

Abstract
Adenosine triphosphate (ATP) synthases (F0F1-ATPases) are crucial for all aerobic organisms. F1, a water-soluble domain, can catalyze both the synthesis and hydrolysis of ATP with the rotation of the central γε rotor inside a cylinder made of α3β3 in three different conformations (referred to as βE, βTP, and βDP). In this study, we determined multiple cryo-electron microscopy structures of bacterial F0F1 exposed to different reaction conditions. The structures of nucleotide-depleted F0F1 indicate that the ε subunit directly forces βTP to adopt a closed form independent of the nucleotide binding to βTP. The structure of F0F1 under conditions that permit only a single catalytic β subunit per enzyme to bind ATP is referred to as unisite catalysis and reveals that ATP hydrolysis unexpectedly occurs on βTP instead of βDP, where ATP hydrolysis proceeds in the steady-state catalysis of F0F1. This indicates that the unisite catalysis of bacterial F0F1 significantly differs from the kinetics of steady-state turnover with continuous rotation of the shaft.
Significance Statement.
The F0F1-ATPase rotates its central axis by continuously changing the structure of the three β subunits upon ATP hydrolysis. Here, we reconstructed cryo-electron microscopy structures under unisite conditions that permit only a single catalytic β subunit per enzyme to bind ATP. The structures indicated that hydrolysis of the first ATP occurs at βTP instead of βDP, where ATP hydrolysis proceeds in the steady-state catalysis of F0F1. This indicates that unisite catalysis is an initial reaction that is distinguished from steady-state rotary catalysis in F0F1.
Introduction
Adenosine triphosphate (ATP) synthases (F0F1) are crucial for aerobic organisms and reside in the inner membranes of mitochondria, plasma membranes of bacteria, and thylakoid membranes of chloroplasts in plants (1–3). F0F1 consists of a hydrophilic F1 domain responsible for ATP hydrolysis or synthesis and a hydrophobic F0 domain housing proton translocation across the membranes. ATP hydrolysis/synthesis in F1 is coupled to proton flow in F0 through the rotation of a common shaft.
F0F1 from the thermophilic bacteria Geobacillus stearothermophilus is one of the best characterized ATP synthases because of its structural stability and simple subunit structure (α3β3γ1ε1δ1a1b2c10; Fig. 1A). In particular, single-molecule rotation experiments using this enzyme enabled direct observation of the rotation of ATP synthase, which considerably improved our understanding of the mechanochemical cycle of F0F1 (4–11).
Fig. 1.
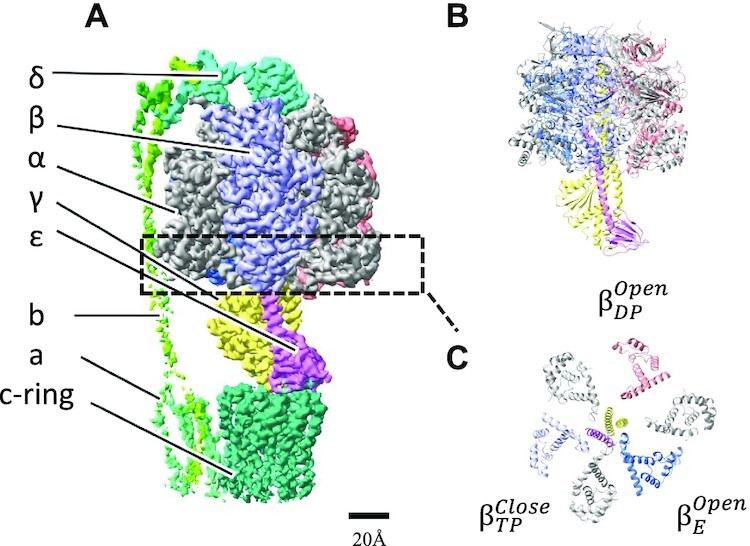
Structure of nucleotide depleted wild-type F0F1 (ND-wt-F0F1) of G. stearothermophilus. Cryo-electron microscopy density map of ND-wt-F0F1 (state 1). (A) Side view of the overall structure of F0F1 as a ribbon model. All α subunits are represented as gray, and other subunits are colored in F1 domain. (B) View of the F0 side of F1 domain at C terminal region. Superscript indicates the closed or open structure of the β subunit. (C) Ribbon representation of view from the F0 side. No nucleotide was found at noncatalytic or catalytic nucleotide-binding sites.
The γε rotor in the F1 domain (α3β3γ1δ1ε1) is surrounded by a cylinder composed of three noncatalytic α and three catalytic β subunits arranged alternately (Fig. 1). The ε subunit modulates ATP hydrolysis activity by the structural change from a contracted to an extended form with a C-terminus helix towards α3β3 (12). Several experimental studies have demonstrated that the extended C-terminus of the ε subunit (ε-CT up form) strongly inhibits ATPase activity of the F1 domain; however, the inhibition mechanism of the ε-CT up form remains elusive.
In F1 domain, six nucleotide-binding sites are located at different interfaces between the α and β subunits; three catalytic sites are located mainly in the β subunit, while the other three sites, called noncatalytic nucleotide-binding sites, are located mainly in the α subunit. According to the binding change mechanism of ATP synthesis, the three catalytic β subunits in F1 are in different conformations (open, loose, and tight); However, they interconvert sequentially between three different conformations as catalysis proceeds (2). Thus, at a given time, all three catalytic β subunits are in different conformations. The crystal structure of F1 from bovine heart mitochondria demonstrated the following asymmetry of catalytic sites: βE, which adopts an open structure with no nucleotide; βTP, which adopts a structure containing ATP analog (AMPPNP); and βDP, which adopts a structure containing ADP (13, 14). The C-terminal region of each β subunit interacts with the γ subunit, and the differences are a consequence of the asymmetric association of the γ subunit with the α3β3 cylinder. Thus, sequential interconversion between three different β subunits drives the rotation of the γ subunit with ATP hydrolysis (4).
The asymmetric structures of α3β3 were found in the F1 structure of other species (15–17) and V-ATPases (16), which is another rotary ATPase that is evolutionarily and structurally related to F0F1. In this study, we refer to the asymmetric architecture as a “Walker structure.”
Assuming that F0F1 adopts the Walker structure during ATP hydrolysis, the alternating participation of β subunits within ATP hydrolysis does not require positive cooperation. Early studies showed that F1-ATPases exhibited multiple Km, likely due to the activation of F1-ATPase by the binding of ATP to the α subunits (18). However, using both bulk and single-molecule experiments, one rotary mechanism was found to govern the entire range of nanomolar to millimolar ATP (19). This indicates that thermophilic F1-ATPase obeys simple Michaelis–Menten kinetics with a single Km value. However, previous experiments using a mitochondrial F1 indicated strong positive cooperation between the catalytic sites. F1-ATPase was compared under unisite and multisite catalytic conditions in which the ATP/enzyme ratio was adjusted to facilitate operation of either one or three sites (20). The multisite/unisite rate enhancement ratio of 106 fold was interpreted as a reflection of the strong positive cooperation among the three catalytic sites. The unisite catalysis experiment also suggests a high affinity of unisite to ATP with a binding constant Kd of ∼10−12 M, which is markedly higher than the Km of ∼10–5 M in bacterial F1 (19, 21). For thermophilic F1-ATPase, similar ATP hydrolysis in a single catalytic site has been reported (21–23). However, the structural basis of the high affinity for ATP observed in unisite catalysis and whether unisite catalysis reflects steady-state activity (multisite catalysis) remains elusive.
In this study, we constructed a mutant F0F1 with a C-terminus-truncated ε subunit and showed that it does not undergo ε inhibition. The structure of this mutant F0F1 was determined using cryo-electron microscopy (cryo-EM) and compared with that of wild-type (wt) F0F1 to understand the structural basis of ε inhibition. Furthermore, we determined the structure of the mutant F0F1 under unisite catalytic conditions to capture the structure after ATP hydrolysis of the β subunit.
Results
Cryo grid preparation of wt-F0F1 and ATPase-active ΔεCT-F0F1
In this study, we used purified F0F1 from G. stearothermophilus expressed in Escherichia coli (24). The ATPase activity of wt-F0F1 is ∼10 s−1 (Supplementary Fig. S1 and Supplementary Table S1), which is considerably lower than that of F1 (α3β3γ, ∼70 s−1) (25). Several studies have indicated that ATP hydrolysis by G. stearothermophilus F0F1 is significantly inhibited when the extended C-terminal region of the ε subunit penetrates the α3β3 cavity (12, 26–28). The initial ATPase activity of wt-F0F1 was very low because of the initial lag (Supplementary Fig. S1). To obtain ATPase-active F0F1, we constructed a mutant F0F1 with a C-terminal-truncated ε subunit (ΔεCT-F0F1). The prepared ΔεCT-F0F1 was subjected to dialysis in phosphate-buffered saline to deplete the bound nucleotide, as described in the Materials and Methods section. For the nucleotide-depleted thermophilic F0F1 (ND-ΔεCT-F0F1), the lag time was shorter than that of wt-F0F1, and the ATPase activity at 1,000 s after the start of reaction was 130 s−1 (Supplementary Table S1), which was comparable to the ATPase activity of wt-F1 without the ε subunit (5, 25).
First, we prepared a cryo grid of nucleotide-depleted wt-F0F1 (ND-wt-F0F1) in the absence or presence of 4 mM ATPγS. In addition, the cryo grid of ΔεCT-F0F1 under unisite conditions (molar ratio of enzyme to ATP is approximately 1:4.) was used for structural analysis. These cryo grids were subjected to cryo-EM image acquisition using a Titan Krios (Thermo Fisher Scientific) equipped with a K3 direct electron detector.
Structure of ND-wt-F0F1
Flowcharts showing the image acquisition and reconstitution of the 3D structure of nucleotide-depleted wt-F0F1 (ND-wt-F0F1) are summarized in Supplementary Fig. S2. We reconstructed three rotational states (state 1: 3.1 Å, state 2: 3.0 Å, and state 3: 3.7 Å resolutions) from the single-particle images of ND-wt-F0F1. The structure of ND-wt-F1 domain is similar to the structure of F1 domain in wt-F0F1 reported in a study by Guo et al.(6N2Y(12)), which adopted the following structures: “open in βE without nucleotide,” “closed in βTP containing ADP,” and “open in βDP without nucleotide.” In fact, the three β subunits in ND-wt-F1 are similar to their counterparts in wt-F0F1 (Supplementary Fig. S3). In the structure of ND-wt-F0F1, the C-terminal helix of the ε subunit (ε-CT) also adopts the up form and penetrates the cavity of the α3β3 cylinder (Fig. 1B). In contrast to the previous structure of wt-F0F1 (12), no nucleotide density was observed at the noncatalytic nucleotide-binding site in the three α subunits or the catalytic site in βTP (Figs. 1C and 3A). Instead, a density likely corresponding to a phosphate was observed at the catalytic site of βTP (Fig. 3A and Supplementary Fig. S4A). This phosphate was possibly derived from the phosphate buffer used for nucleotide depletion.
Fig. 3.
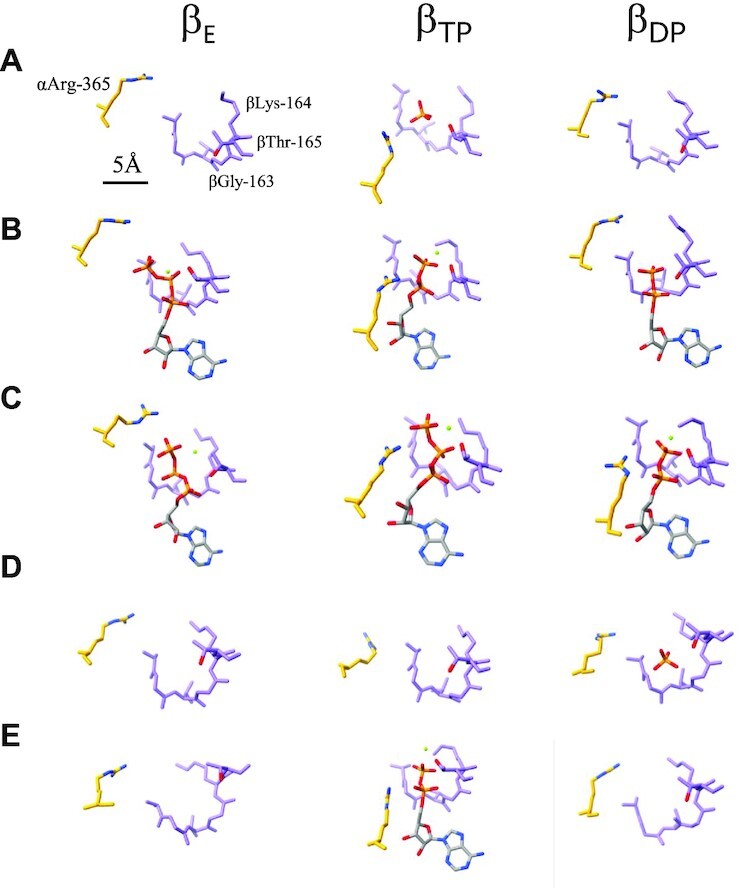
Structure of catalytic sites of wild-type and mutated F0F1 under different condition. Magnified views of the three catalytic sites (βE, βTP, and βDP) in each structure are shown as follows: (A) ND-wt-F0F1, (B) wt-F0F1ε up form with ATPγS, (C) wt-F0F1ε down form with ATPγS, (D) ND-ΔCT-F0F1, and (E) US-ΔCT-F0F1. Bound nucleotides and Mg ions are highlighted as stick and spherical representations, respectively. Scale bar is 5 Å.
Therefore, the structure indicates that the closed structure of βTP is stabilized by the penetration of ε-CT into the cavity between αDP and βTP, which is independent of nucleotide binding to βTP.
Structure of wt-F0F1 exposed to 4 mM ATPγS
Previous studies have indicated that the binding of ATP to an isolated ε subunit induces a conformational change in ε-CT from the “up” to “down” form (25, 29). The ATPase activity of wt-F0F1 gradually accelerated in the presence of 4 mM ATP (Supplementary Fig. S1 and Supplementary Table S1). In this study, we determined the cryo-EM structure of F0F1 exposed to 4 mM ATPγS, which is a slow hydrolyzable ATP analog (30), to prove the conformational change of the ε subunit in the complex. After focused 3D classification using a mask covering the F1 domain (Supplementary Fig. S5B), we obtained the following two structures: F1 with the up form of ε subunit from 526,524 particles at 2.6 Å resolution and F1 with the down form of ε subunit from 39,991 particles at 3.3 Å resolution (Fig. 2 and Supplementary Fig. S5B). In the down form of ε subunit in the complex, the density of bound ATPγS was identified with similar coordination of surrounding amino acid residues to that in the monomeric ε subunit (Supplementary Fig. S6) (31). F0F1 with the down form of ε subunit adopted the following canonical Walker structures: “open in βE with ATPγS,” “closed in βTP with ATPγS,” and “closed in βDP with ADP ” (Figs. 2B and 3C). ATPγS molecules were also identified at the noncatalytic sites of the three α subunits (Fig. 2, lower). In contrast, the structure of F0F1 with the extended ε-CT was similar to the ND-wt-F0F1 structure, except the nucleotide occupancy of “open in βE with ATPγS,” “closed in βTP with ADP,” and “open in βDP with ADP” (Fig. 3B). These results indicate that the extended ε-CT hampers the conformational change of the F1 domain from the inhibitory conformation with “open in βE,” “closed in βTP,” and “open in βDP” to the Walker structure with “open in βE,” “closed in βTP,” and “closed in βDP” during ATP hydrolysis.
Fig. 2.
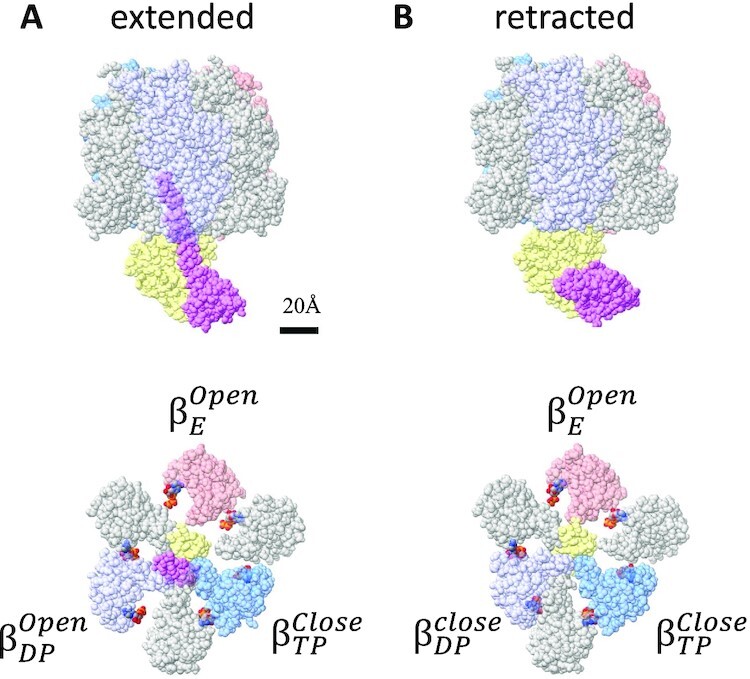
Structure of wild-type F0F1 (wt-F0F1) exposed to 4 mM ATPγS. Structures of wt-F0F1 exposed to 4 mM ATPγS with the retracted (A) or extended (B) ε-subunit viewed from vertical side (upper) and from F1 side (lower). The bound nucleotides are represented as color spheres. All α subunits are represented in gray, and all β subunits are colored.
Structure of ΔεCT-F0F1 under unisite catalysis conditions
To capture the structure of the unisite catalysis of F0F1, 15 µM of ND-ΔεCT-F0F1 was mixed with 4 µM of the ATP-containing regeneration system described in the Materials and Methods section. The mixture was incubated for 120 s at 25°C, then loaded onto a holey grid, and subjected to flash freezing.
We obtained multiple F0F1 structures with and without nucleotides (ND-ΔεCT-F0F1) in the βTP using 418,497 selected particle images of ΔεCT-F0F1 (Supplementary Fig. S7B). For ND-ΔεCT-F0F1, the structures of two rotational states without nucleotides were obtained at the following resolutions: state 1 at 3.6 Å and state 2 at 3.4 Å, with the γ subunit positions differing by 120° in each state. The structure of state 3 without nucleotides was not identified because of the small number of particles. Further, we obtained the F1 parts of states 1 and 2 at resolutions of 3.4 Å and 3.3 Å, respectively, using focused refinement of the F1 domain (Supplementary Fig. S7B).
The structure of ND-ΔεCT-F0F1 significantly differs from the structure of ND-wt-F0F1 in which three catalytic β subunits adopt “open in βE,” “closed in βTP,” and “open in βDP” (Fig. 1C). In contrast, all three β subunits in ND-ΔεCT-F0F1 adopt an almost identical structure of open conformations as follows: “open in βE,” “open in βTP,” and “open in βDP” (Fig. 4A). There was no nucleotide bound to the three open β subunits of ND-ΔεCT-F0F1, but a density corresponding to phosphate was observed in the βDP (Fig. 3D and Supplementary Fig. S4D). This phosphate may be derived from the phosphate buffer used for nucleotide depletion. Our findings suggest that nucleotide depletion from the F1 domain causes all three β subunits to adopt an open conformation. The interaction of the γ subunit with the three open β subunits are shown in Supplementary Fig. S8. The C termini region of closed βTP is in close proximity to the coiled coil of the γ subunit, while the open βTP is in close proximity to the globular domain of the γ subunit (Supplementary Fig. S8C).
Fig. 4.
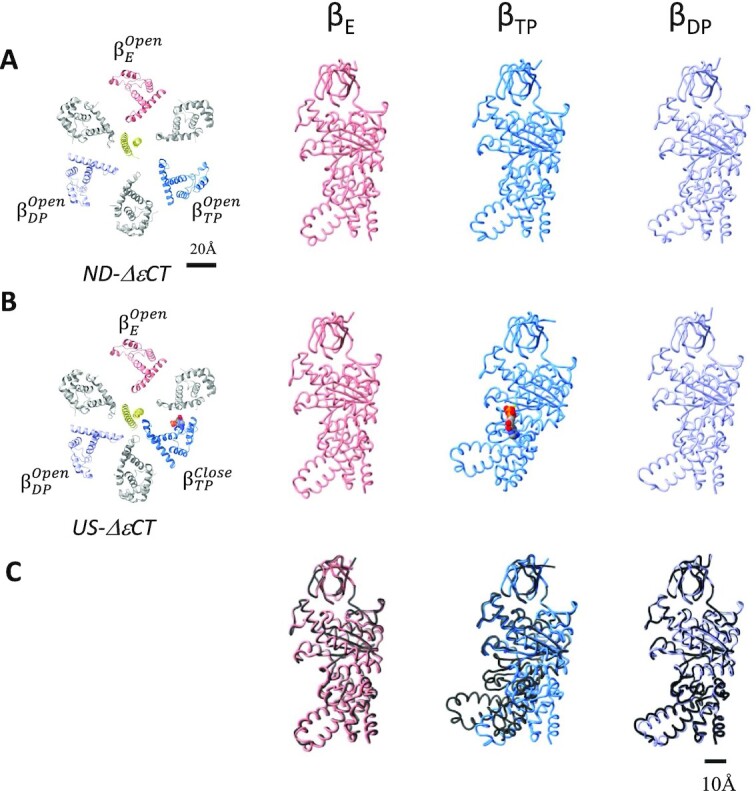
Comparison of the structure of nucleotide-depleted ΔεCT-F0F1 with that of US-ΔεCT-F0F1. Cryo-electron microscopy structures of ND-ΔεCT-F0F1 (A) and ΔεCT-F0F1 under unisite conditions (B). The slice view of the F1 domain is represented in the left panel, and the main chain of each β subunit is lined. Right panels, each β subunits (βTP, βDP, and βE) of ND-ΔεCT-F0F1 or US-ΔεCT-F0F1 is represented as a colored chain. (C) Comparison of the β subunits between ND-ΔεCT-F0F1 and US-ΔεCT-F0F1. Each β subunit (βTP, βDP, and βE) of ND-ΔεCT-F0F1(colored chain) or US-ΔεCT-F0F1 (black chain) is superimposed on the β-barrel domain (1 to 80 amino acids).
In other words, the penetration of extended ε-CT into the α3β3 cavity does not result in the open conformation of βDP but rather forces open βTP without bound nucleotides to the closed conformation (cf. Figs. 1C and 4A).
For ND-ΔεCT-F0F1 containing a nucleotide in βTP, we determined the structures of the following three states: state 1 at 3.2 Å, state 2 at 3.4 Å, and state 3 at 4.0 Å. For state 1, another subclass with nearly identical structure (state 1) was isolated (Supplementary Fig. S6C). Upon focused refinement using an F1 mask, the structure of the F1 domain for each state was obtained (Supplementary Fig. S6B, lower). We refer to the ΔεCT-F0F1 structure containing a nucleotide at the βTP as UniSite-ΔεCT-F0F1 (US-ΔεCT-F0F1). The structure of US-ΔεCT-F0F1 was different from that of ND-ΔεCT-F0F1. The structures of two β subunits, βE and βDP, adopted an open form without nucleotides, whereas βTP adopted a closed form containing nucleotide density (Fig. 3E). These findings indicate that the structure of US-ΔεCT-F0F1 is very similar to that of ND-wt-F0F1. Specifically, F0F1 in both structures adopt “open in βE,” “closed in βTP,” and “open in βDP.” In addition, the relative position of the γ subunit to α3β3 was completely analogous in the two structures (Supplementary Fig. S9). The structure of US-ΔεCT-F0F1 revealed a nucleotide density due to ADP at the catalytic site in βTP, indicating that ATP bound to βTP was already hydrolyzed (Figs. 3E and 4B).
Furthermore, the position of the γ subunit of US-ΔεCT-F0F1 relative to α3β3 is slightly different from that of ND-ΔεCT-F0F1. Superimposition of the two structures with α3β3 shows ∼ 7° rotation of the γ subunit in the hydrolysis direction (Supplementary Fig. S8A). Assuming that the first ATP binds to βTP, the rotation of the γ subunit driven by unisite catalysis is faint compared to the 120° rotation of the γ subunit upon binding and hydrolysis of one ATP molecule under ATP-saturated conditions (multisite conditions).
Discussion
In this study, we reveal that substoichiometric ATP is hydrolyzed at the βTP of ND-ΔεCT-F0F1 under unisite catalysis conditions, where a single catalytic site per enzyme molecule binds ATP.
All three β subunits in ND-ΔεCT-F0F1 adopted an open form, implying that the first ATP can bind to any of the β subunits. Assuming that the first ATP binds to βTP, the conformation change from the open to the closed form of βTP occurs without the 120° rotation of γ subunit (Fig. 5A), which is consistent with previous studies suggesting unisite catalysis without rotation of γ subunit in E. coli F1 (32). In the wt-F0F1 structure, ADP is bound only to βTP, and both βDP and βE are in the open form without nucleotides, which suggests that βTP can easily change to the closed form upon ATP binding, particularly when compared to βDP and βE. βTP bound with ATP immediately changes to the closed form due to the zippering motion by the bound ATP, which likely increases the affinity of βTP for ATP. This is consistent with the high affinity for ATP reported in unisite catalysis experiments (20, 22, 33). Alternatively, it is also possible that the first ATP bound to βE and the γ subunit rotated 120° with a structural change of the ATP-bound βE to βTP, resulting in the structure of US-ΔεCT-F0F1. Although both catalytic pathways (Fig. 5A and B) exhibit high possibilities for unisite catalysis, it is more likely that the first ATP binds to βTP to explain the high affinity of the unisite for ATP.
Fig. 5.
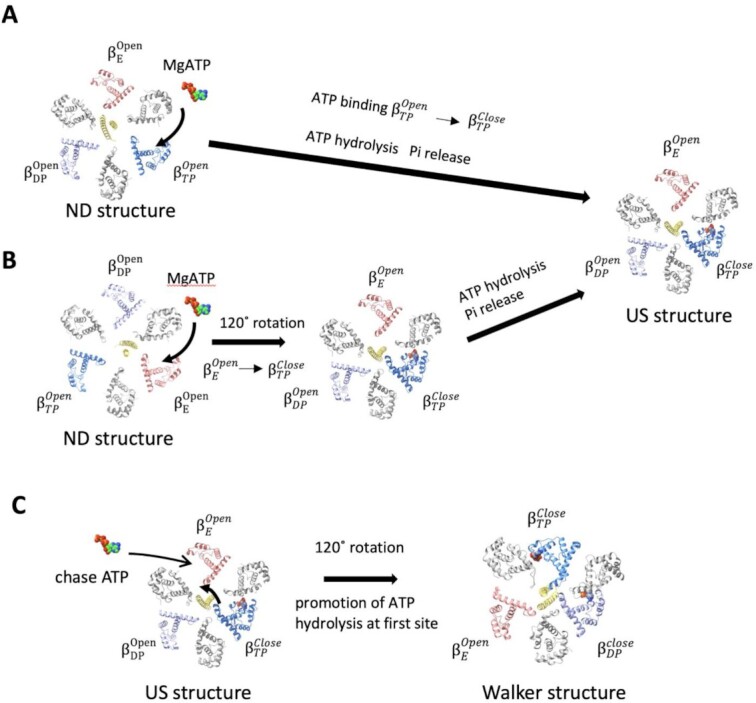
Schematic representation of possible catalytic pathways for unisite catalysis at βTP and promotion of ATP hydrolysis by chase ATP. Three β subunits (βTP, βDP, and βE) and γ subunits are colored in blue, blue–purple, magenta and yellow, respectively. All α subunits are represented as gray. (A) First ATP binds to βTP in the nucleotide-depleted (ND) structure where the three β subunits are open. Then, 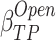 changes to
changes to 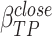 with hydrolysis of bound ATP. The conformation change of βTP induces a 7° rotation of the γ subunit. The phosphate is released spontaneously, resulting in the unisite structure (US). (B) The first ATP binds to βE in the ND structure, followed by structural changes of three β subunits with 120° rotation of the γ subunit. (C) The bound ATP on βTP is slowly hydrolyzed by coordination of catalytic amino acid residues not suitable for hydrolysis of ATP. Binding of the second ATP to βE causes 120° rotation of γ subunit and structural transition of the each of the following β subunits: βE to βTP, βTP to βDP, and βDP to βE. The β subunit to which the first ATP binds becomes βDP, and the hydrolysis of ATP is accelerated.
with hydrolysis of bound ATP. The conformation change of βTP induces a 7° rotation of the γ subunit. The phosphate is released spontaneously, resulting in the unisite structure (US). (B) The first ATP binds to βE in the ND structure, followed by structural changes of three β subunits with 120° rotation of the γ subunit. (C) The bound ATP on βTP is slowly hydrolyzed by coordination of catalytic amino acid residues not suitable for hydrolysis of ATP. Binding of the second ATP to βE causes 120° rotation of γ subunit and structural transition of the each of the following β subunits: βE to βTP, βTP to βDP, and βDP to βE. The β subunit to which the first ATP binds becomes βDP, and the hydrolysis of ATP is accelerated.
Both βDP and βE retain open structures after unisite catalysis at βTP; therefore, the next catalytic event is ATP binding to either βDP or βE. During ATP hydrolysis by F0F1, each conformational change from βE to βTP, βTP to βDP, and βDP to βE occurs with 120° rotation of the γ subunit. Assuming that ATP then binds to βE and induces a conformational change of each β subunit with 120° rotation of the γ subunit, F0F1 adopts the following canonical Walker structure observed in most F1 structures: “open in βE,” “closed in βTP with the second ATP,” and “closed in βDP with the ADP from first ATP” (Fig. 5C). Several studies using various F1-ATPases have reported that hydrolysis of ATP bound to the first catalytic site is markedly accelerated by the addition of an excess ATP (cold chase experiment) (20, 21, 35). In Walker structures, βTP and βDP adopt very similar closed conformations; however, their catalytic sites are not equivalent (Supplementary Fig. S10). Although these differences are relatively small, they have a marked effect on catalysis. ATP in βDP is immediately hydrolyzed, whereas hydrolysis of ATP in βTP proceeds slowly (14, 33). Therefore, most crystal structures of F1 contain ATP analogs in βTP and ADP in βDP. Upon binding of ATP to βE by the addition of excess ATP, the conformational change of βTP to βDP promoted the hydrolysis of ATP at the catalytic site (Fig. 5C).
The F0F1 structures exposed to 4 mM ATPγS provided direct evidence of conformational changes in the ε subunit by ATP binding in the holo-F0F1 complex. The retracted ε subunit in the complex contains ATP with the similar coordination as the monomeric ε subunit (Supplementary Fig. S5) (31). In another wt-F0F1 structure exposed to 4 mM ATPγS concentration, the ε-inhibited structure, which adopted “open in βE,” “closed in βTP,” and “open in βDP,” was maintained, although all nucleotide binding sites were occupied with ADP or ATPγS (Figs. 2 and 3B). This indicates that the inhibited wt-F0F1 structure by the extended ε subunit is not activated by ATP binding to βE or βDP (Fig. 2A), and that the conformational change of the ε subunit by ATP binding is crucial for the activation of wt-F0F1 for ATP hydrolysis activity (Fig. 2B).
In this study, we determined the multiple structures of F0F1 ATPase during catalysis by structural analysis using cryo-EM, which allows the capture of states inaccessible to crystallization. The techniques and approaches used in this study can potentially assist the elucidation of the detailed reaction and regulatory mechanisms of other enzymes.
Materials and methods
Protein purification
Wt-F0F1-ATP synthase from G. stearothermophilus was purified from E. coli DK 8 strain containing an expression vector (pTR19-ASDS) for F0F1, as described previously (24). The expression vector for ΔεCT-F0F1, which lacks the C-terminal of ε subunit (83 to 133 amino acids), was constructed from pTR19-ASDS. Transformed E. coli cells were grown in 2 × YT medium at 37°C for 16 h before harvesting by centrifugation at 5000 × g. The cell pellet was suspended in lysis buffer (50 mM Tris–Cl pH 8.0, 5 mM MgCl2, and 10% [w/v] glycerol), and the cell membranes were collected by ultracentrifugation at 35,000 rpm for 20 min and solubilized by mixing in solubilization buffer (50 mM Tris–Cl, 5 mM MgCl2, 10% [w/v] glycerol, and 2% n-dodecyl-d-maltoside [DDM]) at 4°C for 3 h. The supernatant was then applied to a Ni-NTA column. For bound nucleotide removal, the eluted fractions containing F0F1 were dialyzed against 200 mM sodium phosphate (pH 8.0), 10 mM EDTA, and 0.03% DDM overnight at 25°C and the dialysis buffer was changed thrice. The dialyzed F0F1 fractions were concentrated using ultrafiltration with an Amicon filter (100 KDa cut-off, Amicon corp.) and loaded onto a Superose 6 Increase 10/300 column (Cytiva) equilibrated with gel permeation buffer (20 mM Tris–HCl pH 8.0, 150 mM NaCl, and 0.03% DDM). The peak fractions (6 to 9 mg/mL) were used for the cryo grid preparation or ATPase assay.
Grid preparation
For cryo grid preparation, Quanfifoil R1.2/1.3 Mo grids were glow-discharged for 1 min using an Ion Bombarder (Vacuum Device). Prior to blotting, 3 µL of the samples were placed on a cryo grid and incubated for 15 min. Further, 2.6 to 3.5 µL F0F1 was loaded onto the grid and blotted for 3 to 10 s with a blot force of 10, drain time of 0.5 s, and 100% humidity using a FEI Vitrobot (ThermoFisher). The blotted grid was then plunged into a liquid ethane. For the unisite catalysis condition, 1 µL reaction buffer (0.2 M Tris–Cl pH 8.0, 40 µM ATP, 40 mM PEP, 1 M KCl, and 5 mg/mL pyruvate kinase) was added to 9 µL of the sample. The mixtures were incubated for 60 s at 25°C, followed by blotting and vitrification.
Cryo-EM imaging under ATPγS and US conditions was performed using a Titan Krios (FEI/Thermo Fisher Scientific) operating at 300 kV acceleration voltage and equipped with an electron detector K3 (Gatan) in electron counting mode (CDS). Cryo-EM imaging under wt-ND conditions was performed in CDS using CRYOARM 300 (JEOL) operating at 300 kV and an electron detector K3 (Gatan).
Data collection was performed using SerialEM software with a calibrated magnification of 0.88 Å pixel–1 for the ATPγS and ΔεCT-US conditions and a calibrated magnification of 1.01 Å pixel–1 for the wt-ND condition. Under wt-ND and ATPγS conditions, data were collected at an electron dose of 50.0 e–/Å2 for an exposure time of 5 s, and under unisite conditions, data were collected at an electron dose of 60.0 e–/Å2 for an exposure time of 6 s. The defocus range was 0.8 to 2.0 µm, and data were collected at 50 frames for the wt-ND condition, 55 frames for the ATPγS condition, and 59 frames for the unisite condition.
Image processing
The details of the image-processing procedure for each condition are described in Supplementary Figs. 2, 4, and 6. Image analysis was performed using RELION 4.0β (36) and cryoSPARC v3.2 (37). The file format conversion between RELION and cryoSPARC was performed using the script csparc2star.py in Pyem. The beam-induced drift was corrected using MotionCor2 (38), and the CTF was estimated using CTFFIND 4.1 (39). We analyzed 7,329 movies for the unisite condition, 9,625 movies for the wt-ND condition, and 17,261 movies for the ATPγS condition. Particle picking was performed using Topaz (40). Good particles were selected by 2D classification and trained using 4,000 particles. Autopicking using the trained topaz model yielded 499,788 particles for the unisite condition, 1,381,269 particles for the wt-ND condition, and 1,020,321 particles for the ATPγS condition. Particles picked by Topaz were subjected to 2D classification for further selection of good particles. Then heterogeneous refinements using cryoSPARC were performed to eliminate junk particles. Further, we selected 418,497 particles for unisite conditions, 622,109 particles for wt-ND conditions, and 912,931 particles for ATPγS conditions. Heterogeneous refinement was then used to classify F0F1 into multiple conformational states. These particles were re-extracted at full pixel size and subjected to repeated 3D auto-refinement, CTF refinement, and Bayesian polishing (36). The structures of the three rotational states, including their subclasses, were obtained at 3 to 4.5 Å resolution. All obtained classes of F0F1 were subjected to focused refinement on the F1 part to reduce the resolution loss due to the relative motion of the F0 part with F1. In the analysis of the ATPγS dataset to determine a small percentage of ε-retracted structures, the class of F0F1 of all states were added together to increase the number of particles, and focused refinement was performed on the F1 part. The obtained F1 structure was subjected to focused 3D classification by masking with γε-βDP to detect structural changes between the ε and β subunits. The resolution was estimated using the gold standard Fourier shell correlation (FSC) = 0.143 criterion.
Model building and refinement
We used Phenix real-space refinement (41), ISOLDE (42), and COOT (41) for the atomic model building. The epsilon-extended F1 model was built using PDB 6N2Y as the initial model, and the epsilon-retracted subunit was built using PDB 2E5Y. The initial model, which was a rigid body fitted to the density map by UCSF ChimeraX (43), was first refined by Phenix real-space refinement. Residues that did not fit correctly into the map were manually placed using the COOT and ISOLDE. Refinement and manual modification were repeated until the model parameters were improved. Lastly, the refinement model was evaluated using MolProbity (44) and EMRinger (45).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to all the members of the Yokoyama Lab for their continuous support and technical assistance. We also thank Prof. Allison for his enthusiastic discussion on unisite catalysis long time ago.
Notes
Competing Interest: The authors declare no competing interests.
Contributor Information
Atsuki Nakano, Department of Molecular Biosciences, Kyoto Sangyo University, Kamigamo-Motoyama, Kita-ku, Kyoto 603-8555, Japan.
Jun-ichi Kishikawa, Department of Molecular Biosciences, Kyoto Sangyo University, Kamigamo-Motoyama, Kita-ku, Kyoto 603-8555, Japan; Institute for Protein Research, Osaka University, 3-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Atsuko Nakanishi, Department of Molecular Biosciences, Kyoto Sangyo University, Kamigamo-Motoyama, Kita-ku, Kyoto 603-8555, Japan; Research Center for Ultra-High Voltage Electron Microscopy, Osaka University, 7-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan.
Kaoru Mitsuoka, Research Center for Ultra-High Voltage Electron Microscopy, Osaka University, 7-1 Mihogaoka, Ibaraki, Osaka 567-0047, Japan.
Ken Yokoyama, Department of Molecular Biosciences, Kyoto Sangyo University, Kamigamo-Motoyama, Kita-ku, Kyoto 603-8555, Japan.
Funding
Our research was supported by Grant-in-Aid for Scientific Research (JSPS KAKENHI) Grant Number 20H03231 to K.Y., 20K06514 to J.K., and Grant-in-Aid for JSPS Fellows Grant Number 20J00162 to A.N., and Takeda Science foundation to K.Y. Our research was also supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research [BINDS]) from AMED under Grant Number JP17am0101001 (support number 1312), Grants-in-Aid from “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) to K.M. (Project Number. 12024046), and the Research Program for Next Generation Young Scientists of “Five-star Alliance” in “NJRC Mater. & Dev.” under Grant Number 20215008 to A.N..
Authors’ contributions
K.Y., J.K., A. Nakanishi, and A. Nakano designed, performed, and analyzed the experiments; A. Nakano analyzed the data and contributed to the preparation of the samples; J.K. and K.M. provided technical support and conceptual advice; and K.Y. designed and supervised the experiments and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Data availability
Data is available in the manuscript and supplementary materials. Cryo-EM density maps (.mrc files) and atomic models (.pdb files) obtained in this study were deposited to EMDB and PDB. The accession codes (PDBID and EMDBID) 7XKH, 7XKQ, 7XKR, 7XKO, 7XKP, 33,251, 33,252, 33,253, 33,264, 33,265, 33,266, 33,267, 33,268, 33,277, 33,258, 33,278, 33,259, 33,279, 33,260, 33,280, 33,261, 33,281, 33,269, 33,282, 33,262, 33,283, and 33,263 are summarized in Supplementary Tables S2–S5. The data that support the findings of this study are available from PDB and EMDB.
References
- 1. Kühlbrandt W. 2019. Structure and mechanisms of F-type ATP synthases. Annu Rev Biochem. 88:515–549. [DOI] [PubMed] [Google Scholar]
- 2. Boyer PD. 1997. The ATP synthase—a splendid molecular machine. Annu Rev Biochem. 66:717–749. [DOI] [PubMed] [Google Scholar]
- 3. Yoshida M, Muneyuki E, Hisabori T. 2001. ATP synthase—a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol. 2:669–677. [DOI] [PubMed] [Google Scholar]
- 4. Noji H, Yasuda R, Yoshida M, Kinosita K Jr. 1997. Direct observation of the rotation of F1-ATPase. Nature. 386:299–302. [DOI] [PubMed] [Google Scholar]
- 5. Yasuda R, Noji H, Yoshida M, Kinosita K Jr., Itoh H. 2001. Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase. Nature. 410:898–904. [DOI] [PubMed] [Google Scholar]
- 6. Imamura H, et al. 2003. Evidence for rotation of V1-ATPase. Proc Natl Acad Sci U S A. 100:2312–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yanagisawa S, Frasch WD. 2021. pH-dependent 11°F1F0 ATP synthase sub-steps reveal insight into the F0 torque generating mechanism. eLife. 10:e70016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spetzler D, et al. 2006. Microsecond time scale rotation measurements of single F1-ATPase molecules. Biochemistry. 45:3117–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adachi K, et al. 2007. Coupling of rotation and catalysis in F(1)-ATPase revealed by single-molecule imaging and manipulation. Cell. 130:309–321. [DOI] [PubMed] [Google Scholar]
- 10. Adachi K, Oiwa K, Yoshida M, Nishizaka T, Kinosita K Jr. 2012. Controlled rotation of the F1-ATPase reveals differential and continuous binding changes for ATP synthesis. Nat Commun. 3:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diez M, et al. 2004. Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase. Nat Struct Mol Biol. 11:135–141. [DOI] [PubMed] [Google Scholar]
- 12. Guo H, Suzuki T, Rubinstein JL. 2019. Structure of a bacterial ATP synthase. eLife. 8:e43128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abrahams JP, Leslie AG, Lutter R, Walker JE. 1994. Structure at 2.8-A resolution of F1-ATPase from bovine heart mitochondria. Nature. 370:621–628. [DOI] [PubMed] [Google Scholar]
- 14. Menz RI, Leslie AG, Walker JE. 2001. Structure of bovine mitochondrial F(1)-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell. 106(3):331–341. [DOI] [PubMed] [Google Scholar]
- 15. Suzuki K, et al. 2016. Crystal structures of the ATP-binding and ADP-release dwells of the V(1) rotary motor. Nat Commun. 7:13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arai S, et al. 2013. Rotation mechanism of Enterococcus hirae V1-ATPase based on asymmetric crystal structures. Nature. 493:703–707. [DOI] [PubMed] [Google Scholar]
- 17. Nakanishi A, Kishikawa JI, Tamakoshi M, Mitsuoka K, Yokoyama K. 2018. Cryo EM structure of intact rotary H(+)-ATPase/synthase from Thermus thermophilus. Nat Commun. 9:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jault JM, Allison WS. 1994. Hysteretic inhibition of the bovine heart mitochondrial F1-ATPase is due to saturation of noncatalytic sites with ADP which blocks activation of the enzyme by ATP. J Biol Chem. 269:319–325. [PubMed] [Google Scholar]
- 19. Sakaki N, et al. 2005. One rotary mechanism for F1-ATPase over ATP concentrations from millimolar down to nanomolar. Biophys J. 88:2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grubmeyer C, Cross RL, Penefsky HS. 1982. Mechanism of ATP hydrolysis by beef heart mitochondrial ATPase. Rate constants for elementary steps in catalysis at a single site. J Biol Chem. 257:12092–12100. [PubMed] [Google Scholar]
- 21. Yokoyama K, Hisabori T, Yoshida M. 1989. The reconstituted α3β3δ complex of the thermostable F1-ATPase. J Biol Chem. 264:21837–21841. [PubMed] [Google Scholar]
- 22. Konishi J, Yohda M, Hashimoto T, Yoshida M. 1987. Single site catalysis of the F1-ATPase from Saccharomyces cerevisiae and the effect of inorganic phosphate on it. J Biochem. 102:273–279. [DOI] [PubMed] [Google Scholar]
- 23. Hisabori T, et al. 1992. Single site hydrolysis of 2′,3′-O-(2,4,6-trinitrophenyl)-ATP by the F1-ATPase from thermophilic bacterium PS3 is accelerated by the chase-addition of excess ATP. J Biol Chem. 267:4551–4556. [PubMed] [Google Scholar]
- 24. Suzuki T, Ueno H, Mitome N, Suzuki J, Yoshida M. 2002. F(0) of ATP synthase is a rotary proton channel. Obligatory coupling of proton translocation with rotation of c-subunit ring. J Biol Chem. 277:13281–13285. [DOI] [PubMed] [Google Scholar]
- 25. Kato S, Yoshida M, Kato-Yamada Y. 2007. Role of the epsilon subunit of thermophilic F1-ATPase as a sensor for ATP. J Biol Chem. 282:37618–37623. [DOI] [PubMed] [Google Scholar]
- 26. Shirakihara Y, et al. 2015. Structure of a thermophilic F1-ATPase inhibited by an ε-subunit: deeper insight into the ε-inhibition mechanism. FEBS J. 282:2895–2913. [DOI] [PubMed] [Google Scholar]
- 27. Suzuki T, et al. 2003. F0F1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of epsilon subunit in response to proton motive force and ADP/ATP balance. J Biol Chem. 278:46840–46846. [DOI] [PubMed] [Google Scholar]
- 28. Tsunoda SP, et al. 2001. Large conformational changes of the epsilon subunit in the bacterial F1F0-ATP synthase provide a ratchet action to regulate this rotary motor enzyme. Proc Natl Acad Sci U S A. 98:6560–6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Imamura H, et al. 2009. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci U S A. 106:15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Imamura H, et al. 2005. Rotation scheme of V1-motor is different from that of F1-motor. Proc Natl Acad Sci U S A. 102:17929–17933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yagi H, et al. 2007. Structures of the thermophilic F1-ATPase epsilon subunit suggesting ATP-regulated arm motion of its C-terminal domain in F1. Proc Natl Acad Sci U S A. 104:11233–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. García JJ, Capaldi RA. 1998. Unisite catalysis without rotation of the γ-ε domain inEscherichia coli F1-ATPase. J Biol Chem. 273:15940–15945. [DOI] [PubMed] [Google Scholar]
- 33. Al-Shawi MK, Senior AE. 1988. Complete kinetic and thermodynamic characterization of the unisite catalytic pathway of Escherichia coli F1-ATPase. Comparison with mitochondrial F1-ATPase and application to the study of mutant enzymes. J Biol Chem. 263:19640–19648. [PubMed] [Google Scholar]
- 34. Senior AE. 1992. Catalytic sites of Escherichia coli F1-ATPase. J Bioenerg Biomembr. 24:479–484. [DOI] [PubMed] [Google Scholar]
- 35. Amano T, Tozawa K, Yoshida M, Murakami H. 1994. Spatial precision of a catalytic carboxylate of F1-ATPase beta subunit probed by introducing different carboxylate-containing side chains. FEBS Lett. 348:93–98. [DOI] [PubMed] [Google Scholar]
- 36. Scheres SH. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 180:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. 2017. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods. 14:290–296. [DOI] [PubMed] [Google Scholar]
- 38. Zheng SQ, et al. 2017. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rohou A, Grigorieff N. 2015. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol. 192:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bepler T, et al. 2019. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Methods. 16:1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liebschner D, et al. 2019. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol. 75:861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Croll TI. 2018. Isolde: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D Struct Biol. 74:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pettersen EF, et al. 2021. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barad BA, et al. 2015. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat Methods. 12:943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen VB, et al. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is available in the manuscript and supplementary materials. Cryo-EM density maps (.mrc files) and atomic models (.pdb files) obtained in this study were deposited to EMDB and PDB. The accession codes (PDBID and EMDBID) 7XKH, 7XKQ, 7XKR, 7XKO, 7XKP, 33,251, 33,252, 33,253, 33,264, 33,265, 33,266, 33,267, 33,268, 33,277, 33,258, 33,278, 33,259, 33,279, 33,260, 33,280, 33,261, 33,281, 33,269, 33,282, 33,262, 33,283, and 33,263 are summarized in Supplementary Tables S2–S5. The data that support the findings of this study are available from PDB and EMDB.