaceutical Companies of Johnson & Johnson, Titusville, NJ 08560, United States.
Author Present Address
Self-employed, Wayne, PA, 19087.
Author Present Address
PharmaBlock USA, Hatfield, Pennsylvania, 19440, United States.
Author Present Address
Janssen Biotech, Inc., Malvern, PA 19355, United States.
Author Present Address
Vertex Pharmaceutical, Boston, MA 02210, United States.
Author Present Address
GlaxoSmithKline, plc, Philadelphia, PA 19104, United States.
Author Contributions
The manuscript was prepared by M.E.L. with contributions by all authors to the research.
This work was funded entirely by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
The authors declare no competing financial interest.
Supplementary Material
References
- Soderling S. H.; Bayuga S. J.; Beavo J. A. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 7071–7076. 10.1073/pnas.96.12.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A. T.; Beavo J. A. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006, 58, 488–520. 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Menniti F. S.; Chappie T. A.; Humphrey J. M.; Schmidt C. J. Phosphodiesterase 10A inhibitors: A novel approach to the treatment of the symptoms of schizophrenia. Curr. Opin. Investig. Drugs 2007, 8, 54–59. [PubMed] [Google Scholar]
- Wilson L.; Brandon N. Emerging biology of PDE10A. Curr. Pharm. Des. 2014, 21, 378–388. 10.2174/1381612820666140826114744. [DOI] [PubMed] [Google Scholar]
- Kessler R. C.; Birnbaum H.; Demler O.; Falloon I. R.; Gagnon E.; Guyer M.; Howes M. J.; Kendler K. S.; Shi L.; Walters E.; Wu E. Q. The prevalence and correlates of nonaffective psychosis in the National Comorbidity Survey Replication (NCS-R)). Biol. Psychiatry 2005, 58, 668–676. 10.1016/j.biopsych.2005.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi C.; Uchida H.; Suzuki T.; Watanabe K.; Takeuchi H.; Nakajima S.; Kimura Y.; Tsutsumi Y.; Ishii K.; Imasaka Y.; Kapur S. The evolution of antipsychotic switch and polypharmacy in natural practice - a longitudinal perspective. Schizophrenia Research 2011, 130, 40–46. 10.1016/j.schres.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Menniti F. S.; Chappie T. A.; Schmidt C. J. PDE10A Inhibitors—Clinical Failure or Window Into Antipsychotic Drug Action?. Frontiers in Neuroscience 2021, 14, 600178. 10.3389/fnins.2020.600178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki A. R.; Rodriguez C. G.; Toolan D. M.; Price O.; Henry M.; Forrest G.; Szeto D.; Keohane C. A.; Pan Y.; Smith K. M.; Raheem I. T.; Cox C. D.; Hwa J.; Renger J. J.; Smith S. M. Genetic deletion and pharmacological inhibition of phosphodiesterase 10A protects mice from diet-induced obesity and insulin resistance. Diabetes 2014, 63, 300–311. 10.2337/db13-0247. [DOI] [PubMed] [Google Scholar]
- Gentzel R. C.; Toolan D.; Roberts R.; Koser A. J.; Kandebo M.; Hershey J.; Renger J. J.; Uslaner J.; Smith S. M. The PDE10A inhibitor MP-10 and haloperidol produce distinct gene expression profiles in the striatum and influence cataleptic behavior in rodents. Neuropharmacology 2015, 99, 256–263. 10.1016/j.neuropharm.2015.05.024. [DOI] [PubMed] [Google Scholar]
- Shipe W. D.; Sharik S. S.; Barrow J. C.; McGaughey G. B.; Theberge C. R.; Uslaner J. M.; Yan Y.; Renger J. J.; Smith S. M.; Coleman P. J.; Cox C. D. Discovery and optimization of a series of pyrimidine-based Phosphodiesterase 10A (PDE10A) inhibitors through fragment screening, structure-based design, and parallel synthesis. J. Med. Chem. 2015, 58, 7888–7894. 10.1021/acs.jmedchem.5b00983. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. J. Med. Chem. 2019, 62, 2245–2255. 10.1021/acs.jmedchem.8b01263. [DOI] [PubMed] [Google Scholar]
- Raab C. E.; Butcher J. W.; Connolly T. M.; Karczewski J.; Yu N. X.; Staskiewicz S. J.; Liverton N.; Dean D. C.; Melillo D. G. Synthesis of the first sulfur-35-labeled hERG radioligand. Biorg. Med. Chem. Lett. 2006, 16, 1692–1695. 10.1016/j.bmcl.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Raheem I. T.; Schreier J. D.; Fuerst J.; Gantert L.; Hostetler E. D.; Huszar S.; Joshi A.; Kandebo M.; Kim S. H.; Li J.; Ma B.; McGaughey G.; Sharma S.; Shipe W. D.; Uslaner J.; Vandeveer G. H.; Yan Y.; Renger J. J.; Smith S. M.; Coleman P. J.; Cox C. J. Discovery of pyrazolopyrimidine phosphodiesterase 10A inhibitors for the treatment of schizophrenia. Bioorg. Med. Chem. Lett. 2016, 26, 126–132. 10.1016/j.bmcl.2015.11.013. [DOI] [PubMed] [Google Scholar]
- Mahar Doan K. M.; Humphreys J. E.; Webster L. O.; Wring S. A.; Shampine L. J.; Serabjit-Singh C. J.; Adkison K. K.; Polli J. W. J. Pharmacology and Experimental Therapeutics 2002, 303, 1029–1037. 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- Maligres P.; Li J.; Krska S.; Schreier J.; Raheem I. T. C-O cross-coupling of activated aryl and heteroaryl halides with aliphatic alcohols. Angew. Chem., Int. Ed. 2012, 51, 9071–9074. 10.1002/anie.201203460. [DOI] [PubMed] [Google Scholar]
- Verhoest P. R.; Chapin D. S.; Corman M.; Fonseca K.; Harms J. F.; Hou X.; Marr E. S.; Menniti F. S.; Nelson F.; O’Connor R.; Pandit J.; Proulx-LaFrance C.; Schmidt A. W.; Schmidt C. J.; Suiciak J. A.; Liras S. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]-quinoline (PF-2545920) for the treatment of schizophrenia. J. Med. Chem. 2009, 52, 5188–5196. 10.1021/jm900521k. [DOI] [PubMed] [Google Scholar]
- Cox C. D.; Hostetler E. D.; Flores B. A.; Evelhoch J. L.; Fan H.; Gantert L.; Holahan M.; Eng W.; Joshi A.; McGaughey G.; Meng X.; Purcell M.; Raheem I. T.; Riffel K.; Yan Y.; Renger J. J.; Smith S. M.; Coleman P. J. Discovery of [11C]MK-8193 as a PET tracer to measure target engagement of phosphodiesterase 10A (PDE10A) inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 4893–4898. 10.1016/j.bmcl.2015.05.080. [DOI] [PubMed] [Google Scholar]
- Hostetler E.; Fan H.; Joshi A.; Zeng Z.; Eng W.; Gantert L.; Holahan M.; Meng X.; Miller P.; O’Malley S.; Purcell M.; Riffel K.; Salinas C.; Williams M.; Ma B.; Buist N.; Smith S. M.; Coleman P. J.; Cox C. D.; Flores B. A.; Raheem I. T.; Cook J. J.; Evelhoch J. L. Preclinical characterization of the Phosphodiesterase 10A PET tracer [11C]MK-8193. Molecular Imaging and Biology 2016, 18, 579–587. 10.1007/s11307-015-0910-0. [DOI] [PubMed] [Google Scholar]
- Bradford A. M.; Savage K. M.; Jones D. N. C.; Kalinichev M. Validation and pharmacological characterisation of MK-801-induced locomotor hyperactivity in BALB/C mice as an assay for detection of novel antipsychotics. Psychopharmacology 2010, 212, 155–170. 10.1007/s00213-010-1938-0. [DOI] [PubMed] [Google Scholar]
- Smith S. M.; Uslaner J. M.; Cox C. D.; Huszar S. L.; Cannon C. E.; Vardigan J. D.; Eddins D.; Toolan D. M.; Kandebo M.; Yao L.; Raheem I. T.; Schreier J. D.; Breslin M. J.; Coleman P. J.; Renger J. J. Neuropharmacology 2013, 64, 215–223. 10.1016/j.neuropharm.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Walling D. P.; Banerjee A.; Dawra V.; Boyer S.; Schmidt C. J.; DeMartinis N. Phosphodiesterase 10A Inhibitor Monotherapy Is Not an Effective Treatment of Acute Schizophrenia. J. Clin Psychopharmacol. 2019, 39, 575–582. 10.1097/JCP.0000000000001128. [DOI] [PubMed] [Google Scholar]
- Macek T. A.; McCue M.; Dong X.; Hanson E.; Goldsmith P.; Affinito J.; Mahableshwarkar A. R. A phase 2, randomized, placebo-controlled study of the efficacy and safety of TAK-063 in subjects with an acute exacerbation of schizophrenia. Schizophr Res. 2019, 204, 289–294. 10.1016/j.schres.2018.08.028. [DOI] [PubMed] [Google Scholar]
- Mukai Y.; Lupinacci R.; Marder S.; Mackle M.; Snow-Adami L.; Voss T.; Smith S.; Egan M. P534. Initial Assessment of the Clinical Profile of the PDE10A Inhibitor MK-8189 in Patients With an Acute Episode of Schizophrenia. Biol. Psychiatry 2022, 91, S305. 10.1016/j.biopsych.2022.02.771. [DOI] [Google Scholar]
- Merck Sharp & Dohme LLC , Rahway, NJ, USA, Sponsor. A Phase 2B randomized, double-blind, placebo- and active-controlled trial of the efficacy and safety of MK-8189 in participants experiencing an acute episode of schizophrenia. Identifier NCT04624243, https://clinicaltrials.gov/ct2/show/NCT04624243 (accessed August 31, 2022).
- Mosser S.; Gaul S.; Bednar B.; Koblan K. Automation of in vitro dose-inhibition assays utilizing the tecan genesis and an integrated software package to support the drug discovery process. J. Association for Laboratory Automation 2003, 8, 54–63. 10.1016/S1535-5535(04)00281-3. [DOI] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. 2011, D67, 293–302. 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G.; Blanc E.; Brandl M.; Flensburg C.; Keller P.; Paciorek W.; Roversi P.; Sharff A.; Smart O. S.; Vonrhein C.; Womack T. O.. BUSTER, version 2.11.8; Global Phasing Ltd., Cambridge: 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.