

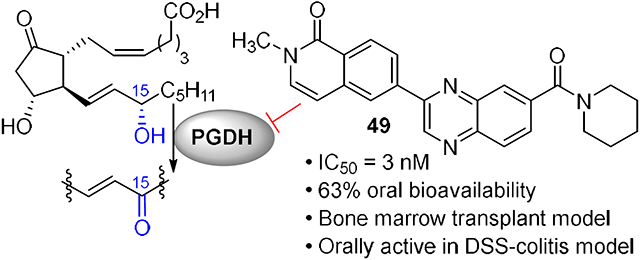
Abstract
15-Prostaglandin dehydrogenase regulates the concentration of prostaglandin E2 in vivo. Inhibitors of 15-PGDH elevate PGE2 levels and promote tissue repair and regeneration. Here we describe a novel class of quinoxaline amides that show potent inhibition of 15-PGDH, good oral bioavailability, and protective activity in mouse models of ulcerative colitis and recovery from bone marrow transplantation.
Graphical Abstract
INTRODUCTION
The enzyme 15-prostaglandin dehydrogenase (15-PGDH) has emerged as an attractive drug target to promote tissue repair and regeneration. It degrades the prostaglandin PGE2 by oxidation of the 15-hydroxyl group to a ketone in an NAD+-dependent manner (Figure 1).1,2,3 PGE2 promotes tissue growth and cell differentiation by activating one of four prostaglandin receptors EP1-4.4 Oxidation of PGE2 to 15-keto-PGE2 by 15-PGDH abrogates binding to EP1-4. Therefore 15-PGDH counteracts the activity of the cyclooxygenase enzymes COX1 and COX2, which, along with microsomal prostaglandin E2 synthase, are involved in the synthesis of PGE2 from arachidonic acid. 5,6 Inhibitors of 15-PGDH would be expected to block the degradation of PGE2, resulting in higher tissue levels of PGE2 and concordant activation of the EP1-4 receptors.
Figure 1.

A. Abbreviated PGE2 pathway. B. Inhibitors of 15-prostaglandin dehydrogenase.
Activation of the PGE2 signaling pathway has proven beneficial in preclinical and clinical settings. For example, 16,16-dimethyl PGE2 (dmPGE2) is more metabolically stable than PGE2 itself, and it has been shown to augment hematopoiesis in zebrafish.7 Ex vivo treatment of bone marrow or cord blood with dmPGE2 was found to enhance their effectiveness in bone marrow transplantation in mice and non-human primates, respectively.8,9,10,11 More recently, dmPGE2 was evaluated in a Phase 1 clinical trial in which human umbilical cord blood was treated ex vivo prior to transplantation. The treatment was safe and showed a modest acceleration of neutrophil recovery.12 In separate studies, PGE2 was found to promote expansion of colonic stem cells in culture13 while dmPGE2 was found to reduce disease severity in the DSS-colitis model in mice.14
Taken together, the results highlighted above illustrate the potential benefit of activating the PGE2 signaling pathways. However, PGE2 itself is a poor drug. Its in vivo half-life is <15 min,15 and it is acutely toxic at high doses.16 While effective in animal models, dmPGE2 has not yet shown clear efficacy in human trials. We and others have therefore considered an alternative strategy – inhibition of 15-PGDH.17,18,19,20 Small molecule inhibitors of 15-PGDH are expected to elevate levels of PGE2 within all tissues within a relevant physiological range. Inhibitors with acceptable half-lives could overcome the metabolic liabilities associated with PGE2 and avoid the Cmax-driven toxicities associated with PGE2 and EP receptor agonists.21,22,23
Several classes of potent small molecule 15-PGDH inhibitors have been reported (Figure 1).24,25,26 Rhodanine alkylidenes such as compound 1 display IC50 values <50 nM and elevate PGE2 levels in the cell culture media of A549 lung cancer cells.27,28 While advancement of this scaffold may be challenging due to the potential PAINS substructure,29 1 did show activity in a cell-based model of wound healing. A group from the National Institutes of Health reported a series of triazoles (e.g., 2) and benzamidazoles (e.g., 3) with IC50’s <25 nM in vitro and activity in cell culture in the sub-μM range.30,31 Poor metabolic stability prevented advancement of these scaffolds.
We recently reported the first inhibitors of 15-PGDH showing in vivo activity.17 SW033291 (4) is a tight-binding inhibitor of 15-PGDH with an apparent Ki of 0.1 nM. In mice sulfoxide 4 doubled the levels of PGE2 in lung, liver, colon and bone marrow 3h after an intraperitoneal (IP) dose of 10 mg/kg. Among other 15-PGDH substrates, we observed smaller increases in PGD2 levels and no impact on PGF2α levels. Additionally, 4 showed beneficial effects in multiple mouse models of human disease when administered IP. In particular, it accelerated recovery following bone marrow transplantation, minimized the severity of colitis in the DSS-colitis model,17 protected from immune-mediated bone marrow failure,32 and minimized the severity of lung fibrosis in the bleomycin-treated mice.33 A recent study from the Blau group demonstrated that 4 could rejuvenate aged muscle mass and strength.34 Chemical optimization of 4 led to imidazole 5, which identified the active enantiomer, replaced the thiophene with a more stable heterocycle, and improved solubility by replacing the phenyl ring with a basic imidazole.35 This analog showed maximal PGE2 induction at a 2.5 mg/kg IP dose and demonstrated additive activity with the current standard of care for bone marrow transplantation, gCSF.36
While our reported sulfoxide inhibitors showed promising activity in multiple murine disease models, IP administration was required owing to modest oral bioavailability. This characteristic was not concerning in the context of treating bone marrow recipients because IV delivery would likely be preferred. However, as 15-PGDH inhibitors show promise in an increasing number of indications, orally available inhibitors would be desirable. Additionally, the stereogenic sulfoxide of 4 and 5 present a synthetic challenge, which required late-stage chromatographic resolution. Herein, we describe the discovery and optimization of an achiral, potent series of 15-PGDH inhibitors showing robust activity in mice when delivered orally.
RESULTS AND DISCUSSION
The same HTS experiment that identified sulfoxide 4 also revealed quinoxaline SW145753 (6) as an inhibitor of 15-PGDH with IC50 = 17 nM.37 Compound 6 was attractive because it was synthetically accessible, achiral, and lacked concerning structural alerts. The absence of the sulfoxide present in 4 and 5 also removed a potential metabolic liability and offered the promise of improved oral bioavailability. We also noticed the presence of the piperidinyl amide in 6, which was shared by previously reported inhibitor 3. Our initial objectives were to improve potency and reduce lipophilicity (CLogP = 5.5) by modifying the amide and pendent aryl rings. Unfortunately, nearly every change to the amide region of the inhibitor resulted in substantial, if not complete loss in potency when tested against recombinant human 15-PGDH (Table 1). For example, a pyrrolidine amide was nearly inactive (7), while the larger azepane amide 8 maintained similar activity as 6. The primary amide 9 and a series of secondary amides 10–16 were all inactive. Returning to the cyclic amides, introduction of a morpholine, piperdine or piperidinone ring abrogated activity (17–19). Fluoro (21) and methyl (23) substituted piperidines were tolerated, but more polar substitution was not. Accordingly, remaining analogs maintained the unsubstituted piperidine ring.
Table 1.
Amide modifications
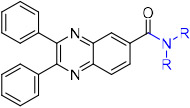
| ||
|---|---|---|
| compound | amide | IC50 (nM)a |
|
||
| 6 | n = 1 | 17 |
| 7 | n = 0 | >2500 |
| 8 | n = 2 | ~20 |
| 9 | −NH2 | >2500 |
| 10 | −NHMe | >2500 |
| 11 | −NHEt | >2500 |
| 12 | −NHnPr | >2500 |
| 13 | −NHnBu | >2500 |
| 14 | −NHnPent | >2500 |
| 15 | −NHcPent | >2500 |
| 16 | −NHPh | >2500 |

|
||
| 17 | X = O | >2500 |
| 18 | X = NBoc | >2500 |
| 19 | X = CO | ~2500 |

|
||
| 20 | X = NHBoc | >2500 |
| 21 | X = F | 34 |
| 22 | X = CH2OH | >2500 |
| 23 | X = CH3 | 37 |
| 24 | X = OMe | ~100 |
IC50 against 5 nM recombinant human 15-PGDH.
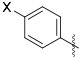
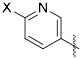
We next modified the quinoxaline substituents in an effort to modulate potency and physicochemical properties (Table 2). Removing one of the phenyl rings (25) improved activity against the enzyme and decreased CLogP to 3.5 from 5.5. Several substituted phenyl rings were explored in a screening assay (26–29) but nothing was found that improved activity substantially. An analog featuring a 3-pyridyl group (31) further doubled the potency such that IC50 = 6 nM while dropping CLogP to 2.3. Building from this discovery, substitution on the pyridine was explored, which showed a wide tolerance for functionality but generally favored a hydrogen bond donor. For example, −NH2 (34), −NHMe (35) and −CO2H (38) groups all improved activity relative to the unsubstituted pyridine. By contrast, the corresponding tertiary amine (36) and methyl ester (37) were similar to the unsubstituted analog 31, while the morpholine 39 was notably less active. Introducing a chloride to the aminopyridine ring (40) further improved this subseries. This analog showed potent enzyme activity, although only modest aqueous solubility. However, the HCl salt was sufficiently soluble to be evaluated in vivo (see below), and mouse liver S9 fraction stability was excellent (t1/2 = 198 min). The pyrimidine 41 lost activity, which was regained with the aminopyrimidine 42, although the latter compound was difficult to work with due to poor solubility.
Table 2.
Variation of the Northwest (Hetero)Aryl Ring
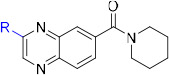
| ||||
|---|---|---|---|---|
| cmpd | R | IC50 (nM)a |
aq. sol. (ug/mL) |
S9c (t1/2, min) |
|
||||
| 25 | X = H | 11 | ||
| 26 | X = C1 | 20-100 | ||
| 27 | X = CO2Me | 4-20 | ||
| 28 | X= CO(morpholine) | 4-20 | ||
| 29 | X = CONH2 | 4-20 | ||
| 30 | 2-Py | 20-100 | ||
| 31 | 3-Py | 5.8 | ||
| 32 | 4-Py | 4-20 | ||
|
||||
| 33 | X = OMe | 3.5 | ||
| 34 | X = NH2 | 3.0 | ||
| 35 | X = NHMe | 2.8 | ||
| 36 | X = NMe2 | 5.6 | ||
| 37 | X = CO2Me | 4-20 | ||
| 38 | X = CO2H | 1.7 | ||
| 39 | X = Morpholine | 20-100 | ||
| 40 |
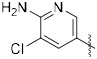
|
2.1 | 2.1, 26b | 198 |
| 41 |
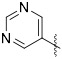
|
4-20 | ||
| 42 |

|
1.6 | ||
| 43 |

|
2.3 | 55 | 124 |
| 44 |
|
1.7 | 14 | 11 |
| 45 |

|
4-20 | ||
| 46 |
|
4-20 | ||
| 47 |

|
2.3 | ||

|
||||
| 48 | X = H | 2.0 | 4 | >240 |
| 49 | X = CH3 | 3.0 | 56 | >240 |
| 50 | X = CH2CO2Et | 4.8 | ||
| 51 |
|
4.4 | ||
| 52 |
|
4.1 | ||
| 53 |
|
20-100 | ||
| 54 |
|
100-500 | ||
Values provided as ranges are derived from a screening assay involving a 5-fold dilution series and single replicates with 5 nM human 15-PGDH. Specific values are derived from a 3-fold dilution series in triplicate with 5 nM human 15-PGDH.
solubility of HCl salt.
Half-life in the presence of mouse liver S9 fractions.
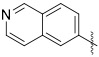
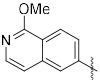
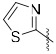
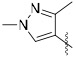
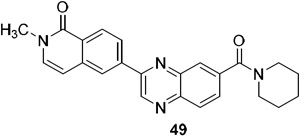
A series of 2-ring heterocycles were explored at the northwest corner of the quinoxaline inhibitors. Quinoline 43 and isoquinoline 44 were both potent inhibitors of 15-PGDH, with IC50 values near half the enzyme concentration, indicating tight binding. Between these two, the isoquinoline was slightly more potent, but less soluble and less stable towards mouse liver S9 fractions. Methoxy groups introduced adjacent to the ring nitrogen compromised activity (45, 46), but quinolone 47 and isoquinolone 48 were both highly active in vitro. In particular, isoquinolone 48 maintained high inhibitory activity and excellent metabolic stability. Solubility of this compound was modest, however, preventing in vivo evaluation. We reasoned that the cyclic secondary amide might form reciprocal hydrogen bonds in the solid state, thereby compromising solubility. Accordingly, N-methyl isoquinolone 49 was synthesized and evaluated. Aqueous solubility was improved relative to 48, and metabolic stability was maintained. In vivo profiling of 49 is described below. The isoquinolone group was found to be superior to the indazole (52), although this ring still maintained potent inhibition. The corresponding thiazole (53) and pyrazole (54) were substantially less active.
A few alternative scaffolds were compared to the disubstituted quinoxalines (Figure 2). The simple analog 55 lost substantial activity. The sulfoxide 56 was envisioned as a hybrid with compound 1, but it proved inactive. Similarly, the quinazolinone 57 was only modestly active. The isomeric quinazoline ring system, as exemplified by 58 – 60 maintained activity, but did not appear to offer an advantage over the quinoxaline scaffold.
Figure 2.

Alternative scaffolds were tested in a screening assay involving a 5-fold dilution experiment with n = 1 at each concentration.
Several of the most promising inhibitors were evaluated in a cell-based assay for their ability to increase PGE2 levels in the cell culture media (Figure 3). In this assay, A549 lung cancer cells were stimulated with IL-1β and treated with compound or DMSO control. PGE2 levels were monitored with a commercially available ELISA assay after 24 h. Optimized sulfoxide inhibitor 5 was included as a positive control. The initial hit in the quinoxaline series, diphenyl quinoxaline 6, only weakly stimulated PGE2 levels at the highest concentrations. Encouragingly, indazole 52 and N-methyl isoquinolone 49 induced PGE2 at 20 nM, while the N-H isoquinolone 48 was active at 4 nM. The aminopyridine 40 appeared to be the most active compound tested, saturating the assay at the lowest concentration tested (4 nM)., The cell based assay allowed us to distinguish among compounds with similar IC50 values in the in vitro enzyme inhibition assay. That assay could not register IC50 values lower than half the enzyme concentration ([15-PGDH] = 5 nM), but differences in potency become evident in the cellular context.
Figure 3.

PGE2 induction in A549 cells. Cells were stimulated with IL-1β and treated with drug for 24 h. PGE2 levels were measured by ELISA and are reported relative to DMSO controls. n = 8 for each data point.
Selected analogs were evaluated in mice for their pharmacokinetic properties (Table 3). 40, 48 and 49 had similar Cmax values, while the AUC values were more variable. Tertiary amide 49 showed a 6-fold lower Cmax value when dosed orally vs. IP, but the AUC was only reduced by half. Oral bioavailability was an encouraging 63%, consistent with its good permeability (Papp,A→B = 45 x 10−6 cm/s; efflux ratio = 0.8). The low solubility of 48 made it difficult to formulate consistently, so in vivo pharmacodynamic experiments were carried out on 40 and 49.
Table 3.
Pharmacokinetic Parameters of Select Analogs in Female CD1 Mice.
| Compound | Dose/route | Cmax (μg/mL) |
AUC (μg*min/mL) |
|---|---|---|---|
| 40·HCl | 10 mg/kg IP | 3.0 | 172 |
| 48 | 10 mg/kg IP | 7.4 | 270 |
| 49 | 10 mg/kg IP | 6.4 | 517 |
| 10 mg/kg PO | 0.95 | 256 (63% Fa) | |
| 5 mg/kg IV | 3.0 | 205b |
Oral bioavailability.
Cl = 30 mL/min/kg
Aminopyridine 40 and isoquinolone 49 and were administered to C57Bl/6 mice using IP administration for convenience during initial in vivo studies. Colon and lung tissue was harvested at various timepoints afterwards, and PGE2 levels were measured by ELISA. After 3h, PGE2 levels had approximately doubled in both tissues in response to 5 mg/kg of both drugs (Figure 4). The 2-fold elevation of PGE2 appears to be maximal as it is also what was observed in the 15-PGDH knockout mouse,17 in mice treated with sulfoxide 6,35 and in mice treated with the higher dose of 10 mg/kg of 40 or 49 (Figure S1A). A trend towards higher activity was observed with 49, particularly at a lower dose of 2.5 mg/kg (Figure S1B). Thus, the higher in vitro activity of 40 appears to be compensated for by the improved pharmacokinetic properties of 49.
Figure 4.

Elevation of PGE2 levels in colon and lung in female C57B1/6 mice following a 5 mg/kg IP dose of the indicated compound.
The pharmacodynamic activity of 49 was further profiled after oral dosing (Figure 5). Mice were administered either a 20 or 40 mg/kg oral suspension. Colons were harvested after 30 or 90 min, and 15-PGDH enzymatic activity was assessed using a tritium-release assay described previously.17 Enzymatic activity was inhibited by >90% in the colon after 30 min at both doses and >80% at 90 min. Similar inhibition was observed in the lung, with the higher 40 mg/kg dose achieving 80% and 70% inhibition of enzymatic activity at 30 and 90 min, respectively (Figure S2).
Figure 5.

Inhibition of 15-PGDH enzymatic activity in the colon following a 20 or 40 mg/kg oral dose of 49.
Based on the ability of 49 to inhibit 15-PGDH in the colon and elevate PGE2 levels, it was evaluated in the DSS-model of ulcerative colitis. Previous work from our laboratories has shown that the 15-PGDH knockout mouse is markedly protected from ulceration and weight loss in this model. Moreover, IP administration of first-generation 15-PGDH inhibitor 4 minimized weight loss, disease severity and colon ulceration.17 Mice were exposed to 2.75% DSS in the drinking water during days 1-7. Isoquinolone 49 was administered by IP (10 mg/kg BID) or PO (40 mg/kg BID) from study day 1 through study day 12 (Figure 6). In the 40 mg/kg PO arm, the body weight of drug-treated mice was significantly higher at every measurement, reaching a maximum difference at day 12. On the last day of the experiment, drug-treated mice had maintained their initial body weight, while the vehicle control mice had lost 20% of their starting body weight. Body weight was also protected in the IP arm, showing statistically significant protection from day 7 through the end of the study. Both IP and PO administration of 49 additionally protected colon lengths. At study day 8, 49-treated mice had 0.9 cm improvement relative to vehicle control, which amounted to a 64% protection. The protection was maintained through day 12, at which point colons from drug-treated mice were still 0.9 cm longer than vehicle controls in the 40 mg/kg PO arm. Protection of colon lengths in the 10 mg/kg IP were less pronounced, although still significant, reaching 0.7 cm at study days 8 and 12.
Figure 6.

15-PGDH inhibitor 49 shows protection in the mouse DSS model of ulcerative colitis. a. Weight change over 12 days. Numbers of mice indicated in each arm. b. Colon length at day 8 and day 12. Naïve, n = 9; day 8, n = 12; day 12, n = 15. c. Colon ulceration at day 8. n = 9 - 12 for each arm. d. Mean total cryptitis score at day 8. n = 9 – 12 for each arm. *p < 0.05.
The protection in colon length afforded by 49 was associated with decreased ulceration in drug-treated animals (Fig. 6c). The 10 mg/kg IP arm showed approximately 50% reduction in ulceration (2.6 vs. 5.2%) while the 40 mg/kg PO arm provided around 80% reduction in ulceration (1.1 vs. 5.5%). Likewise, the cryptitis score, which incorporates measure of colon inflammation, ulceration and mucosal architecture, was improved in both the 10 mg/kg IP and 40 mg/kg PO arms (Fig. 6d).38
We have previously shown that, in addition to protecting from mucosal injury in the colon, 15-PGDH inhibition using (±)-4 and (+)-6 also accelerates hematologic recovery following bone marrow (BM) transplantation.17,36 To evaluate the impact of 49 on early BM transplant recovery, recipient mice were conditioned with total body irradiation followed by the first administration of (+)-4, 49 or vehicle control (Figure 7, S3). Sixteen hours later, mice received 106 BM cells, followed by a second administration of 15-PGDH inhibitor or vehicle control. Recipient animals continued to receive drug or vehicle control twice daily, and neutrophil recovery was analyzed at days 10 and 15. Treatment with 49 resulted in a significant acceleration in the return of peripheral blood neutrophils as evaluated 10 days post-transplant (455±63 versus 303±33 per ul blood; Fig. 7). A similar trend was observed in (+)-4-treated recipients at this time point (416±102 versus 289±26 per μl blood). Importantly, enhanced neutrophil recovery was maintained at day 15 post-transplant (719±58 versus 479±45 per μl blood for 49 versus vehicle; Fig. 7). Acceleration of total white blood cell and red blood cell repopulation was also observed in 49-treated recipients (data not shown). Together, these data demonstrate that 49 treatment improves hematopoietic regeneration following murine BM transplantation.
Figure 7.

15-PGDH inhibitors accelerate recovery following bone marrow transplantation. Mice were irradiated at day-1 and dosing with vehicle or drug was initiated. (+)-4 was dosed in vehicle 1 (10% EtOH, 5% cremophor EL, 85% D5W, pH 7.4. 49 was dosed in vehicle 2 (10% DMSO, 25% PEG 400, 1% Tween-80, 64% D5W, pH 7.6). On day 0, 106 bone marrow cells were injected IV and drug or vehicle administration was continued twice daily. Circulating neutrophils were counted at days 10 and 15 (n =10 – 16 per group). *P < 0.05, **P < 0.005.
The selectivity of inhibitor 49 was determined using several in vitro methods. First, quinoxaline 49 was non-competitive with PGE2, showing similar IC50 values over a PGE2 concentration range of 4 – 40 μM (Figure S4). These data do not exclude the possibility that 49 binds the enzyme active site, however, as PGE2 appears to bind 15-PGDH much less tightly (Km = 7 μM) than 49 (IC50 = 3 nM).39 Under the assay conditions, PGE2 may be unable to displace the quinoxaline from the active site. Second, we confirmed that 49 bound directly to 15-PGDH. Specifically, 49 shifted the melting point of human 15-PGDH by 19.5 °C (50.5 to 71 °C, Figure 8). It appears that 49 binds only to the cofactor:enzyme complex rather than to apo enzyme as evidenced by the lack of melting point shift in the absence of NADH. By contrast, 49 did not shift the melting points of the two most closely related hydrogenases, HSD10 and BDH2 (Figure S5). Third, compound 49 was evaluated against several CYP450 isoforms and the hERG potassium channel. CYP1A, 2B6, 2C8, 2D6 and 3A were inhibited at <10% at 10 μM while CYP2C9 and CYP2C19 showed partial inhibition at this concentration, as did the hERG channel (Table 4). The Eurofins SAFTYscan of 78 receptors, enzymes and channels showed acetylcholinesterase and phosphodiesterase 4D2 were inhibited at low micromolar concentrations, but 49 was clean against the other 76 components. While these data validate 49 as a tool compound to explore the biology of 15-PGDH, they point to areas of emphasis for future optimization efforts.
Figure 8.

Melt curves for 15-PGDH in the presence and absence of NADH and 49. 10 μM of 49, 0.25 μg of human PGDH, and 125 μM NADH.
Table 4.
In vitro properties of 49
| |
|---|---|
| enzyme/channel | % inhibition at 10 μM |
| CYP1A | <10 |
| CYP2B6 | <10 |
| CYP2C8 | <10 |
| CYP2C9 | 37 |
| CYP2C19 | 41 |
| CYP2D6 | <10 |
| CYP3A (midazolam) | <10 |
| CYP3A (testosterone) | <10 |
| hERG | 19 |
| Acetylcholinesterase | IC50 = 5.4 μM |
| Phosphodiesterase 4D2 | IC50 = 1.2 μM |
| 76 components of Eurofins SAFTYscan | IC50 > 10 μM |
| Caco2 PappA→B/PappB→A | 45/34 x10−6 cm/s |
CHEMISTRY
Amide modifications were examined by constructing the diphenyl quinazoline ring system prior to introduction of various amines (Figure 9A). Thus, benzil (61) was condensed with diaminobenzene 62 to provide the carboxylic acid 63. Amidation proceeded smoothly with HATU to yield analogs 6 – 24.
Figure 9.

Synthesis of 15-PGDH inhibitors
Changes to the northwest ring were most expeditiously explored by introducing this group last (Figure 9B). Amide coupling with benzoic acid 64 followed by nucleophilic aromatic substitution with glycine methyl ester formed the nitroarene 65. Reduction with Zn metal led to concurrent cyclization to form lactam 66. Oxidation with MnO2 yielded hydroxy quinoxaline 67, which was then converted to the corresponding triflate 68. Finally, Suzuki coupling with the appropriate aryl boronic acid derivatives generated inhibitors 25 – 55. The structure of compound 49 was confirmed through X-ray crystallography (see supporting information, Figure S6).
Sulfoxide 56 was constructed from butane thiol and 3-chloro-6-nitro aniline (Figure 9C). Substitution followed by reduction and condensation with benzil gave the thioether 70. Oxidation with hydrogen peroxide cleanly formed sulfoxide 56.
Quinazoline analogs were synthesized as outlined in Figure 9D. Aminolysis of ester 71 followed by reduction with Zn metal gave the 2-aminobenzamide 72. Condensation with benzaldehyde and oxidation with iodine formed the quinazolinone 57. Subsequent triflation and Suzuki coupling provided the analogs 58 – 60.
CONCLUSIONS
The quinoxalines described here represent potent and efficacious inhibitors of 15-PGDH, and the first series with reported oral activity in disease models. The scaffold tolerates a wide range of aryl and heteroaryl substitution on the pyrazine ring, whereas substantial changes to the piperidine amide resulted in substantial loss of inhibitory potency. Indeed, all potent 15-PGDH inhibitors that have been reported (see 1 – 5) feature a similarly positioned hydrogen bond acceptor, suggesting that the amide carbonyl may make critical interactions with the binding pocket. In this context, Niesen et al. proposed that the piperidine amide of inhibitor 3 formed hydrogen bonds to the active site Ser38 and Tyr151;30 a similar binding pose could be operative with the quinoxaline scaffold. Thus, our optimization efforts focused on exploration of the quinoxaline substituents. We noted that heterocycles performed better than substituted phenyl rings at the northwest corner of the scaffold, while the southwest ring could be removed without compromising activity. Identification of metabolically stable isoquinolone substituents helped to balance potency, solubility and metabolic stability (Fig S7). A lead compound, 49, displayed excellent oral bioavailability, inhibition of 15-PGDH in vivo, a corresponding elevation of PGE2 in multiple tissues, and protective activity in the mouse DSS-colitis model. These results are consistent with prior studies showing that ablation of 15-PGDH enzyme activity reduces inflammatory cytokines in the colons of DSS treated mice, including TNFα, interferon-γ, interleukin-2, and interleukin 1β.17 The quinoxaline series provides an attractive foundation for the discovery of oral 15-PGDH inhibitors suitable for use in humans.
EXPERIMENTAL SECTION
General.
All tested compounds were >95% pure by HPLC and 1H NMR unless otherwise noted. 15-PGDH inhibitor compounds tested were dissolved in DMSO and used as a 25 mM stock solution, with straight DMSO used for control comparisons. In vivo mouse pharmacokinetic experiments described in this manuscript have been approved and conducted under the oversight of the UT Southwestern Institutional Animal Care and Use Committee. UT Southwestern uses the 'Guide for the Care and Use of Laboratory Animals' when establishing animal research standards. Mouse in vivo pharmacodynamics and efficacy experiments were conducted in the Case Animal Resource Center according to a protocol approved by the Institutional Animal Care Use Committee.
In vitro and cellular testing.
15-PGDH enzyme assay.
Assays of 15-PGDH enzyme activity were carried out at 5 nM 15-PGDH, 150 μM NAD+, 50 mM Tris-HCl, pH 7.5, 0.01% Tween 20, 0.1 mM DTT and 20 μM PGE2 (Sigma, cat. #P5640). Activity was determined as the rate of NADH generation as determined by fluorescence (Ex/Em=340 nM/485 nM) measured every 15 s for 3.5 min as described previously.17 The IC50 values were calculated with GraphPad Prism 7 software (http://www.graphpad.com/scientific-software/prism/).
Elevation of PGE2 level in A549 cells.
The A549 cell line was maintained in F12K medium supplemented with 10% fetal calf serum (FBS) and 50 μg/mL gentamicin in a humidified atmosphere containing 5% CO2 at 37°C. Cells were plated in duplicate in a 24-well plate (1 mL per well) at 1x105 cells per well and grown for 24 h before stimulation with IL-1β (1 ng/mL) overnight (16 h) to induce COX2 expression and PGE2 production. Cells were then treated for an additional 8 hours with fresh medium containing the indicated concentration of compounds. Medium was then collected and the level of PGE2 analyzed using a PGE2 ELISA Kit (R&D System, SKGE004B). Data were collected from four independent experiments. Results were tabulated graphically with error bars corresponding to standard error of the means and compared using 2-tailed t-tests.
15-PGDH thermal denaturation assay:
Thermal denaturation of 15-PGDH was monitored by differential scanning fluorimetry using SYPRO orange dye.30 Briefly, the protein was diluted to a final assay concentration of 10 μM in 100 mM Tris buffer pH 8.0, containing 0.01% Tween-20 and 1:1000 SYPRO orange dye (Sigma S-5692). The final assay volume was 20 μL, with or without 100 μM of NADH. Compound, in assay buffer plus 0.4% (v/v) DMSO, was added to 10 μM final concentration. Heat denaturation curves were recorded using a real-time PCR instrument (CFX-96, BioRad) applying a temperature gradient of 2°C/min. Analysis of the data was performed using default BioRad CFX Manager V3.1 software. Melting temperatures of 15-PGDH were determined by the inflection points of the plots of −d(RFU)/dT. Specificity of interaction of 49 with 15-PGDH was assessed by testing the effect of 49 on melting temperature of HSD10 and BDH2, two short chain dehydrogenases that are both closely structurally related to 15-PGDH. Recombinant HSD10 and BDH2 were purchased from Syd Labs, Boston, MA.
Analytical LC-MS/MS conditions.
Compound levels for in vitro and in vivo pharmacology assays were monitored by LC-MS/MS using an AB Sciex (Framingham, MA) 3200 QTRAP® or 4000 QTRAP® mass spectrometer coupled to a Shimadzu (Columbia, MD) Prominence LC. Analytes were detected with the mass spectrometer in positive MRM (multiple reaction monitoring) mode by following the precursor to fragment ion transition for two daughter ions. Only one parent daughter pair, indicated here, was used for quantitation: 40: 368.100/283.000; 48: 385.300/300.000; 49: 399.123/314.1; 43: 369.300/257.500; 44: 369.300/258.000. An Agilent C18 XDB column (5 micron, 50 X 4.6 mm) was used for chromatography for 40 and 49 with the following conditions: Buffer A: dH20 + 0.1% formic acid, Buffer B: methanol + 0.1% formic acid, 0 - 1.5 min 3% B, 1.5 - 2 min gradient to 100% B, 2 - 3.5 min 100% B, 3.5 - 3.6 min gradient to 3% B, 3.6 - 4.5 3% B. N-benzylbenzamide (transition 212.1 to 91.1 from Sigma (St. Louis, MO) was used as an internal standard (IS). Evaluation of other compounds employed nearly identical conditions except the gradient utilized was 0 - 1.0 min 3% B, 1.0 – 2 min gradient to 100% B, 2.0- 3.5 min 100% B, 3.5 - 3.6 min gradient to 3% B, 3.6 - 4.5 3% B.
Thermodynamic solubility.
Solid compound was added to a glass vial containing dH20 along with a stir bar until compound was visibly not in solution. The vial was covered and left to stir for 18 hours. Solution (1 mL) was then transferred to a Savillex PFA microcentrifuge tube and centrifuged at 16,100 x g for 10 min at room temperature. Being careful to avoid particulate material, the supernatant was removed and centrifuged a second time. Supernatant was removed a second time and either diluted up to 1:1000 or mixed directly 1:2 with methanol containing formic acid (0.1% final) and N-benzylbenzamide (33 ng/mL final). This mixture was vortexed for 15 s, centrifuged 16,100 x g for 5 min and the supernatant analyzed by LC-MS/MS in comparison to a standard curve of each compound made from DMSO standards diluted into a 1:2 mixture of water and methanol containing 0.1% formic acid and 33 ng/mL IS. After correction for dilution, the average concentration was determined.
Mouse liver S9 stability.
Female ICR/CD-1 mouse S9 fractions were purchased from BioIVT (Westbury, NY). S9 protein (1 mg/mL) was added to a glass screw cap tube; 50 mM Tris, pH 7.5 solution, containing the compound of interest was added on ice. The final concentration of compound after addition of all reagents was 2 μM. An NADPH-regenerating system (1.7 mg/ml NADP, 7.8 mg/ml glucose-6-phosphate, 6 U/ml glucose-6-phosphate dehydrogenase in 2% w/v NaHCO3/10 mm MgCl2) was added for analysis of Phase I metabolism. The tube was then placed in a 37 °C shaking water bath. At varying time points after addition of phase I cofactors, the reaction was stopped by the addition of 0.5 ml of methanol containing IS and formic acid such that the final concentration was 0.1%. The samples were incubated 10 min at RT and then spun at 16,100 x g for 5 min in a microcentrifuge at 4 °C. The supernatant was analyzed by LC-MS/MS. The method described in McNaney, et al. was used with modification for determination of metabolic stability half-life by substrate depletion.40 A “% remaining” value was used to assess metabolic stability of a compound over time. The LC/MS/MS peak area of the incubated sample at each time point was divided by the LC/MS/MS peak area of the time 0 (T0) sample and multiplied by 100. The natural Log (LN) of the % remaining of compound was then plotted versus time (in min) and a linear regression curve plotted going through y-intercept at LN(100). The metabolism of some compounds failed to show linear kinetics at later time point, so those time points were excluded. The half-life (t½) was calculated as t½ = 0.693/slope.
Caco2 permeability assays.
Caco2 cells purchased from ATCC were grown in DMEM containing 20% FBS with added glutamine (2 mM), sodium pyruvate (1 mM), non-essential amino acids (1X), hepes (20 mM) and penicillin/streptomycin (100U/mL / 0.2 mg/mL), all from Sigma. Cells were plated at 105 cells/well in 12-well Transwells (Corning). Media was replaced three times per week for three weeks. Prior to analysis, cells were washed three times with Hyclone HBSS with calcium and magnesium (Thermo Scientific) containing 0.5% FBS, and transepithelial resistance (TEER) values measured with a Millipore Epithelial volt-ohmmeter. Cells were used only if TEER values were >800Ωcm2. Compounds were added at 10 μM to the apical or basolateral surface in HBSS containing 0.5% FBS and solution sampled from the basolateral or apical side respectively at 30, 60, 90 and 120 min under sink conditions in which media removed (2/3 of compartment volume) was replaced with fresh compound free HBSS containing 0.5% FBS. Compound concentration was measured by LC-MS/MS and the permeability coefficient (Papp) calculated from the following equation:
Where dQ/dt is the rate of permeation of the drug across the cells, C0 is the donor compartment concentration at time zero and A is the area of the cell monolayer (1.12 for 12 well insert). An efflux ratio is calculated from the mean apical to basolateral (A-B) Papp data and basolateral to apical (B-A) Papp data according to the formula
Assay performance was validated using propranolol as a highly permeable compound (Paap(A-B) >20 x 106), Nadolol as a poorly permeable compound (Paap(A-B)<0.5x 106) and quinidine and cimetidine as Pgp-1 and BCRP1 control substrates respectively (Efflux Ratio >2). TEER values measured after assay completion were within 20% of starting values.
In vivo testing.
Statistical comparisons:
Graphical depictions of data plot mean values of experimental replicates. Error bars depict standard errors of the mean. Statistical comparisons shown in figures and quoted in text are calculated using two tailed t-tests. In figures where groups of comparisons are provided, but only a single * value is indicated, the * is labeled as corresponding to the least significant P value that is <0.05.
Pharmacokinetic studies.
Pharmacokinetic studies were performed by dosing 6-7 week old CD-1 female mice with 40 HCl salt formulated as 5% DMSO, 10% PEG400, 85% HPβCD (30%); 48 formulated as 10% DMSO, 10% Cremophor, and 80% of D5W pH 7.4; and 49 formulated as 10% DMSO, 25% PEG400, 1% Tween-80 and 64% of D5W pH 7.4 at 10 mg/kg IP or PO or 5 mg/kg IV. Animals were sacrificed in groups of three, blood was obtained by cardiac puncture at each time point (0, 10, 30, 90, 180, 360, 960 and 1440 min post dose) using the anticoagulant ACD (acidified citrate dextrose) and plasma isolated by centrifugation. 100 μl of plasma or tissue homogenate was mixed with 200 μl of methanol containing 0.15% formic acid and 37.5 ng/ml N-benzylbenzamide internal standard. The samples were vortexed 15 sec, incubated at room temp for 10' and spun twice at 16,100 x g 4°C in a refrigerated microcentrifuge. The resulting supernatants were evaluated by LC-MS/MS to determine the amount of compound present. Standard curves were generated using blank plasma (Bioreclamation, Westbury, NY) spiked with known concentrations of compound and processed as described above. The concentrations of drug in each time-point sample were quantified using Analyst software (Sciex). A value of 3-fold above the signal obtained from blank plasma was designated the limit of detection (LOD). The limit of quantitation (LOQ) was defined as the lowest concentration at which back calculation yielded a concentration within 20% of theoretical. Cmax and AUC values were calculated using the noncompartmental analysis tool of Phoenix WinNonLin (Certara/Pharsight, Sunnyvale, CA).
Evaluation of PGE2 levels in mouse tissue.
Female C57bl/6J mice 8-9 weeks old (Jackson laboratories) were treated by I.P. injection with indicated compounds at the indicated times and doses in the text. Mice were euthanized by rapid asphyxiation with CO2. Solid tissues were excised, rinsed in ice-cold PBS containing indomethacin (5.6 μgm/ml), and snap frozen in liquid nitrogen. Frozen samples were pulverized over liquid nitrogen. The powder transferred to an Eppendorf tube with 500 μl of cold PBS with indomethacin, and then homogenized using Kimble Kontes Pellet Pestle Cordless Motor (Fisher, K749540-0000) with Kimble Kontes blue pellet pestles (Fisher, K749521-1590) for ~30 s on ice. Then the suspension was sonicated in sonicator containing an ice water bath for 5 min using cycles of 20 seconds of sonication with 20 seconds of cooling, followed by centrifugation for 10 minutes at 12,000 rpm. The PGE2 level of the supernatant was measured using a PGE2 ELISA Kit (Enzo cat. #ADI-931-001) and normalized to protein concentration measured by BCA assay (Thermo Scientific, cat. #23225) and expressed as ng PGE2/mg protein. Each sample was assayed in triplicate.
In vivo enzyme inhibition assay.
8-9 Week old female C57bl/6J mice (Jackson laboratories) were treated by oral gavage with indicated compounds at the indicated times and doses in the text. Compounds were formulated as described in the pharmacokinetics studies. Mice were euthanized by rapid asphyxiation with CO2. Solid tissues were excised, rinsed in ice-cold PBS only and snap frozen in liquid nitrogen. Frozen samples were pulverized over liquid nitrogen. The powder was transferred to an Eppendorf tube with 500 μl of cold lysis buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM DTT, 0.1 mM EDTA) and then homogenized using Kimble Kontes Pellet Pestle Cordless Motor (Fisher, K749540-0000) with Kimble Kontes blue pellet pestles (Fisher, K749521-1590) for ~30s on ice. The suspension was centrifuged for 10 minutes at 12,000 rpm. The enzyme activity was measured by transfer of tritium from a tritiated PGE2 substrate to glutamate by coupling 15-PGDH to glutamate dehydrogenase with 1 mM NAD+, 5 mM NH4Cl, 1mM α-keto-Glutarate (Sigma#75890) and 16 U of Bovine Liver Glutamic dehydrogenase (Sigma, cat. #G-2501) as described previously.17 The activity was normalized to protein concentration measured by BCA assay (Thermo Scientific, cat. #23225) and expressed as CPM/Hour/mg protein.
Dextran Sodium Sulfate (DSS) mouse colitis studies.
DSS colitis studies were performed with female C57BL/6NTac mice (8 weeks of age) obtained from Taconic Bioscience. The animals were housed in standard microisolator cages and maintained on a defined irradiated diet (Prolab Isopro RMH 3000) and autoclaved water. To ensure uniformity of microbiota between cages, bedding from all cages were mixed together and then reintroduced back into the cages a minimum of 3 days prior to the beginning of an experiment. Groups of mice were administered 7 days (Day 1-8) of drinking water containing 2.75% DSS, MW 40-50 kDa (Spectrum Chemical, Cat# DE136-1KGBL), formulated in sterile water, and then switched to regular drinking water for 4 days (Day 8-12). To determine the impact of treatment with compound 49 on DSS-induced colitis, mice were administered either 10 mg/kg 49 twice a day by intraperitoneal administration (I.P.) or 40 mg/kg twice a day by oral administration (P.O.), for the 11 days of the experiment, with control groups being administered vehicle only by I.P or P.O. A naíve group, which were mice maintained on regular water and without 49 or vehicle treatment, was included in each study as a comparator arm. During the study, mice were weighed at Days 1, 3, 5, 7, 8, 10 and 12, and weights graphed as the mean weight with error bars corresponding to standard error of the mean. Different treatment arms were compared across the time course of study using 2-tailed paired t-test. Mice were euthanized either on Day 8 or Day 12 and colon lengths were analyzed. Colons were collected, kept in chilled 1xPBS for a minimum of 5 minutes to ensure recovery of colon length from any potential stretching from the extraction process, then colon length was measured from ascending colon to rectum. Values are expressed as the mean with error bars corresponding to standard error of the mean. After measuring, colons were then flushed, opened by cutting longitudinally, and cut into thirds to produce distal, middle and proximal colon pieces. The tissue pieces were fixed and paraffin embedded in a “petit four” arrangement as previously described.17 Briefly, the tissue sections were laid flat for 30 minutes on Whatman paper soaked with 10% buffered formalin phosphate (NBF). Next, the fixed tissues were layered one level above the next in alternating layers of tissue and agar, solidified by 30 minutes of refrigeration, and then further fixed overnight at 4 °C in 10% NBF. The resultant petit four blocks were embedded in paraffin and cut into 5 mm sections. The slides were stained with hematoxylin and eosin. The Day 8 images were scanned with Axio Scan.Z1 (Zeiss).
Ulcer measurements were obtained from the Day 8 scanned images and lengths calculated using ImageJ freehand line tool with pixel number calibrated for each magnification. Ulcer measurements are summed and a percent of total length which was measured along the muscularis mucosa (% ulceration). Day 8 percent ulceration is graphed for each experimental group as the group mean with error bars corresponding to standard error of the mean.
A colitis-associated crypt abnormality score was assessed in each of the distal, middle, and proximal colon segments. Each segment was divided into 4 roughly equal sections. For each quadrant section, a crypt severity grade was assigned using the grading system as described previously.17 In brief, crypt damage severity grading was scored as: no damage = 0, 1/3 damaged = 1, 2/3 damaged = 2, complete crypt absence with intact surface epithelium = 3, and absent crypts with complete loss of surface epithelium = 4. For each section, the percent of linear measurement affected by crypt damage, termed the linear score, was also scored as: 0 (= not present), 1 (= 1-25%), 2 (= 26-50%), 3 (= 51-75%) and 4 (= 74-100%). A crypt section score was obtained by multiplication of the (severity grade) x (linear score) yielding a section score of 0-16. The segment score was calculated as the average of its component section scores, and the sum of each distal, middle and proximal segment score was tabulated and expressed as the total cryptitis score for each animal. Group wise scores are graphed as the mean with error bars representing standard error of the mean.
Murine bone marrow transplantation:
Recipient mice were conditioned for transplant by 10 Gy irradiation, followed immediately by administration of indicated drug (5 mg/kg) or respective vehicle control by intraperitoneal injection. After 16-18 h, mice received 106 whole bone marrow cells by retroorbital injection, followed immediately by a second administration of drug or respective vehicle control by intraperitoneal injection. Recipient mice received twice daily IP injections with (+)-4, 49, or respective vehicles, spaced by at least 6 hours.
Peripheral blood from mice was collected into Microtainer EDTA tubes (Becton-Dickinson) by submandibular cheek puncture. Blood counts were analyzed using a Hemavet 950 FS hematology analyzer. Values were tabulated graphically with error bars corresponding to standard error of the means.
At 21 days post-bone marrow transplantation, mice were sacrificed via cervical dislocation. Bone marrow cells were obtained by flushing hindlimb bones and filtering the resulting cell suspension through a 40 μm cell strainer. Cellularity was measured following red blood cell lysis. Cells were stained with the antibodies indicated in Table S1 and data was acquired on an LSRII flow cytometer (BD Biosciences). Immunophenotypic analysis was performed on FlowJo software (TreeStar).
Chemical Methods.
General Procedure A: variation through amide coupling (7 – 24).
Step 1. To a solution of benzil (2.1 g, 10 mmol) in MeOH (20 mL) was added 3,4-diaminobenzoic acid (1.52 g, 10 mmol). The slurry was stirred at room temperature for 8 h. The solid was collected by filtration and crystallized by MeOH/EtOAc to afford 2,3-diphenylquinoxaline-6-carboxylic acid as a pale yellow solid (2.93 g, 90% yield). Spectral data were consistent with literature values.41 1H NMR (400 MHz, DMSO-d6) δ 13.31 (s, 1H), 8.64 (d, J = 1.8 Hz, 1H), 8.30 (dd, J = 8.8, 1.8 Hz, 1H), 8.22 (d, J = 8.7 Hz, 1H), 7.50-7.47 (m, 4H), 7.42-7.34 (m, 6H).
Step 2. To a solution of 2,3-diphenylquinoxaline-6-carboxylic acid (33 mg, 0.1 mmol), HATU (42 mg, 0.11 mmol), DIPEA (0.035 mL, 0.2 mmol) in DMF (2 mL) was added the appropriate amine (0.12 mmol). The mixture was stirred at room temperature until the reaction reached completion as monitored by LC/MS. Water was added to the solution, and the mixture was extracted with ethyl acetate, washed with brine, dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The crude material was purified by flash chromatography on silica gel to give the product.
(2,3-Diphenylquinoxalin-6-yl)(pyrrolidin-1-yl)methanone (7) was synthesized according to General Procedure A in 86% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 8.28 (s, 1H), 8.20 (d, J = 8.6 Hz, 1H), 7.94 (d, J = 9.2 Hz, 1H), 7.51 (d, J = 6.8 Hz, 4H), 7.38-7.30 (m, 6H), 3.72 (t, J = 6.9 Hz, 2H), 3.55 (t, J = 6.6 Hz, 2H), 2.04-1.97 (m, 2H), 1.94-1.87 (m, 2H); 13C NMR(101 MHz, Chloroform-d) δ 168.3, 154.4, 154.2, 141.5, 140.4, 138.7, 138.3, 129.8, 129.8, 129.6, 129.1, 129.04, 129.03, 128.3, 127.7, 49.7, 46.4, 26.5, 24.5, (missing 2 signals due to overlap). MS (ES+) m/z = 380 [M+H]+.
Azepan-1-yl(2,3-diphenylquinoxalin-6-yl)methanone (8) was synthesized according to General Procedure A in 68% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 8.21 (d, J = 8.6 Hz, 1H), 8.16 (s, 1H), 7.78 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 6.1 Hz, 4H), 7.39-7.30 (m, 6H), 3.75 (t, J = 5.9 Hz, 2H), 3.45 (t, J = 5.5 Hz, 2H), 1.92-1.86 (m, 2H), 1.70-1.59 (m, 6H); 13C NMR (101 MHz, Chloroform-d) δ 170.3, 154.23, 154.17, 141.1, 140.6, 138.8, 138.7, 138.6, 129.82, 129.76, 129.0, 128.5, 128.3, 126.8, 49.9, 46.4, 29.5, 27.8, 27.3, 26.5, (missing 3 signals due to overlap). MS (ES+) m/z = 408 [M+H]+.
2,3-Diphenylquinoxaline-6-carboxamide (9) was synthesized according to General Procedure A in 57% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.37 (s, 1H), 8.29 (d, J = 8.7 Hz, 1H), 8.18 (d, J = 8.7 Hz, 1H), 7.70 (s, 1H), 7.48 (d, J = 7.2 Hz, 4H), 7.41-7.33 (m, 6H); 13C NMR (101 MHz, DMSO-d6) δ 167.3, 154.7, 154.3, 142.0, 140.3, 138.99, 138.96, 135.8, 130.2, 130.1, 129.5, 129.4, 129.2, 128.6, 128.54, 128.52, (missing 1 signal due to overlap). MS (ES+) m/z = 326.0 [M+H]+.
N-Methyl-2,3-diphenylquinoxaline-6-carboxamide (10) was synthesized according to General Procedure A in 66% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.87-8.84 (m, 1H), 8.63 (s, 1H), 8.26 (d, J = 8.7 Hz, 1H), 8.20 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 7.6 Hz, 4H), 7.42-7.33 (m, 6H), 2.86 (d, J = 3.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 166.0, 154.6, 154.3, 141.9, 140.3, 139.0, 136.0, 130.2, 130.1, 129.5, 129.4, 129.3, 129.1, 128.54, 128.52, 128.1, 27.0, (missing 1 signal due to overlap). MS (ES+) m/z = 340.0 [M+H]+.
N-Ethyl-2,3-diphenylquinoxaline-6-carboxamide (11) was synthesized according to General Procedure A in 67% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (t, J = 5.5 Hz, 1H), 8.66 (s, 1H), 8.27 (d, J = 8.6 Hz, 1H), 8.20 (d, J = 8.8 Hz, 1H), 7.48 (d, J = 7.3 Hz, 4H), 7.42-7.33 (m, 6H), 3.40-3.34 (m, 2H), 1.18 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.3, 154.6, 154.3, 141.9, 140.3, 139.0, 136.2, 130.2, 130.1, 129.5, 129.4, 129.3, 129.2, 128.54, 128.51, 128.1, 34.8, 15.1, (missing 1 signal due to overlap). MS (ES+) m/z = 354 [M+H]+.
2,3-Diphenyl-N-propylquinoxaline-6-carboxamide (12) was synthesized according to General Procedure A in 73% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (d, J = 5.7 Hz, 1H), 8.66 (s, 1H), 8.27 (d, J = 8.6 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.48 (d, J = 7.3 Hz, 4H), 7.42-7.34 (m, 6H), 3.32-3.27 (m, 2H), 1.64-1.54 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.5, 154.6, 154.3, 141.9, 140.2, 138.98, 138.96, 136.2, 130.2, 130.1, 129.5, 129.4, 129.3, 128.5, 128.1, 41.7, 22.8, 12.0, (missing 2 signals due to overlap). MS (ES+) m/z = 368.1 [M+H]+.
N-Butyl-2,3-diphenylquinoxaline-6-carboxamide (13) was synthesized according to General Procedure A in 76% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.85 (t, J = 5.5 Hz, 1H), 8.66 (s, 1H), 8.26 (d, J = 8.9 Hz, 1H), 8.19 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 7.3 Hz, 4H), 7.42-7.33 (m, 6H), 3.35-3.31 (m, 2H), 1.59-1.52 (m, 2H), 1.41-1.31 (m, 2H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.4, 154.6, 154.3, 141.9, 140.2, 138.97, 138.95, 136.2, 130.2, 130.1, 129.5, 129.4, 129.2, 128.5, 128.1, 39.6, 31.6, 20.2, 14.2, (missing 2 signals due to overlap). MS (ES+) m/z = 382 [M+H]+.
N-Pentyl-2,3-diphenylquinoxaline-6-carboxamide (14) was synthesized according to General Procedure A in 76% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (t, J = 5.4 Hz, 1H), 8.66 (s, 1H), 8.27 (d, J = 8.8 Hz, 1H), 8.20 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 7.2 Hz, 4H), 7.42-7.34 (m, 6H), 3.33-3.30 (m, 2H), 1.61-1.54 (m, 2H), 1.34-1.26 (m, 4H), 0.87 (t, J = 6.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.4, 154.6, 154.3, 141.9, 140.2, 138.97, 138.95, 136.2, 130.2, 130.1, 129.5, 129.4, 129.3, 128.5, 128.1, 29.2, 29.1, 22.4, 14.4, (missing 2 signals due to overlap). MS (ES+) m/z = 396 [M+H]+.
N-Cyclopentyl-2,3-diphenylquinoxaline-6-carboxamide (15) was synthesized according to General Procedure A in 86% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.72-8.69 (m, 2H), 8.27 (dd, J = 8.9, 1.9 Hz, 1H), 8.18 (d, J = 8.7 Hz, 1H), 7.49-7.47 (m, 4H), 7.41-7.33 (m, 6H), 4.32-4.25 (m, 1H), 1.97-1.87 (m, 2H), 1.75-1.68 (m, 2H), 1.64-1.50 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 165.2, 154.5, 154.2, 141.9, 140.2, 139.0, 136.2, 130.2, 130.1, 129.5, 129.43, 129.40, 129.1, 128.5, 128.2, 51.7, 32.6, 24.2, (missing 2 signals due to overlap). MS (ES+) m/z = 394 [M+H]+.
N,2,3-Triphenylquinoxaline-6-carboxamide (16) was synthesized according to General Procedure A in 53% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1H), 8.84 (s, 1H), 8.36 (d, J = 8.6 Hz, 1H), 8.25 (d, J = 8.7 Hz, 1H), 7.86 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 7.3 Hz, 4H), 7.42-7.34 (m, 8H), 7.13 (t, J = 7.4 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 164.8, 154.9, 154.5, 142.2, 140.2, 139.4, 138.94, 138.91, 136.3, 130.2, 130.1, 129.54, 129.47, 129.1, 128.8, 128.57, 128.55, 124.4, 120.9, (missing 3 signals due to overlap). MS (ES+) m/z = 402 [M+H]+.
(2,3-Diphenylquinoxalin-6-yl)(morpholino)methanone (17) was synthesized according to General Procedure A as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 8.5 Hz, 1H), 8.15 (s, 1H), 7.86 (d, J = 8.5 Hz, 1H), 7.47 (d, J = 7.0 Hz, 4H), 7.42-7.33 (m, 6H), 3.69-3.58 (m, 6H), 3.42 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 168.3, 154.4, 141.0, 140.2, 138.9, 137.6, 130.2, 130.1, 129.8, 129.5, 129.43, 129.40, 128.5, 127.5, 66.5, 48.2, 42.6, (missing 3 signals due to overlap). MS (ES+) m/z = 396.1 [M+H]+.
tert-Butyl 4**-(2,3-diphenylquinoxaline-6-carbonyl)piperazine-1-carboxylate (18) was synthesized according to General Procedure A in 83% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 8.5 Hz, 1H), 8.15 (s, 1H), 7.86 (d, J = 8.5 Hz, 1H), 7.47 (d, J = 5.7 Hz, 4H), 7.42-7.33 (m, 6H), 3.66 (s, 2H), 3.47-3.36 (m, 6H), 1.40 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 168.4, 154.41, 154.39, 154.2, 141.0, 140.3, 139.0, 137.8, 130.2, 130.1, 129.8, 129.4, 128.5, 127.4, 79.7, 47.4, 42.0, 28.5, (missing 3 signals due to overlap). MS (ES+) m/z = 495.1 [M+H]+.
1-(2,3-Diphenylquinoxaline-6-carbonyl)piperidin-4-one (19) was synthesized according to General Procedure A as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 8.27-8.25 (m, 2H), 7.87 (d, J = 8.5 Hz, 1H), 7.55-7.51 (m, 4H), 7.40-7.33 (m, 6H), 4.13-3.86 (m, 4H), 2.66-2.46 (m, 4H); 13C NMR (101 MHz, Chloroform-d) δ 206.3, 169.6, 154.7, 154.6, 141.6, 140.4, 138.53, 138.51, 136.2, 130.2, 129.82, 129.79, 129.23, 129.21, 128.5, 128.4, 127.6, 46.6, 41.2, (missing 1 signal due to overlap). MS (ES+) m/z = 408 [M+H]+.
tert-Butyl(1-(2,3-diphenylquinoxaline-6-carbonyl)piperidin-4-yl)carbamate (20) was synthesized according to General Procedure A as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 8.19 (d, J = 8.6 Hz, 1H), 8.14 (d, J = 1.9 Hz, 1H), 7.77 (dd, J = 8.6, 1.9 Hz, 1H), 7.51-7.48 (m, 4H), 7.37-7.29 (m, 6H), 4.73-4.64 (m, 2H), 3.80-3.68 (m, 2H), 3.18-3.00 (m, 2H), 2.00-1.93 (m, 2H), 1.47-1.34 (m, 2H), 1.42 (s, 9H); 13C NMR (101 MHz, Chloroform-d) δ 169.1, 155.1, 154.40, 154.36, 141.4, 140.5, 138.7, 138.6, 137.1, 129.9, 129.8, 129.10, 129.08, 128.6, 128.3, 127.3, 79.5, 47.8, 46.7, 41.3, 33.1, 32.2, 28.4, (missing 2 signals due to overlap). MS (ES+) m/z = 509.1 [M+H]+.
(2,3-Diphenylquinoxalin-6-yl)(4-fluoropiperidin-1-yl)methanone (21) was synthesized according to General Procedure A as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 8.22 (d, J = 8.6 Hz, 1H), 8.17 (d, J = 1.8 Hz, 1H), 7.80 (dd, J = 8.5, 1.9 Hz, 1H), 7.53-7.50 (m, 4H), 7.38-7.30 (m, 6H), 5.00-4.84 (m, 1H), 4.13-4.07 (m, 1H), 3.69-3.50 (m, 3H), 1.99-1.80 (m, 4H); 13C NMR (101 MHz, Chloroform-d) δ 169.2, 154.44, 154.39, 141.4, 140.5, 138.7, 138.6, 137.0, 130.0, 129.82, 129.80, 129.1, 129.1, 128.6, 128.3, 127.3, 87.4 (d, J = 171.6 Hz), 43.6, 38.2, 31.6, 31.0, (missing 1 signal due to overlap). MS (ES+) m/z = 412 [M+H]+.
(2,3-Diphenylquinoxalin-6-yl)(4-(hydroxymethyl)piperidin-1-yl)methanone (22) was synthesized according to General Procedure A as a yellow solid containing approximately 10% DMF. 1H NMR (400 MHz, Chloroform-d) δ 8.20 (d, J = 8.6 Hz, 1H), 8.15 (s, 1H), 7.78 (d, J = 8.6 Hz, 1H), 7.56 – 7.46 (m, 4H), 7.40 – 7.27 (m, 6H), 4.86 – 4.69 (m, 1H), 3.91 – 3.76 (m, 1H), 3.47 (bs, 2H), 3.13 – 2.98 (m, 1H), 2.89 – 2.75 (m, 1H), 2.38 - 2.20 (m, 1H), 1.92 – 1.65 (m, 3H), 1.38 – 1.17 (m, 2H). 13C NMR (101 MHz, Chloroform-d) δ 169.0, 154.4, 154.3, 141.3, 140.5, 138.7, 138.6, 137.5, 129.81, 129.80, 129.10, 129.08, 128.7, 128.3, 127.2, 67.0, 47.9, 42.4, 36.5, 29.5, 28.4, (missing 2 signals due to overlap). MS (ES+) m/z = 424 [M+H]+.
(2,3-Diphenylquinoxalin-6-yl)(4-methylpiperidin-1-yl)methanone (23) was synthesized according to General Procedure A as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 8.21 (d, J = 8.5 Hz, 1H), 8.16 (s, 1H), 7.80 (d, J = 8.7 Hz, 1H), 7.52-7.50 (m, 4H), 7.38-7.30 (m, 6H), 4.77-4.73 (m, 1H), 3.82-3.79 (m, 1H), 3.10-2.79 (m, 2H), 1.82-1.58 (m, 3H), 1.31-1.15 (m, 2H), 0.98 (d, J = 6.4 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ 169.0, 154.3, 141.3, 140.5, 138.7, 137.7, 129.82, 129.79, 129.0, 128.8, 128.3, 127.2, 48.2, 42.7, 34.8, 33.9, 31.1, 21.7, (missing 5 signals due to overlap). MS (ES+) m/z = 408 [M+H]+.
(2,3-Diphenylquinoxalin-6-yl)(4-methoxypiperidin-1-yl)methanone (24) was synthesized according to General Procedure A as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 8.20 (d, J = 8.6 Hz, 1H), 8.16 (d, J = 1.9 Hz, 1H), 7.79 (dd, J = 8.5, 1.8 Hz, 1H), 7.51 (d, J = 7.9 Hz, 4H), 7.37-7.29 (m, 6H), 4.10-4.04 (m, 1H), 3.65 (br s, 2H), 3.52-3.47 (m, 1H), 3.37-3.30 (m, 4H), 1.97-1.58 (m, 4H); 13C NMR (101 MHz, Chloroform-d) δ 169.0, 154.32, 154.30, 141.3, 140.5, 138.69, 138.67, 137.4, 129.9, 129.8, 129.1, 128.7, 128.3, 127.3, 75.1, 55.8, 44.8, 39.3, 31.3, 30.2, (missing 3 signals due to overlap). MS (ES+) m/z = 424 [M+H]+.
General Procedure B. Modification through Suzuki Coupling.
Step 1. To a solution of 4-Fluoro-3-nitrobenzoic acid (64, 1.85 g, 10 mmol), HATU (4.2 g, 11 mmol), and i-Pr2NEt (3.5 mL, 20 mmol) in DMF (20 mL) was added piperidine (11 mmol, 0.94 g, 1.1 mL). The mixture was stirred at room temperature for 4 h. Water was added to the solution, and the mixture was extracted with ethyl acetate, washed with brine, dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The crude mixture was purified by flash chromatography on silica gel to give the (4-Fluoro-3-nitrophenyl)(piperidin-1-yl)methanone as yellow oil in approximately 80% purity, which was used without additional purification (2.32 g, 92% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.11 (dd, J = 7.0, 2.1 Hz, 1H), 7.69 (ddd, J = 8.6, 4.2, 2.2 Hz, 1H), 7.34 (dd, J = 10.4, 8.6 Hz, 1H), 3.70 (s, 2H), 3.35 (s, 2H), 1.72-1.56 (m, 6H). MS (ES+) m/z = 253 [M+H]+.
Step 2. To a solution of the amide from step 1 (2.27 g, 9 mmol) in DMSO (18 mL) was added methyl glycinate (801 mg, 9 mmol) and i-Pr2NEt (1.91 mL, 11 mmol) at room temperature. The mixture was then heated at 100 °C for 3 h. The solution was cooled in an ice bath and cold water (30 mL) was added. The precipitate was filtered and washed with water. The resulting solid was dried to obtain the pure product Methyl (2-nitro-4-(piperidine-1-carbonyl)phenyl)glycinate (65) as a yellow solid (2.60 g, 90% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.52 (t, J = 5.9 Hz, 1H), 8.09 (d, J = 2.1 Hz, 1H), 7.54 (dd, J = 8.8, 2.1 Hz, 1H), 6.95 (d, J = 8.9 Hz, 1H), 4.31 (d, J = 5.8 Hz, 2H), 3.69 (s, 3H), 3.44 (s, 4H), 1.60-1.57 (m, 2H), 1.49 (s, 4H). MS (ES+) m/z = 322 [M+H]+.
Step 3. To a solution of Methyl (2-nitro-4-(piperidine-1-carbonyl)phenyl)glycinate (2.57 g, 8 mmol) in Acetone (16 mL) was added Zn powder (2.6 g, 40 mmol), Ammonium Chloride (2.14 g, 40mmol) and water (2 mL). The mixture was refluxed for 3 h, then filtered and concentrated. The residue was diluted with water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4 and concentrated by reduced pressure. The crude was purified by flash chromatography on silica gel to give 7-(Piperidine-1-carbonyl)-3,4-dihydroquinoxalin-2(1H)-one (66) as a white solid (1.7 g, 82% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.52 (t, J = 5.9 Hz, 1H), 8.09 (d, J = 2.1 Hz, 1H), 7.54 (dd, J = 8.8, 2.1 Hz, 1H), 6.95 (d, J = 8.9 Hz, 1H), 4.31 (d, J = 5.8 Hz, 2H), 3.69 (s, 3H), 3.44 (s, 4H), 1.71-1.39 (m, 6H). MS (ES+) m/z = 260 [M+H]+.
Step 4. MnO2 (2.61 g, 30 mmol) was added to a 50-mL flask which was charged with 7-(Piperidine-1-carbonyl)-3,4-dihydroquinoxalin-2(1H)-one (66, 1.55 g, 6 mmol) in dichloromethane (20 mL). The mixture was stirred at room temperature for 4 h, then filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel to give (3-Hydroxyquinoxalin-6-yl)(piperidin-1-yl)methanone (67) as brown oil (1.28 g, 83% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.50 (s, 1H), 8.20 (s, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.26-7.24 (m, 2H), 3.58 (s, 4H), 1.65-1.43 (m, 6H). MS (ES+) m/z = 258 [M+H]+.
Step 5. (3-Hydroxyquinoxalin-6-yl)(piperidin-1-yl)methanone (67, 1.03 g, 4 mmol) was dissolved in dichloromethane (10 mL). The mixture was placed in an ice-bath and Et3N (1.12 mL, 8 mmol) and Tf2O (1.01 mL, 6 mmol) were added to the solution slowly. After the addition of Tf2O, the reaction was stirred at 0 °C for 2 h, and then the ice-bath was removed. The mixture was stirred at room temperature for an additional 3 h. The solvent was removed under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel to give 7-(Piperidine-1-carbonyl)quinoxalin-2-yl trifluoromethanesulfonate (68) as brown oil (949 mg, 61% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.79 (s, 1H), 8.22 (d, J = 8.6 Hz, 1H), 8.04 (s, 1H), 7.85 (d, J = 8.6 Hz, 1H), 3.75 (s, 2H), 3.35 (s, 2H), 1.69 (s, 4H), 1.53 (s, 2H). MS (ES+) m/z = 390 [M+H]+.
Step 6. Under nitrogen atmosphere, boronic acid or boronic ester (0.11 mmol) was added to a dry tube containing 7-(Piperidine-1-carbonyl)quinoxalin-2-yl trifluoromethanesulfonate (68, 39 mg, 0.1 mmol), Pd(dppf)Cl2 (4 mg, 0.5% mmol), K3PO4 (42 mg, 0.2 mmol), 1,4-dioxane (2 mL) and H2O (0.2 mL). The mixture was stirring at 100 °C until the completion of the reaction, which was monitored by HPLC/MS. The reaction mixture was cooled to room temperature, washed with water, extracted with ethyl acetate, dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The crude reaction mixture was purified by flash chromatography on silica gel to give the products as described below.
(3-Phenylquinoxalin-6-yl)(piperidin-1-yl)methanone (25) was synthesized according to General Procedure B in 89% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.36 (s, 1H), 8.22-8.15 (m, 4H), 7.77 (d, J = 8.5 Hz, 1H), 7.61-7.52 (m, 3H), 3.79 (s, 2H), 3.42 (s, 2H), 1.72 (s, 4H), 1.57 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.9, 152.6, 144.1, 141.7, 141.6, 138.2, 136.4, 130.5, 129.7, 129.2, 128.2, 127.6, 48.9, 43.3, 26.6, 25.6, 24.5. (missing 1 signal due to overlap). MS (ES+) m/z = 318 [M+H)+.
(3-(4-Chlorophenyl)quinoxalin-6-yl)(piperidin-1-yl)methanone (26) was synthesized according to General Procedure B in 56% yield as a yellow solid in approximately 90% purity. 1H NMR (400 MHz, Chloroform-d) δ 9.33 (s, 1H), 8.18-8.14 (m, 3H), 7.78 (d, J = Hz, 1H), 7.56 (d, J = 8.2 Hz, 2H), 7.26 (s, 1H), 3.80 (s, 2H), 3.43 (s, 2H), 1.73 (s, 4H), 1.58 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.8, 151.3, 143.6, 141.7, 138.4, 136.9, 134.8, 129.8, 129.5, 129.1, 128.8, 128.4, 127.6, 48.8, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 352 [M+H]+.
Methyl 4-(7-(piperidine-1-carbonyl) quinoxalin-2-yl)benzoate (27) was synthesized according to General Procedure B in 74% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.31 (s, 1H), 8.21 (d, J = 8.4 Hz, 2H), 8.14 (d, J = 8.4 Hz, 2H), 8.11-8.09 (m, 2H), 7.73 (dd, J = 8.7, 1.8 Hz, 1H), 3.89 (s, 3H), 3.73 (s, 2H), 3.37 (s, 2H), 1.66 (s, 4H), 1.51 (s, 2H); 13C NMR (101 MHz, cdcl3) δ 168.6, 166.4, 151.2, 143.9, 141.8, 141.6, 140.3, 138.4, 131.6, 130.3, 129.7, 128.7, 127.6, 127.4, 52.3, 48.8, 43.2, 26.6, 25.6, 24.5. MS (ES+) m/z = 376 [M+H)+.
(3-(4-(Morpholine-4-carbonyl)phenyl)quinoxalin-6-yl)(piperidin-1-yl)methanone (28) was synthesized from ester 27. Hydrolysis with NaOH (5 mL of 1 M) in MeOH (5 mL) followed the same procedure as used to synthesize 38. The intermediate acid (0.1 mmol) was dissolved in DMF (3 mL), and HATU (0.12 mmol), i-Pr2NEt (0.15 mmol) and morpholine (0.12 mmol) were added. After stirring for 3h, water was added, and the resulting solid was recovered by filtration and dried under vacuum overnight to provide 28 in 74% yield as a white solid. 1H NMR (400 MHz, Acetone-d6) δ 9.60 (s, 1H), 8.44 (d, J = 6.8 Hz, 2H), 8.17 (d, J = 8.5 Hz, 1H), 8.11 (s, 1H), 7.83 (d, J = 8.7 Hz, 1H), 7.68 (d, J = 7.9 Hz, 2H), 3.75-3.47 (m, 10H), 2.93 (s, 2H), 1.73-1.60 (m, 6H); 13C NMR (101 MHz, acetone-d6) δ 168.8, 167.9, 151.4, 144.2, 141.7, 141.5, 139.1, 138.0, 137.4, 129.5, 128.5, 128.0, 127.6, 127.3, 66.5, 54.6, 48.3, 42.6, 26.3, 25.4, 24.4. MS (ES+) m/z = 431 [M+H)+.
4-(7-(Piperidine-1-carbonyl)quinoxalin-2-yl)benzamide (29) was synthesized analogously to amide 28 in 77% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.69 (s, 1H), 8.43 (d, J = 8.1 Hz, 2H), 8.19-8.15 (m, 2H), 8.10-8.07 (m, 3H), 7.81 (dd, J = 8.4, 1.8 Hz, 1H), 7.53 (s, 1H), 3.65 (s, 2H), 3.38 (s, 2H), 1.62 (s, 4H), 1.49 (s, 2H); 13C NMR (101 MHz, dmso) δ 168.0, 167.7, 151.4, 145.1, 141.6, 141.3, 139.0, 138.6, 136.3, 129.9, 129.0, 128.7, 127.9, 127.3, 48.5, 42.9, 26.4, 25.7, 24.5. MS (ES+) m/z = 361 [M+H)+.
Piperidin-1-yl(3-(pyridin-2-yl)quinoxalin-6-yl)methanone (30) was synthesized according to General Procedure B in 37% yield as a brown solid. 1H NMR (400 MHz, Chloroform-d) δ 9.99 (s, 1H), 8.79 (d, J = 4.7 Hz, 1H), 8.59 (d, J = 8.0 Hz, 1H), 8.20-8.17 (m, 2H), 7.91 (t, J = 7.7 Hz, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.43 (dd, J = 7.5, 4.9 Hz, 1H), 3.80 (s, 2H), 3.43 (s, 2H), 1.73-58 (m, 6H); 13C NMR (101 MHz, Chloroform-d) δ 168.9, 154.2, 150.9, 149.5, 145.0, 142.6, 141.2, 138.1, 137.2, 129.9, 128.6, 127.7, 124.9, 122.2, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 319 [M+H)+.
Piperidin-1-yl(3-(pyridin-3-yl)quinoxalin-6-yl)methanone (31) was synthesized according to General Procedure B in 46% yield as a brown solid. 1H NMR (400 MHz, Chloroform-d) δ 9.43 (s, 1H), 9.38 (s, 1H), 8.78 (d, J = 4.6 Hz, 1H), 8.54 (d, J = 8.0 Hz,1H), 8.20-8.17 (m, 2H), 7.81 (dd, J = 8.5, 1.8 Hz, 1H), 7.53 (dd, J = 8.0, 4.8 Hz, 1H), 3.80 (s, 2H), 3.43 (s, 2H), 1.74-1.62 (m, 6H); 13C NMR (101 MHz, Chloroform-d) δ 168.7, 151.2, 150.2, 148.7, 143.5, 141.9, 141.8, 138.6, 135.0, 132.2, 129.9, 128.8, 127.6, 124.0, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 319 [M+H)+.
Piperidin-1-yl(3-(pyridin-4-yl)quinoxalin-6-yl)methanone (32) was synthesized according to General Procedure B in 47% yield as a brown solid in approximately 85% purity. 1H NMR (400 MHz, Chloroform-d) δ 9.38 (s, 1H), 8.84 (d, J = 5.3 Hz, 2H), 8.18 (d, J = 9.0 Hz, 1H), 8.17 (s, 1H), 8.09 (d, J = 5.7 Hz, 2H), 7.82 (dd, J = 8.5, 1.8 Hz, 1H), 3.79 (s, 2H), 3.41 (s, 2H), 1.72 (s, 4H), 1.57 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.6, 150.8, 149.9, 143.5, 143.4, 142.4, 141.7, 138.7, 129.9, 129.3, 127.8, 121.4, 48.8, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 319 [M+H)+.
(3-(6-Methoxypyridin-3-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (33). First, (3-(6-Fluoropyridin-3-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone was synthesized according to General Procedure 2 in 52 % yield (IC50 = 3 nM). 1H NMR (400 MHz, Chloroform-d) δ 9.35 (s, 1H), 9.05 (d, J = 2.5 Hz, 1H), 8.68 (td, J = 8.1, 2.6 Hz, 1H), 8.20-8.15 (m, 2H), 7.81 (dd, J = 8.6, 1.8 Hz, 1H), 7.16 (dd, J = 8.6, 3.0 Hz, 1H), 3.80 (s, 2H), 3.42 (s, 2H), 1.74 (s, 4H), 1.58 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.6, 164.7 (d, J = 243.5 Hz), 149.0, 147.0 (d, J = 15.7 Hz), 143.0, 141.8, 141.6, 140.4 (d, J = 8.6 Hz), 138.7, 130.4 (d, J = 4.7 Hz), 129.8, 128.8, 127.5, 110.3 (d, J = 37.7 Hz), 48.8, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 337 [M+H)+.
Next, sodium methoxide (0.3 mmol) was added to a solution of (3-(6-Fluoropyridin-3-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (34 mg, 0.1 mmol) in 1,4-dioxane (2 mL). The mixture was heated at 100 °C until the completion of the reaction, which was monitored by LC/MS, and the solvent was removed by reduced pressure. The resulting residue was purified by flash chromatography on silica gel to give the product methyl ether 33 as a light brown solid in 63% yield. 1H NMR (400 MHz, Chloroform-d) δ 9.30 (s, 1H), 8.98 (d, J = 1.8 Hz, 1H), 8.46 (dd, J = 8.7, 2.5 Hz, 1H), 8.13 (d, J = 8.5 Hz, 1H), 8.10 (d, J = 1.3 Hz, 1H), 7.74 (dd, J = 8.5, 1.8 Hz, 1H), 6.93 (d, J = 9.1 Hz, 1H), 4.03 (s, 3H), 3.78 (s, 2H), 3.41 (s, 2H), 1.72 (s, 4H), 1.56 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.8, 165.5, 150.3, 146.5, 143.2, 141.8, 141.5, 138.4, 137.7, 129.7, 128.1, 127.4, 125.7, 111.7, 53.9, 48.8, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 349.1 [M+H]+.
(3-(6-Aminopyridin-3-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (34) was synthesized analogously to compound 33 in 71% yield using as solution of ammonia in methanol. 1H NMR (400 MHz, Chloroform-d) δ 9.25 (s, 1H), 8.89 (s, 1H), 8.30 (dd, J = 8.7, 2.3 Hz, 1H), 8.10 (d, J = 8.5 Hz, 1H), 8.06 (s, 1H), 7.70 (d, J = 9.3 Hz, 1H), 6.64 (d, J = 8.7 Hz, 1H), 5.00 (s, 2H), 3.78 (s, 2H), 3.40 (s, 2H), 1.71 (s, 4H), 1.55 (s, 2H); 13C NMR (101 MHz, cdcl3) δ 169.0, 159.7, 150.6, 147.8, 143.1, 141.8, 141.2, 138.2, 136.8, 129.7, 127.6, 127.2, 122.5, 108.9, 48.8, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 334 [M+H]+.
(3-(6-(Methylamino)pyridin-3-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (35) was synthesized analogously to compound 33 in 71% yield using as solution of methyl amine in methanol. 1H NMR (400 MHz, Chloroform-d) δ 9.25 (s, 1H), 8.93 (s, 1H), 8.31 (d, J = 8.9 Hz, 1H), 8.08 (d, J = 8.4 Hz, 1H), 8.05 (s, 1H), 7.67 (d, J = 8.5 Hz, 1H), 6.53 (d, J = 8.8 Hz, 1H), 5.15 (d, J = 4.3 Hz, 1H), 3.77 (s, 2H), 3.40 (s, 2H), 3.00 (d, J = 4.3 Hz, 3H), 1.70 (s, 4H), 1.55 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 169.0, 160.4, 150.9, 148.0, 143.1, 141.9, 141.1, 138.1, 136.3, 129.6, 127.3, 127.2, 121.3, 106.7, 48.8, 43.2, 29.0, 26.6, 25.6, 24.5. MS (ES+) m/z = 348 [M+H]+.
(3-(6-(Dimethylamino)pyridin-3-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (36) was synthesized analogously to compound 33 in 74% yield using as solution of dimethyl amine in methanol. 1H NMR (400 MHz, Chloroform-d) δ 9.25 (s, 1H), 8.99 (d, J = 2.2 Hz, 1H), 8.33 (dd, J = 9.0, 2.5 Hz, 1H), 8.06 (d, J = 8.5 Hz, 1H), 8.03 (d, J = 1.6 Hz, 1H), 7.65 (dd, J = 8.5, 1.8 Hz, 1H), 6.64 (d, J = 9.0 Hz, 1H), 3.76 (s, 2H), 3.39 (s, 2H), 3.17 (s, 6H), 1.70 (s, 4H), 1.54 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 169.1, 159.8, 151.0, 147.7, 143.1, 141.9, 141.0, 138.0, 136.0, 129.6, 127.1, 127.1, 119.8, 106.0, 48.8, 43.2, 38.1, 26.6, 25.6, 24.5. MS (ES+) m/z = 362 [M+H]+.
Methyl 5-(7-(piperidine-1-carbonyl)quinoxalin-2-yl)picolinate (37) was synthesized according to General Procedure B in 54% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 9.54 (s, 1H), 8.79 (d, J = 8.2 Hz, 1H), 8.17 (d, J = 8.2 Hz, 1H), 8.13 (d, J = 8.5 Hz, 1H), 8.06 (s, 1H), 7.81 (d, J = 8.5 Hz, 1H), 3.90 (s, 3H), 3.64 (s, 2H), 3.32 (s, 2H), 1.60 (s, 4H), 1.48 (s, 2H); 13C NMR (101 MHz, dmso) δ 167.8, 165.2, 149.2, 149.0, 148.7, 145.0, 141.8, 141.2, 139.1, 136.5, 134.6, 129.8, 129.5, 127.4, 125.4, 53.0, 48.5, 42.9, 26.4, 25.7, 24.5. MS (ES+) m/z = 377 [M+H]+.
5-(7-(Piperidine-1-carbonyl)quinoxalin-2-yl)picolinic acid (38). An aqueous solution of NaOH (5 mL, 1M) was added to a solution of ester 37 in MeOH (5 mL). The solution was stirred at room temperature overnight. The solvent was then removed and the residue was acidified by 1M HCl until pH 2. The precipitate was collected by filtration, and washed with cold water. The resulting solid was dried to obtain the pure product 38 as a white solid (83 mg, 76% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 9.41 (s, 1H), 8.72 (d, J = 7.8 Hz, 1H), 8.20-8.14 (m, 2H), 8.07 (s, 1H), 7.83 (d, J = 8.6 Hz, 1H), 3.65 (s, 2H), 3.31 – 3.18 (m, 2H), 1.63 (s, 4H), 1.49 (s, 2H); 13C NMR (101 MHz, dmso) δ 167.9, 167.8, 158.6, 150.4, 148.0, 145.0, 141.6, 141.3, 139.1, 136.0, 131.6, 129.9, 129.1, 127.2, 124.0, 48.5, 42.9, 26.4, 25.7, 24.5. MS (ES+) m/z = 363 [M+H]+.
(3-(6-Morpholinopyridin-3-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone(39) was synthesized analogously to compound 33 in 78% yield using morpholine. 1H NMR (400 MHz, Chloroform-d) δ 9.27 (s, 1H), 9.00 (d, J = 2.4 Hz, 1H), 8.38 (dd, J = 9.0, 2.4 Hz, 1H), 8.10-8.05 (m, 2H), 7.68 (d, J = 8.5 Hz, 1H), 6.76 (d, J = 8.9 Hz, 1H), 3.83 (t, J = 4.8 Hz, 4H), 3.77 (s, 2H), 3.65 (t, J = 4.9 Hz, 4H), 3.40 (s, 2H), 1.70 (s, 4H), 1.55 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 169.0, 159.8, 150.6, 147.6, 143.1, 141.9, 141.2, 138.2, 136.5, 129.6, 127.4, 127.2, 121.8, 106.7, 66.6, 48.8, 45.1, 43.2, 26.6, 25.6, 24.5. MS (ES+) m/z = 404.1 [M+H]+.
(3-(6-Amino-5-chloropyridin-3-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (40) was synthesized according to General Procedure B in 64% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.26 (s, 1H), 8.80 (d, J = 2.1 Hz, 1H), 8.44 (d, J = 2.0 Hz, 1H), 8.12 (d, J = 8.5 Hz, 1H), 8.07 (d, J = 1.8 Hz, 1H), 7.73 (dd, J = 8.5, 1.8 Hz, 1H), 5.36 (s, 2H), 3.79 (s, 2H), 3.41 (s, 2H), 1.72 (s, 4H), 1.57 (s, 2H); 13C NMR (101 MHz, cdcl3) δ 168.8, 155.9, 149.4, 145.5, 142.8, 141.7, 141.4, 138.4, 135.6, 129.7, 128.0, 127.2, 123.9, 115.7, 48.8, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 368.0 [M+H]+.
Piperidin-1-yl(3-(pyrimidin-5-yl)quinoxalin-6-yl)methanone (41) was synthesized according to General Procedure B in 61% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.55 (s, 2H), 9.38 (s, 2H), 8.22-8.18 (m, 2H), 7.85 (dd, J = 8.6, 1.8 Hz, 1H), 3.80 (s, 2H), 3.42 (s, 2H), 1.73 (s, 4H), 1.59 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.4, 159.6, 155.6, 147.5, 142.8, 142.2, 141.8, 139.0, 130.0, 129.4, 127.7, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 320 [M+H]+.
(3-(2-Aminopyrimidin-5-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (42) was synthesized according to General Procedure B in 52% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.24 (s, 1H), 9.15 (s, 2H), 8.14 (d, J = 8.5 Hz, 1H), 8.10 (s, 1H), 7.75 (d, J = 8.5 Hz, 1H), 5.60 (s, 2H), 3.79 (s, 2H), 3.42 (s, 2H), 1.72 (s, 4H), 1.58 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.8, 163.4, 157.6, 148.5, 142.3, 141.7, 141.6, 138.5, 129.8, 128.1, 127.3, 120.6, 48.8, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 335 [M+H]+.
Piperidin-1-yl(3-(quinolin-6-yl)quinoxalin-6-yl)methanone (43) was synthesized according to General Procedure B in 77% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.52 (s, 1H), 9.00 (s, 1H), 8.67 (s, 1H), 8.61 (dd, J = 8.8, 2.0 Hz, 1H), 8.34 (d, J = 8.3 Hz, 1H), 8.30 (d, J = 8.9 Hz, 1H), 8.20 (s, 1H), 8.18 (d, J = 5.6 Hz, 1H), 7.80 (d, J = 8.7 Hz, 1H), 7.51 (dd, J = 8.3, 4.1 Hz, 1H), 3.80 (s, 2H), 3.44 (s, 2H), 1.73 (s, 4H), 1.58 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.8, 151.7, 151.6, 149.0, 144.0, 141.8, 141.7, 138.5, 137.0, 134.5, 130.6, 129.8, 128.6, 128.1, 127.6, 127.4, 122.0, 121.9, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 369 [M+H]+.
(3-(Isoquinolin-6-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (44) was synthesized according to General Procedure B in 76% yield as a yellow solid in approximately 90% purity. 1H NMR (400 MHz, Chloroform-d) δ 9.51 (s, 1H), 9.35 (s, 1H), 8.63 (s, 1H), 8.62 (d, J = 6.1 Hz, 1H), 8.49 (dd, J = 8.6, 1.7 Hz, 1H), 8.20-8.16 (m, 3H), 7.83-7.80 (m, 2H), 3.80 (s, 2H), 3.43 (s, 2H), 1.73 (s, 4H), 1.58 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.7, 152.5, 151.4, 144.1, 144.0, 141.9, 141.7, 138.6, 138.0, 135.8, 129.8, 128.92, 128.85, 128.7, 127.7, 126.02, 125.98, 121.1, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 369 [M+H]+.
(3-(2-Methoxyquinolin-6-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (45). NaOMe (27 mg, 0.5 mmol) was added to a solution of (3-(2-Chloroquinolin-6-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (51, 40 mg, 0.1 mmol) in 1,4-dioxane (2 mL). The mixture was heated at 100 °C for 24 h. The solvent was removed under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel to give the product 45 as a pale yellow solid (32% yield). 1H NMR (400 MHz, Chloroform-d) δ 9.48 (s, 1H), 8.57 (s, 1H), 8.50 (d, J = 8.8 Hz, 1H), 8.18-8.13 (m, 3H), 8.03 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 8.9 Hz, 1H), 4.12 (s, 3H), 3.80 (s, 2H), 3.44 (s, 2H), 1.73 (s, 4H), 1.59 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.9, 163.4, 152.0, 147.9, 144.0, 141.8, 141.5, 139.3, 138.3, 131.9, 129.8, 128.3, 128.2, 128.2, 127.5, 127.1, 125.1, 114.1, 53.7, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 399.0 [M+H]+.
(3-(1-Methoxyisoquinolin-6-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (46). First, (3-(1-Chloroisoquinolin-6-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone was synthesized according to General Procedure B in 75 % yield as a yellow solid. (IC50 = 6.4 nM) 1H NMR (400 MHz, Chloroform-d) δ 9.52 (s, 1H), 8.67 (s, 1H), 8.58-8.51 (m, 2H), 8.38 (d, J = 5.6 Hz, 1H), 8.22-8.20 (m, 2H), 7.84 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 5.6 Hz, 1H), 3.81 (s, 2H), 3.44 (s, 2H), 1.74 (s, 4H), 1.59 (s, 2H); 13C NMR (101 MHz, cdcl3) δ 168.6, 151.6, 150.7, 143.8, 142.4, 141.9, 141.7, 138.7, 138.7, 137.8, 129.8, 129.0, 127.7, 127.5, 127.3, 127.2, 126.2, 121.3, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 403 [M+H]+.
Next, the chloroisoquinoline was converted to methyl ether 46 analogously to the synthesis of 45 as a pale yellow solid in 35% yield. 1H NMR (400 MHz, Chloroform-d) δ 9.50 (s, 1H), 8.56 (s, 1H), 8.44 (d, J = 8.7 Hz, 1H), 8.39 (d, J = 8.7 Hz, 1H), 8.21-8.19 (m, 2H), 8.10 (d, J = 5.8 Hz, 1H), 7.82 (d, J = 8.7 Hz, 1H), 7.38 (d, J = 5.9 Hz, 1H), 4.18 (s, 3H), 3.81 (s, 2H), 3.44 (s, 2H), 1.74 (s, 4H), 1.58 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.8, 161.0, 151.7, 144.1, 141.9, 141.7, 140.7, 138.5, 138.1, 138.0, 129.8, 128.7, 127.7, 125.6, 125.4, 125.3, 120.4, 115.4, 53.9, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 399 [M+H]+.
6-(7-(Piperidine-1-carbonyl)quinoxalin-2-yl)quinolin-2(1H)-one (47) was synthesized according to General Procedure B in 61% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 12.00 (s, 1H), 9.63 (s, 1H), 8.68 (s, 1H), 8.47 (d, J = 8.6 Hz, 1H), 8.13 (d, J = 8.5 Hz, 1H), 8.04 (d, J = 8.9 Hz, 1H), 8.02 (s, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.47 (d, J = 8.7 Hz, 1H), 6.58 (d, J = 9.5 Hz, 1H), 3.64 (s, 2H), 3.32 (s, 2H), 1.61 (s, 4H), 1.48 (s, 2H); 13C NMR (101 MHz, dmso) δ 168.0, 162.4, 151.4, 144.8, 141.3, 141.2, 140.9, 140.9, 138.9, 129.8, 129.7, 129.7, 128.4, 127.9, 127.0, 123.1, 119.8, 116.5, 48.5, 42.9, 26.4, 25.7, 24.5. MS (ES+) m/z = 385 [M+H]+.
6-(7-(Piperidine-1-carbonyl)quinoxalin-2-yl)isoquinolin-1(2H)-one (48) was synthesized according to General Procedure B in 60% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.40 (s, 1H), 9.73 (s, 1H), 8.64 (s, 1H), 8.41 (d, J = 7.7 Hz, 1H), 8.36 (d, J = 8.4 Hz, 1H), 8.20 (d, J = 8.5 Hz, 1H), 8.11 (s, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.26 (t, J = 6.4 Hz, 1H), 6.71 (d, J = 7.2 Hz, 1H), 3.66 (s, 2H), 3.27 (s, 2H), 1.62 (s, 4H), 1.49 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 167.9, 162.0, 151.4, 145.2, 141.7, 141.3, 139.5, 139.1, 138.8, 130.3, 129.9, 129.2, 128.1, 127.5, 127.3, 126.1, 125.4, 105.3, 48.5, 42.9, 26.4, 25.7, 24.5. MS (ES+) m/z = 385 [M+H]+.
2-Methyl-6-(7-(piperidine-1-carbonyl)quinoxalin-2-yl)isoquinolin-1(2H)-one (49) was synthesized according to General Procedure B in 82% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 8.66 (d, J = 1.6 Hz, 1H), 8.43 (dd, J = 8.5, 1.7 Hz, 1H), 8.40 (d, J = 8.4 Hz, 1H), 8.20 (d, J = 8.5 Hz, 1H), 8.11 (d, J = 1.8 Hz, 1H), 7.83 (dd, J = 8.5, 1.8 Hz, 1H), 7.57 (d, J = 7.3 Hz, 1H), 6.78 (d, J = 7.3 Hz, 1H), 3.66 (s, 2H), 3.54 (s, 3H), 3.34 (s, 2H), 1.71 – 1.56 (m, 4H), 1.49 (s, 2H). 13C NMR (101 MHz, CDCl3 δ 168.7, 162.3, 151.6, 143.9, 141.8, 141.7, 139.5, 138.6, 137.7, 133.4, 129.8, 128.9, 128.8, 127.7, 127.1, 125.5, 125.3, 106.1, 48.9, 43.3, 37.2, 26.6, 25.6, 24.5.MS (ES+) m/z = 399.1 [M+H]+. A single crystal suitable for X-ray diffraction was grown by dissolving ca. 5 mg of 49 in 0.5 mL of EtOH with heating. The hot solution was filtered then allowed to cool to rt and stand at that temperature for 12 h. The solution was moved to a cold room (4 °C), which resulted in the formation of single crystals. CCDC 2179639.
Ethyl 2-(1-oxo-6-(7-(piperidine-1-carbonyl)quinoxalin-2-yl)isoquinolin-2(1H)-yl)acetate (50). To a suspension of isoquinolin-1(2H)-one 48 (76 mg, 0.2 mmol) in acetone were added K2CO3 (139 mg, 1 mmol) and ethyl 2-bromoacetate (0.03 mL, 0.3 mmol) successively at r.t. The mixture was stirred at r.t. overnight, and the solid was removed by filtration and washed with acetone. The filtrate was concentrated under reduced pressure, and the corresponding crude mixture was purified by flash chromatography on silica gel to obtain the product 50 (77 mg, 82% yield) as a light-yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.45 (s, 1H), 8.60 (d, J = 8.4 Hz, 1H), 8.37 (s, 1H), 8.31 (d, J = 8.6 Hz, 1H), 8.20-8.15 (m, 2H), 7.81 (d, J = 8.8 Hz, 1H), 7.10 (d, J = 7.3 Hz, 1H), 6.69 (d, J = 7.3 Hz, 1H), 4.75 (s, 2H), 4.27 (q, J = 7.1 Hz, 2H), 3.80 (s, 2H), 3.43 (s, 2H), 1.73 (s, 4H), 1.58 (s, 2H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, cdcl3) δ 168.7, 168.0, 161.9, 151.4, 144.0, 141.9, 141.7, 140.0, 138.5, 137.8, 132.7, 129.8, 129.1, 128.9, 127.7, 126.9, 125.7, 125.3, 106.5, 61.9, 50.3, 48.9, 43.3, 26.6, 25.6, 24.5, 14.1. MS (ES+) m/z = 399 [M+H]+.
(3-(2-Chloroquinolin-6-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (51) was synthesized according to General Procedure B in 77% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.50 (s, 1H), 8.67 (s, 1H), 8.64 (d, J = 9.0 Hz, 1H), 8.29 (d, J = 8.5 Hz, 1H), 8.23-8.19 (m, 3H), 7.81 (d, J = 8.9 Hz, 1H), 7.49 (d, J = 8.6 Hz, 1H), 3.81 (s, 2H), 3.44 (s, 2H), 1.74 (s, 4H), 1.59 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.7, 152.0, 151.1, 148.6, 143.8, 141.7, 141.7, 139.5, 138.6, 134.8, 129.8, 129.6, 129.2, 128.7, 127.6, 127.0, 126.8, 123.3, 48.9, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 403 [M+H]+.
(3-(1H-Indazol-5-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (52) was synthesized according to General Procedure B in 51% yield as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 13.33 (s, 1H), 9.68 (d, J = 1.5 Hz, 1H), 8.81 (s, 1H), 8.37 (d, J = 8.9 Hz, 1H), 8.25 (s, 1H), 8.13 (d, J = 7.9 Hz, 1H), 8.03 (s, 1H), 7.76-7.72 (m, 2H), 3.65 (s, 2H), 3.33 (s, 2H), 1.61 (s, 4H), 1.48 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 168.1, 152.7, 145.1, 141.4, 141.1, 141.1, 138.8, 135.4, 129.7, 128.8, 128.2, 127.0, 125.9, 123.8, 121.4, 111.4, 48.5, 42.9, 26.4, 25.7, 24.5. MS (ES+) m/z = 358 [M+H]+.
Piperidin-1-yl(3-(thiazol-2-yl)quinoxalin-6-yl)methanone (53) was synthesized according to General Procedure B in 45% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.77 (s, 1H), 8.18 (d, J = 8.5 Hz, 1H), 8.13 (s, 1H), 8.07 (d, J = 2.9 Hz, 1H), 7.80 (d, J = 8.5 Hz, 1H), 7.60 (d, J = 3.0 Hz, 1H), 3.80 (s, 2H), 3.42 (s, 2H), 1.73 (s, 4H), 1.59 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.6, 166.6, 146.5, 144.8, 143.4, 142.8, 141.2, 138.6, 130.0, 129.0, 127.3, 123.1, 48.9, 43.3, 29.7, 26.6, 24.5. MS (ES+) m/z = 325 [M+H]+.
(3-(1,3-Dimethyl-1H-pyrazol-4-yl)quinoxalin-6-yl)(piperidin-1-yl)methanone (54) was synthesized according to General Procedure B in 38% yield as a yellow solid. 1H NMR (400 MHz, Chloroform-d) δ 9.02 (s, 1H), 8.06 (d, J = 8.3 Hz, 1H), 8.02 (s, 1H), 8.00 (s, 1H), 7.66 (d, J = 8.3 Hz, 1H), 3.94 (s, 3H), 3.77 (s, 2H), 3.40 (s, 2H), 2.67 (s, 3H), 1.71 (s, 4H), 1.55 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 169.1, 149.1, 148.7, 144.4, 141.9, 140.4, 138.1, 131.1, 129.6, 127.1, 127.0, 117.7, 48.8, 43.2, 39.0, 26.6, 25.6, 24.5, 14.5. MS (ES+) m/z = 336 [M+H]+.
Piperidin-1-yl(quinoxalin-6-yl)methanone (55). To a solution of 7-(piperidine-1-carbonyl)quinoxalin-2-yl trifluoromethanesulfonate (68, 39 mg, 0.1 mmol) in MeOH (2 mL) was added palladium on carbon (4 mg, 10% wt.%). The mixture was stirred at r.t. under hydrogen atmosphere for 2 h. The Pd/C was removed by filtration, and the resulting filtrate was concentrated under reduced pressure. The crude residue was purified by flash chromatography on silica gel to provide the product 55 (14 mg, 58% yield) as colorless oil. 1H NMR (400 MHz, Chloroform-d) δ 8.89 (d, J = 1.3 Hz, 2H), 8.16 (d, J = 8.6 Hz, 1H), 8.12 (d, J = 1.8 Hz, 1H), 7.81 (ddJ = 8.6, 1.8 Hz, 1H), 3.79 (s, 2H), 3.40 (s, 2H), 1.72 (s, 4H), 1.56 (s, 2H); 13C NMR (101 MHz, cdcl3) δ 143.1, 142.5, 138.1, 130.1, 128.8, 127.6, 48.8, 43.3, 26.6, 25.6, 24.5. MS (ES+) m/z = 242 [M+H]+.
6-(Butylsulfinyl)-2,3-diphenylquinoxaline (56). Step 1. To a solution of butane-1-thiol (990 mg, 11 mmol) in DMF (20 mL) was added NaH (480 mg, 12 mmol, 60% in mineral oil) slowly at 0 °C. The mixture was stirred at 0 °C for 30 min and 5-chloro-2-nitroaniline (1.73 g, 10 mmol) was added to the solution. The reaction was heated at 40 °C for an additional 2.5 h. The mixture was cooled to room temperature and quenched with water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4 and concentrated by reduced pressure. The crude mixture was purified by flash chromatography on silica gel to give the 5-(Butylthio)-2-nitroaniline (69) as a brown solid (1.97 g, 87% yield). MS (ES+) m/z = 227 [M+H]+.
Step 2. To a solution of nitroarene 69 (1.81 g, 8 mmol) in acetone (16 mL) was added Zn powder (2.6 g, 40 mmol), ammonium chloride (2.14 g, 40 mmol) and water (2 mL). The mixture was stirred at reflux for 30 min, then filtered and concentrated. The crude mixture was dissolved in MeOH (16 mL), and benzil (1.68 g, 8 mmol) was added. The mixture was stirred at room temperature for 5 h. The solvent was removed, and the residue was diluted with ethyl acetate, washed with water, brine and dried over Na2SO4 and concentrated under reduced pressure. The crude mixture was purified by flash chromatography on silica gel to give the quinoxaline 70 as a yellow solid (1.88 g, 64% yield). IC50 > 2500 nM. 1H NMR (400 MHz, Chloroform-d δ 8.03 (d, J = 8.8 Hz, 1H), 7.91 (s, 1H), 7.62 (dd, J = 8.8, 2.0 Hz, 1H), 7.51-7.48 (m, 4H), 7.37-7.30 (m, 6H), 3.12 (t, J = 7.4 Hz, 2H), 1.81-1.73 (m, 2H), 1.56-1.48 (m, 2H), 0.97 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ 152.3, 141.7, 141.4, 139.5, 139.1, 139.0, 130.2, 129.78, 129.76, 129.0, 128.8, 128.7, 128.3, 128.2, 123.7, 32.1, 30.6, 22.1, 13.7, (1 signal not observed). MS (ES+) m/z = 371 [M+H]+.
Step 3. Butyl(6,7-diphenylnaphthalen-2-yl)sulfane (70, 37 mg, 0.1 mmol) was dissolved in chloroform (2 mL). H2O2 (3.7 μL, 0.12 mmol) and AcOH (0.5 mL) were added. The reaction was monitored by LC/MS. After the completion of the reaction, NaHCO3 aq was added to quench the reaction. The mixture was extracted with dichloromethane, washed with brine, dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The crude mixture was purified by flash chromatography on silica gel to give the product 56 as a yellow solid (35 mg, 90% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.42 (s, 1H), 8.32 (d, J = 8.7 Hz, 1H), 7.98 (d, J = 8.7 Hz, 1H), 7.54-7.52 (m, 4H), 7.41-7.32 (m, 6H), 2.97-2.90 (m, 2H), 1.86-1.75 (m, 1H), 1.67-1.55 (m, 1H), 1.53-1.39 (m, 2H), 0.92 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, Chloroform-d) δ 154.9, 154.7, 145.8, 142.1, 140.6, 138.5, 138.4, 130.7, 129.8, 129.3, 128.38, 128.36, 125.8, 124.0, 56.7, 23.9, 21.9, 13.7, (missing 2 signals due to overlap). MS (ES+) m/z = 387 [M+H]+.
2-Phenyl-7-(piperidine-1-carbonyl)quinazolin-4(3H)-one (57). Step 1. To a solution of 4-(methoxycarbonyl)-3-nitrobenzoic acid (2.25 g, 10 mmol), HATU (4.2 g, 11 mmol), and i-Pr2NEt (3.5 mL, 20 mmol) in DMF (20 mL) was added Piperidine (1.09 mL, 11 mmol). The mixture was stirred at room temperature for 4 h. Water was added to the solution, and the mixture was extracted with ethyl acetate, washed with brine, dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The crude mixture was purified by flash chromatography on silica gel to give methyl 2-nitro-4-(piperidine-1-carbonyl)benzoate (71) as yellow oil (2.63 g, 90% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.94 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 3.94 (s, 3H), 3.72 (s, 2H), 3.32 (s, 2H), 1.71 (s, 4H), 1.54 (s, 2H). MS (ES+) m/z = 293 [M+H]+.
Step 2. To a solution of ester 71 (2.34 g, 8 mmol) in MeOH (18 mL) was added NH3 in MeOH (7N, 3.4 mL, 24 mmol), and the mixture was then heated at 100 °C for 12 h. The solvent was removed, the residue was dissolved in acetone (20 mL), and Zn powder (2.6 g, 40 mmol), ammonium chloride (2.14 g, 40 mmol) and water (2 mL) were added. The mixture was stirred at reflux for 1 h, then filtered and concentrated. The resulting residue was diluted with water and extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4 and concentrated by reduced pressure. The crude mixture was purified by flash chromatography on silica gel to give the 2-amino-4-(piperidine-1-carbonyl)benzamide (72) as brown oil (889 mg, 45% yield). MS (ES+) m/z = 248 [M+H]+.
Step 3. To a solution of amide 72 (247 mg, 1 mmol) in EtOH (5 mL) was added benzaldehyde (0.11 mL, 1.1 mmol) and I2 (279 mg, 1.1 mmol). The mixture was stirred at reflux for 5 h, and the solution was allowed to cool to room temperature. Sodium thiosulfate aqueous solution was added to quench excess I2’ and the resulting mixture was extracted with ethyl acetate. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4 and concentrated by reduced pressure. The crude was purified by flash chromatography on silica gel to give the 57 as a white solid (100 mg, 30% yield). 1H NMR (400 MHz, Chloroform-d) δ 12.10 (s, 1H), 8.35 (d, J = 8.1 Hz, 1H), 8.30-8.27 (m, 2H), 7.80 (d, J = 1.5 Hz, 1H), 7.62-7.57 (m, 3H), 7.49 (dd, J = 8.1, 1.5 Hz, 1H), 3.76 (s, 2H), 3.36 (s, 2H), 1.70 (s, 4H), 1.55 (s, 2H); 13C NMR (101 MHz, Chloroform-d) δ 168.7, 163.6, 152.6, 149.5, 142.9, 132.5, 131.9, 129.0, 127.6, 127.0, 125.8, 125.0, 121.1, 48.7, 43.2, 26.6, 25.6, 24.5. MS (ES+) m/z = 334 [M+H]+.
Step 4. 2-Phenyl-7-(piperidine-1-carbonyl)quinazolin-4-yl trifluoromethanesulfonate. To a solution of 2-phenyl-7-(piperidine-1-carbonyl)quinazolin-4(3H)-one (57, 333 mg, 1 mmol) in DCM (2 mL) were added DIPEA (0.26 mL, 1.5 mmol) and triflic anhydride (0.1 mL, 1.2 mmol) at 0 °C. The reaction mixture was stirred at this temperature for 7 h, and the crude solution was loaded to the column directly and purified by flash chromatography on silica gel to give the triflate as yellow oil (260 mg, 56% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.56 (dd, J = 6.1, 2.6 Hz, 2H), 8.14 (d, J = 8.6 Hz, 1H), 8.12 (s, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.57-7.53 (m, 3H), 3.80 (s, 2H), 3.37 (s, 2H), 1.74 (s, 4H), 1.58 (s, 2H).
(2-Phenylquinazolin-7-yl)(piperidin-1-yl)methanone (58). To a solution of 2-Phenyl-7-(piperidine-1-carbonyl)quinazolin-4-yl trifluoromethanesulfonate(46 mg, 0.1 mmol) in ethyl acetate (5 mL) were added palladium on carbon (5 mg, 10% wt.%) and Et3N (0.2 mL). The mixture was stirred at room temperature under a hydrogen atmosphere for 2 h, and then the Pd/C was removed by filtration. The resulting filtrate was concentrated under reduced pressure and the resulting residue was purified by flash chromatography on silica gel to provide the product quinazoline 58 (15 mg, 48% yield). 1H NMR (400 MHz, Chloroform-d) δ 9.49 (s, 1H), 8.62 (d, J = 7.5 Hz, 2H), 8.05 (s, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.63 (d, J = 8.2 Hz, 1H), 7.54 (d, J = 6.6 Hz, 3H), 3.79 (s, 2H), 3.39 (s, 2H), 1.73 (s, 4H), 1.57 (s, 2H); 13C NMR (101 MHz, cdcl3) δ 168.6, 161.8, 160.4, 150.4, 142.0, 137.7, 130.9, 128.7, 128.6, 127.8, 126.3, 125.9, 123.5, 48.8, 43.2, 26.6, 25.6, 24.5. MS (ES+) m/z = 318 [M+H]+.
(2-Phenyl-4-(pyridin-3-yl)quinazolin-7-yl)(piperidin-1-yl)methanone (59). Under nitrogen atmosphere, 3-pyridine boronic acid pinacol ester (0.11 mmol) was added to a dry tube containing 2-phenyl-7-(piperidine-1-carbonyl)quinazolin-4-yl trifluoromethanesulfonate (46 mg, 0.1 mmol), Pd(dppf)Cl2 (4 mg, 0.5% mmol), K3PO4 (42 mg, 0.2 mmol), 1,4-dioxane (2 mL) and H2O (0.2 mL). The mixture was stirring at 100 °C until the completion of the reaction, which was monitored by LC/MS. The reaction mixture was cooled to room temperature, washed with water, extracted with ethyl acetate, dried over anhydrous Na2SO4 and concentrated by rotary evaporation. The crude residue was purified by flash chromatography on silica gel to give 59 in 56% yield as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 8.61-8.58 (m, 1H), 8.68 – 8.52 (m, 2H), 8.35 (d, J = 7.7 Hz, 1H), 8.13 (d, J = 8.5 Hz, 1H), 8.06 (s, 1H), 7.71-7.65 (m, 2H), 7.59-7.56 (m, 3H), 3.65 (s, 2H), 3.31 (s, 2H), 1.62 (s, 4H), 1.48 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 167.6, 166.1, 160.1, 151.5, 151.3, 150.6, 142.8, 138.1, 137.5, 133.1, 131.6, 129.2, 128.7, 127.8, 126.8, 126.3, 124.2, 121.6, 48.4, 42.8, 26.4, 25.6, 24.4. MS (ES+) m/z = 395 [M+H]+.
(2-Phenyl-4-(pyridin-4-yl)quinazolin-7-yl)(piperidin-1-yl)methanone (60) was synthesized analogously to pyridine 59 in 64% yield as a white solid. 1H NMR (400 MHz, Chloroform-d) δ 8.89 (s, 2H), 8.68-8.65 (m, 2H), 8.14 (s, 1H), 8.07 (d, J = 8.5 Hz, 1H), 7.76 (d, J = 4.8 Hz, 2H), 7.60 (d, J = 8.5 Hz, 1H), 7.56-7.52 (m, 3H), 3.79 (s, 2H), 3.41 (s, 2H), 1.73 (s, 4H), 1.57 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 168.4, 165.8, 161.0, 151.7, 150.3, 144.7, 141.9, 137.4, 131.1, 128.7, 128.7, 127.1, 126.8, 126.1, 124.3, 121.1, 48.8, 43.2, 26.6, 25.6, 24.5. MS (ES+) m/z = 395 [M+H]+.
Supplementary Material
Funding Sources
Funding provided by the Welch Foundation (I-1612, J.M.R.), NIH (R01CA216863 RM1GM142002 to J.M.R, S.D.M; R35CA197442 to S.D.M.; R50CA211119 to S.F.).
ABBREVIATIONS
- AUC
area under the curve
- BID
bis in die (twice-daily dosing)
- BM
bone marrow
- cLogP
calculated log(partition coefficient)
- Cmax
maximum plasma concentration
- COX1/2
cyclooxygenase 1/2
- dmPGE2, 16
16-dimethyl prostaglandin E2
- DMSO
dimethyl sulfoxide
- DSS
dextran sodium sulfate
- ELISA
enzyme-linked immunosorbent assay
- HATU
hexafluorophosphate azabenzotriazole tetramethyl uronium
- HTS
high throughput screening
- IP
intraperitoneal
- IV
intravenous
- NAD+
nicotinamide adenine dinucleotide
- 15-PGDH
15-prostagandin dehydrogenase
- PGE2
prostaglandin E2
- PO
per os (oral dosing)
Footnotes
Case-Western Reserve and UT Southwestern hold joint patents on the compounds described herein, which have been licensed to Rodeo Therapeutics. B.H., M.A., A.D., S.D.M., and J.M.R. have financial interests in Rodeo Therapeutics.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Spectra and supplementary figures (PDF)
Molecular Forma String (CSV)
X-ray structure of 49 (cif)
REFERENCES
- (1).Hansen HS 15-Hydroxyprostaglandin dehydrogenase. A review. Prostaglandins 1976, 12, 647–679. [DOI] [PubMed] [Google Scholar]
- (2).Tai H-H; Ensor CM; Tong M; Zhou H; Yan F Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002, 68–69, 483–493. [DOI] [PubMed] [Google Scholar]
- (3).Tai HH; Cho H; Tong M; Ding Y NAD+-linked 15-hydroxyprostaglandin dehydrogenase: Structure and biological functions. Curr. Pharm. Des 2006, 12, 955–962. [DOI] [PubMed] [Google Scholar]
- (4).Kalish BT; Kieran MW; Puder M; Panigrahy D The growing role of eicosanoids in tissue regeneration, repair, and wound healing. Prostaglandins Other Lipid Mediat. 2013, 104–105, 130–138. [DOI] [PubMed] [Google Scholar]
- (5).Funk CD Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875.. [DOI] [PubMed] [Google Scholar]
- (6).Dey I; Lejeune M; Chadee K Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br. J. Pharmacol 2006, 149, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Goessling W; North TE; Loewer S; Lord AM; Lee S; Stoick-Cooper CL; Weidinger G; Puder M; Daley GQ; Moon RT; Zon LI Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009, 136, 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).North TE; Goessling W; Walkley CR; Lengerke C; Kopani KR; Lord AM; Weber GJ; Bowman TV; Jang I-H; Grosser T; FitzGerald GA; Daley GQ; Orkin SH; Zon LI Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007, 447, 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hoggatt J; Singh P; Sampath J; Pelus LM Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood 2009, 113, 5444–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Goessling W; Allen RS; Guan X; Jin P; Uchida N; Dovey M; Harris JM; Metzger ME; Bonifacino AC; Stroncek D; Stegner J; Armant M; Schlaeger T; Tisdale JF; Zon LI; Donahue RE; North TE Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell 2011, 8, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Pelus LM; Hoggatt J; Singh P Pulse exposure of haematopoietic grafts to prostaglandin E2 in vitro facilitates engraftment and recovery. Cell Proliferation 2011, 44, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cutler C; Multani P; Robbins D; Kim HT; Le T; Hoggatt J; Pelus LM; Desponts C; Chen Y-B; Rezner B; Armand P; Koreth J; Glotzbecker B; Ho VT; Alyea E; Isom M; Kao G; Armant M; Silberstein L; Hu P; Soiffer RJ; Scadden DT; Ritz J; Goessling W; North TE; Mendlein J; Ballen K; Zon LI; Antin JH; Shoemaker DD Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood 2013, 122, 3074–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jung P; Sato T; Merlos-Suarez A; Barriga FM; Iglesias M; Rossell D; Auer H; Gallardo M; Blasco MA; Sancho E; Clevers H; Batlle E Isolation and in vitro expansion of human colonic stem cells. Nat. Med 2011, 17, 1225–1227. [DOI] [PubMed] [Google Scholar]
- (14).Kabashima K; Saji T; Murata T; Nagamachi M; Matsuoka T; Segi E; Tsuboi K; Sugimoto Y; Kobayashi T; Miyachi Y; Ichikawa A; Narumiya S The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J. Clin. Invest 2002, 109, 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hamberg M; Samuelsson B On the metabolism of prostaglandins E1 and E2 in man. J. Biol. Chem 1971, 246, 6713–6721. [PubMed] [Google Scholar]
- (16).Ganesan PA; Karim SM Acute toxicity of prostaglandins E2, F2a and 15-(S)-15 methyl prostaglandin E2 methyl ester in the baboon. Prostaglandins 1974, 7, 215–221. [DOI] [PubMed] [Google Scholar]
- (17).Zhang Y; Desai A; Yang SY; Bae KB; Antczak MI; Fink SP; Tiwari S; Willis JE; Williams NS; Dawson DM; Wald D; Chen W-D; Wang Z; Kasturi L; Larusch GA; He L; Cominelli F; Di Martino L; Djuric Z; Milne GL; Chance M; Sanabria J; Dealwis C; Mikkola D; Naidoo J; Wei S; Tai H-H; Gerson SL; Ready JM; Posner B; Willson JKV; Markowitz SD Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science 2015, 348, aaa2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Markowitz S; Willson JKV; Posner BA; Ready J; Zhang Y; Tai H-H; Moss M; Antczak M Compositions and Methods of Modulating 15-Hydroxy-Prostaglandin Dehydrogenase. WO2013158649A1, 2013.
- (19).Markowitz S; Willson JKV; Posner B; Ready J; Antczak M; Zhang Y; Desai A; Gerson S; Greenlee W Preparation of Thienopyridines and Similar Bicyclic Heterocyclic Compounds as Modulators of 15-Hydroxyprostaglandin Dehydrogenase Type I for Therapy. WO2015065716A1, 2015.
- (20).Markowitz SD; Ready J; Zhang Y; Antczak M; Willson JKV; Posner BA; Greenlee W Compositions and Methods of Modulating Short-Chain Dehydrogenase Activity. WO2016168472A1, 2016.
- (21).Robert A; Schultz JR; Nezamis JE; Lancaster C Gastric antisecretory and antiulcer properties of PGE2, 15-methyl PGE2, and 16,16-dimethyl PGE2: Intravenous, oral and intrajejunal administration. Gastroenterology 1976, 70, 359–370.. [PubMed] [Google Scholar]
- (22).Nakase H; Fujiyama Y; Oshitani N; Oga T; Nonomura K; Matsuoka T; Esaki Y; Murayama T; Teramukai S; Chiba T; Narumiya S Effect of EP4 agonist (ONO-4819cd) for patients with mild to moderate ulcerative colitis refractory to 5-aminosalicylates: A randomized phase ii, placebo-controlled trial. Inflamm. Bowel Dis 2009, 16, 731–733. [DOI] [PubMed] [Google Scholar]
- (23).Nakatsuji M; Minami M; Seno H; Yasui M; Komekado H; Higuchi S; Fujikawa R; Nakanishi Y; Fukuda A; Kawada K; Sakai Y; Kita T; Libby P; Ikeuchi H; Yokode M; Chiba T EP4 receptor–associated protein in macrophages ameliorates colitis and colitis-associated tumorigenesis. PLOS Genetics 2015, 11, e1005542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Michelet JF; Bernard B; Rozot R; Boulle C Composition Comprising at Least one 15-PGDH Inhibitor. US20080206320 A1, 2008.
- (25).Rozot R; Boulle C; Dalko M Care/Makeup Compositions Comprising a 2-Alkylideneaminooxyacetamide Compound for Stimulating the Growth of the Hair or Eyelashes and/or Slowing Loss Thereof. US7396525 B2, 2008.
- (26).Cho H; Tai HH Inhibition of NAD+-dependent 15-hydroxyprostaglandin dehydrogenase (15-PGDH) by cyclooxygenase inhibitors and chemopreventive agents. Prostag. Leukotr. ESS 2002, 67, 461–465. [DOI] [PubMed] [Google Scholar]
- (27).Wu Y; Karna S; Choi CH; Tong M; Tai H-H; Na DH; Jang CH; Cho H Synthesis and biological evaluation of novel thiazolidinedione analogues as 15-hydroxyprostaglandin dehydrogenase inhibitors. J. Med. Chem 2011, 54, 5260–5264. . [DOI] [PubMed] [Google Scholar]
- (28).Choi D; Piao YL; Wu Y; Cho H Control of the intracellular levels of prostaglandin E2 through inhibition of the 15-hydroxyprostaglandin dehydrogenase for wound healing. Bioorg. Med. Chem 2013, 21, 4477–4484. [DOI] [PubMed] [Google Scholar]
- (29).Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- (30).Niesen FH; Schultz L; Jadhav A; Bhatia C; Guo K; Maloney DJ; Pilka ES; Wang M; Oppermann U; Heightman TD; Simeonov A High-affinity inhibitors of human NAD+-dependent 15-hydroxyprostaglandin dehydrogenase: Mechanisms of inhibition and structure-activity relationships. PLoS One 2010, 5, e13719.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Duveau DY; Yasgar A; Wang Y; Hu X; Kouznetsova J; Brimacombe KR; Jadhav A; Simeonov A; Thomas CJ; Maloney DJ Structure–activity relationship studies and biological characterization of human NAD+-dependent 15-hydroxyprostaglandin dehydrogenase inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Smith J; Otegbeye F; Jogasuria A; Christo K; Antczak M; Ready J; Gerson SL; Markowitz SD; Desai A Inhibition of 15-PGDH protects mice from immune-mediated bone marrow failure. Biol. Blood Marrow Transplant 2020, 26, 1552–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bärnthaler T; Theiler A; Zabini D; Trautmann S; Stacher-Priehse E; Lanz I; Klepetko W; Sinn K; Flick H; Scheidl S; Thomas D; Olschewski H; Kwapiszewska G; Schuligoi R; Heinemann A Inhibiting eicosanoid degradation exerts antifibrotic effects in a pulmonary fibrosis mouse model and human tissue. J. Allergy Clin. Immunol 2020, 145, 818–833.e811. [DOI] [PubMed] [Google Scholar]
- (34).Palla AR; Ravichandran M; Wang YX; Alexandrova L; Yang AV; Kraft P; Holbrook CA; Schürch CM; Ho ATV; Blau HM Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science 2020, eabc8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Antczak MI; Zhang Y; Wang C; Doran J; Naidoo J; Voruganti S; Williams NS; Markowitz SD; Ready JM Inhibitors of 15-prostaglandin dehydrogenase to potentiate tissue repair. J Med. Chem 2017, 60, 3979–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Desai A; Zhang Y; Park Y; Dawson DM; Larusch GA; Kasturi L; Wald D; Ready JM; Gerson SL; Markowitz SD A second-generation 15-PGDH inhibitor promotes bone marrow transplant recovery independently of age, transplant dose and granulocyte colony-stimulating factor support. Haematologica 2018, 103, 1054–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Markowitz SD; Yuan Y; Zhang Y; Ready J; Hu B Preparation of Pyrazolopyrimidine and Quinoxaline Dderivatives for Modulating Short-Chain Dehydrogenase Activity. WO2018145080A1, 2018.
- (38).Erben U; Loddenkemper C; Doerfel K; Spieckermann S; Haller D; Heimesaat MM; Zeitz M; Siegmund B; Kühl AA A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int. J. Clin. Exp. Pathol 2014, 7, 4557–4576. [PMC free article] [PubMed] [Google Scholar]
- (39).Cho H; Tai HH Threonine 11 of human NAD+-dependent 15-hydroxyprostaglandin dehydrogenase may interact with NAD+ during catalysis. Prostag. Leukotr. Ess 2002, 66, 505–509. [DOI] [PubMed] [Google Scholar]
- (40).McNaney CA; Drexler DM; Hnatyshyn SY; Zvyaga TA; Knipe JO; Belcastro JV; Sanders M An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. Assay Drug Dev. Technol 2008, 6, 121–129. [DOI] [PubMed] [Google Scholar]
- (41).Rong E; Chow S; Yan S; Larson G; Hong Z; Wu J Structure–activity relationship (SAR) studies of quinoxalines as novel HCV NS5B RNA-dependent RNA polymerase inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 1663–1666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.