

Abstract
BACKGROUND
The DNA-repair enzyme Artemis is essential for rearrangement of T- and B-cell receptors. Mutations in DCLRE1C, which encodes Artemis, cause Artemis-deficient severe combined immunodeficiency (ART-SCID), which is poorly responsive to allogeneic hematopoietic-cell transplantation.
METHODS
We carried out a phase 1–2 clinical study of the transfusion of autologous CD34+ cells, transfected with a lentiviral vector containing DCLRE1C, in 10 infants with newly diagnosed ART-SCID. We followed them for a median of 31.2 months.
RESULTS
Marrow harvest, busulfan conditioning, and lentiviral-transduced CD34+ cell infusion produced the expected grade 3 or 4 adverse events. All the procedures met pre-specified criteria for feasibility at 42 days after infusion. Gene-marked T cells were detected at 6 to 16 weeks after infusion in all the patients. Five of 6 patients who were followed for at least 24 months had T-cell immune reconstitution at a median of 12 months. The diversity of T-cell receptor β chains normalized by 6 to 12 months. Four patients who were followed for at least 24 months had sufficient B-cell numbers, IgM concentration, or IgM isohemagglutinin titers to permit discontinuation of IgG infusions. Three of these 4 patients had normal immunization responses, and the fourth has started immunizations. Vector insertion sites showed no evidence of clonal expansion. One patient who presented with cytomegalovirus infection received a second infusion of gene-corrected cells to achieve T-cell immunity sufficient for viral clearance. Autoimmune hemolytic anemia developed in 4 patients 4 to 11 months after infusion; this condition resolved after reconstitution of T-cell immunity. All 10 patients were healthy at the time of this report.
CONCLUSIONS
Infusion of lentiviral gene-corrected autologous CD34+ cells, preceded by pharmacologically targeted low-exposure busulfan, in infants with newly diagnosed ART-SCID resulted in genetically corrected and functional T and B cells. (Funded by the California Institute for Regenerative Medicine and the National Institute of Allergy and Infectious Diseases; ClinicalTrials.gov number, NCT03538899.)
Severe combined immunodeficiency (SCID), characterized by a lack of T lymphocytes with a lack of or nonfunctional B lymphocytes, is genetically heterogeneous: it is caused by a defect in any one of approximately 20 genes.1,2 Population-based newborn screening has established that SCID occurs in approximately 1 in 65,000 births (95% confidence interval, 1 in 51,000 to 1 in 90,000) in the United States.3,4 Artemis-deficient SCID (ART-SCID), resulting from mutations in the gene DCLRE1C (DNA cross-link repair 1C), constitutes 2 to 3% of all SCID. It has a high incidence among persons of Navajo or Apache descent and in certain consanguineous populations and shows poor response to treatment by allogeneic hematopoietic-cell transplantation (HCT).5–7 In the absence of normal levels of functional Artemis protein, the repair of double-strand DNA breaks is compromised, which results in a T-cell–negative, B-cell–negative, natural killer (NK) cell–positive immunophenotype — arising from arrested V(D)J recombination of T and B lymphocytes — and increased systemic sensitivity to alkylating agents typically used as pre-HCT conditioning.8
Treatment of ART-SCID by HCT, even with an HLA-matched sibling, is less likely to achieve T- and B-cell reconstitution than are other SCID genotypes treated by HCT.7,9 HCT from unrelated or haplocompatible related donors also has an elevated risk of graft rejection and graft-versus-host disease (GVHD) and a decreased likelihood of B-cell reconstitution among persons with ART-SCID. Although high-dose alkylators improve engraftment and immune reconstitution, they often cause late adverse effects in patients with ART-SCID, including short stature, dental maldevelopment, endocrinopathies, and premature death.10 The administration of gene-transduced autologous CD34+ hematopoietic stem cells (HSCs), which eliminates the risk of graft rejection and GVHD, is therefore a compelling alternative to allogeneic HCT.
Studies of gene therapy for X-linked SCID and SCID associated with adenosine deaminase deficiency have shown that some conditioning is necessary to open marrow niches for engraftment of gene-corrected HSCs.11–14 In mouse models of ART-SCID, conditioning to open marrow niches was similarly necessary for engraftment,15 but low-dose busulfan was sufficient.16
We previously showed in Artemis-deficient mice that physiologic levels of Artemis protein were critical for cell survival.17 DCLRE1C complementary DNA (cDNA) that was expressed under control of a strong promoter had less efficacy with respect to immune reconstitution than expression under the physiologic human Artemis promoter.17 Therefore, our new lentiviral construct, AProArt, contains the human DCLRE1C cDNA driven by a 1-kb endogenous DCLRE1C promoter sequence.15,18 This use of an autologous promotor and reduced exposure to busulfan are key characteristics of this study of AProArt-transduced autologous CD34+ bone marrow cells to treat infants with newly diagnosed ART-SCID.
METHODS
PATIENTS AND CLINICAL INTERVENTION
The protocol for this phase 1–2, nonrandomized, single-center clinical study was approved by the Food and Drug Administration and the University of California, San Francisco (UCSF), institutional review board and is available with the full text of this article at NEJM.org. Written informed consent was obtained from the parents of each child. Ten infants with newly diagnosed ART-SCID who lacked an HLA-matched sibling donor were enrolled and treated at UCSF between June 2018 and September 2021. Three older patients with insufficient immunity after allogeneic HCT were also treated; data from these patients are being analyzed.
Care of patients was managed in the UCSF Benioff Children’s Hospital Blood and Marrow Transplant Unit and followed standard guidelines for autologous HCT. After the manufacture, cryopreservation, and quality-control testing of the gene-transduced autologous CD34+ cells, patients received intravenous busulfan targeted to a blood cumulative area under the curve (AUC) of 20 mg × hour per liter administered over a period of 2 days. The first dose was calculated from a pharmacokinetic model based on age and weight.19 First-dose pharmacokinetic data were used to adjust the second dose. Pharmacokinetic samples after the second dose allowed calculation of each patient’s cumulative AUC. One day later, the gene-corrected cells were thawed and infused.
Measurements of immune reconstitution and gene marking began 4 weeks after infusion, with T-cell receptor (TCR) diversity and vector insertion-site analysis beginning 12 weeks after infusion. Follow-up evaluations occurred monthly through month 6, every 3 months through month 24, and less often subsequently. Results that were obtained through June 30, 2022, are reported here. The authors vouch for the accuracy and completeness of the data and for the fidelity of the study to the protocol.
PRODUCT MANUFACTURING
The AProArt vector, described previously,15 was produced at the University of Indiana Vector Production Facility. Bone marrow (20 ml per kilogram of body weight) that was collected while the patient was under general anesthesia was enriched for CD34+ cells (CliniMACS machine, Miltenyi Biotec) according to the manufacturer’s instructions. In accordance with the protocol, patients received a transfusion of red cells before marrow harvest for a hemoglobin level of less than 10 g per deciliter, and all the patients received a transfusion (from the same donor) after marrow harvest. The CD34+ cells were cultured overnight in RetroNectin (Takara Bio)–coated plates with recombinant human thrombopoietin, human stem-cell factor, human Fms-like tyrosine kinase receptor 3 ligand, and interleukin-3. The cells were transduced with AProArt for 6 to 8 hours on each of the following 2 days, after which they were cryopreserved in liquid nitrogen, with aliquots reserved for testing.
PATIENT CELL LINEAGE SEPARATION
Blood samples were prepared as described previously.15 They were sorted by means of flow cytometry (JAZZ sorter, Becton Dickinson) into lineages of myeloid cells (CD14+/CD15+), T cells (CD3+), B cells (CD19+), and NK cells (CD3−/CD19−/CD56+).
VECTOR COPY NUMBER AND TRANSDUCTION EFFICIENCY
DNA from lineage-isolated cells was analyzed by means of droplet digital polymerase-chain-reaction (PCR) assay with primers specific for the AProArt vector and a region of the albumin gene to generate mean vector copy number (VCN) per cell.15 AProArt-transduced CD34+ cells that were cultured in myeloid colony-forming methylcellulose were pooled for VCN determination, and 100 14-day colonies were assayed individually to calculate the percentage positive for the vector sequence (transduction efficiency).15
IMMUNE RECONSTITUTION
We defined immune reconstitution as the meeting of three of four criteria: at least 1000 CD3+ cells per cubic millimeter, at least 500 CD4+ helper T cells per cubic millimeter, a percentage of naive CD4+ cells of at least 20% of total CD4+ cells, and a lymphocyte proliferative response to phytohemagglutinin of at least 50% of the lower limit of the reference range. We assayed, by means of flow cytometry, T- and B-cell subsets and the proliferative responses of CD3+ and CD45+ cells to phytohemagglutinin. B-cell function was determined by serum IgM concentration, IgM isohemagglutinin titers, and, after discontinuation of immune globulin infusions, specific antibody titers after vaccinations. The diversity of TCR β-chain (TCRβ) repertoire and number of vector insertion sites were determined through deep DNA sequencing (see the Supplementary Appendix, available at NEJM.org).
STUDY DESIGN
The first (stage 1) measure of treatment success was the safe administration of busulfan and transduced cells, determined at 42 days after infusion. We assessed feasibility, safety, post-conditioning recovery, and evidence of early engraftment of transduced cells without adverse events of grade 3 or higher (adapted National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0). Feasibility was defined as having at least 2×106 infused AProArt-transduced CD34+ cells per kilogram with a mean VCN of at least 0.2 and less than 10 copies per cell; an absolute neutrophil count of more than 500; a platelet count of more than 20,000 with no clinical bleeding and without platelet transfusion; and no grade 3 or 4 adverse events not associated with busulfan treatment, collection of cells, or infusion. The second (stage 2) measure of treatment success was functional T-cell reconstitution at month 12.
Results
PATIENTS
The median follow-up for the 10 patients was 31.2 months (range, 10.0 to 48.9) (Table 1). There were no deaths. Four patients were of Navajo or Apache descent and homozygous for the founder DCLRE1C mutation (c.597C→A; p.Tyr199X).5 Six patients had combinations of previously reported missense and gross deletion mutations (Table 1 and Table S1 in the Supplementary Appendix).20,21 Patients ART003 and ART012 had novel heterozygous variants of unproven significance in addition to heterozygous variants on opposite parental alleles previously reported to be pathogenic; however, skin fibro-blasts from each patient had a radiation sensitivity that was rescued after AProArt transduction, findings that support that the variants were causal (data not shown).15
Table 1.
Patient Characteristics and Disease Course.*
| Variable | Patient ART001 | Patient ART002 | Patient ART003 | Patient ART007 | Patient ART008 | Patient ART009 | Patient ART010 | Patient ART011 | Patient ART012 | Patient ART013 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Female | Male | Female | Male | Male | Male | Female | Male |
| Race or ethnic group† | Navajo | Hispanic | Hispanic/White | White | Navajo | Navajo | Vietnamese | Hispanic/Japanese | Lebanese | Apache/Navajo |
| DCLRE1C mutation, chromosome 10p13 | Hom c.597C→A (p.Tyr199X) | Hom del exons 1–(82 kb) | Del exons 1–3‡ | Del exons 1–5 (82 kb); c.406G→A (p.Asp136Asn) | Hom c.597C→A (p.Tyr199X) | Hom c.597C→A (p.Tyr199X) | Del exons 1–3; c.47T→C (p.Ile16Thr) | Del exon 3; del exons 1–3 | c.161+2T→G (splice); c.346T→C (p.Cys116Arg) | Hom c.597C→A (p.Tyr199X) |
| Clinical findings before GT | ||||||||||
| NBS | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes |
| SCID neutron-penia§ | Yes | Yes | Yes | No | Yes | Yes | No | Yes | No | Yes |
| Other | Rhinovirus; noninfectious oral ulcers | Noninfectious oral ulcers | Maternal chimerism with GVHD of skin¶ | CMV viremia | Preterm birth at 31 wk of gestation; grade 1 bilateral intraventricular bleeding | Maternal chimerism with GVHD of skin and liver¶ | Maternal chimerism with GVHD of skin¶ | |||
| Clinical findings after GT | Noninfectious oral ulcers; central-catheter bacteremia; Cd and norovirus enteritis; all resolved | CCMV treated with valganciclovir; RVE; acalculous cholangitis treated with CCE; all resolved | None | None | Noninfectious oral ulcers, resolved; behavior suggestive of autism | Persistent maternal GVHD, resolved | CMV encephalitis treated with multiple antiviral agents; non-infectious oral ulcers; all resolved | None | Persistent maternal GVHD; noninfectious oral ulcers; Cd colitis; intestinal bleeding from sirolimus; all resolved | Persistent maternal GVHD, resolved |
| Age at cell infusion (mo) | 2.6 | 3.7 | 3.0 | 2.3 | 2.5 | 2.4 | 2.5 | 3.8 | 13.3 | 2.3 |
| Busulfan cAUC (mg × hr/liter) | 17.6 | 19.9 | 18.8 | 19.7 | 18.6 | 18.9 | 19.2 | 20.2 | 19.4 | 18.9 |
| Busulfan dose (mg/kg) | 2.5 | 4.0 | 3.7 | 3.5 | 4.1 | 3.8 | 2.9 | 2.8 | 2.0 | 4.1 |
| CD34+ cell dose (×10−6/kg) | 6.6 | 3.9 | 6.0 | 10.6 | 6.1 | 12.1 | 10.4 | 9.9 | 2.2 | 8.8 |
| Mean VCN (copies/cell) | 1.9 | 1.9 | 3.9 | 2.3 | 1.8 | 3.2 | 2.9 | 3.4 | 1.6 | 2.6 |
| TDE TDE(%) | 68 | 66 | 84 | 81 | 65 | 81 | 81 | 82 | 62 | 74 |
| Postbusulfan lowest ANC (cells/mm3)‖ | 1370 | 1020 | 170 | 260 | 490 | 350 | 190 | 450 | 140 | 80 |
| Time of postbulsulfan lowest ANC (days)‖ | 13 | 19 | 19; G | 18; G | 14 | 18 | 14; G | 20; G | 21; G | 14; G |
| Follow-up after GT (mo) | 48.9 | 45.2 | 43.9 | 34.5 | 33.4 | 28.9 | 12** | 17.8 | 14.5 | 10.0 |
None of the patients had acute toxic effects related to gene therapy (GT) within 42 days after the infusion. ANC denotes absolute neutrophil count, cAUC cumulative area under the curve, CCE cholecystectomy, Cd Clostridioides difficile, CMV cytomegalovirus, hom homozygous, NBS positive newborn screening for severe combined immunodeficiency (SCID), RVE rotavirus enteritis, TDE transduction efficiency, and VCN vector copy number.
Race or ethnic group was reported by the parents.
The other mutation was c.492_504delins13, and the amino acid change was p.Thr167_Phe168delins→MetLeu.
SCID neutropenia refers to neutropenia in patients with SCID that has no other cause and that is responsive to granulocyte colony-stimulating factor.
Graft-versus-host disease (GVHD) was treated with topical or oral prednisone, sirolimus, and multiple small doses (1 to 2.5 mg per kilogram) of rabbit antithymocyte globulin (rATG) in all three patients before and after infusion of gene-transduced cells to maintain the maternal T-cell count as low as possible and control the GVHD; Patient ART012 also received a single dose of alemtuzumab as initial therapy before admission for gene therapy. The rATG infusions were stopped before T cells appeared (determined by the monitoring of gene marking), and the last dose was at 11 weeks after infusion in one patient (ART009) and at 8 weeks in the other two patients. There was no apparent effect from the rATG on T-cell reconstitution in any of these patients.
Patients ART008 and ART011 each received one transfusion of red cells on day 13 and day 18, respectively, for busulfan-related anemia. None of the patients received platelet transfusions related to busulfan. “Days” indicates days after cell infusion. G indicates that granulocyte colony-stimulating factor was given to stimulate neutrophil recovery.
Follow-up was censored at the second infusion (12 months after the first infusion).
The sex, race, and ethnic group of the patients (with a predominance of Navajo or Apache descent) were representative of persons affected by this very rare autosomal recessive disorder (Table S2). Nine infants received a diagnosis after newborn screening for SCID; 8 were free of infections before gene therapy (Table 1). Illnesses included transplacental maternal T-cell chimerism with grade 2 or 3 GVHD that involved the liver or skin and warranted systemic immunosuppression (Patients ART009, ART012, and ART013) and perinatally acquired cytomegalovirus (CMV) infection (Patient ART010), which progressed to encephalitis and warranted multiple antiviral medications and twice-weekly infusions of third-party CMV-specific cytotoxic T cells (provided by Richard O’Reilly, M.D., Memorial Sloan Kettering Cancer Center). The CMV in Patient ART010 cleared as T-cell immunity reconstituted. However, subsequent waning of T-cell numbers with recrudescence of CMV viremia led to a second treatment with autologous AProArt-transduced CD34+ cells 12 months after the initial treatment (Fig. S1). Data from this patient were censored from further analysis.
STUDY END POINTS
A total of 10 patients met our criteria for feasibility of the procedure at 42 days after infusion. Of the 9 patients with at least 12 months of follow-up, 4 met our criteria for T-cell immune reconstitution at 12 months.
INFUSIONS AND ACUTE TOXIC EFFECTS
The median age at infusion of gene-transduced cells was 2.7 months (range, 2.3 to 13.3) (Table 1). The median busulfan cumulative AUC was 19.0 mg × hour per liter (range, 17.6 to 20.2). The median CD34+ cell dose was 7.7×106 per kilogram (range, 2.2 to 12.1), and the median VCN in the delivered cells was 2.5 copies per cell (range, 1.6 to 3.9). The median transduction efficiency was 78% (range, 62 to 84). Busulfan toxicity manifested as transient blood cytopenias only; nadir neutrophil counts occurred at 13 to 21 days (Table 1 and Fig. S2). Consistent with noninfectious mucosal ulcerations known to occur in ART-SCID,22 four patients (two of Navajo descent) had oral ulcers that appeared before or more than 42 days after busulfan conditioning; the ulcers responded to topical glucocorticoids and resolved after T-cell immunity was restored. Six patients had neutropenia (as seen in other genetic forms of SCID13), which responded to granulocyte colony-stimulating factor and resolved on immune reconstitution.
IMMUNE RECONSTITUTION
Gene-marked (VCN, ≥0.01) CD3+ T cells were detected at a median of 12 weeks (range, 6 to 16) in all 10 patients, and 5 of 6 patients followed for at least 24 months had cellular immune reconstitution at a median of 12 months (range, 6 to 24) (Fig. 1 and Table 2).1,7,13,24 Levels of CD3+, CD4+, and CD8+ T cells, naive CD4+ T cells, and regulatory T cells (Tregs) increased over a period of 9 months after infusion (Fig. 1).7,23 T-cell–receptor excision circles (a product of the recombination of TCR genes, which takes place during normal T-cell differentiation) were detected 3 to 6 months after infusion, and their numbers increased in parallel with naive T cells (Fig. S3). Lymphocyte proliferation in response to phytohemagglutinin normalized by 9 months in all the patients except Patient ART010, who had ongoing CMV infection (Fig. S4). Improvements in TCRβ diversity were evident (Fig. 2 and Fig. S5), with scores on the Shannon index and equitability index and scores for information density stabilizing by 6 to 12 months after infusion at levels similar to those of healthy adults.25
Figure 1. Lymphocytes and Lymphocyte Subsets after Infusion of Autologous Lentiviral-Transduced CD34+ Cells.
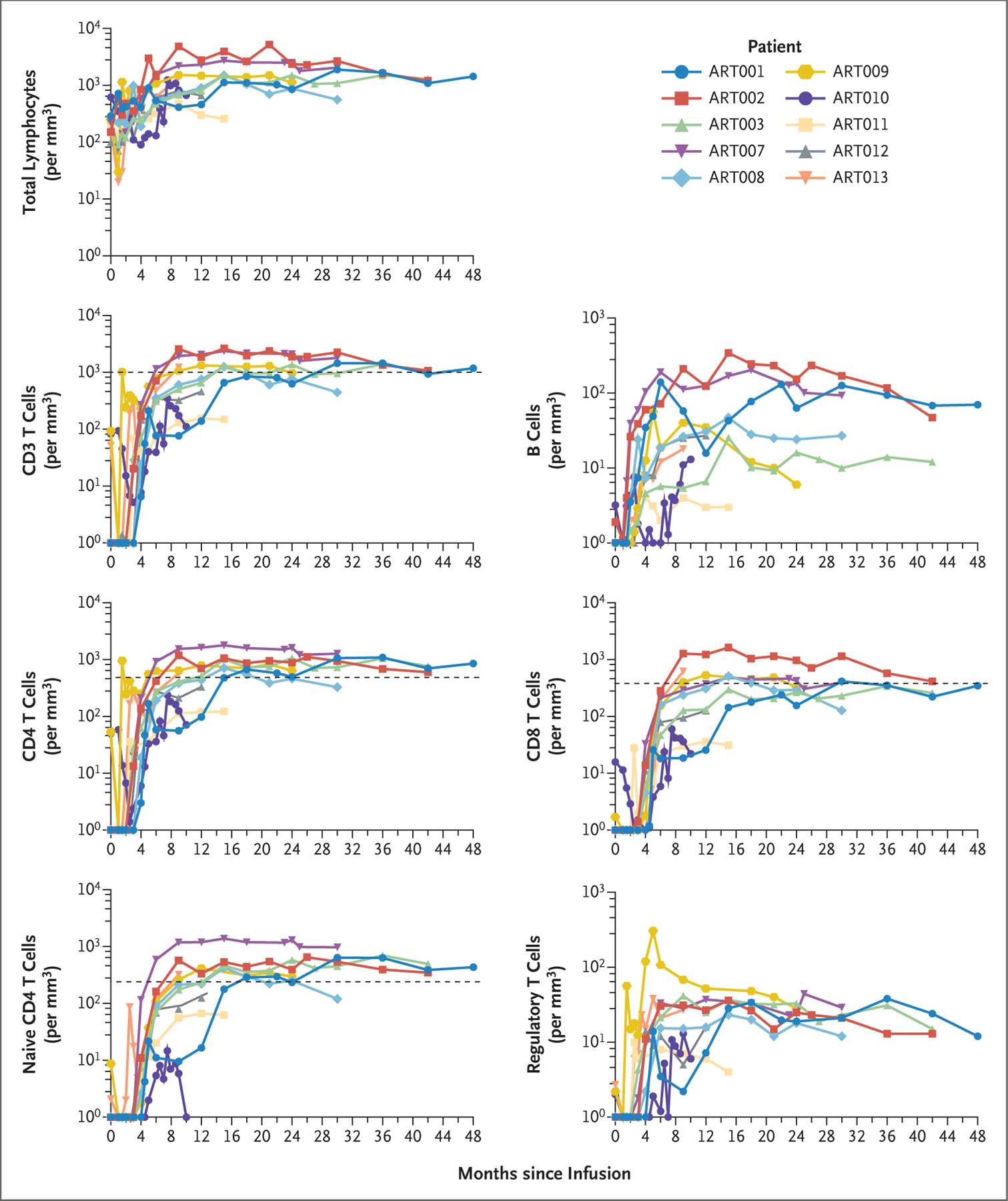
Shown are cells per cubic millimeter of peripheral blood. The values that were used to define T-cell immune reconstitution (dashed lines) are as follows: CD3 T cells, more than 1000 per cubic millimeter; CD4 T cells, more than 500 per cubic millimeter; CD8 T cells, more than 300 per cubic millimeter; and naive CD4 T cells, more than 200 per cubic millimeter.7,13 The mean number of regulatory T cells for children 10 to 48 months of age is 60 per cubic millimeter (range, not available).23
Table 2.
Immune Status at Last Follow-up after Infusion of Lentiviral-Transduced Hematopoietic Stem Cells.*
| Variable | Patient ART001 | Patient ART002 | Patient ART003 | Patient ART007 | Patient ART008 | Patient ART009 | Patient ART010 | Patient ART011 | Patient ART012 | Patient ART013 |
|---|---|---|---|---|---|---|---|---|---|---|
| Most recent laboratory follow-up (mo after cell infusion) | 48 | 43 | 42 | 30 | 31 | 25 | 12† | 18 | 13 | 10 |
| Lymphocyte subsets | ||||||||||
| CD3 >1000 cells/mm3 | Yes | Yes | Yes | Yes | No | Yes | No | No | No | Yes |
| CD4 >500 cells/mm3 | Yes | Yes | Yes | Yes | No | Yes | No | No | No | Yes |
| CD19 cells detectable (cells/mm3) | Yes (128) | Yes (73) | Yes (11) | Yes (144) | Yes (34) | Yes (8) | Yes (20) | Yes (2) | Yes (40) | Yes (28) |
| Naive CD4+ cells >20% of CD4+ cells (%)‡ | Yes (49) | Yes (59) | Yes (68) | Yes (77) | Yes (37) | Yes (45) | No (16) | Yes (51) | Yes (34) | Yes (61) |
| PHA response >100% LLN | ||||||||||
| CD45+ lymphocytes | Yes§ | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes |
| CD3+ T cells | Yes§ | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes |
| IgA (mg/dl) | 27 | 55 | 36 | 44 | <7 | <7 | <7 | <7 | 24 | <7 |
| IgM (mg/dl) | 59 | 29 | 18 | 90 | 152 | 26 | 6 | 15 | 65 | 16 |
| IgM IHG titer¶ | 1:16 | 1:32 | 1:2 | <1:2 | 1:32 | 1:2 | 1:1 | Not tested‖ | <1:2 | 1:2 |
| IgG infusions stopped (mo)** | Yes (32); K | Yes (19); K, L | No | Yes (20); K, L | Yes (30) | No | No | No | No | No |
| Infections, response | CMV after treatment, resolved | Persistent CMV from before treatment | ||||||||
| AIHA (mo that treatment stopped) | Yes (32) | Yes; never treated | No | No | No | Yes (11) | Yes | No | No | No |
AIHA denotes autoimmune hemolytic anemia, IHG isohemagglutinin, LLN lower limit of the normal range, and PHA phytohemagglutinin.
Shown are the results after the initial infusion of lentiviral-transduced cells. Results after the second infusion are shown in Figure S1.
Naive CD4+ refers to CD3+CD4+CD45RA+CCR7+; the range for the percentage of naive CD4+ cells in healthy control participants is 16 to 80%.24
The most recent lymphocyte proliferation was performed at 36 months after infusion.
Shown is the highest IgM titer to blood group antigen A or B.
The blood type was AB.
K indicates protective titers to killed vaccines: tetanus, diphtheria, polio, Streptococcus pneumoniae (23 serotypes), hepatitis A and B, and Haemophilus influenzae type B. L indicates protective titers to live vaccines: varicella and measles, mumps, and rubella.
Figure 2. Diversity of T-Cell Receptor β-Chain Variable Sequences at Baseline and after Infusion of CD34+ Gene-Corrected Cells.
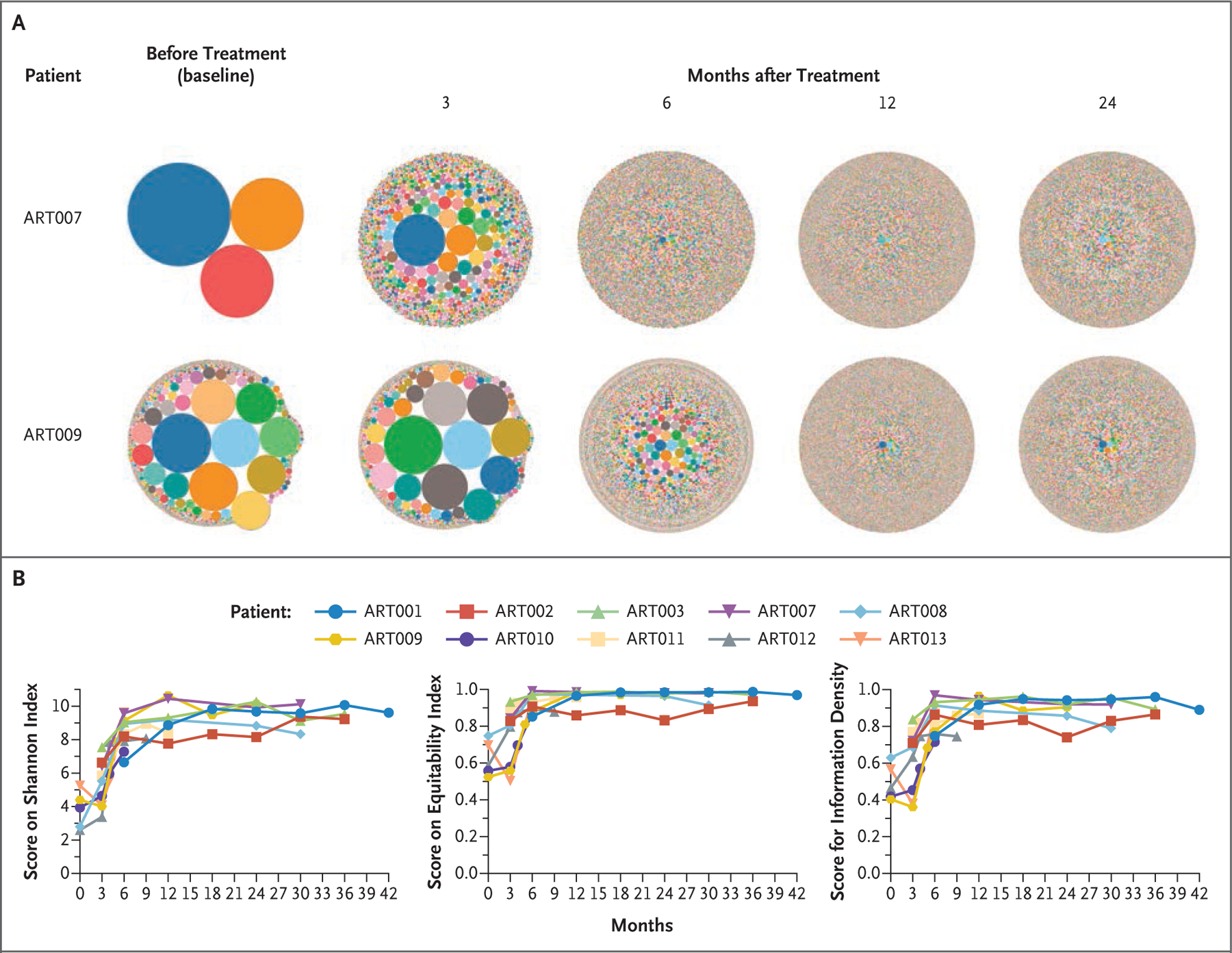
Panel A shows hierarchical tree maps of sequence diversity in Patients ART007 and ART009 from baseline to 36 months after infusion. Panel B shows serial measurements of scores on the Shannon index (the range includes all numbers >0), scores on the equitability index (range, 0 to 1), and scores for information density (range, 0 to 1); for all three scales, higher values indicate greater diversity. Scores on all three scales increased during the first 6 to 12 months after infusion, and these increases were then sustained. Details are provided in Figure S5 in the Supplementary Appendix.
B cells were detected by means of flow cytometry and gene marking (VCN, ≥0.01) in all 10 patients at a median of 6 weeks (range, 4 to 10); in 1 patient (ART009), the levels subsequently declined (Table 2). Three patients (ART001, ART002, and ART007) had normal IgM concentrations and responses to immunizations at 24 months after infusion. At the time of this report, a fourth patient (ART008) had normal IgM and isohemagglutinin titers, no longer received IgG infusions, and had started immunizations (Fig. 1 and Table 2). Three additional patients (ART003, ART009, and ART012) had pre-B cells, immature B cells, and plasma-blasts (Fig. S6).
Table 2 shows the status of the 10 patients at their last evaluation (with data on Patient ART010 censored at 12 months). Nine had 34 to 77% CD3+/CD4+/CD45RA+/CCR7+ naive T cells and normal lymphocyte proliferative responses. All 10 patients had IgM in their blood; 4 had normal levels for their age.
GENE MARKING AND INSERTION-SITE ANALYSIS
Gene-marked peripheral-blood mononuclear cells were identified 1 month after infusion in all 10 patients (data not shown). In 8 patients, the VCN in T and B cells was 3 to 5 copies per cell by 6 months (Fig. 3). The mean VCN per cell in other lineages varied from 0.05 to 0.2 in myeloid (CD14+/15+) cells and 0.05 to 0.3 in NK cells (Fig. 3).
Figure 3. Gene Marking in Peripheral Blood Cells after Infusion of Autologous Lentiviral-Transduced CD34+ Cells.
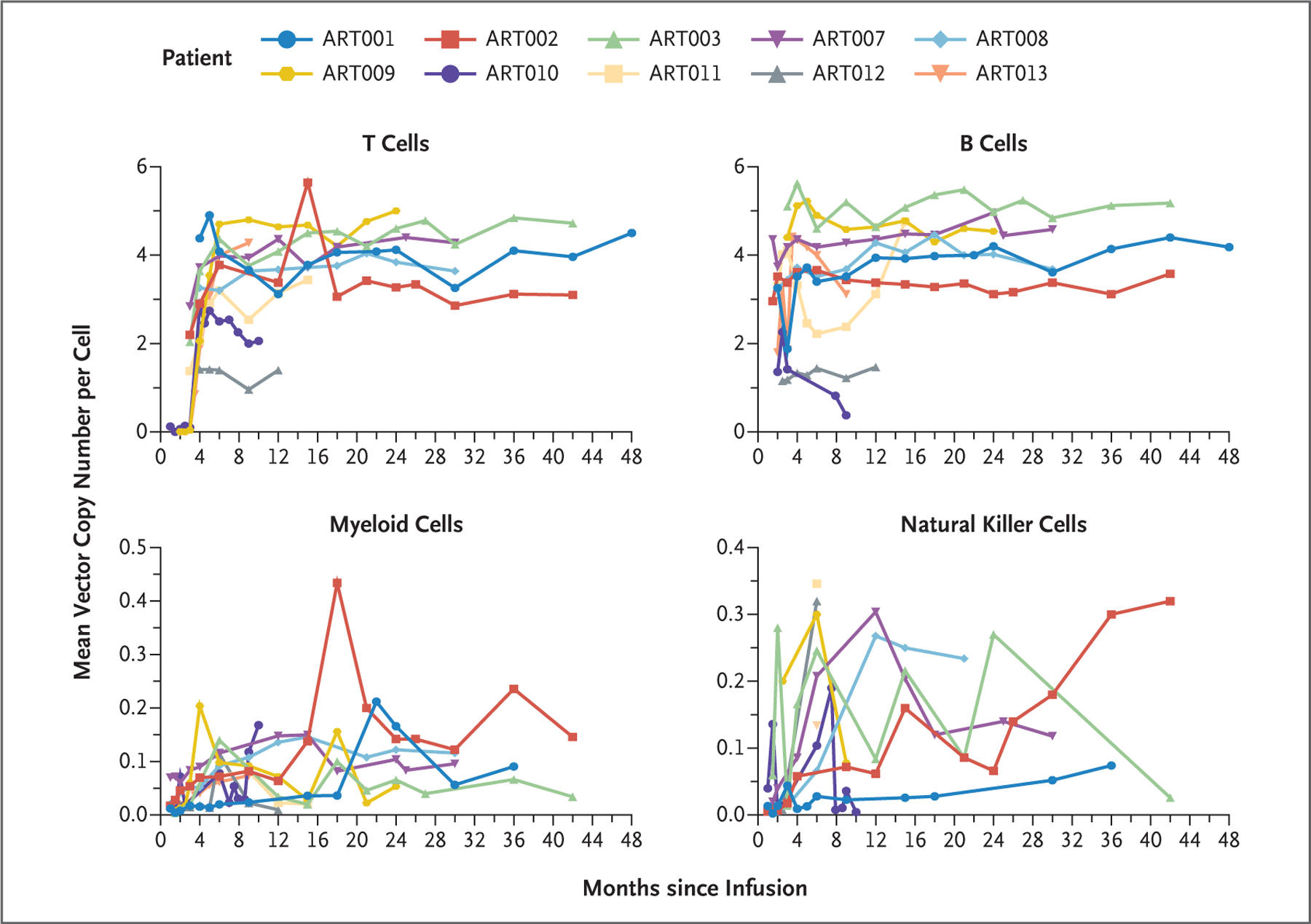
Subpopulations of peripheral-blood mononuclear cells were sorted after staining for the following cell-surface markers: T cells, CD3+; B cells, CD19+; myeloid cells, CD14+/CD15+; and natural killer cells, CD3−/CD19−/CD56+.
The distributions of vector insertion sites in six patients at least 12 months after infusion showed no evidence of clonal expansion in sorted subset populations (Fig. S7). Insertion sites did not cluster near reported oncogenes,26–28 and Venn diagrams revealed insertion sites common to both myeloid and lymphoid lineages, findings consistent with transduction of hematopoietic stem cells.
ADVERSE EVENTS
Serious adverse events are shown in Table S3. A total of 40 nonserious adverse events were considered by the investigators to be possibly, probably, or definitely related to the study treatment (Table S4); 23 of them were of grade 1 or 2. Of the 17 grade 3 or 4 adverse events, 16 were cytopenias, the timing of which supports marrow harvest or busulfan as being causal; 1 was auto-immune hemolytic anemia (AIHA). AIHA developed 4 to 11 months after infusion in four of the nine patients evaluable at least 12 months after infusion. All had IgM warm antibodies and had positive results on Coombs’ antiglobulin testing. Patient ART001 was hospitalized, whereas Patients ART009 and ART010 received immuno-suppression on an outpatient basis, and Patient ART002 recovered without treatment. CMV infection was associated with AIHA in Patients ART002 and ART010.
DISCUSSION
The lentiviral construct that was used in this gene-insertion therapy included the natural human Artemis promoter to avoid toxic overexpression of Artemis17,18 and yet yield sufficient, therapeutic levels of Artemis protein.29 Results show strong T-cell numbers and function and TCRβ diversity in 5 of 6 patients at least 24 months after infusion. Moreover, B cells developed in all 10 patients, permitting the discontinuation of immune globulin infusions in 4 of the 6 patients followed for at least 24 months. Three patients who were followed for fewer than 24 months had B cells at all maturational stages. In a previous study, only 3 of 16 patients with ART-SCID who received allogeneic HCT had re-constitution of B-cell immunity.9 B-cell counts in all 16 patients at last follow-up were less than 1 per cubic millimeter to 15 per cubic millimeter.
Infused CD34+ cells had mean VCNs of 1.6 to 3.9 copies per cell and generally higher VCNs in differentiated T lymphocytes (1.5 to 5.0 copies per cell) and B lymphocytes (1.5 to 5.2 copies per cell) (Table 1 and Fig. 3), findings consistent with a requirement for Artemis expression for antigen receptor recombination and T- and B-cell maturation. However, it may be that not every inserted vector copy is active, and the study was too small and follow-up time too short to permit meaningful tests of correlation between infused CD34+ cell numbers or transduction efficiency–specific and lineage-specific VCN and immune recovery. The vector insertion sites were diverse, across cells and patients, without evidence of clonal expansion or cancer.
The low mean VCN in myeloid and NK cells as compared with that in T and B cells probably reflects the fact that low-exposure busulfan was not myeloablative and that a large component of pretreatment, nontransduced cells remained, which diluted the VCN in myeloid and NK cells. Another factor is that in contrast to T- and B-cell lineages, which require Artemis protein for re-arrangement of T- and B-cell receptors, gene-corrected myeloid and NK lineages have little or no selective advantage.
Of the six patients whom we followed for at least 24 months after infusion, four met criteria for stopping immune globulin infusions. Three have been fully immunized and produced protective antibodies, and the fourth has started immunizations. All four had higher B-cell numbers than the two patients still receiving immune globulin infusions. Moreover, each of the four had B cells in various stages of maturation, whereas those receiving immune globulin infusions did not. However, the patients were too few and the follow-up too short to reveal possible relationships between gene marking in non-lymphoid lineages and B-cell reconstitution. There was no correlation between B-cell reconstitution and treatment-related variables — for example, CD34+ cell dose, busulfan exposure, graft VCN, or the number of transduced cells.
Pharmacologically targeted busulfan at approximately 25% of the standard exposure appeared to be safe, causing only expected transient blood cytopenias in this radiation-sensitive SCID, which is fortunate, because opening marrow niches is essential to achieve multilineage engraftment and full immune reconstitution.11,13,14 Longer follow-up to assess dental development, growth, and endocrinopathies is planned. In addition, on the basis of the four patients with B-cell reconstitution, it appears that engraftment of fewer than 15% gene-corrected hematopoietic stem cells (as estimated by mean VCN in myeloid cells) can produce T- and B-cell reconstitution (Fig. 3). T and B cells developed in all the patients in this study, and all the patients survived, whereas not all patients with ART-SCID who receive alternative-donor or matched-sibling allogeneic HCT survive.7,9
Patient ART010, who did not have reconstitution of adaptive immunity despite receiving 10.4×106 CD34+ cells per kilogram (VCN, 2.9 copies per cell), had CMV infection before infusion that progressed to CMV encephalitis while receiving anti-CMV therapy. High-dose ganciclovir30 and CMV infection31 may have hindered immune reconstitution, warranting a second infusion of gene-transduced CD34+ cells. At the last follow-up, the patient was not receiving anti-CMV therapy and had unquantifiable CMV on PCR assay, rising T- and B-cell numbers, normal T-cell function, and a stable mean VCN in T and B cells. In one trial of lentiviral gene therapy for newly diagnosed X-linked SCID, the only patient who received a second treatment also presented with CMV.13
AIHA developed in 4 of 9 participants at least 1 year after infusion; the early appearance of B cells before T-cell reconstitution including Tregs may be responsible, although the study was too small for a definitive assessment. AIHA is a known complication of allogeneic HCT.32 In addition, AIHA was reported in 3 of 19 previously treated patients and in 2 of 24 previously untreated patients with X-linked SCID undergoing autologous gene therapy (Malech H, National Institutes of Health, and Mamcarz E, St. Jude Children’s Hospital: personal communications). Although the incidence appears high in our study, 2 of the 4 patients had CMV infection when AIHA developed,33 1 of whom warranted no immunosuppression and 1 of whom was treated with sirolimus. Further follow-up with a larger sample size will be necessary to characterize AIHA in this patient population.
In this study, we treated 10 patients with newly diagnosed ART-SCID by infusing autologous lentiviral-transduced CD34+ cells after conditioning with pharmacologically targeted low-exposure busulfan. We found that this approach restored immunity and was safe (within the context of the disease and alternative approaches) and conclude that further studies are warranted.
Supplementary Material
Acknowledgments
Supported by grants from the California Institute for Re-generative Medicine (CLIN2–10830, to Drs. Cowan and Puck) and the National Institute of Allergy and Infectious Diseases (NIAID) (U54-AI082973, to Drs. Cowan, Dvorak, and Puck) and intramural NIAID (to Dr. Malech). Drs. Cowan and Puck receive support from the UCSF Smith Cardiovascular Research Institute, and Dr. Puck receives support from the NIAID (P01-AI138962) and the Lisa and Douglas Goldman Fund.
We thank Matthew Kan, M.D., Ph.D., for his advice regarding T-cell receptor β-chain analysis; Farhan Murshed, M.D., for clinical management of patient care after treatment; Xin-Hua Chen, L.C.S.W., for logistic and emotional support of patients and their families; Aidan Rossiter, B.S., for technical assistance; Sherman Bakabak, C.L.S., Erskine Apao, C.L.S., Joseph Ong, C.L.S., and Suan Teoh, C.L.S., for technical assistance with the transduction protocol; the attending physicians and nurses for the care that they provided to the patients; and the parents and caregivers of the patients. We recognize the contributions of our deceased colleague, Brian P. Sorrentino, M.D., who provided valuable guidance during the preclinical phase of this work.
APPENDIX
The authors’ affiliations are as follows: the Departments of Pediatrics (M.J.C., J.Y., J.F., C.F.-B., U.S., M.K., J.D., J.L.-B., W.C., S.C., R.C., C.C.D., J.M.P.) and Epidemiology and Biostatistics (J.F.H.), the Smith Cardiovascular Research Institute (M.J.C., J.M.P.), and the School of Pharmacy (J.L.-B.), University of California, San Francisco (UCSF), and UCSF Benioff Children’s Hospital (M.J.C., J.F., J.D., J.L.-B., J.O., C.C.D., J.M.P.), San Francisco, the Department of Pediatrics, University of California, San Diego, and Rady Children’s Hospital, San Diego (L.B.), and the Department of Pediatrics, UCLA Mattel Children’s Hospital, Los Angeles (C.Y.K.) — all in California; the Department of Pediatrics, Johns Hopkins All Children’s Hospital, St. Petersburg, FL (D.C.); the Department of Pediatrics, Sainte-Justine University Hospital Center, University of Montreal, Montreal (H.D.); Tuba City Regional Health Care, Tuba City (C.G., D.H.), and Phoenix Children’s Hospital, Phoenix (H.K.M.) — both in Arizona; the Department of Pediatrics, University of Washington Seattle Children’s Hospital, Seattle (A.P.); Clinical Development, Roche Diagnostics Solutions, Singapore (D.P.); the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD (H.L.M.); and the Department of Genetics, Cell Biology, and Development, University of Minnesota, Minneapolis (R.S.M.).
Footnotes
References
- 1.Shearer WT, Dunn E, Notarangelo LD, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol 2014;133:1092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dvorak CC, Haddad E, Buckley RH, et al. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010–2018). J Allergy Clin Immunol 2019; 143:405–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwan A, Abraham RS, Currier R, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014; 312:729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amatuni GS, Currier RJ, Church JA, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California, 2010–2017. Pediatrics 2019;143(2):e20182300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L, Moshous D, Zhou Y, et al. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol 2002;168:6323–9. [DOI] [PubMed] [Google Scholar]
- 6.Dvorak CC, Cowan MJ. Radiosensitive severe combined immunodeficiency disease. Immunol Allergy Clin North Am 2010;30:125–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haddad E, Logan BR, Griffith LM, et al. SCID genotype and 6-month post-transplant CD4 count predict survival and immune recovery. Blood 2018;132:1737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Pluth JM, Cooper PK, Cowan MJ, Chen DJ, Yannone SM. Artemis deficiency confers a DNA double-strand break repair defect and Artemis phosphorylation status is altered by DNA damage and cell cycle progression. DNA Repair (Amst) 2005;4:556–70. [DOI] [PubMed] [Google Scholar]
- 9.O’Marcaigh AS, DeSantes K, Hu D, et al. Bone marrow transplantation for T-Bsevere combined immunodeficiency disease in Athabascan-speaking Native Americans. Bone Marrow Transplant 2001;27:703–9. [DOI] [PubMed] [Google Scholar]
- 10.Schuetz C, Neven B, Dvorak CC, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood 2014;123:281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohn DB, Booth C, Shaw KL, et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N Engl J Med 2021;384:2002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanco E, Izotova N, Booth C, Thrasher AJ. Immune reconstitution after gene therapy approaches in patients with X-linked severe combined immunodeficiency disease. Front Immunol 2020;11:608653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mamcarz E, Zhou S, Lockey T, et al. Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N Engl J Med 2019;380:1525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Ravin SS, Wu X, Moir S, et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immuno-deficiency. Sci Transl Med 2016;8:335ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Punwani D, Kawahara M, Yu J, et al. Lentivirus mediated correction of Artemis-deficient severe combined immuno-deficiency. Hum Gene Ther 2017;28:112–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc Natl Acad Sci U S A 2006; 103:16406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Multhaup M, Karlen AD, Swanson DL, et al. Cytotoxicity associated with Artemis overexpression after lentiviral vector-mediated gene transfer. Hum Gene Ther 2010;21:865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Multhaup MM, Gurram S, Podetz-Pedersen KM, et al. Characterization of the human Artemis promoter by heterologous gene expression in vitro and in vivo. DNA Cell Biol 2011;30:751–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Savic RM, Cowan MJ, Dvorak CC, et al. Effect of weight and maturation on busulfan clearance in infants and small children undergoing hematopoietic cell transplantation. Biol Blood Marrow Transplant 2013;19:1608–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pannicke U, Ma Y, Hopfner K-P, Niewolik D, Lieber MR, Schwarz K. Functional and biochemical dissection of the structure-specific nuclease ARTEMIS. EMBO J 2004;23:1987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pannicke U, Hönig M, Schulze I, et al. The most frequent DCLRE1C (ARTEMIS) mutations are based on homologous recombination events. Hum Mutat 2010;31: 197–207. [DOI] [PubMed] [Google Scholar]
- 22.Kwong PC, O’Marcaigh AS, Howard R, Cowan MJ, Frieden IJ. Oral and genital ulceration: a unique presentation of immunodeficiency in Athabascan-speaking American Indian children with severe combined immunodeficiency. Arch Dermatol 1999;135:927–31. [DOI] [PubMed] [Google Scholar]
- 23.Holcar M, Goropevšek A, Ihan A, Avčin T. Age-related differences in percentages of regulatory and effector T lymphocytes and their subsets in healthy individuals and characteristic STAT1/STAT5 signalling response in helper T lymphocytes. J Immunol Res 2015;2015:352934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoshino A, Boutboul D, Zhang Y, et al. Gain-of-function IKZF1 variants in humans cause immune dysregulation associated with abnormal T/B cell late differentiation. Sci Immunol 2022;7(69):eabi7160. [DOI] [PubMed] [Google Scholar]
- 25.Basharin GP. On a statistical estimate for the entropy of a sequence of independent random variables. Theory Probab Appl 1959;4:333–6. [Google Scholar]
- 26.Ronen K, Negre O, Roth S, et al. Distribution of lentiviral vector integration sites in mice following therapeutic gene transfer to treat β-thalassemia. Mol Ther 2011;19:1273–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang GP, Levine BL, Binder GK, et al. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol Ther 2009;17:844–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biffi A, Bartolomae CC, Cesana D, et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood 2011;117:5332–9. [DOI] [PubMed] [Google Scholar]
- 29.Multhaup MM, Podetz-Pedersen KM, Karlen AD, et al. Role of transgene regulation in ex vivo lentiviral correction of Artemis deficiency. Hum Gene Ther 2015; 26:232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Battiwalla M, Wu Y, Bajwa RPS, et al. Ganciclovir inhibits lymphocyte proliferation by impairing DNA synthesis. Biol Blood Marrow Transplant 2007;13:765–70. [DOI] [PubMed] [Google Scholar]
- 31.Rashidi A, Luo X, Cooley S, et al. The association of CMV with NK-cell reconstitution depends on graft source: results from BMT CTN-0201 samples. Blood Adv 2019;3:2465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horn B, Viele M, Mentzer W, Mogck N, DeSantes K, Cowan M. Autoimmune hemolytic anemia in patients with SCID after T cell-depleted BM and PBSC transplantation. Bone Marrow Transplant 1999; 24:1009–13. [DOI] [PubMed] [Google Scholar]
- 33.Kruizinga MD, van Tol MJD, Bekker V, et al. Risk factors, treatment, and immune dysregulation in autoimmune cytopenia after allogeneic hematopoietic stem cell transplantation in pediatric patients. Biol Blood Marrow Transplant 2018;24:772–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.