

Abstract
The incidence of infections from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiologic agent for coronavirus disease 2019 (COVID-19), has dramatically escalated following the initial outbreak in China, in late 2019, resulting in a global pandemic with millions of deaths. Although the majority of infected patients survive, and the rapid advent and deployment of vaccines have afforded increased immunity against SARS-CoV-2, long-term sequelae of SARS-CoV-2 infection have become increasingly recognized. These include, but are not limited to, chronic pulmonary disease, cardiovascular disorders, and proinflammatory-associated neurological dysfunction that may lead to psychological and neurocognitive impairment. A major component of cognitive dysfunction is operationally categorized as “brain fog” which comprises difficulty concentrating, forgetfulness, confusion, depression, and fatigue. Multiple parameters associated with long-term neuropsychiatric sequelae of SARS-CoV-2 infection have been detailed in clinical studies. Empirically elucidated mechanisms associated with the neuropsychiatric manifestations of COVID-19 are by nature complex, but broad-based working models have focused on mitochondrial dysregulation, leading to systemic reductions of metabolic activity and cellular bioenergetics within the CNS structures. Multiple factors underlying the expression of brain fog may facilitate future pathogenic insults, leading to repetitive cycles of viral and bacterial propagation. Interestingly, diverse neurocognitive sequelae associated with COVID-19 are not dissimilar from those observed in other historical pandemics, thereby providing a broad and integrative perspective on potential common mechanisms of CNS dysfunction subsequent to viral infection. Poor mental health status may be reciprocally linked to compromised immune processes and enhanced susceptibility to infection by diverse pathogens. By extrapolation, we contend that COVID-19 may potentiate the severity of neurological/neurocognitive deficits in patients afflicted by well-studied neurodegenerative disorders, such as Alzheimer's disease and Parkinson’s disease. Accordingly, the prevention, diagnosis, and management of sustained neuropsychiatric manifestations of COVID-19 are pivotal health care directives and provide a compelling rationale for careful monitoring of infected patients, as early mitigation efforts may reduce short- and long-term complications.
Keywords: Central nervous system, neuroinflammation, neuropsychiatric disease, mitochondria, microglia, SARS-CoV-2, COVID-19, long COVID, cognitive impairment, brain fog, depression, anxiety
1. INTRODUCTION
1.1. Clinical Signs and Symptoms
While the respiratory system is the predominant entry route of severe acute respiratory syndrome coronavirus (2 SAR-CoV-2) infection, extensive evidence strongly suggests that COVID-19 is a progressive systemic disease [1]. A wide range of COVID-19 symptoms are reported in clinical studies that implicate infective processes in multiple organ systems that include central nervous system (CNS) structures [2, 3]. Indeed, alterations in brain function can result from infection with SARS-CoV-2; in one study, up to 80% of hospitalized patients experienced neurological symptoms [4]. The most characteristic neurological finding associated with COVID-19 is “brain fog,” a broad term that describes fatigue, confusion, delirium and an impaired ability to concentrate, leading to decreased memory, cognition and executive function [5]. This phenomenon appears distinct from acutely altered cognition in the setting of severe diseases, such as those experienced by hospitalized patients with or without respiratory compromise [6]. It is of major concern that symptoms of brain fog may persist several months after the initial resolution of symptoms [7].
Other common neuropsychiatric findings in COVID-19 patients include functional behavioral deficits associated with depressive disorders, delirium, mania, autism spectrum disorders and psychosis [8-12]. Post-infectious syndromes associated with COVID-19 include acute demyelinating encephalomyelitis, necrotizing encephalopathy, generalized myoclonus and acute transverse myelitis [13]. Atypical Bickerstaff's encephalitis, a rare post-infectious neurological syndrome characterized by impaired consciousness, ataxia, areflexia, and extensor plantar responses, has also been reported as a potential functional consequence of COVID-19 [14].
A diagnosis of neuropsychiatric complications of comorbid COVID-19 is determined by careful clinical history and examination. Modern neuroimaging modalities may assist in establishing objective neurological changes subsequent to viral infection. Brain imaging by fluid-attenuated inversion recovery magnetic resonance imaging (FLAIR-MRI) in some COVID-19 patients with neurological involvement has shown neuroradiological patterns in the medial temporal lobe, multifocal lesions in the cerebral white matter, and microhemorrhages [15]. Some studies have also found evidence of ischemic vascular damage, leptomeningeal enhancement, and encephalitis [16, 17]. Involvement of the olfactory bulb, amygdala, entorhinal area, temporal and frontal neocortex, and dorsal medulla abnormalities, has also been reported in rare instances [18]. Importantly, significant cases of neurocognitive and neuropsychiatric disease may be present in the absence of abnormal neuroimaging patterns. Moreover, clear evidences of classic neuropathological signs of viral central nervous system (CNS) infections, including lymphocytic leptomeningitis, microglial nodules, pronounced or frequent perivascular lymphocytic cuffing, focal demyelination or viral inclusions, are not frequently encountered in even severe COVID-19 cases [19, 20]. Thus, controversies surround the utility of neuroimaging in the diagnosis and correlation of neurological findings in patients with COVID-19, even in those with severe symptoms.
The difficulties in the objective measurement of neuropsychiatric manifestations of COVID-19 are sharply contrasted by those established for loss of pulmonary function and multiple organ failure associated with COVID-19. For example, COVID-19 cases with significant respiratory symptoms, acute respiratory distress syndrome with diffuse alveolar damage, diffuse thrombotic alveolar microvascular occlusion, and inflammatory mediator-associated airway inflammation, are easily identifiable by evidence-based radiographical and histological criteria [21]. These findings aid in the rapid diagnosis and implementation of therapies to improve pulmonary function. However, neuropsychiatric symptoms are often refractory to characterization by established diagnostic criteria and may delay detailed analyses of this vital aspect of the disease course of COVID-19.
A diagnosis of direct neuropsychiatric manifestations of COVID-19 is potentially confounded by non-selective or generalized effects of the pandemic on mental health. Enforced social isolation, limited social gatherings, and self-quarantine protocols, deemed necessary to reduce the spread of the virus have the potential to cause or exacerbate serious mental illnesses, including depression, anxiety, and sleep disorders [9]. There is also a component of post-traumatic stress disorder (PTSD), which was prevalent after SARS-CoV-1 [22, 23], and has been proposed to have a strong effect on the impaired neurocognitive function of SARS-CoV-2 patients (Fig. 1).
Fig. (1).
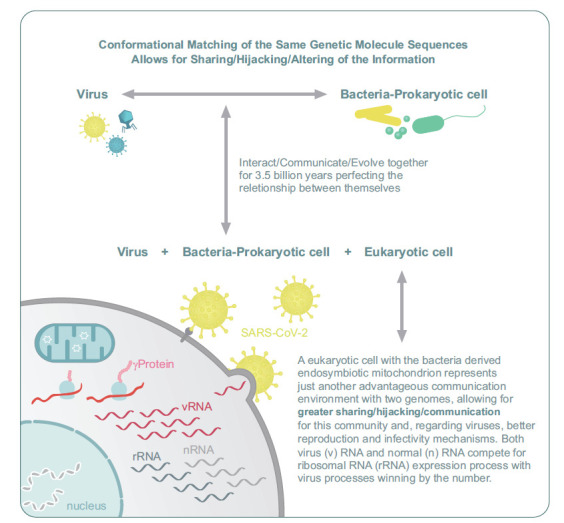
Theoretical pathways developed by eminent theoretical biologists strongly suggest that ssDNA and dsDNA genomes have evolved from primordial RNA constructs in protocellular and protoviral packages. Within the primordial RNA world, we surmise that the persistence of protocellular and protoviral genetic material was insured by packaging into protective phospholipid-and protein-based membrane vesicles. Accordingly, within the realm of the last universal common ancestor (LUCA) theory, co-evolution of DNA viruses within archaea and prokaryotes and RNA viruses within eukaryotes likely emerged due to extensive adaptive complexity or differentiation of viral membranes [49, 50]. In simple terms, viral-mediated horizontal transfer of genetic information may be physically dependent on conformational matching or shape recognition processes driving high-affinity binding interactions of complementary viral and extracellular host membrane domains. Thus, given over 3.5 billion years of viral and cellular co-evolution, it is not surprising that intracellular communication between infective and replicating viruses and host mitochondria is extensive due to the endosymbiont origin of these eukaryotic organelles. Currently, these strong evolutionary considerations underlie the existential role of SARS-CoV-2 to effectively hijack host mitochondrial energy production to drive replicative processes. COVID-19 may also result in chronic alteration of brain metabolism resulting in long-term cognitive and affective behavioral deficits.
2. NEUROPSYCHIATRIC SEQUELAE OF COVID-19: A COMPARATIVE ANALYSIS
To fully elucidate the long-term neurological manifestations of COVID-19, it is important to establish an understanding of the epidemiology and medical history of significant past viral pandemics and other endemics with regard to documented neurocognitive outcomes [12]. Over the past century, the major viral pandemics have included the influenza pandemic of 1889 and 1892 (Russian flu), the Spanish flu pandemic (1918-1920), and the SARS pandemic. During the Russian flu, documented long-term neurological effects included neuralgia, neurasthenia, neuritis, nerve exhaustion, gripped catalepsy, psychosis, prostration, inertia, anxiety and paranoia [24].
The long-term neurological effects of the Spanish flu pandemic included Parkinsonism, catatonia, and encephalitis lethargica, a syndrome that involves clinical presentation of meningitis and delirium [25, 26]. While direct causality was not established between the Spanish flu and encephalitis lethargica, isolated regions of the world that were not exposed to the pandemic demonstrated little to no cases of this syndrome, whereas areas significantly associated with viral infection experienced increasing cases, coinciding with peak episodes of the pandemic and declining rapidly thereafter [25]. Other localized epidemics with neurological symptoms mimicking COVID-19 include the 1935 outbreak of “atypical poliomyelitis,” which predominantly afflicted healthcare providers at the Los Angeles County [27, 28]. The affected hospital staff demonstrated symptoms ranging from severe headache, painful oculomotion, to gastrointestinal symptoms and brain involvement characterized by dullness and an inability to concentrate [27, 28]. There was a high incidence of diphtheria at the time of this outbreak, and some of these patients had positive diphtheria culture along with symptoms of pharyngitis. Distinguishing this entity from poliomyelitis, cerebrospinal fluid findings of the majority of patients with atypical poliomyelitis were normal.
Perhaps the most striking post-viral syndrome that mimics the general characteristics of COVID-19 is the so-called benign myalgic encephalomyelitis [29]. This clinical entity was first described in the 1950s and was initially considered to be a variant of poliomyelitis. Strikingly, the symptoms of headache, myalgia, malaise, and paresis were more pronounced in females, and were not associated with fever or death even when symptoms persisted for several years. Similar to COVID-19, it was not unusual for a significant proportion of patients who did not achieve complete remission of symptoms within 3 months of onset to have a fluctuating clinical course, with remissions and flares of depression, emotional lability, and a lack of concentration [29]. Interestingly, the etiologic cause of benign myalgic encephalomyelitis was never identified. A more common, generalizable extension of benign myalgic encephalomyelitis is post-infectious fatigue syndrome, which is a subtype of chronic fatigue syndrome [30]. Indeed, diagnostic criteria for post-infectious fatigue syndrome are identical with those of chronic fatigue syndrome, except that those symptoms associated with the former must, by definition, be preceded by a known infection.
Notably, whereas chronic fatigue syndrome and related post-infectious fatigue syndromes are more prevalent in females, short-term COVID-19 severity is associated with poorer outcomes in males as compared to females [31, 32]. By contrast, recent studies suggest that long-COVID symptoms, including delayed neurological sequelae, may follow reverse female sex bias compared to the systemic manifestations of the disease [33].
3. PATHOGENESIS
How COVID-19 leads to neurocognitive dysfunction and psychiatric incidence remains largely unknown. Undoubtedly, some of the acute manifestations of anxiety in some settings of high-risk patients may be potentially attributed to heightened attention and publicity of a novel disease at a time where societies are beleaguered by disinformation and current incomplete understanding of a rapidly evolving pandemic. As previously described, limited social events, changes in routines, and implementation of isolation protocols for infected individuals likely exacerbate pre-existing mental health challenges in our society. However, the presence of persistent sequelae and the evolution of the disease in previously healthy individuals, coupled with ample evidence of post-infectious neuropsychiatric syndromes, raise the possibility of clear viral pathogenesis that warrants detailed understanding. In this regard, the potential mechanisms, operationally dichotomized into direct and indirect etiologies, are discussed.
3.1. Direct Mechanisms
The underlying rationale for direct functional linkage of COVID-19 psychological and neurological dysfunctions is based on long-held views that coronaviruses are neurotropic [34]. This may be supported by the empirical demonstration of the presence of SARS-CoV-2 genomic RNA in human CNS tissues and cerebrospinal fluid [35]. Indeed, the putative receptor for the SARS-CoV-2 spike (S) protein, angiotensin-converting enzyme 2 (ACE2), is widely expressed in neurons and non-neuronal brain cells, including microvascular endothelial cells and oligodendrocytes, thereby supporting direct neuro-invasion via receptor-mediated cellular entry [36-38]. Empirical studies have demonstrated that the severe neurological sequelae of rabies virus (RABV) infections are mediated by neuroinvasion or CNS transport of virus tightly bound to the nicotinic acetylcholine receptor (nAChR) [39, 40]. A similar mechanism of direct neuroinvasion has been proposed to explain the severe neurological deficits associated with SARS-CoV-2 infections via the formation of SARS-CoV-2/nAChR complexes in primary olfactory nerve terminals and/or peripheral trigeminal sensory terminal structures located within the olfactory epithelium followed by orthograde or retrograde transport into the CNS. Interestingly, snake venom toxins have been demonstrated to be high-affinity competitive antagonists of the nAChR and homologous regions of 4-5 amino acids within the SARS-CoV-2 spike protein, and aligned sequences in alpha-bungarotoxin and alpha-cobratoxin have been identified that suggest a common binding affinity to a7-nACh receptors [39, 40]. Accordingly, these defined pathways may directly lead to tropism and viral-induced pathologies [5].
Further evidence for direct mechanisms of SARS-CoV-2 neuropathology is provided by post-mortem studies of COVID-19 patients and the use of advanced 3D microfluid models of the human blood brain barrier (BBB). These studies reveal three essential features [36, 41]. First, ACE2 is widely expressed, not only in brain microvascular endothelial cells but also in other components of the BBB, including astrocytes and pericytes. Indeed, astrocytic and neuronal injury in COVID-19 has been demonstrated by observing elevated levels of glial fibrillary acidic protein (GFAP) [42]. Second, the S protein can directly damage the integrity of the BBB to varying degrees [36, 41]. Third, the S protein can induce an inflammatory response and transcriptional reprogramming of cellular components of the BBB, even when evidence of direct infection by the virus is absent [36, 41]. These observations are confirmed and expanded by a clinical study employing molecular profiling of 65,309 single-nucleus transcriptomes from frontal cortex and choroid plexus samples obtained from post-mortem brain of 8 patients with COVID-19 in comparison to those obtained from 14 control individuals [43]. First, differential alterations in the expression of functional genes were observed in choroid plexus cells that relay peripheral inflammatory signals to activated brain microglia and astrocyte subpopulations associated with COVID-19. Consistent with previous reports of dysregulation of numerous brain and choroid plexus cell types in COVID-19 patients, these findings also suggest that severe disruption of the BBB may significantly facilitate direct access of the virus to cortical neurons involved in the complex processing of cognitive information. These contentions are consistent with the observation of functionally disruptive gene expression changes linked to synaptic deficits in cortical L2/3 excitatory neurons and resident VIP-expressing interneurons from COVID-19 patient samples, which indicate dysfunction in upper-layer cortical circuitry associated with processing and integration of complex cognitive behaviors [43].
In addition to direct CNS entry via compromised BBB cells, SARS-CoV-2 may translocate to the brain via other routes. These include trans-synaptic, optic and olfactory neurons, and vascular endothelial cells [39, 44]. Given that similar receptor interactions are required for cellular entry, it is perhaps unsurprising that previous studies have confirmed similar routes of infection by the SARS-CoV-1. Studies confirm the direct replication of SARS-CoV-1 in brain tissue, and the pathological assessment of the brains of patients with SARS-CoV-1 showed that the virus was present in the cytoplasm of cortical and hypothalamic neurons [45]. The pathological changes of brain tissue from patients with SARS-CoV-1 associated encephalitis 1 also included neuronal necrosis, glial cell hyperplasia, and infiltration of monocytes and T cells [35]. SARS-CoV-1 was also shown to directly cause the death of cerebral neurons in the absence of encephalitis in an ACE2-expressing transgenic mouse model [36]. These mechanisms support the appearance of neurological symptoms, the formation of fatal microthrombi, and even the occurrence of encephalitis associated with COVID-19, providing pathologic correlates to the neuroimaging findings previously noted and discussed.
3.1.1. Mitochondria Hijacking
The potential molecular mechanisms of COVID-19-associated neuropathology appear to be critically linked to the “parasitic” hijacking of mitochondrial bioenergetics in order to support its own biosynthetic replicative processes. In addition to its existential role in providing the cell with requisite ATP production, the mitochondrion maintains complex intracellular regulatory processes that are functionally associated with innate immune responses. Following a pathogenic assault by viral or bacterial agents, mitochondrial-mediated signaling is also functionally synchronized with the activation of classes of leukocytes involved in adaptive immune responses [46] (Fig. 1). As noted, immune energy processes would be a sensitive and an ideal target for viruses, in general, in their strategy for reproduction, that is, hijacking the energy producing system to complement the host genome take-over. The generation of ATP is fundamental for neurotransmission and generation of membrane potential along neuronal axons. Moreover, ATP is required for metabolic clearance of pathological deposits, including amyloid-beta plaque of Alzheimer' disease. Consequently, recent studies have shown that pathways that impair the metabolic fitness not only of neurons but other supportive cells, including microglia, compromise neurological function [47, 48].
One possible mechanism for long-term neurological sequelae of SARS-CoV-2 is the direct infection of mitochondria, leading to the integration of the viral genome, potentially directly impairing mitochondrial energy metabolism via targeted action on oxygen availability and utilization [51]. A computational modeling approach has observed a localized enrichment of genomic and subgenomic SARS-CoV-2 5′ and 3′untranslated RNA sequences within host mitochondrial matrix and nucleolar structures. This potential for host mitochondrial residency and integration into the host genome portends direct co-option of the metabolic center of the cell, leading to the production of a favorable metabolic support system to the virus for replication and other metabolically demanding cell cycle functions [52-54] (Fig. 1).
The hijacking of the cellular metabolic hub provides numerous benefits to the virus. This includes activation of inflammatory pathways, including inflammasomes, which may inadvertently suppress host innate and adaptive immune responses [54-56]. Viral metabolic control may also account for long-term neurological dysfunction. Where microglia are involved, this may lead to impaired metabolic fitness, which leads to impaired autophagy and metabolic support of basic function, including the clearance of pathologic plaques and deposits. Ultimately, this could promote neurocognitive decline, which is an emerging concept of Alzheimer’s disease pathobiology [48, 57]. Secondly, mitochondria-induced neuroinflammation may drive Alzhei- mer’s disease via mechanisms similar to those described for other neuroinflammatory pathways [58]. Interestingly, parallels in the pathological processes of HIV and SARS-CoV-2 may be found, indicating these processes to emerge over evolutionary time and settled on common critical substrates [59]. It is important to note that viral hijacking of cellular metabolic function is not unique to SARS-CoV-2 or coronaviruses. Such a mechanism has been proposed for other infections, such as the Ebola, Zika, and influenza A viruses [55].
3.1.2. Autophagy
To achieve maximized replicative processes, viruses conform to simple survival laws. It is in the interest of the pathogen to maintain the integrity of the infected host cells, at least throughout the incubation phase of the virus. Thus, processes that initiate apoptosis may be halted to guarantee the formation of the greatest numbers of viral progenies possible [60]. However, the spread or persistence of the virus also involves the induction of apoptotic cell death of host cells to trigger the release of newly synthesized viral particles [61]. Thus, a fine balance between timing and extent of host cellular death must be navigated by the virus to optimize infectivity, replication, and spread. Infected host cells accumulate autophagosomes to activate autophagy-linked apoptosis, aiming to cut off the loop of virus replication. Therefore, it is in the interest of a virus to delay aggregation of autophagosomes in the infected host cells at the beginning phase of infection, while promoting this process in the later life cycle may facilitate spread outside the host [60]. Although the relationship between SARS-CoV-2 and autophagy currently remains unclear, possible mutually beneficial interactions cannot be completely ruled out.
3.2. Indirect Mechanisms
3.2.1. Collateral Brain Injury from Hypoxia
The lung has been established as the major entry route of COVID-19 into the systemic circulation. COVID-19-associated pneumonia may result in acute respiratory distress syndrome, hypoxemia and acidosis [21, 62]. As oxygenation and ventilation are essential for vital organ function, the degree of initial lung damage is predictably associated with short and long-term effects of COVID-19 on cardiovascular and neurological systems [62]. Under hypoxic conditions, neurons with the highest oxygen demand become dysfunctional given their high metabolic demand. Thus, even brief episodes of poor respiratory impairment may lead to cognitive impairment. The limitation for this potential mechanism lies in the fact that several patients who develop long-term neurocognitive decline demonstrate complete recovery of their initial course, and sometimes do not present with severe symptoms at the time of diagnosis [63]. Thus, alternative possibilities must account for these disparate results.
3.2.2. Cytokine Storm
The systemic increase in inflammatory mediators as a potential host defense response to pathogens, termed the ‘cytokine storm,’ may explain the multi-organ damage found in some patients with COVID-19 [64] (Fig. 2). This phenomenon increases vascular permeability, abnormal blood coagulation, and multiple-organ inflammatory damage, leading to organ failure [64]. A similar effect may occur in the CNS, where these cytokines may also augment microvascular permeability and facilitate the entry of SARS-CoV-2 through the impaired BBB [36]. The ‘cytokine storm’ may also promote the formation of microthrombi by aberrant activation of coagulation processes [64]. Susceptibility of the brain to the potentially debilitating effects of ‘cytokine storm’ is facilitated by a compromised BBB. In this regard, we note a similar phenomenon in the potential involvement of the vascular system as an important etiological factor in Alzheimer’s Disease progression via a cascading proinflammatory response associated with constitutive nitric oxide impairment [65]. Interestingly, constitutive nitric oxide is directly involved in modulating mitochondrial metabolic metabolism [66].
Fig. (2).
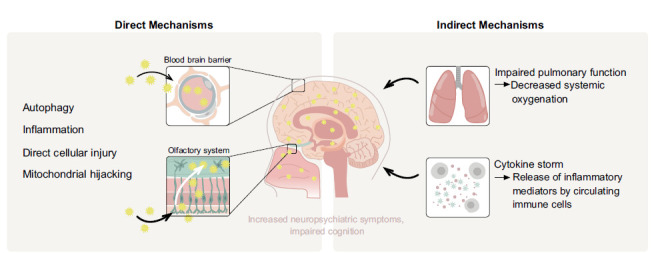
A summary of the direct and indirect mechanisms by which SARS-CoV-2 affects the CNS, leading to neuropsychiatric symptoms; direct routes of access to the brain, disruption of the blood brain barrier, trans-synaptic, optic and olfactory nerve channels. Once these entry routes are breached, the virus may directly infect neurons and other supportive non-neuronal cells. SARS-CoV-2 then co-opt mitochondrial function, disrupting autophagy to facilitate propagation. Alternatively, even brief episodes of systemic hypoxia alter mitochondrial function leading to direct impairment of susceptible, metabolically-demanding organs, including the brain. The release of cytokines in response to overwhelming infection may trigger uncontrolled and cascading inflammation that may disrupt the BBB and lead to neurocognitive symptoms.
The concept of ‘cytokine storm’ is widely appealing, as it provides a fundamental link between inflammation, infection and tissue damage that lead to severely compromised organ function associated with debilitating long-term physiological and neurological sequelae. The manifestation of chronic physiological and neurological deficits in the post-infectious setting most likely applies not only to COVID-19 but to prior infections by diverse viral pathogens (Fig. 2). This notion may also provide a rationale why certain tissues with limited ACE2 expression may still experience complications of COVID-19, as the release of cytokines may reach several targets not directly infected by the virus [67]. In this regard, persistent elements or remnants of SARS-CoV-2 acute infection may be involved in Long-COVID [52]. We have recently evidenced that SARS-CoV-2-reactive T memory cells occur in unexposed healthy individuals due to previous infections by endemic coronaviruses that cause the ‘common cold’ [68]. Accordingly, expression of adaptive SARS-CoV-2-reactive T memory cells in unexposed healthy individuals may be due to multiple cross-reactive viral protein targets following previous exposure to endemic human coronavirus infections. In this regard, it appears that further investigation is required to evaluate whether T memory cells arising from prior ‘common cold’ coronavirus infections provide additive or synergistic cellular immune responses following exposure to SARS-CoV-2, with particular reference to components of the associated ‘cytokine storm’.
4. EFFECTS OF NEUROPSYCHIATRIC MANIFESTATIONS ON VIRAL PROPAGATION AND SURVIVAL
We surmise that given the 3.5-billion-year evolutionary history of viral-bacterial interaction and the resulting co-evolution of these entities, specific molecular points/targets of genetic sensitivity exist for this core-informational interaction [69] (Fig. 1). Furthermore, because accumulated evidence supports the bacterial endosymbiont origin of the mitochondrion, this existential organelle represents an evolutionary conserved viral target for subjugation or hijacking a critically needed energy source for their reproduction. Importantly, many biomedical and popular science publications have provided recent estimates of 40 trillion bacteria and 400 trillion viral particles contained within the human microbiomes and viromes, respectively. By comparison, the human body contains approximately 10 trillion eukaryotic cells [69]. Thus, our bodies represent a privileged environment whereby bacteria and viruses continue their evolutionary evolvement/communication [70] (Fig. 1). Within this functional context, concerted dysbiosis of commensal bacterial and viral species is predicted to differentially contribute to the etiology and sustained pathophysiology of diverse medical syndromes affecting human populations. By logistical criteria, the human host may in part be viewed as an ideal vehicle for viral spreading based on its large capacity for viral replication and subsequent transmission. Diminished cognitive abilities required for requisite executive functions and critical decision-making behaviors have been associated with severe neuropsychiatric disorders. Similar cognitive deficits have been observed in cohorts of patients afflicted with COVID-19 patients, presumably due to global brain hypoxia [62, 63]. Whether diminished or significantly compromised cognitive processes facilitate the capabilities of infective viral pathogens to hijack cellular bioenergtics awaits further empirical investigation. Accordingly, the potential of cognitively impaired human hosts to transmit viral pathogens with enhanced efficiency appears to be beyond dispute and may be functionally associated with distinct evolutionary advantages [52, 71].
Taken together, cognitive impairments induced by SARS-CoV-2 infection, such as brain fog, may trigger behavioral changes that favor viral survival and propagation [71]. Specifically, brain fog may compromise self-control and self-care practices while promoting a lack of insight into the danger posed by the virus [9, 71]. This could lead both to increased vulnerability to SARS-CoV-2 infection and secondary complications, as well as progress of viral spread within the population by increasing exposure to others. Thus, viral induction of these behaviors may represent an evolutionary advantage for survival [71]. The cellular mechanisms, other than those presented earlier, by which virus-induced behavioral disorders provide benefit to viral propagation remained elusive until recently.
5. LESSONS FROM LUNG INJURY SEQUELAE
In 2003, SARS-CoV-1 infection led to more than 8000 cases and 900 deaths worldwide. A 1-year follow-up of 97 SARS-CoV-1 survivors identified abnormalities on chest X-ray, evidence of functional impairment, and decreased overall quality of life, compared to age-matched individuals not previously infected by the virus [73]. This worrisome finding persisted two years post-infection, and for up to 15 years of follow-up [74, 75]. Similar to SARS-CoV-1, SARS-CoV-2 directly binds to ACE2 distributed on the extracellular surface of pulmonary type 2 pneumocytes [76]. ACE2 represents a key regulatory enzyme of the renin-angiotensin system and catalyzes the formation of the vasorelaxant peptide angiotensin (1-7) from angiotensin II (Ang II). SARS-CoV-2-induced downregulation of cell surface distributions of ACE2 has been proposed to occur by concurrent internalization of viral-enzyme complexes or by proteolytic processing and/or conversion of the extracellular globular domain of the enzyme into a soluble blood-borne protein [76]. Accordingly, the consequences of viral infection are predicted to include diminished ACE2 activity that is coupled to enhanced concentrations of Ang II, a major vasoconstrictive peptide that is functionally linked to potentiation of pulmonary tissue damage. The magnitude of cell damage may depend not only on the effects of viral replication, but also on the release of pro-inflammatory cytokines, resulting in impaired function of type 2 pneumocytes [76]. These effects result in impaired cell function, followed by cell death (necrosis) or apoptosis, exudates, desquamation of pneumocytes, and formation of hyaline membranes, which are characteristic of diffuse alveolar damage. If direct infectivity by neurotropism is true, then a similar pathway would predict long-term impaired function of affected neurons, leading to irrecoverable neurodegeneration.
5.1. Looking at the Future - Long Term Neurocognitive Outcomes after COVID-19 Infection
The potential for direct CNS infectivity via ACE2 receptors, via mitochondria metabolic function coupled with inflammatory ‘cytokine storm’, offers an attractive potential mechanism for inducing long-term symptoms of COVID-19. However, whether these mechanisms are sufficient to induce or accelerate the premature occurrence of neurodegenerative diseases, Alzheimer's, Parkinson's, and multiple sclerosis, remain an intriguing area that warrants serious investigation. In this regard, multiple sclerosis (MS) is associated with focal gray and white matter demyelination and diffuse neurodegeneration of the brain caused by inflammation [77]. Some of the neurological changes caused by SARS-CoV-2 share similarities with those found in MS. In both disease processes, pro-inflammatory ‘cytokine storm’ is an important contributory initiating factor for the CNS neuroinflammatory damage [78]. Second, SARS-CoV-2 can cause demyelination in the brain and spinal cord [79]. Third, a limited but increasing number of reports show cases of SARS-CoV-2 infection with associated signs and symptoms identical to those of MS [80, 81]. Indeed, previous studies have shown an association between coronavirus infection and the onset of MS [35]. If an association between SARS-CoV-2 infection and demyelinating neurological disease is further established over time, careful consideration is warranted regarding the use of immunotherapies, which may themselves trigger collateral demyelinating neuropathies [82].
A hallmark feature of Parkinson's disease (PD) is the slow progressive decline in movement, muscle control and balance, secondary to degenerative changes predominantly occurring in substantia nigra and other dopaminergic regions of the brain [83]. However, similar to SARS-CoV-2, patients with PD also demonstrate impairment of cognitive and memory functions [84-86]. Moreover, hyposmia and anosmia, early symptoms in COVID-19, are also precursor clinical symptoms of PD [87]. While there is currently limited evidence from clinical studies to support the onset of PD as a late complication of SARS-CoV-2 infection, the wide expression of ACE2 at different areas in the CNS provides a molecular basis for SARS-CoV-2 to mediate or accelerate the occurrence of PD. Interestingly, a recent case described a COVID-19 patient with encephalopathy and neuroimaging evidence of hemorrhaging injury involving the bilateral basal ganglia [88].
Alzheimer's disease is the leading cause of neurocognitive impairment in the elderly population.
While the etiologic agent for AD remains unknown, neuroinflammation plays a critical role [65]. The simultaneous expression of ACE2 in glutamatergic and GABAergic neurons indicates that SARS-CoV-2 infection can affect the balance of both signaling pathways in the CNS. Moreover, neuroinflammation, synaptic pruning, and neuron loss occurring in SARS-CoV-2 share commonalities with AD [83, 89].
5.2. Pharmacological Considerations in the Design of Efficacious Anti-viral Agents
Currently, the development of small molecule anti-SARS-CoV-2 drugs is focused on the inhibition of replicative viral genome production catalyzed by RNA-dependent RNA polymerase. Alternatively, relatively limited clinical studies have focused on repurposing of anti-retroviral drugs clinically employed in anti-HIV1 therapeutic regimens as anti-replicative SARS-CoV-2 agents [90]. Mixtures of anti-retroviral drugs typically include chymotrypsin-like protease inhibitors, such as lopinavir and/or darunavir, that block intracellular post-translational processing of key viral proteins, in combination with the CYP 3A4 inhibitor ritonavir that effectively increases blood concentrations and bioavailability of the protease inhibitors. Although chronic usage of anti-retroviral cocktails has been associated with adverse neurological and chemosensory side effects, a major clinical concern directly relates to pharmacological inhibition of CYP 3A4-mediated hepatic metabolism of multiple psychotropic drugs commonly used in clinical psychiatry. Accordingly, further exploration of potential beneficial effects of anti-retroviral therapeutic regimens on COVID-19 progression should proceed with caution due to confounding effects on the management of diverse psychiatric disorders [90].
From a different perspective, clinical intervention may be required to effectively manage well-documented memory deficits, cognitive impairment, delirium and manic episodes, following short term administration of high potency glucocorticoid agents, such as dexamethasone, often in combination with the anti-inflammatory antibiotic azithromycin, for treatment of COVID-19 [91, 92]. Interestingly, more than 50% of patients on or following chemotherapy develop symptoms indicative of cognitive dysfunction similar to those described above for long-COVID. This neurological condition is termed “chemofog” or “chemobrain,” and has been associated with clinical usage of anti-proliferative cancer drugs, such as doxorubicin, methotrexate, lenalidomide, rituximab, and trastuzumab, some of which have also been employed in treatment strategies for diverse autoimmune syndromes [93]. Accordingly, clinicians should proceed with caution in employing off-label applications of potent anti-inflammatory agents due to potential long-term neurological/neuropsychiatric complications associated with brain fog. It remains to be seen if we are able to exquisitely determine reliable clinical predictors for people at higher risk for long-term neurological/neuropsychiatric sequelae of COVID-19.
CONCLUSION
Emerging studies, as noted earlier, indicate that a substantial number of individuals who survive COVID-19 portray long-term symptoms, including psychiatric disorders and neurocognitive decline, initially manifesting as brain fog. Direct and indirect mechanisms may contribute to the development of these symptoms. As was shown in the prior coronavirus epidemic, patients with SARS-CoV-2 may be at heightened risk of post-traumatic stress disorder, further compounding neuropsychiatric complications [22, 23]. These manifestations present a pressing source of the debilitating long-term consequences of SARS-CoV-2 infection. They may also play a direct role in promoting viral survival and propagation by eliciting behavioral changes in patients that decrease awareness and implementation of preventive lifestyle. Importantly, the neuropsychiatric manifestations of COVID-19 may confer increased additional risk for long-term neurocognitive diseases, including Alzheimer's disease, as they share common mechanistic features. To this end, close monitoring of patients following the acute stage of infection with SARS-CoV-2 is essential to provide a rationale for the prevention, diagnosis, and management of these potential long-term sequelae. Efforts to increase mental health awareness would aid in the diagnosis of neuropsychiatric complications of COVID-19 while assisting in overcoming the stigma associated with neuropsychiatric disorders. These efforts, along with investments into mental health research and community-based initiatives, may prove to be useful avenues that not only help in the management of short- and long-term complications but also potentially play a vital role in reducing the spread of COVID-19.
Fig. (3).
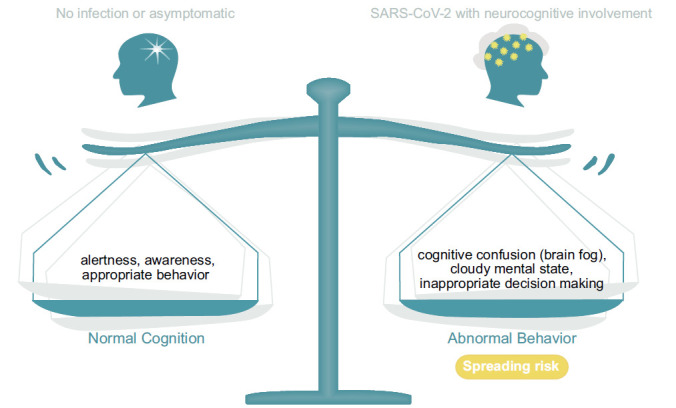
The diagram illustrates a balance between what is regarded as normal behavior and which is multifaceted and emerges as appropriate behavior given a dynamic input and appropriate output. In this process, for the most part, the mind is self-aware, instituting conscious decision-making. As discussed in the text, viral infections, such as SARS-CoV-2, have the potential to disrupt these processes and alter a person’s mental state. The altered mental state still allows the person to be functional, but with reduced behavioral efficacy. In part, we surmise this is due to altering mitochondrial energy processes since maintaining a normal mental state requires a high level of energy availability. Significantly, a similar alteration of a normal mental state may occur with the use of antibiotics, which target bacteria as well as the “power house of the cell”, i.e., mitochondria given their prokaryotic origin [72]. This further demonstrates the susceptibility and sensitivity of cognition to its energy requirements (Fig. 3). Thus, it is surmised that viral-induced modifications of energy availability benefit viral survival because carefully thought-out preventative behaviors may be compromised.
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
P.B. is a PhD candidate at the Dept of Psychiatry, Charles University, Prague, Czech Republic.
REFERENCES
- 1.Ramos-Casals M., Brito-Zerón P., Mariette X. Systemic and organ-specific immune-related manifestations of COVID-19. Nat. Rev. Rheumatol. 2021;17(6):315–332. doi: 10.1038/s41584-021-00608-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H., Xiao X., Zhang J., Zafar M.I., Wu C., Long Y., Lu W., Pan F., Meng T., Zhao K., Zhou L., Shen S., Liu L., Liu Q., Xiong C. Impaired spermatogenesis in COVID-19 patients. EClinicalMedicine. 2020;28:100604. doi: 10.1016/j.eclinm.2020.100604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singal C.M.S., Jaiswal P., Seth P. SARS-CoV-2, More than a respiratory virus: its potential role in neuropathogenesis. ACS Chem. Neurosci. 2020;11(13):1887–1899. doi: 10.1021/acschemneuro.0c00251. [DOI] [PubMed] [Google Scholar]
- 4.Chou S.H., Beghi E., Helbok R., Moro E., Sampson J., Altamirano V., Mainali S., Bassetti C., Suarez J.I., McNett M., GCS-Neuro COVID Consortium and ENERGY Consortium Global incidence of neurological manifestations among patients hospitalized with COVID-19-a report for the GCS-Neuro COVID consortium and the ENERGY consortium. JAMA Netw. Open. 2021;4(5):e2112131. doi: 10.1001/jamanetworkopen.2021.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang F., Kream R.M., Stefano G.B. Long-term respiratory and neurological sequelae of COVID-19. Med. Sci. Monit. 2020;26:e928996. doi: 10.12659/MSM.928996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong D.M., Zhou Y.T., Wang Y.W. COVID-19-associated acute brain dysfunction related to sepsis. J. Clin. Med. Res. 2021;13(2):82–91. doi: 10.14740/jocmr4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carfì A., Bernabei R., Landi F., Gemelli Against C-P-A.C.S.G., Gemelli Against COVID-19 Post-Acute Care Study Group Persistent symptoms in patients after acute COVID-19. JAMA. 2020;324(6):603–605. doi: 10.1001/jama.2020.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Correa-Palacio A.F., Hernandez-Huerta D., Gómez-Arnau J., Loeck C., Caballero I. Affective psychosis after COVID-19 infection in a previously healthy patient: a case report. Psychiatry Res. 2020;290:113115. doi: 10.1016/j.psychres.2020.113115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ptacek R., Ptackova H., Martin A., Stefano G.B. Psychiatric manifestations of COVID-19 and their social significance. Med. Sci. Monit. 2020;26:e930340. doi: 10.12659/MSM.930340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epstein D., Andrawis W., Lipsky A.M., Ziad H.A., Matan M. Anxiety and suicidality in a hospitalized patient with COVID-19 infection. Eur. J. Case Rep. Intern. Med. 2020;7(5):001651. doi: 10.12890/2020_001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mawhinney J.A., Wilcock C., Haboubi H., Roshanzamir S. Neurotropism of SARS-CoV-2: COVID-19 presenting with an acute manic episode. BMJ Case Rep. 2020;13(6):e236123. doi: 10.1136/bcr-2020-236123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stefano G.B. Historical insight into infections and disorders associated with neurological and psychiatric sequelae similar to long COVID. Med. Sci. Monit. 2021;27:e931447. doi: 10.12659/MSM.931447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maury A., Lyoubi A., Peiffer-Smadja N., de Broucker T., Meppiel E. Neurological manifestations associated with SARS-CoV-2 and other coronaviruses: A narrative review for clinicians. Rev. Neurol. (Paris) 2021;177(1-2):51–64. doi: 10.1016/j.neurol.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llorente Ayuso L., Torres Rubio P., Beijinho do Rosário R.F., Giganto Arroyo M.L., Sierra-Hidalgo F. Bickerstaff encephalitis after COVID-19. J. Neurol. 2021;268(6):2035–2037. doi: 10.1007/s00415-020-10201-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kremer S., Lersy F., de Sèze J., Ferré J.C., Maamar A., Carsin-Nicol B., Collange O., Bonneville F., Adam G., Martin-Blondel G., Rafiq M., Geeraerts T., Delamarre L., Grand S., Krainik A., Caillard S., Constans J.M., Metanbou S., Heintz A., Helms J., Schenck M., Lefèbvre N., Boutet C., Fabre X., Forestier G., de Beaurepaire I., Bornet G., Lacalm A., Oesterlé H., Bolognini F., Messié J., Hmeydia G., Benzakoun J., Oppenheim C., Bapst B., Megdiche I., Henry Feugeas M.C., Khalil A., Gaudemer A., Jager L., Nesser P., Talla Mba Y., Hemmert C., Feuerstein P., Sebag N., Carré S., Alleg M., Lecocq C., Schmitt E., Anxionnat R., Zhu F., Comby P.O., Ricolfi F., Thouant P., Desal H., Boulouis G., Berge J., Kazémi A., Pyatigorskaya N., Lecler A., Saleme S., Edjlali-Goujon M., Kerleroux B., Zorn P.E., Matthieu M., Baloglu S., Ardellier F.D., Willaume T., Brisset J.C., Boulay C., Mutschler V., Hansmann Y., Mertes P.M., Schneider F., Fafi-Kremer S., Ohana M., Meziani F., David J.S., Meyer N., Anheim M., Cotton F. Brain MRI findings in severe COVID-19: a retrospective observational study. Radiology. 2020;297(2):E242–E251. doi: 10.1148/radiol.2020202222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katal S., Balakrishnan S., Gholamrezanezhad A. Neuroimaging and neurologic findings in COVID-19 and other coronavirus infections: A systematic review in 116 patients. J. Neuroradiol. 2021;48(1):43–50. doi: 10.1016/j.neurad.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kremer S., Lersy F., Anheim M., Merdji H., Schenck M., Oesterlé H., Bolognini F., Messie J., Khalil A., Gaudemer A., Carré S., Alleg M., Lecocq C., Schmitt E., Anxionnat R., Zhu F., Jager L., Nesser P., Mba Y.T., Hmeydia G., Benzakoun J., Oppenheim C., Ferré J.C., Maamar A., Carsin-Nicol B., Comby P.O., Ricolfi F., Thouant P., Boutet C., Fabre X., Forestier G., de Beaurepaire I., Bornet G., Desal H., Boulouis G., Berge J., Kazémi A., Pyatigorskaya N., Lecler A., Saleme S., Edjlali-Goujon M., Kerleroux B., Constans J.M., Zorn P.E., Mathieu M., Baloglu S., Ardellier F.D., Willaume T., Brisset J.C., Caillard S., Collange O., Mertes P.M., Schneider F., Fafi-Kremer S., Ohana M., Meziani F., Meyer N., Helms J., Cotton F. Neurologic and neuroimaging findings in patients with COVID-19: A retrospective multicenter study. Neurology. 2020;95(13):e1868–e1882. doi: 10.1212/WNL.0000000000010112. [DOI] [PubMed] [Google Scholar]
- 18.Serrano G.E., Walker J.E., Arce R., Glass M.J., Vargas D., Sue L.I., Intorcia A.J., Nelson C.M., Oliver J., Papa J., Russell A., Suszczewicz K.E., Borja C.I., Belden C., Goldfarb D., Shprecher D., Atri A., Adler C.H., Shill H.A., Driver-Dunckley E., Mehta S.H., Readhead B., Huentelman M.J., Peters J.L., Alevritis E., Bimi C., Mizgerd J.P., Reiman E.M., Montine T.J., Desforges M., Zehnder J.L., Sahoo M.K., Zhang H., Solis D., Pinsky B.A., Deture M., Dickson D.W., Beach T.G. Mapping of SARS-CoV-2 brain invasion and histopathology in COVID-19 disease. medRxiv. 2021 doi: 10.1101/2021.02.15.21251511. 2021.02.15.21251511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Solomon I.H., Normandin E., Bhattacharyya S., Mukerji S.S., Keller K., Ali A.S., Adams G., Hornick J.L., Padera R.F., Jr, Sabeti P. Neuropathological Features of Covid-19. N. Engl. J. Med. 2020;383(10):989–992. doi: 10.1056/NEJMc2019373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Remmelink M., De Mendonça R., D’Haene N., De Clercq S., Verocq C., Lebrun L., Lavis P., Racu M.L., Trépant A.L., Maris C., Rorive S., Goffard J.C., De Witte O., Peluso L., Vincent J.L., Decaestecker C., Taccone F.S., Salmon I. Unspecific post-mortem findings despite multiorgan viral spread in COVID-19 patients. Crit. Care. 2020;24(1):495. doi: 10.1186/s13054-020-03218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calabrese F., Pezzuto F., Fortarezza F., Hofman P., Kern I., Panizo A., von der Thüsen J., Timofeev S., Gorkiewicz G., Lunardi F. Pulmonary pathology and COVID-19: lessons from autopsy. The experience of European Pulmonary Pathologists. Virchows Arch. 2020;477(3):359–372. doi: 10.1007/s00428-020-02886-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu K.K., Chan S.K., Ma T.M. Posttraumatic stress after SARS. Emerg. Infect. Dis. 2005;11(8):1297–1300. doi: 10.3201/eid1108.041083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park H.Y., Park W.B., Lee S.H., Kim J.L., Lee J.J., Lee H., Shin H.S. Posttraumatic stress disorder and depression of survivors 12 months after the outbreak of Middle East respiratory syndrome in South Korea. BMC Public Health. 2020;20(1):605. doi: 10.1186/s12889-020-08726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honigsbaum M., Krishnan L. Taking pandemic sequelae seriously: from the Russian influenza to COVID-19 long-haulers. Lancet. 2020;396(10260):1389–1391. doi: 10.1016/S0140-6736(20)32134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ravenholt R.T., Foege W.H. 1918 influenza, encephalitis lethargica, parkinsonism. Lancet. 1982;2(8303):860–864. doi: 10.1016/S0140-6736(82)90820-0. [DOI] [PubMed] [Google Scholar]
- 26.Reid A.H., McCall S., Henry J.M., Taubenberger J.K. Experimenting on the past: the enigma of von Economo’s encephalitis lethargica. J. Neuropathol. Exp. Neurol. 2001;60(7):663–670. doi: 10.1093/jnen/60.7.663. [DOI] [PubMed] [Google Scholar]
- 27.Meals R.W., Hauser V.F., Bower A.G. Poliomyelitis-the Los Angeles epidemic of 1934: part II. Cal. West. Med. 1935;43(3):215–222. [PMC free article] [PubMed] [Google Scholar]
- 28.Meals R.W., Hauser V.F., Bower A.G. Poliomyelitis-the Los Angeles epidemic of 1934 : part I. Cal. West. Med. 1935;43(2):123–125. [PMC free article] [PubMed] [Google Scholar]
- 29.Acheson E.D. The clinical syndrome variously called benign myalgic encephalomyelitis, Iceland disease and epidemic neuromyasthenia. Am. J. Med. 1959;26(4):569–595. doi: 10.1016/0002-9343(59)90280-3. [DOI] [PubMed] [Google Scholar]
- 30.Sharpe M.C., Archard L.C., Banatvala J.E., Borysiewicz L.K., Clare A.W., David A., Edwards R.H., Hawton K.E., Lambert H.P., Lane R.J., et al. A report-chronic fatigue syndrome: guidelines for research. J. R. Soc. Med. 1991;84(2):118–121. doi: 10.1177/014107689108400224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vahidy F.S., Pan A.P., Ahnstedt H., Munshi Y., Choi H.A., Tiruneh Y., Nasir K., Kash B.A., Andrieni J.D., McCullough L.D. Sex differences in susceptibility, severity, and outcomes of coronavirus disease 2019: Cross-sectional analysis from a diverse US metropolitan area. PLoS One. 2021;16(1):e0245556. doi: 10.1371/journal.pone.0245556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi T., Ellingson M.K., Wong P., Israelow B., Lucas C., Klein J., Silva J., Mao T., Oh J.E., Tokuyama M., Lu P., Venkataraman A., Park A., Liu F., Meir A., Sun J., Wang E.Y., Casanovas-Massana A., Wyllie A.L., Vogels C.B.F., Earnest R., Lapidus S., Ott I.M., Moore A.J., Shaw A., Fournier J.B., Odio C.D., Farhadian S., Dela Cruz C., Grubaugh N.D., Schulz W.L., Ring A.M., Ko A.I., Omer S.B., Iwasaki A., Iwasaki A., Yale IMPACT Research Team Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature. 2020;588(7837):315–320. doi: 10.1038/s41586-020-2700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sudre C.H., Murray B., Varsavsky T., Graham M.S., Penfold R.S., Bowyer R.C., Pujol J.C., Klaser K., Antonelli M., Canas L.S., Molteni E., Modat M., Jorge Cardoso M., May A., Ganesh S., Davies R., Nguyen L.H., Drew D.A., Astley C.M., Joshi A.D., Merino J., Tsereteli N., Fall T., Gomez M.F., Duncan E.L., Menni C., Williams F.M.K., Franks P.W., Chan A.T., Wolf J., Ourselin S., Spector T., Steves C.J. Attributes and predictors of long COVID. Nat. Med. 2021;27(4):626–631. doi: 10.1038/s41591-021-01292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desforges M., Le Coupanec A., Dubeau P., Bourgouin A., Lajoie L., Dubé M., Talbot P.J. Human coronaviruses and other respiratory viruses: underestimated opportunistic pathogens of the central nervous system? Viruses. 2019;12(1):E14. doi: 10.3390/v12010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arbour N., Day R., Newcombe J., Talbot P.J. Neuroinvasion by human respiratory coronaviruses. J. Virol. 2000;74(19):8913–8921. doi: 10.1128/JVI.74.19.8913-8921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pezzini A., Padovani A. Lifting the mask on neurological manifestations of COVID-19. Nat. Rev. Neurol. 2020;16(11):636–644. doi: 10.1038/s41582-020-0398-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen R., Wang K., Yu J., Howard D., French L., Chen Z., Wen C., Xu Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 2021;11:573095. doi: 10.3389/fneur.2020.573095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrantes F.J. Central nervous system targets and routes for SARS-CoV-2: current views and new hypotheses. ACS Chem. Neurosci. 2020;11(18):2793–2803. doi: 10.1021/acschemneuro.0c00434. [DOI] [PubMed] [Google Scholar]
- 39.Stefano M.L., Kream R.M., Stefano G.B. A novel vaccine employing non-replicating rabies virus expressing chimeric SARS-CoV-2 spike protein domains: functional inhibition of viral/nicotinic acetylcholine receptor complexes. . Med. Sci. Monit. 2020;26:e926016. doi: 10.12659/MSM.926016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farsalinos K., Eliopoulos E., Leonidas D.D., Papadopoulos G.E., Tzartos S., Poulas K. Nicotinic cholinergic system and COVID-19: in silico identification of an interaction between SARS-CoV-2 and nicotinic receptors with potential therapeutic targeting implications. Int. J. Mol. Sci. 2020;21(16):E5807. doi: 10.3390/ijms21165807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai L.K., Hsieh S.T., Chang Y.C. Neurological manifestations in severe acute respiratory syndrome. Acta Neurol. Taiwan. 2005;14(3):113–119. [PubMed] [Google Scholar]
- 42.Kanberg N., Ashton N.J., Andersson L.M., Yilmaz A., Lindh M., Nilsson S., Price R.W., Blennow K., Zetterberg H., Gisslén M. Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology. 2020;95(12):e1754–e1759. doi: 10.1212/WNL.0000000000010111. [DOI] [PubMed] [Google Scholar]
- 43.Yang A.C., Kern F., Losada P.M., Agam M.R., Maat C.A., Schmartz G.P., Fehlmann T., Stein J.A., Schaum N., Lee D.P., Calcuttawala K., Vest R.T., Berdnik D., Lu N., Hahn O., Gate D., McNerney M.W., Channappa D., Cobos I., Ludwig N., Schulz-Schaeffer W.J., Keller A., Wyss-Coray T. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature. 2021;595(7868):565–571. doi: 10.1038/s41586-021-03710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bilinska K., Jakubowska P., Von Bartheld C.S., Butowt R. Expression of the SARS-CoV-2 entry proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: Identification of cell types and trends with age. ACS Chem. Neurosci. 2020;11(11):1555–1562. doi: 10.1021/acschemneuro.0c00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding Y., Wang H., Shen H., Li Z., Geng J., Han H., Cai J., Li X., Kang W., Weng D., Lu Y., Wu D., He L., Yao K. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J. Pathol. 2003;200(3):282–289. doi: 10.1002/path.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esch T., Stefano G.B., Ptacek R., Kream R.M. Emerging roles of blood-borne intact and respiring mitochondria as bidirectional mediators of pro- and anti-inflammatory processes. Med. Sci. Monit. 2020;26:e924337. doi: 10.12659/MSM.924337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z., Okamoto K., Hayashi Y., Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 48.Ulland T.K., Song W.M., Huang S.C., Ulrich J.D., Sergushichev A., Beatty W.L., Loboda A.A., Zhou Y., Cairns N.J., Kambal A., Loginicheva E., Gilfillan S., Cella M., Virgin H.W., Unanue E.R., Wang Y., Artyomov M.N., Holtzman D.M., Colonna M. TREM2 maintains microglial metabolic fitness in alzheimer’s disease. Cell. 2017;170(4):649–663.e13. doi: 10.1016/j.cell.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krupovic M., Dolja V.V., Koonin E.V. The LUCA and its complex virome. Nat. Rev. Microbiol. 2020;18(11):661–670. doi: 10.1038/s41579-020-0408-x. [DOI] [PubMed] [Google Scholar]
- 50.Krupovic M., Dolja V.V., Koonin E.V. Origin of viruses: primordial replicators recruiting capsids from hosts. Nat. Rev. Microbiol. 2019;17(7):449–458. doi: 10.1038/s41579-019-0205-6. [DOI] [PubMed] [Google Scholar]
- 51.Wu K.E., Fazal F.M., Parker K.R., Zou J., Chang H.Y. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst. 2020;11(1):102–108.e3. doi: 10.1016/j.cels.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stefano G.B., Büttiker P., Weissenberger S., Martin A., Ptacek R., Kream R.M. Editorial: the pathogenesis of long-term neuropsychiatric COVID-19 and the role of microglia, mitochondria, and persistent neuroinflammation: a hypothesis. Med. Sci. Monit. 2021;27:e933015. doi: 10.12659/MSM.933015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stefano G.B., Kream R.M. Mitochondrial DNA heteroplasmy as an informational reservoir dynamically linked to metabolic and immunological processes associated with COVID-19 neurological disorders. Cell. Mol. Neurobiol. 2021 doi: 10.1007/s10571-021-01117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh K.K., Chaubey G., Chen J.Y., Suravajhala P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 2020;319(2):C258–C267. doi: 10.1152/ajpcell.00224.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dutta S., Das N., Mukherjee P. Picking up a fight: fine tuning mitochondrial innate immune defenses against RNA viruses. Front. Microbiol. 2020;11:1990. doi: 10.3389/fmicb.2020.01990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elesela S., Lukacs N.W. Role of mitochondria in viral infections. Life (Basel) 2021;11(3):232. doi: 10.3390/life11030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stefano G.B., Esch T., Ptacek R., Kream R.M. Dysregulation of nitric oxide signaling in microglia: multiple points of functional convergence in the complex pathophysiology of alzheimer’s disease. Med. Sci. Monit. doi: 10.12659/MSM.927739. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng J., Dong Y., Ma J., Pan R., Liao Y., Kong X., Li X., Li S., Chen P., Wang L., Yu Y., Yuan Z. Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer’s disease pathology. Sci. Adv. 2021;7(35):eabe3600. doi: 10.1126/sciadv.abe3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stefano G.B., Bilfinger T.V., Fricchione G.L. The immune-neuro-link and the macrophage: postcardiotomy delirium, HIV-associated dementia and psychiatry. Prog. Neurobiol. 1994;42(4):475–488. doi: 10.1016/0301-0082(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 60.Shojaei S., Suresh M., Klionsky D.J., Labouta H.I., Ghavami S. Autophagy and SARS-CoV-2 infection: Apossible smart targeting of the autophagy pathway. Virulence. 2020;11(1):805–810. doi: 10.1080/21505594.2020.1780088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richard A., Tulasne D. Caspase cleavage of viral proteins, another way for viruses to make the best of apoptosis. Cell Death Dis. 2012;3:e277. doi: 10.1038/cddis.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L., Tai Y., Bai C., Gao T., Song J., Xia P., Dong J., Zhao J., Wang F.S. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8(4):420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Graham E.L., Clark J.R., Orban Z.S., Lim P.H., Szymanski A.L., Taylor C., DiBiase R.M., Jia D.T., Balabanov R., Ho S.U., Batra A., Liotta E.M., Koralnik I.J. Persistent neurologic symptoms and cognitive dysfunction in non-hospitalized Covid-19 “long haulers”. Ann. Clin. Transl. Neurol. 2021;8(5):1073–1085. doi: 10.1002/acn3.51350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jose R.J., Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir. Med. 2020;8(6):e46–e47. doi: 10.1016/S2213-2600(20)30216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de la Torre J.C., Stefano G.B. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res. Brain Res. Rev. 2000;34(3):119–136. doi: 10.1016/S0165-0173(00)00043-6. [DOI] [PubMed] [Google Scholar]
- 66.Stefano G.B., Mantione K.J., Capellan L., Casares F.M., Challenger S., Ramin R., Samuel J.M., Snyder C., Kream R.M. Morphine stimulates nitric oxide release in human mitochondria. J. Bioenerg. Biomembr. 2015;47(5):409–417. doi: 10.1007/s10863-015-9626-8. [DOI] [PubMed] [Google Scholar]
- 67.Yang L., Xie X., Tu Z., Fu J., Xu D., Zhou Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021;6(1):255. doi: 10.1038/s41392-021-00679-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stefano G.B., Kream R.M. Convalescent memory T cell immunity in individuals with mild or asymptomatic SARS-CoV-2 infection may result from an evolutionarily adapted immune response to coronavirus and the ‘common cold’. Med. Sci. Monit. 2020;26:e929789. doi: 10.12659/MSM.929789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dance A. The Incrediible diversity of viruses. Nature. 2021;595:22–25. doi: 10.1038/d41586-021-01749-7. [DOI] [PubMed] [Google Scholar]
- 70.Büttiker P., Weissenberger S., Stefano G.B., Kream R.M., Ptacek R. SARS-CoV-2, trait anxiety, and the microbiome. Front. Psychiatry. 2021;12:720082. doi: 10.3389/fpsyt.2021.720082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stefano G.B., Ptacek R., Ptackova H., Martin A., Kream R.M. Selective neuronal mitochondrial targeting in SARS-CoV-2 infection affects cognitive processes to induce ‘brain fog’ and results in behavioral changes that favor viral survival. Med. Sci. Monit. 2021;27:e930886. doi: 10.12659/MSM.930886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stefano G.B., Samuel J., Kream R.M. Antibiotics may trigger mitochondrial dysfunction inducing psychiatric disorders. Med. Sci. Monit. 2017;23:101–106. doi: 10.12659/MSM.899478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aarts C.E.M., Hiemstra I.H., Béguin E.P., Hoogendijk A.J., Bouchmal S., van Houdt M., Tool A.T.J., Mul E., Jansen M.H., Janssen H., van Alphen F.P.J., de Boer J.P., Zuur C.L., Meijer A.B., van den Berg T.K., Kuijpers T.W. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv. 2019;3(22):3562–3574. doi: 10.1182/bloodadvances.2019031609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ngai J.C., Ko F.W., Ng S.S., To K.W., Tong M., Hui D.S. The long-term impact of severe acute respiratory syndrome on pulmonary function, exercise capacity and health status. Respirology. 2010;15(3):543–550. doi: 10.1111/j.1440-1843.2010.01720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang P., Li J., Liu H., Han N., Ju J., Kou Y., Chen L., Jiang M., Pan F., Zheng Y., Gao Z., Jiang B. Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: a 15-year follow-up from a prospective cohort study. Bone Res. 2020;8:8. doi: 10.1038/s41413-020-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bourgonje A.R., Abdulle A.E., Timens W., Hillebrands J.L., Navis G.J., Gordijn S.J., Bolling M.C., Dijkstra G., Voors A.A., Osterhaus A.D., van der Voort P.H., Mulder D.J., van Goor H. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020;251(3):228–248. doi: 10.1002/path.5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lassmann H. Multiple sclerosis pathology. Cold Spring Harb. Perspect. Med. 2018;8(3):a028936. doi: 10.1101/cshperspect.a028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kempuraj D., Selvakumar G.P., Ahmed M.E., Raikwar S.P., Thangavel R., Khan A., Zaheer S.A., Iyer S.S., Burton C., James D., Zaheer A. COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist. 2020;26(5-6):402–414. doi: 10.1177/1073858420941476. [DOI] [PubMed] [Google Scholar]
- 79.Zanin L., Saraceno G., Panciani P.P., Renisi G., Signorini L., Migliorati K., Fontanella M.M. SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir. (Wien) 2020;162(7):1491–1494. doi: 10.1007/s00701-020-04374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Palao M., Fernández-Díaz E., Gracia-Gil J., Romero-Sánchez C.M., Díaz-Maroto I., Segura T. Multiple sclerosis following SARS-CoV-2 infection. Mult. Scler. Relat. Disord. 2020;45:102377. doi: 10.1016/j.msard.2020.102377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moore L., Ghannam M., Manousakis G. A first presentation of multiple sclerosis with concurrent COVID-19 infection. eNeurologicalSci. 2021;22:100299. doi: 10.1016/j.ensci.2020.100299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Maleissye M.F., Nicolas G., Saiag P. Pembrolizumab-induced demyelinating polyradiculoneuropathy. N. Engl. J. Med. 2016;375(3):296–297. doi: 10.1056/NEJMc1515584. [DOI] [PubMed] [Google Scholar]
- 83.Beitz J.M. Parkinson’s disease: a review. Front. Biosci. (Schol. Ed.) 2014;6:65–74. doi: 10.2741/S415. [DOI] [PubMed] [Google Scholar]
- 84.Das T., Hwang J.J., Poston K.L. Episodic recognition memory and the hippocampus in Parkinson’s disease: A review. Cortex. 2019;113:191–209. doi: 10.1016/j.cortex.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bezdicek O., Ballarini T., Buschke H., Růžička F., Roth J., Albrecht F., Růžička E., Mueller K., Schroeter M.L., Jech R. Memory impairment in Parkinson’s disease: The retrieval versus associative deficit hypothesis revisited and reconciled. Neuropsychology. 2019;33(3):391–405. doi: 10.1037/neu0000503. [DOI] [PubMed] [Google Scholar]
- 86.Krajcovicova L., Klobusiakova P., Rektorova I. Gray matter changes in parkinson’s and alzheimer’s disease and relation to cognition. Curr. Neurol. Neurosci. Rep. 2019;19(11):85. doi: 10.1007/s11910-019-1006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ponsen M.M., Stoffers D., Booij J., van Eck-Smit B.L., Wolters E.Ch., Berendse H.W. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann. Neurol. 2004;56(2):173–181. doi: 10.1002/ana.20160. [DOI] [PubMed] [Google Scholar]
- 88.Haddadi K., Ghasemian R., Shafizad M. Basal ganglia involvement and altered mental status: a unique neurological manifestation of coronavirus disease 2019. Cureus. 2020;12(4):e7869. doi: 10.7759/cureus.7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bernaus A., Blanco S., Sevilla A. Glia crosstalk in neuroinflammatory diseases. Front. Cell. Neurosci. 2020;14:209. doi: 10.3389/fncel.2020.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abers M.S., Shandera W.X., Kass J.S. Neurological and psychiatric adverse effects of antiretroviral drugs. CNS Drugs. 2014;28(2):131–145. doi: 10.1007/s40263-013-0132-4. [DOI] [PubMed] [Google Scholar]
- 91.Fardet L., Flahault A., Kettaneh A., Tiev K.P., Généreau T., Tolédano C., Lebbé C., Cabane J. Corticosteroid-induced clinical adverse events: frequency, risk factors and patient’s opinion. Br. J. Dermatol. 2007;157(1):142–148. doi: 10.1111/j.1365-2133.2007.07950.x. [DOI] [PubMed] [Google Scholar]
- 92.Sirois F. [Delirium associated with azithromycin administration]. Can. J. Psychiatry. 2002;47(6):585–586. doi: 10.1177/070674370204700622. [DOI] [PubMed] [Google Scholar]
- 93.Moore H.C. An overview of chemotherapy-related cognitive dysfunction, or ‘chemobrain’. Oncology (Williston Park) 2014;28(9):797–804. [PubMed] [Google Scholar]