

Abstract
Background
Muscle mitochondrial decline is associated with aging‐related muscle weakness and insulin resistance. FoxO transcription factors are targets of insulin action and deletion of FoxOs improves mitochondrial function in diabetes. However, disruptions in proteostasis and autophagy are hallmarks of aging and the effect of chronic inhibition of FoxOs in aged muscle is unknown. This study investigated the role of FoxOs in regulating muscle strength and mitochondrial function with age.
Methods
We measured muscle strength, cross‐sectional area, muscle fibre‐type, markers of protein synthesis/degradation, central nuclei, glucose/insulin tolerance, and mitochondrial bioenergetics in 4.5‐month (Young) and 22–24‐month‐old (Aged) muscle‐specific FoxO1/3/4 triple KO (TKO) and littermate control (Ctrl) mice.
Results
Lean mass was increased in Aged TKO compared with both Aged Ctrl and younger groups by 26–33% (P < 0.01). Muscle strength, measured by max force of tibialis anterior (TA) contraction, was 20% lower in Aged Ctrl compared with Young Ctrls (P < 0.01) but was not decreased in Aged TKOs. Increased muscle strength in Young and Aged TKO was associated with 18–48% increased muscle weights compared with Ctrls (P < 0.01). Muscle cross‐sectional analysis of TA, soleus, and plantaris revealed increases in fibre size distribution and a 2.5–10‐fold increase in central nuclei in Young and Aged TKO mice, without histologic signs of muscle damage. Age‐dependent increases in Gadd45a and Ube4a expression as well accumulation of K48 polyubiquitinated proteins were observed in quad and TA but were prevented by FoxO deletion. Young and Aged TKO muscle showed minimal changes in autophagy flux and no accumulation of autophagosomes compared with Ctrl groups. Increased strength in Young and Aged TKO was associated with a 10–20% increase in muscle mitochondrial respiration using glutamate/malate/succinate compared with controls (P < 0.05). OXPHOS subunit expression and complex I activity were decreased 16–34% in Aged Ctrl compared with Young Ctrl but were prevented in Aged TKO. Both Aged Ctrl and Aged TKO showed impaired glucose tolerance by 33% compared to young groups (P < 0.05) indicating improved strength and mitochondrial respiration are not due to improved glycemia.
Conclusions
FoxO deletion increases muscle strength even during aging. Deletion of FoxOs maintains muscle strength in part by mild suppression of atrophic pathways, including inhibition of Gadd45a and Ube4a expression, without accumulation of autophagosomes in muscle. Deletion of FoxOs also improved mitochondrial function by maintenance of OXPHOS in both young and aged TKO.
Keywords: FoxO, Aging, Mitochondrial function, Insulin resistance, Muscle hypertrophy, Glucose tolerance
Introduction
Aging‐related muscle atrophy is a significant contributor to disability and mortality. In humans, muscle mass declines by around 1% per year starting in the fourth decade, 1 and can accelerate 2‐ to 4‐fold by age 70. 2 Muscle strength is an independent predictor of mortality in older adults. 3 Anabolic resistance, or the blunted response of muscle protein synthesis to ingested amino acids/protein, is a major contributor to muscle loss with aging and is often associated with physical inactivity, inflammation, and insulin resistance. 4 , 5 Although anabolic resistance is a significant contributor to muscle loss with aging, recent studies indicate that resistance exercise may not influence anabolic resistance. 6 Unfortunately, there are currently no therapies beyond exercise and nutritional supplementation for muscle atrophy, in part because the underlying mechanisms are not fully elucidated.
Proteostasis, or the balance between protein synthesis and breakdown, is important to maintaining muscle mass. Basal muscle protein breakdown is not changed during aging, but the suppression of protein breakdown by modest doses of insulin is blunted in older individuals. 7 Forkhead box O, or FoxO, transcription factors are downstream targets of the insulin/PI3K/AKT pathway and are major regulators of acute muscle atrophy. Mammals have three FoxOs expressed in skeletal muscle: FoxO1, FoxO3, and FoxO4. 8 FoxOs mediate muscle atrophy in response to various stimuli such as uncontrolled diabetes, 9 starvation and denervation, 10 or deletion of insulin/IGF‐1 receptors. 11 We showed deletion of FoxOs in skeletal muscle can lead to muscle hypertrophy and increased metabolic rate, which mitigates fat gain and increases lean mass in response to high fat diet. 12 In response to acute atrophic stimuli, FoxOs regulate expression of protein degradation genes, including Fbxo32 (Atrogin‐1), Trim63 (MuRF‐1), and autophagy genes. 10 , 13 However, studies in mice and humans with chronic muscle atrophy show no changes/decreases in Fbxo32 and Trim63. 14 , 15 One might speculate that decreased autophagy or protein degradation would be detrimental to muscle function during aging. However, unlike deletion of critical autophagy genes such as ATG7 which causes muscle atrophy, 16 deletion of FoxOs in muscle does not inhibit basal autophagy and actually improves muscle size and strength, indicating FoxOs play a larger role in upregulating autophagy in acute atrophy and other aspects of FoxOs may influence muscle function during aging.
Mitochondrial dysfunction is observed in many tissues during aging. Mitochondrial dysfunction can be defined as either a reduction in transmembrane potential, electron transport chain malfunction, or impaired transport of critical metabolites into mitochondria. Some studies show aging contributes to declines in both quantity and function of skeletal muscle mitochondria, as measured by phospho‐creatine recovery. 17 Furthermore, mitochondrial ATP synthesis rates and expression of mitochondrial mRNA decreases with age in muscle, leading to oxidative capacity in aged populations that can be 50% lower than younger populations. 17 , 18 However, other studies show that exercise training can increase mitochondrial function in skeletal muscle from older individuals indicating that aging per se may not be the primary driver of mitochondrial dysfunction. 19
Insulin resistance and muscle mitochondrial dysfunction are associated with aging‐related muscle atrophy, 1 , 20 but studies are mixed as to whether either is causal. Some rodent and human studies show a decrease in mitochondrial enzyme activities, and reduced mitochondrial number and size with diet‐induced obesity (DIO), 21 , 22 whereas others indicate that DIO is less important than loss of insulin action to induce mitochondrial abnormalities. 23 Still others argue that these are not mitochondrial intrinsic defects, and merely reflect decreases in mitochondrial content per muscle. 24 Interestingly, a study in obese patients with/without type 2 diabetes that received gastric bypass surgery showed that muscle mitochondrial respiratory capacity is not correlated with marked improvements in whole body insulin sensitivity. 25 Despite these controversies, mouse models show that insulin action can directly regulate muscle mitochondrial function in part via FoxOs. 26 , 27 Although FoxOs play a major role in acute muscle atrophy in response to diabetes or starvation, the roles of FoxOs in age‐related muscle strength and mitochondrial function are unknown.
We hypothesize that muscle‐specific FoxO deletion will increase muscle size and improve mitochondrial function even with aging. To test this, we measured muscle strength, mitochondrial bioenergetics, and performed histologic and biochemical analyses on 4.5‐month (Young) and 22–24‐month‐old (Aged) muscle‐specific FoxO1/3/4 TKO mice.
Materials and methods
Animal Studies
Animal studies were approved by the Institutional Animal Care and Use Committee at the University of Iowa. Male mice were used for all studies. Muscle‐specific FoxO1, FoxO3, and FoxO4 triple knockout (TKO) mice were generated using Acta1‐Cre as previously described. 12 Littermate controls were used for all experiments as the mice were on a mixed background containing C57Blk6, C57Blk6J, and 129 strains. Bodyweight and body composition were measured by nuclear magnetic resonance (NMR) using an LF‐50 NMR. All animals were maintained on a 2920× phytoestrogen free chow. Unless otherwise specified, all animals were allowed ad libitum access to food and water.
See Supplemental Methods for Grip strength test, Exercise tolerance test (ETT), In vivo muscle contractile function, indirect calorimentry, protein synthesis assays, and autophagy flux analysis.
Glucose Tolerance Test (GTT) and Insulin Tolerance Test (ITT)
Mice were fasted overnight (16 h) prior to GTTs. Whole blood glucose levels were measured at indicated times after an IP injection of 2 mg dextrose/g body weight. Mice were fasted 2 h prior to ITTs. Whole blood glucose levels were measured at indicated times after injection with 1 mU/g body weight of regular human insulin (Novolin brand).
Histology
For cross sectional area (CSA), immunofluorescence staining of laminin in plantaris, TA, and soleus was performed with DAPI to stain nuclei. See Table S1 for antibodies. For quantification, single channel fluorescence images were segmented using Cellpose (cellpose.org) as previously described. 28 Briefly, Cellpose segmented images were fed to the Fiji: ImageJ plugin LabelsToRois together with the original image. Ninety to 450 fibres were counted per image. The resulting ROIs were eroded by three pixels. Fibres along the edges were excluded because they were fragmented. Labelled areas under 50 μm2 were excluded as these structures were not muscle fibres. For LC3 vesicle density, TA and soleus sections were stained with LC3A and quantified as previously described, 11 with the following modifications: vesicles were normalized to CSA quantified using Cellpose as described above. See Supplemental Methods for IgG and IgM Immunofluorescence.
Frozen cross sections of plantaris muscle were stained for succinate dehydrogenase (SDH) in buffer containing PBS with 0.5 mol/L disodium succinate, 20 mmol/L MgCl2, and 0.5 mg/mL nitro blue tetrazolium for 15 min at 37°C and quantified as previously described. 29 Briefly, 300–900 plantaris fibres were manually counted and normalized to total number of fibres in the field of interest.
Myosin fibre‐typing was performed in plantaris, TA, and soleus sections co‐stained with myosin I and IIa. Unstained fibres were designated as IIb/IIx and fibre‐type distribution was quantified as previously described. 30 See Table S1 for antibodies used.
Transmission Electron Microscopy (TEM)
TEM images from plantaris were obtained and mitochondrial area was quantified using a grid system as described previously 26 using 3–5 images from each mouse. Cristae were graded on 3–5 images per mouse as previously described. 31 Briefly a 5‐grade scoring system for cristae morphology in individual mitochondria was created: 0 = no defined cristae, 1 = >50% of mitochondrial area without cristae, 2 = >25% of mito without cristae, 3 = many irregular cristae, and 4 = many regular cristae.
Mitochondrial Function Assays
Mitochondrial function and ATP production were measured in permeabilized plantaris fibres or isolated muscle mitochondria using an Oroboros Oxygraph as previously published. 26 Muscle mitochondria was isolated by differential centrifugation from one quadricep and one gastrocnemius muscle as previously described. 26 , 32 See Supplemental Methods for full description.
Citrate synthase, Complex I&II, and Proteasome Activity assays
TA was homogenized in buffer containing 250 mM sucrose, 20 mM Tris, 40 mM KCl, and 2 mM EGTA at a pH of 7.4 to measure citrate synthase, complex I, and complex II activity, as previously described. 26 Proteasome activity was determined in 20 μg of quadriceps muscle homogenates. See Supplemental Methods for full description.
qPCR and Western Analysis
Quad or TA was homogenized in RIPA buffer (Millipore) with protease and phosphate A and B inhibitors (Bimake). Lysates were subjected to SDS‐PAGE and blotted using antibodies listed in Table S1. Quantitative RT‐PCR was performed as described 26 with primers listed in Table S2.
Statistics
All data are presented as mean ± SEM. Two‐way ANOVA with Tukey's test for multiple comparison was performed using GraphPad Prism software to determine significance, with P < 0.05 being significant. If the Tukey's test for multiple comparisons did not show any P < 0.05, then ANOVA main effects are presented with a line over all data to indicate individual groups were not statistically different than each other. Indirect claorimetry was analysed using CalR (See Supplemental Methods).
Results
Muscle‐specific FoxO deletion increases muscle size and prevents loss of muscle strength with aging
To determine the impact of FoxO deletion on age‐related changes in muscle, we measured body composition and muscle size/function in TKO and control (Ctrl) mice as they age. FoxO isoform knockout was confirmed by quantitative RT‐PCR (Figure S1a). We observed no differences in body weight between Young (4.5‐month‐old) Ctrl and TKO mice (Figure 1A). By 22–24 months, Ctrl and TKOs weighed more than young groups, but Aged TKOs also weighed more than Aged Ctrl. Aged Ctrl and Aged TKO showed more total fat mass than their young counterparts and Aged TKO had significantly more total lean mass than all other groups (Figure 1A). Percent fat mass was significantly increased in Aged Ctrl compared with Young Ctrl, and percent lean mass was higher in Aged TKO compared with Aged Ctrl (Figure S1b).
Figure 1.
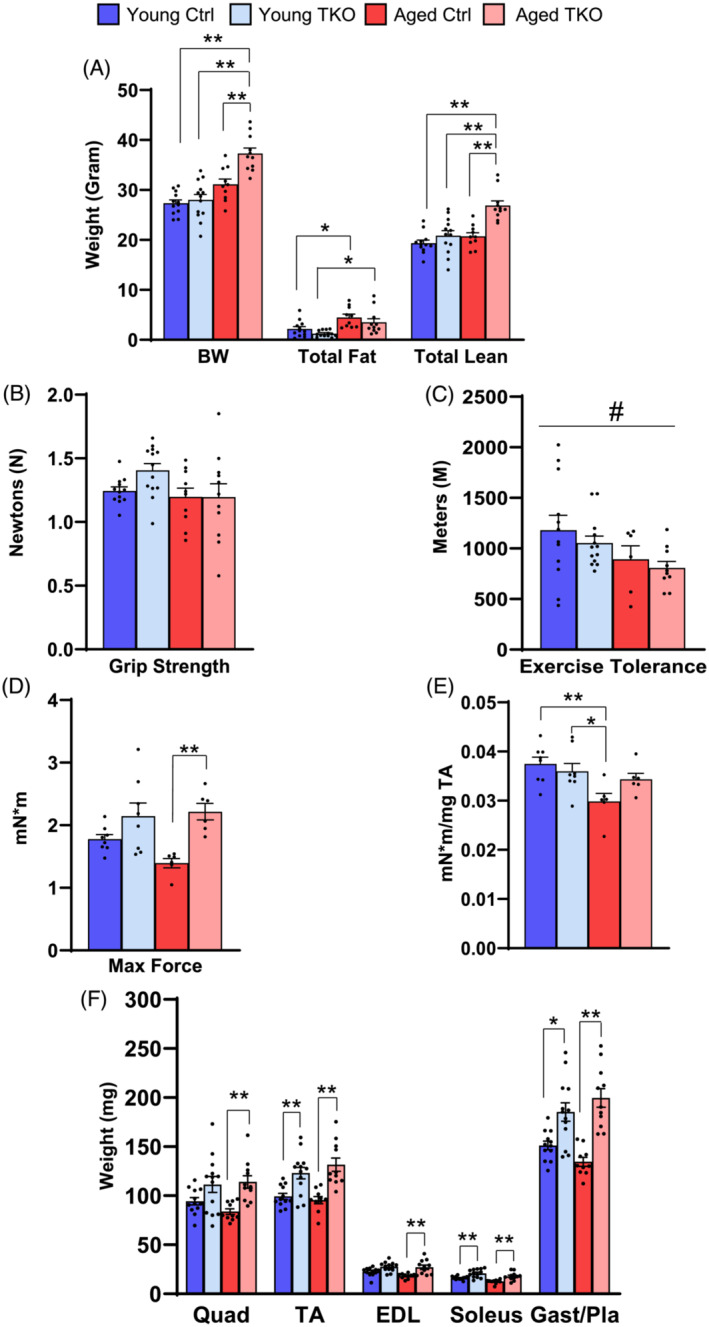
Muscle‐specific FoxO deletion increases muscle size and prevents loss of muscle strength with aging. Body composition measured by NMR showing body weight (BW), total fat, and total lean mass in Young (4.5‐month‐old) Ctrl, Young TKO, Aged (22‐ to 24‐month‐old) Ctrl, and Aged TKO mice (A). Force generated during grip strength test (B) (n = 10–14 per group). Total distance run during an exercise tolerance test (C) (n = 10–14 per group). Total muscle force generation (D) and muscle force normalized to TA weight (E) was measured via max isometric force test (see In vivo Muslc Contractile Function in Supplemental methods) in TA muscle (n = 6–8 per group). Dissected muscle weights of Young and Aged TKO and littermate control mice (F) (n = 10–14 per group). Only one quad and one gast/pla muscle from each mouse was weighed for analysis but both TAs, EDLs, and soleus muscles were weighed for analysis. *P < 0.05, **P < 0.01 as indicated by 2‐way ANOVA (Quad, quadriceps; TA, tibialis anterior; EDL, extensor digitorum longus; and Gast/Pla, gastrocnemius and plantaris).
Grip strength (Figure 1B) was unchanged between all groups, and total distance run on an exercise tolerance test was mildly decreased in aged groups compared with young (Figure 1C). Maximal isometric torque ankle dorsiflexors in tibialis anterior (TA) muscle was lowest in Aged Ctrl mice and significantly increased in Aged TKO mice, exerting 60% more force than littermate Aged Ctrl (Figure 1D). When max force was normalized to TA weight, we observed a 20% decrease in Aged Ctrl compared with Young Ctrl, but max force/TA mass was maintained in Aged TKO relative to younger groups (Figure 1E), suggesting FoxO deletion prevents loss of muscle strength with aging.
Muscle weights in Young TKO mice were 18–29% larger than Young Ctrl (Figure 1F). Aged Ctrl muscles showed a trend toward decreased mass but was not significant by ANOVA. The differences in muscle mass between Ctrl and TKO increased with age, such that Aged TKO muscles were 36–48% larger (Figure 1F). Liver weight in Aged TKO was ~30% larger than the Aged Ctrl and heart weight was increased in both aged groups compared with young groups (Figure S1c–d). Perigonadal white adipose tissue (pgWAT) accumulated in the aged groups, with the Aged TKO showing 50% larger pgWAT than Young TKO (Figure S1e). Although the study was not designed to test survival, an analysis of the aged cohorts used in this study revealed no significant change in survival between Ctrl and TKO (Figure S1f).
FoxO deletion in muscle increases fibre diameter in TA and soleus from aging mice, and dramatically increases central nuclei in young and aged TKO muscle
In TA, average cross sectional area (CSA) tended to increase in TKO groups. Myofibre distribution showed significant increases in the percent of fibres with CSA > 6000 μm2 with decreases in fibres 2000–4000 μm2 in both Young and Aged TKO compared with controls (Figure 2A–C). A similar pattern was seen in plantaris with milder changes (Figure S2a,b). Soleus muscle showed more dramatic increases in average CSA and in large fibres (>4000) in Aged TKO compared with other groups, with a shift toward smaller fibres in Aged Ctrl (Figure 2E–G). Quantification of fibres with central nuclei in all three muscle groups showed a 2.5–10‐fold increase in central‐nucleated fibres in Young TKO and Aged TKO compared with age‐matched controls (Figure 2D,H, Figure S2c). Total nuclei in plantaris was slightly increased in both Aged Ctrl and Aged TKO groups, but increases were minor in comparison with central nucleated fibres (Figure S2d). Central nuclei are typically a marker of regeneration after myopathy with infiltration of inflammatory cells. However, H&E stain of TA and soleus muscles did not show signs of dystrophy (Figure S2e), and the mice showed no signs of distress. Anti‐mouse IgG and IgM staining in TA and soleus muscle confirmed that there was no myofibre necrosis (Figure S3f). Thus, the increased size of TKO muscles correlates with increased strength and is likely a combination of increases in fibre hypertrophy and possibly myogenesis at young and old ages.
Figure 2.
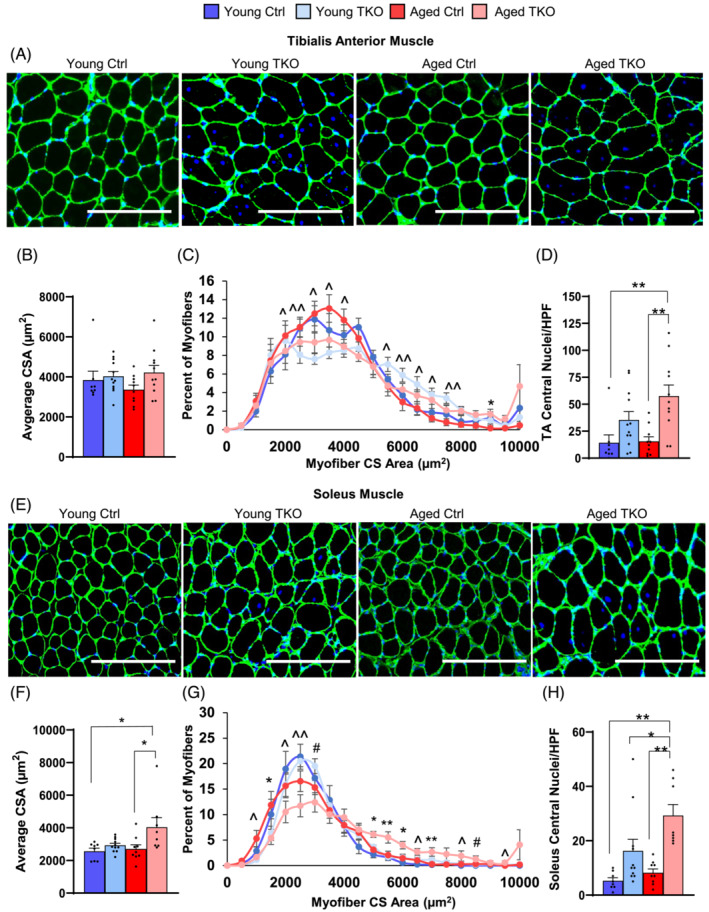
FoxO deletion in muscle mildly increases fibre size and markedly increases central nuclei in TKO muscle. Laminin and DAPI stain of tibialis anterior (TA) cross sections (scale bar 250 μm) (n = 8–13 per group) (A). Average cross sectional area (CSA) (B) and distribution of CSA (C) from TA. Quantification of fibres with central nuclei of TA muscle are shown (D). Laminin and DAPI stain of soleus cross sections (scale bar 250 μm) (n = 8–13 per group) (E). Average CSA (F) and distribution of myofibre CSA (G) from soleus. Quantification of central nucleated fibres in soleus (H). HPF, high power field. *P < 0.05; **P < 0.01 Aged Ctrl vs. Aged TKO or as indicated; ^P < 0.05, ^^P < 0.01 genotype main effect and #P < 0.05 age main effect by 2‐way ANOVA.
Deletion of FoxOs prevents age‐related increases in Gadd45a and Ube4a and suppresses FoxO‐ dependent ubiquitin‐proteasome genes
Acute induction of ubiquitin‐proteasome and autophagy mRNAs is dependent on FoxOs, 10 but their regulation with aging is less clear. Quantitative RT‐PCR of FoxO‐dependent genes in our cohorts reveal distinct expression patterns across age in quad, TA, and soleus muscles. In all three muscle groups Eif4ebp1 mRNA was decreased in both young and aged TKO groups, relative to controls (Figure 3A). Gadd45a, a critical mediator of muscle atrophy, 33 was increased in Quad and TA from Aged Ctrl compared with Young Ctrl, and this increase was prevented in Aged TKO. Another FoxO target, p27 Kip1, was decreased in both aged groups relative to young mice (Figure 3A). Measurement of a subset of ubiquitin‐proteasome mRNAs showed most were unchanged or decreased in Quad and TA from Aged Ctrl relative to Young Ctrl, but nearly all were decreased in TKO groups (Figure 3B,C). Quad and TA muscle also showed that Ube4a mRNA levels were significantly increased in Aged Ctrl compared with Young Ctrl, and deletion of FoxOs in Aged TKO mice prevented increases in Ube4a (Figure 3B,C), like Gadd45a. By contrast, only Trim63 was decreased in soleus from Aged TKO relative to young controls (Figure 3D).
Figure 3.
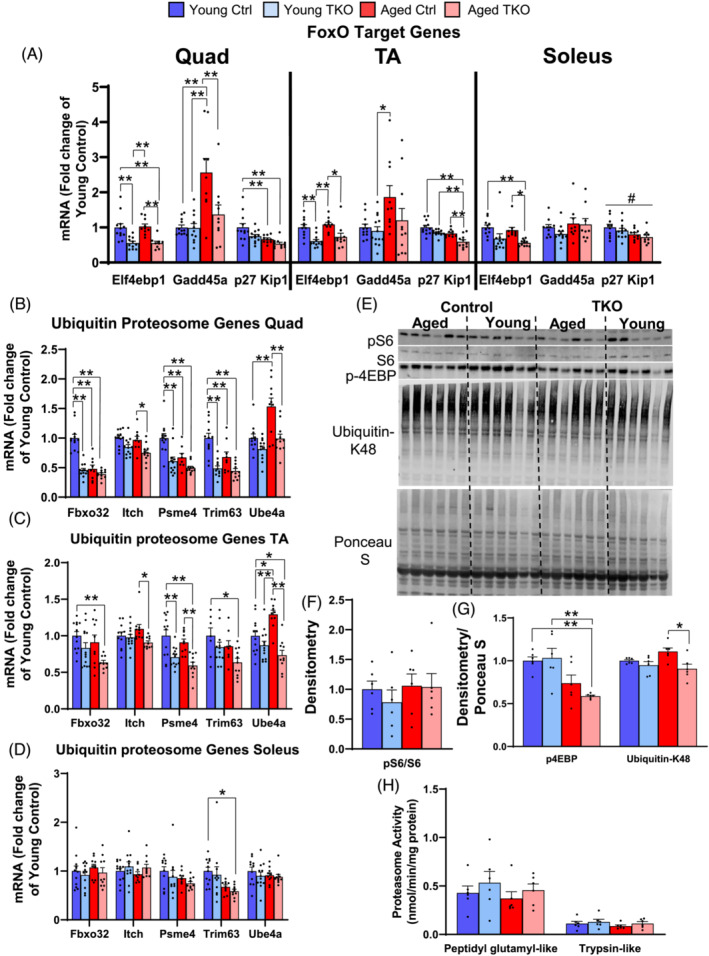
Age‐related increases in Gadd45a and Ube4a are prevented with FoxO deletion whereas other ubiquitin proteasome genes are decreased in Young and Aged TKO muscle. Quantitative RT‐PCR of FoxO‐target genes (A), from Young Ctrl, Young TKO, Aged Ctrl, and Aged TKO quadriceps (Quad), TA, and soleus muscle. (n = 9–13 per group). Quantitative RT‐PCR of ubiquitin‐proteasome genes in Quad (B), TA (C), and soleus (D) muscle. (n = 9–13 per group). Expression of TBP was used as a housekeeping normalizer. Western blot (E) and densitometry (F, G) for p‐S6, total S6, p‐4EBP, and K48 polyubiquitinated proteins in quad muscle (n = 6 per group). Proteasome activity in quad lysates (H) measured by breakdown of fluorescently labelled peptidyl glutamyl‐like (LLE) or trypsin‐like (LSTR) substrates (n = 6 per group). *P < 0.05, **P < 0.01 as indicated and #P < 0.05 age main effect by 2‐way ANOVA.
In quad, phosphorylation of S6 showed no differences in any of the groups (Figure 3E,F). Despite decreased Eif4ebp mRNA in young and Aged TKO, phosphorylation of 4EBP, a downstream mediator of mTOR, was decreased in Aged TKO and mildly decreased in Aged Ctrl, compared with young groups (Figure 3G). K48‐polyubiquitinated proteins were minimally increased in Aged Ctrl Quad, but were decreased in Aged TKO. We also measured proteasome activity in Quad muscle given decreases in Psme4 and in K48 ubiquitin, but found no differences between groups (Figure 3H). Overall, these data show that deletion of FoxOs decreased expression of many ubiquitin‐proteasome genes in predominantly glycolytic muscles regardless of age, and that age‐related increases in Gadd45a and Ube4a were prevented by FoxO deletion.
Loss of FoxOs does not alter basal autophagy in muscle during aging
TA muscle showed small decreases in mRNA levels of autophagy genes including Ctsl, LC3B, and Ulk1 in Aged TKO (Figure 4A). Soleus muscle showed no changes in autophagy gene expression between TKO and Ctrl groups (Figure 4E). To determine if loss of FoxOs causes autophagosome accumulation in Aged muscle, we measured LC3 vesicles in our study cohorts. LC3A vesicle number in TA showed no significant changes across groups but soleus did show increased LC3A vesicles per fibre (Figure 4B,C,F,G), likley due to increased fibre size. When normalized to area, neither TA nor soleus showed changes in LC3A vesicles (Figure 4D,H). In Quad, autophagy genes including Bnip3, Bnip3L, Gabarapl and LC3B, were decreased in Young TKO, but the changes were less robust in aged groups (Figure S3a). Ulk1 and LC3B transcript and protein levels were ~30–40% lower in the TKO groups compared with controls (Figure S3a–d).
Figure 4.
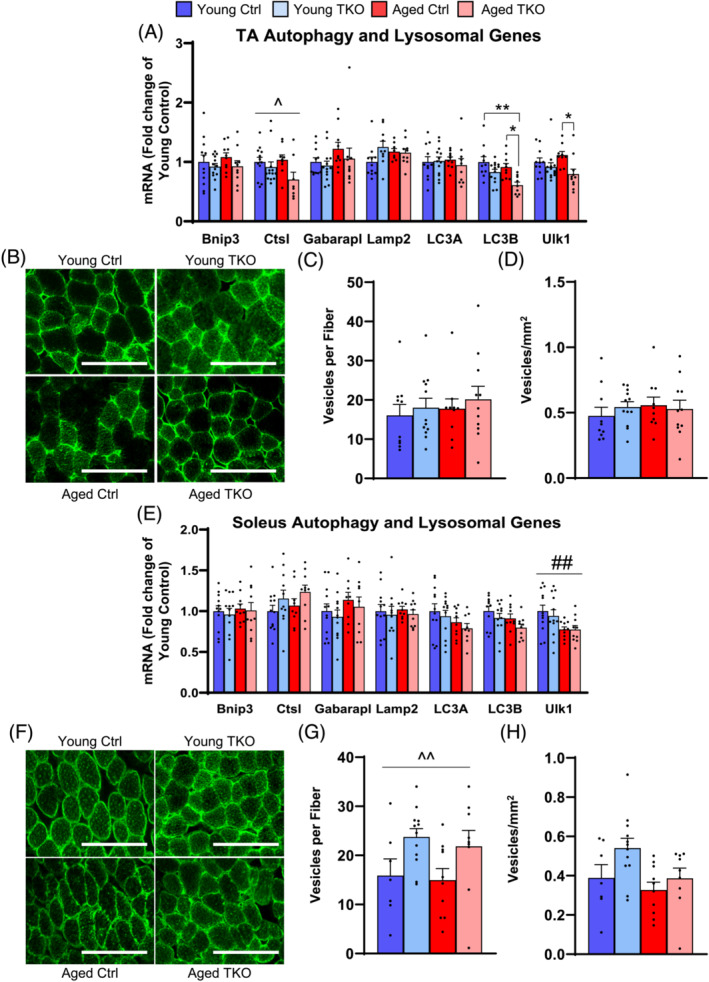
Autophagy‐lysosome genes are minimally decreased and LC3A vesicles do not accumulate in TA or soleus from Young and Aged TKO. Quantitative RT‐PCR of autophagy lysosomal genes in TA (A) and soleus (E) muscle from Young Ctrl, Young TKO, Aged Ctrl, and Aged TKO (n = 9–13 per group). TBP was used as a housekeeping normalizer. LC3A stained cross section of TA (B) and soleus muscle (F) (scale bar 200 μm). Quantification of vesicles per fibre in TA (C) and soleus (G). Quantification of vesicle per mm2 in TA (D) and soleus (H) (n = 10–11 per group). *P < 0.05, **P < 0.01 as indicated, ^P < 0.05, ^^P < 0.01 genotype main effect, ##P < 0.01 age main effect by 2‐way ANOVA.
FoxOs are required for the increased autophagy under pathologic conditions, such as diabetes, 9 but deletion of FoxOs in muscle does not seem to affect basal proteostasis. We directly measured protein synthesis using the SUnSET method and autophagy flux by treatment with/without colchicine. These studies require treatments prior to euthanasia so we restricted these studies to Young TKO and Ctrl groups, which show similar changes in central nuclei, fibre size, and proteostasis markers compared with aged groups. Protein synthesis measured by puromycin incorporation was unchanged between Young Ctrl and TKO mice after a 6 h post‐prandial fast (Figure S4a–f), although the colchicine treated Ctrl and TKO groups did show a mild decrease in quad and soleus. Bnip3 expression, a marker of mitophagy, increased after colchicine treatment in quad from Ctrl and TKO groups, whereas TA and soleus showed no differences. Levels of LC3‐II and LC3‐II/LC3‐I ratio showed significant increases in all three muscle groups from Ctrl and TKO mice when treated with colchicine for 2 days indicating basal autophagy was preserved in TKO (Figure S4g–i).
Age‐related increases in fasting glucose are not prevented in Aged TKO mice
To determine the role of muscle‐specific FoxO deletion on glucose homeostasis as mice age we measured fasting glycaemia and preformed glucose tolerance tests (GTT) and insulin tolerance tests (ITT). Fasted overnight glucose levels were 16% higher in the Aged Ctrl mice compared with the Young Ctrl, and 39% higher in Aged TKO mice compared with Young TKO (Figure 5A). Aged TKO mice showed impaired glucose clearance during GTT (Figure 5B). Two‐hour fasted glucose levels prior to ITT were elevated in aged groups compared with young (Figure 5C), but glycaemic excursions during ITT were not different between TKO or control groups at either age (Figure 5D). Randomly‐fed blood glucose levels were ~0.667 mmol/L higher in both aged control and TKO mice compared with young mice (Figure 5E) with no differences in insulin levels between any of the four groups (Figure 5F).
Figure 5.
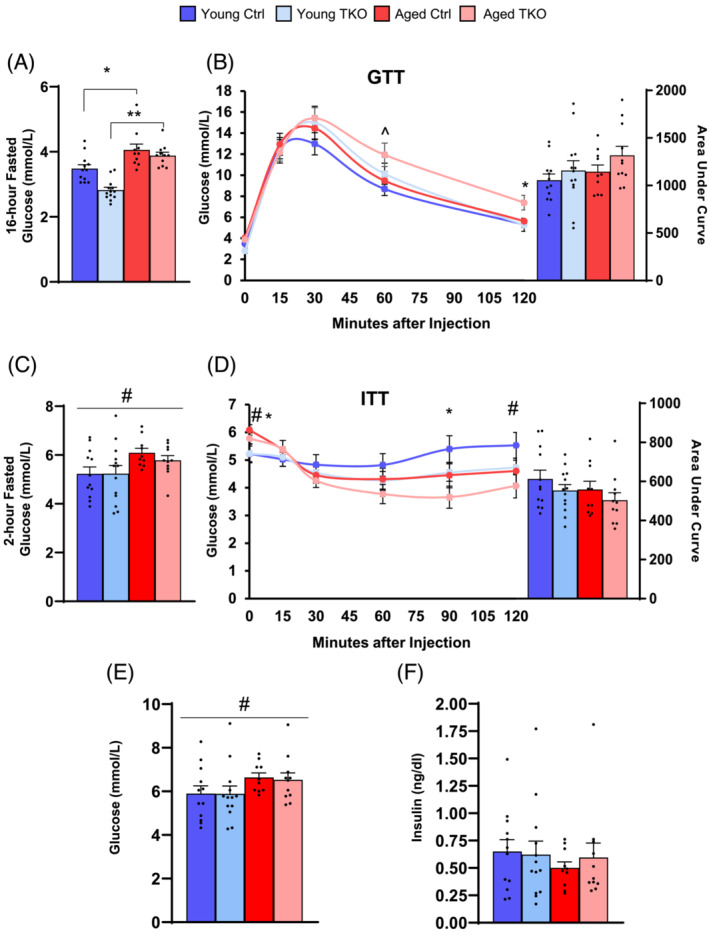
Age‐related increases in fasting glucose are not prevented in aged TKO mice. Overnight (16‐hour) fasted glucose levels in Young and Aged TKO mice and littermate controls (A). Glucose excursions and area under the curve for GTT (B) (n = 10–14 per group). Two‐hour fasted glucose prior to ITT (C). Glucose excursions and area under the curve for ITT (D) (n = 10–14 per group). Ad libitum fed glucose levels (E) and serum insulin concentrations (F) at the time of sacrifice (n = 10–13 per group). *P < 0.05, **P < 0.01 as indicated. ^P < 0.05 genotype main effect, #P < 0.05 age main effect by 2‐way ANOVA.
Maximal mitochondrial respiration is increased in muscle from FoxO TKO mice
Muscle mitochondrial respiration can influence basal metabolism, but indirect calorimetry showed no differences in energy expenditure between Aged Ctrl and Aged TKO (Figure S5a and Table S3). Respiratory exchange ratio trended lower in TKO mice compared with controls but was not significant (Figure S5b). Total distance in cage was mildly decreased in Aged TKO mice and average food intake trended lower in the TKO mice (Figure S5c,d).
Mitochondrial function was measured in saponin‐permeabilized plantaris fibres using several different substrates. Unstimulated respiration in plantaris fibres with glutamate/malate (Glu/M) showed decreased basal respiration in Aged groups compared with Young (Figure 6A). After addition of 1 mM ADP, we observed no differences in respiration with Glu/M. However, after the addition of succinate, maximal respiration was increased 10–20% in TKO groups compared with Ctrl groups and remained increased after addition of rotenone (Figure 6A). After addition of Oligomycin (complex V inhibitor) or FCCP (uncoupler), the differences between young and aged mice predominated with decreased respiration in Aged TKO and Aged Ctrl relative to young groups. Interestingly, ATP production with Glu/M under physiologic 75 μM ADP or maximal 500 μM ADP was unchanged among the four groups, nor was it different after addition of succinate (Figure 6B).
Figure 6.
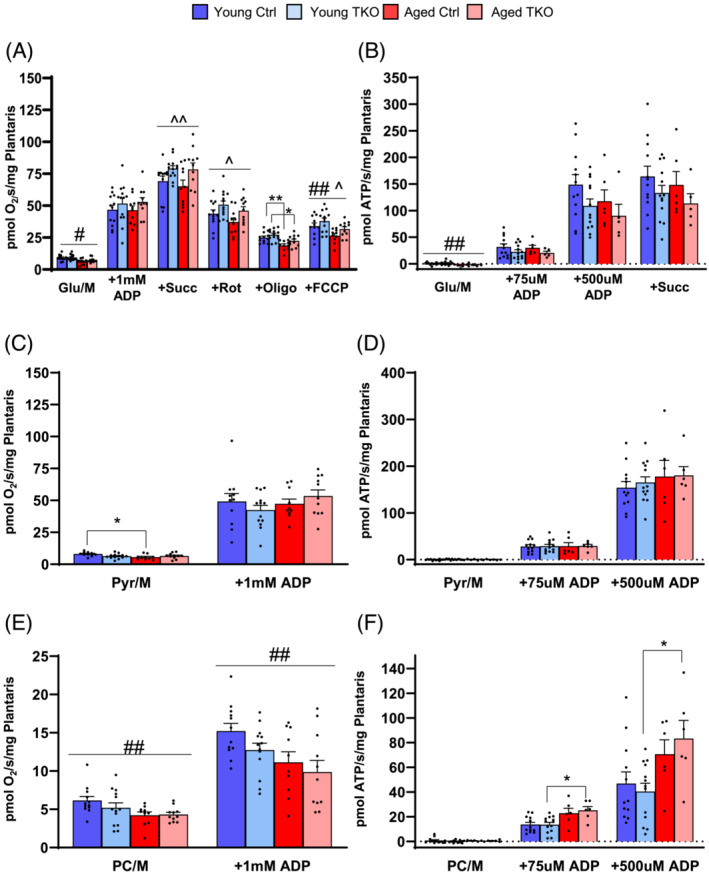
Maximal mitochondrial respiration is increased in plantaris from FoxO TKO mice using combined complex I and II substrates. Basal and maximal (+1 mM ADP) respiration in saponin‐permeabilized plantaris fibres using glutamate/malate (Glu/M) substates, then sequential addition of succinate (Succ), rotenone (Rot), oligomycin (Oligo), and FCCP (A). ATP production rates in permeabilized plantaris fibres using Glu/M and subsequent addition of succinate (B). Basal and maximal respiration in permeabilized plantaris fibres using pyruvate/malate (Pyr/M) substrates (C). ATP production rates in permeabilized plantaris fibres using Pyr/M (D). Basal and maximal respiration in permeabilized plantaris fibres using palmitoyl‐carnitine/Malte (PC/M) substrates (E). ATP production in permeabilized plantaris fibres using PC/M (F). (n = 5–13 per group). *P < 0.05 as indicated. ^P < 0.05, ^^P < 0.01 genotype main effect. #P < 0.05, ##P < 0.01 age main effect by 2‐way ANOVA.
Basal respiration with pyruvate/malate (Pyr/M) in Aged Ctrl was significantly lower than Young Ctrl, but after addition of 1 mM ADP there was no difference between the four groups (Figure 6C). Likewise, there were no differences in ATP production with Pyr/M after physiologic or maximal ADP was added (Figure 6D). Using a fat‐derived substrate, we found respiration was decreased in aged group with palmitoyl‐carnitine/malate (PC/M), both before and after maximal ADP were added (Figure 6E). ATP production with PC/M was increased in aged Ctrl and Aged TKO mice when compared with the young TKO mice at both physiologic and maximal ADP levels, but without differences between Aged Ctrl and Aged TKO mice (Figure 6F). It is important to note that TKO mice have muscle hypertrophy and these mitochondrial measurements are normalized to muscle weight, indicating mitochondrial function with Pyr/M and PC/M is at least proportionally increased with muscle size. These results indicated that deletion of FoxOs in muscle can mildly increase maximal respiration in plantaris fibres and maintain mitochondrial function in the presence of muscle hypertrophy.
Oxygen consumption and ATP production in isolated mitochondria is decreased in Aged‐control mice, but less‐severely affected in Aged‐FoxO TKO mice
To determine FoxO effects in other muscle groups, we also measured respiration and ATP production in isolated mitochondria from a mixture of one quadriceps and one gastrocnemius. Aged Ctrl mitochondrial respiration and ATP production with Glu/M was decreased ~50% compared with Young Ctrl after addition of low physiologic (5–10 μM) or after maximal (1000 μM) ADP, but Aged TKO respiration trended slightly higher than Aged Ctrl (Figure S6a,b). Maximal mitochondrial respiration with glutamate/malate/succinate tended to be higher in Aged TKO compared with Aged controls (Figure 7A). After addition of rotenone, respiration in Aged TKO mitochondria was significantly higher than Aged Ctrl, although both remained lower than Young groups. Finally, ATP production with glutamate/malate/succinate and maximal ADP was decreased in aged Ctrl compared with young Ctrl, but aged TKOs were not significantly different than young groups (Figure 7B).
Figure 7.
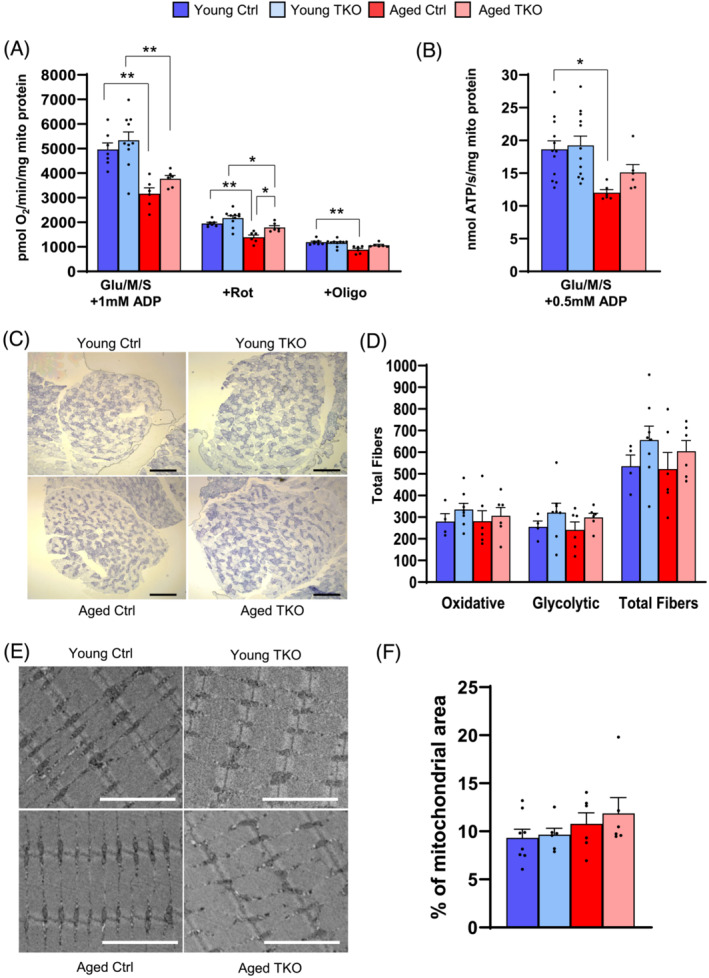
Oxygen consumption and ATP production in isolated mitochondria is decreased in aged mice, but partially rescued in Aged TKO. Maximal respiration of isolated mitochondria from mixed quad/gastroc with glutamate/malate/succinate (Glu/M/S) substrates, followed by addition of rotenone (Rot), and oligomycin (Oligo) (A). Maximal ATP production of isolated mitochondria with Glu/M/S substrates (B) (n = 5–13 per group). Images (C) and quantification (D) of total muscle fibres from succinate dehydrogenase (SDH) stained plantaris muscles (n = 4–8 per group, scale bar: 500 μm). Images (E) and quantification (F) of mitochondrial area on TEM images from plantaris muscle (n = 6–10 per group, scale bar 3 μm). *P < 0.05, **P < 0.01 as indicated by 2‐way ANOVA.
Because we saw a decrease in oxygen consumption, and ATP production in aging animals we quantified oxidative/glycolytic fibre content with succinate dehydrogenase staining in plantaris muscle. Oxidative/glycolytic fibre content was unchanged between groups, although there was a trend toward increased total fibres in TKO groups (Figure 7C,D and Figure S7a). Decreased mitochondrial mass can influence mitochondrial function in muscle, 24 but quantification of mitochondrial area in TEM images from plantaris showed no differences across groups (Figure 6E,F). Changes in cristae morphology can also be associated with mitochondrial dysfunction, 31 but cristae grading showed no differences between groups (Figure S7b,c). Quantification of fibre types in plantaris muscle showed a small increase in the percent of myosin type I fibres in aged TKO mice compared with Aged Controls, but no differences were seen in myosin IIa or IIb/IIx, which comprised most of the fibres (Figure S7d,e). Fibre typing in the deep portion of TA muscle from Young and Aged TKO showed mild increases in type IIa fibres with corresponding decreases in IIb/IIx (Figure S8a,b). In the soleus, the percent of type I fibres was increased in both aged groups when compared with their young counterparts. Percent of type IIa and IIb/IIx were lower in both aged groups compared with young groups (Figure S8c,d). Age‐related atrophy has greater effects on type I fibre size. Type I CSA in soleus showed no differences across groups, whereas type IIa fibres showed a 25% increase in Aged TKO compared with Aged Ctrl (Figure S8e,f). Thus, although small changes in myosin fibre types were observed in each muscle, quantification of mitochondrial area, cristae morphology or fibre typing did not explain the mild preservation of mitochondrial function in Young and Aged TKO mice.
Deletion of FoxOs in muscle maintains OXPHOS subunit expression and activity with age
Western blot of quadriceps muscle showed subunits of complex I (Ndufb8), complex II (Sdhb), and Complex IV (Mtco2) were decreased 12–34% in the Aged Ctrl compared with Young Ctrl, but levels of these subunits were preserved in Aged TKOs (Figure 8A,B). Uqcrc2 from complex III was increased in TKO groups compared with controls, whereas Atp5a from complex V showed an increase in both aged groups (Figure 8A,B). Ndufs1 protein, another complex I subunit, was increased in Aged TKO (Figure S9a,b), and VDAC tended to decrease in Aged Ctrl, but it did not reach significance (Figure S9c). In TA, Ndufb8 protein was significantly increase in Aged TKO compared with all other groups whereas other subunits showed less change, but similar patterns to Quad (Figure S9d,e). Transcriptional regulation of OXPHOS subunits by insulin action is mediated by FoxOs, 26 , 27 so we measured mRNA levels of several OXPHOS subunits in our young and aged groups. Ndufs2, Ndufb8, and Sdhb were significantly decreased in Aged Ctrl Quad when compared with Young Ctrl, but Aged TKO showed no changes. mRNA levels of other subunits were not changed (Figure 8C).
Figure 8.
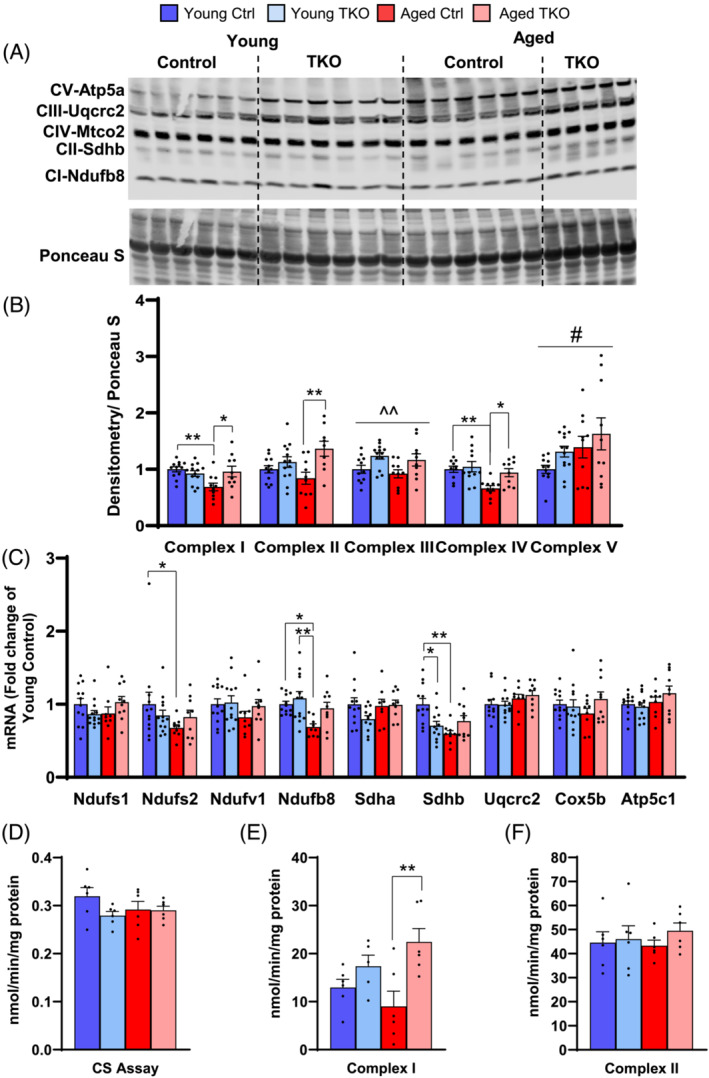
Deletion of FoxOs preserves OXPHOS subunit levels and activity in muscle with age. OXPHOS subunits measured by western blot in quad muscle (A). Densitometry of indicated subunit from complexes I–V (B) shown in panel A (n = 9–13 per group). Quantitative RT‐PCR of OXPHOS subunit genes in quad muscle (C) (n = 9–13 per group). Citrate synthase (CS) (D) complex I (E), and complex II (F) enzyme activities measured in tibialis anterior muscle from Young and Aged TKO and Ctrl mice (n = 6 per group). *P < 0.05, **P < 0.01 as indicated, ^^P < 0.01 genotype main effect, #P < 0.05 age main effect by 2‐way ANOVA.
Biochemical analysis of mitochondrial enzymes showed citrate synthase activity was not different between the groups (Figure 8D). Complex I activity tended to decrease in Aged Ctrl compared with young groups but was increased 70% in Aged TKO compared with Ctrl (Figure 8E). Complex II activity was not different between groups (Figure 8F). These data show that deletion of FoxOs in muscle helps maintain expression of OXPHOS subunits with age.
Discussion
Muscle weakness due to age‐related sarcopenia is associated with mitochondrial impairments and remains a critical health problem without effective treatments. Given that FoxOs are critical regulators of muscle atrophy and transcriptionally regulate OXPHOS, 26 the aim of this study was to determine if deletion of FoxOs could increase muscle strength and mitochondrial function even in aged mice. Given that anabolic resistance and impaired autophagy are observed in aged muscle, one might speculate that deletion of FoxOs would be detrimental in aging. We found FoxO deletion leads to increases in muscle size by 20–30% in young mice and by 36–48% in aged mice. Importantly, when normalized to muscle size, the 20% decrease in max force of the Aged Ctrl was prevented in Aged TKO. This was associated with mild hypertrophy of muscle fibres in each muscle group tested and with increased mitochondrial respiration and OXPHOS levels in Aged TKO muscle. Thus, loss of FoxOs can increase muscle strength and mitochondrial respiration through the aging process.
Anabolic resistance is the predominant defect seen in aging studies that contributes to muscle loss. 4 , 5 However, muscle proteostasis is the balance between protein breakdown and synthesis. Although increases in protein breakdown are not observed with aging, a blunted suppression of protein degradation in response to low‐dose insulin has been observed. 7 Some of the molecular markers of acute muscle atrophy overlap with sarcopenia, such as increased expression of Trim63 and FoxO3. 34 The current study shows that ubiquitin ligase expression was decreased in Quad and TA when FoxOs were deleted in both young and aged mice, although many ubiquitin‐proteasome and autophagy‐lysosome genes remained unchanged. This fits with previous observations indicating deletion of FoxOs is not detrimental to basal muscle function and only appears to inhibit Ubiquitin Proteasome System (UPS) or autophagy activation in response to chronic stress, such as diabetes, denervation or starvation. 9 , 10 However, Gadd45a expression increased by 2–2.5‐fold in Quad and TA from Aged Ctrl compared with young groups, and this increase was prevented in Aged TKO. Gadd45a expression is typically low in muscle but highly inducible by several transcriptional regulators (ATF4, HDAC4, and/or FoxOs). 33 Gadd45a can cause atrophy upon a wide variety of both physiologic and pathologic stimuli, 35 and Gadd45a is sufficient to cause muscle atrophy via interaction with MEKK4. 33 These data provide evidence that suppression of FoxO activation may help to maintain muscle mass and strength via mechanisms beyond simple regulation of ubiquitin ligase expression.
The increase in central nuclei in TKO groups was striking at 2.5–10‐fold. In healthy skeletal muscle, nuclei are located at the periphery of myofibres. Centrally positioned nuclei have routinely been used as a pathological marker in order to differentiate muscle disorders from neurological disorders. 36 This is interesting because TKO mice appear normal with normal exercise tolerance, grip strength, muscle hypertrophy, improved force generation and histology that appears grossly normal, without fibre necrosis. Furthermore, protein synthesis and autophagy flux were normal under post‐prandial 6‐hour fasting in Young TKO mice. We did perform exercise tolerance and grip strength 1–2 weeks prior to muscle harvest which may stimulate myogenesis in TKOs. The increase in central nuclei may represent increased myotube formation which would fit with the trend toward increased number of total fibres in the plantaris. Recent studies indicate that a skeletal muscle specific protein, skCIP, is important for myonuclei localization, 37 and future studies will be important to discern whether FoxOs directly regulate myogenesis and/or interact with skCIP.
Aging, obesity, and diabetes have been associated with muscle mitochondrial dysfunction in some cases. 1 , 18 Mitochondrial respiration was increased in FoxO TKO mice and associated with preserved expression of OXPHOS subunits. Interestingly, this did not change whole body metabolism measured by indirect calorimetry. Furthermore, mitochondrial improvements coincided with impaired glucose tolerance in Aged TKO, which may be related to larger body mass and higher amount of glucose given during the GTT, because we did not see similar effects on ITT. Although increased mitochondrial content can be due to increase oxidative fibre density, we saw minimal changes in SDH‐positive fibres, type I fibres, and mitochondrial area/cristae in TKO plantaris. FoxOs can regulate OXPHOS subunits in muscle in response to diabetes, 26 , 27 and our study indicates that the decreases in OXPHOS subunits that occur with aging are reversed with FoxO deletion. Thus, the maintenance of mitochondrial respiration in Aged TKO mice may be due to reversal of transcriptional repression of OXPHOS.
Study limitations
This work is not without limitations. First, Aged Ctrl showed mild differences compared with Young Ctrl, despite aging 22–24 months. This makes it difficult to fully assess the role of FoxOs in age‐related sarcopenia. This mild aging phenotype may be due to the mixed genetic background that we described in detail previously, 27 which is a major challenge when using mice to study aging‐related muscle defects. Nonetheless, we do see important decreases in max force generation that are prevented in Aged TKO, which is an important measure of muscle strength in aging. The muscle hypertrophy, suppression of UPS genes, and enhanced mitochondrial respiration present in the TKO mice may have masked small differences between Aged Ctrl vs. Young Ctrl when compared by ANOVA. Another limitation is that the study was designed to measure mitochondrial function, and we could not directly measure muscle protein synthesis or autophagy flux in Aged Ctrl or Aged TKO muscle. In young TKO mice, basal protein synthesis and autophagy flux were unchanged compared with Young Ctrl, but whether this is true in Aged TKO is unknown. Lastly, although we do see preservation of UPS and OXPHOS mRNAs in Aged TKO, they are mild and the definitive mechanisms by which FoxOs mediate the effects on muscle function may go beyond these mild transcriptional changes. The specific FoxO targets and interacting proteins that mediate these effects will require further investigation.
Conclusions
In conclusion, deletion of FoxOs in skeletal muscle increases muscle strength even during aging. This was associated with mild muscle hypertrophy and prevention of aged‐related inductions of Gadd45a and Ube4a. FoxO deletion also enhanced mitochondrial respiration in the absence of improved glucose or insulin tolerance. Suppression of FoxO activation in non‐terminally differentiated tissues can lead to cell proliferation and cancer. 38 It is therefore critical to identify FoxO targets in muscle that increase strength and mitigate mitochondrial abnormalities in hopes of preventing age‐related muscle atrophy.
Author Contributions
C.M.P. designed the study, researched data, and wrote the manuscript. G.B., C.J.N., C.U.B., J.J., C.K.B., T.L.J., and J.D.F. researched data, helped design experiments and helped to write the manuscript. V.A.L. provided reagents, helped design experiments and helped to write the manuscript. B.T.O. designed the study and helped to write the manuscript.
Conflict of interest
The authors declare no conflict of interest regarding the present work.
Supporting information
Data S1. Supporting information
Acknowledgements
This work was supported by VA Merit Review Award Number lO1 BXOO4468 (to B.T.O.) from the US Department of Veterans Affairs Biomedical Laboratory R&D (BLRD) Service and by startup funds from the Fraternal Order of Eagles Diabetes Research (to B.T.O. and V.A.L.) This work was also supported by the National Institutes of Health R56AG063820 (to V.A.L.) We also thank the Fraternal Order of Eagles Diabetes Research Center Metabolic Phenotyping Core Facility for assistance with all the body composition, metabolic cage, respiration, and ATP production data. We thank the Microscopy Core at the University of Iowa for help with histology and TEM. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.
Penniman C. M., Bhardwaj G., Nowers C. J., Brown C. U., Junck T. L., Boyer C. K., Jena J., Fuqua J. D., Lira V. A., and O'Neill B. T. (2023) Loss of FoxOs in muscle increases strength and mitochondrial function during aging, Journal of Cachexia, Sarcopenia and Muscle, 14, 243–259, 10.1002/jcsm.13124
References
- 1. Peterson CM, Johannsen DL, Ravussin E. Skeletal muscle mitochondria and aging: a review. J Aging Res 2012;2012:194821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, et al. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 2006;61:1059–1064. [DOI] [PubMed] [Google Scholar]
- 3. Newman AB, Kupelian V, Visser M, Simonsick EM, Goodpaster BH, Kritchevsky SB, et al. Strength, but not muscle mass, is associated with mortality in the health, aging and body composition study cohort. J Gerontol A Biol Sci Med Sci 2006;61:72–77. [DOI] [PubMed] [Google Scholar]
- 4. Paulussen KJM, McKenna CF, Beals JW, Wilund KR, Salvador AF, Burd NA. Anabolic Resistance of Muscle Protein Turnover Comes in Various Shapes and Sizes. Front Nutr 2021;8:615849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morton RW, Traylor DA, Weijs PJM, Phillips SM. Defining anabolic resistance: implications for delivery of clinical care nutrition. Curr Opin Crit Care 2018;24:124–130. [DOI] [PubMed] [Google Scholar]
- 6. Moro T, Brightwell CR, Deer RR, Graber TG, Galvan E, Fry CS, et al. Muscle Protein Anabolic Resistance to Essential Amino Acids Does Not Occur in Healthy Older Adults Before or After Resistance Exercise Training. J Nutr 2018;148:900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilkes EA, Selby AL, Atherton PJ, Patel R, Rankin D, Smith K, et al. Blunting of insulin inhibition of proteolysis in legs of older subjects may contribute to age‐related sarcopenia. Am J Clin Nutr 2009;90:1343–1350. [DOI] [PubMed] [Google Scholar]
- 8. Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 2013;14:83–97. [DOI] [PubMed] [Google Scholar]
- 9. O'Neill BT, Bhardwaj G, Penniman CM, Krumpoch MT, Suarez Beltran PA, Klaus K, et al. FoxO Transcription Factors Are Critical Regulators of Diabetes‐Related Muscle Atrophy. Diabetes 2019;68:556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Neill BT, Lee KY, Klaus K, Softic S, Krumpoch MT, Fentz J, et al. Insulin and IGF‐1 receptors regulate FoxO‐mediated signaling in muscle proteostasis. J Clin Invest 2016;126:3433–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Penniman CM, Suarez Beltran PA, Bhardwaj G, Junck TL, Jena J, Poro K, et al. Loss of FoxOs in muscle reveals sex‐based differences in insulin sensitivity but mitigates diet‐induced obesity. Mol Metab 2019;30:203–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007;6:458–471. [DOI] [PubMed] [Google Scholar]
- 14. Suetta C, Frandsen U, Jensen L, Jensen MM, Jespersen JG, Hvid LG, et al. Aging affects the transcriptional regulation of human skeletal muscle disuse atrophy. PLoS ONE 2012;7:e51238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bertaggia E, Coletto L, Sandri M. Posttranslational modifications control FoxO3 activity during denervation. Am J Physiol Cell Physiol 2012;302:C587–C596. [DOI] [PubMed] [Google Scholar]
- 16. Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 2013;19:83–92. [DOI] [PubMed] [Google Scholar]
- 17. Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol 2000;526:203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Short KR, Bigelow ML, Kahl J, Singh R, Coenen‐Schimke J, Raghavakaimal S, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A 2005;102:5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Broskey NT, Greggio C, Boss A, Boutant M, Dwyer A, Schlueter L, et al. Skeletal muscle mitochondria in the elderly: effects of physical fitness and exercise training. J Clin Endocrinol Metab 2014;99:1852–1861. [DOI] [PubMed] [Google Scholar]
- 20. Metter EJ, Lynch N, Conwit R, Lindle R, Tobin J, Hurley B. Muscle quality and age: cross‐sectional and longitudinal comparisons. J Gerontol A Biol Sci Med Sci 1999;54:B207–B218. [DOI] [PubMed] [Google Scholar]
- 21. Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet‐induced insulin‐resistant mice. J Clin Invest 2008;118:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyle KE, Canham JP, Consitt LA, Zheng D, Koves TR, Gavin TP, et al. A high‐fat diet elicits differential responses in genes coordinating oxidative metabolism in skeletal muscle of lean and obese individuals. J Clin Endocrinol Metab 2011;96:775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franko A, von Kleist‐Retzow JC, Böse M, Sanchez‐Lasheras C, Brodesser S, Krut O, et al. Complete failure of insulin‐transmitted signaling, but not obesity‐induced insulin resistance, impairs respiratory chain function in muscle. J Mol Med (Berl) 2012;90:1145–1160. [DOI] [PubMed] [Google Scholar]
- 24. Rabol R, Boushel R, Dela F. Mitochondrial oxidative function and type 2 diabetes. Appl Physiol Nutr Metab 2006;31:675–683. [DOI] [PubMed] [Google Scholar]
- 25. Lund MT, Larsen S, Hansen M, Courraud J, Floyd AK, Støckel M, et al. Mitochondrial respiratory capacity remains stable despite a comprehensive and sustained increase in insulin sensitivity in obese patients undergoing gastric bypass surgery. Acta Physiol (Oxf) 2018;223:e13032. [DOI] [PubMed] [Google Scholar]
- 26. Bhardwaj G, Penniman CM, Jena J, Suarez Beltran PA, Foster C, Poro K, et al. Insulin and IGF‐1 receptors regulate complex‐I dependent mitochondrial bioenergetics and supercomplexes via FoxOs in muscle. J Clin Invest 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhardwaj G, Penniman CM, Klaus K, Weatherford ET, Pan H, Dreyfuss JM, et al. Transcriptomic Regulation of Muscle Mitochondria and Calcium Signaling by Insulin/IGF‐1 Receptors Depends on FoxO Transcription Factors. Front Physiol 2021;12:779121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waisman A, Norris AM, Elías Costa M, Kopinke D. Automatic and unbiased segmentation and quantification of myofibers in skeletal muscle. Sci Rep 2021;11:11793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Neill BT, Lauritzen HPMM, Hirshman MF, Smyth G, Goodyear LJ, Kahn CR. Differential Role of Insulin/IGF‐1 Receptor Signaling in Muscle Growth and Glucose Homeostasis. Cell Rep 2015;11:1220–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sawano S, Komiya Y, Ichitsubo R, Ohkawa Y, Nakamura M, Tatsumi R, et al. A One‐Step Immunostaining Method to Visualize Rodent Muscle Fiber Type within a Single Specimen. PLoS ONE 2016;11:e0166080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eisner V, Cupo RR, Gao E, Csordás G, Slovinsky WS, Paillard M, et al. Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc Natl Acad Sci U S A 2017;114:E859–E868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2007;2:287–295. [DOI] [PubMed] [Google Scholar]
- 33. Bullard SA, Seo S, Schilling B, Dyle MC, Dierdorff JM, Ebert SM, et al. Gadd45a Protein Promotes Skeletal Muscle Atrophy by Forming a Complex with the Protein Kinase MEKK4. J Biol Chem 2016;291:17496–17509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Raue U, Slivka D, Jemiolo B, Hollon C, Trappe S. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J Gerontol A Biol Sci Med Sci 2007;62:1407–1412. [DOI] [PubMed] [Google Scholar]
- 35. Moskalev AA, Smit‐McBride Z, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, et al. Gadd45 proteins: relevance to aging, longevity and age‐related pathologies. Ageing Res Rev 2012;11:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Folker ES, Baylies MK. Nuclear positioning in muscle development and disease. Front Physiol 2013;4:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu J, Huang ZP, Nie M, Wang G, Silva WJ, Yang Q, et al. Regulation of myonuclear positioning and muscle function by the skeletal muscle‐specific CIP protein. Proc Natl Acad Sci U S A 2020;117:19254–19265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, et al. FoxOs are lineage‐restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007;128:309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting information