

Abstract
Proteins are the key biological actors within cells, driving many biological processes integral to both healthy and diseased states. Understanding the depth of complexity represented within the proteome is crucial to our scientific understanding of cellular biology and to provide disease specific insights for clinical applications. Mass spectrometry-based proteomics is the premier method for proteome analysis, with the ability to both identify and quantify proteins. Although proteomics continues to grow as a robust field of bioanalytical chemistry, advances are still necessary to enable a more comprehensive view of the proteome. In this review, we provide a broad overview of mass spectrometry-based proteomics in general, and highlight four developing areas of bottom-up proteomics: 1) protein inference, 2) alternative proteases, 3) sample-specific databases and 4) post-translational modification discovery.
Overview of Mass Spectrometry-Based Proteomics
Proteins are central to nearly all major biological processes within the cell, acting as a bridge between genotype and phenotype.1 Comprehensive characterization of the proteome would deepen our understanding of diseases and complex biological processes, and is an ongoing goal of mass spectrometry-based proteomics. However, this is not trivial, as the proteome is not only highly complex but also dynamic in nature. Therefore, proteomic analysis must not only seek to identify which proteins are present, but also their abundance and modification status.
Initially, it was thought that a single gene was transcribed to a single RNA transcript, which was then translated into a single protein. However, this single gene to single protein hypothesis has since been abandoned. Instead, it is now understood that the proteome is incredibly diverse, with numerous protein products, or proteoforms, coming from a single gene (see Figure 1). A proteoform is defined as the distinct molecular form of a protein, with a specific amino acid sequence and set of post-translational modifications.2 The depth of proteoform complexity within the proteome is not yet fully understood, but this immense diversity can stem from numerous sources such as mutations at the gene-level, variants or alternative splicing at the transcript-level, and post-translational modifications or cleavage events at the protein-level.2–6 This depth of complexity further supports the importance of characterizing the proteome, because analysis of the genome and transcriptome alone cannot fully account for the complex phenotypes observed in healthy and disease states.
Figure 1:
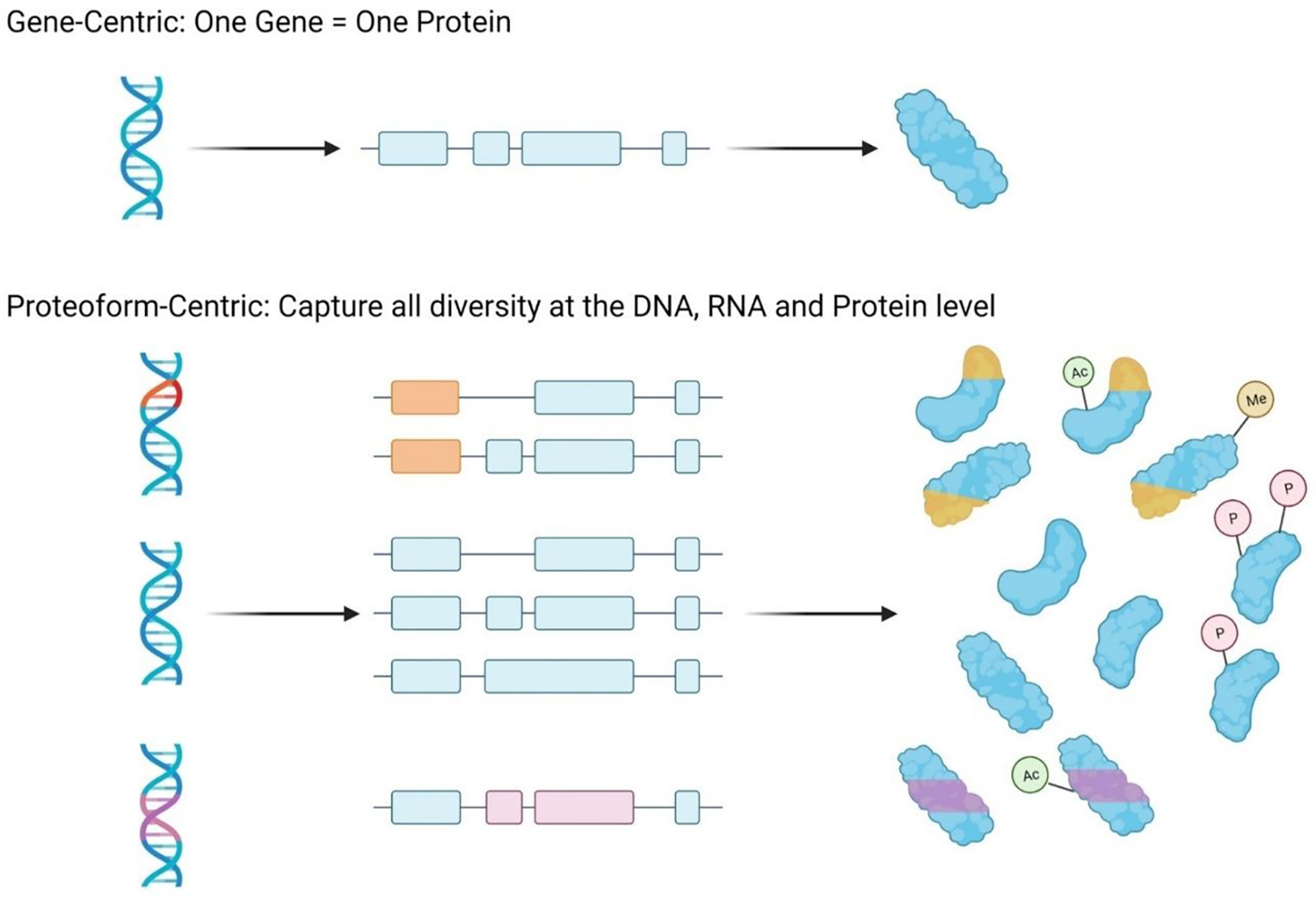
Sources of proteome complexity. Proteoforms provide a depth of complexity to the proteome which would not be possible if a gene only led to the production of a single protein product. Instead, mutations at the gene-level, variants or alternative splicing at the transcript-level, and post-translational modifications or cleavage events at the protein-level contribute to a still undefined number of proteoforms, which are the functional units of the proteome.
Mass spectrometry-based proteomics has quickly become the most high-throughput, reliable, and sensitive method for the characterization of the proteome.7–10 The principle of applying tandem mass spectrometry to the study of proteins is quite simple. In the initial MS1 spectra, the intact mass of a peptide or proteoform analyte is determined by measuring its mass-to-charge (m/z) ratio and using the observed charge state (z). In the subsequent MS2 scan, the intact peptide or proteoform is fragmented, generating product ions whose m/z values enable amino acid sequence determination leading to identifications.
Beyond identification, mass spectrometry-based proteomics can also facilitate the quantification of peptide or proteoform analytes. Approaches to mass spectrometric-based quantification can be divided into two categories, stable isotopic labeling, and label-free quantification. In isotopic labeling-based approaches, a mass tag which can then be recognized by the mass spectrometer is associated with the analyte of interest and is used to provide its relative abundance.11–13 In label-free approaches, no mass tags are utilized, and the intensity value of the peptide or protein of interest is used to determine its relative abundance.14,15
Mass spectrometry-based proteomics can be divided into two different approaches, bottom-up and top-down. The key difference between these two approaches is the analyte, which is either a peptide or a proteoform, respectively (see Figure 2). The vast majority of proteomics experiments utilize the bottom-up approach. In bottom-up, or shotgun proteomics, proteins are digested into peptides which are then analyzed via LC-MS/MS.16 Peptides are ideal analytes for mass spectrometry-based proteomics because they are easy to solubilize, separate and ionize. Since peptides are the observed unit in bottom-up proteomics, but protein-level information is still the desired outcome, peptides must act as proxies for their proteins or proteoforms of origin. All information regarding the presence and abundance of proteins in the sample are inferred from the peptides identified. The assumption that peptides are ideal proxies for the proteins or proteoforms in the sample is somewhat faulty. When proteoforms are digested into peptides, they lose their connectivity to their proteoforms of origin, which not only complicates the process of protein identification but also prevents the determination of which proteoforms are present in the sample. When reconstructing proteins from peptides, it is impossible to completely reconstruct the complexity of the proteome at the proteoform-level.
Figure 2:
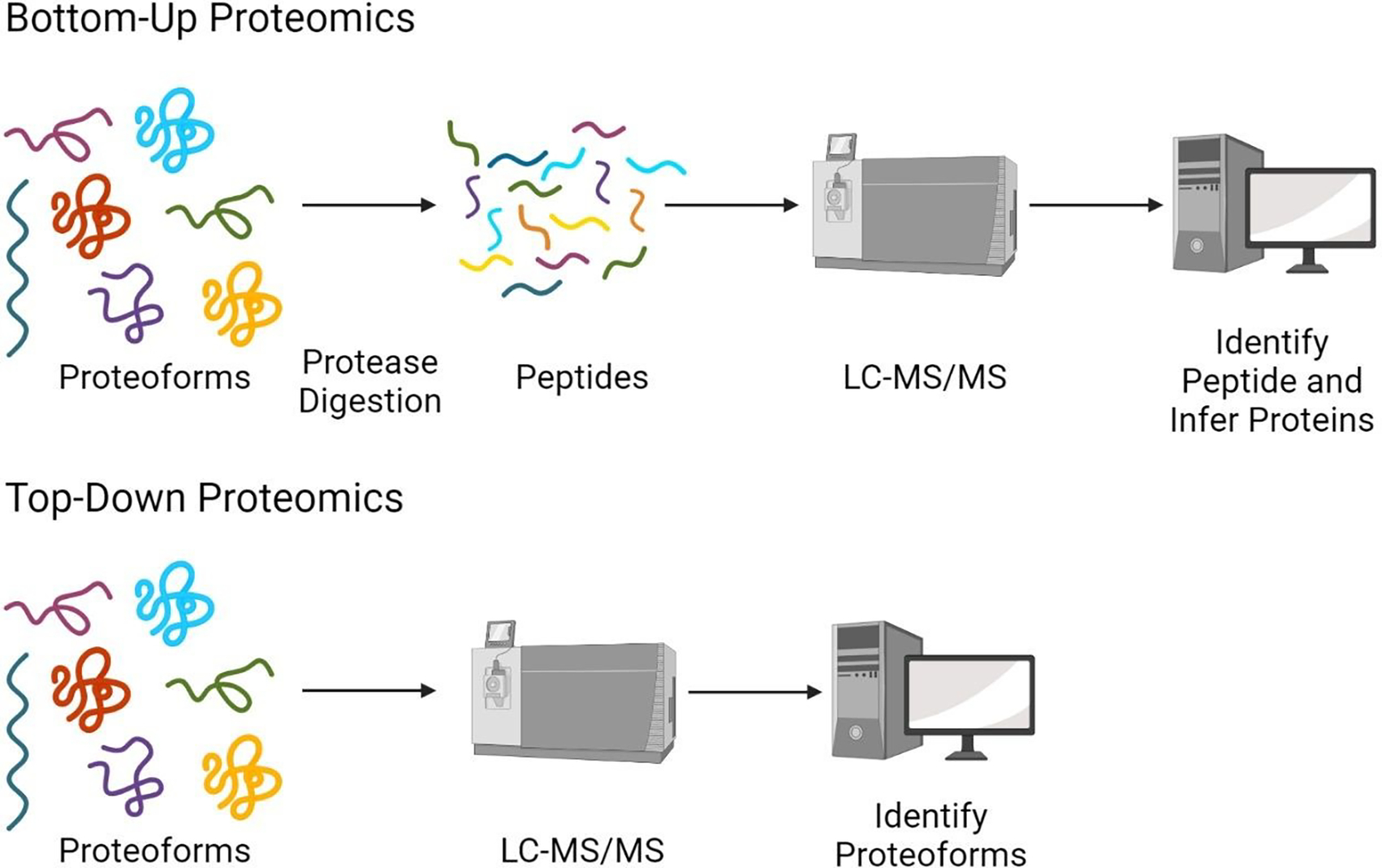
Experimental workflows for bottom-up and top-down proteomic approaches.
In top-down proteomics, intact proteins/proteoforms are analyzed via tandem mass spectrometry.17–22 Here intact proteoforms are directly being observed, and the relationship between the base amino acid sequence and the post-translational modifications on the proteoform are preserved. Therefore, no proxies are required in top-down proteomics. However, top-down analysis is very complicated and there are many challenges that must be overcome including but not limited to the low abundance of many proteoforms, the low signal-to-noise ratio of large molecular weight proteoforms, and low solubility of intact proteoforms.23–25 Additionally, for proteoforms, the sequence coverage of fragment ions is drastically reduced relative to peptides. Generally, in top-down proteomics, most fragment ions identified are localized near the N- and C- termini, leaving large portions of the middle of the proteoform without fragment ion coverage. This reduced sequence coverage can make localization of PTMs difficult creating ambiguity in the identification of proteoforms.26 Currently, the sensitivity of top-down proteomics is quite restricted compared to that of bottom-up proteomics. Top-down proteomics is limited to those proteins with high abundance and low molecular weight, with sensitivity diminishing drastically for proteins with a mass above 30 kDa.23
Although bottom-up proteomics is quite robust, there are many places within the conventional workflow where improvements can be made to enhance the characterization of the proteome. In this review we will discuss four areas in which the development of new tools and methods can further improve proteome characterization via mass spectrometry-based proteomics: 1) the process of protein inference, 2) the use of alternative proteases, 3) the use of sample-specific databases and 4) the discovery and validation of post-translational modifications.
The Process of Protein Inference
In bottom-up proteomics, as discussed in the previous section, peptides are the analyte. Proteins within a sample are digested into peptides, which are then analyzed via tandem mass spectrometry. Although peptides are directly observed, more often than not, protein-level identifications and abundance measures are desired.27 Therefore, the observed peptides serve as an intermediate to the desired protein-level results, making it necessary to reconstruct the original proteins in the sample. This reconstruction process is called protein inference and is often quite complicated and imperfect. The process of protein inference is convoluted by the existence of “shared peptides”, which are peptide sequences that could result from the digestion of multiple proteins present in the sequence database.27,28 The identification of these peptides generates ambiguity in the protein-level results, because it is impossible to distinguish the peptide’s protein of origin. Conversely, there are “unique peptides”, which are peptides distinct to a single protein within the sequence database, and the identification of such a peptide can confidently identify a single protein.27,28 The more shared peptides identified, the more complicated the process of protein inference becomes. Shared peptides are increasingly prevalent in higher order eukaryotic organisms where there is a greater degree of sequence homology resulting from related protein families, paralogous genes and complex alternative splicing.27,29,30 Various models exist to address the protein inference problem, most of which differ from each other in their approach to handling the complications arising from shared peptides.
Algorithms for protein inference can be broadly grouped into three categories: 1) optimistic, 2) statistical and 3) parsimonious. In optimistic algorithms, all possible proteins which could exist, based on the peptides identified, are considered detected. The underlying assumption made when utilizing this approach, is that the sample contains a large number of homologous proteins.27 Optimistic algorithms tend to be the simplest approach to protein inference, since there is no effort to reduce the ambiguity conferred by shared peptides. This also makes these algorithms the easiest to follow and comprehend for the end user. However, the increased ambiguity present in these algorithms is also why this model for protein inference is not widely utilized. Instead, statistical and parsimonious approaches have been and continue to be heavily favored. One example of optimistic inference is the original algorithm employed in DTASelect.31
Statistical approaches assemble evidence from the peptide identifications to estimate the probability a given protein is present in the sample. Typically, these algorithms utilize peptide posterior error probability (PEP) values, or other peptide scoring metrics to calculate protein-level probabilities.10,27,28 Statistical protein inference algorithms can be further sub-classified into non-parametric or parametric models. Non-parametric, or distribution free methods, make few to no assumptions regarding the probability distributions of the data being assessed.27 Due to this, these methods are easier to use and are generally more robust. One of the most well-known and utilized non-parametric statistical protein inference algorithms is ProteinProphet.32 Conversely, parametric models assume that the data used to generate the model comes from a probability distribution, and also makes assumptions regarding the parameters of said distribution.27 Due to the increased number of assumptions made in parametric models, they tend to produce more accurate protein probability estimates than non-parametric models, when the assumptions made are accurate. A major limitation to statistical approaches to protein inference is the inaccessibility of the logic underlying the algorithm. It can be unclear to the end user why certain proteins are weighted more heavily than others.
Parsimonious approaches to protein inference seek to apply the principle of Occam’s razor, which states the simplest answer is most likely the correct answer, to handle the problem of shared peptides.27 The goal of these approaches is to establish the minimum set of proteins which can explain all the identified peptides. The complexity of parsimony is equivalent to the computationally prohibitive NP-hard set cover problem.27 Therefore, to be able to “solve” what the minimum set of proteins are in the sample, heuristics and assumptions must be established, enforcing the simplest answer is likely to be the correct answer. Several statistical approaches have principles of parsimonious algorithms at their core.10,33 The discarding of putative proteins when alternative protein identifications have more support is a major limitation of parsimonious approaches, because these removed proteins could be present in the sample.28 Additionally, the heuristics and assumptions that are central to the algorithm may not be clear to the end user, making it difficult to understand the end protein list, and why some proteins are absent.33 Examples of parsimonious protein inference algorithms can be found within the search software programs Andromeda34 and MetaMorpheus35, as well as in standalone tools such as IDPicker36.
The problem of protein inference and how to handle shared peptides is not yet solved, and new algorithms are still being developed.33 One method for improving the quality of protein inference results, outside of continued algorithm development, is the curation of peptide identifications used as input for the inference algorithm. All assumptions regarding the presence or absence of a protein are based on the peptides used within the inference process. Increasing the depth and quality of the peptide identifications will in turn also increase the depth and quality of the inferred protein identifications. One approach to increasing the quality of inferred proteins is to increase the stringency of applied peptide filters.37 If false positive peptide identifications are incorporated for inference, they can lead to identifications that are not reflective of the sample’s proteome. However, being overly conservative can result in the loss of valuable true positives.37 In the process of protein inference, the more quality peptide identifications utilized, the better. Towards this end, several studies have shown protein inference results can be improved through the aggregation of peptide identifications across multiple search engines prior to inference.38 Another approach to improve protein inference, is the use of peptide identifications from multiple orthogonal proteolytic digests.35 Leveraging these peptide identifications from alternate proteases increases sequence coverage of the proteome, and the number of unique peptides identified, both of which have a positive impact on the accuracy of protein inference results.35
The Value of Alternative Proteases
For bottom-up proteomics, the serine protease trypsin is used almost exclusively. Trypsin is robust, reliable, and affordable.39–41 Cleaving after lysine or arginine residues, trypsin generates small peptides with a charged residue at the C-terminal position, ideal for collision-induced dissociation (CID) fragmentation.39–41 However, the near ubiquitous utilization of trypsin provides a tunnel-like view of the proteome.39,42 Trypsin alone is incapable of producing peptide identifications sufficient for the comprehensive characterization of the proteome. One factor contributing to this lack of comprehension is the mismatch of the peptide length distribution between those produced by tryptic digest and those identified via mass spectrometry (see Figure 3). Most peptides identified by mass spectrometry are between 7–35 amino acids in length. Nearly one-third of the peptides theoretically produced by tryptic digestion of the human proteome are under 6 amino acids in length and are too small for MS/MS based identification. This can lead to regions of proteins which are intractable to tryptic peptides. There are also entire classes of proteins and post-translational modifications (PTMs) which are difficult to characterize with tryptic digests.42 One such class of proteins are membrane proteins, whose transmembrane domains are composed mainly of hydrophobic amino acids, with very few lysine or arginine residues. Digestion of these transmembrane proteins generate very long and very hydrophobic peptides which are difficult to solubilize and ionize for mass spectrometry-based proteomics.
Figure 3:
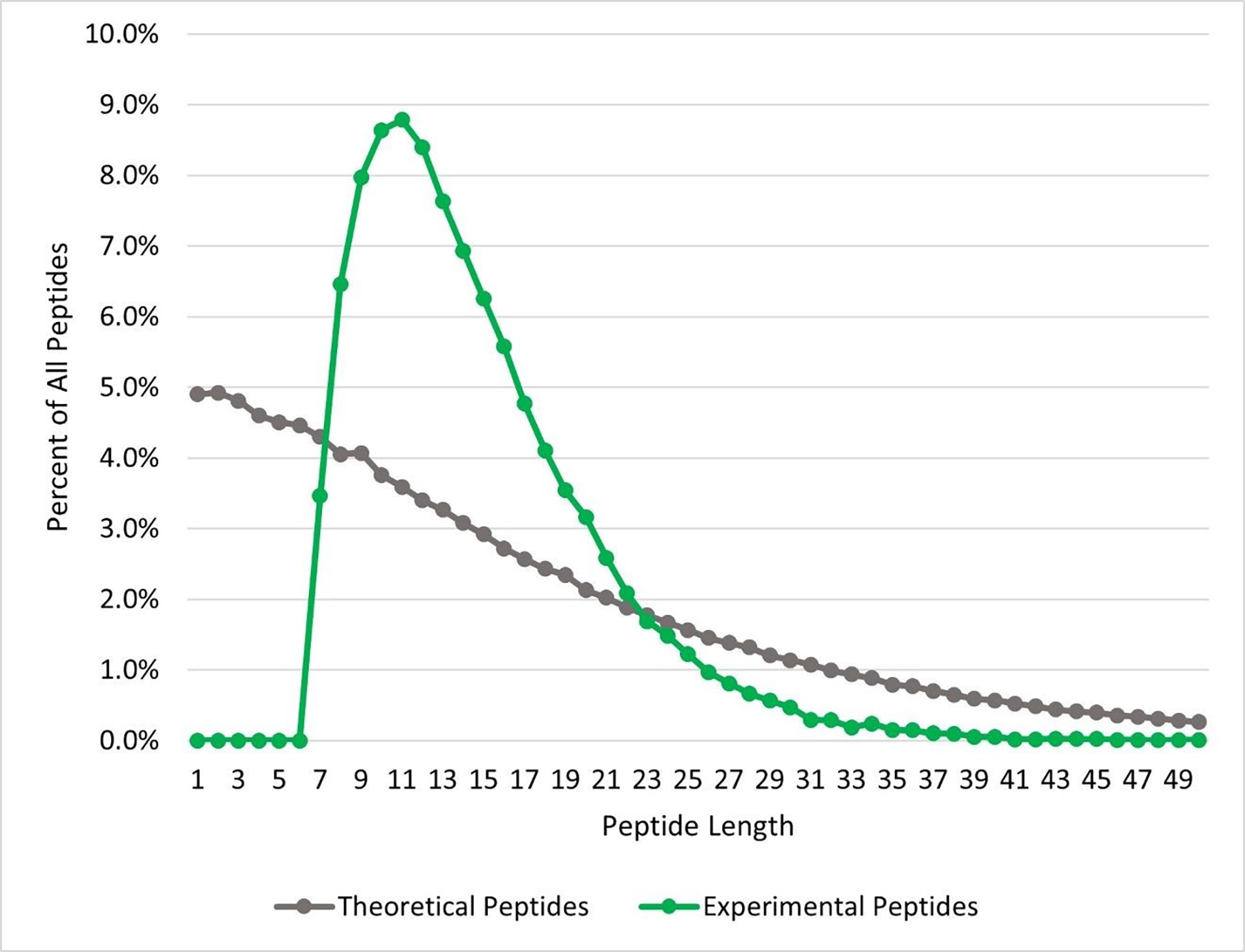
Comparison of the theoretical and experimental length distribution of tryptic peptides. The length distribution of in silico digested tryptic peptides (grey), as determined by ProteaseGuru, is compared to the length distribution of peptides experimentally identified from MetaMorpheus analysis of the tryptic data Miller et. al. (green).35 Most experimentally identified peptides are between 7–35 amino acids in length, whereas the theoretical tryptic digest favors the generation of shorter peptides.
Additionally, tryptic digestion may elicit an inherit bias in the proteomic results obtained. Examples of this are 1) phosphoproteome analysis, 2) the identification of splice junction peptides and 3) quantitative proteomic experiments. In phosphoproteome analysis, when negatively charged phosphorylated serine or threonine residues are adjacent to arginine or lysine residues, cleavage with trypsin can be inhibited. This results in longer peptides, with higher charge states that are not as amenable to identification with CID or higher-energy C-trap dissociation (HCD) fragmentation.39,41 This can result in biased phosphoproteome results, missing key phosphorylation sites, and lacking coverage in some of the most important regulatory regions throughout the proteome.42,43 For the identification of splice junction peptides, trypsin can also provide incomplete and therefore biased results. Surrounding exon boundaries, there are evolutionary preferred nucleotides which increase the occurrence of lysine and arginine coding triplets.44 Due to this, most identifiable tryptic peptides flank splice junctions, and the peptides crossing the junction are too small to be identified. This is problematic for the characterization of proteome-wide alternative splicing, where identifying splice junction peptides are critical. The use of trypsin alone can also introduce bias in protein quantification results. Studies have shown that protein quantification values differ based on the protease used for analysis, and that the pooling of data from multiple proteases can provide the best estimate for accurate protein abundance values.45,46
To overcome these pitfalls of trypsin, alternative proteases can be considered. The use of an alternative protease or multiple proteases has been shown to increase protein sequence coverage, the number of post-translational modifications identified, and the number of splice junctions covered. 35,39,40,42,44,45,47 Different proteases have various strengths which may make them ideal for different proteomic applications.
Chymotrypsin, Glu-C and Lys-C, like trypsin, are all serine proteases and can be utilized for high-throughput proteomic analysis.39 Chymotrypsin cleaves after tyrosine, phenylalanine and tryptophan residues, and is favored for proteins with long stretches of hydrophobic amino acids. As an alternative protease, chymotrypsin produces peptides which are generally considered to be the most orthogonal to those obtained by tryptic digests.39 Lys-C has strict specificity, cleaving only after lysine residues, and can produce longer peptides than trypsin.39 Lys-C is often paired with trypsin to improve the efficiency of cleavage after lysine residues. Glu-C which cleaves after glutamic acid, and also after aspartic acid when in phosphate buffers, is ideal for the digestion of heavily glycosylated proteins.39 Since the side chains of both glutamic and aspartic acid cannot be glycosylated, the modification will not inhibit cleavage of the proteins to peptides. Glu-C has also been heavily utilized for plasma proteomic applications.39,48
There are also proteases which cleave N-terminally, or before their triggering amino acids. Asp-N cleaves before aspartic acid residues. One distinct advantage of Asp-N is its compatibility with detergents during the digestion process. Asp-N has been noted as an especially valuable alternative protease for sensitive targeted proteomic applications such as selected reaction monitoring (SRM) analyses.39,41,45 Lys-N, which cleaves before lysine residues, has high resistance to both denaturants and temperatures up to 70 °C.39 Peptide products of Lys-N digestion, when paired with electron transfer dissociation (ETD) fragmentation, can provide exceptional product ion coverage which in many cases could even enable facile de novo sequencing of the peptides.39
Arg-C, which cleaves after arginine residues, is another valuable alternative protease. Unlike with trypsin, the presence of a proline residue adjacent to an arginine residue does not prevent cleavage when using Arg-C.39 Arg-C, like Lys-C, produces longer peptides than what is achieved with trypsin. Arg-C is typically utilized alongside other proteases in a multi-protease approach to help characterize and map post-translational modifications as well as increase protein sequence coverage.
Protease discovery and optimization is an on-going area of research and interest. One of the newer proteases is Proalanase, which cleaves after proline and alanine residues in highly acidic conditions.49 Proalanase enables the digestion of proline-rich proteins, such as collagen, and enables phospho-site profiling. It has been shown to be heavily orthogonal to tryptic digestion, providing valuable complementary coverage of the proteome.49
The use of multiple proteases, or alternative proteases, is crucial for the comprehensive characterization of the proteome. There are barriers that exist preventing widespread adoption of multiple, or alternative proteases. One such hurdle is the determination of which proteases are most beneficial to specific applications. This hurdle can be addressed using an in silico digestion tool to aid in experimental planning. Tools like this can be utilized to determine which proteases provide adequate or unique sequence coverage of target proteins, or sufficient PTM coverage. Another, more critical hurdle, is the increased time and sample requirements needed for multi-protease approaches. Towards addressing this concern, the Swaney group at University of California- San Francisco has developed a method which enables the pooling of peptides from multiple proteolytic digestions prior to data independent acquisition (DIA) analysis.50 Advances such as this are key to the future of comprehensive bottom-up proteomics leveraging multiple proteases.
The Importance of Sample-Specific Databases
Protein sequence databases are critical for high-throughput proteomic data analysis. Within search programs for bottom-up proteomics, protein sequence databases are digested in silico to generate a pool of candidate theoretical peptides. For each theoretical peptide, theoretical fragment ion m/z values are generated. These theoretical peaks are then compared to those experimentally observed in the MS2 spectra to determine peptide identifications. Without protein databases, peptide identifications would necessitate the use of de novo sequencing, or more recently spectral library searching. De novo search approaches take significantly longer than database searching methods, and generally tend to have higher false positive rates.
Typically, for many model organisms, there are reference protein databases (UniProt, Ensembl, RefSeq) which can be utilized for proteomic analysis.51,52 These reference protein databases seek to broadly represent all proteins present. While these reference databases are useful starting points, it is known that even within the same species, protein sequences can vary between individuals, tissues, and cell lines. Therefore, reference databases may be incomplete and fail to represent each individual sample. If the protein database used for proteomic analysis is not concordant with the sample being analyzed, the accuracy of the proteomic results is detrimentally impacted, and the biological conclusions drawn from the results may be inaccurate. In many cases, the reference database may not only lack sequence variants, but may lack entire protein isoform sequences for a given gene. When the reference database is incomplete in this manner, peptides containing these variants, or that are unique to missing isoforms cannot be identified. Peptides shared between the missing isoforms and those present in the sequence database will be incorrectly parsed resulting in inaccurate protein inference results. It is also possible the sample may express a subset of the protein isoforms present in the reference database. In this case, protein-level results can have false positive identifications, or an inflated level of protein ambiguity.
One approach to dealing with this database-sample discordance is the generation of sample-specific databases. This idea spawns from the sub-field of proteomics called proteogenomics, which seeks to integrate transcriptomic and proteomic data.53,54 For the specific application of sample-specific database generation, RNA-sequencing data can be translated in silico to construct a protein sequence database. Since this database is based on the RNA transcripts which function as protein precursors, the generated database is likely more accurate to the proteins and protein isoforms present in the sample than the reference. However, these constructed sample-specific databases still are subject to several limitations such as the sensitivity and specificity of the RNA-sequencing technologies utilized. Also, not all transcripts carry equivalent coding potential, and select protein isoforms, although translated, may not be stable.
Initially utilizing proteogenomics, reference databases were supplemented with peptide sequences containing variants or alternative splice junctions, as identified from short-read RNA-sequencing technology.55–60 These augmented databases represented the first attempts to generate a sample-specific search space. However these databases could become rather large, containing many sequences within reference proteins that were not relevant to the actual sample.54,61 To address this, tools such as Spritz were created to generate entire sample-specific protein databases by reconstructing full transcripts from short-read RNA-sequencing followed by in silico translation.62 Short-read RNA-sequencing has many parallels to bottom-up proteomics, in that the transcripts within the samples are fragmented to form short RNA oligonucleotides. These RNA fragments are then sequenced and mapped back to a reference genome to reconstruct RNA transcripts, much in the way that peptides are mapped to proteins through the protein inference process. Just like protein inference is imperfect, the process of reconstructing full transcripts from short-read RNA-sequencing is also imperfect. Short-read RNA-sequencing excels at the identification of sequence variants but can fall short in the reconstruction of alternatively spliced transcripts, just as bottom-up proteomics cannot reliably identify proteoforms (see Figure 4).
Figure 4:
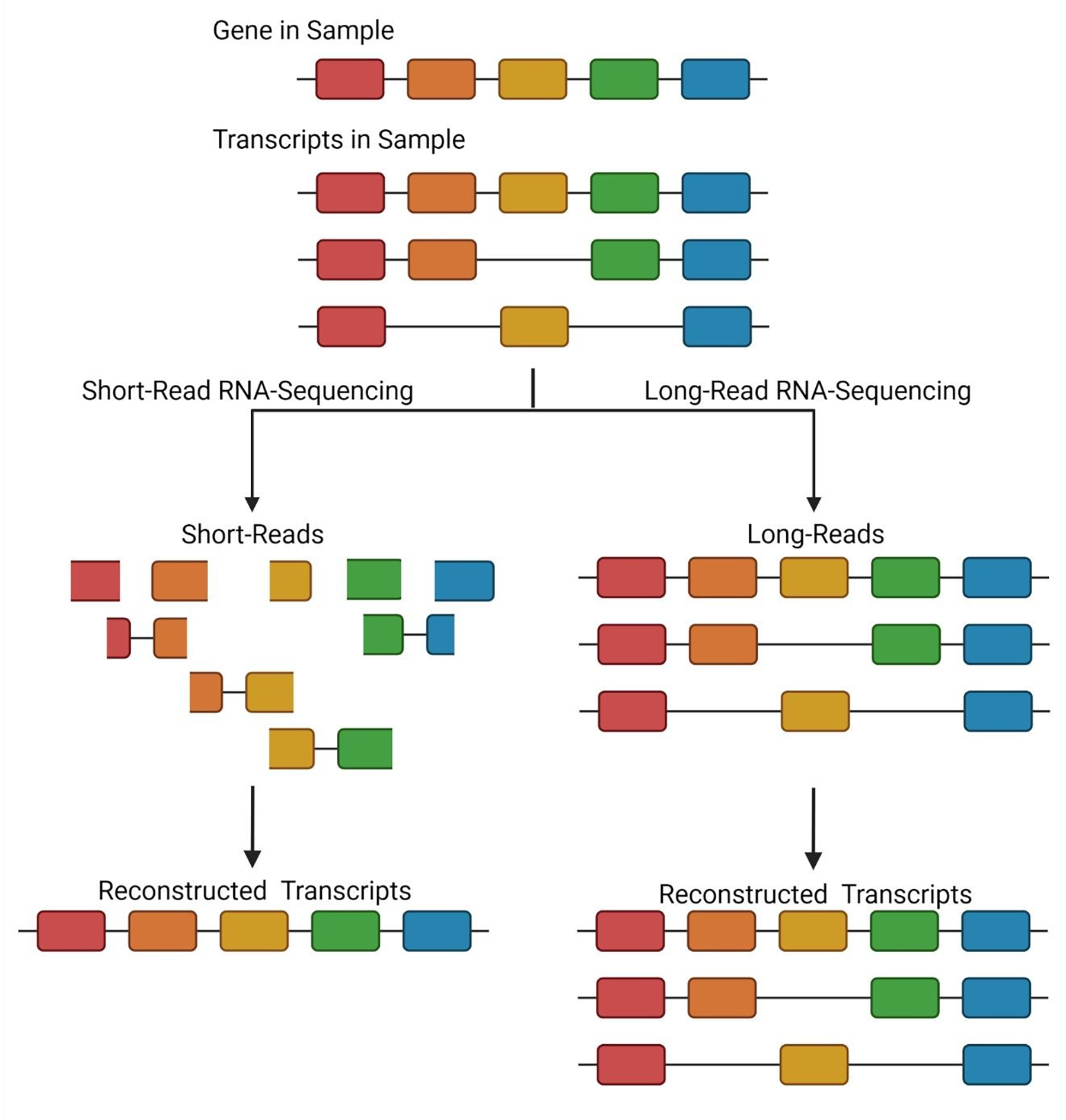
Comparison of short- and long-read sequencing for the reconstruction of transcript isoforms. In short-read RNA-sequencing approaches, RNA fragments are generated from which full-length transcripts must be reconstructed. Depending on the coverage of alternative splice junctions, incorrect transcript inference can be achieved. In this example, based on the fragments identified, a single transcript is reconstructed. Therefore, the two additional transcript isoforms are missed. In long-read RNA-sequencing, full-length transcripts are sequenced, and no reconstruction is required. Therefore, in the provided example, all three transcript isoforms expressed in the sample are identified.
To overcome complications arising from the inaccurate parsing of RNA fragments into full-length transcripts, long-read RNA-sequencing technologies can be utilized. In recent years, technology platforms from Pacific Biosciences and Oxford Nanopore, have become more prevalent in the transcriptomic community due to their ability to sequence full-length RNA transcripts with increasing accuracy.63–66 Specifically, for PacBio, technology has been developed to provide greater than 99% accuracy for the sequencing of single RNA transcripts.67 Sequencing of intact, full-length RNA transcripts eliminates the read parsing issues of short-read sequencing approaches and enables a more comprehensive view of the transcript isoform landscape of the sample (see Figure 4). This can provide an even more precise sample-specific database than those constructed using short-read RNA-sequencing data, especially for protein isoforms.
The Analysis and Discovery of Post-Translational Modifications
As powerful as proteogenomic approaches are for the generation of sample-specific databases, no transcriptional information can inform on the presence of post-translational modifications (PTMs). Post-translational modifications represent a critical layer of proteome diversity and are central to many important biological processes. The presence or absence of PTMs impact the function of proteoforms, contribute to signaling cascades and regulate diverse cellular functions.68–74 Mass spectrometry-based proteomics has quickly become the premier tool for the proteome-wide analysis of post-translational modifications. Using mass spectrometry-based proteomics for PTM mapping provides high sensitivity and throughput, as well as the ability to localize PTMs to a single amino acid residue using fragment ions. Localization of PTMs is of critical importance, especially for the investigation of functional significance. Unlike other PTM mapping approaches, such as antibody-based methods, proteomic analysis of PTMs is flexible in terms of the PTMs being analyzed and is not limited to a single modification at a time.9
As the field of proteomics has evolved, the ability to characterize PTMs reliably and accurately has grown in its importance. Ignoring modified peptides or proteins leads to a vast under sampling of the proteome. Some modification sites are very well established and may be included as annotated modifications in the reference database (UniProt XML). However, these annotations are nowhere near complete, and proteins with unannotated PTMs are a large contributor to the dark proteome.75–79
Methods for the discovery of PTMs not present in the protein database have evolved over time, giving greater PTM coverage. Initially, an approach called variable modification searching was applied.80 In this strategy, a selected PTM is allowed to occur on any amino acid residue fitting the modification motif in the search space. For example, for phosphorylation, theoretical peptides with phosphorylation at all serine, threonine or tyrosine residues are generated, as well as the unmodified theoretical peptides. This approach greatly expands the search space, increases search time, and introduces bias in the peptide-level false discovery rate (FDR) calculations.79 The bias in FDR calculations leads to a dramatic increase in the false positive rate for modified peptides. This approach is most valid when the variable modification being considered is widespread or enriched in the sample.79 Variable modification searching should only be applied for a small number of modifications at a time, as the negative repercussions of inflated false positive rates and increased database size compound with increasing numbers of modifications.79 These limitations make variable modification searching incompatible with reliable proteome-wide PTM discovery. To overcome many of the limitations of variable modification searching, Chick et. al. proposed a flexible method for PTM discovery and coined it “open search” or “open mass search”.77 In open searching, a large precursor mass tolerance is permitted. Therefore, the precursor mass of the experimental peptide can vary from the unmodified theoretical mass of the peptide and still be considered a match. The difference in mass observed can be accounted for by the mass(es) of unannotated PTMs. For the purpose of the Chick et. al foundational study, a mass difference up to 500 Da between the experimental and theoretical peptides was permitted.77 In the open search approach, the product mass tolerance applied for the search remains narrow, requiring high-mass accuracy for fragment ion matches.77 Therefore, a quality sequence tag can be utilized to identify the peptide’s amino acid sequence in question, and the difference between the experimental precursor and theoretical peptide mass could be used to identify a PTM, or combination of PTMs. This process eliminates the database size issues of variable modification searching and maintains an accurate FDR rate for modified peptides. However, there are still several limitations with the open search approach, first of which is the high computational and time requirements necessary to complete this kind of search.75,76,79 Second, the difference in mass between the experimental and theoretical peptide may not always be easily identifiable as a PTM or combination of PTMs, leaving confusion and ambiguity. Third, this method fails to permit the identification of fragment ions containing the modified amino acid residue.77 This becomes problematic if many of the potentially identifiable product ions contain the modified residue, making the modified peptide intractable to identification. Examples of search software programs which implement an open search strategy include Open-pFind81 and MS-Fragger82. Both tools have implemented approaches which seek to address the excessive time requirements of the traditional open search approach. The fragment ion index approach of MSFragger greatly reduces the overall search time relative to traditional open search approaches.82. In Open-pFind, a two-stage search is implemented consisting of an open sequence-tag-based search followed by a restricted search. The tag-index technique is essential in reducing the overall search time for the open search stage of Open-pFind, while the database reduction implemented in the restricted search further reduces overall search times.81 Additionally, MSFragger includes a location-aware algorithm which has addressed the limitation of excluding fragment ions containing the modified residue. This not only permits PTM localization, but also increases the sensitivity of the search.82
To further build on the open search approach, and address its downfalls, the Smith group invented global post-translational modification discovery (GPTMD), a multi-notch search approach for global discovery of PTMs.75,76 GPTMD searches for putative modifications found with an initial search using a multi-notch approach. This PTM discovery approach enables the identification of a large variety of PTMs while maintaining high confidence. The process of GPTMD has two main steps: 1) a multi-notch initial search to augment the protein database with putative PTMs and 2) a narrow precursor mass search using the augmented database to confidently identify PTM modified peptides.76 The multi-notch search is an extension of a narrow-precursor mass search enabling the inclusion of a variety of specific mass differences, or notches, between the precursor and theoretical masses. This approach improves upon the advantages of the open search approach, enabling the discovery of PTMs, without generating identifications with incomprehensible mass shifts.76 GPTMD also reduces the search time and increases the accuracy of modified peptide identifications relative to open search approaches.76 Using GPTMD, users define the mass notches they are willing to accept by selecting a list of modifications they are interested in discovering. A notch is generated for each mass shift associated with a PTM. Then, for each theoretical peptide, only experimental spectra with precursor masses that correspond to the unmodified peptide, or that differ by one of the defined notches are considered. These candidate spectra are then investigated for fragment ions matching the theoretical peptide. If a spectrum could correspond to a modified peptide, the corresponding PTM for the given notch is added to the augmented database. Once this augmented GPTMD database containing putative PTMs is generated, a final narrow-precursor mass search is completed to generate high confidence peptide identifications for both modified and unmodified peptides.76 This approach can be used to consistently identify PTM modified peptides which are not present in the sequence database.76
References
- (1).Aebersold R; Mann M Mass Spectrometry-Based Proteomics. Nature 2003, 422 (6928), 198–207. 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- (2).Smith LM; Kelleher NL Proteoform: A Single Term Describing Protein Complexity. Nat. Methods 2013, 10 (3), 186–187. 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schlüter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlén M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B How Many Human Proteoforms Are There? Nat. Chem. Biol. 2018, 14 (3), 206–214. 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Smith LM; Kelleher NL Proteoforms as the next Proteomics Currency. Science 2018, 359 (6380), 1106–1107. 10.1126/science.aat1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Uhlén M; Fagerberg L; Hallström BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson Å; Kampf C; Sjöstedt E; Asplund A; Olsson I; Edlund K; Lundberg E; Navani S; Szigyarto CA-K; Odeberg J; Djureinovic D; Takanen JO; Hober S; Alm T; Edqvist P-H; Berling H; Tegel H; Mulder J; Rockberg J; Nilsson P; Schwenk JM; Hamsten M; von Feilitzen K; Forsberg M; Persson L; Johansson F; Zwahlen M; von Heijne G; Nielsen J; Pontén F Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347 (6220), 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- (6).Gaudet P; Michel P-A; Zahn-Zabal M; Britan A; Cusin I; Domagalski M; Duek PD; Gateau A; Gleizes A; Hinard V; Rech de Laval V; Lin J; Nikitin F; Schaeffer M; Teixeira D; Lane L; Bairoch A The NeXtProt Knowledgebase on Human Proteins: 2017 Update. Nucleic Acids Res. 2017, 45 (D1), D177–D182. 10.1093/nar/gkw1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Han X; Aslanian A; Yates JR Mass Spectrometry for Proteomics. Curr. Opin. Chem. Biol. 2008, 12 (5), 483–490. 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Catherman AD; Skinner OS; Kelleher NL Top Down Proteomics: Facts and Perspectives. Biochem. Biophys. Res. Commun. 2014, 445 (4), 683–693. 10.1016/j.bbrc.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang Y; Fonslow BR; Shan B; Baek M-C; Yates JR Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113 (4), 2343–2394. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Noble W; Serang O A Review of Statistical Methods for Protein Identification Using Tandem Mass Spectrometry. Stat. Interface 2012, 5 (1), 3–20. 10.4310/SII.2012.v5.n1.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bantscheff M; Schirle M; Sweetman G; Rick J; Kuster B Quantitative Mass Spectrometry in Proteomics: A Critical Review. Anal. Bioanal. Chem. 2007, 389 (4), 1017–1031. 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- (12).Pappireddi N; Martin L; Wühr M A Review on Quantitative Multiplexed Proteomics. ChemBioChem 2019, 20 (10), 1210–1224. 10.1002/cbic.201800650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schubert OT; Röst HL; Collins BC; Rosenberger G; Aebersold R Quantitative Proteomics: Challenges and Opportunities in Basic and Applied Research. Nat. Protoc. 2017, 12 (7), 1289–1294. 10.1038/nprot.2017.040. [DOI] [PubMed] [Google Scholar]
- (14).Wong JWH; Cagney G An Overview of Label-Free Quantitation Methods in Proteomics by Mass Spectrometry. In Proteome Bioinformatics; Hubbard SJ, Jones AR, Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2010; Vol. 604, pp 273–283. 10.1007/978-1-60761-444-9_18. [DOI] [PubMed] [Google Scholar]
- (15).Lai X; Wang L; Witzmann FA Issues and Applications in Label-Free Quantitative Mass Spectrometry. Int. J. Proteomics 2013, 2013, 1–13. 10.1155/2013/756039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Link AJ; Eng J; Schieltz DM; Carmack E; Mize GJ; Morris DR; Garvik BM; Yates JR Direct Analysis of Protein Complexes Using Mass Spectrometry. Nat. Biotechnol. 1999, 17 (7), 676–682. 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- (17).Siuti N; Kelleher NL Decoding Protein Modifications Using Top-down Mass Spectrometry. Nat. Methods 2007, 4 (10), 817–821. 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen B; Brown KA; Lin Z; Ge Y Top-Down Proteomics: Ready for Prime Time? Anal. Chem. 2018, 90 (1), 110–127. 10.1021/acs.analchem.7b04747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cai W; Tucholski TM; Gregorich ZR; Ge Y Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert Rev. Proteomics 2016, 13 (8), 717–730. 10.1080/14789450.2016.1209414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Toby TK; Fornelli L; Kelleher NL Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. Palo Alto Calif 2016, 9 (1), 499–519. 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Armirotti A; Damonte G Achievements and Perspectives of Top-down Proteomics. PROTEOMICS 2010, 10 (20), 3566–3576. 10.1002/pmic.201000245. [DOI] [PubMed] [Google Scholar]
- (22).Gregorich ZR; Ge Y Top-down Proteomics in Health and Disease: Challenges and Opportunities. Proteomics 2014, 14 (10), 1195–1210. 10.1002/pmic.201300432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Compton PD; Zamdborg L; Thomas PM; Kelleher NL On the Scalability and Requirements of Whole Protein Mass Spectrometry. Anal. Chem. 2011, 83 (17), 6868–6874. 10.1021/ac2010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Moore SM; Hess SM; Jorgenson JW Extraction, Enrichment, Solubilization, and Digestion Techniques for Membrane Proteomics. J. Proteome Res. 2016, 15 (4), 1243–1252. 10.1021/acs.jproteome.5b01122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schaffer LV; Millikin RJ; Miller RM; Anderson LC; Fellers RT; Ge Y; Kelleher NL; LeDuc RD; Liu X; Payne SH; Sun L; Thomas PM; Tucholski T; Wang Z; Wu S; Wu Z; Yu D; Shortreed MR; Smith LM Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19 (10), e1800361. 10.1002/pmic.201800361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smith LM; Thomas PM; Shortreed MR; Schaffer LV; Fellers RT; LeDuc RD; Tucholski T; Ge Y; Agar JN; Anderson LC; Chamot-Rooke J; Gault J; Loo JA; Paša-Tolić L; Robinson CV; Schlüter H; Tsybin YO; Vilaseca M; Vizcaíno JA; Danis PO; Kelleher NL A Five-Level Classification System for Proteoform Identifications. Nat. Methods 2019, 16 (10), 939–940. 10.1038/s41592-019-0573-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Huang T; Wang J; Yu W; He Z Protein Inference: A Review. Brief. Bioinform. 2012, 13 (5), 586–614. 10.1093/bib/bbs004. [DOI] [PubMed] [Google Scholar]
- (28).Nesvizhskii AI; Aebersold R Interpretation of Shotgun Proteomic Data: The Protein Inference Problem. Mol. Cell. Proteomics MCP 2005, 4 (10), 1419–1440. 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- (29).Rappsilber J; Mann M What Does It Mean to Identify a Protein in Proteomics? Trends Biochem. Sci. 2002, 27 (2), 74–78. 10.1016/S0968-0004(01)02021-7. [DOI] [PubMed] [Google Scholar]
- (30).Black DL Protein Diversity from Alternative Splicing: A Challenge for Bioinformatics and Post-Genome Biology. Cell 2000, 103 (3), 367–370. 10.1016/S0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- (31).Tabb DL; McDonald WH; Yates JR DTASelect and Contrast: Tools for Assembling and Comparing Protein Identifications from Shotgun Proteomics. J. Proteome Res. 2002, 1 (1), 21–26. 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nesvizhskii AI; Keller A; Kolker E; Aebersold R A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75 (17), 4646–4658. 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- (33).Pfeuffer J; Sachsenberg T; Dijkstra TMH; Serang O; Reinert K; Kohlbacher O EPIFANY: A Method for Efficient High-Confidence Protein Inference. J. Proteome Res. 2020, 19 (3), 1060–1072. 10.1021/acs.jproteome.9b00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann M Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10 (4), 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- (35).Miller RM; Millikin RJ; Hoffmann CV; Solntsev SK; Sheynkman GM; Shortreed MR; Smith LM Improved Protein Inference from Multiple Protease Bottom-Up Mass Spectrometry Data. J. Proteome Res. 2019, 18 (9), 3429–3438. 10.1021/acs.jproteome.9b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ma Z-Q; Dasari S; Chambers MC; Litton MD; Sobecki SM; Zimmerman LJ; Halvey PJ; Schilling B; Drake PM; Gibson BW; Tabb DL IDPicker 2.0: Improved Protein Assembly with High Discrimination Peptide Identification Filtering. J. Proteome Res. 2009, 8 (8), 3872–3881. 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Claassen M; Reiter L; Hengartner MO; Buhmann JM; Aebersold R Generic Comparison of Protein Inference Engines *. Mol. Cell. Proteomics 2012, 11 (4). 10.1074/mcp.O110.007088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Audain E; Uszkoreit J; Sachsenberg T; Pfeuffer J; Liang X; Hermjakob H; Sanchez A; Eisenacher M; Reinert K; Tabb DL; Kohlbacher O; Perez-Riverol Y In-Depth Analysis of Protein Inference Algorithms Using Multiple Search Engines and Well-Defined Metrics. J. Proteomics 2017, 150, 170–182. 10.1016/j.jprot.2016.08.002. [DOI] [PubMed] [Google Scholar]
- (39).Tsiatsiani L; Heck AJR Proteomics beyond Trypsin. FEBS J. 2015, 282 (14), 2612–2626. 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- (40).Swaney DL; Wenger CD; Coon JJ Value of Using Multiple Proteases for Large-Scale Mass Spectrometry-Based Proteomics. J. Proteome Res. 2010, 9 (3), 1323–1329. 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vandermarliere E; Mueller M; Martens L Getting Intimate with Trypsin, the Leading Protease in Proteomics. Mass Spectrom. Rev. 2013, 32 (6), 453–465. 10.1002/mas.21376. [DOI] [PubMed] [Google Scholar]
- (42).Giansanti P; Tsiatsiani L; Low TY; Heck AJR Six Alternative Proteases for Mass Spectrometry–Based Proteomics beyond Trypsin. Nat. Protoc. 2016, 11 (5), 993–1006. 10.1038/nprot.2016.057. [DOI] [PubMed] [Google Scholar]
- (43).Schlosser A; Vanselow JT; Kramer A Mapping of Phosphorylation Sites by a Multi-Protease Approach with Specific Phosphopeptide Enrichment and NanoLC–MS/MS Analysis. Anal. Chem. 2005, 77 (16), 5243–5250. 10.1021/ac050232m. [DOI] [PubMed] [Google Scholar]
- (44).Wang D; Eraslan B; Wieland T; Hallström B; Hopf T; Zolg DP; Zecha J; Asplund A; Li L-H; Meng C; Frejno M; Schmidt T; Schnatbaum K; Wilhelm M; Ponten F; Uhlen M; Gagneur J; Hahne H; Kuster B A Deep Proteome and Transcriptome Abundance Atlas of 29 Healthy Human Tissues. Mol. Syst. Biol. 2019, 15 (2), e8503. 10.15252/msb.20188503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Guo X; Trudgian DC; Lemoff A; Yadavalli S; Mirzaei H Confetti: A Multiprotease Map of the HeLa Proteome for Comprehensive Proteomics. Mol. Cell. Proteomics MCP 2014, 13 (6), 1573–1584. 10.1074/mcp.M113.035170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Peng M; Taouatas N; Cappadona S; van Breukelen B; Mohammed S; Scholten A; Heck AJ R. Protease Bias in Absolute Protein Quantitation. Nat. Methods 2012, 9 (6), 524–525. 10.1038/nmeth.2031. [DOI] [PubMed] [Google Scholar]
- (47).Lanigan LT; Mackie M; Feine S; Hublin J-J; Schmitz RW; Wilcke A; Collins MJ; Cappellini E; Olsen JV; Taurozzi AJ; Welker F Multi-Protease Analysis of Pleistocene Bone Proteomes. J. Proteomics 2020, 228, 103889. 10.1016/j.jprot.2020.103889. [DOI] [PubMed] [Google Scholar]
- (48).Fossati A; Richards AL; Chen K-H; Jaganath D; Cattamanchi A; Ernst JD; Swaney DL Toward Comprehensive Plasma Proteomics by Orthogonal Protease Digestion. J. Proteome Res. 2021. 10.1021/acs.jproteome.1c00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Samodova D; Hosfield CM; Cramer CN; Giuli MV; Cappellini E; Franciosa G; Rosenblatt MM; Kelstrup CD; Olsen JV ProAlanase Is an Effective Alternative to Trypsin for Proteomics Applications and Disulfide Bond Mapping. Mol. Cell. Proteomics 2020, 19 (12), 2139–2157. 10.1074/mcp.TIR120.002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Richards AL; Chen K-H; Wilburn DB; Stevenson E; Polacco BJ; Searle BC; Swaney DL Data-Independent Acquisition Protease-Multiplexing Enables Increased Proteome Sequence Coverage Across Multiple Fragmentation Modes. J. Proteome Res. 2022, 21 (4), 1124–1136. 10.1021/acs.jproteome.1c00960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).The UniProt Consortium. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45 (D1), D158–D169. 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Aken BL; Achuthan P; Akanni W; Amode MR; Bernsdorff F; Bhai J; Billis K; Carvalho-Silva D; Cummins C; Clapham P; Gil L; Girón CG; Gordon L; Hourlier T; Hunt SE; Janacek SH; Juettemann T; Keenan S; Laird MR; Lavidas I; Maurel T; McLaren W; Moore B; Murphy DN; Nag R; Newman V; Nuhn M; Ong CK; Parker A; Patricio M; Riat HS; Sheppard D; Sparrow H; Taylor K; Thormann A; Vullo A; Walts B; Wilder SP; Zadissa A; Kostadima M; Martin FJ; Muffato M; Perry E; Ruffier M; Staines DM; Trevanion SJ; Cunningham F; Yates A; Zerbino DR; Flicek P Ensembl 2017. Nucleic Acids Res. 2017, 45 (D1), D635–D642. 10.1093/nar/gkw1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Nesvizhskii AI Proteogenomics: Concepts, Applications and Computational Strategies. Nat. Methods 2014, 11 (11), 1114–1125. 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Wang X; Liu Q; Zhang B Leveraging the Complementary Nature of RNA-Seq and Shotgun Proteomics Data. Proteomics 2014, 14 (23–24), 2676–2687. 10.1002/pmic.201400184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Sheynkman GM; Shortreed MR; Frey BL; Smith LM Discovery and Mass Spectrometric Analysis of Novel Splice-Junction Peptides Using RNA-Seq. Mol. Cell. Proteomics MCP 2013, 12 (8), 2341–2353. 10.1074/mcp.O113.028142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Sheynkman GM; Shortreed MR; Frey BL; Scalf M; Smith LM Large-Scale Mass Spectrometric Detection of Variant Peptides Resulting from Nonsynonymous Nucleotide Differences. J. Proteome Res. 2014, 13 (1), 228–240. 10.1021/pr4009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sheynkman GM; Johnson JE; Jagtap PD; Shortreed MR; Onsongo G; Frey BL; Griffin TJ; Smith LM Using Galaxy-P to Leverage RNA-Seq for the Discovery of Novel Protein Variations. BMC Genomics 2014, 15, 703. 10.1186/1471-2164-15-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ty L; S van H.; H van den T.; P G; A C; P T; S S; N H; B van B.; S M; E C; Aj H; V G Quantitative and Qualitative Proteome Characteristics Extracted from In-Depth Integrated Genomics and Proteomics Analysis. Cell Rep. 2013, 5 (5). 10.1016/j.celrep.2013.10.041. [DOI] [PubMed] [Google Scholar]
- (59).Ning K; Nesvizhskii AI The Utility of Mass Spectrometry-Based Proteomic Data for Validation of Novel Alternative Splice Forms Reconstructed from RNA-Seq Data: A Preliminary Assessment. BMC Bioinformatics 2010, 11 Suppl 11, S14. 10.1186/1471-2105-11-S11-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Evans VC; Barker G; Heesom KJ; Fan J; Bessant C; Matthews DA De Novo Derivation of Proteomes from Transcriptomes for Transcript and Protein Identification. Nat. Methods 2012, 9 (12), 1207–1211. 10.1038/nmeth.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Jeong K; Kim S; Bandeira N False Discovery Rates in Spectral Identification. BMC Bioinformatics 2012, 13 (16), S2. 10.1186/1471-2105-13-S16-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Cesnik AJ; Miller RM; Ibrahim K; Lu L; Millikin RJ; Shortreed MR; Frey BL; Smith LM Spritz: A Proteogenomic Database Engine. J. Proteome Res. 2021, 20 (4), 1826–1834. 10.1021/acs.jproteome.0c00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kasianowicz JJ; Brandin E; Branton D; Deamer DW Characterization of Individual Polynucleotide Molecules Using a Membrane Channel. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (24), 13770–13773. 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Jain M; Koren S; Miga KH; Quick J; Rand AC; Sasani TA; Tyson JR; Beggs AD; Dilthey AT; Fiddes IT; Malla S; Marriott H; Nieto T; O’Grady J; Olsen HE; Pedersen BS; Rhie A; Richardson H; Quinlan AR; Snutch TP; Tee L; Paten B; Phillippy AM; Simpson JT; Loman NJ; Loose M Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36 (4), 338–345. 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).van Dijk EL; Jaszczyszyn Y; Naquin D; Thermes C The Third Revolution in Sequencing Technology. Trends Genet. TIG 2018, 34 (9), 666–681. 10.1016/j.tig.2018.05.008. [DOI] [PubMed] [Google Scholar]
- (66).Sharon D; Tilgner H; Grubert F; Snyder M A Single-Molecule Long-Read Survey of the Human Transcriptome. Nat. Biotechnol. 2013, 31 (11), 1009–1014. 10.1038/nbt.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Wenger AM; Peluso P; Rowell WJ; Chang P-C; Hall RJ; Concepcion GT; Ebler J; Fungtammasan A; Kolesnikov A; Olson ND; Töpfer A; Alonge M; Mahmoud M; Qian Y; Chin C-S; Phillippy AM; Schatz MC; Myers G; DePristo MA; Ruan J; Marschall T; Sedlazeck FJ; Zook JM; Li H; Koren S; Carroll A; Rank DR; Hunkapiller MW Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37 (10), 1155–1162. 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Doerr A Making PTMs a Priority. Nat. Methods 2012, 9 (9), 862–863. 10.1038/nmeth.2153. [DOI] [PubMed] [Google Scholar]
- (69).Deribe YL; Pawson T; Dikic I Post-Translational Modifications in Signal Integration. Nat. Struct. Mol. Biol. 2010, 17 (6), 666–672. 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- (70).Sirover MA Subcellular Dynamics of Multifunctional Protein Regulation: Mechanisms of GAPDH Intracellular Translocation. J. Cell. Biochem. 2012, 113 (7), 2193–2200. 10.1002/jcb.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Gould N; Doulias P-T; Tenopoulou M; Raju K; Ischiropoulos H Regulation of Protein Function and Signaling by Reversible Cysteine S-Nitrosylation. J. Biol. Chem. 2013, 288 (37), 26473–26479. 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Cousin C; Derouiche A; Shi L; Pagot Y; Poncet S; Mijakovic I Protein-Serine/Threonine/Tyrosine Kinases in Bacterial Signaling and Regulation. FEMS Microbiol. Lett. 2013, 346 (1), 11–19. 10.1111/1574-6968.12189. [DOI] [PubMed] [Google Scholar]
- (73).Doll S; Burlingame AL Mass Spectrometry-Based Detection and Assignment of Protein Posttranslational Modifications. ACS Chem. Biol. 2015, 10 (1), 63–71. 10.1021/cb500904b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Olsen JV; Mann M Status of Large-Scale Analysis of Post-Translational Modifications by Mass Spectrometry. Mol. Cell. Proteomics MCP 2013, 12 (12), 3444–3452. 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Li Q; Shortreed MR; Wenger CD; Frey BL; Schaffer LV; Scalf M; Smith LM Global Post-Translational Modification Discovery. J. Proteome Res. 2017, 16 (4), 1383–1390. 10.1021/acs.jproteome.6b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Solntsev SK; Shortreed MR; Frey BL; Smith LM Enhanced Global Post-Translational Modification Discovery with MetaMorpheus. J. Proteome Res. 2018, 17 (5), 1844–1851. 10.1021/acs.jproteome.7b00873. [DOI] [PubMed] [Google Scholar]
- (77).Chick JM; Kolippakkam D; Nusinow DP; Zhai B; Rad R; Huttlin EL; Gygi SP A Mass-Tolerant Database Search Identifies a Large Proportion of Unassigned Spectra in Shotgun Proteomics as Modified Peptides. Nat. Biotechnol. 2015, 33 (7), 743–749. 10.1038/nbt.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Skinner OS; Kelleher NL Illuminating the Dark Matter of Shotgun Proteomics. Nat. Biotechnol. 2015, 33 (7), 717–718. 10.1038/nbt.3287. [DOI] [PubMed] [Google Scholar]
- (79).Shortreed MR; Wenger CD; Frey BL; Sheynkman GM; Scalf M; Keller MP; Attie AD; Smith LM Global Identification of Protein Post-Translational Modifications in a Single-Pass Database Search. J. Proteome Res. 2015, 14 (11), 4714–4720. 10.1021/acs.jproteome.5b00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Eng JK; McCormack AL; Yates JR An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. J. Am. Soc. Mass Spectrom. 1994, 5 (11), 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- (81).Chi H; Liu C; Yang H; Zeng W-F; Wu L; Zhou W-J; Wang R-M; Niu X-N; Ding Y-H; Zhang Y; Wang Z-W; Chen Z-L; Sun R-X; Liu T; Tan G-M; Dong M-Q; Xu P; Zhang P-H; He S-M Comprehensive Identification of Peptides in Tandem Mass Spectra Using an Efficient Open Search Engine. Nat. Biotechnol. 2018, 36 (11), 1059–1061. 10.1038/nbt.4236. [DOI] [PubMed] [Google Scholar]
- (82).Yu F; Teo GC; Kong AT; Haynes SE; Avtonomov DM; Geiszler DJ; Nesvizhskii AI Identification of Modified Peptides Using Localization-Aware Open Search. Nat. Commun. 2020, 11 (1), 4065. 10.1038/s41467-020-17921-y. [DOI] [PMC free article] [PubMed] [Google Scholar]