

Abstract
Summary
The management of raw nanopore sequencing data poses a challenge that must be overcome to facilitate the creation of new bioinformatics algorithms predicated on signal analysis. SquiggleKit is a toolkit for manipulating and interrogating nanopore data that simplifies file handling, data extraction, visualization and signal processing.
Availability and implementation
SquiggleKit is cross platform and freely available from GitHub at (https://github.com/Psy-Fer/SquiggleKit). Detailed documentation can be found at (https://psy-fer.github.io/SquiggleKitDocs/). All tools have been designed to operate in python 2.7+, with minimal additional libraries.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Nanopore sequencers, such as those manufactured by Oxford nanopore technologies (ONT), generate long sequencing reads by measuring disruptions in ionic current as biopolymers, such as DNA or RNA, transit through a nanopore (Garalde et al., 2018; Laszlo et al., 2014). The resulting signal is then de-convoluted into nucleotide sequence through probabilistic models, which often introduce errors given sampling stochasticity and imperfect models (Rang et al., 2018; Schreiber and Karplus, 2015).
When a strand of nucleotides is sequenced with an ONT device, a data file is generated containing the raw signal and machine metadata. Sequencing runs can produce upwards of 100 million reads, which are grouped into a multitude of agglomerative binary files (.fast5). The yield of ONT sequencers is steadily increasing, and storing large amounts of data is resource intensive. Therefore, discarding the raw data once it has been converted into bases is a practical solution for many labs. However, the current base calling algorithms are limited in that they are error prone and do not include all known nucleotide analogues (Jonkhout et al., 2017). This means that stored raw nanopore files can be revisited and mined to extract additional information as new and more advanced tools become available (Rand et al., 2017; Simpson et al., 2017).
In addition to storing the data for future use, users also need to process a vast quantity of files and extracting pertinent information from the binary file format can be a significant hurdle to overcome when analyzing nanopore data. Once the files are processed and the data are extracted, nanopore signal data remain difficult to navigate given the unfamiliar and noisy nature of single-molecule sensing. Altogether, these challenges hinder the full utilization of nanopore data, the development of new bioinformatics tools and the training of more accurate machine learning algorithms.
Although existing software applications contain methods for file management, data extraction, plotting, segmentation and signal to base comparison, none encapsulate all of these applications (a detailed comparison of these tools is presented in the Supplementary Material). Furthermore, some of these tools are outdated, unsupported, or are limited to specific use cases, such as modification detection or real-time analysis, highlighting the need for a modular, general use toolkit for nanopore signal data mining.
Here, we describe SquiggleKit, a unified toolkit for extracting, processing and plotting raw nanopore sequencing signal data. In addition to data mining applications, SquiggleKit can be used to manage the extensive number of data files generated during nanopore sequencing, as well as a starting point for developing new tools predicated on nanopore signal data.
2 The toolkit
File management and processing: Fast5_fetcher extracts individual fast5 files from an index based on a list of reads of interest, thus reducing both search time and storage space. SquigglePull opens fast5 files, extracts the embedded signal data and converts it into a tab separated (.tsv) format.
Visualization: SquigglePlot is a command line visualization tool for signal data.
Targeting regions of interest in raw signal data: Segmenter identifies the boundaries of relatively long regions of signal attenuation, such as adapter stalls and homopolymer stretches, by measuring the difference in the average signal over a minimal distance, with error tolerance for signal noise. MotifSeq identifies raw signal traces that correspond to a given nucleotide sequence, such as an adapter, barcode, or motif of interest. MotifSeq takes a query nucleotide sequence as input, converts it to a normalized signal trace (i.e. ‘events’), then performs signal-level local alignment using a dynamic programming algorithm. MotifSeq outputs the location of a matching target in the raw signal with an associated P-value (Supplementary Material).
3 Practical example
SquiggleKit can be used to facilitate data management, to generate fine-tuned datasets for machine learning, to visualize signal, to validate de-multiplexing results and to identify motifs of interest without base calling, amongst other applications.
In the following example, we demonstrate how SquiggleKit can be used to validate the 3’ end of a terminal exon using raw nanopore signal data from a cDNA run. Specifically, a cDNA read that aligned to two distinct isoforms (A and B) from a reference transcriptome (Fig. 1A). We will interrogate the raw signal to identify the 3’ end of one of the reference isoforms.
Fig. 1.
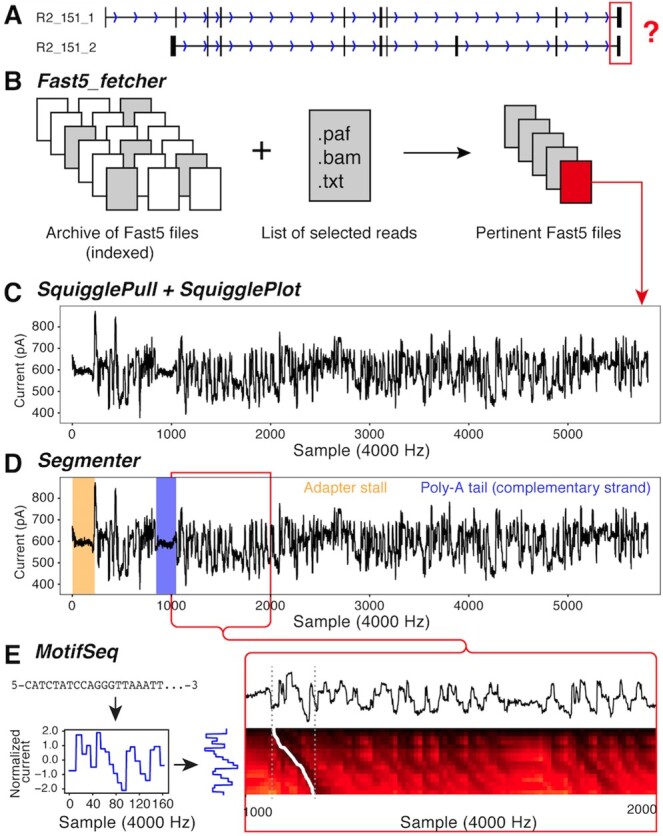
(A) Two similar reference transcripts with similar mapping. (B) Using fast5_fetcher to extract raw files. (C) SquigglePull and SquigglePlot converting to.tsv and plotting the signal. (D) Adapter stalls and poly-A tail identified using Segmenter. (E) Converting last 20 nt of the 3’ end to a signal using a pore current model and aligning this synthetic signal to the empirical signal (dashed grey lines) by backtracking through a dynamic programming matrix (white trace)
First, a list of all read identifiers that mapped to isoform A is extracted from the alignment output into a .txt file. This list is used as input for Fast5_fetcher (Fig. 1B) together with an index of the archived raw data files (.fast5.tar). Fast5_fetcher outputs a subset of extracted .fast5 files instead of all 2 710 372 files that are present in the full dataset. This decreases computational load and processing times of the subsequent steps.
Next, the raw ionic signal for the read of interest is extracted with SquigglePull and plotted with SquigglePlot (Fig. 1C). The .tsv output of SquigglePull is then used as input for Segmenter to identify the adapter stall and poly-A homopolymer sequence (Fig. 1D). The raw signal directly downstream of the poly-A signal is then extracted with Segmenter, thus selecting the 3’ end of the terminal exon in the signal data.
Finally, the nucleotide sequence corresponding to the last 20 nucleotides of the 3’ end of isoform A is converted into normalized signal space. The resulting theoretical current trace is aligned to the empirical signal from Segmenter using MotifSeq (Fig. 1E), returning the position of the sequence in the terminus of the raw signal, a match score of 39.70 and associated P-value of 0.0169 (z-test), thus confirming the exon boundary.
4 Conclusion
SquiggleKit helps researchers extract more information from their data by making raw signal analysis more accessible. We anticipate that this toolkit will facilitate the development of future bioinformatics tools and help create more accurate probabilistic models for nanopore sequencing data analysis, in a timely and user friendly manner.
Supplementary Material
Acknowledgements
The authors would like to thank Shaun Carswell, Hasindu Gamaarachchi, Kai Martin and Tansel Ersavas for testing the toolkit and providing valuable feedback. Many thanks to Tim Mercer who kindly provided the synthetic RNA controls used in the practical example and to Kirston Barton for preparing the samples, sequencing them and being awesome.
Funding
This work was supported by a philanthropic program grant from The Kinghorn Foundation (to M.S.). Computing hardware was supported by a Cancer Institute NSW research equipment grant [REG181268 to M.S.].
Conflict of Interest: J.F. and M.S. have received travel and accommodation expenses to speak at Oxford Nanopore Technologies conferences and are listed as inventors on patent PCT/AU2018/050265 relating to signal alignment.
Contributor Information
James M Ferguson, Kinghorn Centre for Clinical Genomics, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia.
Martin A Smith, Kinghorn Centre for Clinical Genomics, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia; St Vincent's Clinical School, Faculty of Medicine, UNSW Sydney, NSW, Australia.
References
- Garalde D.R. et al. (2018) Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods, 15, 201–206. [DOI] [PubMed] [Google Scholar]
- Jonkhout N. et al. (2017) The RNA modification landscape in human disease. RNA, 23, 1754–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo A.H. et al. (2014) Decoding long nanopore sequencing reads of natural DNA. Nat. Biotechnol., 32, 829–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand A.C. et al. (2017) Mapping DNA methylation with high-throughput nanopore sequencing. Nat. Methods, 14, 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang F.J. et al. (2018) From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol., 19, 90.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber J., Karplus K. (2015) Analysis of nanopore data using hidden Markov models. Bioinformatics, 31, 1897–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J.T. et al. (2017) Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods, 14, 407.. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.