

Summary:
We report the systematic development of an electrooxidative methodology that translates stoichiometric C-H fluorination reactivity of an isolable CuIII fluoride complex into a catalytic process. The critical challenges of electrocatalysis with a highly reactive CuIII species were addressed by the judicious selection of electrolyte, F− source, and sacrificial electron acceptor. Catalyst-controlled C-H fluorination occurs with a preference for hydridic C-H bonds with high bond dissociation energies over weaker but less hydridic C-H bonds. The selectivity is driven by an oxidative asynchronous proton-coupled elelctron transfer (PCET) at an electrophilic CuIII-F complex. We further demonstrate that the asynchronicity factor of hydrogen atom transfer η can be used as a guideline to rationalize the selectivity of C-H fluorination.
Graphical Abstract
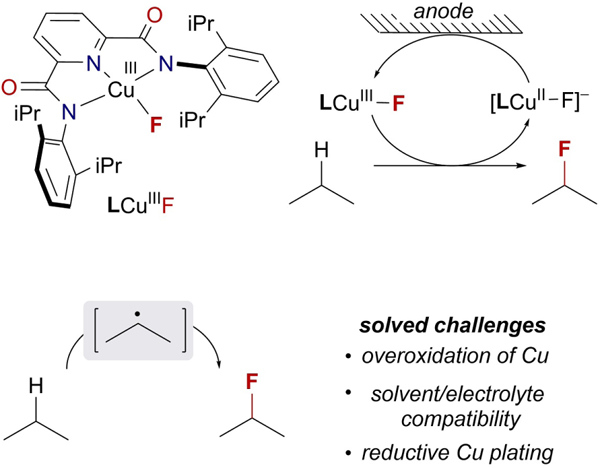
eTOC
We report the systematic development of an electrochemical method that translates stoichiometric C-H fluorination reactivity of a copper(III) fluoride complex into a catalytic process. The challenges associated with a highly reactive copper(III) species were addressed by the judicious selection of electrolyte, fluoride source, and sacrificial electron acceptor. The catalyst-controlled C-H fluorination occurs with a preference for hydridic C-H bonds with high bond dissociation energies over weaker but less hydridic C-H bonds, due to the oxidative asynchronous nature of proton-coupled electron transfer at an electrophilic CuIII-F complex.
INTRODUCTION
Carbon-fluorine bonds endow highly desirable chemical and physical properties to materials, pharmaceuticals, and commodity chemicals.1 An ideal approach to synthesizing fluorinated organic molecules is to directly replace ubiquitous C−H bonds with C−F bonds. The extensive efforts devoted to developing this transformation by the groups of Lectka,2–7 Baran,8 Cook,9,10 Sorensen,11 Stahl,12,13 and others14–18 using thermal, photochemical, or electrochemical approaches underscore the importance of the aliphatic C-H bond fluorination (Figure 1A, top path). However, these methodologies all rely on expensive electrophilic F+ reagents such as Selectfluor or N-fluorobenzenesulfonimide (NFSI). In contrast, Csp3-H bond fluorination reactions with abundant sources of nucleophilic fluoride (KF, CsF, TBAF, Et3N(HF)3) remain rare and underdeveloped.
Figure 1.
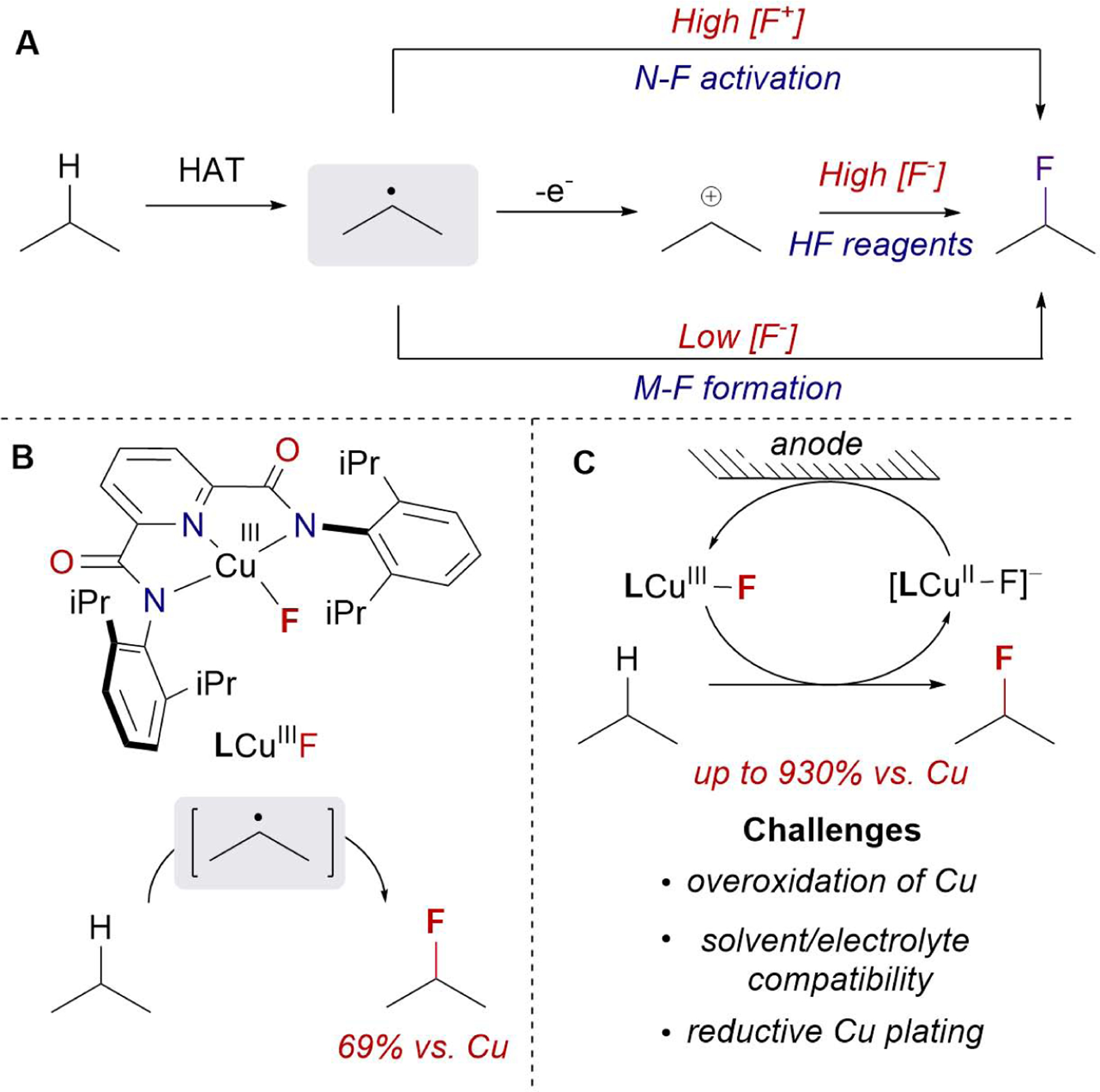
Overview of C-H fluorination. (A) Strategies for C-H fluorination with electrophilic N-F reagents, HF, or nucleophilic F− sources. (B) C-H fluorination with LCuIII-F complex. (C) LCu-catalyzed C-H fluorination enabled by electrochemistry and associated challenges.
The groups of Doyle19 and Musacchio20 recently reported elegant strategies for oxidative C-H fluorination with nucleophilic fluorides. These methods rely on hydrogen atom transfer (HAT) to form a carbon-centered radical that is subsequently oxidized to a carbonium ion by a photoredox catalyst (Figure 1A, middle path). This strategy of radical-polar crossover (RPC) merges two one-electron oxidation steps to form C-F bonds with simple fluoride nucleophiles. However, efficient capture of the carbonium intermediates requires a high concentration of solvated F−, which is achieved with superstoichiometric quantities of soluble Et3N(HF)3. The limitation of these methodologies to hazardous F− sources can be circumvented by combining transition metal catalysts with benign but less soluble fluoride salts. The high affinity of transition metals for F− facilitates the formation of transition metal fluoride complexes even at low concentrations of solvated fluoride from inexpensive salts like KF, CsF, or AgF (Figure 1A, bottom path).21–26 For example, Groves et al. reported a rare example of catalytic C-H fluorination using a bioinspired Mn porphyrin catalysts.27–31
We previously identified a formal copper(III) fluoride complex (LCuIIIF, L = pyridinebiscarboxamide) that performs both HAT and fluorine atom transfer (Figure 1B).32 Despite the relatively mild potentials at which LCuIIIF can be formed from the oxidation of [LCuII-F]− precursor (0.47 V vs. Ag+/Ag), the C-H fluorination reactions were limited to stoichiometric quantities of LCuIIIF complex, and no turnover was observed in the presence of exogenous chemical oxidants. The inability to translate the reactivity of discrete CuIII complexes to catalytic reactions is a general challenge. Although CuIII intermediates are frequently proposed in catalytic organic transformations, most isolable CuIII complexes are not active for catalysis and are only employed as mechanistic probes for understanding stoichiometric reactions, e.g. proton-coupled electron transfer/HAT33–36 and C-X bond formation processes.37–40 If the gap between stoichiometric and proposed catalytic reactivity of Cu(III) complexes can be bridged, the well-characterized reactivities of CuIII species can serve as blueprints for developing desirable C-H bond functionalization methodologies.
Herein, we report the systematic development of an electrooxidative methodology that translates the stoichiometric C-H fluorination reactivity of an isolable CuIII fluoride complex into a catalytic process while addressing critical limitations of electrocatalysis with Cu (Figure 1C). This strategy leverages electrochemistry as a means of controlling potentials and rates of oxidation to establish a rare example of molecular catalysis for electrochemical C-H fluorination (ECF) with simple fluoride salts.41 Translating the reactivity of a well-defined complex to catalysis allows predictable regioselectivity based on the calculation of the HAT asynchronicity factor η. Analogous to polarity matching effects,42–44 Cu-controlled C-H fluorination occurs by an oxidative asynchronous mechanism that is selective for strong hydridic C-H bonds over weaker C-H bonds that are less hydridic.
RESULTS AND DISCUSSION
We envisioned a catalytic ECF process that involves (i) electrooxidation of [LCuII-F]− to LCuIII-F, (ii) fluorination of C-H substrates according to the established 2:1 Cu:substrate stoichiometric reactivity,32 and (iii) regeneration of the [LCuII-F]− from a metal fluoride salt (Figure 1C). Establishing this electrocatalytic methodology requires solutions to several challenges. First, the regeneration of [LCuII-F]− from LCuII and a metal fluoride must be favorable and should occur at a high rate. High concentrations of [LCuII-F]− would ensure productive anodic reactions and mitigate oxidative reactions of other Cu complexes that lead to decomposition. Second, the reaction medium must be conducive to electrolysis but inert to highly reactive LCuIII-F intermediates and basic metal fluorides. Third, the reduction of LCuII at the cathode must be circumvented to avoid Cu0 deposition in an undivided cell.45
LCuIII-F Stability and Electrogeneration.
The key LCuIIIF complex that mediates C-H bond fluorination was previously prepared by sequential reaction of LCuII(MeCN) with tetrabutylammonium fluoride (TBA)F(H2O)3 and [NAr3]PF6 (Ar = 4-bromophenyl, E = 0.66 V vs. Ag/AgNO3) in dichloromethane (DCM).32 However, electrolysis of such solutions is problematic because DCM is resistive even with high loadings of supporting electrolytes, and most ammonium electrolytes (including TBA+) decompose in the presence of anhydrous fluoride to release amine byproducts that undergo competitive electrooxidation. As such, our initial studies focused on identifying electrochemically-applicable solvents in which the LCuIIIF complex is both long-lived and readily formed from a LCuII precursor, a metal fluoride, and a chemical oxidant (Figure 2).
Figure 2.
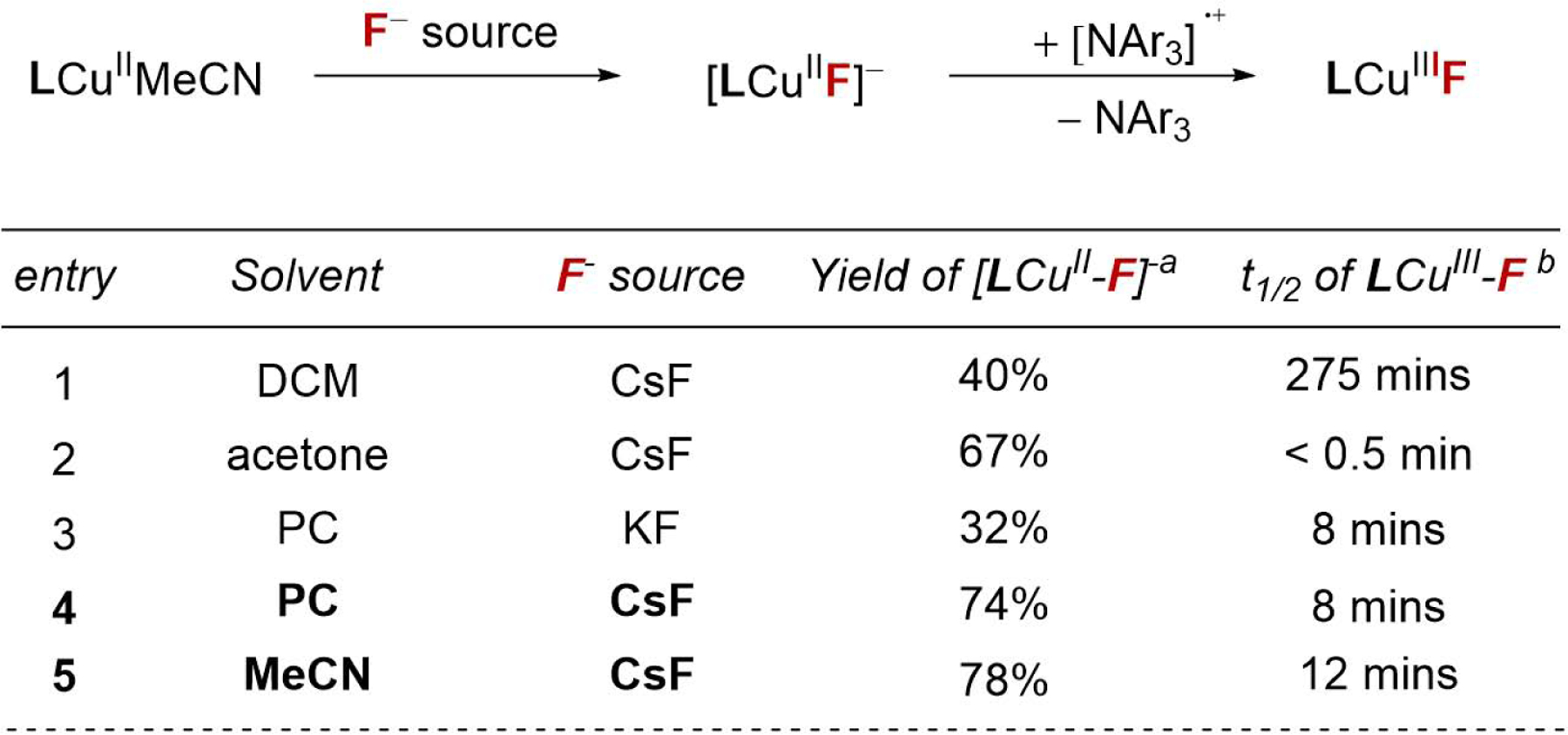
The yield and stability of copper fluoride complexes under various conditions. a Yield after stirring LCuIIMeCN with 10 eq F− sources for 3 min. b Measured at 22 °C. See also Figure S19–S23.
We first quantified the extent of fluoride ligation to form [LCuIIF]− after just 3 minutes of stirring a combination of [LCuII], metal fluoride salt (KF or CsF), and solvent (Figure 2). Ligation reactions with CsF formed the targeted complex [LCuIIF]− in highest yields, despite the poor solubility of the fluoride source. The [LCuIIF]− complex is typically formed in good yield after the short reaction time in polar aprotic solvents (entries 2, 4, 5), while yields are lower in less polar solvents like dichloromethane (DCM, entry 1). However, the persistence of the high-valent LCuIIIF intermediate is highest in the less polar solvents where fluoride ligation is slow. As examples, the LCuIIIF half-life in DCM is nearly two orders of magnitude longer than in acetone (entries 1 vs 2). Propylene carbonate (PC) and acetonitrile (MeCN) are the only two solvents that support both in situ formation of [LCuIIF]− and long-lived LCuIIIF intermediates (t1/2 = ca. 12 mins). Together, these experiments suggest that the combination of CsF with PC or MeCN are good starting conditions for developing an electrocatalytic C-H fluorination.
ECF Reaction Development.
The identified conditions for the formation of a persistent LCuIIIF complex were next subjected to electrolysis in the presence of organic substrates (Figure 3). Reactions containing the ligated Cu precursor LCuIIMeCN, CsF, and THF as the C-H substrate were electrolyzed in undivided cells at a constant voltage of 3.3 V with TBAClO4 as a supporting electrolyte in MeCN. Even with this high applied potential, the current rapidly decreased to just 0.2 mA after 20 min, resulting in just 40% yield of fluorinated product relative to added Cu (entry 1). The cell resistance in other solvents compatible with LCuIIIF was even higher (PC, entry 2), which allowed for only 1.7 e− equiv. relative to Cu to be passed. The fluorinated product formed in only 15% yield along with a significant quantity of C-H fluorinated PC as a competing product. The combination of MeCN and PC completely inhibited ECF of PC and resulted in slightly higher yields than either solvent alone but still failed to sustain currents (entry 3).
Figure 3.

ECF reaction development. See also Figure S1–S3 and Table S1–S3.
The surprisingly high resistance of these reactions was found to result from the irreversible consumption of the redox-active Cu complex by Cu plating and deposition of insoluble materials at the cathode (Figure S4). To address these limitations, we evaluated reactions with redox mediators or sacrificial electron acceptors that undergo reduction in preference to Cu complexes without interfering with the anodic reactions.45 While reactions with ferrocenyl mediators operated at high currents (>3 mA), no product was detected (entries 4, 5; see Figure S3 for a full list of mediators). Reactions with sacrificial e− acceptors (e.g., C6Br6, C6Cl6, DCM) similarly resulted in low yields (entries 6, 7, 8). The Br− or Cl− ions released during the reduction of these additives were found to bind LCu in preference to F−, and the unreactive [LCuIICl]− complex could be identified by its characteristic green color during electrolysis with DCM.32
These results guided us to evaluate fluorinated e− acceptors that could decompose to generate additional fluoride. While hexabromo- or hexachlorobenzene completely inhibited the reaction (entries 7, 8), reactions with hexafluorobenzene formed the desired product in 560% yield relative to Cu. These results constitute the first evidence of electrocatalysis. Analysis of the reaction mixture after electrolysis by 19F NMR spectroscopy revealed that hexafluorobenzene was converted to a mixture of pentafluorobenzene (57% yield vs. Cu) and 2,3,4,5-tetrafluorobenzene (35% yield vs. Cu) through electroreduction and subsequent protodefluorination. We simultaneously evaluated buffered acid-base combinations that could promote the desired cathodic reactions and improve the overall conductivity of the reaction medium. Reactions performed with a collidinium-TFA (TFA = trifluoroacetic acid) mixture formed the targeted product in 930% relative to Cu (entry 13). Finally, control reactions demonstrated that CsF, electrochemistry, and the LCuII (MeCN) complex are necessary for C-H fluorination (entries 14–17). Collectively, these results underscore the counterintuitive challenges of Cu-catalyzed electrosynthesis. Specifically, the development of this oxidative reaction primarily focused on challenges associated with cathodic chemistry.
Substrate Scope.
We next applied the developed conditions to a range of substrates to evaluate the reactivity of the system. While reactions with THF proved high-yielding, the expected trends associated with radical-based reactivity were not immediately apparent (Figure 4). In particular, substrates with weak CH bonds that are prototypical targets for radical C-H fluorination methods often resulted in lower yields than substrates with stronger C-H bonds (e.g., 8 vs 1, 87 vs. 92 kcal/mol). The chemoselectivity of this reaction is demonstrated by the fluorination reaction of 2-(4-ethylbenzyl)tetrahydrofuran. Fluorination occurs exclusively at the position alpha to oxygen (12 and 13) rather than at the benzylic site (14 and 15). This selectivity diverges from other known C-H fluorination methods that preferentially functionalize benzylic C-H bonds.21–26,28 Intermolecular competition experiments between THF and tetrahydronaphthalene revealed similar selectivity trends where the strong C-H bonds alpha to oxygen were preferentially functionalized over the weaker benzylic C-H bonds (1 vs. 11).
Figure 4.
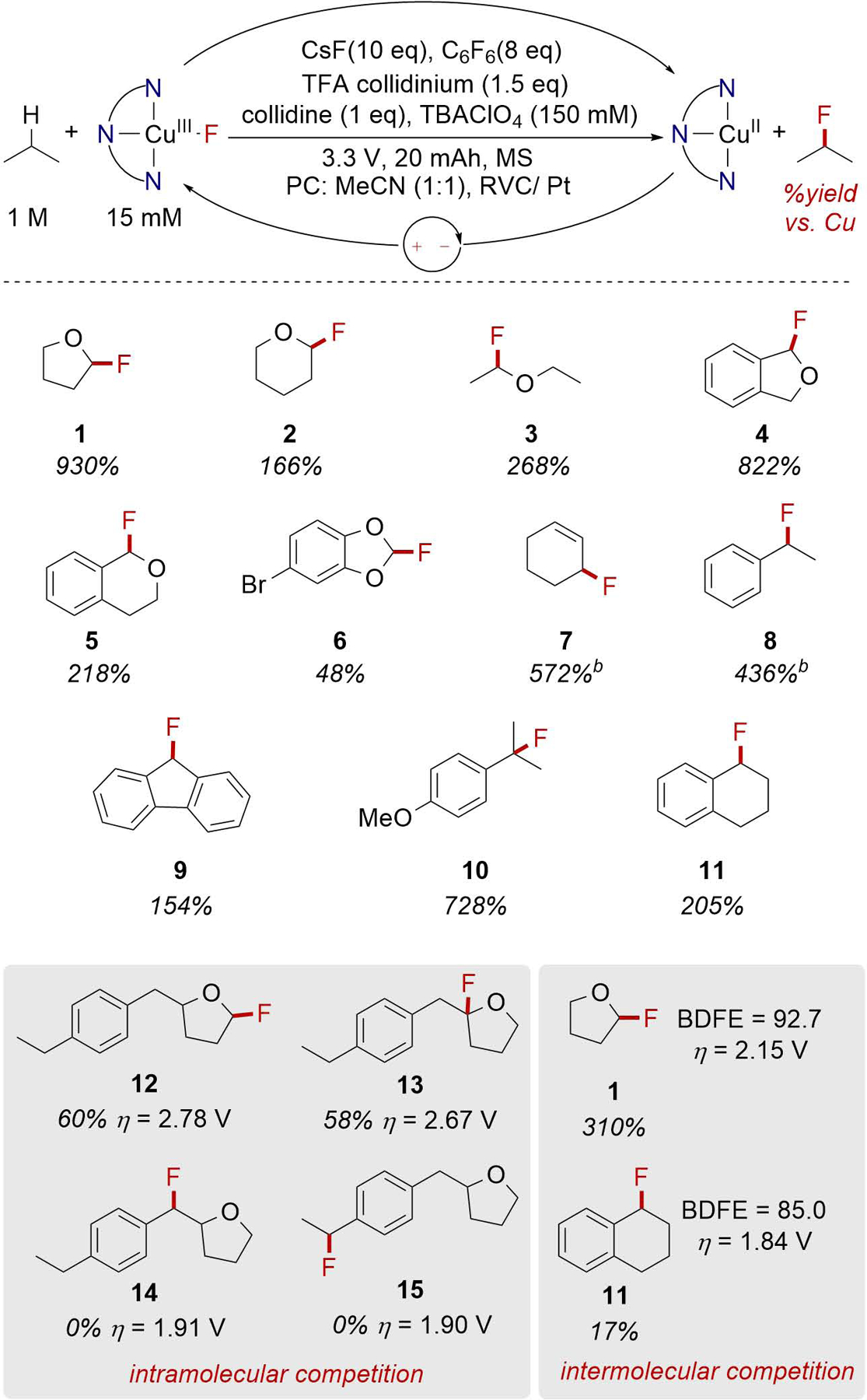
ECF substrate scope. aYields determined by 19F NMR with fluorobenzene as an internal standard. bconditions: 9 equiv. C6F6, 15 equiv. CsF, no collidine, TFA collidinium and MS, acetone instead of PC:MeCN. c50 mAh instead of 20 mAh.
Regioselectivity of C-H fluorination.
The rare access to an isolable and well-characterized Cu(III) complex that is the exact intermediate of a Cu-catalyzed fluorination reaction serves as a blueprint for understanding the regioselectivity in Figure 4. We hypothesized that the observed selectivities in both inter- and intramolecular competition experiments highlighted in Figure 4 are due to the high rates of HAT at C-H bonds alpha to oxygen. Accordingly, we measured the HAT rates from various organic substrates by monitoring the consumption of LCuIIIF (Figure 5). The logarithm of resulting secondary HAT rates log(kHAT), spreading over four orders of magnitude, were plotted against the computed driving forces of electron transfer (ΔGoET, Figure S24), proton transfer (ΔGoPT, Figure S25), and proton-coupled electron transfer (ΔGoPCET, Figure 2A). The correlations between log(kHAT) with ΔGoPT and ΔGoET are poor (R2 = 0.2184 and 0.5227). Additionally, direct electron transfer and proton transfer are both highly endergonic (ΔGoET > 0.65 eV, ΔGoPT > 2.42 eV, Table S4), suggesting that stepwise ET-PT and PT-ET pathways are unlikely.46–48
Figure 5.
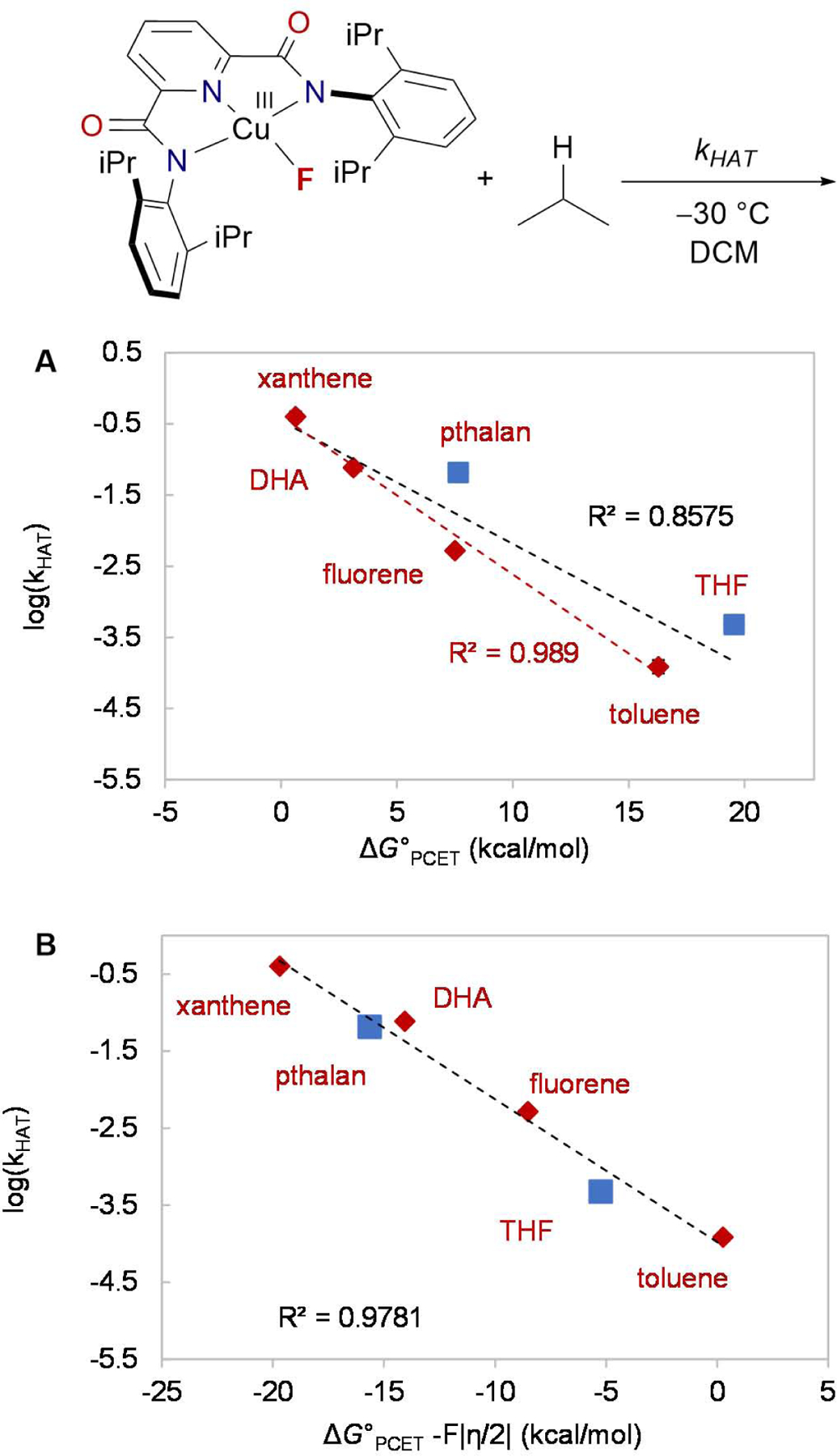
Plot of the log of second order rate constant kHAT of the reaction between LCuIIIF and C-H substrates with (A) the driving force of PCET, ΔGoPCET (R2=0.858) and (B) ΔGoPCET – F|η|/2. The R2 value of the first plot improves to 0.989, if THF and pthalan are excluded. Error bars are smaller than the site of the data points. See also Figure S6–S18, Table S4–S8.
In contrast, a linear correlation between log(kHAT) vs. ΔGoPCET was observed (R2 = 0.858, Figure 2A). However, THF and pthalan are clearly outliers. If they are excluded from the plot, the R2 value of the plot improves to 0.989. We also attempted to correlate the rates of C-H activation using a linear combination of ΔGPCET, ΔGPT, and ΔGET using semiempirical models developed by Anderson and Borovik (see Figure S26, S27).49,50 However, the correlations do not improve to more than R2 = 0.858.
Notably, although the BDFE of THF is higher than toluene by 3 kcal/mol, LCuIII-F reacts faster with THF compared to toluene, suggesting that the rate of C-H activation does not follow the classical trend predicted by the Bell-Evans-Polanyi principle.51 C-H bond cleavage by high valent metal complexes is often described as PCET. However, recent work by Borovik,52 Anderson,53 Tolman,34 and Kojima54 suggests that the basicity or redox potential of the metal complex can play a dominant role in the rate of C-H bond cleavage. Accordingly, we rationalize the higher-than-expected rates of THF and pthalan with asynchronous transition states of HAT, which could be dominated by electron transfer (oxidative asynchronous pathway) or proton transfer (basic asynchronous pathway, Scheme 1). HAT processes with greater asynchronicity are predicted to have lower activation barriers.55
Scheme 1.
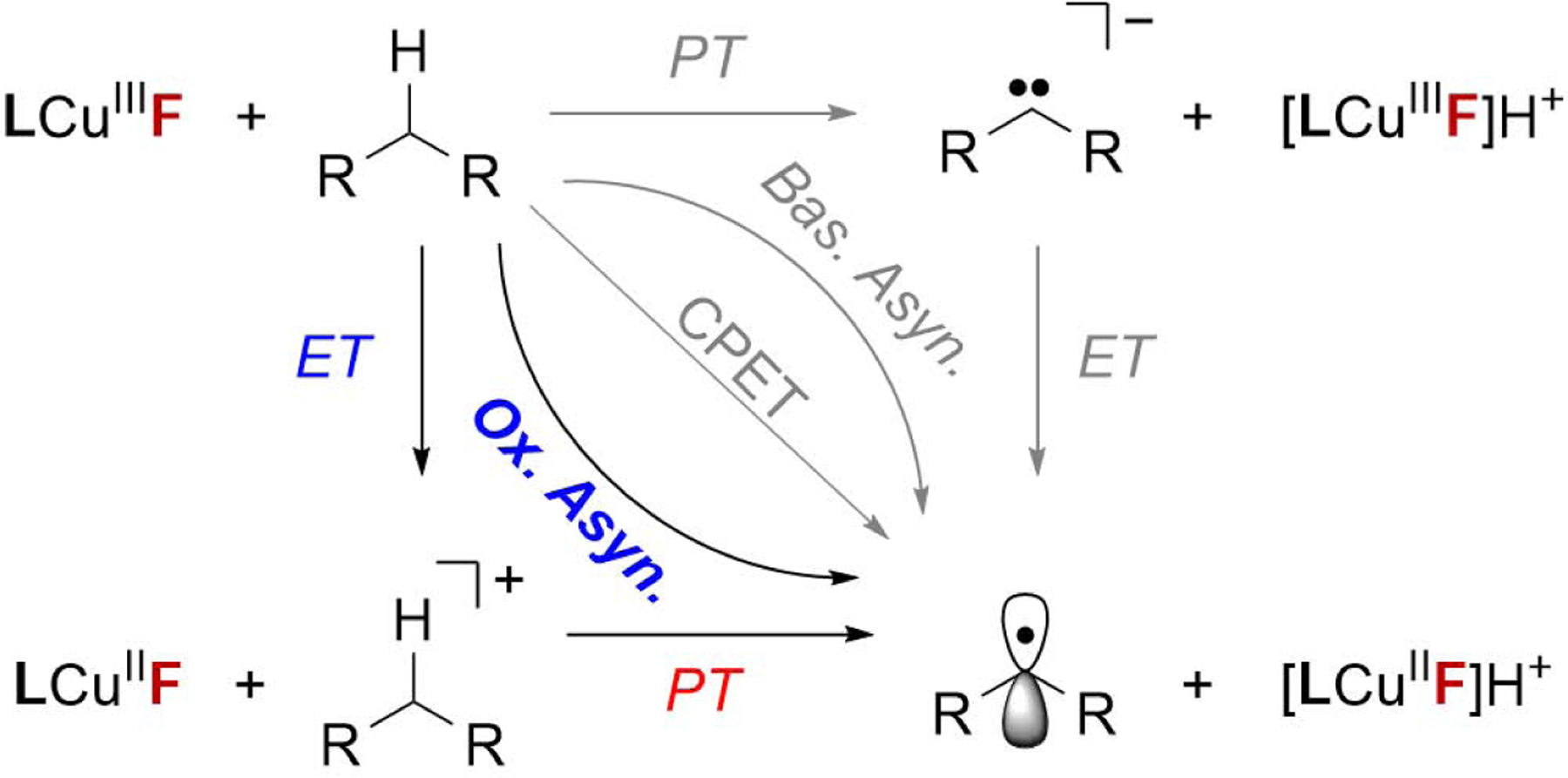
Mechanisms of PCET.
Work by Tolman and Cramer et al. demonstrated that LCuIIIO2CR complexes activate C-H bonds through an oxidative asynchronous process with an ET-dominated transition state (Scheme 1).34 Given the similar E1/2 of LCuIIIF and LCuIII(O2CAr), HAT by LCuIIIF is expected to be oxidative synchronous as well. To consider the combined effects of classical thermodynamics of HAT and propensity for asynchronicity, Srnec argues that the barrier of asynchronous HAT process is a function of ΔGoPCET – F|η|/2, where ΔGoPCET is the driving force of HAT/PCET, F is the Faraday constant and η is the asynchronicity factors. Indeed, we found that the plot of log(kHAT) vs. ΔGoPCET – F|η|/2 shows a linear correlation with R2 = 0.9781 (Figure 2B). With this plot, the ether and benzylic substrates follow a unified trend. An F-test with a null hypothesis that the model with ΔGoPCET only is better shows a p-value of 0.02208, suggesting the strong correlation between log(kHAT) and ΔGoPCET – F|η|/2 is statistically significant.49
Due to the oxidative asynchronous nature of HAT by LCuIII species,34 the reactions of LCuIIIF with the easily oxidized ether substrates are enhanced by virtue of high η values. For example, ECF of chroman occurs exclusively at the more asynchronous α-ethereal position (ηether = 2.69 V, ηbenzylic = 2.21 V), which is the opposite of conventional selectivity predicted by BDFE values (BDFEether = 94.4, BDFEbenzylic = 84.8 kcal/mol, Figure 5). This approach can also be applied to rationalize the intermolecular selectivity of tetralin vs. THF. Although tetralin has a lower BDFE than THF (85.0 vs. 92.7 kcal/mol), C-H fluorination occurs preferentially at the more asynchronous substrate (ηtetraline = 1.84 V, ηTHF = 2.15 V). While leveraging asynchronous effects has been touted as an appealing tool for reversing the selectivity of C-H functionalization, our work is the first demonstration of this principle. Unlike the empirical polarity matching effect,42–44 asynchronicity calculation has quantitative prediction ability and can potentially be applied to a broader range of systems, including transition metal.
CONCLUSION
In summary, we developed an electrooxidative methodology that translates the stoichiometric C-H fluorination reactivity of a well-characterized CuIII complex into a catalytic process. Through systematic studies of isolable CuII/CuIII intermediates, we identify conditions that allow electrochemical generation of highly reactive LCuIII-F complex at the anode while preventing its decomposition at the cathode. The use of a molecular copper catalyst allows access to F radicaltype reactivity at a much lower potential (3.3 V in an undivided cell) compared to previous ECF reactions. The mild electrolysis conditions enable selective monofluorination of strong electron-rich C-H bonds over weak benzyl C-H bonds. For the first time, we demonstrate the use of asynchronous effect of HAT to override the traditional C-H activation selectivity that often trends with bond dissociation energies. The selectivity of C-H functionalization can be easily predicted by using the computed η values as a guideline. Ongoing work aims to further leverage LCuIII complexes for electrochemical C-H functionalization reactions.
Experimental Procedures
Resource Availability
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shiyu Zhang (zhang.8941@osu.edu).
Materials Availability:
All unique reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and Code Availability
All other data supporting the findings of this study are available within the article and the supplemental information or from the lead contact upon reasonable request.
Supplementary Material
Data S2. NMR spectra.
Data S3. Computational coordinates.
Highlights.
Electrochemistry is used to translate a stoichiometric reaction to a catalytic process
The fluorination reaction is selective for electron-rich C-H bonds over weak ones
The selectivity is rationalized by the asynchronous effect of PCET
Bigger picture statement.
Although copper(III) intermediates are frequently proposed in catalytic organic transformations, most isolable copper(III) complexes are not active for catalysis and are only employed as mechanistic probes for understanding stoichiometric reactions. We demonstrated that electrochemistry could be used to translate the stoichiometric C-H fluorination reactivity of an isolable CuIII fluoride complex into a catalytic process. This strategy leverages control of potentials and rates of oxidation to establish a rare example of molecular catalysis for electrochemical C-H fluorination with simple fluoride salts. The Cu-mediated C-H fluorination occurs by an oxidative asynchronous proton-coupled electron transfer that is selective for strong hydridic C-H bonds over weaker C-H bonds that are less hydridic.
Funding Sources
This material is based on work supported by the National Institutes of Health under award no. R01-GM145746 (S.Z.). J.K.B. was supported by a Presidential Fellowship from The Ohio State University Graduate School. The authors thank The Ohio State University Department of Chemistry and Biochemistry for additional financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
The authors declare no competing financial interest.
REFERENCES
- 1.Müller K, Faeh C, and Diederich F (2007). Fluorine in pharmaceuticals: Looking beyond intuition. Science 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 2.Bloom S, Pitts CR, Miller DC, Haselton N, Holl MG, Urheim E, and Lectka T (2012). A polycomponent metal-catalyzed aliphatic, allylic, and benzylic fluorination. Angew. Chem., Int. Ed 51, 10580–10583. 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]
- 3.Pitts CR, Bloom S, Woltornist R, Auvenshine DJ, Ryzhkov LR, Siegler MA, and Lectka T (2014). Direct, catalytic monofluorination of sp3 C–H Bonds: A radical-based mechanism with ionic selectivity. J. Am. Chem. Soc 136, 9780–9791. 10.1021/ja505136j. [DOI] [PubMed] [Google Scholar]
- 4.Bloom S, Pitts CR, Woltornist R, Griswold A, Holl MG, and Lectka T (2013). Iron(II)-catalyzed benzylic fluorination. Org. Lett 15, 1722–1724. 10.1021/ol400424s. [DOI] [PubMed] [Google Scholar]
- 5.Capilato JN, Pitts CR, Rowshanpour R, Dudding T, and Lectka T (2020). Site-Selective Photochemical Fluorination of Ketals: Unanticipated Outcomes in Selectivity and Stability. J. Org. Chem 85, 2855–2864. [DOI] [PubMed] [Google Scholar]
- 6.Bloom S, McCann M, and Lectka T (2014). Photocatalyzed benzylic fluorination: shedding “light” on the involvement of electron transfer. Org. Lett 16, 6338–6341. 10.1021/ol503094m. [DOI] [PubMed] [Google Scholar]
- 7.Bloom S, Knippel JL, and Lectka T (2014). A photocatalyzed aliphatic fluorination. Chem. Sci 5, 1175–1178. 10.1039/C3SC53261E. [DOI] [Google Scholar]
- 8.Takahira Y, Chen M, Kawamata Y, Mykhailiuk P, Nakamura H, Peters BK, Reisberg SH, Li C, Chen L, Hoshikawa T, et al. (2019). Electrochemical C(sp3)-H Fluorination. Synlett 30, 1178–1182. 10.1055/s-0037-1611737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groendyke BJ, AbuSalim DI, and Cook SP (2016). Iron-catalyzed, fluoroamide-directed C–H fluorination. J. Am. Chem. Soc 138, 12771–12774. 10.1021/jacs.6b08171. [DOI] [PubMed] [Google Scholar]
- 10.Pinter EN, Bingham JE, Abusalim DI, and Cook SP (2020). N-Directed fluorination of unactivated Csp3-H bonds. Chem. Sci 11, 1102–1106. 10.1039/c9sc04055b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.West JG, Bedell TA, and Sorensen EJ (2016). The Uranyl Cation as a Visible-Light Photocatalyst for C(sp3)−H Fluorination. Angew. Chem., Int. Ed 55, 8923–8927. 10.1002/anie.201603149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasilopoulos A, Golden DL, Buss JA, and Stahl SS (2020). Copper-Catalyzed C–H Fluorination/Functionalization Sequence Enabling Benzylic C–H Cross Coupling with Diverse Nucleophiles. Org. Lett 22, 5753–5757. 10.1021/acs.orglett.0c02238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buss JA, Vasilopoulos A, Golden DL, and Stahl SS (2020). Copper-Catalyzed Functionalization of Benzylic C–H Bonds with N-Fluorobenzenesulfonimide: Switch from C–N to C–F Bond Formation Promoted by a Redox Buffer and Brønsted Base. Org. Lett 22, 5749–5752. 10.1021/acs.orglett.0c02239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amaoka Y, Nagatomo M, and Inoue M (2013). Metal-free fluorination of C(sp3)–H bonds Using a catalytic N-oxyl radical. Org. Lett 15, 2160–2163. 10.1021/ol4006757. [DOI] [PubMed] [Google Scholar]
- 15.Xia J-B, Zhu C, and Chen C (2013). Visible light-promoted metal-free C–H activation: Diarylketone-catalyzed selective benzylic mono- and difluorination. J. Am. Chem. Soc 135, 17494–17500. 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halperin SD, Fan H, Chang S, Martin RE, and Britton R (2014). A Convenient Photocatalytic Fluorination of Unactivated C-H Bonds. Angew. Chem., Int. Ed 53, 4690–4693. 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]
- 17.Xia J-B, Zhu C, and Chen C (2014). Visible light-promoted metal-free sp3-C–H fluorination. Chem. Commun 50, 11701–11704. 10.1039/C4CC05650G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kee CW, Chin KF, Wong MW, and Tan C-H (2014). Selective fluorination of alkyl C–H bonds via photocatalysis. Chem. Commun 50, 8211–8214. 10.1039/C4CC01848F. [DOI] [PubMed] [Google Scholar]
- 19.Leibler INM, Tekle-Smith MA, and Doyle AG (2021). A general strategy for C(sp3)–H functionalization with nucleophiles using methyl radical as a hydrogen atom abstractor. Nat. Commun 12, 6950. 10.1038/s41467-021-27165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Fitzpatrick NA, Das M, Bedre IP, Yayla HG, Lall MS, and Musacchio PZ (2022). A photoredox-catalyzed approach for formal hydride abstraction to enable Csp3–H functionalization with nucleophilic partners (F, C, O, N, and Br/Cl). Chem Catal [Google Scholar]
- 21.Braun M-G, and Doyle AG (2013). Palladium-catalyzed allylic C–H fluorination. J. Am. Chem. Soc 135, 12990–12993. 10.1021/ja407223g. [DOI] [PubMed] [Google Scholar]
- 22.McMurtrey KB, Racowski JM, and Sanford MS (2012). Pd-catalyzed C–H fluorination with nucleophilic fluoride. Org. Lett 14, 4094–4097. 10.1021/ol301739f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto K, Li J, Garber JAO, Rolfes JD, Boursalian GB, Borghs JC, Genicot C, Jacq J, van Gastel M, Neese F, et al. (2018). Palladium-catalysed electrophilic aromatic C–H fluorination. Nature 554, 511–514. 10.1038/nature25749. [DOI] [PubMed] [Google Scholar]
- 24.Eunsung L, S., K.A., C., P.D., N., N.C., B., B.G., Takeru F, C., C.D., M., H.J., and Tobias R (2011). A Fluoride-Derived Electrophilic Late-Stage Fluorination Reagent for PET Imaging. Science 334, 639–642. 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Börgel J, and Ritter T (2017). Carbon–Fluorine Reductive Elimination from Nickel(III) Complexes. Angew. Chem., Int. Ed 56, 6966–6969. 10.1002/anie.201701552. [DOI] [PubMed] [Google Scholar]
- 26.Hull KL, Anani WQ, and Sanford MS (2006). Palladium-Catalyzed Fluorination of Carbon−Hydrogen Bonds. J. Am. Chem. Soc 128, 7134–7135. 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Liu W, Ren H, Neelamegam R, Hooker JM, and Groves JT (2014). Late stage benzylic C–H fluorination with [18F]fluoride for PET imaging. J. Am. Chem. Soc 136, 6842–6845. 10.1021/ja5039819. [DOI] [PubMed] [Google Scholar]
- 28.Liu W, Huang X, Cheng M-J, Nielsen RJ, Goddard WA, and Groves JT (2012). Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 337, 1322 LP – 1325. 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- 29.Liu W, Huang X, Placzek MS, Krska SW, McQuade P, Hooker JM, and Groves JT (2018). Site-selective 18F fluorination of unactivated C–H bonds mediated by a manganese porphyrin. Chem. Sci 9, 1168–1172. 10.1039/C7SC04545J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu W, and Groves JT (2013). Manganese-catalyzed oxidative benzylic C–H fluorination by fluoride ions. Angew. Chem., Int. Ed 52, 6024–6027. 10.1002/anie.201301097. [DOI] [PubMed] [Google Scholar]
- 31.Liu W, and Groves JT (2015). Manganese catalyzed C–H halogenation. Acc. Chem. Res 48, 1727–1735. 10.1021/acs.accounts.5b00062. [DOI] [PubMed] [Google Scholar]
- 32.Bower JK, Cypcar AD, Henriquez B, Stieber SCE, and Zhang S (2020). C(sp3)–H Fluorination with a Copper(II)/(III) Redox Couple. J. Am. Chem. Soc 142, 8514–8521. 10.1021/jacs.0c02583. [DOI] [PubMed] [Google Scholar]
- 33.Dhar D, and Tolman WB (2015). Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J. Am. Chem. Soc 137, 1322–1329. 10.1021/ja512014z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandal M, Elwell CE, Bouchey CJ, Zerk TJ, Tolman WB, and Cramer CJ (2019). Mechanisms for Hydrogen-Atom Abstraction by Mononuclear Copper(III) Cores: Hydrogen-Atom Transfer or Concerted Proton-Coupled Electron Transfer? J. Am. Chem. Soc 141, 17236–17244. 10.1021/jacs.9b08109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dhar D, Yee GM, Spaeth AD, Boyce DW, Zhang H, Dereli B, Cramer CJ, and Tolman WB (2016). Perturbing the Copper(III)–Hydroxide Unit through Ligand Structural Variation. J. Am. Chem. Soc 138, 356–368. 10.1021/jacs.5b10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donoghue PJ, Tehranchi J, Cramer CJ, Sarangi R, Solomon EI, and Tolman WB (2011). Rapid C–H bond activation by a monocopper(III)–hydroxide complex. J. Am. Chem. Soc 133, 17602–17605. 10.1021/ja207882h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huffman LM, Casitas A, Font M, Canta M, Costas M, Ribas X, and Stahl SS (2011). Observation and Mechanistic Study of Facile C-O Bond Formation between a Well-Defined Aryl–Copper(III) Complex and Oxygen Nucleophiles. Chem. – A Eur. J 17, 10643–10650. 10.1002/chem.201100608. [DOI] [PubMed] [Google Scholar]
- 38.Huffman LM, and Stahl SS (2011). Mechanistic analysis of trans C–N reductive elimination from a square-planar macrocyclic aryl-copper(iii) complex. Dalt. Trans 40, 8959–8963. 10.1039/C1DT10463B. [DOI] [PubMed] [Google Scholar]
- 39.King AE, Huffman LM, Casitas A, Costas M, Ribas X, and Stahl SS (2010). Copper-Catalyzed Aerobic Oxidative Functionalization of an Arene C−H Bond: Evidence for an Aryl-Copper(III) Intermediate. J. Am. Chem. Soc 132, 12068–12073. 10.1021/ja1045378. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Yao B, Zhao L, Wang D-X, Xu B-Q, and Wang M-X (2014). Direct Synthesis of High-Valent Aryl–Cu(II) and Aryl–Cu(III) Compounds: Mechanistic Insight into Arene C–H Bond Metalation. J. Am. Chem. Soc 136, 6326–6332. 10.1021/ja412615h. [DOI] [PubMed] [Google Scholar]
- 41.Fuchigami T, and Inagi S (2020). Recent Advances in Electrochemical Systems for Selective Fluorination of Organic Compounds. Acc. Chem. Res 53, 322–334. 10.1021/acs.accounts.9b00520. [DOI] [PubMed] [Google Scholar]
- 42.Tedder JM (1982). Which Factors Determine the Reactivity and Regioselectivity of Free Radical Substitution and Addition Reactions? Angew. Chem. Int. Ed. Engl 21, 401–410. 10.1002/anie.198204011. [DOI] [Google Scholar]
- 43.Roberts BP (1999). Polarity-reversal catalysis of hydrogen-atom abstraction reactions: Concepts and applications in organic chemistry. Chem. Soc. Rev 28, 25–35. 10.1039/a804291h. [DOI] [Google Scholar]
- 44.Le C, Liang Y, Evans RW, Li X, and MacMillan DWC (2017). Selective sp3 C–H alkylation via polarity-match-based cross-coupling. Nature 547, 79–83. 10.1038/nature22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker BR, Manabe S, Brusoe AT, and Sevov CS (2021). Mediator-Enabled Electrocatalysis with Ligandless Copper for Anaerobic Chan–Lam Coupling Reactions. J. Am. Chem. Soc 143, 6257–6265. 10.1021/jacs.1c02103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyazaki S, Kojima T, Mayer JM, and Fukuzumi S (2009). Proton-Coupled Electron Transfer of Ruthenium(III)−Pterin Complexes: A Mechanistic Insight. J. Am. Chem. Soc 131, 11615–11624. 10.1021/ja904386r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kotani H, Kaida S, Ishizuka T, Sakaguchi M, Ogura T, Shiota Y, Yoshizawa K, and Kojima T (2015). Formation and characterization of a reactive chromium(v)-oxo complex: mechanistic insight into hydrogen-atom transfer reactions. Chem. Sci 6, 945–955. 10.1039/C4SC02285H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morimoto Y, Park J, Suenobu T, Lee Y-M, Nam W, and Fukuzumi S (2012). Mechanistic Borderline of One-Step Hydrogen Atom Transfer versus Stepwise Sc3+-Coupled Electron Transfer from Benzyl Alcohol Derivatives to a Non-Heme Iron(IV)-Oxo Complex. Inorg. Chem 51, 10025–10036. 10.1021/ic3016723. [DOI] [PubMed] [Google Scholar]
- 49.Schneider JE, Goetz MK, and Anderson JS (2021). Statistical analysis of C-H activation by oxo complexes supports diverse thermodynamic control over reactivity. Chem. Sci 12, 4173–4183. 10.1039/d0sc06058e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barman SK, Yang MY, Parsell TH, Green MT, and Borovik AS (2021). Semiempirical method for examining asynchronicity in metal-oxido-mediated C-H bond activation. Proc. Natl. Acad. Sci. U. S. A 118, e2108648118. 10.1073/pnas.2108648118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salamone M, Galeotti M, Romero-Montalvo E, van Santen JA, Groff BD, Mayer JM, DiLabio GA, and Bietti M (2021). Bimodal Evans–Polanyi Relationships in Hydrogen Atom Transfer from C(sp3)–H Bonds to the Cumyloxyl Radical. A Combined Time-Resolved Kinetic and Computational Study. J. Am. Chem. Soc 143, 11759–11776. 10.1021/jacs.1c05566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.K., B.S., Meng-Yin Y, H., P.T., T., G.M., and S., B.A. (2021). Semiempirical method for examining asynchronicity in metal–oxido-mediated C–H bond activation. Proc. Natl. Acad. Sci 118, e2108648118. 10.1073/pnas.2108648118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goetz MK, and Anderson JS (2019). Experimental Evidence for pKa-Driven Asynchronicity in C–H Activation by a Terminal Co(III)–Oxo Complex. J. Am. Chem. Soc 141, 4051–4062. 10.1021/jacs.8b13490. [DOI] [PubMed] [Google Scholar]
- 54.Kojima T, Kotani H, Shimomura H, Ikeda K, Ishizuka T, Shiota Y, and Yoshizawa K (2020). Mechanistic insight into concerted proton-electron transfer of a Ru(IV)-oxo complex: A possible oxidative asynchronicity. J. Am. Chem. Soc 142, 16982–16989. 10.1021/jacs.0c05738. [DOI] [PubMed] [Google Scholar]
- 55.Bím D, Maldonado-Domínguez M, Rulísek L, and Srnec M (2018). Beyond the classical thermodynamic contributions to hydrogen atom abstraction reactivity. Proc. Natl. Acad. Sci. U. S. A 115, E10287–E10294. 10.1073/pnas.1806399115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S2. NMR spectra.
Data S3. Computational coordinates.
Data Availability Statement
All other data supporting the findings of this study are available within the article and the supplemental information or from the lead contact upon reasonable request.