

ABSTRACT
Rotavirus (RV), the most common cause of gastroenteritis in children, carries a high economic and health burden worldwide. RV encodes six structural proteins and six nonstructural proteins (NSPs) that play different roles in viral replication. NSP4, a multifunctional protein involved in various viral replication processes, has two conserved N-glycosylation sites; however, the role of glycans remains elusive. Here, we used recombinant viruses generated by a reverse genetics system to determine the role of NSP4 N-glycosylation during viral replication and pathogenesis. The growth rate of recombinant viruses that lost one glycosylation site was as high as that of the wild-type virus. However, a recombinant virus that lost both glycosylation sites (glycosylation-defective virus) showed attenuated replication in cultured cell lines. Specifically, replications of glycosylation-defective virus in MA104 and HT29 cells were 10- and 100,000-fold lower, respectively, than that of the wild-type, suggesting that N-glycosylation of NSP4 plays a critical role in RV replication. The glycosylation-defective virus showed NSP4 mislocalization, delay of cytosolic Ca2+ elevation, and less viroplasm formation in MA104 cells; however, these impairments were not observed in HT29 cells. Further analysis revealed that assembly of glycosylation-defective virus was severely impaired in HT29 cells but not in MA104 cells, suggesting that RV replication mechanism is highly cell type dependent. In vivo mouse experiments also showed that the glycosylation-defective virus was less pathogenic than the wild-type virus. Taken together, the data suggest that N-glycosylation of NSP4 plays a vital role in viral replication and pathogenicity.
IMPORTANCE Rotavirus is the main cause of gastroenteritis in young children and infants worldwide, contributing to 128,500 deaths each year. Here, we used a reverse genetics approach to examine the role of NSP4 N-glycosylation. An N-glycosylation-defective virus showed attenuated and cell-type-dependent replication in vitro. In addition, mice infected with the N-glycosylation-defective virus had less severe diarrhea than mice infected with the wild type. These results suggest that N-glycosylation affects viral replication and pathogenesis. Considering the reduced pathogenicity in vivo and the high propagation rate in MA104 cells, this glycosylation-defective virus could be an ideal live attenuated vaccine candidate.
KEYWORDS: rotavirus, NSP4, N-glycosylation, viral replication
INTRODUCTION
Rotavirus (RV) is the main cause of gastroenteritis, especially in children under 5 years of age; indeed, RV is the cause of 39% of all gastroenteritis hospitalizations every year, along with 128,500 deaths (1–3). RV vaccines have been introduced globally and show high efficacy (4–8); however, they show lower efficacy in middle- to low-income countries, especially in Southeast Asia and sub-Saharan Africa (9–13). Therefore, it is important to understand the RV replication process to generate future vaccine candidates and antivirals.
RV is a triple-layered virus with an 11 segmented genome that encodes six structural proteins (VP1 to VP4, VP6, and VP7) and six nonstructural proteins (NSP1 to NSP6). Upon viral entry, infectious virions called triple-layered particles (TLPs) transform into double-layered particles (DLPs) (14). The transcriptionally competent RNA inside DLPs replicates the viral mRNA, followed by translation of the viral proteins. NSP2 and NSP5 play roles in formation of viral inclusions (called viroplasm), in which VP1, VP2, VP3, VP6, and segments of the viral RNA genome are assembled to form new DLPs, with VP6 as the outer layer at this stage. Then, NSP4 translocates DLPs to the endoplasmic reticulum (ER) through interaction with its DLP-binding domain. Inside the ER, DLPs acquire another layer comprising VP4 and VP7; this yields new TLPs that are released by either cell lysis or exocytosis (15).
NSP4, encoded by segment 10 of the RV genome, is a multifunctional protein involved in viroporin and enterotoxin activity (16–18). RV NSP4 is comprised of 175 amino acids that form three hydrophobic domains, a coiled-coil domain, and a DLP-binding domain. The hydrophobic domain that spans amino acids 47 to 90 act as a viroporin, which causes Ca2+ leakage from stores within the ER, resulting in higher cytosolic Ca2+ concentrations in infected cells (19, 20). The coiled-coil domain binds to several receptors on the cell membrane, including integrin α1β1 and α2β1, which triggers increases in intracellular Ca2+ concentrations, resulting in vomiting and diarrhea (21–23). The DLP-binding domain is located at the C terminus of NSP4, suggesting its importance during viral assembly (24–27).
At the N terminus of NSP4, mammalian RV group A has two conserved N-glycosylation sites on amino acids 8 and 18. These N-glycosylation sites are highly conserved, regardless of the main host species (28–30). Despite their conservation, the role of these N-glycosylation sites remains elusive. The major obstacle facing those engaged in studies of RV replication mechanisms was the lack of a reverse genetics system. Recently, an entirely plasmid-based RV reverse genetics system was established for simian and human strains of RV and even more recently for murine-like RV strains (31–35). Here, we aimed to dissect the role of NSP4 N-glycosylation using a reverse genetics system.
RESULTS
Generation of recombinant RVs with glycosylation sites on NSP4.
First, we generated recombinant viruses harboring N-glycosylation sites on NSP4 using the reverse genetics system for simian RV strain SA11, as previously described (31). N-glycosylation has the motif of asparagine (N)-X-serine (S)/threonine (T), where X could be any amino acid except proline (P). A recombinant virus in which both N-glycosylation sites were disrupted was rescued by mutating the tyrosine (Y) at position 9 to P and the N at position 18 to alanine (A); the mutant was named rsSA11-Y9P-N18A (Fig. 1A). Other attempts were made to generate glycosylation-defective recombinant virus by introducing another combination of mutations at the two N-glycosylation sites of NSP4; however, only recombinant virus harboring Y9P and N18A mutations could be rescued. We also rescued recombinant viruses in which a single N-glycosylation site was disrupted (these viruses were designated rsSA11-Y9P and rsSA11-N18A) (Fig. 1A). The NSP4 glycosylation patterns of these viruses in HT29 cells were determined by Western blotting (Fig. 1B). The molecular weight of the NSP4 protein of rsSA11-Y9P and rsSA11-N18A viruses was lower than that of the wild-type recombinant strain SA11 (rsSA11) (23 kDa versus ~27 kDa, respectively). Moreover, the molecular weight of rsSA11-Y9P-N18A NSP4 (~20 kDa) was even lower than that of the other recombinant viruses and the wild type (Fig. 1B). To further investigate the glycosylation pattern of these viruses, infected cell lysates were treated with Endo-H, which removes N-glycans. NSP4 from rsSA11, rsSA11-Y9P, and rsSA11-N18A deglycosylated by Endo-H showed a molecular weight similar to that of NSP4 from rsSA11-Y9P-N18A, confirming that the recombinant viruses lost their N-glycosylations due to mutations at amino acid positions 9 and 18. These results also confirm those of a previous study showing that N-glycosylations at both sites are the high-mannose type, which is processed only in the ER without further processing in the Golgi (28, 36). A similar result was obtained using MA104 (see Fig. S1 in the supplemental material).
FIG 1.
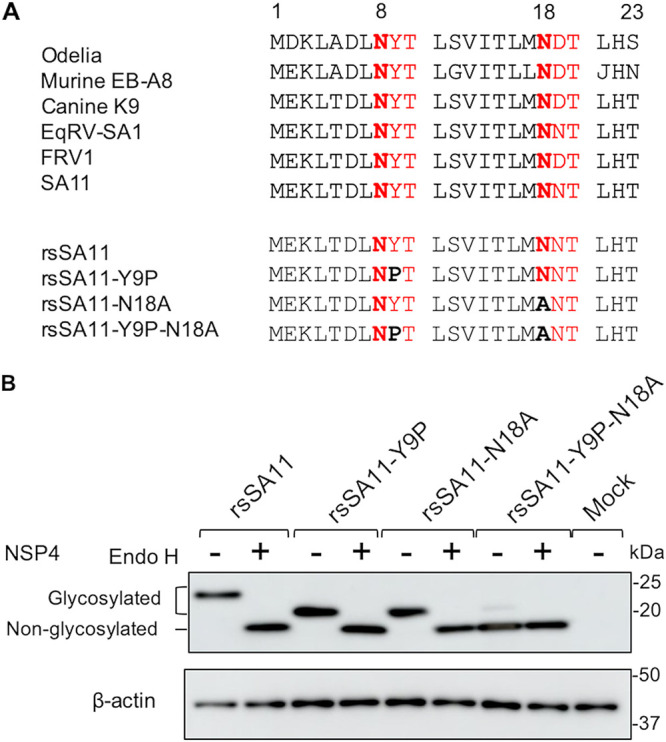
Generation of recombinant NSP4 N-glycosylation-defective viruses. (A) Comparison of RV NSP4 N-glycosylations in multiple host species. The sequences of human (Odelia), murine (murine EB-A8), canine (canine K9), equine (EqRV-SA1), feline (FRV1), and simian (SA11) RVs are shown above. The sequences of NSP4 N-glycosylation-defective recombinant viruses are shown below. Amino acid numbers are shown above the sequences. N-glycosylation sites are highlighted in red. Asparagine residues are highlighted in red bold, and mutations are highlighted in black boldface. The UniProt accession numbers for SA11, Odelia, murine EB-A8, canine K9, EqRV-SA1, and FRV1 are AIC34764, BAF80160, AHN05774, ACH97437, AEX49922, and BAA24414, respectively. (B) N-glycosylation patterns of recombinant NSP4. HT29 cells were infected with rsSA11, rsSA11-Y9P, rsSA11-N18A, or rsSA11-Y9P-N18A at an MOI of 5 in the absence of trypsin. Cell lysates were collected at 18 hpi, followed by treatment with Endo-H or distilled water.
N-glycosylation of NSP4 affects viral replication.
To understand the importance of NSP4 N-glycosylation sites during viral replication, we compared the replication kinetics of rsSA11, rsSA11-Y9P, rsSA11-N18A, and rsSA11-Y9P-N18A in cell lines MA104, CV1, HT29, and PC3 (all infected at a multiplicity of infection [MOI] of 0.01). These cell lines are reported to support robust RV replication (31). The replication rate of rsSA11-Y9P and rsSA11-N18A was similar to that of rsSA11 in all cell lines (Fig. 2A). However, that of rsSA11-Y9P-N18A in MA104 cells at 72 h postinfection (hpi) was ~10-fold lower than that of the other viruses (Fig. 2A). These differences were even more apparent in CV1, HT29, and PC3 cells. The rsSA11-Y9P-N18A titer reached its peak at early time points; however, replication then plummeted, with a 100- to 100,000-fold reduction at 72 hpi compared to that of rsSA11 (Fig. 2A).
FIG 2.

Effect of NSP4 N-glycosylation on viral replication. (A) Replication kinetics of NSP4 N-glycosylation-defective recombinant viruses in multiple cell lines. MA104, CV1, HT29, and PC3 cells were infected with rsSA11, rsSA11-Y9P, rsSA11-N18A, or rsSA11-Y9P-N18A at an MOI of 0.01. Cells were collected at 0, 12, 24, 48, and 72 hpi and freeze-thawed three times at −80°C. IECs derived from human iPSCs were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 0.005. Cells were collected at 0, 24, 48, and 72 hpi and freeze-thawed three times at −80°C. The virus titer was determined in a focus assay using MA104 cells. Foci were detected using antiserum against NSP4. Significant differences in replication between rsSA11 and rsSA11-Y9P-N18A were determined by two-way analysis of variance (ANOVA). *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B) Confluent MA104 cells were infected with the indicated viruses and subjected to a plaque assay. Plaque size (n = 7) was quantified in ImageJ by comparing the pixel size of the plaques with the dish size. The data are expressed as means ± the SD. The significance of the differences in plaque size between rsSA11 and rsSA11-Y9P-N18A was determined using a Mann-Whitney U test. ***, P < 0.001.
Induced-pluripotent stem cells (iPSCs) could be a good model for viral infection due to their versatility and resemblance to human tissue (37–40). Therefore, we used intestinal epithelial cells (IECs) derived from human iPSCs (41) to examine replication of rsSA11 and rsSA11-Y9P-N18A. Consistent with the results from the cell lines, replication of rsSA11-Y9P-N18A was 10-fold lower than that of rsSA11 (Fig. 2A).
A plaque assay using MA104 cells was performed to investigate and compare the infectivity of rsSA11-Y9P-N18A to that of the wild-type. The results showed that rsSA11-Y9P-N18A formed significantly smaller plaques than rsSA11 (Fig. 2B). This is consistent to the result showing that the glycosylation-defective virus exhibited 10-fold lower replication in MA104 cells (Fig. 2A). Taken together, these results suggest that N-glycosylation of NSP4 plays a vital role during viral replication in vitro.
N-glycosylation affects NSP4 localization, cytosolic Ca2+ levels, and viroplasm formation in MA104 cells.
The reduced replication of glycosylation-defective virus was cell-type dependent. Thus, we first analyzed MA104 cells infected with either wild-type or glycosylation-defective viruses since this is the most commonly used cell line for RV replication studies. First, we examined localization of NSP4 because N-glycosylation is reportedly involved in protein localization in other viruses (42). As reported previously, NSP4 from wild-type virus is diffusely localized, with clearly formed puncta surrounding viroplasm (43). Meanwhile, NSP4 from glycosylation-defective virus was aggregated (Fig. 3A). The NSP4 protein colocalized with the ER, with no clear difference between the wild-type and glycosylation-defective viruses (see Fig. S2). These results suggest that N-glycosylation is important for localization of NSP4, and NSP4 is inserted into the ER irrespective of glycosylation status.
FIG 3.
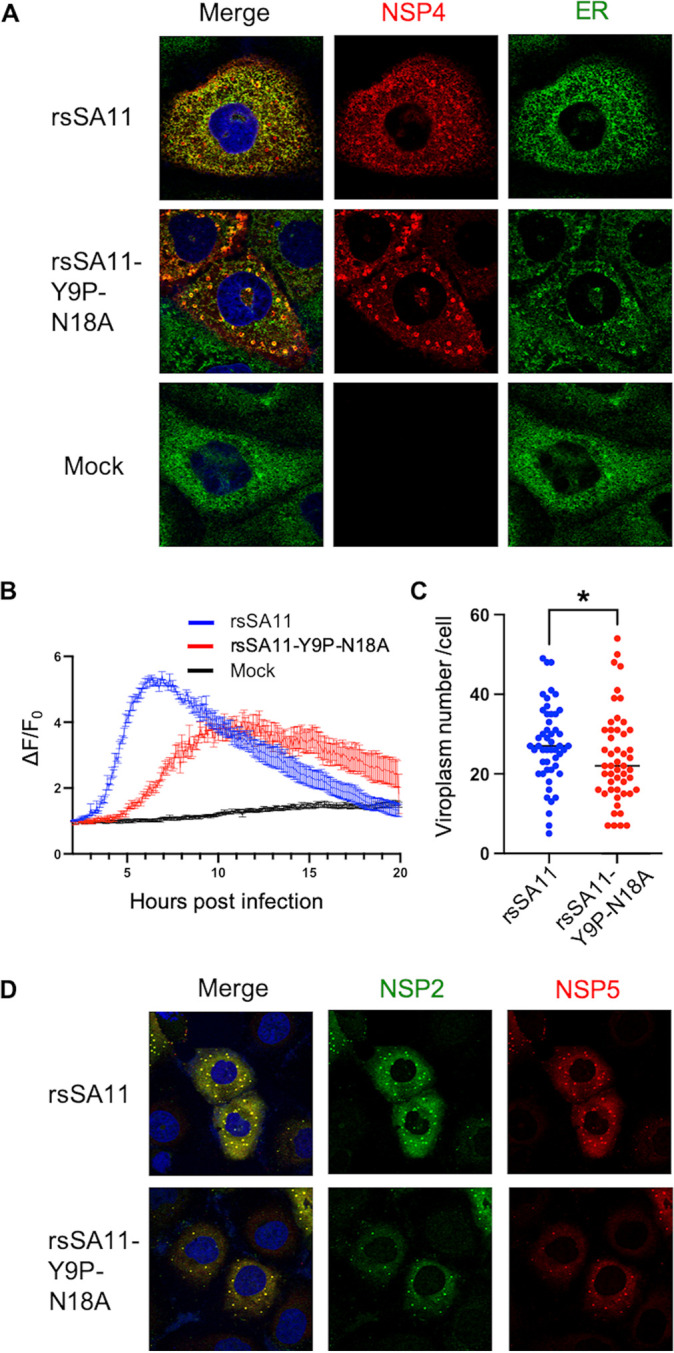
N-glycosylation affects NSP4 localization, cytosolic Ca2+ levels, and viroplasm formation in MA104 cells. (A) Colocalization of NSP4 and the ER. MA104 cells were infected with viruses at an MOI of 1. At 8 hpi, the cells were fixed and stained with anti-NSP4 and anti-ER antibodies. (B) Cytosolic Ca2+ kinetics. MA104 stably expressing GCaMP6s was infected with viruses at an MOI of 5. The fluorescence intensity was measured every 5 min at 37°C and 5% CO2. The Ca2+ kinetics are expressed as relative fluorescence intensity compared to that at 2 hpi (ΔF/F0). (C and D) Viroplasm quantification and imaging. MA104 cells were infected with viruses at an MOI of 1. At 8 hpi, the cells were fixed and stained with anti-NSP2 and anti-NSP5 antibodies. Viroplasm numbers were counted manually under a confocal microscope. The dot-like structures stained by both anti-NSP2 and anti-NSP5 are regarded as viroplasm. Each dot represents the number of viroplasm in each infected cell (n = 50). Statistical differences were determined using the Mann-Whitney U test, with P < 0.05 considered significant (*, P < 0.05).
This mislocalization of NSP4 may affect the function of NSP4 involved in viral replication. NSP4 is a viroporin that elevates cytosolic Ca2+ levels (19). Therefore, we examined cytosolic Ca2+ concentrations in cells stably expressing genetically encoded calcium indicators (i.e., GCaMP6s), as described previously (44). The rsSA11 strain increased cytosolic Ca2+ levels as early as 4 hpi, peaking at 7 hpi; these data are in agreement with a previous report (44) (Fig. 3B). Meanwhile, rsSA11-Y9P-N18A showed delayed Ca2+ elevation, as well as lower peak Ca2+ levels, indicating that N-glycosylation is important for viroporin activity.
Viroplasm is a key structure involved in RV replication. Viroplasm formation is Ca2+-dependent since the process is inhibited in Ca2+-starved cells (45). In addition, a study shows that small interfering RNA (siRNA)-mediated knockdown of NSP4 reduces the amount of viroplasm (25). Therefore, we measured the amount of viroplasm in infected cells using an indirect immunofluorescence assay with anti-NSP2 and anti-NSP5 antibodies. As expected, we found significantly less viroplasm in MA104 cells infected with rsSA11-Y9P-N18A than in cells infected with rsSA11 (Fig. 3C and D). Taken together, the data suggest that N-glycosylation is important for NSP4 localization, viroporin activity, and viroplasm formation in MA104 cells. These impairments may reduce viral replication by 10-fold (Fig. 2A).
N-glycosylation does not affect NSP4 localization, cytosolic Ca2+ elevation, or viroplasm formation in HT29 cells.
Replication of the glycosylation-defective virus in HT29 cells was 100,000-fold lower than that of the wild-type virus; this difference is much bigger than that observed in MA104 cells. Thus, we expect that NSP4 localization, Ca2+ elevation, and viroplasm formation were severely impaired in HT29 cells. However, there was no apparent difference of NSP4 localization between rsSA11 and rsSA11-Y9P-N18A (Fig. 4A). In addition, cytosolic Ca2+ elevation and the amount of viroplasm were also unchanged (Fig. 4B to D). These results suggest that the function of N-glycosylation is cell type dependent, and the 100,000-fold reduction in viral replication is due to other mechanisms.
FIG 4.
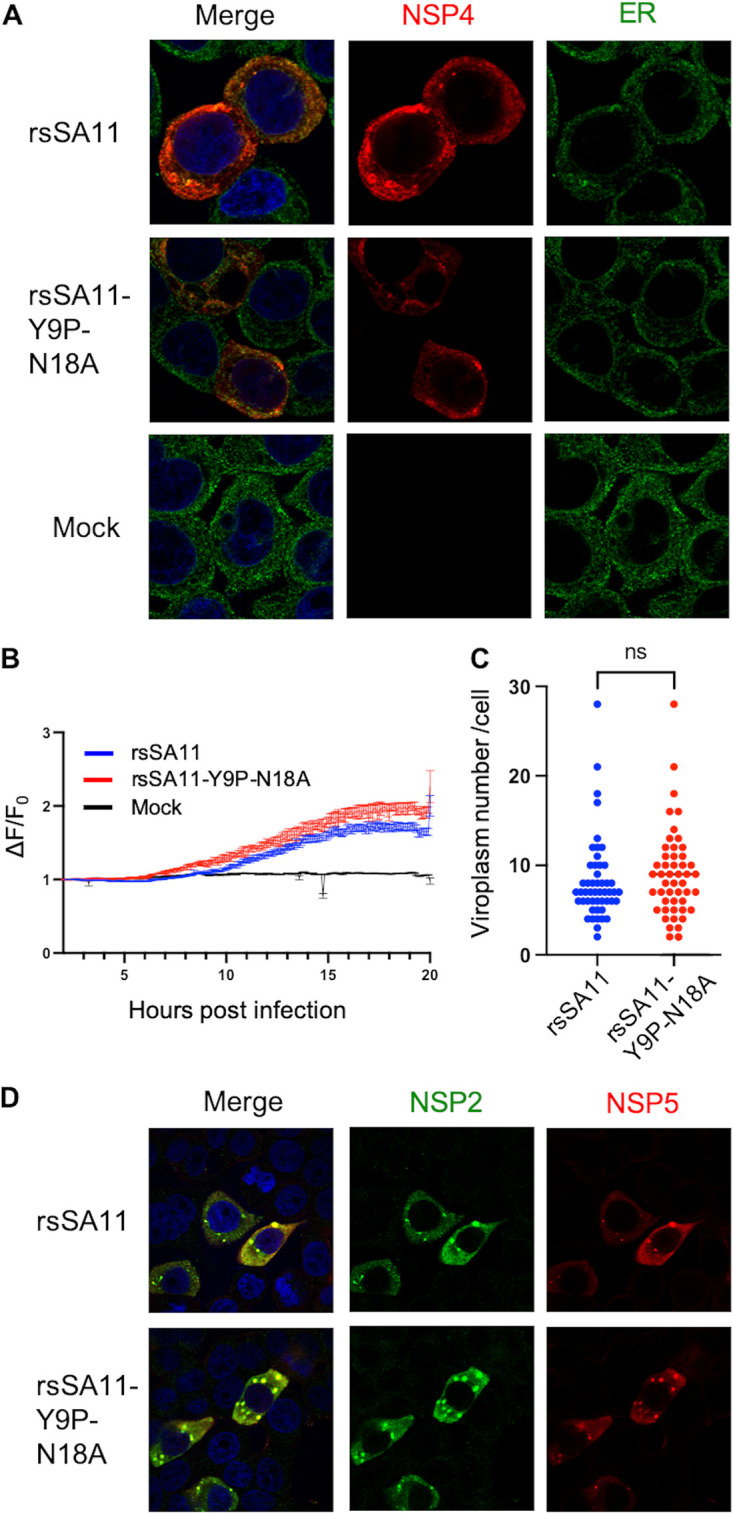
N-glycosylation does not affect NSP4 localization, cytosolic Ca2+ elevation, or viroplasm formation in HT29 cells. (A) Colocalization of NSP4 and the ER. HT29 cells were infected with viruses at an MOI of 1. At 8 hpi, the cells were fixed and stained with anti-NSP4 and anti-ER antibodies. (B) Cytosolic Ca2+ kinetics. HT29 cells stably expressing GCaMP6s were infected with viruses at an MOI of 5. The fluorescence intensity was measured every 5 min at 37°C and 5% CO2. The Ca2+ kinetics are expressed as the relative fluorescence intensity compared to that at 2 hpi (ΔF/F0). (C and D) Viroplasm quantification and imaging. HT29 cells were infected with viruses at an MOI of 1. At 8 hpi, the cells were fixed and stained with anti-NSP2 and anti-NSP5 antibodies. Viroplasm numbers were counted manually under a confocal microscope. The dot-like structures stained by both anti-NSP2 and anti-NSP5 are regarded as viroplasm. Each dot represents the number of viroplasm in each infected cell (n = 50). Statistical differences were determined using the Mann-Whitney U test, with P < 0.05 considered significant (*, P < 0.05).
TLP assembly is impaired by mutation of NSP4 N-glycosylation sites.
NSP4 localizes inside the ER and plays a role in TLP maturation by bridging DLP and the outer viral proteins (25). To investigate whether N-glycosylation of NSP4 plays a critical role in viral assembly, we subjected MA104 and HT29 cells infected with rsSA11 or rsSA11-Y9P-N18A to CsCl gradient centrifugation to confirm the ability of recombinant viruses to generate TLP virions. MA104 cells infected with rsSA11 or rsSA11-Y9P-N18A showed no apparent difference in DLP and TLP formation since both fractions were detected after centrifugation (Fig. 5A). This result was confirmed by silver staining; the VP7 protein was detected in the TLP fractions but not in the DLP fraction (Fig. 5B). Meanwhile, in HT29 cells, rsSA11 formed both the TLP and DLP fractions, while rsSA11-Y9P-N18A only formed the DLP fraction (Fig. 5C and D), suggesting that TLP assembly is inhibited in rsSA11-Y9P-N18A. Silver staining confirmed the absence of TLP; VP7 was undetectable in the sample collected from where TLP is supposed to reside. Taken together, these results imply that NSP4 glycosylation plays an important role in TLP assembly in HT29 cells.
FIG 5.

N-glycosylation affects TLP assembly. (A) Isopycnic CsCl gradient used to isolate TLPs from MA104 infected with rsSA11 or rsSA11-Y9P-N18A. The cells were infected with viruses at an MOI of 1. (B) Silver staining of the TLP and DLP fractions from MA104 cells. Predicted proteins (based on their molecular weight) are indicated on the right. (C and D) Isopycnic CsCl gradient and silver staining of rsSA11 and rsSA11-Y9P-N18A from HT29 cells. (E) Transmission electron microscopy images showing HT29 cells infected with rsSA11 (left) or rsSA11-Y9P-N18A (right). Viroplasm is marked as “V,” and viral particles are marked by arrows.
To further validate the impaired viral assembly, we examined HT29 cells infected with viruses under a transmission electron microscope (TEM). Interestingly, TEM analysis showed that viral particles were present only in cells infected with rsSA11, not cells infected with rsSA11-Y9P-N18A (Fig. 5E). These results confirmed that viral maturation from DLP to TLP was disrupted by the N-glycosylation-defective mutation, at least in HT29 cells.
RNA transcription, protein expression, interferon responses, and NSP4-VP6 interaction were unaffected by mutation of N-glycosylation sites.
To further determine the role of NSP4 N-glycosylation sites in viral replication, as well as the mechanism underlying reduced viral replication in HT29 cells, we compared each step of the RV replication cycle between rsSA11 and rsSA11-Y9P-N18A in HT29 cells. The series of experiments was conducted in HT29 cells because this cell line showed a clear difference in growth between rsSA11 and rsSA11-Y9P-N18A. First, we examined viral RNA transcription kinetics by quantitative reverse transcription-PCR (qRT-PCR). We found that the levels of rsSA11 and rsSA11-Y9P-N18A RNA were similar at 0 to 24 hpi, suggesting that mutation of N-glycosylation sites does not affect transcription of viral RNA (Fig. 6A). Furthermore, observation of similar RNA levels at 0 hpi suggests that viral attachment is unaffected by these mutations (Fig. 6A). Protein expression was also examined by Western blotting at different times postinfection. There was no apparent difference in expression of NSP2 and VP6, also suggesting that the mutations do not affect expression of viral proteins (Fig. 6B).
FIG 6.

RNA transcription, protein translation, interferon responses, and NSP4-VP6 interactions are unaffected by N-glycosylations. (A) Viral RNA kinetics in cells infected with rsSA11 or rsSA11-Y9P-N18A. HT29 cells were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 5. Cell lysates were collected at 0, 4, 8, 12, and 24 hpi. RNA was extracted and subjected to qRT-PCR. The data are expressed as means ± the SD of triplicate samples. (B) Serial Western blots. HT29 cells were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 5, and lysates were collected at the designated time points. The molecular weight (kDa) is shown on the right. (C) IFN responses. HT29 cells were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 5. At 8 hpi, cell lysates were collected and subjected to qRT-PCR. Relative mRNA levels (compared to mock samples) are shown (mock sample was set as 1). The data are expressed as means ± the SD of triplicate samples. Statistical significance was determined using a t test, with P < 0.05 considered significant (*, P < 0.05). (D) NSP4-VP6 interaction. HT29 cells were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 5. At 18 hpi, cell lysates were collected and subjected to coimmunoprecipitation with an anti-NSP4 antibody. The molecular weight is shown on the right (IP, immunoprecipitation; WCL, whole-cell lysate).
Next, we examined the interferon (IFN) response; this is because IFN inhibits several steps of viral replication, including viral assembly (46). RV suppresses the innate immune response; thus, IFN-β stimulation by simian RV infection is almost undetectable in MA104 cells (47). However, it is still detectable in HT29 cells (48), prompting us to hypothesize that the drastic reduction in replication of rsSA11-Y9P-N18A in HT29 is due to an innate immune response. Therefore, we measured the levels of type I and III interferons (IFN-β and IFN-λ) and interferon-stimulated genes (ISGs; IFIT1 and MX1) by qRT-PCR. These genes were upregulated in infected cells compared to mock-infected cells (Fig. 6C). However, there was no significant difference in response between cells infected with rsSA11 and cells infected with rsSA11-Y9P-N18A.
Next, we examined the interaction between NSP4 and VP6, a component of DLP, by coimmunoprecipitation (co-IP) because this is a critical step involved in viral assembly (24). After boiling lysates and co-IP products at 60°C, VP6 was still detected as a trimer. NSP4 could still interact with trimerized VP6 irrespective of glycosylation status (Fig. 6D). This was expected because two N-glycosylation sites, both located in the N-terminal region, are structurally distant from the DLP-binding domain located at the C-terminal region (19, 24). Thus, even though viral attachment, RNA replication, protein expression, IFN responses, and NSP4-VP6 interactions were unchanged, there were still marked differences between the viruses with respect to production of infectious virions.
N-glycosylation is required for interaction with calnexin; however, it does not affect viral replication in HT29 cells.
One of the important functions of N-glycosylation is quality control and protein folding via interaction with ER chaperons, including calnexin (49, 50). NSP4 interacts with calnexin and is folded in an N-glycosylation-dependent manner (51, 52). In addition, knockdown of calnexin reduces virus replication in MA104 cells (52). To investigate whether the NSP4-calnexin interaction is involved in the attenuation of glycosylation-defective virus, we performed a co-IP assay using infected HT29 cell lysates and an anti-calnexin antibody. The results revealed a strong interaction between calnexin and the NSP4 protein of rsSA11, but a weak (almost nonexistent) interaction with that of rsSA11-Y9P-N18A, confirming that N-glycosylation of NSP4 is required for interaction with calnexin (Fig. 7A). The result was confirmed using the N-glycosylation inhibitor, tunicamycin. Exposure of rsSA11 to tunicamycin abolished the interaction with calnexin (Fig. 7A). A reciprocal co-IP experiment (pulldown with anti-NSP4 antibody) further validated this result (see Fig. S3). A similar experiment was performed in MA104 cells, and the results were the same as those in HT29 cells (see Fig. S4).
FIG 7.

N-glycosylation is required for interaction with calnexin; however, it does not affect viral replication in HT29 cells. (A) NSP4-calnexin interaction. HT29 cells were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 5 in the presence or absence of tunicamycin. Cell lysates were collected at 18 hpi and subjected to coimmunoprecipitation with an anti-calnexin antibody. The molecular weight (kDa) is shown on the right. IP, immunoprecipitation; WCL, whole-cell lysate; CNX, calnexin. (B) UPR-related genes. HT29 cells were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 5. At 8 hpi, cell lysates were collected and subjected to qRT-PCR. The relative mRNA levels (compared to mock samples) are shown (mock sample was set as 1). The data are expressed as means ± the SD of triplicate samples. The statistical significance was determined using a t test, with P < 0.05 considered significant (*, P < 0.05). (C and D) siRNA-mediated knockdown of calnexin. HT29 cells were transfected with siRNA targeting calnexin (CNX) or GFP (irrelevant control, Irr). At 48 h posttransfection, the cells were infected with viruses at an MOI of 0.01. At 24 hpi, cells were freeze-thawed and subjected to viral titration. The mock-infected cells were subjected to Western blotting. Relative intensity of calnexin bands was measured using imageJ software. Expression of calnexin was normalized to that of β-actin. The data are expressed as means ± the SD of triplicate samples. Statistical significance was determined using a t test, with P < 0.05 considered significant (**, P < 0.01).
Considering that the calnexin-glycan interaction is essential for glycoprotein folding (49, 50) and that a previous study shows that the NSP4-calnexin interaction is involved in NSP4 protein folding (52), we speculated that glycosylation-defective NSP4 is not properly folded, which may result in deficient viral replication. To validate this hypothesis, we measured expression of the unfolded protein response (UPR)-associated genes sXBP1, CHOP, and GADD34, all of which are upregulated in response to unfolded protein accumulation (53). These genes were upregulated in infected cells compared with mock cells (Fig. 7B). However, there was no significant difference in UPR upregulation between rsSA11 and rsSA11-Y9P-N18A, indicating no differences in terms of unfolded protein accumulation.
In addition, we knocked down calnexin using siRNA. However, knockdown of calnexin did not cause any significant change in replication of either rsSA11 or rsSA11-Y9P-N18A in HT29 cells (Fig. 7C and D). These results suggest that the NSP4-calnexin interaction does not affect viral replication, at least in HT29 cells. Taken together, calnexin and/or misfolding of NSP4 cannot explain the difference in viral replication between rsSA11 and rsSA11-Y9P-N18A in HT29 cells. The mechanism underlying the marked reduction of glycosylation-defective virus replication in HT29 cell remains unclear.
N-glycosylation-defective recombinant viruses are less pathogenic in vivo.
Finally, we conducted mouse challenge experiments to examine whether N-glycosylation affects pathogenicity. Three-day-old BALB/c pups were infected with 2 × 106 focus-forming units (FFU) of rsSA11 or rsSA11-Y9P-N18A via the oral inoculation route. Diarrhea and body weight were observed daily. In agreement with the in vitro results, pups infected with rsSA11-Y9P-N18A showed less severe diarrhea than those infected with rsSA11 (Fig. 8A). Although there was no significant difference in diarrhea levels during the early days of infection, the rsSA11-Y9P-N18A-infected group recovered faster than the rsSA11 group. Moreover, rsSA11-Y9P-N18A-infected pups gained significantly more body weight than rsSA11-infected pups, suggesting that rsSA11-Y9P-N18A is less pathogenic than rsSA11 (Fig. 8B). Thus, N-glycosylation of NSP4 is important for RV pathogenesis in vivo.
FIG 8.

The N-glycosylation-defective recombinant virus is less pathogenic in vivo. (A) Two groups of 3-day-old mouse pups were infected with rsSA11 (n [total] = 14) or rsSA11-Y9P-N18A (n [total] = 10) in a blinded manner. Severity of diarrhea in each mouse was examined daily. The data are presented as the percentages of mice (per group) that had diarrhea. (B) Individual mice in each group were weighed daily and changes in body weight presented as a percentage of the initial weight. The data are expressed as means ± the SD. Statistical differences in diarrhea severity and body weight between rsSA11- and rsSA11-Y9P-N18A-infected mice were determined using two-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
NSP4 is a multifaceted RV protein that plays various roles in the virus life cycle; thus, it has been studied thoroughly. However, previous studies were based mostly on recombinant proteins and siRNA rather than on recombinant viruses. Furthermore, the function of NSP4 N-glycosylation remains unknown. Here, we used a reverse genetics system to generate N-glycosylation-defective recombinant viruses and demonstrated that N-glycosylation of RV NSP4 affects viral replication and pathogenesis. In addition, we demonstrated that the RV replication mechanism is highly cell type dependent.
Replication of the fully N-glycosylation-defective virus was lower than that of the wild-type or single N-glycosylation-defective recombinant viruses in cell lines and iPSC-derived IECs. Interestingly, the difference in viral replication between wild-type and glycosylation-defective virus in MA104 cells was small (~10-fold), while that in the other cell line was large (100- to 100,000-fold) (Fig. 2A). In MA104 cells, which are used commonly for RV replication studies, NSP4 mislocalization, delayed Ca2+ elevation, and less viroplasm formation were observed in cells infected with rsSA11-Y9P-N18A (Fig. 3). However, TLP assembly itself was relatively unaffected (Fig. 5). These results are consistent with those of previous studies showing that (i) NSP4 has viroporin activity (19), (ii) NSP4 siRNA reduces viroplasm formation but not TLP formation in MA104 cells (25, 54), and (iii) lack of cytosolic Ca2+ disrupts viroplasm formation (45). Thus, N-glycosylation is important for the function of NSP4 in MA104 cells. Taken together, our data suggest that loss of N-glycosylation causes NSP4 mislocalization, which may impair viroporin activity and cytosolic Ca2+ elevation. Attenuated cytosolic Ca2+ elevation reduces the amount of viroplasm, which in turn reduces viral replication.
Meanwhile, in HT29 cells in which replication of glycosylation-defective virus was reduced by 100,000-fold, there was no difference in NSP4 localization, cytosolic Ca2+ levels, or amount of viroplasm between cells infected with rsSA11 and cells infected with rsSA11-Y9P-N18A. This indicates that the mechanism underlying RV replication is different in MA104 and HT29 cells. In addition, this also indicates that the function of N-glycosylation is cell-type dependent. Only MA104 cells showed a relatively mild reduction in viral replication compared with other epithelial cell lines (HT29, CV1, and PC3). MA104 is the most commonly used cell line for RV replication studies because it supports robust RV replication (55). Our results suggest that the findings in MA104 cannot be simply translated to other cell lines, including HT29. In addition, our data demonstrate that iPSC-derived IECs could be a good model for studying the RV replication machinery (Fig. 2B).
Because the mutant virus lacks the NSP4-calnexin interaction (Fig. 7A), we initially hypothesized that glycosylation-defective NSP4 is misfolded, resulting in impaired viral assembly. However, this hypothesis could not explain the marked attenuation of glycosylation-defective virus in HT29 cells. Unexpectedly, knockdown of calnexin did not affect viral replication in HT29 cells (Fig. 7C and D). This contradicts a previous report showing that knockdown of calnexin in MA104 cells mildly, but significantly, reduces virus replication (by ~2-fold) (52). Again, this may be due to differences in cell type (MA104 versus HT29 cells). Thus, the exact mechanism underlying attenuation in HT29 cells remains elusive. Nevertheless, we (at least in part) clarified that the mechanism underlying impaired viral assembly is not related to cytosolic Ca2+ concentrations, IFN responses, the NSP4-VP6 interactions, or the NSP4-calnexin interaction. Further investigations, including recruitment of VP7 to form TLP (which is also facilitated by NSP4), are needed (56) to identify the differences between MA104 and HT29 with respect to the viral replication machinery.
Mice infected with rsSA11-Y9P-N18A showed milder symptoms than those infected with the wild-type rsSA11, making the former a potential candidate for live attenuated RV vaccine design. The high titer in MA104 cells (~107 FFU/mL), coupled with less severe pathogenicity in vivo, supports this notion. Although we confirmed that the N-glycosylation-defective recombinant virus triggers similar levels of IFN and ISGs production in vitro (Fig. 6C), the immunogenicity of the virus in vivo remains to be determined. In addition, the pathogenicity of a glycosylation-defective virus needs to be examined by comparing its effects with those in mock-infected pups. Furthermore, introduction of N-glycosylation-defective mutations into murine-like RV is warranted to examine the effects on pathogenicity (34, 35).
In summary, we demonstrated that N-glycosylation of RV NSP4 affects viral replication and pathogenicity. N-glycosylation-defective RV showed attenuated and cell-type-dependent replication in vitro. In MA104 cells, the glycosylation-defective virus led to NSP4 mislocalization, delayed Ca2+ elevation, and less viroplasm, which in turn reduced viral replication slightly. Meanwhile, in HT29 cells, viral assembly was severely impaired, although the underlying mechanism remains elusive. Also, the N-glycosylation-defective virus is less pathogenic than the wild-type in mice pups. This study also highlighted differences in the RV replication machinery in different cell lines. Rather than relying on MA104 cells, using various cell lines and/or human iPSC-derived IECs is ideal for examining RV replication. Further research into the detailed mechanisms that determine how N-glycosylation of NSP4 affects viral assembly in HT29 will be of interest.
MATERIALS AND METHODS
Cells and viruses.
Monkey kidney epithelial MA104 cells, monkey kidney CV1 cells, human colon cancer HT29 cells, and human prostate cancer PC3 cells were cultured in Dulbecco modified Eagle medium (DMEM) (Nacalai Tesque) supplemented with 5% fetal bovine serum (FBS; Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Nacalai Tesque). Baby hamster kidney fibroblast cells stably expressing T7 RNA polymerase (BHK-T7) (31) were cultured in DMEM supplemented with 5% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 4 μg/mL puromycin (Sigma-Aldrich). IECs derived from the human iPSC line, TkDN4-M, were cultured as previously described (37, 41). Briefly, IECs were cultured as organoids in BD Matrigel Matrix (BD Biosciences) and culture medium (Advanced DMEM/F-12 [Gibco], supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM GlutaMAX [Gibco], 10 mM HEPES [Gibco], 1× B-27 supplement [Gibco], 50 ng/mL recombinant murine EGF [Peprotech], 10 μM SB202190 [Sigma], 500 nM A83-01 [Tocris], 200 ng/mL recombinant mouse Wnt3a [R&D], 750 ng/mL recombinant human R-Spondin1 [R&D], and 100 ng/mL recombinant human Noggin [R&D]). To differentiate IECs as a monolayer, 2 × 104 cells were seeded into 96-well plates precoated with 2.5% Matrigel. The culture medium was replaced with differentiation medium (Advanced DMEM/F-12, supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM GlutaMAX, 10 mM HEPES, 1× B-27 supplement, 50 ng/mL recombinant murine EGF, 500 nM A83-01, 375 ng/mL recombinant human R-Spondin1, and 50 ng/mL recombinant human Noggin) on days 2 and 4 postseeding. Monolayered IECs were used for experiments 6 days after seeding. Recombinant strain SA11 (rsSA11) was generated by reverse genetics, as previously described (31). Simian RV strain SA11 was propagated in MA104 cells cultured in DMEM supplemented with 0.5 μg/mL trypsin (Sigma-Aldrich). Viruses were inoculated apically unless stated otherwise.
Antibodies.
A monoclonal antibody specific for NSP4 was generated. Briefly, recombinant baculovirus expressing SA11 NSP4 was generated using the Bac-to-Bac system (Thermo Fisher Scientific). Purified NSP4 conjugated to an Alhydrogel adjuvant (Invivogen) was injected into the footpads of ICR mice (Japan CLEA). Immunization was repeated six times at 5-day intervals. At 2 days after the final immunization, lymphocytes isolated from popliteal lymph nodes were fused with PAI myeloma cells using 50% polyethylene glycol (Hybri-Max; Sigma-Aldrich). Hybridoma cells were cultured in RPMI 1640 supplemented with 10% FBS and 1× HAT medium (Hybri-Max). The monoclonal antibody was purified from culture supernatant of a single hybridoma clone and used for the experiments. Rabbit anti-NSP2 antiserum was raised against a synthetic SA11 NSP2 peptide spanning amino acid residues 299 to 312 (Eurofins Genomics). Rabbit anti-NSP4 antiserum was raised against a synthetic SA11 NSP4 peptide spanning amino acid residues 158 to 171 (Eurofins Genomics). Guinea pig anti-NSP5 antiserum was raised against a synthetic SA11 NSP5 peptide spanning amino acid residues 48 to 66 (Eurofins Genomics). Mouse anti-VP6 antiserum was generated as previously described (57). Other antibodies were anti-KDEL (ER marker; Enzo), anti-calnexin (Abcam), anti-β-actin (Sigma-Aldrich), and anti-tubulin (Sigma-Aldrich).
Plaque assay and focus assay.
The plaque assay was performed as previously described (33). Plaque size was determined using ImageJ. A monolayer of MA104 cells or iPSC-derived IECs seeded into 96-well-plates (1.5 × 104 or 2 × 104 cells/well, respectively) was infected with recombinant virus for 18 h. The cells were then fixed for 30 min in absolute ethanol. Then, the cells were incubated for 1 h at 37°C with antiserum specific for RV-NSP4 (1:3,000), followed by CF488A goat anti-rabbit IgG (Biotium; 1:2,000). The number of FFU was counted manually under an Axio Observer 7 fluorescence microscope (Zeiss). The virus titer was expressed as the number of FFU/mL. The MOI was calculated as FFU/cell.
Plasmid construction.
The pT7-NSP4-Y9P, pT7-NSP4-N18A, and pT7-NSP4-Y9P-N18A plasmids were generated by PCR mutagenesis; nucleotide positions 66 to 68 were mutated from TAT (Y) to CAC (P) and/or nucleotide positions 93 to 95 from AAC (N) to GCC (A), using pT7-NSP4SA11 as the template (31). Plasmid sequences were confirmed by DNA sequencing.
Reverse genetics system.
Recombinant viruses were recovered from cells following plasmid transfection, as previously described (31, 57). In short, a subconfluent monolayer of BHK-T7 cells in a 12-well-plate (2 × 105 cells/well) was transfected with 11 plasmids encoding each gene segment of strain SA11 (0.25 μg of each); expression plasmids pCAG-NSP2SA11, pCAG-NSP5SA11, pCAG-D1R, and pCAG-D12L (0.25 μg of each); and pCAG-FAST (0.005 μg), using 2 μL of TransIT-LT1 (Mirus) per microgram of plasmid. After 48 h, MA104 cells (5 × 104 cells/well) were added to the transfected BHK-T7 cells and incubated for 3 days in the presence of 0.5 μg/mL trypsin (Sigma-Aldrich). The cells were lysed by freeze/thawing and then transferred to fresh MA104 cells. To generate N-glycosylation recombinant viruses rsSA11-Y9P, rsSA11-N18A, and rsSA11-Y9P-N18A, the pT7-NSP4SA11 plasmid was replaced with pT7-NSP4-Y9P, pT7-NSP4-N18A, and pT7-NSP4-Y9P-N18A, respectively. Recombinant virus sequences were confirmed by Sanger sequencing.
Endo-H treatment.
MA104 and HT29 cells were infected with viruses at MOIs of 10 and 5, respectively. The infected cells were harvested from MA104 and HT29 cells at 10 and 18 hpi, respectively. The cells were lysed and deglycosylated using Endo-H (NEB). The treated lysates were subjected to immunoblotting as described below.
Immunoblot analysis.
MA104 or HT29 cells were washed with ice cold phosphate-buffered saline (PBS). The cells were lysed for 30 min on ice using radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) and then mixed with 2× Laemmli sample buffer (50 mM Tris-HCl [pH 6.8], 2% SDS, 6% 2-mercaptoethanol, 10% glycerol, and 0.01% bromophenol blue) and boiled at 60°C for 5 min unless stated otherwise. Proteins were separated by SDS-PAGE in 10% acrylamide gels (TGX FastCast Acrylamide Solutions; Bio-Rad), according to the manufacturer’s instructions. Proteins were transferred to an Immobilon-P polyvinylidene difluoride membrane (Merck) and then incubated with appropriate primary antibodies, followed by an appropriate horseradish peroxidase-conjugated secondary antibody. The immune complex was visualized using Chemi-Lumi One Ultra (Nacalai Tesque), according to the manufacturer’s instructions. Images were acquired by an Amersham Imager 600 (GE Healthcare Life Sciences) or Amersham ImageQuant 800 (Cytiva).
Growth kinetics.
A monolayer of MA104, CV1, HT29, or PC3 cells in a 24-well-plate (1 × 105, 1 × 105, 2 × 105, and 1 × 105 cells/well, respectively) was infected with virus at an MOI of 0.01 (titrated on MA104 cell). A monolayer of iPSC-derived IECs in 96-well plates (2 × 104 cells/well) was infected with virus at an MOI of 0.005 (titrated on iPSC-derived IECs). This method was used because IECs are less susceptible to virus infection. After 1 h, the cells were washed twice with PBS and cultured in DMEM supplemented with 0.5 μg/mL trypsin. At the indicated time points, cells were frozen at −80°C. After freeze/thawing twice, the virus titer was determined in a focus-forming assay.
Indirect immunofluorescence assay.
A monolayer of MA104 or HT29 cells was established on 24-well-plates (1 × 105 cells and 2 × 105 cells/well, respectively) containing glass coverslips. Cells were infected with virus at an MOI of 1.0. At 8 hpi, cells were fixed with 3.4% formaldehyde (Nacalai Tesque), permeabilized for 15 min with 0.5% Triton X-100, and then blocked with 3% skim milk for 1 h. The primary antibody was diluted to 1:1,000 in 3% skim milk and added for 1 h. After washing, the secondary antibody was diluted to 1:1,000 in 3% skim milk and added for 1 h. Nuclei were stained using Hoechst (Thermo Fisher). The glass coverslips were mounted with VectaMount AQ aqueous mounting medium (Vector) and samples were visualized under a C2+Eclipse Ti2 confocal microscope (Nikon). Colocalization was analyzed using NIS-Elements software (Nikon).
Cytosolic Ca2+ quantification assay.
MA104 and HT29 cells stably expressing a calcium indicator were generated as previously described (44). pLVX-puro_GCaMP6s was a gift from Joseph Hyser (Addgene plasmid 164589; RRID: Addgene_164589). Briefly, a monolayer of MA104 or HT29 cells was seeded into a 6-well plate (~4 × 104 cells/well) and transduced with lentivirus containing pLVX-puro_GCaMP6s. Two days after transduction, the MA104 and HT29 cells were treated with puromycin at concentrations of 5 and 1 μg/mL, respectively, for selection. Surviving cells were subjected to cell sorting and clonal selection using a FACSAria II (BD). A monolayer of MA104-GCaMP6S and HT29-GCaMP6S was cultured in 8-well cover glass chamber slides. Cells were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 5. Fluorescence intensity was quantified at 37°C and 5% CO2 using a C2+Eclipse Ti2 confocal microscope (Nikon) and a stage-top incubator (Tokai Hit). For each experiment, five positions per group were selected and measured every 5 min for 20 h. Fluctuations in Ca2+ were expressed as relative fluorescence intensity compared to that at 2 hpi (F/F0) (44).
Isopycnic gradient centrifugation.
MA104 or HT29 cells were infected with rsSA11 or rsSA11-Y9P-N18A at an MOI of 1. At 1 hpi, cells were washed with PBS to remove unattached viruses, followed by addition of DMEM containing 0.5 μg/mL trypsin. Samples were freeze-thawed once at 2 or 3 days postinfection. The supernatant was centrifuged at 153,700 × g for 90 min at 12°C in a 32Ti rotor using a Optima XE-90 ultracentrifuge (Beckman Coulter) to pellet the virus. The pellet was resuspended in PBS, overlaid onto 40 and 55% CsCl (wt/vol) dissolved in PBS, and centrifuged at 148,900 × g for 17 h at 12°C in a 55Ti rotor. The TLP and DLP fractions were extracted and dialyzed using Slide-A-Lyzer Dialysis Cassette A (Thermo Scientific). For HT29 cells, the TLP fraction of the glycosylation-defective virus was invisible. Thus, the sample was collected from where TLP is supposed to reside. The samples were then analyzed using SDS-PAGE and silver staining (EZStainSilver; Atto).
Transmission electron microscopy.
HT29 cells were cultured on a polystyrene cover slip (Cell Desk; Sumitomo Bakelite). Then, the cells were infected with viruses at an MOI of 5 and fixed at 6 hpi with 2% formaldehyde and 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.4). The cells were washed three times for 5 min in the same buffer and postfixed for 1 h with 1% osmium tetroxide and 1% potassium ferrocyanide in 0.1 M sodium phosphate buffer (pH 7.4). Samples were then dehydrated in a graded series of ethanol solutions, embedded in Epon812 (TAAB), cut into 80-nm ultrathin sections, and stained with saturated uranyl acetate and lead citrate solution. Electron micrographs were obtained under a JEM-1400plus TEM (JEOL).
RNA quantification.
A monolayer of HT29 cells in a 24-well-plate (2 × 105 cells/well) was infected with virus at an MOI of 5 at 4°C. After 1 h, the cells were washed three times with PBS and cultured at 37°C with DMEM (designated as 0 hpi). At the designated time points, the supernatant was collected, cells were washed using PBS, and 500 μL of Sepasol (Nacalai Tesque) was added, followed by RNA extraction. RNA extracted from cell lysates was subjected to DNase treatment using DNase RT-grade (Nippon Gene). The RNA was subjected to qRT-PCR to measure expression of viral RNA and human mRNA. Viral RNA was detected using a Thunderbird one-step kit (Toyobo). Human genes, including GAPDH, IFN-related genes, and UPR-related genes, were quantified using a iTaq Universal SYBR Green One-Step Reaction kit (Bio-Rad). The levels of viral RNA and human mRNA were normalized to that of GAPDH. The primers and probes used are available upon request.
Coimmunoprecipitation assay.
MA104 and HT29 cells infected with viruses at an MOI of 5 were washed with ice cold PBS at 10 and 18 hpi, respectively. Then, TMGK buffer (20 mM Tris [pH 8], 20 mM MgCl2, 110 mM KCl, 1% Triton X-100) was added on ice for 30 min. Coimmunoprecipitation was performed using Dynabeads protein G (Thermo Fisher Scientific) or protein G-Sepharose (Cytiva). Eluted samples were mixed with Laemmli sample buffer, followed by Western blotting (see above).
Tunicamycin treatment.
A monolayer of HT29 cells was seeded into 6-well plates (8 × 105 cells/well) and inoculated with rsSA11 or rsSA11-Y9P-N18A at an MOI of 5. At 1 hpi, the medium was removed and cells were washed with PBS prior to addition of FBS-free medium supplemented with 1.5 μg/mL tunicamycin (Sigma-Aldrich) or dimethyl sulfoxide. Cells were collected at 18 hpi for coimmunoprecipitation.
siRNA transfection.
The siRNAs targeting calnexin and GFP (irrelevant control) were described in a previous study (52). HT29 cells were seeded in 24-well plates (1 × 105 cells/well). On the next day, the cells were transfected with 20 pmol of siRNA using Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific). At 48 h posttransfection, the cells were infected with virus at an MOI of 0.01 for 1 h at 37°C. The cells were then washed with PBS, followed by addition of DMEM containing 0.5 μg/mL trypsin. The cells were incubated for a further 24 h at 37°C, freeze-thawed three times, and subjected to viral titration. At the same time, mock-infected cells were lysed with RIPA buffer and subjected to Western blotting to confirm knockdown efficiency. The band intensity was quantified by ImageJ software. The expression level of calnexin was normalized to that of β-actin.
In vivo experiments.
All animal experiments were approved by the institutional animal experiment committee (approval BidouR03-10-0). Pregnant BALB/cAJcl mice were purchased from CLEA Japan, Inc. Three days after delivery, pups were randomized into four groups and infected orally with either 2 × 106 FFU of rsSA11 or rsSA11-Y9P-N18A (infection was blinded). The percentage of pups with diarrhea was determined as previously described (58); briefly, severity of diarrhea was scored as follows: 0 = no diarrhea, 1 = pasty, 2 = semiliquid, and 3 = liquid. A score ≥2 was considered “diarrhea.” At the end of the experiment, the average percentage of pups with diarrhea in each group was calculated. Body weight was also measured daily; changes in individual mice were expressed as a relative increase compared with the initial weight.
Statistical analysis.
Data analysis was performed using Prism 9 (GraphPad Software, Inc.). Data are expressed as means ± the standard deviations (SD) of at least two independent experiments (each with three biological replicates unless stated otherwise). P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We thank Y. Maeda (Laboratory of Viral Dynamism Research, International Research Center for Infectious Diseases, Osaka University) for fruitful discussion, M. Onishi for technical assistance, M. Yoshida for secretarial work, Y. Kabumoto for technical assistance with cell sorting, and H. Oomori for technical assistance with transmission electron microscopy.
This study was supported in part by AMED grants JP21fk0108018 and JP21fk0108122, by KAKENHI grants JP18H02663, JP21K19379, and JP21H02739, and by JST Moonshot R&D-MILLENNIA program grant JPMJMS2025.
Footnotes
Supplemental material is available online only.
Contributor Information
Yuta Kanai, Email: y-kanai@biken.osaka-u.ac.jp.
Takeshi Kobayashi, Email: tkobayashi@biken.osaka-u.ac.jp.
Susana López, Instituto de Biotecnologia/UNAM.
REFERENCES
- 1.Munos MK, Walker CL, Black RE. 2010. The effect of rotavirus vaccine on diarrhoea mortality. Int J Epidemiol 39(Suppl 1):i56–i62. 10.1093/ije/dyq022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Troeger C, Khalil IA, Rao PC, Cao S, Blacker BF, Ahmed T, Armah G, Bines JE, Brewer TG, Colombara DV, Kang G, Kirkpatrick BD, Kirkwood CD, Mwenda JM, Parashar UD, Petri WA, Jr, Riddle MS, Steele AD, Thompson RL, Walson JL, Sanders JW, Mokdad AH, Murray CJL, Hay SI, Reiner RC, Jr.. 2018. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 years. JAMA Pediatr 172:958–965. 10.1001/jamapediatrics.2018.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Estes M, Greenberg H. 2013. Rotaviruses, p 1347–1401. In Fields virology, 6th ed. Lippincott/Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Rha B, Tate JE, Payne DC, Cortese MM, Lopman BA, Curns AT, Parashar UD. 2014. Effectiveness and impact of rotavirus vaccines in the United States: 2006–2012. Expert Rev Vaccines 13:365–376. 10.1586/14760584.2014.877846. [DOI] [PubMed] [Google Scholar]
- 5.Jonesteller CL, Burnett E, Yen C, Tate JE, Parashar UD. 2017. Effectiveness of rotavirus vaccination: a systematic review of the first decade of global postlicensure data, 2006–2016. Clin Infect Dis 65:840–850. 10.1093/cid/cix369. [DOI] [PubMed] [Google Scholar]
- 6.Leshem E, Moritz RE, Curns AT, Zhou F, Tate JE, Lopman BA, Parashar UD. 2014. Rotavirus vaccines and health care utilization for diarrhea in the United States (2007–2011). Pediatrics 134:15–23. 10.1542/peds.2013-3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gray J. 2011. Rotavirus vaccines: safety, efficacy and public health impact. J Intern Med 270:206–214. 10.1111/j.1365-2796.2011.02409.x. [DOI] [PubMed] [Google Scholar]
- 8.Tate JE, Haynes A, Payne DC, Cortese MM, Lopman BA, Patel MM, Parashar UD. 2013. Trends in national rotavirus activity before and after introduction of rotavirus vaccine into the national immunization program in the United States, 2000 to 2012. Pediatr Infect Dis J 32:741–744. 10.1097/INF.0b013e31828d639c. [DOI] [PubMed] [Google Scholar]
- 9.Phua KB, Lim FS, Lau YL, Nelson EA, Huang LM, Quak SH, Lee BW, Teoh YL, Tang H, Boudville I, Oostvogels LC, Suryakiran PV, Smolenov IV, Han HH, Bock HL. 2009. Safety and efficacy of human rotavirus vaccine during the first 2 years of life in Asian infants: randomised, double-blind, controlled study. Vaccine 27:5936–5941. 10.1016/j.vaccine.2009.07.098. [DOI] [PubMed] [Google Scholar]
- 10.Armah GE, Sow SO, Breiman RF, Dallas MJ, Tapia MD, Feikin DR, Binka FN, Steele AD, Laserson KF, Ansah NA, Levine MM, Lewis K, Coia ML, Attah-Poku M, Ojwando J, Rivers SB, Victor JC, Nyambane G, Hodgson A, Schodel F, Ciarlet M, Neuzil KM. 2010. Efficacy of pentavalent rotavirus vaccine against severe rotavirus gastroenteritis in infants in developing countries in sub-Saharan Africa: a randomised, double-blind, placebo-controlled trial. Lancet 376:606–614. 10.1016/S0140-6736(10)60889-6. [DOI] [PubMed] [Google Scholar]
- 11.Madhi SA, Cunliffe NA, Steele D, Witte D, Kirsten M, Louw C, Ngwira B, Victor JC, Gillard PH, Cheuvart BB, Han HH, Neuzil KM. 2010. Effect of human rotavirus vaccine on severe diarrhea in African infants. N Engl J Med 362:289–298. 10.1056/NEJMoa0904797. [DOI] [PubMed] [Google Scholar]
- 12.Zaman K, Dang DA, Victor JC, Shin S, Yunus M, Dallas MJ, Podder G, Vu DT, Le TP, Luby SP, Le HT, Coia ML, Lewis K, Rivers SB, Sack DA, Schodel F, Steele AD, Neuzil KM, Ciarlet M. 2010. Efficacy of pentavalent rotavirus vaccine against severe rotavirus gastroenteritis in infants in developing countries in Asia: a randomised, double-blind, placebo-controlled trial. Lancet 376:615–623. 10.1016/S0140-6736(10)60755-6. [DOI] [PubMed] [Google Scholar]
- 13.Desselberger U. 2017. Differences of rotavirus vaccine effectiveness by country: likely causes and contributing factors. Pathogens 6:65. 10.3390/pathogens6040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chemello ME, Aristimuno OC, Michelangeli F, Ruiz MC. 2002. Requirement for vacuolar H+-ATPase activity and Ca2+ gradient during entry of rotavirus into MA104 cells. J Virol 76:13083–13087. 10.1128/jvi.76.24.13083-13087.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desselberger U. 2014. Rotaviruses. Virus Res 190:75–96. 10.1016/j.virusres.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 16.Au KS, Chan WK, Burns JW, Estes MK. 1989. Receptor activity of rotavirus nonstructural glycoprotein NS28. J Virol 63:4553–4562. 10.1128/JVI.63.11.4553-4562.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang M, Zeng CQ, Morris AP, Estes MK. 2000. A functional NSP4 enterotoxin peptide secreted from rotavirus-infected cells. J Virol 74:11663–11670. 10.1128/jvi.74.24.11663-11670.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ball JM, Mitchell DM, Gibbons TF, Parr RD. 2005. Rotavirus NSP4: a multifunctional viral enterotoxin. Viral Immunol 18:27–40. 10.1089/vim.2005.18.27. [DOI] [PubMed] [Google Scholar]
- 19.Hyser JM, Collinson-Pautz MR, Utama B, Estes MK. 2010. Rotavirus disrupts calcium homeostasis by NSP4 viroporin activity. mBio 1:e00265-10. 10.1128/mBio.00265-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pham T, Perry JL, Dosey TL, Delcour AH, Hyser JM. 2017. The rotavirus NSP4 viroporin domain is a calcium-conducting ion channel. Sci Rep 7:43487. 10.1038/srep43487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong X, Hu Y, Liu C, Li X. 2017. Rotavirus NSP4(86–175) interacts with H9c2(2–1) cells in vitro, elevates intracellular Ca2+ levels and can become cytotoxic: a possible mechanism for extra-intestinal pathogenesis. Virus Genes 53:179–189. 10.1007/s11262-016-1419-6. [DOI] [PubMed] [Google Scholar]
- 22.Sastri NP, Viskovska M, Hyser JM, Tanner MR, Horton LB, Sankaran B, Prasad BV, Estes MK. 2014. Structural plasticity of the coiled-coil domain of rotavirus NSP4. J Virol 88:13602–13612. 10.1128/JVI.02227-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seo NS, Zeng CQ, Hyser JM, Utama B, Crawford SE, Kim KJ, Höök M, Estes MK. 2008. Integrins α1β1 and α2β1 are receptors for the rotavirus enterotoxin. Proc Natl Acad Sci USA 105:8811–8818. 10.1073/pnas.0803934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer JC, Bergmann CC, Bellamy AR. 1989. Interaction of rotavirus cores with the nonstructural glycoprotein NS28. Virology 171:98–107. 10.1016/0042-6822(89)90515-1. [DOI] [PubMed] [Google Scholar]
- 25.Silvestri LS, Tortorici MA, Vasquez-Del Carpio R, Patton JT. 2005. Rotavirus glycoprotein NSP4 is a modulator of viral transcription in the infected cell. J Virol 79:15165–15174. 10.1128/JVI.79.24.15165-15174.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergmann CC, Maass D, Poruchynsky MS, Atkinson PH, Bellamy AR. 1989. Topology of the nonstructural rotavirus receptor glycoprotein NS28 in the rough endoplasmic reticulum. EMBO J 8:1695–1703. 10.1002/j.1460-2075.1989.tb03561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor JA, Meyer JC, Legge MA, O’Brien JA, Street JE, Lord VJ, Bergmann CC, Bellamy AR. 1992. Transient expression and mutational analysis of the rotavirus intracellular receptor: the C-terminal methionine residue is essential for ligand binding. J Virol 66:3566–3572. 10.1128/JVI.66.6.3566-3572.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Storey SM, Gibbons TF, Williams CV, Parr RD, Schroeder F, Ball JM. 2007. Full-length, glycosylated NSP4 is localized to plasma membrane caveolae by a novel raft isolation technique. J Virol 81:5472–5483. 10.1128/JVI.01862-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagashima S, Kobayashi N, Ishino M, Alam MM, Ahmed MU, Paul SK, Ganesh B, Chawla-Sarkar M, Krishnan T, Naik TN, Wang YH. 2008. Whole genomic characterization of a human rotavirus strain B219 belonging to a novel group of the genus Rotavirus. J Med Virol 80:2023–2033. 10.1002/jmv.21286. [DOI] [PubMed] [Google Scholar]
- 30.Lin SL, Tian P. 2003. Detailed computational analysis of a comprehensive set of group A rotavirus NSP4 proteins. Virus Genes 26:271–282. 10.1023/A:1024451314534. [DOI] [PubMed] [Google Scholar]
- 31.Kanai Y, Komoto S, Kawagishi T, Nouda R, Nagasawa N, Onishi M, Matsuura Y, Taniguchi K, Kobayashi T. 2017. Entirely plasmid-based reverse genetics system for rotaviruses. Proc Natl Acad Sci USA 114:2349–2354. 10.1073/pnas.1618424114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komoto S, Fukuda S, Kugita M, Hatazawa R, Koyama C, Katayama K, Murata T, Taniguchi K. 2019. Generation of infectious recombinant human rotaviruses from just 11 cloned cDNAs encoding the rotavirus genome. J Virol 93:e02207-18. 10.1128/JVI.02207-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawagishi T, Nurdin JA, Onishi M, Nouda R, Kanai Y, Tajima T, Ushijima H, Kobayashi T. 2020. Reverse genetics system for a human group A rotavirus. J Virol 94:e00963-19. 10.1128/JVI.00963-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez-Tacuba L, Feng N, Meade NJ, Mellits KH, Jais PH, Yasukawa LL, Resch TK, Jiang B, Lopez S, Ding S, Greenberg HB. 2020. An optimized reverse genetics system suitable for efficient recovery of simian, human, and murine-like rotaviruses. J Virol 94:e01294-20. 10.1128/JVI.01294-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hou G, Zeng Q, Matthijnssens J, Greenberg HB, Ding S. 2021. Rotavirus NSP1 contributes to intestinal viral replication, pathogenesis, and transmission. mBio 12:e03208-21. 10.1128/mBio.03208-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ericson BL, Graham DY, Mason BB, Estes MK. 1982. Identification, synthesis, and modifications of simian rotavirus SA11 polypeptides in infected cells. J Virol 42:825–839. 10.1128/JVI.42.3.825-839.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato S, Hisaie K, Kurokawa S, Suzuki A, Sakon N, Uchida Y, Yuki Y, Kiyono H. 2019. Human norovirus propagation in human induced pluripotent stem cell-derived intestinal epithelial cells. Cell Mol Gastroenterol Hepatol 7:686–688.e685. 10.1016/j.jcmgh.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peischard S, Ho HT, Piccini I, Strutz-Seebohm N, Ropke A, Liashkovich I, Gosain H, Rieger B, Klingel K, Eggers B, Marcus K, Linke WA, Muller FU, Ludwig S, Greber B, Busch K, Seebohm G. 2020. The first versatile human iPSC-based model of ectopic virus induction allows new insights in RNA-virus disease. Sci Rep 10:16804. 10.1038/s41598-020-72966-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imamura K, Sakurai Y, Enami T, Shibukawa R, Nishi Y, Ohta A, Shu T, Kawaguchi J, Okada S, Hoenen T, Yasuda J, Inoue H. 2021. iPSC screening for drug repurposing identifies anti-RNA virus agents modulating host cell susceptibility. FEBS Open Bio 11:1452–1464. 10.1002/2211-5463.13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez-Bermejo JA, Kang S, Rockwood SJ, Simoneau CR, Joy DA, Silva AC, Ramadoss GN, Flanigan WR, Fozouni P, Li H, Chen PY, Nakamura K, Whitman JD, Hanson PJ, McManus BM, Ott M, Conklin BR, McDevitt TC. 2021. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci Transl Med 13:eabf7872. 10.1126/scitranslmed.abf7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takahashi Y, Sato S, Kurashima Y, Yamamoto T, Kurokawa S, Yuki Y, Takemura N, Uematsu S, Lai CY, Otsu M, Matsuno H, Osawa H, Mizushima T, Nishimura J, Hayashi M, Yamaguchi T, Kiyono H. 2018. A refined culture system for human induced pluripotent stem cell-derived intestinal epithelial organoids. Stem Cell Rep 10:314–328. 10.1016/j.stemcr.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erickson BR, Deyde V, Sanchez AJ, Vincent MJ, Nichol ST. 2007. N-linked glycosylation of Gn (but not Gc) is important for Crimean Congo hemorrhagic fever virus glycoprotein localization and transport. Virology 361:348–355. 10.1016/j.virol.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 43.Crawford SE, Hyser JM, Utama B, Estes MK. 2012. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication. Proc Natl Acad Sci USA 109:E3405–W3413. 10.1073/pnas.1216539109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perry JL, Ramachandran NK, Utama B, Hyser JM. 2015. Use of genetically-encoded calcium indicators for live cell calcium imaging and localization in virus-infected cells. Methods 90:28–38. 10.1016/j.ymeth.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sen A, Sen N, Mackow ER. 2007. The formation of viroplasm-like structures by the rotavirus NSP5 protein is calcium regulated and directed by a C-terminal helical domain. J Virol 81:11758–11767. 10.1128/JVI.01124-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao Y, Goraya MU, Yuan X, Zhang B, Chiu SH, Chen JL. 2019. Functional involvement of interferon-inducible transmembrane proteins in antiviral immunity. Front Microbiol 10:1097. 10.3389/fmicb.2019.01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holloway G, Truong TT, Coulson BS. 2009. Rotavirus antagonizes cellular antiviral responses by inhibiting the nuclear accumulation of STAT1, STAT2, and NF-κB. J Virol 83:4942–4951. 10.1128/JVI.01450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arnold MM, Patton JT. 2011. Diversity of interferon antagonist activities mediated by NSP1 proteins of different rotavirus strains. J Virol 85:1970–1979. 10.1128/JVI.01801-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danilczyk UG, Cohen-Doyle MF, Williams DB. 2000. Functional relationship between calreticulin, calnexin, and the endoplasmic reticulum luminal domain of calnexin. J Biol Chem 275:13089–13097. 10.1074/jbc.275.17.13089. [DOI] [PubMed] [Google Scholar]
- 50.Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, Helenius A. 2004. Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol Cell 13:125–135. 10.1016/s1097-2765(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 51.Mirazimi A, Nilsson M, Svensson L. 1998. The molecular chaperone calnexin interacts with the NSP4 enterotoxin of rotavirus in vivo and in vitro. J Virol 72:8705–8709. 10.1128/JVI.72.11.8705-8709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maruri-Avidal L, Lopez S, Arias CF. 2008. Endoplasmic reticulum chaperones are involved in the morphogenesis of rotavirus infectious particles. J Virol 82:5368–5380. 10.1128/JVI.02751-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trujillo-Alonso V, Maruri-Avidal L, Arias CF, López S. 2011. Rotavirus infection induces the unfolded protein response of the cell and controls it through the nonstructural protein NSP3. J Virol 85:12594–12604. 10.1128/JVI.05620-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopez T, Camacho M, Zayas M, Najera R, Sanchez R, Arias CF, Lopez S. 2005. Silencing the morphogenesis of rotavirus. J Virol 79:184–192. 10.1128/JVI.79.1.184-192.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Estes MK, Graham DY, Gerba CP, Smith EM. 1979. Simian rotavirus SA11 replication in cell cultures. J Virol 31:810–815. 10.1128/JVI.31.3.810-815.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martínez JL, Arnoldi F, Schraner EM, Eichwald C, Silva-Ayala D, Lee E, Sztul E, Burrone ÓR, López S, Arias CF. 2019. The guanine nucleotide exchange factor GBF1 participates in rotavirus replication. J Virol 93:e01062-19. 10.1128/JVI.01062-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanai Y, Kawagishi T, Nouda R, Onishi M, Pannacha P, Nurdin JA, Nomura K, Matsuura Y, Kobayashi T. 2019. Development of stable rotavirus reporter expression systems. J Virol 93:e01774-18. 10.1128/JVI.01774-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li B, Ding S, Feng N, Mooney N, Ooi YS, Ren L, Diep J, Kelly MR, Yasukawa LL, Patton JT, Yamazaki H, Shirao T, Jackson PK, Greenberg HB. 2017. Drebrin restricts rotavirus entry by inhibiting dynamin-mediated endocytosis. Proc Natl Acad Sci USA 114:E3642–E3651. 10.1073/pnas.1619266114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4. Download jvi.01861-22-s0001.pdf, PDF file, 0.8 MB (777.5KB, pdf)