

Abstract
The outer membrane of Gram-negative bacteria has an external leaflet that is largely composed of lipopolysaccharide, which provides a selective permeation barrier, particularly against antimicrobials1. The final and crucial step in the biosynthesis of lipopolysaccharide is the addition of a species-dependent O-antigen to the lipid A core oligosaccharide, which is catalysed by the O-antigen ligase WaaL2. Here we present structures of WaaL from Cupriavidus metallidurans, both in the apo state and in complex with its lipid carrier undecaprenyl pyrophosphate, determined by single-particle cryo-electron microscopy. The structures reveal that WaaL comprises 12 transmembrane helices and a predominantly α-helical periplasmic region, which we show contains many of the conserved residues that are required for catalysis. We observe a conserved fold within the GT-C family of glycosyltransferases and hypothesize that they have a common mechanism for shuttling the undecaprenyl-based carrier to and from the active site. The structures, combined with genetic, biochemical, bioinformatics and molecular dynamics simulation experiments, offer molecular details on how the ligands come in apposition, and allows us to propose a mechanistic model for catalysis. Together, our work provides a structural basis for lipopolysaccharide maturation in a member of the GT-C superfamily of glycosyltransferases.
The external leaflet of the outer membrane of Gram-negative bacteria is composed mainly of lipopolysaccharide (LPS), which makes it a robust permeability barrier. LPS is critical for bacterial fitness during infection, can affect pathogenicity and serves as one of the conserved microorganism-associated molecular patterns that are recognized by the mammalian innate immune system1,3,4. LPS is made up of a lipid anchor (termed lipid A), an oligosaccharide core and an O-antigen, which is a highly variable polysaccharide composed of repeating sequences of three to six sugar moieties3,5,6. The O-antigen domain of LPS has been shown to contribute to bacterial evasion of complement-mediated killing, affect host autoimmunity through molecular mimicry and alter bacterial adherence to host tissue7–9. Within Escherichia coli alone, more than 187 different O-antigens have been identified10, and many of the corresponding O-antigen structures have been determined11.
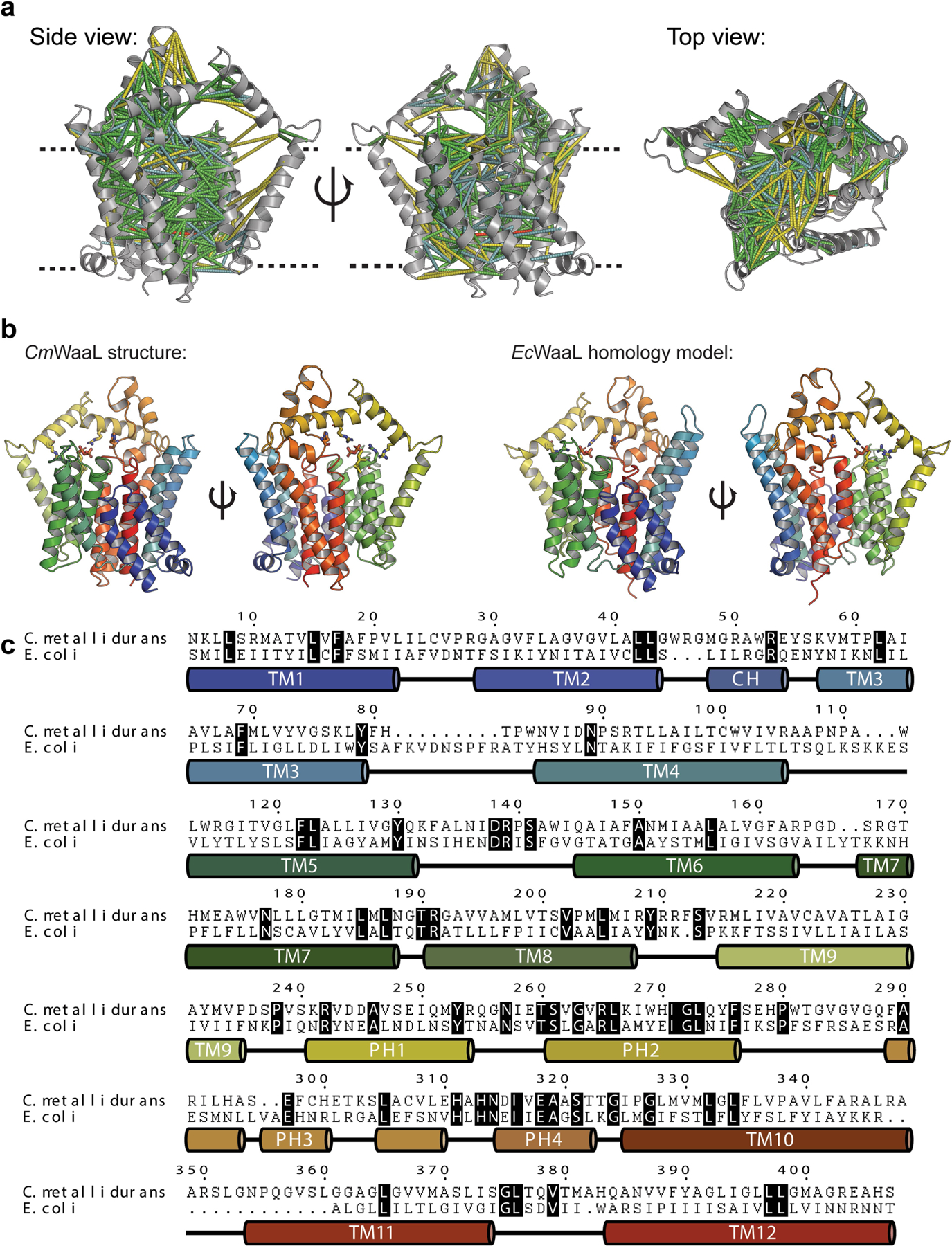
The polysaccharide repeats that constitute the O-antigen are synthesized in the bacterial cytoplasm by a series of glycosyltransferases, and then attached to the lipid carrier undecaprenyl pyrophosphate (Und-PP), before being flipped to the periplasmic leaflet of the inner membrane3,12. This occurs through the Wzy polymerase pathway, the ABC transporter pathway or the less well-characterized synthase-dependent pathway, according to the composition of the O-antigen and the nature of transport and elongation13 (Extended Data Fig. 1a). In all cases, the final step is the addition of O-antigen to the lipid A core oligosaccharide, which is performed by the O-antigen ligase WaaL, through a metal-independent glycosyltransferase reaction2,14 (Fig 1a, Extended Data Fig. 1b). Mutations within the gene that encodes WaaL prevent this ligation step, and result in a lack of O-antigen and the accumulation of Und-PP-linked precursors in the periplasm15. WaaL is predicted to have 12 transmembrane (TM) helices, and a periplasmic region between TM helix 9 and TM helix 105,16,17 has been shown to be essential for the formation of mature LPS1,7.
Fig. 1 |. The central role of WaaL in LPS biosynthesis in bacteria and the structure of CmWaaL.
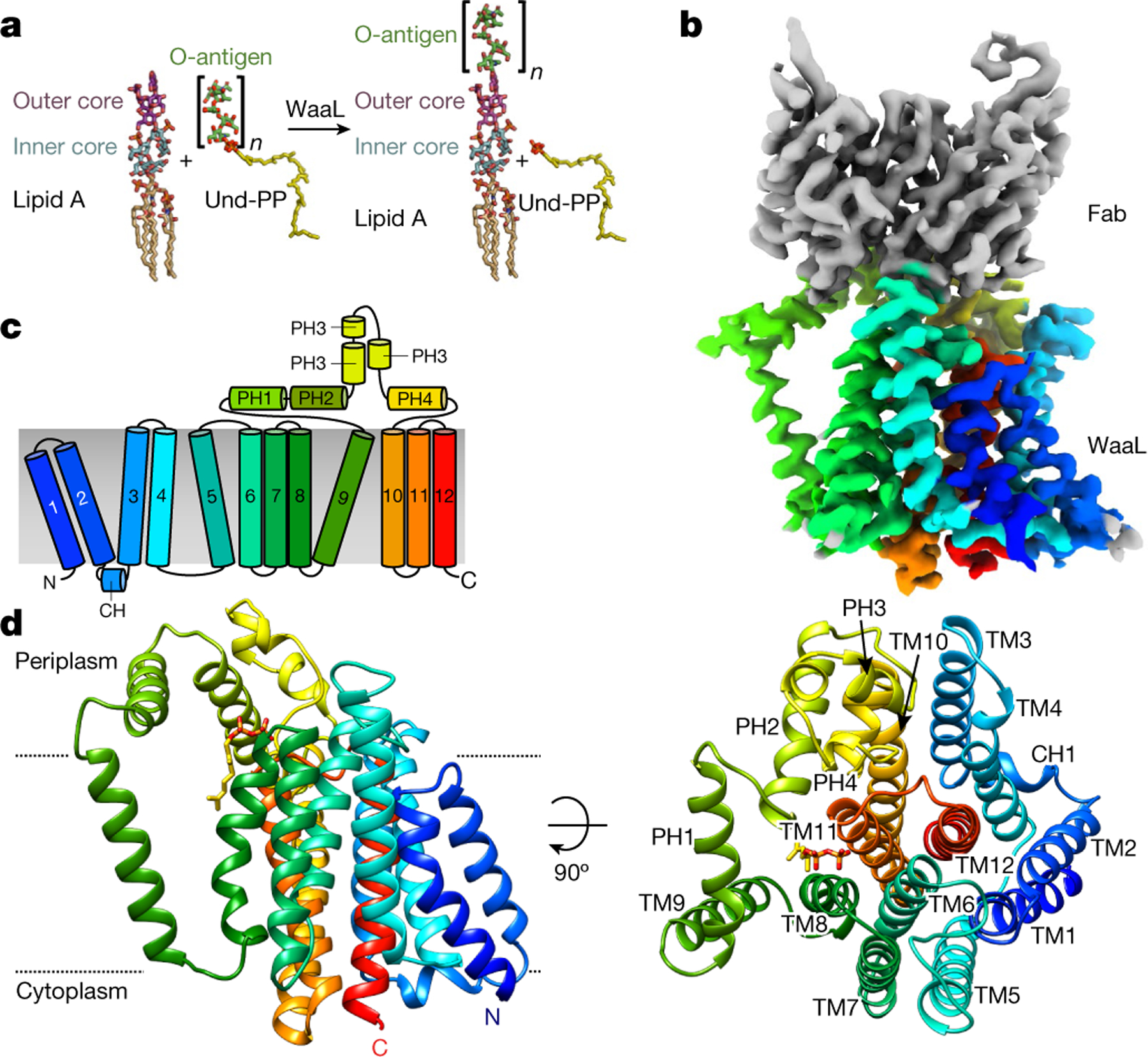
a, Atomic representation of an O1 O-antigen ligation to the R1 lipid A core oligosaccharide. The O-antigen is transferred to the lipid A core oligosaccharide by WaaL, forming the mature LPS. After ligation, Und-PP is recycled. b, Cryo-EM density map of the Und-PP-bound CmWaaL–Fab complex. Density corresponding to the variable region of the Fab (vFAB) is shown in grey and CmWaaL in rainbow colour. Representation is in rainbow colour from the N terminus (blue) to the C terminus (red). c, Schematic diagram showing the topology of WaaL, consisting of 12 TM helices and a large periplasmic loop between TM9 and TM10. d, The 3.2 Å cryo-EM structure of Und-PP-bound WaaL shown in two different orientations, with the 12 TM helices coloured as in c. The N and C termini are labelled. The right-hand side shows a 90° rotation with helices numbered, as viewed from the top. Und-PP is shown as sticks (in gold). Approximate membrane boundaries are represented with dotted lines.
To investigate the molecular details of the final assembly step in LPS biosynthesis, we determined structures of WaaL by single-particle cryo-electron microscopy (cryo-EM), in its ligand-bound and apo states. By combining structural information with genetics, biochemical assays and molecular dynamics (MD) simulations, we provide a rationale for substrate binding as well as a hypothesis for the reaction mechanism.
Structure determination of WaaL
We screened WaaL orthologues from around 200 different species for expression and stability in detergent to select suitable candidates for structural studies. Out of nineteen shortlisted candidates, WaaL from C. metallidurans (CmWaaL; 44 kDa) was identified as the most promising. We confirmed that CmWaaL is an O-antigen ligase by deleting the gene that encodes CmWaaL, and showing a loss of O-antigen ligase activity that could be restored when CmWaaL was expressed in trans (Extended Data Fig. 1c). CmWaaL was purified in detergent and reconstituted into lipid-filled nanodiscs for structure determination by cryo-EM (Extended Data Fig. 1d, e). To overcome the present size limitations for this technique and to provide fiducials for particle alignment18, we screened a synthetic phage display library to select for recombinant antigen-binding fragments (Fabs) against CmWaaL19. Seven high-affinity Fab candidates were screened for complex formation with CmWaaL, and WaB10 was chosen owing to its high binding affinity (Extended Data Fig. 1f–h). Data processing of particles picked from 2,378 micrographs allowed us to separate them into 2 distinct classes, which resulted in density maps to 3.5 Å and 3.2 Å resolution (Extended Data Table 1, Extended Data Fig. 2). We first built residues 5–407 of the 413 amino acids of CmWaaL into the higher-resolution map (Fig. 1b, Extended Data Fig. 3), and then adapted the model to fit the density of the second map.
Structure of Und-PP-bound CmWaaL
The structure of CmWaaL derived from the 3.2 Å map shows a monomer with twelve TM helices and one short cytoplasmic helix (CH) that connects TM2 and TM3 (Fig. 1c, d). Both the N and the C terminus are on the cytoplasmic side of the membrane. There is also a region of around 90 amino acids between TM9 and TM10 on the periplasmic side; this is mainly α-helical and is divided into four short α-helical segments (periplasmic helix (PH) 1–PH4), two of which (PH1 and PH2) are amphipathic and parallel to the membrane (Fig. 1c, d). The Fab required for structure determination binds to this periplasmic region (Fig. 1b, Extended Data Fig. 4a, b).
The overall TM core of the protein is tightly packed and comprises TM1–TM8 and TM10–TM12. By contrast, TM9 protrudes from the TM core (Fig. 1c, d), and forms a conduit to a central cavity (cavity 1) that is lined with conserved charged and polar residues at the periplasmic surface, and hydrophobic residues within the membrane core (Fig. 2a, Extended Data Figs. 4c, 5a–c). A cluster of four conserved arginine residues—R139, R191, R242 and R265—provide a positive net electro-static charge within this region (Fig. 2a, Extended Data Fig. 5a). Here, we observed clear non-protein density, which we modelled as two isoprenyl groups and a pyrophosphate, and interpreted it as being a partially ordered Und-PP (Fig. 2b). The presence and identity of Und-PP, which co-purified with the protein, was confirmed by thin-layer chromatography (TLC) (Extended Data Fig. 5d). R191 and R265 coordinate the diphosphate moiety of Und-PP (Fig. 2b) and the polyprenyl tail appears to interact with several conserved hydrophobic residues, including I373, L376, T377 (TM11) and V195 (TM8) (Extended Data Fig. 5e).
Fig. 2 |. Key structural features of CmWaaL.
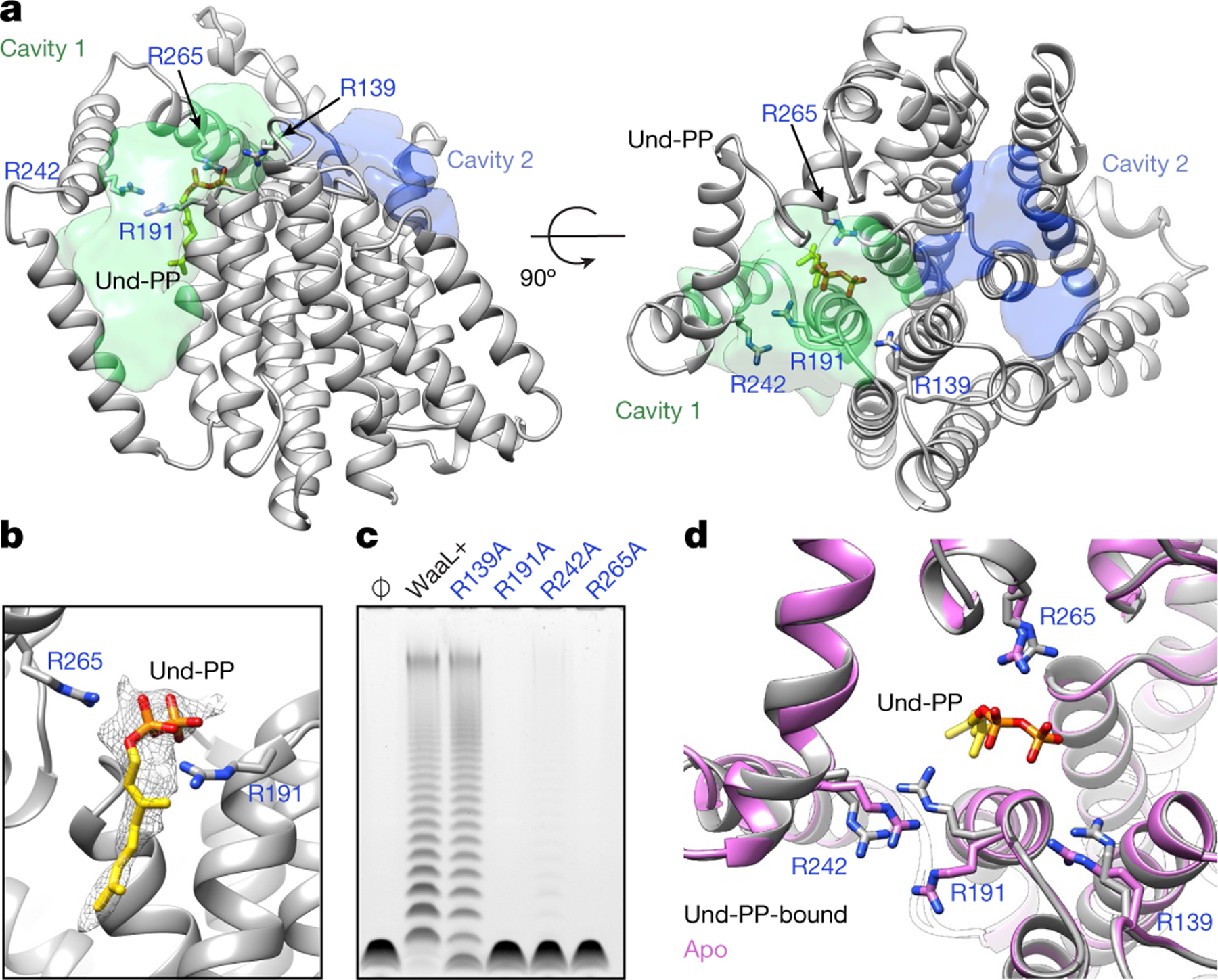
a, Cavities within the CmWaaL structure. CmWaaL is shown in ribbon representation in grey, and the two cavities as a semi-transparent surface, with cavity 1 in green and cavity 2 in blue. Volumes were calculated using the Voss Volume Voxelator (3V) server30, using probes with 10 Å and 2 Å radii, corresponding to the outer and the inner probe, respectively. Und-PP (in gold) and the key arginine (R191, R242, R265) residues for CmWaaL are shown as sticks. b, Magnified view of R191 and R265 coordinating the Und-PP (in gold) within the binding pocket. The cryo-EM density assigned to bound Und-PP is displayed as a semi-transparent grey mesh. c, Functional analysis of CmWaaL ligase activity in whole cells. LPS gel showing O-antigen extension in C. metallidurans expressing CmWaaL variants (mutated CmWaaL residues are shown in blue). WaaL+ indicates expression of wild-type (WT) CmWaaL and Ø indicates empty plasmid. d, Magnified view of the Und-PP-binding pocket in the apo structure (pink) and the Und-PP-bound structure (grey), superimposed, showing R139, R191, R242 and R265 in sticks with CPK colouring.
The complete Und-PP was modelled by superposition onto the two isoprenyl groups and the pyrophosphate coordinates described above. This WaaL–Und-PP complex was subjected to MD simulations within a lipid membrane. The simulations are consistent with the density map, illustrating that the portion of Und-PP that is visible in the structure is stabilized in the cavity, whereas the remainder of the undecaprenyl tail is highly mobile within the membrane (Extended Data Fig. 5f).
To study the effect on enzymatic activity of the four conserved arginine residues and other potentially functionally important residues in the Und-PP-binding cavity, we shifted our attention to E. coli K12, as this organism is more amenable to genetic manipulation and functional characterization. WaaL from E. coli (EcWaaL) could not be cross-complemented by CmWaaL, which is likely to reflect differences in the chemical composition of the lipid A core oligosaccharide, which aids in determining WaaL specificity5,15. We therefore generated a homology model of EcWaaL based on the CmWaaL structure—guided by co-evolutionary analysis and the coordinates of a structure modelled by AlphaFold20 (Extended Data Fig. 6)—and used this to select mutants to test functionally in a ligation assay. We observed that the ligase activity is abolished for EcWaaL when R161, R215, R265 and R288 (corresponding to R139, R191, R242 and R265 in CmWaaL) are mutated to alanine (Extended Data Fig. 7a–c).
We next evaluated the role of these four arginine residues in CmWaaL ligase activity directly in C. metallidurans. In this organism, when the WaaL knockout (ΔwaaL) was complemented with R191A and R265A in trans, ligase activity was abolished and almost abolished in R242A, whereas when ΔwaaL was complemented with R139A, activity appeared to be retained (Fig. 2c, Extended Data Fig. 7d).
Structure of apo-CmWaaL
Analysis of the lower-resolution (3.5 Å) map showed no evidence of density corresponding to Und-PP (Extended Data Fig. 8a), which is likely to represent the apo state of the enzyme. The structure of CmWaaL shows a similar overall fold in both states, with the exception of TM9 (Extended Data Fig. 8b, c), which is poorly resolved in the apo state, probably owing to its flexibility in the absence of Und-PP. R191 and R242 also appear to change conformation (Fig. 2d). The density of R265 together with a nearby cluster of conserved histidine residues (H311, H313 and H383) is weaker, probably because of the more dynamic nature of the apo state (Extended Data Fig. 8d).
The structures in the apo and Und-PP-bound states of CmWaaL, coupled with functional experiments, suggest that R191 and R242 (EcR215 and EcR265) might be involved in shuttling the Und-PP in and/or out of its binding pocket. Once the substrate is in place, R191 (EcR215), together with R265 (EcR288) appear to have a dominant role in coordinating it.
CmR139 and the equivalent EcR161 could instead have different roles in the two orthologues. CmR139 is in a flexible linker between TM5 and TM6, and is the most distant of the four arginine residues from the bound Und-PP. In the model, EcR161 is directed towards EcD389, which has previously been shown to be important for function2. EcD389 is likely to correspond to CmQ378, which when mutated to alanine does not affect activity3 (Extended Data Fig. 7e), suggesting that there are inter-species differences that may correlate with diversity in the chemical composition of LPS.
Putative lipid A-binding site
A second cavity (cavity 2) is located between the first three TM helices and extends over the periplasmic surface of the enzyme to cavity 1 (Fig. 2a). We hypothesize that this may be the binding site for lipid A. By performing a focused classification around cavity 2, we identified density consistent with lipid within the proposed lipid A-binding site (Fig. 3a). In this region of the density map, we also observed a membrane deformation in the periplasmic leaflet adjacent to TM1–TM3 (Fig. 3a). Membrane deformation was also observed in MD simulations, with and without bound Kdo2-lipid A (lipid A with two 3-deoxy-d-manno-octulonsonic acid (Kdo) sugars of the inner core). This appears to be induced by the presence of two short TM helices (1 and 2) in conjunction with the buried basic residues, R28 and R92. In these simulations, the phospholipid head groups deform by around 8.5 Å towards both arginine residues to accommodate the hydrophobic mismatch that is induced by the two short TM helices (Fig. 3b–d). The thinning of the membrane could favour the capture of the shorter, C10 to C14, acyl tails of lipid A, in comparison to the longer, inner membrane phospholipid species. This is comparable to the membrane thinning—from a thickness of 38 Å to 33 Å—that is observed around outer membrane proteins when embedded and simulated in model phospholipid membranes21. In addition, the distance between the two arginine CZ atoms, at approximately 8 Å, is ideally positioned to dynamically coordinate the phosphate groups of lipid A, which are around 10 Å apart. Furthermore, both residues are also located in the same plane of the membrane.
Fig. 3 |. A putative binding site for lipid A.
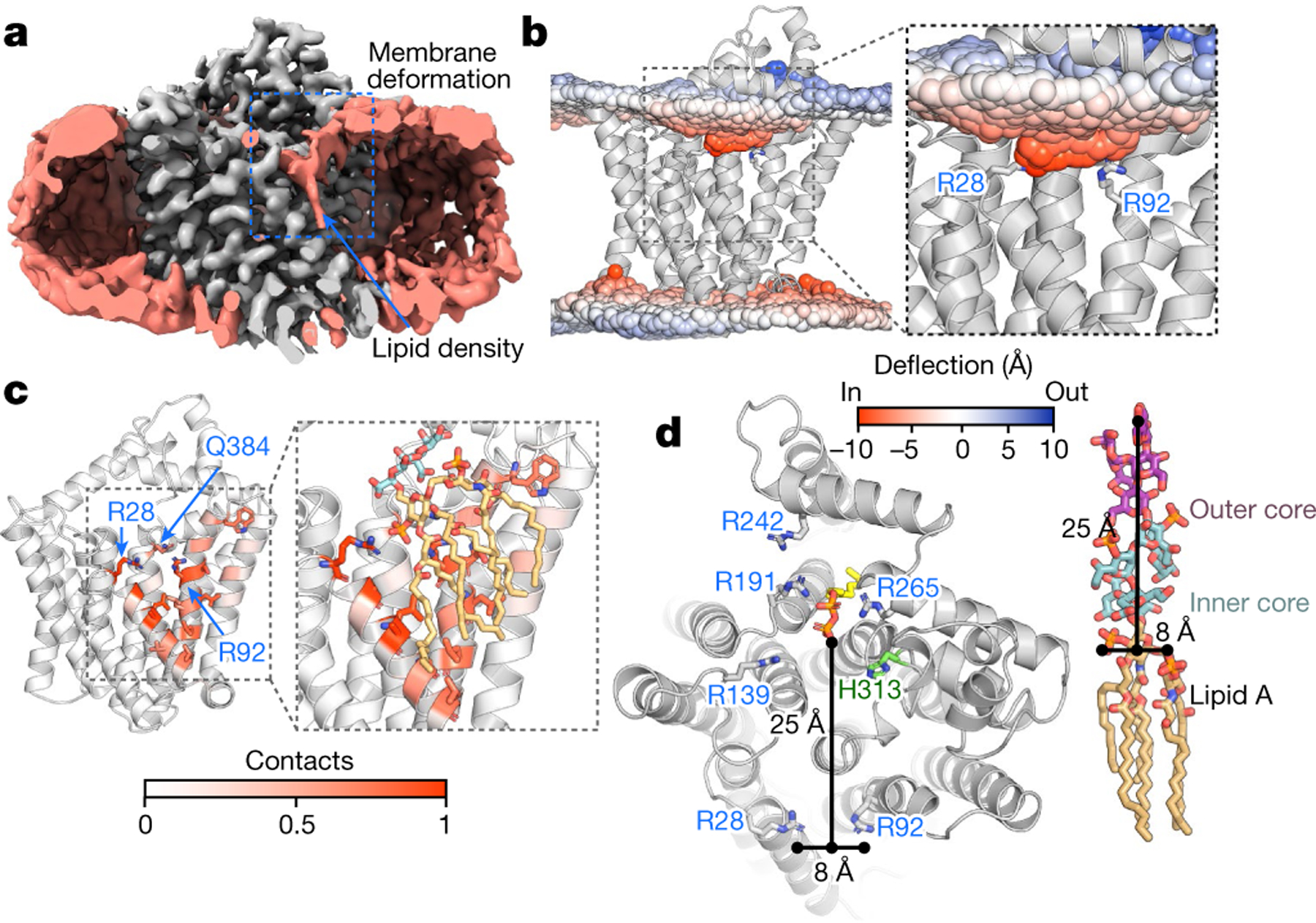
a, Cryo-EM density of WaaL (grey) incorporated into a nanodisc (salmon), showing a slice through the nanodisc in which membrane deformation can be observed (outlined with a dashed blue box). Also in salmon colour, a lipid tail density is seen in the putative lipid A-binding site. The density for the Fab is not included. b, The mean phosphate atom positions, highlighting a strong thinning of the membrane of around 8.5 Å around the two short helices (TM1 and TM2) in the apo-CmWaaL MD simulations. The mean phosphate position is coloured according to the deviation from the mean (blue, outward; red, inward). c, Contact of lipid A phosphates to CmWaaL in MD simulations. Lipid A is coloured orange and the two Kdo sugars are coloured cyan with CPK colouring. Throughout the MD simulations, the phosphate groups of lipid A remain in contact with R28, and to a lesser extent with R92 and Q384. The residue contacts with threshold above 0.5 are shown as sticks. d, Left, top view of CmWaaL, in ribbon, highlighting conserved residues, shown in sticks. The distances between R28 and R92 and between the phosphate groups of the lipid A core and the Und-PP are shown as black lines. Right, the chemical structure of lipid A core oligosaccharide, with distances marked with black lines.
The two short TM helices and the basic residues in CmWaaL are also conserved in EcWaaL (K45 and K111). However, alanine mutations of these residues in EcWaaL did not abolish O-antigen ligation (Extended Data Fig. 7a–c), although the CmWaaL R92A mutant does show a slight decrease in activity (Extended Data Fig. 7d, e), as does the double mutant K45A and K111A in EcWaaL (Extended Data Fig. 7a–c). Tryptophan mutations on residues at the interface between cavity 1 and cavity 2 were also carried out to monitor their effect on ligase activity. T168W and A394W did not abolish O-antigen ligation, whereas T170W did (Extended Data Fig. 7a–c, f). This could be due to the T170W mutant having a steric effect on Und-PP binding (Extended Data Fig. 7g). We also expect that the activity of WaaL could partly be retained owing to the large size and complexity of lipid A and the nature of the multifaceted binding interface between the lipid A core oligosaccharide and WaaL. Alternatively, lipid A could bind in cavity 1, near Und-PP (Extended Data Fig. 7h)—a hypothesis that we cannot exclude a priori. However, this could result in steric clashes between both substrates. Furthermore, the geometry of the two ligands would be likely to be somewhat constrained, with the groups to be ligated positioned at an acute angle, when compared with the more linear arrangement observed if the substrates are modelled bound to two distinct sites.
Proposed mechanism of action
The reaction catalysed by WaaL is known to be independent of ATP and metal ions2, and the energy required to drive it has been suggested to result from cleavage of the bond between the distal phosphate group of Und-PP and its O-antigen cargo22. This is analogous to classical glycosyltransferases in which a nucleotide diphosphate sugar is the substrate23. In inverting glycosyltransferases, nucleophilic attack by the acceptor hydroxy group leads to an inversion of stereochemistry at the anomeric centre of the donor substrate. These enzymes typically use a catalytic base and a bound metal ion that stabilizes the leaving phosphate groups. However, metal-ion-independent inverting glycosyltransferases also exist and use basic amino acids such as arginine, lysine or histidine to perform a similar function to that of the metal ion2,16.
We propose a mechanism for EcWaaL in which the absolutely conserved EcH338 residue abstracts a proton from the leading hydroxyl group of the terminal outer core of the lipid A core oligosaccharide. This would enable the oxygen to perform a nucleophilic attack at the C1 carbon of the Und-PP-linked O-antigen sugar ring and induce cleavage of the sugar-phosphate bond (Fig. 4a, b). EcH338 appears to be stabilized by a network of hydrogen bonds involving nearby EcH336 and EcE343. The enzyme would then reset as the proton moves from EcH338 to the pyrophosphate of Und-PP. The Und-PP moiety is held in position by EcR215 and EcR288, as detailed above (Fig. 4b). The mutation EcH338A abolishes enzymatic activity (Fig. 4c), as does mutagenesis of the corresponding residue in CmWaaL, CmH313A (Extended Data Fig. 7d, e). The residues EcH336 and EcE343, which are adjacent to EcH338 in the structure, also substantially reduce overall activity when mutated to alanine. We speculate that they might stabilize the position of EcH338, to allow the mechanism to proceed (Fig. 4c, d). Inspection of the structure shows that the highly conserved residues EcN339 and EcE340 are also in the vicinity of the active site; however, alanine mutants of these residues showed no change in activity compared to the wild-type enzyme and thus are not included in our proposed mechanism (Fig. 4c, d).
Fig. 4 |. Mechanism of catalysis for WaaL.
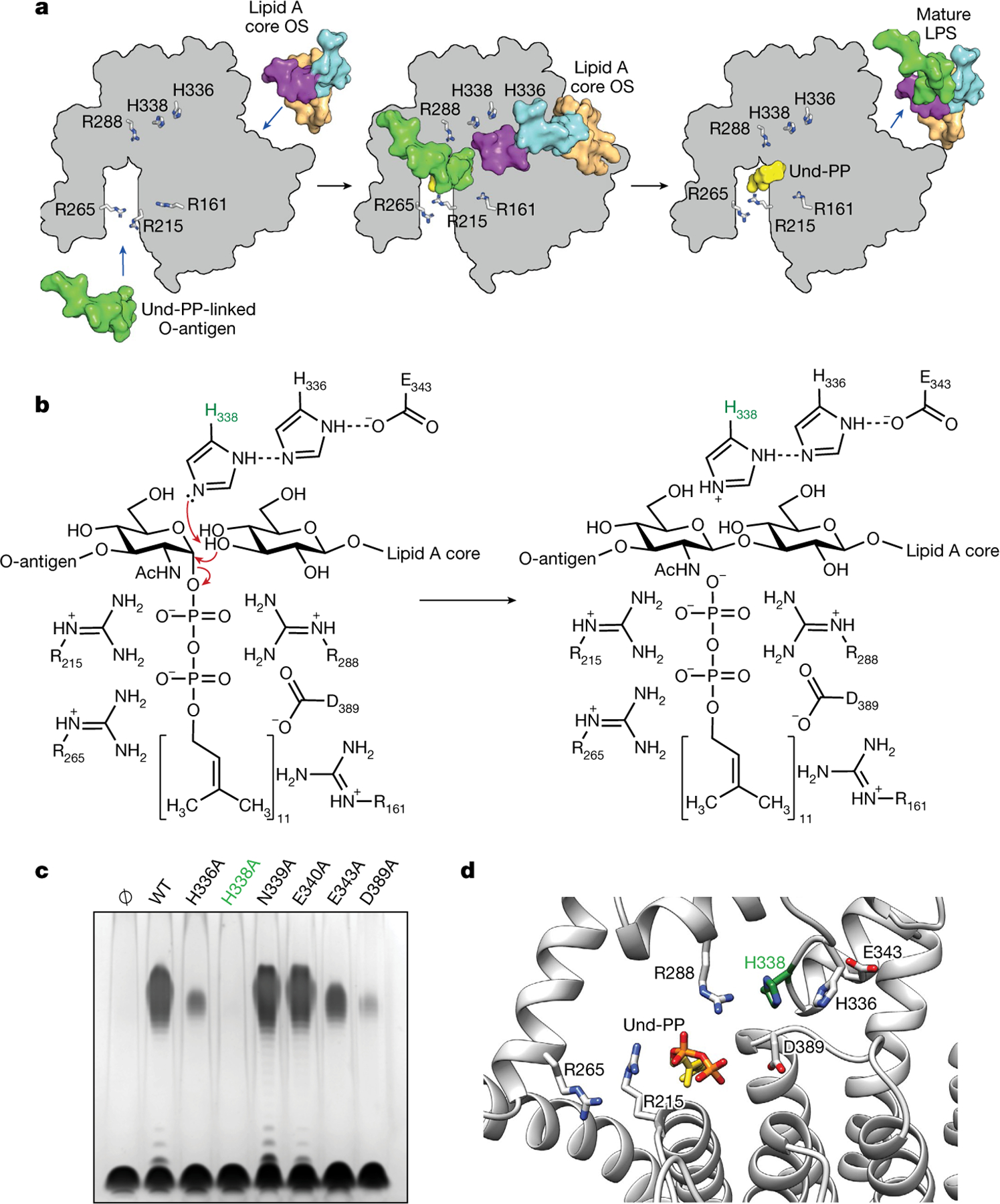
a, Schematic representation of the ligation of the O-antigen-linked Und-PP to the lipid A core oligosaccharide (OS) in EcWaaL, viewed from the periplasmic side of the membrane. In all cases, key arginine (R161, R215, R265, R288) and histidine (H338, H336) residues are shown as grey sticks. Left, a surface representation for both lipid A (peach) with its inner (cyan) and outer (purple) core oligosaccharide and the O-antigen (green)-linked Und-PP (yellow) approaching their binding sites on the apo state of WaaL. Middle, the coordination of both O-antigen-linked Und-PP and lipid A core oligosaccharide in both sites. Right, the mature LPS, with the Und-PP (yellow) product still bound, as shown in the cryo-EM structure. b, Residues within the active site of EcWaaL are shown around the Und-PP-linked O1 O-antigen N-acetylglucosamine (GlcNAc) and terminal R1 outer core glucose. H338 (highlighted in green) is coordinated by a hydrogen-bond network between H336 and E343, which permits the abstraction of a proton from the terminal hydroxyl group of the R1 outer core glucose to protonate H338. The deprotonated oxygen may then perform a nucleophilic attack on the C1 of the O1 GlcNAc. This allows cleavage of the GlcNAc–phosphate bond. To reset the enzyme, H338 will deprotonate, with the proton possibly transferring to the phosphate group of Und-PP, which then leaves the active site. c, Functional analysis of EcWaaL ligase variants in whole cells by LPS gel analysis. WaaL proteins were expressed from plasmid pWSK29 in the W3110 ΔwaaL strain. Ø indicates empty plasmid. WT, wild type. d, Key residues involved in the putative ligation mechanism and ligand coordination within EcWaaL. The EcWaaL homology model is based on the CmWaaL structure and shown as cartoon. Key residues R215, R265, R288, E343, H336, H338 and D389 are shown as sticks.
In addition to MD simulations with bound Und-PP, we also performed MD simulations of the apo enzyme (Extended Data Fig. 9a). On the basis of these experiments, we observed TM9 in two discrete states, which we defined as the closed and the open state (Extended Data Fig. 9b). In the absence of Und-PP, an outward movement of TM9 is observed, thereby transitioning from a closed to an open state. This appears to be driven by an electrostatic repulsion of CmR242 (EcR265) from the adjacent basic groups of the active site, in particular CmR191 (EcR215) (Extended Data Fig. 9b).
Features of GT-C enzymes that use Und-PP
The structure of WaaL reveals that it shares an overall similar fold to that of T. thermophilus (Tt) RodA24, with a Cα root mean square deviation (RMSD) of 4.73 Å over 248 residues (Extended Data Fig. 10a). RodA is a transglycosylase of the shape, elongation, division and sporulation (SEDS) family of proteins25, and is critically required for the formation of peptidoglycan cell walls. Mechanistically, WaaL is a transferase whereas RodA is a processive polymerase. Both use Und-PP substrates and appear to share a similar helical structural arrangement with conserved arginine residues lining the binding pocket. Indeed, R242 in CmWaaL and R203 in TtRodA are both highly conserved and could adopt the same mechanism of shuttling the Und-PP into or out of the active site (Extended Data Fig. 10b). The major structural distinction between WaaL and RodA is that the latter lacks the first two TM helices, which we propose are integral to binding the lipid A core oligosaccharide (Extended Data Fig. 10a).
We also noticed structural and topological similarities between WaaL, PglB and ArnT glycosyltransferases26–28 (Extended Data Fig. 10b). ArnT catalyses the transfer of a sugar moiety via a undecaprenyl-phosphate (Und-P) carrier to lipid A23, whereas the metal-dependent PglB is a protein glycosyltransferase28. For all three enzymes, residues in the periplasmic loops have been shown to be critical for substrate recognition and catalysis2,23,26, suggesting commonalities in structure, function and mechanism.
Discussion
We have determined the structures of Und-PP-bound and apo-CmWaaL by cryo-EM, to 3.2 Å and 3.5 Å resolution, respectively. The structure reveals two major cavities on the periplasmic side, which we suggest are the binding sites for Und-PP and lipid A, respectively There are conserved residues bridging these two cavities that have previously been suggested to be involved in the ligation of the O-antigen to the lipid A core oligosaccharide2,5,7,17.
We have combined structural evidence, MD simulations and biochemical experiments to propose a reaction mechanism in which the O-antigen linked Und-PP is shuttled into and coordinated within the binding pocket by a set of conserved arginine residues. Subsequently, the O-antigen is transferred from the Und-PP onto the lipid A core oligo-saccharide through a reaction that is carried out by a highly conserved histidine residue, and in turn coordinated by another histidine and a glutamate. WaaL shares features with metal-ion-independent inverting glycosyltransferases to bring about catalysis, but uses a membrane embedded Und-PP substrate as opposed to the soluble nucleotide diphosphate moiety of classical nucleoside glycosyltransferases2,29. In both cases—Und-PP-linked, or nucleotide-diphosphate-linked sugars—the leaving group upon glycosyl transfer is a diphosphate molecule and the proximal sugar to the distal phosphate is O-linked to the terminal sugar of the acceptor9,23.
We propose that the lipid A-binding site is on the opposite side of the structure from the location of the bound Und-PP. This site is coordinated by two basic residues and is shaped by two unique short TM helices (1 and 2). These helices induce a membrane deformation that appears to draw the phospholipids towards two basic residues, CmR28 and CmR92, which are appropriately spaced to coordinate the phosphate groups of the lipid A moiety of LPS. The distance between this binding site and that of the Und-PP is around 25 Å, which is comparable to the dimensions of the core oligosaccharide of LPS. This suggests that carbohydrates of the immature LPS might rest on the periplasmic surface of WaaL to position the terminal outer core sugar for the attachment of O-antigen.
The structure of CmWaaL in its Und-PP-bound state, along with the previously published structures of RodA–PBP2, PglB and ArnT, has allowed us to identify structural similarities24,26,28. In all of these cases, we observe a TM helix protruding from the helical bundle, and a short amphipathic helix parallel to the membrane that connects the protruding helix back to the helical bundle, creating a binding cavity for the ligand24 (Extended Data Fig. 10). This suggests an evolutionary relationship between these enzymes, which are all members of the GT-C family of glycosyltransferases23.
The structure of CmWaaL provides a framework for understanding how O-antigen ligases perform the final, critical step in LPS maturation in Gram-negative bacteria, and offers a structural basis for designing inhibitors of this process in the biosynthesis of bacterial cell envelopes.
Methods
Target identification and cloning
Nineteen putative WaaL genes corresponding to a wide genomic background were identified using a bioinformatics approach, as previously described39. Ligation-independent cloning (LIC) was used to clone these 19 orthologues into 5 different LIC-adapted expression vectors (pNYCOMPS-Nterm, pNYCOMPS-Cterm, pMCSG7, pNYCOMPS-N23 and pNYCOMPS-C23), all bearing protease-cleavable decahistidine tags for metal-affinity-chromatography-based purification. Small-scale expression and purification tests were performed as previously described40, to identify 11 unique targets as ‘expression-positive’ from the small-scale expression tests. These 11 candidates were carried forward for mid-scale expression, purification and detergent screens as previously described40, for the selection of C. metallidurans (Cm) as the best candidate (protein accession code: WP_011517284.1). All cloning and initial protein characterization were performed at the protein production core facility of the former New York Consortium on Membrane Protein Structure (NYCOMPS)41, now the Center on Membrane Protein Production and Analysis (COMPPÅ).
Protein expression and purification in detergent
WaaL from C. metallidurans (CmWaaL), cloned in the pNYCOMPS-Cterm vector, was used to transform BL21(DE3)pLysS E. coli competent cells, and grown overnight in 2xYT medium supplemented with 100 μg ml−1 ampicillin and 35 μg ml−1 chloramphenicol at 37 °C with shaking (240 rpm.). The next day, 800 ml (large scale for protein expression) or 8 ml (small scale to test expression) of the same medium was inoculated with the starter culture at a 1:100 ratio, and left to grow at 37 °C with shaking (240 rpm.), until the optical density at 600 nm (OD600 nm) reached 0.8–1.2. The temperature was then reduced to 22 °C, protein expression was induced with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and the culture was incubated overnight with shaking (240 rpm). Cells were collected by centrifugation (3,700 rpm for 15 min) at 4 °C, washed once with phosphate-buffered saline (PBS) and centrifuged again to produce a solid pellet that was stored at −80 °C, until further use. For large-scale purification of CmWaaL, cell pellets were resuspended in lysis buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, 20 mM MgSO4, 10 μg ml−1 DNase I, 8 μg ml−1 RNase A, 1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP), 1 mM PMSF, and Complete Mini EDTA-free protease inhibitor cocktail (Roche) used according to the manufacturer’s instructions. Cells were lysed with an Emulsiflex C3 homogenizer (Avestin) and the lysate was solubilized for 2 h with n-dodecyl-β-d-maltopyranoside (DDM; Anatrace) added to a final concentration of 1% (w/v), in a volume of approximately 40 ml per cell pellet from 800 ml culture. Insoluble material was removed by ultracentrifugation at 34,000 rpm for 30 min at 4 °C and the protein was purified from the supernatant by metal-affinity chromatography using Ni-NTA agarose beads (Qiagen). The supernatant after addition of 40 mM imidazole was incubated with pre-equilibrated Ni-NTA agarose beads (0.7 ml per pellet from an 800 ml culture) overnight. The beads were then loaded on a column and washed with 10 column volumes of 20 mM HEPES pH 7.5, 500 mM NaCl, 75 mM imidazole and 0.03% (w/v) DDM. Protein was eluted with 4 column volumes of 20 mM HEPES pH 7.0, 150 mM NaCl, 300 mM imidazole and 0.03% (w/v) DDM. Imidazole was removed from the eluted protein by exchanging buffer to 20 mM HEPES pH 7.0, 200 mM NaCl and 0.03% (w/v) DDM (final protein buffer) using a PD-10 desalting column (GE Healthcare).
Nanodisc incorporation after detergent purification
The imidazole-containing buffer of the protein eluted from metal-affinity chromatography was desalted using a PD-10 column into the final protein buffer. CmWaaL was incorporated into lipid nanodiscs with a 1:300:5 molar ratio of protein:1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (POPG):membrane scaffold protein 1E3D1 (MSP1E3D1)42,43. This mixture was incubated at 4 °C for 2 h with gentle agitation. Reconstitution was initiated by removing detergent with the addition of Bio-beads (Bio-Rad) at 4 °C overnight with constant rotation. Bio-beads were removed and the nanodisc reconstitution mixture was bound again to Ni2+-NTA resin at 4 °C for 2 h to remove empty nanodiscs. The resin was washed with 10 column volumes of wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl and 60 mM imidazole) followed by 4 column volumes of elution buffer (20 mM HEPES pH 7.0, 150 mM NaCl and 300 mM imidazole). Subsequently, the protein was incubated with TEV protease44 to cleave the decahistidine tag (around 0.5 mg TEV protease added per pellet equivalent from 800 ml of initial bacterial culture) overnight at 4 °C, while exchanging into an imidazole-free buffer using 3.5 K cut-off Slide-A-Lyzer Dialysis Cassettes (Thermo Fisher Scientific). The sample was then passed through a column containing Ni2+-NTA beads to separate the cleaved decahistidine tag, the TEV protease and the uncleaved protein. The cleaved protein was further purified by loading onto a Superdex 200 Increase 10/300 GL size-exclusion column (GE Healthcare Life Sciences) in gel filtration buffer (20 mM HEPES pH 7.0 and 150 mM NaCl). Protein typically eluted as a sharp monodispersed peak, observed by monitoring the absorbance at 280 nm (Extended Data Fig. 1d).
Phage display to identify CmWaaL-specific Fab fragments (WaB10)
CmWaaL was reconstituted into nanodiscs formed using chemically biotinylated MSP1E3D1, which were prepared as previously described19. Biotinylation efficiency was evaluated by a pull-down assay using streptavidin-coated paramagnetic particles (Promega). Phage displayed synthetic antigen binder (sAB) Library E36,37 (prepared by S. Mukherjee) was used as the naive starting pool. Library E and the target were both diluted into selection buffer (20 mM HEPES, pH 7.4, 200 mM NaCl and 1% BSA) and selections were performed using a protocol adapted from previous publications45,46. In round one, biopanning was performed manually using 300 nM of CmWaaL immobilized onto magnetic beads. Following a one-hour incubation, beads were washed three times with selection buffer and then used to subsequently infect log-phase E. coli XL-1 Blue cells. Infected cells were used to inoculate 30-ml cultures containing ampicillin (100 μg ml−1) and M13-K07 helper phage (109 plaque-forming units (pfu) per ml), and phage were amplified overnight. Four additional rounds of biopanning were performed by stepwise reduction of the target concentration to increase the stringency of selection. Rounds 2–5 were performed semi-automatically using a Kingfisher Beads Handling Robot (Thermo Fisher Scientific), and phage from each preceding round were amplified and used as the input pools. In addition, the amplified phage pool was pre-cleared using 200 μl of streptavidin beads, and in all rounds 1.5 μM of empty MSP1E3D1 nanodiscs were used as competitors in solution. Finally, in the last four rounds, phage were eluted from magnetic beads using a 15-min incubation with 1% Fos-choline 12 (Anatrace).
Single-point phage enzyme-linked immunosorbent assay
E. coli XL-1 Blue cells were infected with phage from the fourth- and fifth-round pools and plated on LB-agar supplemented with 100 μg ml−1 ampicillin. The following day, individual colonies containing phagemids were used to inoculate 400 μl of 2xYT medium supplemented with ampicillin (100 μg ml−1) and M13-K07 helper phage (109 pfu per ml). Phage were amplified overnight with shaking (280 rpm). Single-point phage enzyme-linked immunosorbent assay (ELISA) was subsequently performed as previously described45,46 (Supplementary Table 1). All amplified phage were tested against CmWaaL in MSP1E3D1 nanodiscs (30 nM), empty 1E3D1 nanodiscs (50 nM) or buffer alone to determine specific target binding. Bound phage particles were detected by a colorimetric assay using an HRP-conjugated anti-M13 monoclonal antibody (GE Healthcare). Binders with high target and low non-specific signal were chosen for subsequent experiments.
Surface antibody cloning, expression and purification
All specific binders were sequenced and then cloned into the surface antibody (sAB) expression vector RH2.2 (a gift from S. Sidhu) as previously described46. All phage and expression vector subclones were sequence-verified at the University of Chicago Comprehensive Cancer Center DNA sequencing facility. sABs were expressed and purified as previously described46 and subsequently dialysed overnight in buffer with 20 mM HEPES, pH 7.5 and 150 mM NaCl.
Multi-point sAB ELISA
To estimate the apparent binding affinity of each sAB, multi-point ELISA was performed in triplicate as previously described45,46 for each unique sAB. All sABs were again verified for specific binding by testing against CmWaaL in MSP1E3D1 nanodiscs (30 nM), empty MSP1E3D1 nanodiscs (50 nM) or buffer alone. The amount of bound sAB was measured by a colorimetric assay using an HRP-conjugated anti-Fab monoclonal antibody (Jackson ImmunoResearch). Measured absorbance (A450) values were plotted against the log sAB concentration, and estimated binding affinities (half-maximum effective concentration; EC50) were determined using a variable slope model with sigmoidal dose response in GraphPad Prism (Supplementary Table 1).
CmWaaL complex formation with the Fab WaB10
TEV-cleaved CmWaaL incorporated into nanodiscs was incubated with the WaB10 Fab on ice for 1 h in a 1:2 molar ratio of protein to Fab. The CmWaaL–Fab complex was concentrated and filtered, and then loaded on a Superdex 200 Increase 10/300 GL size-exclusion column in gel filtration buffer (20 mM HEPES pH 7.0 and 150 mM NaCl).
Single-particle cryo-EM vitrification and data acquisition
The purified CmWaaL–Fab complex was concentrated to 1.2 mg ml−1 using a 50-kDa concentrator (Amicon). Three microlitres of sample was added to a plasma-cleaned (Gatan Solarus) 1.2/1.3-μm (1.2 μm circular holes and a spacing of 1.3 μm between the holes) holey gold grid (Quantifoil UltrAuFoil) and blotted using filter paper on one side for 3.5 s using the Vitrobot (Thermo Fisher Scientific) with a blot force of 3 and a wait time of 30 s, before plunging immediately into liquid ethane for vitrification. The plunger was operating at 4 °C with greater than 90% humidity to minimize evaporation and sample degradation. Images were recorded using a Titan Krios electron microscope (FEI), at the Simons Electron Microscopy Center, equipped with an energy filter and a K2 direct electron detector (Gatan K3-BioQuantum) using a 1.061 Å pixel size. An energy filter slit width of 20 eV was used during the collection and was aligned automatically every hour using Leginon47. Data collection was performed using a dose of around 70.15 e− per Å2 across 50 frames (200 ms per frame) at a dose rate of around 7.0 e− per pixel per second, using a set defocus range of −1.3 μm to −2.8 μm. A 100 μm objective aperture was used. In total, 2,378 micrographs were recorded over a single two-day collection using an image beam shift data collection strategy. Ice thickness was monitored after every fourth exposure using the Leginon zero-loss peak (ZLP) algorithm and was determined to be 23.1 ± 9.1 nm (s.d.).
Data processing
Contrast transfer function (CTF) estimation was performed using Patch CTF as implemented in cryoSPARC v.2.8. Blob picker in cryoSPARC v.2.8 was used to pick particles and inspect picks was used to curate the picks. This resulted in 844,438 particles, which were then subjected to two-dimensional (2D) classification in cryoSPARC v.2.8. A total of 132,664 particles were selected for further processing from 2D classes with well-defined high-resolution features. One round of ab initio reconstruction was performed in cryoSPARC v.2.8 using four classes, with a maximum resolution set at 7 Å and an initial resolution at 9 Å; the best class was selected, resulting in a stack of 46,362 particles. Heterogeneous refinement was carried out on this particle stack, after which this stack was then extracted with a 320-pixel size box. This particle stack was subjected to non-uniform refinement in cryoSPARC v.2.8, resulting in a 3.9 Å reconstruction. Using a mask covering CmWaaL and the variable region of the Fab, local refinement using non-uniform regularization was performed in cryoSPARC v.3.2, resulting in a 3.2 Å density map (Extended Data Fig. 2a).
On initial observation it was hypothesized that a mixed population of bound and unbound WaaL particles contributed to the 46,362-particle stack, which was mainly due to the density that we attributed to the geranyl diphosphate part of the Und-PP and the density for TM9. We observed that depending on the particles that were selected from the initial 132,664 particles, the density of the Und-PP and TM9 had either weak or strong density, indicating that a mixed population of Und-PP bound and unbound may be present within the 132,664-particle stack. To address this, three-dimensional (3D) classification was then performed using the initial 132,664 particles, which were extracted with a 400-pixel box size for further processing. These extracted particles were then imported into RELION v.3.1.1 for 3D classification using a mask that only included TM9–TM11 as well as PH1–PH4, helices that surrounded the Und-PP. In total, six classes were obtained, with the initial low pass set at 20 Å and the T value set at 40; the two best classes were then selected, resulting in a stack of 39,844 and 30,514 particles. Both particle stacks were imported back into cryoSPARC v.3.2 and using a mask covering CmWaaL and the variable region of the Fab for the 39,844-particle stack, and a mask covering CmWaaL for the 30,514-particle stack, local refinement using non-uniform regularization was performed, resulting in a 3.23 Å density map for the 39,844-particle stack and 3.5 Å density map for the 30,514-particle stack (Extended Data Fig. 2a).
A 3D classification followed by a focused refinement was performed to obtain the lipid density in our proposed lipid A-binding site (Fig. 3a). The initial 132,664 particles, which were extracted with a 400-pixel size box, were imported into RELION v.3.1.1 for 3D classification using a mask surrounding the proposed lipid A site, including residues 29–79 and 315–352. In total, six classes were obtained, with the initial low pass set at 20 Å and the T value set at 40; the best class containing 50,857 particles was selected. The particle stack was imported into cryoSPARC v.3.2 and using a mask covering CmWaaL and the variable region of the Fab, a local refinement using non-uniform regularization was performed, resulting in a density map at 3.22 Å resolution that has a clear lipid tail density within the binding cleft of cavity 2 (Fig. 3a).
Structural model building and refinement
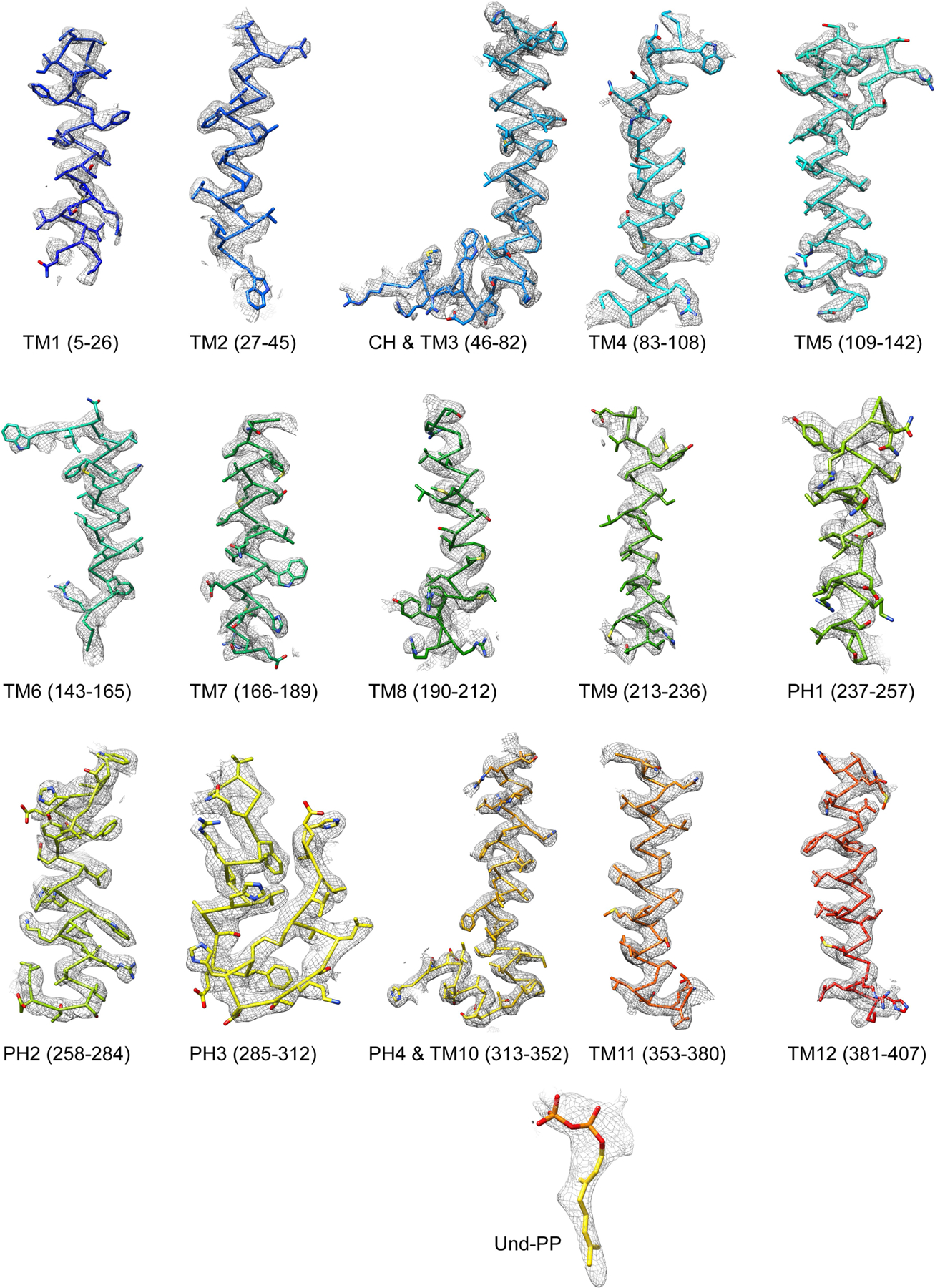
To build the CmWaaL model, Coot was used for manual model building. We observed extra density within the map of the 39,844-particle stack and were able to fit two isoprenyl groups and a pyrophosphate for the Und-PP (PubChem ID 5280604) into the density. The map obtained from the 39,844 stack was the local refinement with the CmWaaL and the variable region of the Fab (vFAB) mask created that gave the 3.23 Å map that was used to build the backbone and most of the side chains. The map from the 30,514-particle stack was a local refinement map created from the CmWaaL mask that yielded the 3.5 Å map. CmWaaL was modelled de novo in Coot using secondary structure predictions from the XtalPred server48 as a guide. Subsequent model refinement and adjustment was performed in Coot49–51 and PHENIX52,53 iteratively.
Model analysis
A cavity search using the Solvent Extractor from Voss Volume Voxelator server30 was performed using an outer-probe radius of 10 Å and inner-probe radius of 2 Å. Chimera54 and ChimeraX55 were used to visualize the structures in the figures.
Mutagenesis
Mutations of the E. coli K12 EcWaaL (protein accession code: WP_001395405.1), in pWSK29 vector, were generated with an in-house method using KOD polymerase and custom primers.
Expression test of E. coli K12 EcWaaL mutants
WaaL (wild type and all mutants) from E. coli K12 (EcWaaL), cloned in the pWSK29 vector, were used to transform BL21(DE3)pLysS E. coli competent cells, and grown overnight in 2xYT medium supplemented with 100 μg ml−1 ampicillin and 35 μg ml−1 chloramphenicol at 37 °C with shaking (240 rpm). The next day, 50 ml of the same medium was inoculated with the starter culture at a 1:100 ratio, and left to grow at 37 °C with shaking (240 rpm), until the OD600 nm reached 0.8–1.2. The temperature was then reduced to 22 °C, protein expression was induced with 0.2 mM IPTG, and the culture was incubated overnight with shaking (240 rpm). Cells were collected by centrifugation (3,700 rpm for 15 min) at 4 °C, washed once with PBS and centrifuged again to produce a solid pellet. Cell pellets were resuspended in 2 ml lysis buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, 20 mM MgSO4, 10 μg ml−1 DNase I, 8 μg ml−1 RNase A, 1 mM TCEP, 1 mM PMSF and Complete Mini EDTA-free protease inhibitor cocktail (Roche) used according to the manufacturer’s instructions. Cells were lysed by sonication and the lysate was solubilized for two hours with DDM, added to a final concentration of 1% (w/v). The solubilized material was clarified by centrifugation for 30 min (13,000 rpm) at 4 °C, and the supernatant was mixed with Ni-NTA agarose beads (Qiagen) and binding allowed to proceed overnight in the presence of 40 mM imidazole. The beads were then loaded on a column and washed with 5 column volumes of 20 mM HEPES pH 7.5, 500 mM NaCl, 75 mM imidazole and 0.03% (w/v) DDM. Protein was eluted with two column volumes of 20 mM HEPES pH 7.0, 150 mM NaCl, 300 mM imidazole and 0.03% (w/v) DDM (100 μl). Imidazole was removed from the eluted protein by exchanging buffer to 20 mM HEPES pH 7.0, 200 mM NaCl and 0.03% (w/v) DDM by concentrating the protein (Amicon Ultra 0.5 ml; 50 kDa cut-off) and washing the sample through with 5 column volumes of buffer (500 μl), and repeating this 5 times, until the final step in which the samples were concentrated to 20 μl. The eluates were then separated on a 14% SDS–PAGE gel, to confirm expression (Extended Data Fig. 7c).
Preparation of Und-PP (C55H101N3O7P2)
The procedure was modified from a previous publication56. Prenol composed of 11 isoprenoid units (C55H90O) and trichloroacetonitrile was dissolved in dichloromethane and added to tetra-n-butylammonium dihydrogen phosphate and the mixture was stirred for 90 min at room temperature. The solvent was evaporated, and the residue was dissolved in tetrahydrofuran. An ammonia solution (25%) was added to the mixture to precipitate the inorganic phosphate, and left overnight at 4 °C. The following day, the supernatant was removed, and the pellet was washed twice with tetrahydrofuran. The tetrahydrofuran extracts were pooled and evaporated. The residue was dissolved in a chloroform:methanol mixture (2:1 v/v) and applied onto a DEAE-Sephadex A-25 (acetal form) column. Any unreacted undecaprenol was eluted with a chloroform:methanol mixture (2:1 v/v). The final product was eluted with increasing concentrations of ammonium acetal (0–55 mM). Fractions containing Und-PP were collected and analysed by TLC. The solvent was then evaporated, and the residues were dissolved in a mixture of chloroform:methanol (2:1 v/v) with the addition of a 3% ammonia solution. The Und-PP was stored at −20 °C until further use.
TLC analysis of Und-PP
TLC analysis was used to identify the ligand, as mass spectrometry was not successful. Lipids were extracted from detergent-purified CmWaaL by adding chloroform:methanol (2:1) in a 1:1 ratio. The solvent for the mobile phase of the TLC on silica gel plates (Millipore; TLC Silica gel 60 F254) was a solution of chloroform, methanol and water (65:25:4 per volume ratio). TLC plates were dried for five minutes and placed in an enclosed container along with iodine crystals for staining.
CRISPR–Cas9 gene editing in C. metallidurans
To allow for efficient genetic manipulation in C. metallidurans, we adapted our recently optimized single plasmid CRISPR–Cas9/lambda red recombineering system57–63. We used a pBBR1MCS2 origin of replication, Streptococcus cas9 controlled by an araBAD promoter; a guide RNA specific to the CmWaaL gene controlled by a pTAC promoter for constitutive expression, a 764 base pair (bp) area of homology to WaaL, the lambda red recombineering genes to improve the efficiency of homologous recombination, and an ampicillin resistance cassette60,64,65. For the knock-out, we first analysed the wild-type CmWaaL sequence via the CRISPR direct website to identify an appropriate N20 sequence, which was incorporated into CmWaaL sgRNA. The homology was engineered to contain a 183-bp deletion surrounding the cas9 cut site. The CmWaaL specific sgRNA and homology cassettes were cloned into the pBBR1_CRISPR vector. The sequence confirmed plasmid was inserted into the CH34 C. metallidurans isolate by electroporation and appropriate transformants were identified through colony PCR. Transformants were grown at 30 °C under ampicillin selection in tryptic soy broth (TSB) and induced with 0.2% arabinose after two hours. After 12–24 h of induction, the cultures were diluted 1:100 and plated on tryptic soy agar (TSA) + ampicillin at 100 mg ml−1 and 0.2% arabinose. Appropriate mutants were identified by colony PCR and Sanger sequencing (Genewiz). Mutants were cured of the CRISPR plasmid with serial passage on non-selective medium TSA.
Expression test of wild-type and mutant CmWaaL
Wild-type and mutant CmWaaL, which was cured of the CRISPR plasmid and transformed with pBBR1_WaaL complementation plasmids—all bearing a C-terminal Flag tag fused to CmWaaL wild-type and mutant constructs for detection purposes—were grown in 5 ml TSB for 3–4 days and supplemented with 100 μg ml−1 tetracycline at 30 °C while shaking (80 rpm). The next day, 50 ml of the same medium was inoculated with the starter culture at a 1:100 ratio, and left to grow at 30 °C with shaking (80 rpm), for 3–4 days. Cells were collected by centrifugation (3,700 rpm for 15 min) at 4 °C to produce a solid pellet. Cell pellets were resuspended in 2 ml lysis buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, 20 mM MgSO4, 10 μg ml−1 DNase I, 8 μg ml−1 RNa-seA, 1 mM TCEP, 1 mM PMSF and Complete Mini EDTA-free protease inhibitor cocktail (Roche) according to the manufacturer’s instructions. Cells were lysed using a sonicator and the lysate was solubilized for two hours with DDM added to a final concentration of 1% (w/v). The solubilized material was then spun on a tabletop centrifuge for 30 min (13,000 rpm) and a portion of the supernatant was loaded on to a 14% SDS–PAGE gel. Protein expression was confirmed by western blot analysis (Extended Data Fig. 7d). Proteins from SDS–PAGE gels were transferred electrophoretically onto nitrocellulose membrane overnight at 200 mA. The membrane was blocked for one hour with 5% BSA and immobilized proteins were probed with an anti-Flag mouse antibody (Sigma’s Anti-FLAG M2 Antibody: 8146) used at a 1:5,000 dilution. Immobilized protein–Flag antibody complexes were detected using a secondary IR-labelled, goat anti-mouse green antibody (IRDye 800CW secondary antibodies: 926–32210) diluted 1:10,000. The immunoblot was developed and quantified using an Odyssey system (LI-COR Biosciences, Lincoln, NE) (Extended Data Fig. 7d).
Functional analysis of WaaL variants by LPS gel
The E. coli W3110 ΔwaaL mutant was generated by P1 vir phage transduction66 using the waaL::km mutant from the Keio collection67 and the strain confirmed by PCR. E. coli cells were grown in lysogeny broth (LB) at 37 °C and C. metallidurans in TSB at 30 °C supplemented with antibiotics. Cells were collected at an OD600 nm of 1.0 and resuspended in 100 μl 1× LDS sample buffer (Novex), containing 4% 2-mercaptoethanol (Sigma). Whole cells were treated with proteinase K as previously described68. LPS samples were separated by 4–12% NUPAGE Bis-Tris gel and visualized by silver staining (for E. coli samples)68 or using ProQ Emerald 300 from Molecular Probes (for C. metallidurans). For E. coli work only, all strains also contained plasmid pMF19 for expression of WbbL, a rhamonsyltransferease required for O-antigen synthesis, as the wbbL gene in K12 strains contain an IS5 insertion mutation.
Statistics and reproducibility
For reproducibility, all O-antigen ligase assay gels were run in triplicate (Figs. 2c, 4c, Extended Data Figs. 1c, 7a, e) and all SDS page gels and westerns were run in duplicate (Extended Data Figs. 1e, 7c, d). The initial screen single-point ELISA to identify potential binders was performed once (Extended Data Fig. 1g), and the multi-point sAB ELISA on the initial hits was run in triplicate (Extended Data Fig. 1h).
Bioinformatics sequence analysis and homology modelling of EcWaaL
WaaL homologues were identified using HHblits69, from the UniClust UniRef30 sequence database70. The resulting sequence alignment was illustrated using WebLogo371. Co-evolution analysis for WaaL was performed using MapPred72 and visualized with PyMOL. The HHblits sequence alignment, in combination with the model of EcWaaL from the AlphaFold database20, was used to create and refine a pairwise sequence alignment between CmWaaL and EcWaaL. The sequence alignment was visualized using ESPript73. Modeller 9.2474 was used to generate the EcWaaL homology model, with the co-evolutionary data used to evaluate the pairwise residue distances in the resulting structure.
Set-up of MD simulations
All simulations were run using GROMACS 202075. The Martini 2.2 force field75 was used to run a coarse-grained MD simulation to permit the assembly and equilibration of a (POPG) and palmitoyl-oleoyl-phosphatidylethanolamine (POPE) (1:4 mole ratio) bilayer around CmWaaL76 in the apo, Und-PP-bound, Und-PP- and lipid A core oligosaccharide-bound, or WaB10 Fab-bound states. The initial glycerophospholipid bilayer was created using the ‘insane’ Python script77 (https://github.com/Tsjerk/Insane) after which the CmWaaL binding partners were added manually. An elastic network of 1,000 kJ mol−1 nm−2 was applied between all backbone beads between 0.5 and 1 nm. Electrostatics were described using the reaction field method, with a cut-off of 1.1 nm using the potential shift modifier, and the Van der Waals interactions were shifted between 0.9–1.1 nm. The systems were first energy minimized by steepest descent algorithm to 1,000 kJ mol−1 nm−2 and then simulated for a total of 200 ns. Temperature and pressure were kept constant throughout the simulation at 310 K and 1 bar respectively, with protein, lipids and water/ions coupled individually to a temperature bath by the V-rescale method78 and a semi-isotropic Parrinello–Rahman barostat79. The final snapshots from the coarse-grained simulations were then converted back to an atomistic description with CG2AT2, using the protein-aligned method80 (https://github.com/owenvickery/CG2AT/).
Atomistic MD simulations
All ionizable groups were simulated with default protonation states, unless otherwise mentioned. The CHARMM36m forcefield81 was used, with the use of a 2-fs time step during the simulations. Electrostatics were described using the particle mesh Ewald (PME) method, with a cut-off of 1.2 nm, and the van der Waals interactions were shifted between 1 and 1.2 nm. The TIP3P water model was used and water bond angles and distances were constrained by SETTLE82. All other bonds were constrained using the LINCS algorithm83. The systems were then equilibrated for an additional 1 ns using a 2-fs timestep, with positional restraints of 1,000 kJ mol−1 nm−2 on the protein heavy atoms, in an NPT ensemble, with temperature V-rescale coupling at 310 K with protein, lipids and water/ions coupled individually78 and a semi-isotropic Parrinello–Rahman barostat at 1 bar79. Where present during the equilibration, the non-hydrogen atoms of the first isoprenyl unit and pyrophosphate were additionally restrained with positional restraints of 1,000 kJ mol−1 nm−2 applied to the coordinates taken from the cryo-EM structure. The parameters for Und-PP are part of the CHARMM36m force field and the Kdo2-lipidA parameters were generated using CHARMM-GUI81,84. The simulations for the four states were performed without position restraints for a total of 500 ns and run in triplicate.
MD simulation analysis
RMSD and root mean square fluctuation (RMSF) calculations were performed on the backbone of CmWaaL using the GROMACS gmx rms and gmx rmsf tools, respectively. The dynamics of Und-PP over a total of 1.5 μs were calculated on a per-atom basis from the three replicates using the gmx rmsf tool75. The TM9 movement was calculated via the distance between the geometric centres of residues 184–295 and 229–245 using the PLUMED v.2.5 software package85. Membrane deformation was calculated by mapping the xy coordinates for each phosphate group over a total of 1.5 μs of simulation onto a 1 Å resolution grid. The grid represented by beads is coloured according to the deviation from the average membrane phosphate z coordinate for each leaflet.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Extended Data
Extended Data Fig. 1 |. Functional validation of CmWaaL, identification of WaaL-specific Fabs and preparation of a nanodisc-reconstituted WaaL–Fab complex for structural analysis.
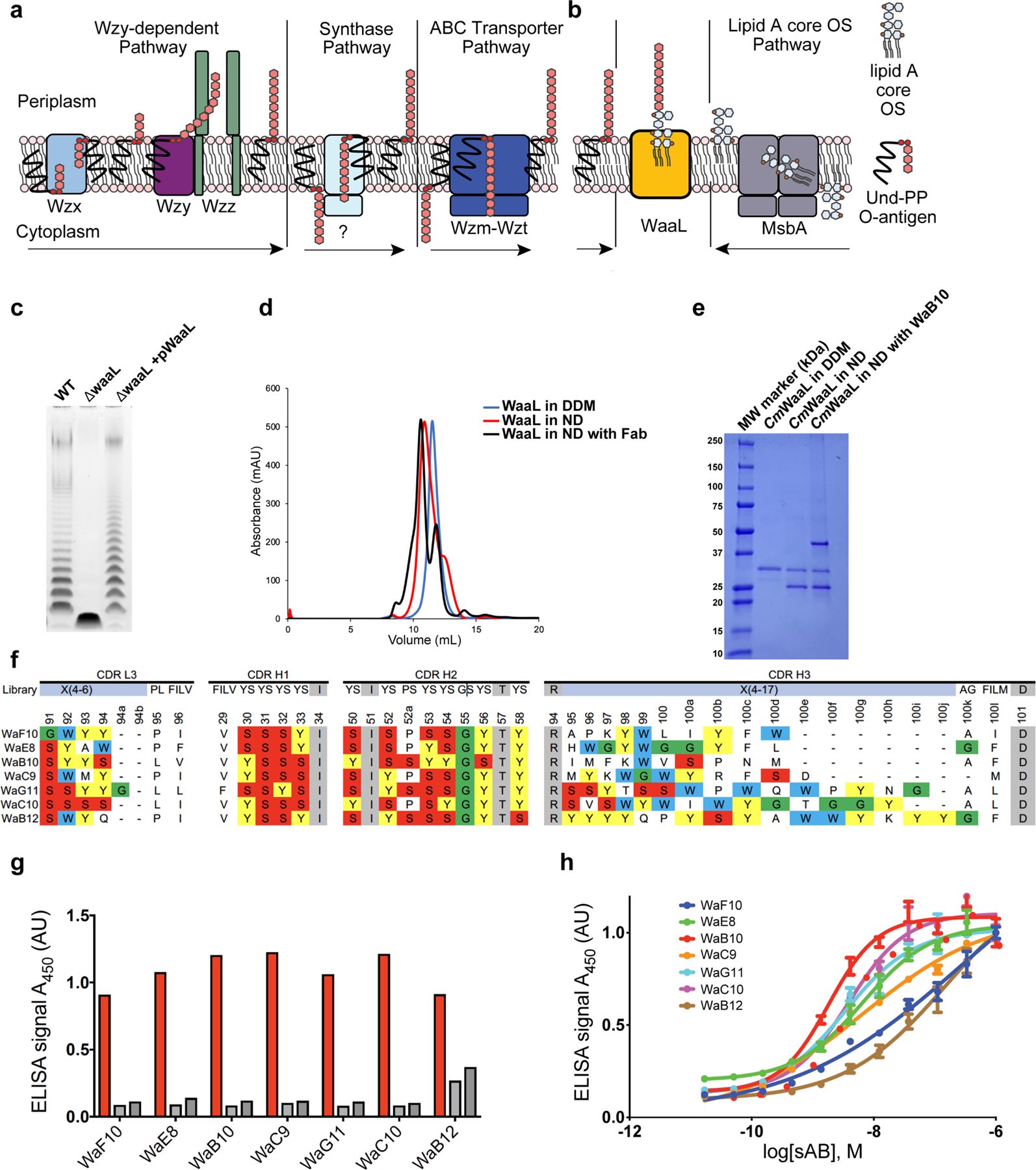
a, Schematic representation of O-antigen synthesis and transfer to the periplasmic leaflet of the inner membrane by the three different pathways, the arrows represent the direction of the Und-PP linked O-antigen takes in each pathway. Individual lipid-linked O-antigen repeat units are ligated to the lipid carrier Und-PP by glycosyltransferase enzymes. In the Wzy-dependent pathway, the O units are transported into the periplasm by the flippase Wzx. Wzy then catalyses the polymerization of O-antigen repeats, and Wzz controls the preferred modal length of the final O-antigen polymer9. The synthase dependent pathway is the least well characterized pathway31, the O-antigen is assembled at the cytoplasmic face of the inner membrane by a synthase that is also involved in its transportation across the membrane. In the ABC-dependent pathway, the polymerized Und-PP-O-antigen molecule is flipped to the periplasmic face of the inner membrane by an ABC transporter, Wzm-Wzt flippase31–34. It is important to note that the chemical composition of the C. metallidurans O-antigen is unknown. b, Schematic of WaaL function. On the right, the lipid A core oligosaccharide is synthesized in the cytoplasm and flipped to the periplasm via MsbA35. On the left, the lipid A core oligosaccharide and the O-antigen, irrespective of the pathway of origin, are ligated by WaaL. c, Functional analysis of CmWaaL ligase activity in whole cells. LPS gel showing that O-antigen ligase activity is abolished when Cm waaL is deleted, and activity is restored by plasmid complementation. d, Size-exclusion chromatography elution profiles of purified CmWaaL in detergent (blue), CmWaaL incorporated into a nanodisc (red), and CmWaaL incorporated into a nanodisc with Fab (WaB10) bound (black). e, SDS-PAGE gel of CmWaaL purification. First lane is CmWaaL purified in DDM, second lane is CmWaaL reconstituted into nanodiscs (MSP1E3D1 and POPG), and third lane is CmWaaL reconstituted into nanodiscs (MSP1E3D1 and POPG) with Fab (WaB10) bound. f, Complementarity-determining region (CDR) sequences of unique synthetic antigen binders (sABs) from biopanning against CmWaaL in MSPE3D1 nanodiscs. sABs were selected following multiple rounds of phage display starting from Fab Library E36,37. Enriched YSGW residues are highlighted by coloured boxes (yellow, red, green, and blue, respectively). YSGW residues have been previously shown to play dominant roles in highly specific and high affinity antigen recognition38. g, Single-point ELISA measuring the binding of phage-displayed sABs to CmWaaL in MSP1E3D1 nanodiscs (red), empty nanodiscs (light grey), or buffer (empty wells, dark grey). ELISA signal measured at 450 nm absorbance, see Supplementary Table 1. h, Multi-point sAB ELISA: EC50 estimation for purified sAB binding to CmWaaL incorporated into MSP1E3D1 nanodiscs, showing high affinity binding of WaE8 (green, 6.6 ± 0.045 nM), WaB10 (red, 1.87 ± 0.07 nM), WaC9 (orange, 6.26 ± 0.18 nM), WaG11 (cyan, 3.31 ± 0.06 nM), and WaC10 (magenta, 3.90 ± 0.09 nM), and modest affinity binding of WaF10 (blue, 279.5 ± 0.68 nM) and WaB12 (brown, 154 ± 0.11 nM), see Supplementary Table 1. EC50 values represent the mean of three independent experiments +/− standard error (n = 3).
Extended Data Fig. 2 |. Cryo-EM analysis of the CmWaaL–Fab complex.
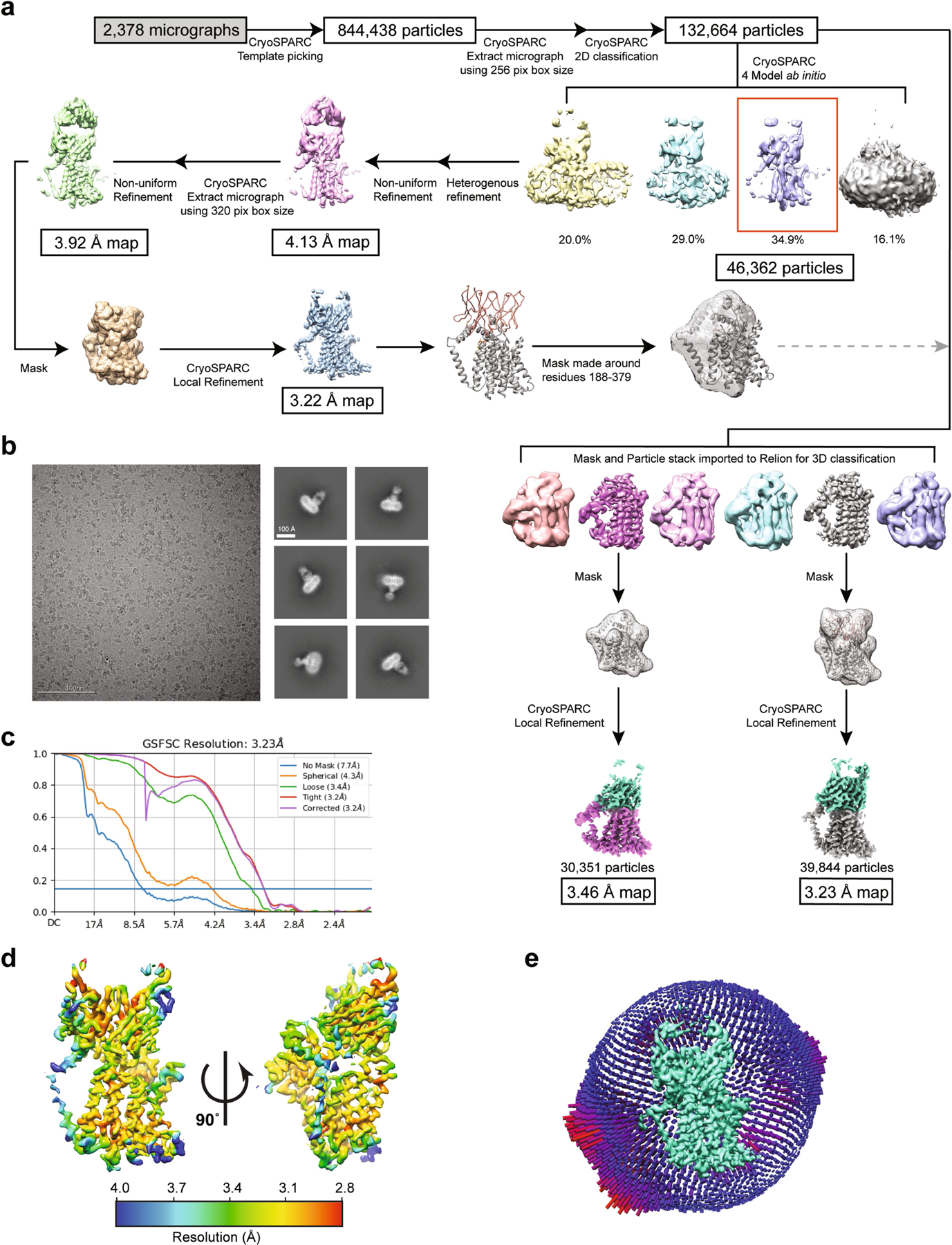
a, Flow chart outlining cryo-EM data processing and refinement performed to obtain a structure of a nanodisc-reconstituted CmWaaL with the Fab WaB10, both for the apo and the Und-PP-bound structures. b, On the left, representative micrograph (2.44 μm defocus). On right, representative two-dimensional class averages from CryoSPARC two-dimensional classification38,86. c, Fourier shell correlation (FSC) curves for the Und-PP-bound CmWaaL–Fab complex. d, Local resolution display of unsharpened reconstructions of Und-PP-bound CmWaaL in complex with the WaB10 Fab, in orthogonal views. e, Euler angle distribution of all Und-PP-bound particles used in the final map reconstruction. Final map shown in green. Each orientation is represented by a cylinder, with each cylinder’s height and colour (from blue to red) proportional to the number of particles for that specific direction.
Extended Data Fig. 3 |. Fit of cryo-EM density with model.
Cryo-EM densities (mesh) are superimposed on TM and PH helices of the CmWaaL model. The model is rendered as sidechain, coloured in rainbow, as in Fig. 1d. Und-PP (gold) is shown as sticks.
Extended Data Fig. 4 |. Interaction of CmWaaL and Fab in the complex, and sequence alignment WebLogo.
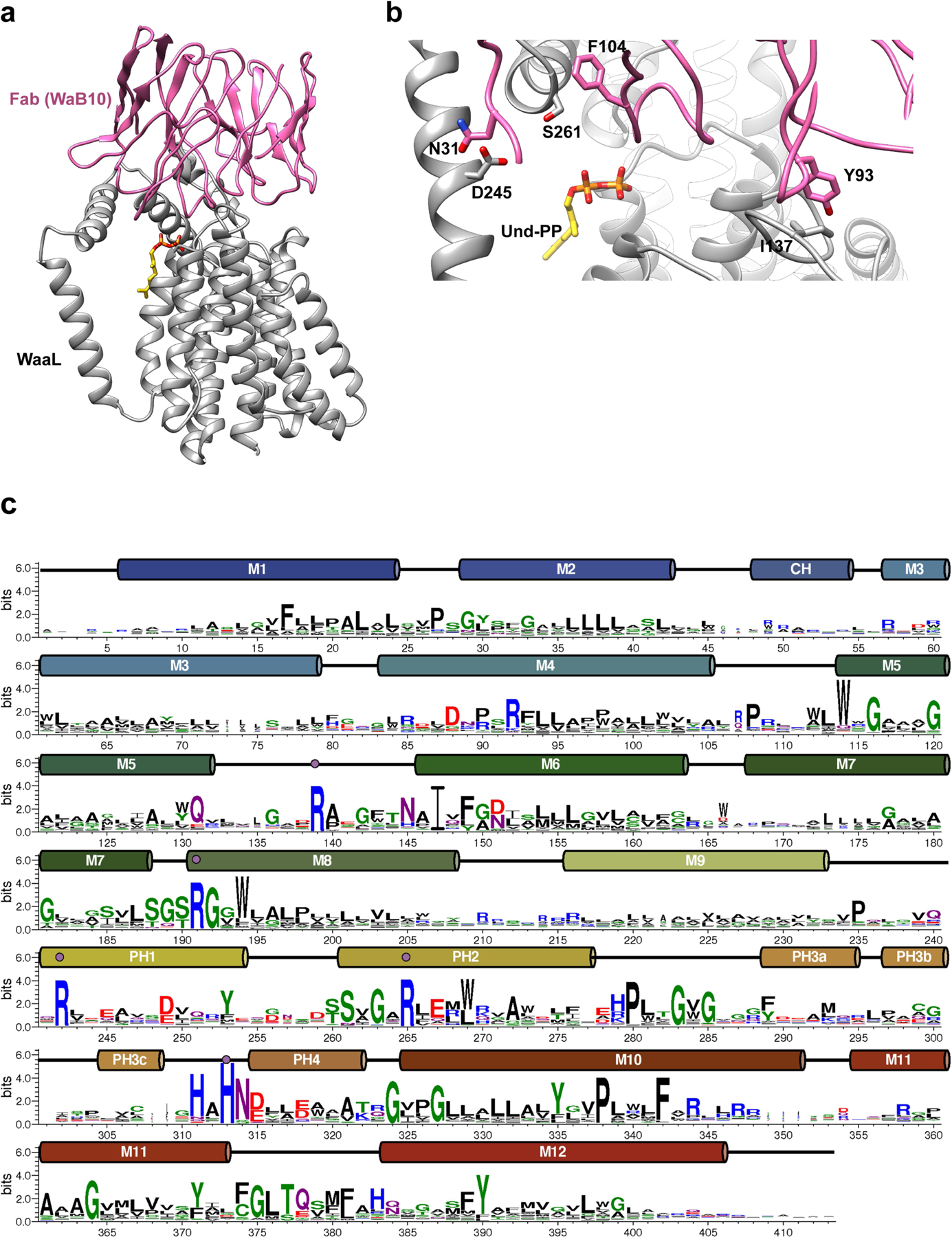
a, CmWaaL-WaB10 complex structure shown in ribbon with WaaL in grey and WaB10 in pink. Only the variable domain of WaB10 was modelled into the map. b, Interface between CmWaaL (grey) and WaB10 (pink). Residues shown in sticks (I137, D245, S261 for CmWaaL, and N31, Y93, F104 for Fab). c, A WebLogo for orthologues of CmWaaL annotated with TM and soluble helices. The numbering for CmWaaL is shown.
Extended Data Fig. 5 |. Structural features and analysis of CmWaaL.
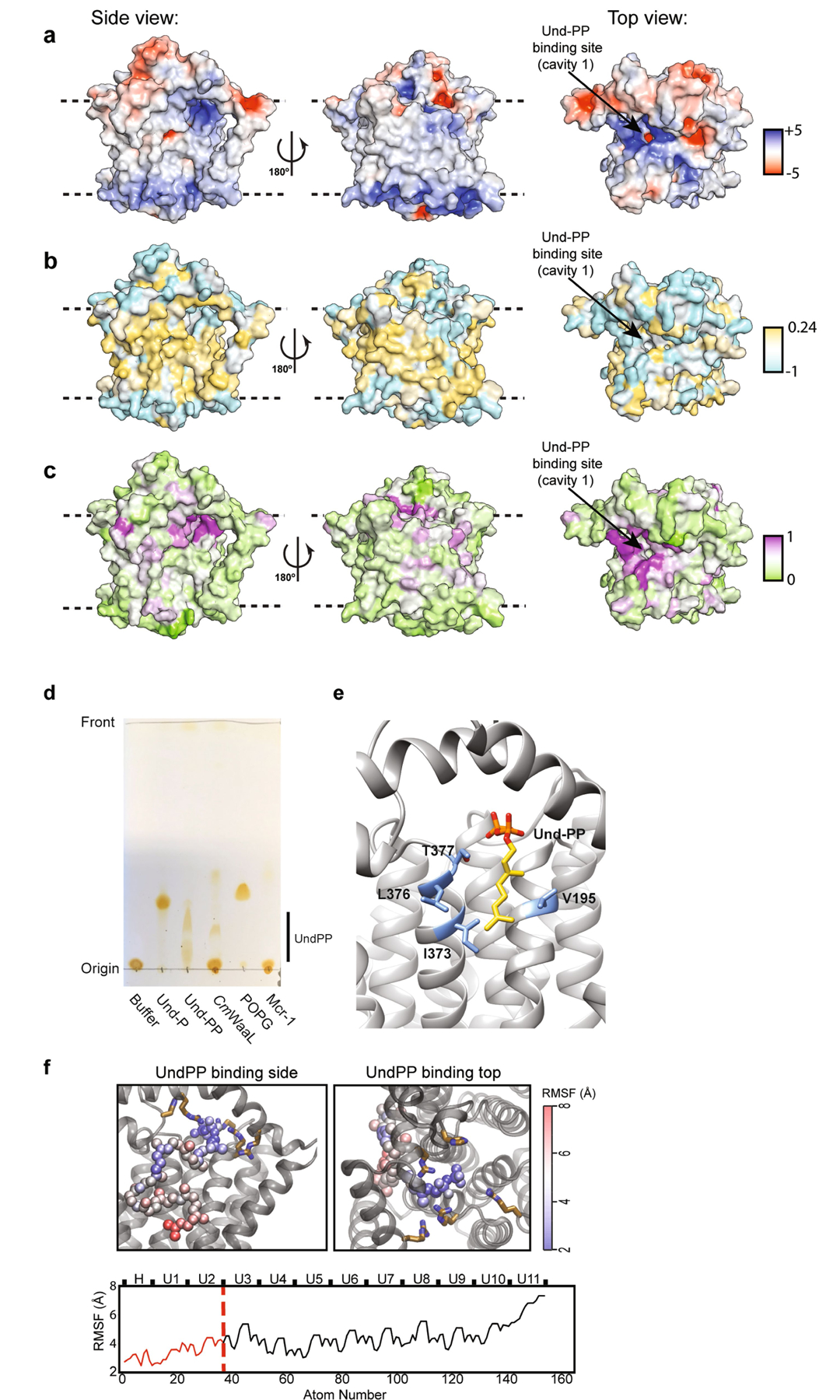
a, CmWaaL rendered in surface representation coloured by electrostatic potential on a range of ±5 kBT/e. b, CmWaaL surface coloured by Wimley-White hydrophobicity, on a cyan (very hydrophilic) to gold (very hydrophobic) scale. c, CmWaaL surface coloured by residue conservation on a green (no conservation) to purple (absolute conservation) scale. d, TLC analysis of detergent purified CmWaaL. CmWaaL was purified in detergent and run on the TLC plate after organic-phase extraction of lipids. POPG, Und-P, Und-PP and an unrelated control protein expressed in E. coli were run in separate lanes as standards. MCR-1 was chosen as it uses a lipid donor (phosphatidylethanolamine) to modify the lipid A domain of LPS, but does not use Und-PP. e, Und-PP-binding site. Residues coordinating the first two isoprenyl groups in the Und-PP tail, shown as sticks (blue). Und-PP shown as sticks coloured golden. f, (Top) MD simulations showing the Und-PP binding to CmWaaL from two views, and the flexibility of the Und-PP tail beyond the first two isoprenyl groups. The Und-PP shows increasing mobility away from the pyrophosphate. Here, the root mean squared fluctuation (RMSF) of Und-PP is shown for 3 repeats of 500 ns simulation. The non-hydrogen atoms of Und-PP are coloured by RMSF from blue to red. (Bottom) The RMSF of all atoms of the Und-PP are shown. The portion of the Und-PP resolved in the cryo-EM density map is highlighted in red.
Extended Data Fig. 6 |. EcWaaL homology model.
a, Co-evolutionary analysis for CmWaaL calculated using MapPred and mapped onto the cryo-EM structure of CmWaaL, using a threshold of 0.272. Predicted contacts between Cα atoms of given residues are shown as dashes for distances less than 10 Å (green), between 10 and 12 Å (cyan), between 12 and 20 Å (yellow), and above 20 Å (red). The CmWaaL structure is shown as a grey ribbon representation. b, Comparison of the CmWaaL structure with the homology model of EcWaaL, both coloured in rainbow from N to C termini. Und-PP is shown bound to both structures and coloured in gold, as well as the key arginine residues that surround Und-PP, show as sticks. c, Sequence alignment and secondary structure of CmWaaL and EcWaaL. Conserved residues are highlighted in black.
Extended Data Fig. 7 |. Analysis of the ligase activity of CmWaaL and EcWaaL.
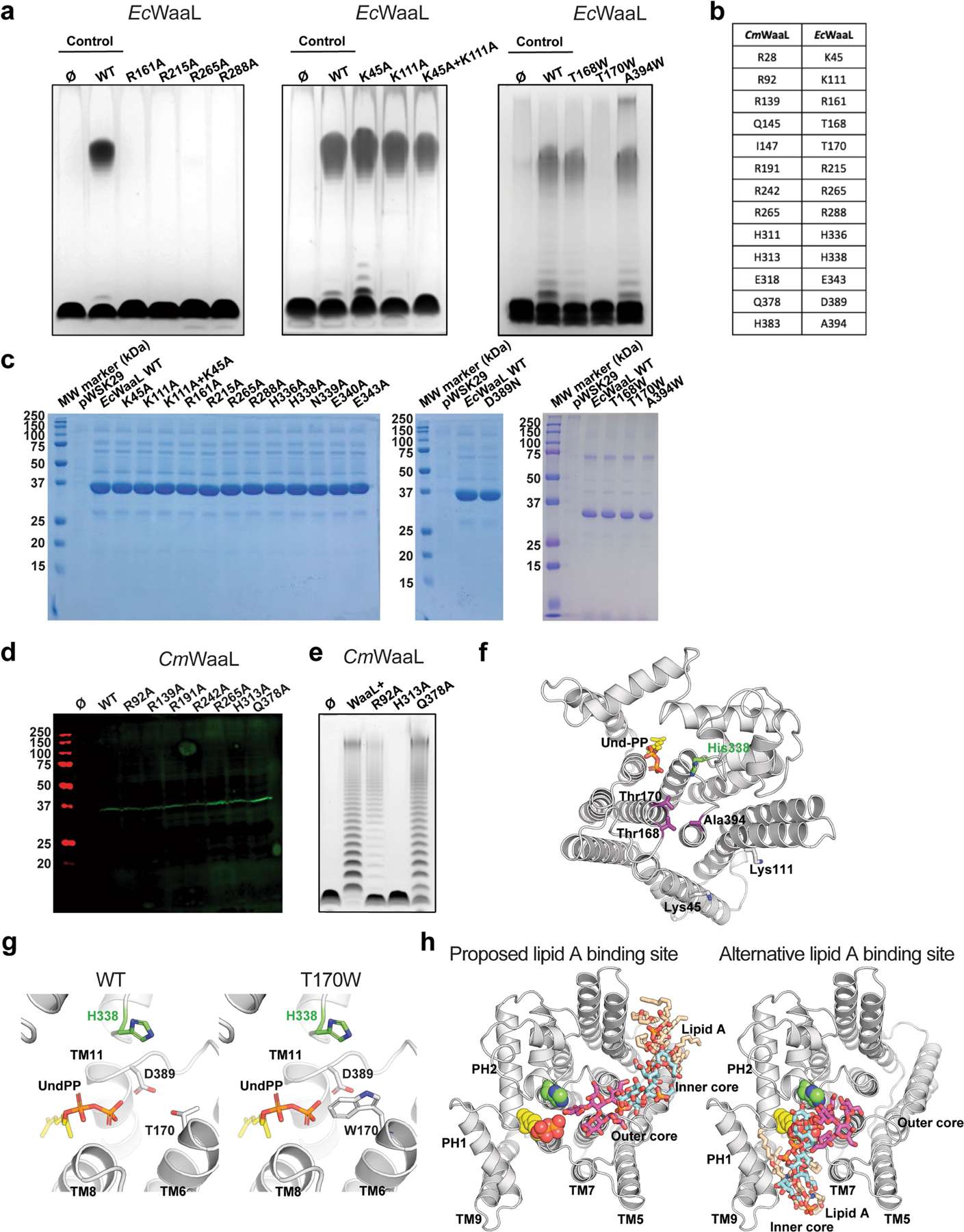
a, Functional analysis of EcWaaL ligase activity in whole cells by LPS gel analysis. Ec LPS profile. W3110 ΔwaaL containing either empty vector pWSK29 (Ø), pWSK29::EcWaaL (WT) or pWSK19::EcWaaL-variants87, 101 was evaluated for O-antigen extension. W3110 EcWaaL point mutations that cause loss of ligase activity. b, Table showing key residues in CmWaaL and their corresponding residues in EcWaaL. c, SDS-PAGE gel of all EcWaaL mutants that were purified to verify expression. d, Western blot analysis, using a mouse monoclonal anti-Flag antibody, of Flag purified WT CmWaaL and mutants, grown in C. metallidurans. e, Functional analysis of CmWaaL ligase activity in whole cells by LPS gel analysis C. metallidurans ΔwaaL containing either empty vector pBBR1(Ø), pBBR1:CmWaaL (WT) or pBBR1:CmWaaL-variants was evaluated for O-antigen extension. f, Top view of EcWaaL showing the residues mutated in the two right panels in panel a. g, Top view of the EcWaaL model, highlighting T170 (left panel) and when mutated to Trp (right panel). h, Representative views of lipid A bound to EcWaaL within the interface of cavity 2 (left panel) and an alternative binding site within the Und-PP pocket of cavity 1 (right panel). The lipid A core oligosaccharide is shown as sticks, Und-PP (gold) and H338 are shown as spheres.
Extended Data Fig. 8 |. Comparison of the Und-PP-bound and the apo-CmWaaL structures.
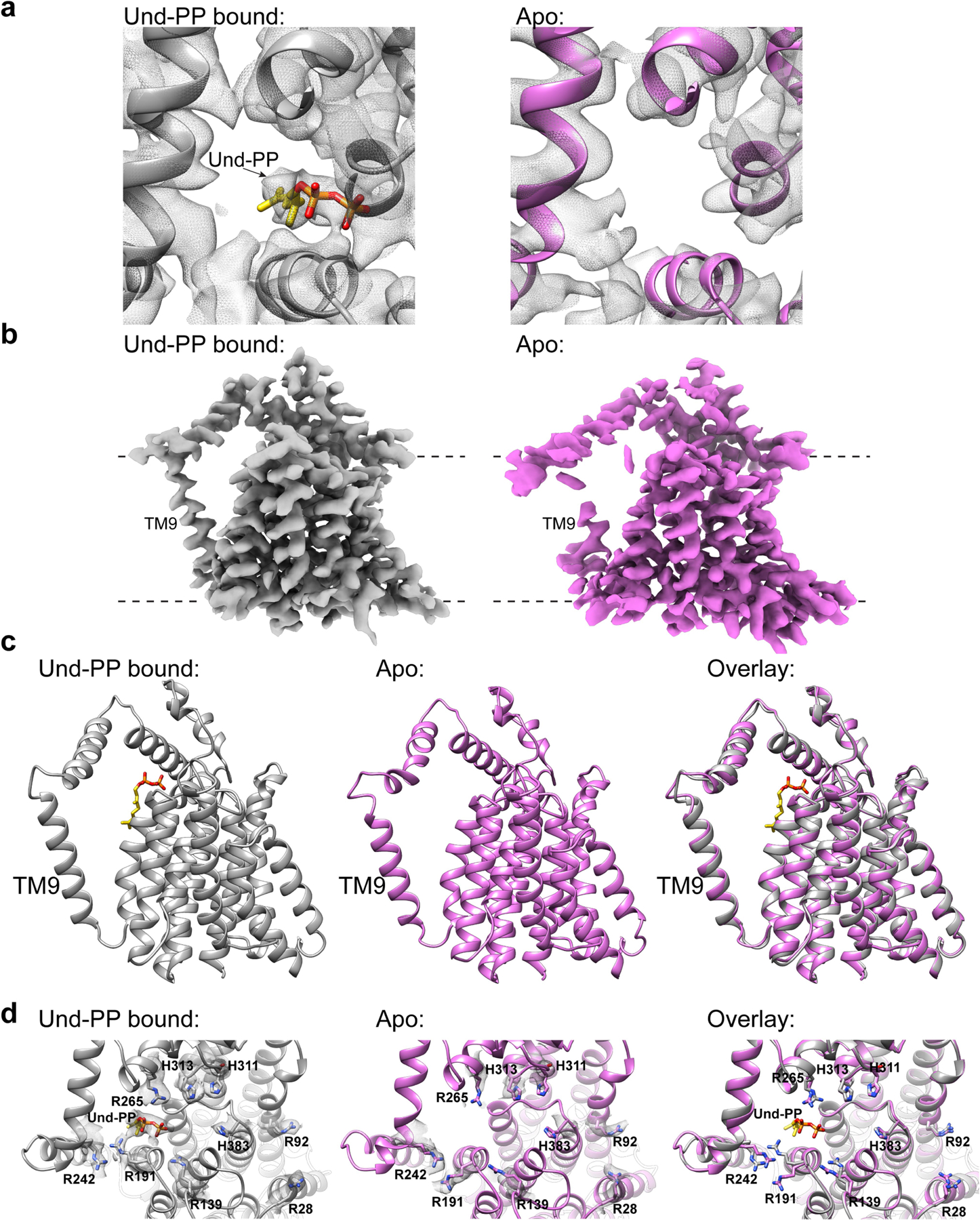
a, Top Views of the Und-PP bound (grey) and the apo (pink) CmWaaL structures, showing the density (mesh) of the Und-PP (yellow) in the ligand-bound structure in comparison to the apo structure. b, Cryo-EM density maps for the Und-PP-bound (grey) and the apo (pink) CmWaaL. Density maps were prepared in chimeraX55, by deleting any density within a 4 Å radius of the Fab in the final model. c, Side views of the Und-PP-bound (grey) and the apo (pink) CmWaaL shown as ribbon. The Und-PP (yellow) is shown as sticks in the bound structure. d, Top views of the Und-PP-bound (grey) and the apo (pink) CmWaaL showing the key residues that we hypothesize play a role in either binding/shuttling or ligation of the substrates. The density for the selected residues is shown as grey mesh. On the right an overlay of bound and apo states are shown with highlighted residues shown in stick representation.
Extended Data Fig. 9 |. CmWaaL MD simulations.
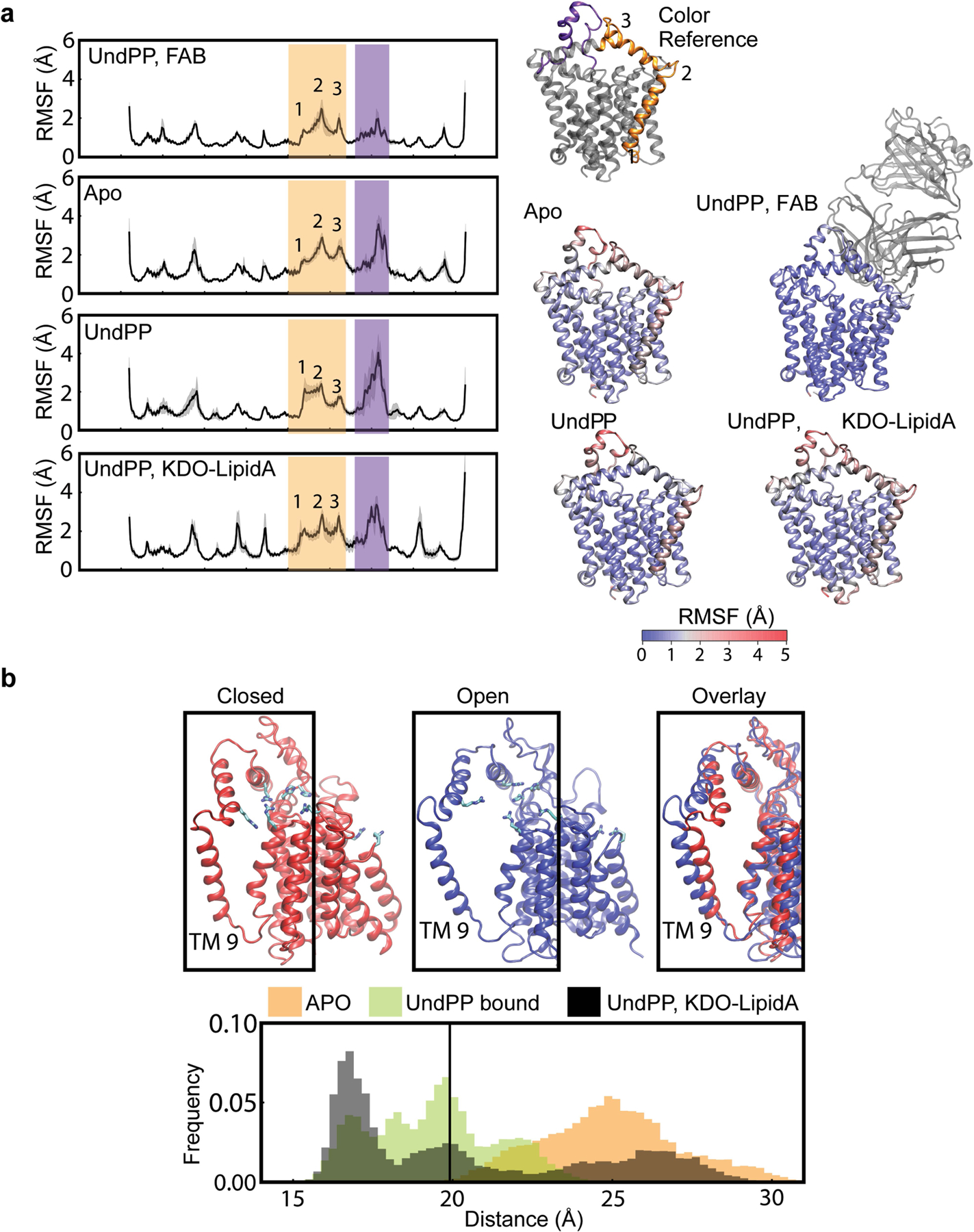
a, The RMSF of the backbone of CmWaaL. The RMSF measurements were averaged across 3 repeats of 500 ns simulation. The grey shading refers to the standard deviation across the repeats. Two highly mobile domains are highlighted in yellow (TM9 and PH1) and purple (PH3). These domains have been highlighted on the ribbon structure for reference. The ribbon structures shown are coloured by their respective RMSF from blue to red. b, Representative frames for the closed (red) and open (blue) states are shown, derived from the simulations. The mobility of TM9 is demonstrated by the histograms of the distance between the geometric centres of residues 229–244 and 184–195. The simulations of CmWaaL with Und-PP bound show a stabilization of the closed state, while TM9 separates from the core of CmWaaL in the apo state, opening an access channel to the active site. The initial distance from the cryo-EM structure is highlighted by a black line.
Extended Data Fig. 10 |. A putative common shuttling mechanism between CmWaaL, RodA, ArnT and PglB.
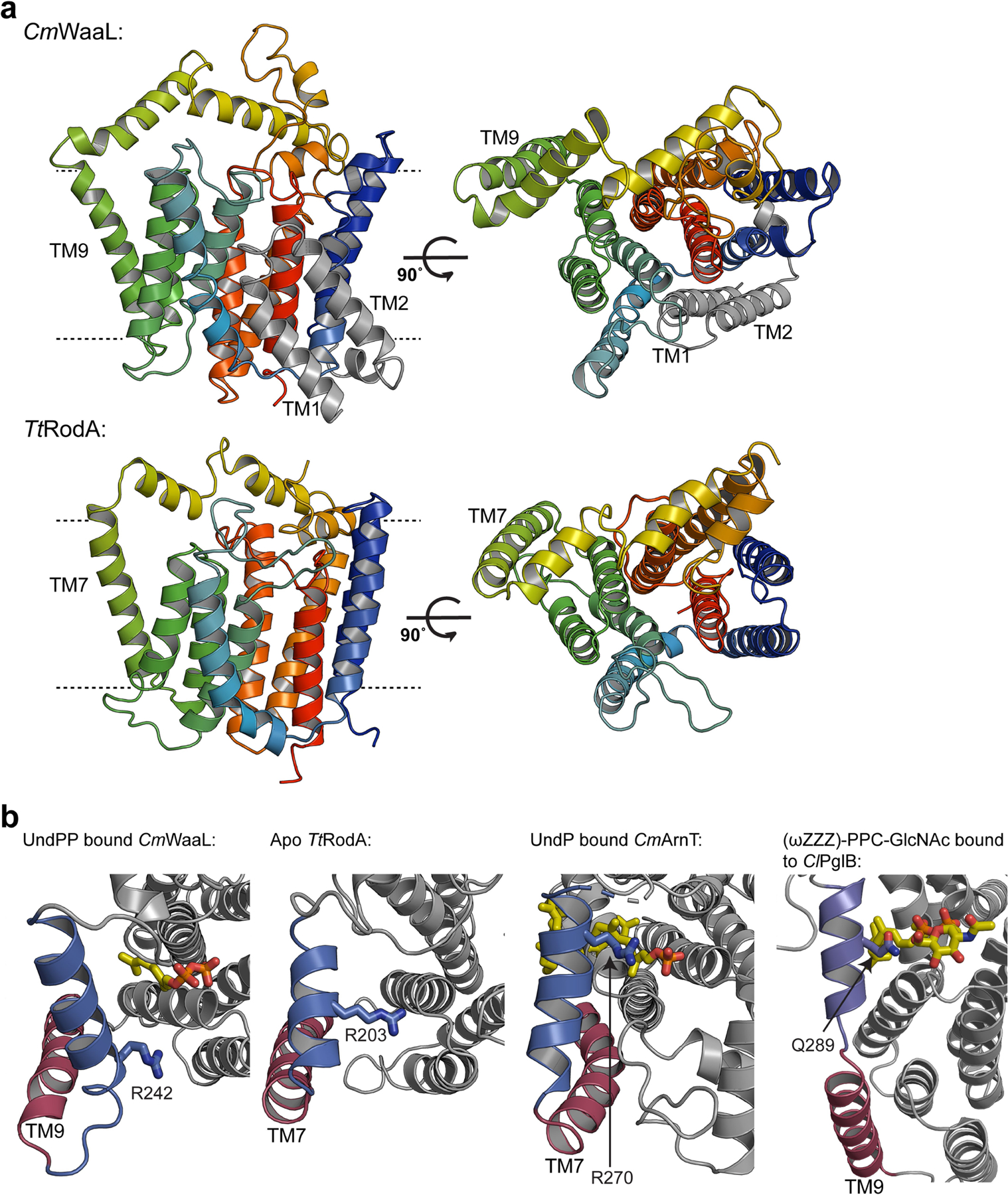
a, Structural comparison between CmWaaL and TtRodA (PDB ID 6PL5), coloured on a blue to red rainbow from N to C terminus. The two additional helices (TM1 and TM2) of CmWaaL are in grey. b, A comparison of the putative access pathway for polyprenyl-linked-phosphate containing ligands for CmWaaL, TtRodA, CmArnT (PDB ID 5F15) and ClPglB (PDB ID 5OGL). The equivalent TM helix is shown in red and periplasmic helix in blue. Conserved residues, that may be involved in the shuttling and/or coordination of polyprenyl-phosphate-containing ligands in CmWaaL, TtRodA, CmArnT and ClPglB are shown in sticks and highlighted in blue.
Extended Data Table 1 |.
Cryo-EM data collection, refinement and validation statistics
| WaaL-WaB10 (Und-PP bound) (EMD-26054) (PDB-7TPG) |
Waal-WaB10 (apo) (EMD-26057) (PDB-7TPJ) |
|
|---|---|---|
| Data collection and processing | ||
| Magnification | 130,000 | 13,000 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e-/Å2) | 70 | 70 |
| Exposure time (s) | 2.5 | 2.5 |
| Dose rate (e-/pixel/s) | 16 | 16 |
| Nominal defocus range (μm) | 1–2 | 1–2 |
| Defocus range (μm) | 0.8–1.2 | 0.8–1.2 |
| Pixel size (Å) | 1.061 | 1.061 |
| Symmetry imposed | C1 | C1 |
| Number of micrographs | 2,378 | 2,378 |
| Initial particle imagers (no.) | 844,438 | 844,438 |
| Final particle images (no.) | 39,844 | 30,514 |
| Map resolution (Å) | 3.23 | 3.46 |
| FSC threshold | 0.143 | 0.143 |
| Sphericity of 3DFSC | 0.944 | 0.944 |
| Refinement | ||
| Map sharpening B factor (Å2) | −24.07 | −12.22 |
| Reside range | 5–407 (CmWaal) 2–105 and 5–123 (Fab) |
|
| Model composition | ||
| Non-hydrogen atoms | 4778 | 4759 |
| Protein residues | 626 | 626 |
| Ligands | 1 | 0 |
| B factors (Å2) | ||
| Protein | 43.04 | 35.95 |
| Ligand | 61.05 | ---- |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.002 | 0.004 |
| Bond angles (°) | 0.621 | 0.700 |
| Validation | ||
| MolProbity score | 1.63 | 1.83 |
| Clashscore | 7.19 | 9.09 |
| Poor rotamers (%) | 0.20 | 0.00 |
| EM-Ringer Score | 2.45 | 1.60 |
| Ramachandran plot | ||
| Favored (%) | 96.45 | 95.00 |
| Allowed (%) | 3.55 | 5.00 |
| Disallowed | 0.00 | 0.00 |
Supplementary Material
Acknowledgements
We gratefully acknowledge the assistance of members of the laboratory of F.M., and of the Columbia University cryo-EM facility. We thank G. Davies for his input on the glycosyl transferase mechanism. This work was funded by NIH grants GM132120 (to F.M.), AI150098, AI138576 and AI129940 (to M.S.T.), GM117372 (to A.A.K.), GM116799 (to W. A. Hendrickson), U54 DK104309 (to A.-C.U.), and T32 AI100852 and K08 AI146284 (to T.H.M.). Research in the laboratory of P.J.S. was funded by Wellcome (208361/Z/17/Z), the MRC (MR/S009213/1) and the BBSRC (BB/P01948X/1, BB/R002517/1 and BB/S003339/1). This project made use of time on ARCHER and JADE granted via the UK High-End Computing Consortium for Biomolecular Simulation, HECBioSim (https://www.hecbiosim.ac.uk), supported by the EPSRC (grant no. EP/R029407/1). P.J.S. acknowledges Athena and Sulis at HPC Midlands+, which were funded by the EPSRC on grants EP/P020232/1 and EP/T022108/1, and the University of Warwick Scientific Computing Research Technology Platform for computational access. C.L.B.G. is funded by BBSRC studentship grant BB/M01116X/1 and D.I.R. is funded by a Schaefer Research Scholars Program Awards to Columbia University and MRC grant MR/N002679/1. Some of the work was performed at the Center for Membrane Protein Production and Analysis (COMPPÅ) and at the National Resource for automated Molecular Microscopy at the National Resource for Molecular Microscopy at the Simons Electron Microscopy Center, both located at the New York Structural Biology Center.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-04555-x.
Competing interests The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-04555-x.
Peer review information Nature thanks Russell Bishop and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
All raw movie frames have been deposited into the Electron Microscopy Public Image Archive (EMPIAR), with accession code EMPIAR-10938. The density maps have been deposited into the Electron Microscopy Data Bank (EMDB), with accession code EMD-26054 for the Und-PP-bound CmWaaL and EMD-26057 for the apo CmWaaL. Both models have been deposited in the Protein Data Bank (PDB), with accession code 7TPG for the Und-PP-bound and 7TPJ for the apo-CmWaaL model. All raw gels are available in the Supplementary Information.
References
- 1.Valvano MA Export of O-specific lipopolysaccharide. Front. Biosci 8, s452–s471 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Ruan X, Loyola DE, Marolda CL, Perez-Donoso JM & Valvano MA The WaaL O-antigen lipopolysaccharide ligase has features in common with metal ion-independent inverting glycosyltransferases. Glycobiology 22, 288–299 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Whitfield C & Trent MS Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem 83, 99–128 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Woodward L & Naismith JH Bacterial polysaccharide synthesis and export. Curr. Opin. Struct. Biol 40, 81–88 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Kaniuk NA, Vinogradov E & Whitfield C Investigation of the structural requirements in the lipopolysaccharide core acceptor for ligation of O antigens in the genus Salmonella: WaaL “ligase” is not the sole determinant of acceptor specificity. J. Biol. Chem 279, 36470–36480 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Raetz CR, Reynolds CM, Trent MS & Bishop RE Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem 76, 295–329 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong Y & Reeves PR Model for the controlled synthesis of O-antigen repeat units involving the WaaL ligase. mSphere 1, e00074–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lundstedt E, Kahne D & Ruiz N Assembly and maintenance of lipids at the bacterial outer membrane. Chem. Rev 121, 5098–5123 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitfield C, Williams DM & Kelly SD Lipopolysaccharide O-antigens—bacterial glycans made to measure. J. Biol. Chem 295, 10593–10609 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu B et al. Structure and genetics of Escherichia coli O antigens. FEMS Microbiol. Rev 44, 655–683 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feldman MF et al. The activity of a putative polyisoprenol-linked sugar translocase (Wzx) involved in Escherichia coli O antigen assembly is independent of the chemical structure of the O repeat. J. Biol. Chem 274, 35129–35138 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Bertani B & Ruiz N Function and biogenesis of lipopolysaccharides. EcoSal Plus 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmid J, Sieber V & Rehm B Bacterial exopolysaccharides: biosynthesis pathways and engineering strategies. Front. Microbiol 6, 496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruan X & Valvano MA in Glycosyltransferases (ed. Brockhausen I) 185–197 (Springer, 2013). [Google Scholar]
- 15.Abeyrathne PD, Daniels C, Poon KK, Matewish MJ & Lam JS Functional characterization of WaaL, a ligase associated with linking O-antigen polysaccharide to the core of Pseudomonas aeruginosa lipopolysaccharide. J. Bacteriol 187, 3002–3012 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pérez JM, McGarry MA, Marolda CL & Valvano MA Functional analysis of the large periplasmic loop of the Escherichia coli K‐12 WaaL O‐antigen ligase. Mol. Microbiol. 70, 1424–1440 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Islam ST, Taylor VL, Qi M & Lam JS Membrane topology mapping of the O-antigen flippase (Wzx), polymerase (Wzy), and ligase (WaaL) from Pseudomonas aeruginosa PAO1 reveals novel domain architectures. mBio 1, e00189–00110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nygaard R, Kim J & Mancia F Cryo-electron microscopy analysis of small membrane proteins. Curr. Opin. Struct. Biol 64, 26–33 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dominik PK & Kossiakoff AA in Methods in Enzymology Vol. 557 (ed. Shukla AK) 219–245 (Elsevier, 2015). [DOI] [PubMed] [Google Scholar]
- 20.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newport TD, Sansom MSP & Stansfeld PJ The MemProtMD database: a resource for membrane-embedded protein structures and their lipid interactions. Nucleic Acids Res 47, D390–D397 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazarus MB, Nam Y, Jiang J, Sliz P & Walker S Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 469, 564–567 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valvano MA in Recent Trends in Carbohydrate Chemistry (eds Rauter AP et al) 37–49 (Elsevier, 2020). [Google Scholar]
- 24.Sjodt M et al. Structural coordination of polymerization and crosslinking by a SEDS–bPBP peptidoglycan synthase complex. Nature Microbiol 5, 813–820 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meeske AJ et al. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537, 634–638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petrou VI et al. Structures of aminoarabinose transferase ArnT suggest a molecular basis for lipid A glycosylation. Science 351, 608–612 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tavares-Carreón F, Fathy Mohamed Y, Andrade A & Valvano MA ArnT proteins that catalyze the glycosylation of lipopolysaccharide share common features with bacterial N-oligosaccharyltransferases. Glycobiology 26, 286–300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Napiórkowska M et al. Molecular basis of lipid-linked oligosaccharide recognition and processing by bacterial oligosaccharyltransferase. Nat. Struct. Mol. Biol 24, 1100–1106 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Ruan X, Monjarás Feria J, Hamad M & Valvano MA Escherichia coli and Pseudomonas aeruginosa lipopolysaccharide O‐antigen ligases share similar membrane topology and biochemical properties. Mol. Microbiol 110, 95–113 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Voss NR & Gerstein M 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res 38, W555–W562 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitney J & Howell P Synthase-dependent exopolysaccharide secretion in Gram-negative bacteria. Trends Microbiol 21, 63–72 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitfield C Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu. Rev. Biochem 75, 39–68 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Cuthbertson L, Kos V & Whitfield C ABC transporters involved in export of cell surface glycoconjugates. Microbiol. Mol. Biol. Rev 74, 341–362 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pérez-Burgos M et al. Characterization of the exopolysaccharide biosynthesis pathway in Myxococcus xanthus. J. Bacteriol 202, e00335–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mi W et al. Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 549, 233–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rizk SS et al. Allosteric control of ligand-binding affinity using engineered conformation-specific effector proteins. Nat. Struct. Mol. Biol 18, 437 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller KR et al. T cell receptor-like recognition of tumor in vivo by synthetic antibody fragment. PLoS ONE 7, e43746 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fellouse FA et al. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol 373, 924–940 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Punta M et al. Structural genomics target selection for the New York consortium on membrane protein structure. J. Struct. Funct. Genomics 10, 255–268 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mancia F & Love J High-throughput expression and purification of membrane proteins. J. Struct. Biol 172, 85–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mancia F & Love J High throughput platforms for structural genomics of integral membrane proteins. Curr. Opin. Struct. Biol 21, 517–522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bayburt TH, Grinkova YV & Sligar SG Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2, 853–856 (2002). [Google Scholar]
- 43.Denisov IG, Grinkova YV, Lazarides AA & Sligar SG Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J. Am. Chem. Soc 126, 3477–3487 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Kapust RB, Tözsér J, Copeland TD & Waugh DS The P1′ specificity of tobacco etch virus protease. Biochem. Biophys. Res. Commun 294, 949–955 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Dominik PK et al. Conformational chaperones for structural studies of membrane proteins using antibody phage display with nanodiscs. Structure 24, 300–309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J et al. Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature 576, 315–320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suloway C et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Slabinski L et al. XtalPred: a web server for prediction of protein crystallizability. Bioinformatics 23, 3403–3405 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casañal A, Lohkamp B & Emsley P Current developments in Coot for macromolecular model building of electron cryo‐microscopy and crystallographic data. Protein Sci 29, 1055–1064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Afonine PV et al. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr. D 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Afonine PV et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettersen EF et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Goddard TD et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Danilov L, Druzhinina T, Kalinchuk N, Maltsev S & Shibaev V Polyprenyl phosphates: synthesis and structure-activity relationship for a biosynthetic system of Salmonella anatum O-specific polysaccharide. Chem. Phys. Lipids 51, 191–203 (1989). [DOI] [PubMed] [Google Scholar]
- 57.Jiang W, Bikard D, Cox D, Zhang F & Marraffini LA RNA-guided editing of bacterial genomes using CRISPR–Cas systems. Nat. Biotechnol 31, 233–239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang Y et al. Multigene editing in the Escherichia coli genome via the CRISPR–Cas9 system. Appl. Environ. Microbiol 81, 2506–2514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Selle K & Barrangou R Harnessing CRISPR–Cas systems for bacterial genome editing. Trends Microbiol 23, 225–232 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Jiang X et al. Vector promoters used in Klebsiella pneumoniae. Biotechnol. Appl. Biochem 63, 734–739 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Zhao D et al. Development of a fast and easy method for Escherichia coli genome editing with CRISPR/Cas9. Microb. Cell Fact 15, 205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y et al. CRISPR-Cas9 and CRISPR-assisted cytidine deaminase enable precise and efficient genome editing in Klebsiella pneumoniae. Appl. Environ. Microbiol 84, e01834–01818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McConville TH et al. CrrB positively regulates high-level polymyxin resistance and virulence in Klebsiella pneumoniae. Cell Rep 33, 108313 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mijnendonckx K et al. Characterization of the survival ability of Cupriavidus metallidurans and Ralstonia pickettii from space-related environments. Microb. Ecol 65, 347–360 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Schmidt C, Schwarzenberger C, Große C & Nies DH FurC regulates expression of zupT for the central zinc importer ZupT of Cupriavidus metallidurans. J. Bacteriol 196, 3461–3471 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66..Sambrook J & Rusell D Molecular Cloning: A Laboratory Manual 3rd edn (Cold Spring Harbor Laboratory Press, 2001). [Google Scholar]
- 67.Baba T et al. Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: the Keio collection. Mol. Syst. Biol 2, 2006.0008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hitchcock PJ & Brown TM Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol 154, 269–277 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Remmert M, Biegert A, Hauser A & Söding J HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175 (2012). [DOI] [PubMed] [Google Scholar]
- 70.Mirdita M et al. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res 45, D170–D176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crooks GE, Hon G, Chandonia J-M & Brenner SE WebLogo: a sequence logo generator. Genome Res 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Q et al. Protein contact prediction using metagenome sequence data and residual neural networks. Bioinformatics 36, 41–48 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robert X & Gouet P Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Šali A & Blundell TL Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- 75.Abraham MJ et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015). [Google Scholar]
- 76.Vogeley L et al. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 351, 876–880 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Wassenaar TA, Ingólfsson HI, Böckmann RA, Tieleman DP & Marrink SJ Computational lipidomics with insane: a versatile tool for generating custom membranes for molecular simulations. J. Chem. Theory Comput 11, 2144–2155 (2015). [DOI] [PubMed] [Google Scholar]
- 78.Bussi G, Donadio D & Parrinello M Canonical sampling through velocity rescaling. J. Chem. Phys 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Parrinello M & Rahman A Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys 52, 7182–7190 (1981). [Google Scholar]
- 80.Stansfeld PJ & Sansom MS From coarse grained to atomistic: a serial multiscale approach to membrane protein simulations. J. Chem. Theory Comput 7, 1157–1166 (2011). [DOI] [PubMed] [Google Scholar]
- 81.Huang J et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miyamoto S & Kollman PA Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem 13, 952–962 (1992). [Google Scholar]
- 83.Hess B, Bekker H, Berendsen HJC & Fraaije JGEM LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem 18, 1463–1472 (1997). [Google Scholar]
- 84.Jo S, Kim T, Iyer VG & Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Bonomi M et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 16, 670–673 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Rubinstein JL & Brubaker MA Alignment of cryo-EM movies of individual particles by optimization of image translations. J. Struct. Biol 192, 188–195 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Vinés ED, Marolda CL, Balachandran A & Valvano MA Defective O-antigen polymerization in tolA and pal mutants of Escherichia coli in response to extracytoplasmic stress. J. Bacteriol 187, 3359–3368 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw movie frames have been deposited into the Electron Microscopy Public Image Archive (EMPIAR), with accession code EMPIAR-10938. The density maps have been deposited into the Electron Microscopy Data Bank (EMDB), with accession code EMD-26054 for the Und-PP-bound CmWaaL and EMD-26057 for the apo CmWaaL. Both models have been deposited in the Protein Data Bank (PDB), with accession code 7TPG for the Und-PP-bound and 7TPJ for the apo-CmWaaL model. All raw gels are available in the Supplementary Information.