

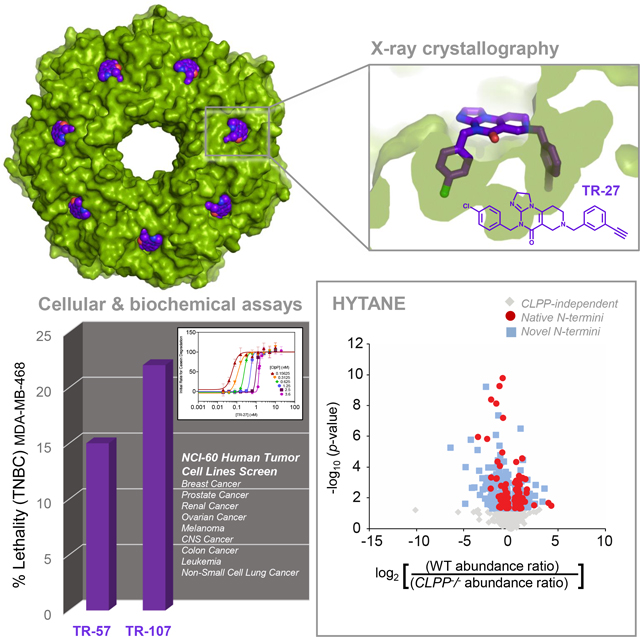
SUMMARY
The mitochondrial ClpP protease is responsible for mitochondrial protein quality control through specific degradation of proteins involved in several metabolic processes. ClpP overexpression is also required in many cancer cells to eliminate ROS-damaged proteins and to sustain oncogenesis. Targeting ClpP to dysregulate its function using small molecule agonists is a recent strategy in cancer therapy. Here, we synthesized imipridone-derived compounds and related chemicals, which we characterized using biochemical, biophysical, and cellular studies. Using X-ray crystallography, we found that these compounds have enhanced binding affinities due to their greater shape and charge complementarity with the surface hydrophobic pockets of ClpP. N-terminome profiling of cancer cells upon treatment with one of these compounds revealed the global proteomic changes that arise and identified the structural motifs preferred for protein cleavage by compound-activated ClpP. Together, our studies provide the structural and molecular bases by which dysregulated ClpP affects cancer cell viability and proliferation.
Keywords: ClpP protease, ClpP agonist, Drug design, Imipridones, TR compounds, Mitochondria, N-terminome, Cancer, X-ray crystallography, HYTANE, Mass spectrometry
Graphical Abstract
eTOC
Mabanglo et al. describe compounds, named TR, targeting human mitochondrial ClpP protease with nM affinity. Cocrystal structures with ClpP demonstrate excellent shape and charge complementarity of TR compounds with their binding site on ClpP. The compounds activate ClpP resulting in unspecific protein degradation leading to antiproliferative activity on cancer cells.
INTRODUCTION
In Eukaryotes, cellular energy in the form of ATP is generated during glycolysis in the cytoplasm and oxidative phosphorylation (OXPHOS) in the mitochondria. Within the mitochondria, many enzymes catalyze metabolic processes that yield small molecules and reducing equivalents needed to sustain the electron transport chain of OXPHOS. The native states and concentrations of these enzymes need to be continually preserved to maintain mitochondrial integrity in the background of very high redox activities. Because overall cell health is dependent on functional mitochondria, intricate protein quality control (PQC) mechanisms exist, carried out by molecular chaperones that help fold and repair misfolded proteins, and proteases that degrade and recycle those in excess or that are damaged beyond repair1,2. The ClpXP complex is one of the major PQC enzymes in the mitochondria3.
ClpP is the proteolytic component of the ClpXP complex whose active form is a tetradecameric, cylindrical chamber composed of two stacked heptameric rings4. Each ClpP subunit contains a catalytic serine-histidine-aspartate triad (S153, H178, D227 in human ClpP) used for peptide bond cleavage5. Encapsulation of catalytic sites within the chamber prevents spurious protein degradation and constitutes the basic level of ClpP regulation4. The chamber, with 14 catalytic sites, also enables tandem cleavage of unfolded polypeptides that ensure their rapid turnover. Crystal structures of ClpP show a highly conserved architecture composed of flexible N-terminal loops that regulate the chamber’s axial entrance pores, core domains that contain the catalytic triads and oligomerization sensor residues (E225 and R226 in human ClpP), and handle domains composed of a β-strand and a long α-helix that interdigitate to form the tetradecamer5–8. Deep hydrophobic pockets (H-sites) on the apical surfaces of ClpP are formed by two adjacent subunits and serve as docking sites for ClpP’s binding chaperone, ClpX5,6. ClpX is the unfoldase component of ClpXP that is responsible for selecting proteins to be degraded by ClpP9. Its active form is a hexamer with subunits that spiral around a central axis. ClpX docks on ClpP H-sites to form the functional complex. Human ClpX also has known functions of its own such as promoting heme biosynthesis by activation of δ-aminolevulinate synthase (ALAS), which catalyzes the first step of heme synthesis10–12.
The substrate pool of human ClpXP is large and encompasses proteins involved in several mitochondrial processes3. ClpXP functions in mitophagy, heme biosynthesis, and the mitochondrial unfolded protein response1,10,11,13–15. Abnormalities in ClpXP function or level are, therefore, associated with various diseases of mitochondrial etiology, including cancer3. Notably, ClpP is overexpressed in various human cancers, suggesting its specific role in oncogenesis and tumor metastasis16–18.
Small molecules that target ClpP either by inhibiting or dysregulating (activating) its function have thus been explored as potential anticancer drugs6–8,19–21. For instance, we identified one of the first small molecule agonists found to activate human ClpP and induce caspase-dependent apoptosis in mammalian cells, which are the acyldepsipeptides (ADEPs)6. In vitro, ADEP binds to ClpP H-sites and dissociates the ClpXP complex at substoichiometric concentrations, leading to ClpP activation independent of ClpX6. Recently, other agonists having imipridone derived scaffolds, such as ONC201, were found to activate ClpP in several cancer cells, causing the reduction in levels of respiratory chain components that leads to mitochondrial damage and, eventually, cell death8,22–24.
Since the discovery of ONC201, it has been investigated as a potential anticancer molecule in the lab and in over twenty clinical trials25–28. For example, ONC201 has shown promise in treating H3 K27M mutant gliomas28, but is considered to be an unoptimized agent. Herein, we describe the synthesis and characterization of imipridone-derived analogs and related chemicals, all of which are referred to here as TR compounds. We present X-ray structures of the compounds in complex with human ClpP and provide the structural basis for their enhanced binding affinities over the parent molecule. Using hydrophobic tagging-assisted N-termini enrichment (HYTANE)29, we identified the proteomic changes that occur in mammalian cells in response to ClpP activation by the TR compounds. We also determined the primary and secondary structural specificity of TR-activated ClpP complexes as revealed by N-terminome analysis. The TR compounds, therefore, augment the list of known small molecule effectors of mitochondrial energetics and apoptotic pathways with the potential to treat human cancers.
RESULTS
Measuring the antiproliferative effects and ClpP affinity of the TR compounds
The TR compounds were synthesized using procedures described in the Methods. The chemical strategy involved optimizing residues on the two phenyl moieties of the imipridone chemical scaffold (Fig. 1A). This led to the imipridone-based series of the TR compounds exemplified by TR-27 and TR-65 (Fig. 1A). TR-65 was found to be the most potent imipridone identified in this series (see below). Once we established that small electron withdrawing groups were preferred for both phenyl moieties, one featuring a para group and the other a meta, the use of halogen and nitrile moieties were selected to evaluate alternative chemical cores. In this regard, we favored simplification of the chemical core in our quest of designing a clinical agent. These studies led to the identification of the pyrimidone-based agents exemplified by TR-107 and TR-133, and pyrimidinedione-based agents exemplified by TR-57.
Figure 1. Growth inhibition profiling of TR compounds.
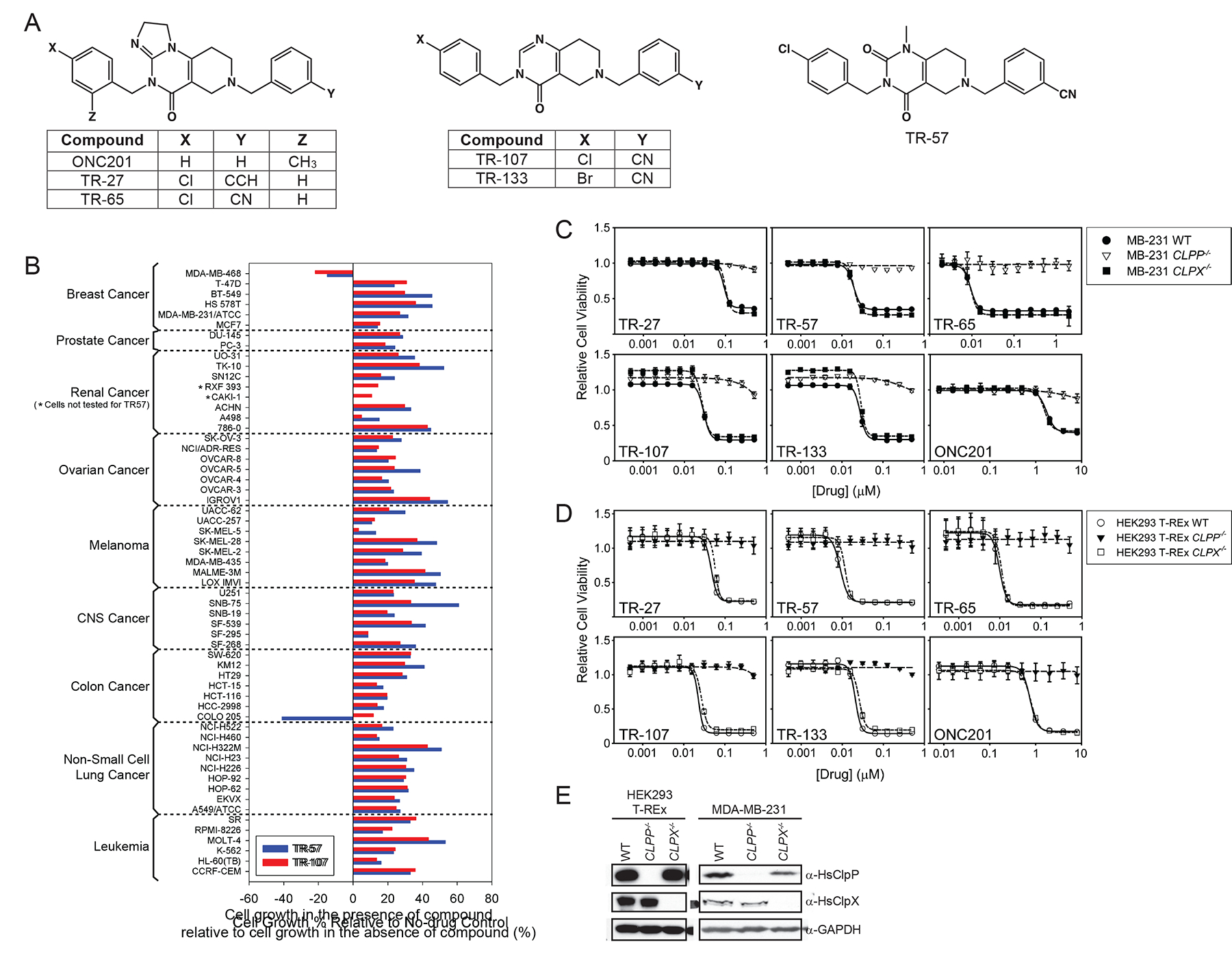
(A) Chemical structures of TR compounds used in this study.
(B) NCI-60 screening data for TR-57 and TR-107 at 10 μM. Drug-treated cell viability for each cell line is expressed as percent of cell growth in the presence of compound relative to cell growth in the absence of compound (see Methods).
(C,D) Growth inhibition profiles of TR compounds on MDA-MB-231 or HEK293 T-REx WT, CLPP−/−, and CLPX−/− cells. Error bars represent standard deviations from three biological replicates. IC50 values are also listed in Table 1.
(E) Null mutants of CLPP (CLPP−/−) and CLPX (CLPX−/−) generated with CRISPR-Cas9 in HEK293 T-REx and MDA-MB-231 were confirmed by western blotting for the two proteins. GAPDH was also blotted as loading control.
Initially, a general assessment of the anti-cancer properties of two analogues, TR-57 and TR-107, was carried out using the one-dose NCI-60 human tumor cell line screen that covers 60 cell lines categorized into 9 cancer panels30. The results are shown in Figure 1B. Both TR compounds inhibited the growth of the cell lines tested across all 9 panels by various degrees from 39% to 97% inhibition using a single dose of 10 μM drug for a 48-hour exposure period (refer to Methods for definition of % cell growth on the x-axis). Notably, both TR-57 and TR-107 induced cell death by 15% and 22% lethality, respectively, in the triple-negative breast cancer (TNBC) cell line MDA-MB-468. Similarly, TR-57 induced 41% lethality in the colon cancer cell line COLO205. Notably, both TR-57 and TR-107 can effectively suppress the growth of various types of cancer cell lines.
Next, we proceeded to analyze the growth inhibition/cytotoxicity of the five TR analogues in greater detail by profiling the growth of the TNBC cell line MDA-MB-231 in the presence of these compounds (Fig. 1C). MDA-MB-231 was chosen because of our general interest in targeting breast cancers and because this cell line is a commonly used model of TNBC that is easily maintained and whose growth is effectively suppressed by both TR-57 (68% suppression) and TR-107 (73% suppression) (Fig. 1B). The HEK293 T-REx cell line was also used (Fig. 1D). Cells were cultured and incubated with the five TR compounds for 72 hours at serially diluted concentrations. Cell viability was assessed with the sulforhodamine B (SRB) assay (see Methods). For comparison, the original imipridone ONC201 that targets ClpP and activates it was also included8. Cell lines with null mutants of the CLPP or CLPX genes were generated (Fig. 1E) and utilized to determine their role in cell viability in the presence of the TR compounds. All five TR compounds induced potent growth inhibition in both cell lines with IC50 values measured in the nM range (Table 1). Specifically, TR-65 (Figure 1C,D) was the most potent, followed by TR-57, TR-133, TR-107, with TR-27 being the weakest of the five compounds (Table 1). By comparison, ONC201 (Figure 1C,D) was significantly less potent than TR-27. Hence, the TR compounds exhibited at least 20-fold improvement in growth inhibition over the parent molecule ONC201 in both MDA-MB-231 and HEK293 T-REx cells (Table 1). All compounds exerted their growth inhibition effects in a ClpP-dependent manner, as deletion of CLPP conferred strong drug resistance. In contrast, ClpX did not appear to contribute to TR growth inhibition as shown for the CLPX null cells (Fig. 1C,D and Table 1).
Table 1.
IC50 values and affinity to ClpP for each TR compound.
| IC50 (nM), MDA-MB-231 | IC50 (nM), HEK293 T-REx | Casein-FITC degradation | ||||||
|---|---|---|---|---|---|---|---|---|
| Compound | WT | CLPP −/− | CLPX −/− | WT | CLPP −/− | CLPX −/− | Kd, app (nM) | h (at 2.5 μM ClpP) |
| TR-27 | 90.7 ± 1.7 | > 1000 | 98.9 ± 1.7 | 46.1 ± 1.3 | > 1000 | 57.7 ± 3.4 | 23.7 ± 10.0 | 8.1 ± 1.2 |
| TR-57 | 19.3 ± 0.2 | > 1000 | 19.9 ± 0.3 | 8.9 ± 0.2 | > 1000 | 11.8 ± 0.4 | 15.5 ± 12.0 | 9.4 ± 7.8 |
| TR-65 | 9.4 ± 0.2 | > 1000 | 9.4 ± 0.2 | 9.8 ± 0.3 | > 1000 | 11.0 ± 0.2 | 23.3 ± 9.1 | 10.9 ± 2.5 |
| TR-107 | 29.4 ± 0.2 | > 1000 | 27.4 ± 0.8 | 22.8 ± 0.7 | > 1000 | 27.0 ± 2.0 | 32.2 ± 6.3 | 5.5 ± 0.7 |
| TR-133 | 27.8 ± 0.4 | > 1000 | 29.5 ± 0.7 | 20.7 ± 0.7 | > 1000 | 26.0 ± 2.0 | 29.1 ± 7.8 | 6.2 ± 1.3 |
| ONC201 | 1720 ± 10 | > 10000 | 1530 ± 20 | 750 ± 10 | > 10000 | 790 ± 10 | 3556 ± 130 | 2.1 ± 0.2 |
Errors represent standard deviations from three experiments.
Subsequently, the binding affinity of the compounds to ClpP was measured using activity assays. Briefly, the degradation of the model substrate casein-FITC was carried out at varied concentrations of either compound or ClpP (Fig. 2A–C). Initial velocities (V0) were determined from kinetic traces collected in triplicate (Fig. 2A). Nonlinear regression analysis was then performed on the initial velocities vs. compound concentration at each ClpP concentration using the Hill equation to obtain the EC50 values (Fig. 2B). EC50 values obtained from these plots varied linearly with ClpP concentration (Fig. 2C). Hence, the change in EC50 values as a function of ClpP concentrations allowed the determination of the apparent Kd (Kd, app) (binding constant obtained when the fitted straight line in Figure 2C is extrapolated to zero ClpP concentration - see Methods). The Kd, app obtained for the TR compounds was in the nM range (Table 1). In comparison, the binding of ONC201 to ClpP was significantly weaker with a Kd, app of at least 100-fold greater than that of the TR compounds (Table 1). Additionally, the Hill coefficients (h) values for the non-linear regression fits in Figure 2B for all compounds tested were greater than 1, indicating cooperative ligand binding of the TR compounds to ClpP, as seen for other small molecule ClpP activators31. The h values are given in Table 1 for each compound assayed with 2.5 μM ClpP protomer. It should be noted that the cell-based IC50 for the TR compounds generally agreed with the Kd, app obtained from the protein degradation activity assays using purified protein (Table 1).
Figure 2. Measurement of the affinity of TR compound to ClpP by casein-FITC degradation.
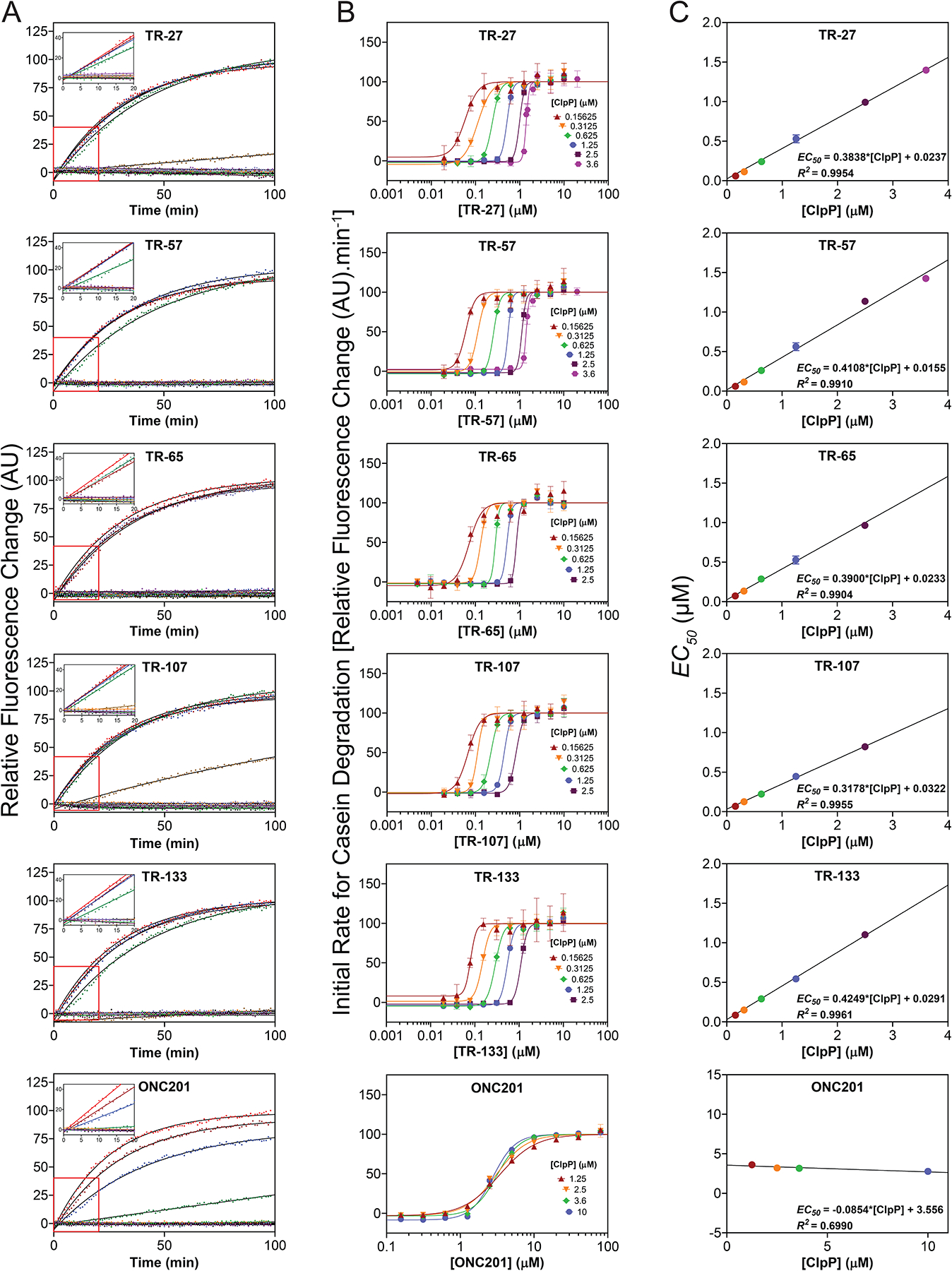
(A) Shown are representative curves of casein-FITC degradation by 2.5 μM ClpP in the presence of different concentrations of each compound from 0–10 μM for the TR compounds and 0–80 μM for ONC201. Data were normalized to the highest drug concentration reaction. Experiments were repeated at least three times. Solid black lines show fits to single exponentials. Insets show the linear fits from 0 to 20 min.
(B) EC50 for TR compound binding to ClpP were determined by fitting the change in initial degradation velocity of casein-FITC by compound-activated ClpP as a function of compound concentration to the Hill equation (see Methods). The data and fits are shown as semilog plots. Legends indicate the ClpP concentration for each fit. The data points in the absence of TR compound are not shown. Error bars represent the standard deviations from the average of three repeats.
(C) Kd, app for TR compound binding to ClpP were determined by fitting the EC50 obtained from B as a function of ClpP concentration to a straight line (see Methods). The Y-intercept of each plot represents the Kd, app value. The equations and R2 values are shown. Error bars represent the standard deviations from the average of three repeats. Kd,app values are also listed in Table 1.
Surface plasmon resonance (SPR) technique was also used to obtain a direct measure of the ClpP-compound interaction (Fig. S1A). The TR compounds, except for TR-27 and TR-133, exhibited appreciably tighter binding to ClpP than ONC201 (Fig. S1B). However, the Kd, app values obtained from SPR were at least 10-fold greater than those obtained by the activity assays described above for all tested compounds (Table 1). This large difference in Kd, app obtained by activity assays vs. SPR might indicate the importance of allosteric changes in ClpP that are needed to cause protease activation, which could be restricted when ClpP is crosslinked onto the SPR chip32. The harsh conditions required to crosslink the protein onto the chip could also be an issue. Alternatively, the assumption of a direct correlation between the affinity of the compound to ClpP and the ability of the compound to activate the protease might not strictly hold in this case.
Crystal structures of human ClpP-TR complexes
To determine the structural basis for the enhanced activation of ClpP by TR compounds, we determined the co-crystal structures of the ClpP-TR complexes for all five compounds. Crystallographic data collection and refinement statistics are shown in Table S1. In the crystals, TR-bound ClpP was found to be in the compact conformation as was observed when the structure of the protease was solved in the presence of ADEP-28 (PDB id 6BBA), ONC201 (PDB id 6DL7), and D9 (PDB id 6H23) (mean r. m. s. d. of 0.3106 Å for protein atoms)6–8. This compact state is achieved by partial distortion of the handle domains in which the β-strands are completely disordered and the αE helices have lost two N-terminal helical turns (Fig. 3A, left panel). The compaction also distorted the oligomeric sensor interactions and the catalytic triad geometry but brought the catalytic H178 and D227 residues of opposing subunits within interaction distance (Fig. 3A, right panel). Since the extended state of ClpP possesses ordered handle domains, oligomeric sensors, and catalytic triads, it is often regarded as the active form of the enzyme4. The observation that small molecule-activated complexes of ClpP can exist in the compact state with structural distortions in these parts has been interpreted by us to mean that ClpP itself may undergo a series of conformational transitions during its functional cycle6. This hypothesis is supported by the presence of N-terminal loop ordering and consequent axial pore widening in the compact state, a structural feature of the active state required for substrate entry (Fig. 3A,B)4,33.
Figure 3. Crystal structures of TR-bound human ClpP.
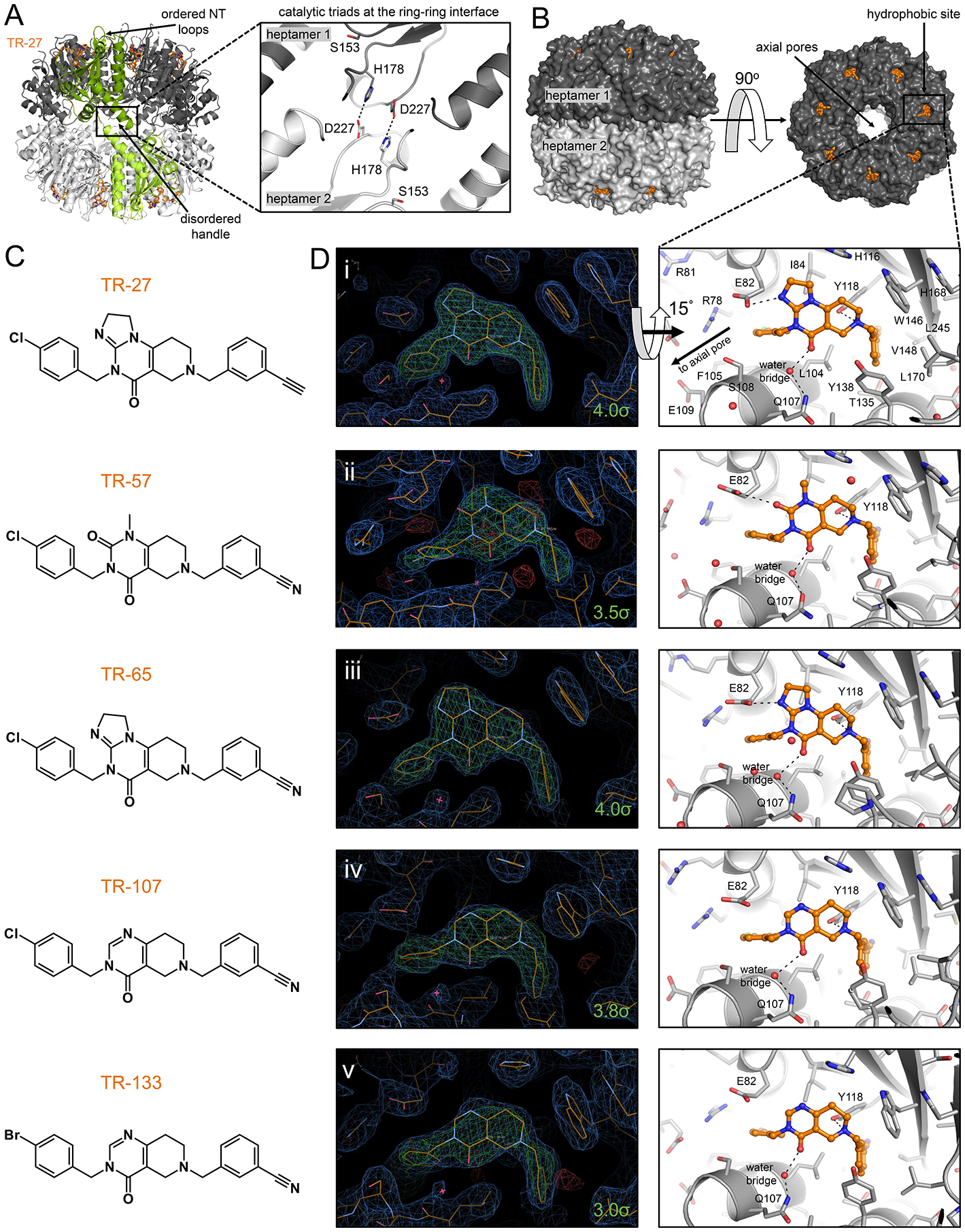
(A) The compact conformation of TR-bound ClpP featured organized N-terminal loops characteristic of the active form but shortened αE helices in the equatorial region. The catalytic triad residues H178 and D227 shown on the left as sticks formed hydrogen bonds with equivalent residues in a subunit of the opposite heptameric ring, helping stabilize the compact structure. Bound TR-27 is shown as orange sticks, and two opposing subunits are colored in green for emphasis. The two heptameric rings of human ClpP are colored in different shades of gray. The boxed area shows the general locations of the oligomerization sensors and the catalytic triads, the latter contained within. The oligomerization sensor residues E225 and R226 were disordered in the model and are not shown.
(B) Shown are side and top views of ClpP with the H-sites occupied by TR compounds drawn in orange sticks.
(C) Chemical structures of TR compounds.
(D) Left panels show electron density maps around the TR compounds that bind human ClpP H-sites in a characteristic pincer-like topology. 2Fo-Fc maps are shown in blue and are contoured at 1.0 σ, while composite omit maps are shown in green and are contoured at the indicated σ level for each molecule. Sequestered water molecules are represented by a red asterisk. Right panels are stick representations of the crystal structures, tilted by 15° along the x-axis, and show the various hydrophobic, π-stacking, and hydrogen bonding interactions with the TR compounds. Only the right panel of D(i) has the protein residues fully labeled. H-site residues that form important hydrophobic and van der Waals interactions with the TR compounds are shown as grey sticks, while those that form hydrogen bonds are indicated with black broken lines connecting to the small molecule.
The chemical structures of individual TR analogues and electron density maps showing their binding configurations are shown (Fig. 3C,D). The TR compounds bound H-sites with a conserved pincer topology similar to the crystallographic binding pose of the parent molecule ONC201 (Fig. 3D and Fig. 4A)8. This binding pose was stabilized by non-covalent interactions that arose from shape complementarity with the H-site (Fig. 4A). TR binding covered an average contact surface area of ~ 530 Å2, greater than that calculated for ONC201 (495 Å2) and buried an average of 83.5% of the TR compound’s total surface area, compared with 81.5% for ONC201 (Table S2). Thus, TR compounds have enhanced binding affinities relative to ONC201 partly due to increased van der Waals footprints and shape complementarity with H-sites.
Figure 4. Comparison of the ClpP binding modes of TR compounds, ONC201, ADEP-28, ZG111, and D9.
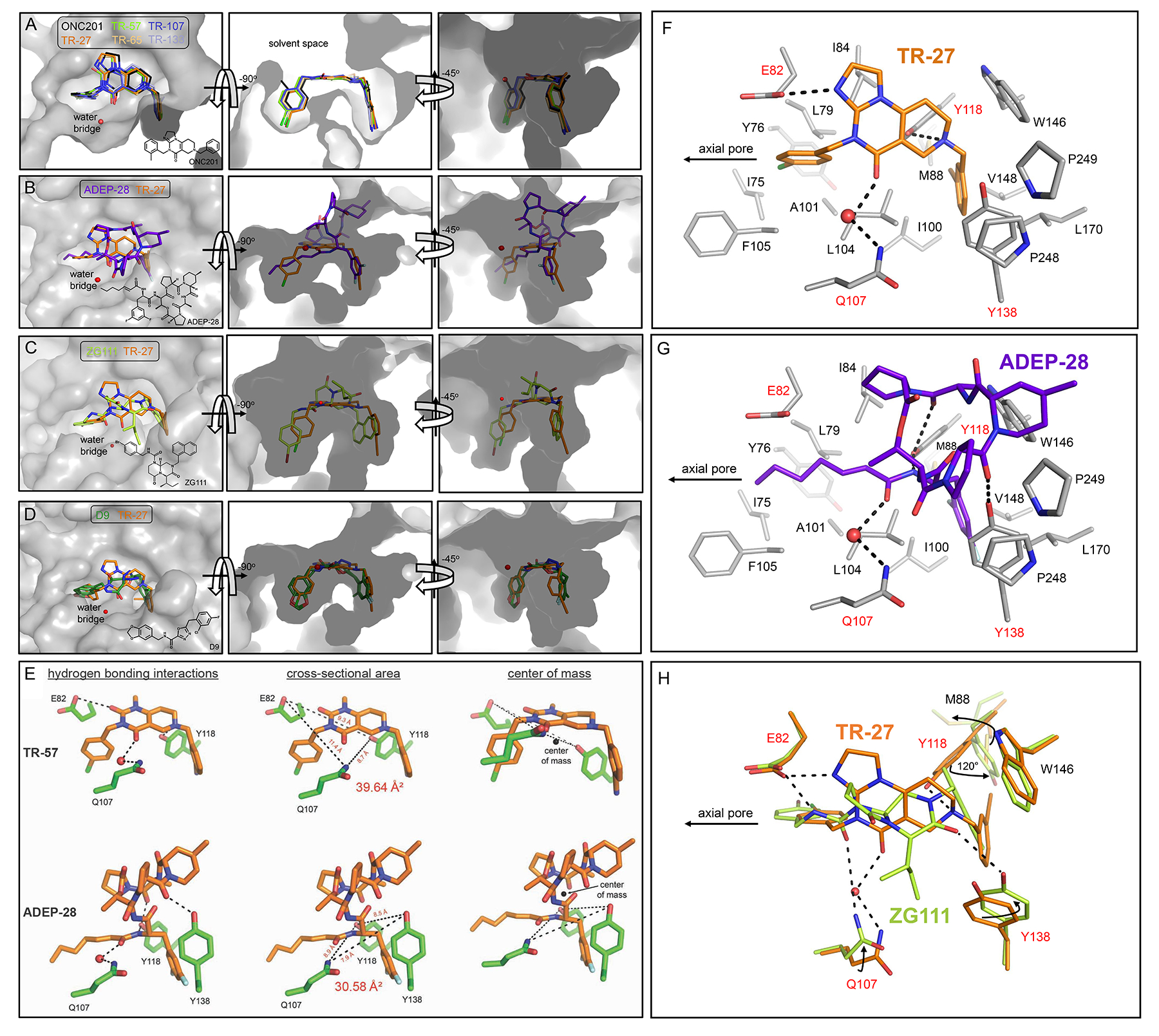
(A) Shown is an overlay of the five TR compounds and ONC201 bound to the H-site. The conserved water molecule that acts as a bridge between Q107 and the TR molecules is shown as a red sphere. The chemical structure of ONC201 is shown in the left panel.
(B) Shown is an overlay of ADEP-28 (purple sticks) and TR-27 (orange sticks) bound to the H-site. The bridging water molecule important for small molecule stabilization within the H-site is shown as a red sphere and is conserved for both TR compounds and ADEP-28. The chemical structure of ADEP-28 is shown in the left panel.
(C) Shown is an overlay of ZG111 (yellow green sticks, chemical structure shown on the left panel) and TR-27 (orange sticks) showing a similar pincer topology when bound to the H site. The red sphere indicates a bridging water molecule between TR-27 and a H site residue of ClpP (Q107, not shown). This “water bridge” is not observed for ZG11; rather its hydrogen bonding interaction with Q107 is direct (see Fig. 4H).
(D) Shown is an overlay of D9 (green sticks) and TR-27 (orange sticks) bound to the H-sites. D9 was observed in two major poses that flipped the orientation of its central carbonyl group between poses, and the water-bridged hydrogen bond interaction was not seen in the human ClpP-D9 complex. Also, the D9 complex structure was determined using the Y118A ClpP mutant protein. A steric clash with the chlorofluorophenyl ring moiety of D9 would occur with Y118 in the native protein if D9 was bound in the same poses observed in the crystal structure. The chemical structure of D9 is shown in the left panel.
(E) Shown on the top panel are the three hydrogen bonds formed by TR-57 (orange sticks) with E82, Q107, and Y118 (green sticks). On the bottom panel, the three hydrogen bonds formed by Q107, Y118, and Y138 (green sticks) and ADEP-28 (orange sticks) are shown. The dimensions of the triangle whose vertices are the hydrogen bond donor atoms in the interaction are also shown. The centers of mass (COM) of TR-57 and ADEP-28 were calculated using PyMOL and are indicated by black spheres. For TR-57, the hydrogen bonding atom of the amino acid residues were located above (E82, Q107) and below (Y118) the COM (right panel). For ADEP-28, the hydrogen bonding atom of all three residues (Q107, Y118, Y138) were located below the COM.
(F,G,H) Detailed view of the interactions of TR-27 (orange sticks), ADEP-28 (purple sticks), and ZG111 (yellow green sticks) in the ClpP binding pocket. Hydrophobic residues are labeled black, while polar/charged hydrogen bonding residues are labeled red. The conserved bridging water molecule is shown as a red sphere. In H, the binding modes of TR-27 and ZG111 are overlaid. Black arrows indicate movement of residue side chains to accommodate the two different activators.
In addition to the hydrophobic π-stacking interactions between the TR compounds’ di-substituted phenyl rings and protein side chains (E82, H116, Y118, Y138, and W146), three hydrogen bonds stabilized the binding (Fig. 3D, right panels). First, the N atom of the compounds’ non-planar ring formed a hydrogen bond with the hydroxyl group of Y118. This interaction occurred close to the exterior rim of ClpP. Second, near the rim of the axial pore, a water-mediated hydrogen bond was formed between a carbonyl group of the bicyclic core and the side chain amino group of Q107. An additional hydrogen bond stabilized the binding of TR-27, TR-57, and TR-65 formed by the carboxylate group of E82 and the imidazole group N atom of TR-27 and TR-65, or a second carbonyl group on the bicyclic core of TR-57 (Fig. 3D). The same interaction was observed for ONC201, but not for TR-107 and TR-133, due to the absence of a nearby hydrogen bond acceptor8.
In comparing the binding data for TR-27 and TR-65 (Table 1 and Fig. S1), it appears that a polar nitrile group is preferred over the nonpolar ethynyl group for binding the small cavity in ClpP (Fig. 3C). This might be due to the peptide bonds of the residues that form the walls of the largely hydrophobic binding pocket, across which they distribute patches of both positive and negative surface potentials (Fig. S2). While the hydrogen bond with E82 enhanced the overall binding affinity of TR compounds, the current chemical modifications associated with this interaction did not appear to have a significant effect on binding affinity given the similar dissociation constants for TR-57, TR-65, and TR-107 (Table 1 and Fig. S1).
Influence of shape and charge complementarity and noncovalent interactions on compound binding affinity is evident when comparing binding poses of the TR and ADEP-28 (Fig. 4B), the TR and ZG111 (Fig. 4C), or the TR and D9 (Fig. 4D) in human ClpP. TR and ADEP-28 differed in the extent of their van der Waals footprints on H-sites and in the arrangement of their non-covalent interactions. In our previous work, we reported ADEP-28 to have K0.5 = 0.12 μM for human ClpP6, and, hence, has lower affinity for ClpP compared to the TR compounds. TR compounds were buried within H-sites to a greater degree than ADEP-28 (Table S2). The pincer topology of a bound TR compound was more complementary in shape with the two pockets in the H-site than the T shape topology of ADEP-28 (Fig. 4B, middle and right panels). In addition, the 7-carbon aliphatic chain of ADEP-28 has greater rotational freedom around single carbon-carbon bonds than the corresponding phenyl ring moiety of TR analogs (see chemical structure in Fig. 4B, left panel). In fact, the electron density of this aliphatic chain is often weak at its last carbon atom found closest to the axial pore. The aliphatic chain is also less compatible with the polar and charged binding pocket than the halide-substituted phenyl ring moieties of the TR compounds (Fig. S2). Finally, although both compound classes were stabilized by three hydrogen bonding protein residues, including the conserved Q107 and Y118 (Fig. 4E), for TR analogues these residues were distributed across a larger trigonal cross-section, above and below the molecules’ center of mass, to bind the ligands (Fig. 4E). For ADEP-28, the hydrogen bonding residues covered a smaller cross-section and were located below its center of mass, resulting in less effective binding compared to TR compounds. Since the TR compounds are smaller in size than ADEP-28, resulting in a larger footprint area-to-mass ratio, they are in effect held by non-covalent interactions to a greater extent than ADEP-28 (Fig. 4E).
Figures 4F,G provide a more detailed comparative view of the binding mode between a TR compound, namely TR-27, and ADEP-28. As can be seen, the H-site of human ClpP is replete with lipophilic residues that largely drive small molecule binding. For TR-27 (Fig. 4F), hydrophobic, van der Waals, and π interactions contribute to the binding affinity of TR-27, in addition to hydrogen bonding interactions with E82, Q107, and Y118 (broken lines). The eastern fragment of TR-27 occupies a pocket formed by residues M88, I100, Y118, Y138, W146, V148, L170, P248, and P249. The western fragment of TR-27 occupies a pocket formed by residues I75, Y76, L79, I84, A101, L104, and F105. These terminal rings anchor the central 6,6,5 imipridone core to the hydrophobic pocket. In effect, TR-27 straddles the ClpP H-site using two terminal rings that are each cradled by surrounding hydrophobic residues. The small size of TR-27 relative to the H-site effectively buries >80% of the molecule’s surface area (see Table S2). Substitution with electron-withdrawing groups (chlorine or ethynyl for TR-27) at any position of the terminal rings enables anion- π interactions with nearby Tyr or Phe residues. For instance, an anion-π interaction exists between the western p-chlorophenyl ring of TR-27 and nearby E82. The electronegative Cl atom of the phenyl ring can likewise form an anion-π interaction with the phenyl ring of F105 located 4.3 Å away (nearest Cγ atom), although the geometry is not optimal. The eastern m-ethynyl-substituted phenyl ring is also sandwiched between the two π systems of Y118 and W146, although the distance and geometry for π stacking are not optimal.
ADEP-28 is sequestered in the H-site using similar hydrophobic, van der Waals, and π-interactions with protein residues, in addition to hydrogen bonds (Fig. 4G). The eastern dichlorophenyl ring of ADEP-28 is stacked between the two π systems of Y118 and W146, similar to the eastern fragment of TR-27. The 3,5 chlorine substitution on the phenyl ring fills the binding pocket. The western 7-carbon aliphatic chain is stabilized by hydrophobic interactions within the small pocket and extends towards the axial pore at a smaller angle relative to the western fragment of TR-27 (see Fig. 4B). Thus, substitution of the Cβ atom of TR-27’s western fragment and of the other TR compounds to form a branched moiety that engages both the lower and upper portions of the cavity, might further enhance binding affinity. One of the proline groups of the solvent exposed ADEP-28 macrocylic ring has close van der Waals interactions with P248 and P249 of the ClpP C-terminus. Furthermore, a portion of the macrocyclic ring sits on top of Y138 and W146 making polar and van der Waals contacts, respectively. These or similar interactions are not seen with the smaller TR-27 bound deeper in the pocket. Hydrogen bonds with Q107 and Y118 are conserved with TR-27. The hydrogen bond between Y138 and a carbonyl group of the macrocyclic ring is unique for ADEP-28 and is not possible for the TR compounds due to the latter’s smaller size that prevents reaching the solvent space to the same extent as ADEP-28. On the other hand, there is no hydrogen bonding interaction between E82 and ADEP-28, which is important for binding TR-27, TR-57, and TR-65 (see Fig. 3D).
In comparing the binding modes of TR-27 and ZG111, a human ClpP activator with therapeutic activity against pancreatic ductal adenocarcinoma34, we found that both molecules bind with the same pincer topology and are buried to similar extents within H sites (Fig. 4C, Table S2). Conserved stabilizing interactions between the two activators occur with human ClpP residues E82 and Q107 (Fig. 4H). Moreover, π-π stacking interactions form between W146 and the naphthyl group of ZG111 and the m-substituted phenyl ring of TR-27. However, unlike TR-27 but like ADEP-14, ZG111 forms a hydrogen bond using a ring carbonyl group with Y138 of human ClpP (Fig. 4H). In addition, unlike with TR-27, residue Y118 does not form a hydrogen bond with ZG111 and is rather moved 120° to avoid steric clash with the activator’s naphthyl group34. This Y118 rotamer shift then pushes the side chain of M88 away from its observed position in the human ClpP-TR-27 complex. These observations indicate that larger eastern fragments that increase van der Waals footprints of TR compounds might be accommodated by side chain rearrangements within their binding pocket.
While D9 bound human ClpP Y118A mutant with a similar pincer topology that is observed for TR compounds (Fig. 4D), the observed crystallographic poses might not occur at all with the native ClpP due to predicted steric clash between the Y118 side chain and the chlorofluorophenyl moiety of D97. Therefore, it was difficult to specify its non-covalent interactions with the native enzyme. In addition, the poses suggest compound mobility within the H-sites. This agrees with the Kd, app of 2.1 μM reported for D9 in a casein-FITC degradation assay7, which is much higher than that for the TR compounds (Table 1).
Evaluation of the proteomic changes caused by TR-induced activation of ClpP
The ClpP-dependent growth inhibition/cytotoxicity upon its dysregulation has been linked to specific proteomic changes that impair various mitochondrial functions and other biological processes8,19,22,35. To identify the specific changes induced by the TR compounds, we utilized HYTANE29 to analyze the proteomic N-terminal profiles of ClpP-expressing MDA-MB-231 wild-type (WT) cells and the corresponding genetic CLPP knockout (CLPP−/−), treated with either 1 μM TR compound or DMSO (no-drug control) for 24 hr. Using this process, the peptides that correspond to the N-termini of proteins and protein fragments generated via proteolysis were enriched for subsequent analysis and for construction of the N-terminome profiles associated with TR-27 treatment. To preclude caspase-dependent proteolysis that might complicate N-terminome analysis, the TR compound with the weakest affinity for ClpP (Table 1 and Fig. S1), TR-27, was selected for this analysis.
Prior to the HYTANE experiment, both WT and CLPP−/− MDA-MB-231 cells were examined for signs of apoptotic onset after they were drug (or DMSO) treated for 24 hours – significantly shorter than the 72-hour exposure used previously to initiate apoptosis in HEK293 cells6, which may obscure proteolysis associated with TR activation of ClpP. Specifically, both light and fluorescence microscopy (DAPI-staining of nuclei) confirmed that all cells shared the same morphology and nuclear integrity, respectively, after 24-hour exposure to 1 μM TR-27 or DMSO (Fig. S3A). Furthermore, western blotting clearly showed that, while ClpX level was reduced as a result of drug-induced ClpP expression as observed before6, neither Mcl1 nor caspases 9, 3, or 8 exhibited any alteration in expression or degradation (Fig. S3B). These results indicated the absence of significant apoptosis in these cells. As a control, treatment of cells with 2 μM of doxorubicin for 72 hrs clearly showed caspase 9 cleavage (Fig. S3C).
Details of the HYTANE experiments and subsequent data processing are given in Figure S4. 1161 unique N-terminal peptides present in both WT and CLPP−/− cells were identified and mapped to 1011 proteins (798 proteins yielded one peptide each; 213 yielded two or more peptides each) with high confidence (see Data S1). Among them, 829 displayed a change in abundance in TR-27-treated WT cells that were not statistically significant (p ≥ 0.05) compared to TR-27-treated CLPP−/− and thus were considered CLPP-independent (Fig. 5A, grey points; Fig. S4D, green data points in panels i and ii). For the remaining 332 peptides showing significant ClpP-dependent changes in abundance in TR-27 treated WT cells (p < 0.05) (Fig. S4D, red data points in panels i and iii), 236 had an N-terminal residue that either fell within the first three residues of the originating protein or was part of a known pro-peptide, transit peptide, or signal peptide that was cleaved off during maturation36 (Fig. 5A, blue points). The remaining 96 peptides do not fall into these categories (Fig. 5A, red points) and, hence, were considered novel N-termini generated as a result of TR-27-mediated ClpP activation either by direct action of activated ClpP or indirectly by another protease.
Figure 5. HYTANE analysis of the effects of TR-27-induced ClpP-dependent proteolysis on the N-terminome of the proteome in MDA-MB-231 cells.
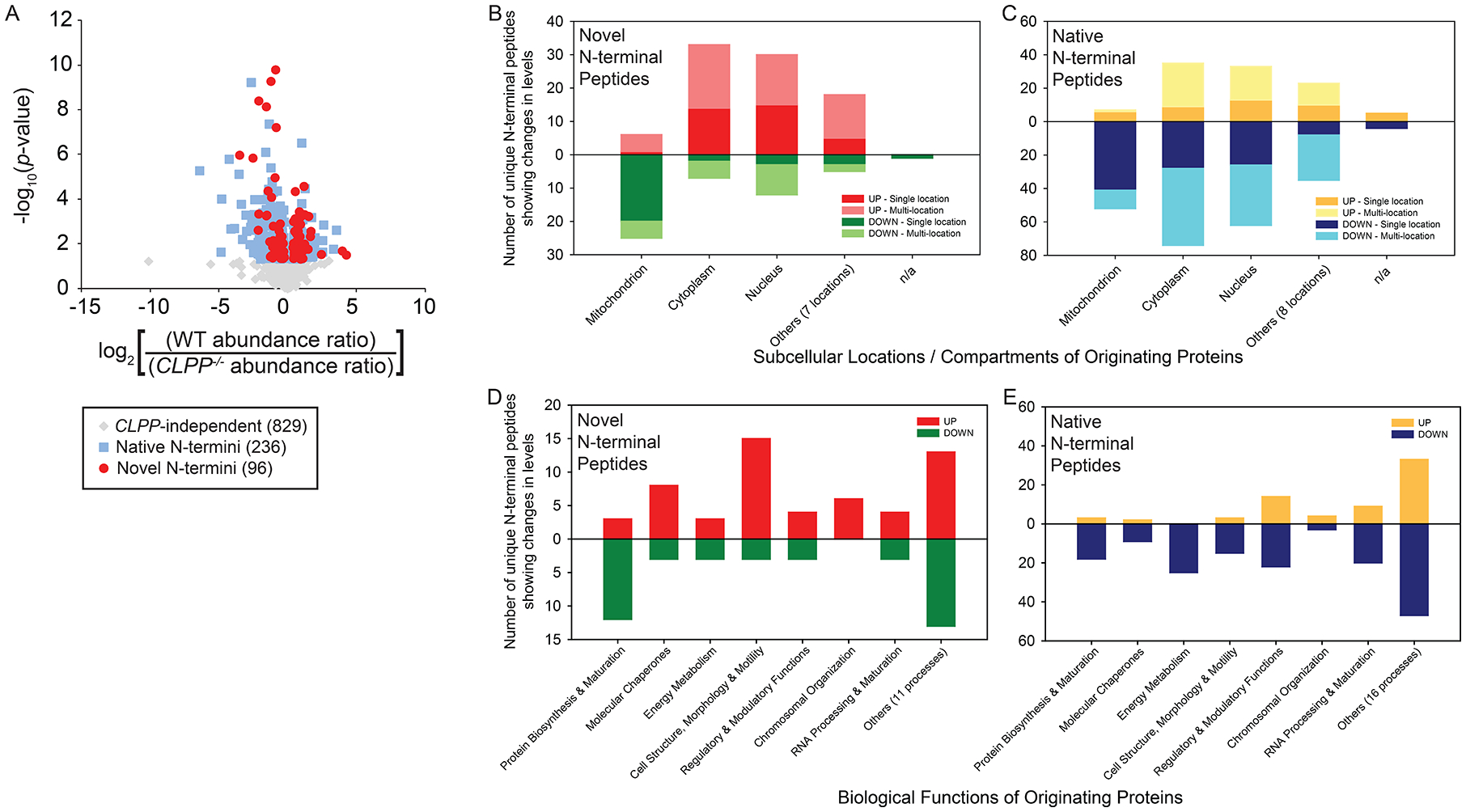
(A) Volcano plot of processed HYTANE data corresponding to quantified abundance changes in the levels of novel N-termini (red circles), native N-termini (blue squares) and CLPP-independent N-termini (grey diamonds) in the presence of TR27. p-values were calculated for the change in abundance for each N-terminal peptide and are considered statistically significant if p < 0.05.
(B,C) Changes in abundance of novel or native N-terminal peptides categorized by the subcellular localization of their originating proteins. Subcellular locations other than mitochondrion, cytoplasm and nucleus are grouped under “Others”, while cases of unknown or un-annotated localization are categorized under “n/a”. “UP” refers to the accumulation of a given peptide and “DOWN” refers to its depletion; “Single location” refers to the peptide’s originating protein localizing to a specific subcellular location; “Multi-location” refers to the originating protein localizing to more than one location.
(D,E) Changes in abundance of novel or native N-terminal peptides categorized by biological functions based on annotated functional descriptions of their originating proteins. Functions other than the categories shown are grouped under “Others”. “UP” refers to the accumulation of a given peptide and “DOWN” refers to its depletion.
Both native and novel N-termini were subsequently analyzed with respect to the subcellular localization and function of their parent proteins (Fig. 5B-E). Notably, novel N-terminal peptides originating from mitochondrial proteins were largely depleted, whereas N-termini originating from proteins localized to the cytoplasm, nucleus or seven other subcellular locations mostly accumulated (Fig. 5B). In contrast, the majority of native N-terminal peptides were depleted, regardless of subcellular localization of their originating proteins (Fig. 5C).
Among the novel mitochondrial N-terminal peptides, 25 were depleted (Fig. 5B). Ten of these originated from various subunits of the mitochondrial ribosome, two from Mortalin (mt-Hsp70), two from subunits of ETC complexes, one from a subunit of MPP (mitochondrial-processing peptidase), one from TOM40 of the TOM translocase complex, and 9 others involved in different amino acid or lipid metabolic processes (Fig. 5B,D). Six mitochondrial novel peptides accumulated (Fig. 5B). These included three from mitochondrion-associated molecular chaperones, one from the ATP-AMP transphosphorylase 2, one from L-lactate dehydrogenase subunit B and one from the RNA cytosine C(5)-methyltransferase Nsun2 (Fig. 5B,D).
There were 53 mitochondrial native N-terminal peptides that were depleted (Fig. 5C,E). Among these, 19 originated from ETC complex subunits or TCA cycle components. The others included four that originated from mitochondrial ribosomal subunits or other proteins involved in mitochondrial translation or post-translational modification, three from mitochondrial molecular chaperones, four from lipid metabolic enzymes, three from amino acid metabolic enzymes, three from apoptotic effectors or modulators, plus 17 others that originated from proteins that perform various functions, such as stress response, nucleic acid metabolism, transport across mitochondrial membranes, RNA processing and maturation, transcription/gene expression and specific processes involving regulatory proteins (Fig. 5C,E). Seven mitochondrial native peptides accumulated (Fig. 5C,E). These included two from the signal transduction protein 14–3-3 protein zeta/delta, one from ferredoxin-2, one from the inner membrane transporter ODC and three others from enzymes for glycerol or amino acid metabolism (Fig. 5E).
Based on the results of Figure 5D,E, these changes in mitochondrial N-termini profiles highlighted the following: (1) suppression of mitochondrial de novo protein biosynthesis and/or increased degradation of existing mitochondrial proteins; (2) reduced presence of molecular chaperones and auxiliary proteins required for mitochondrial protein maturation; and (3) general downregulation of key biological processes such as mitochondrial respiration, the metabolism of specific amino acids, lipids and other metabolites which reflected the reduced biosynthesis of mitochondrial proteins, leading to organelle dysfunction.
In contrast to their mitochondrial counterparts, novel N-terminal peptides originating from cytoplasmic and nuclear proteins mostly accumulated, whereas native ones were mostly depleted (Fig. 5B,C). Furthermore, the biological functions associated with these peptides were diverse (Fig. 5D,E). Except for the 14 depleted cytoplasmic peptides derived from ribosomal subunits or related to protein translation and those originating from molecular chaperones/co-chaperones associated with de novo protein biosynthesis, most of the cytoplasmic and nuclear proteins identified did not share any obvious functional relationship with the mitochondrial proteins showing depletion of N-terminal peptides. This further suggested that changes in cytoplasmic and nuclear proteomes, as revealed by the accumulation or removal of associated N-terminal peptides, likely occurred indirectly in response to the TR-27-induced ClpP activation that directly impacted the mitochondrion and its associated functions.
To validate the HYTANE results, we assessed the change in levels of the mitochondrial transcriptional factor A (TFAM) and mitochondrial stress-70 protein (Grp75 or mitochondrial Hsp70) upon compound treatment. The native N-terminus of TFAM and a novel N-terminus of Grp75 were found to be depleted according to HYTANE (Data S1). In agreement, western blot analysis of WT cells treated with TR-27 using the same growth conditions as HYTANE showed the levels of both proteins to be reduced upon addition of TR-27 (Fig. S5A). The levels of both proteins were unchanged in CLPP−/− cells (Fig. S5A).
In vitro degradation assays were then performed using purified TFAM (lacking the MTS) and ClpP proteins that clearly demonstrated the degradation of TFAM by TR-activated ClpP in a time-dependent manner (Fig. S5B). TR-65 was used in these experiments rather than TR-27. To investigate the cleavage site(s) of TFAM, the concentrations of ClpP and TFAM were increased to differentiate the generated TFAM fragments (Fig. S5C). Two bands at ~15 kDa and ~9 kDa (at 2 hrs) were selected for N-terminal sequencing, which allowed us to identify two cleavage sites in TFAM: between residues 127 and 128, and between 167 and 168 (Fig. S5D). TFAM contains two high mobility group (HMG)-box domains (HMG-box A and HMG-box B) separated by a helical linker region; it also has an N-terminal MTS and a C-terminal segment (Fig. S5D). One cut site lies within the linker region of TFAM, while the other cut site is at the N-terminus of the HMG-box B domain (Fig. S5D,E).
Primary and secondary structure characteristics of the cleavage sites generated by TR-induced activation of ClpP
Subsequently, we carried out further analysis on the novel N-terminal peptides that were generated by ClpP-dependent proteolysis. Both the primary and secondary structure of the 15 residues flanking the cleavage site (i.e. 15 on each side) of each peptide were analyzed to decipher potential sequence and structural motifs that promoted proteolysis. The sequence of residues flanking both sides of the cleavage site was identified for each peptide, using the annotated amino acid sequence of the originating protein from UniProt37 or other public databases. The reconstructed sequences were aligned by their proteolytic sites, and then categorized based on subcellular localization. Sequences of accumulated peptides were analyzed separately from depleted ones. The results are shown in Figure 6.
Figure 6. Characterization of the sequence and structural properties of the novel N-terminomes generated upon TR-27-induced ClpP-dependent proteolysis of cellular proteins.
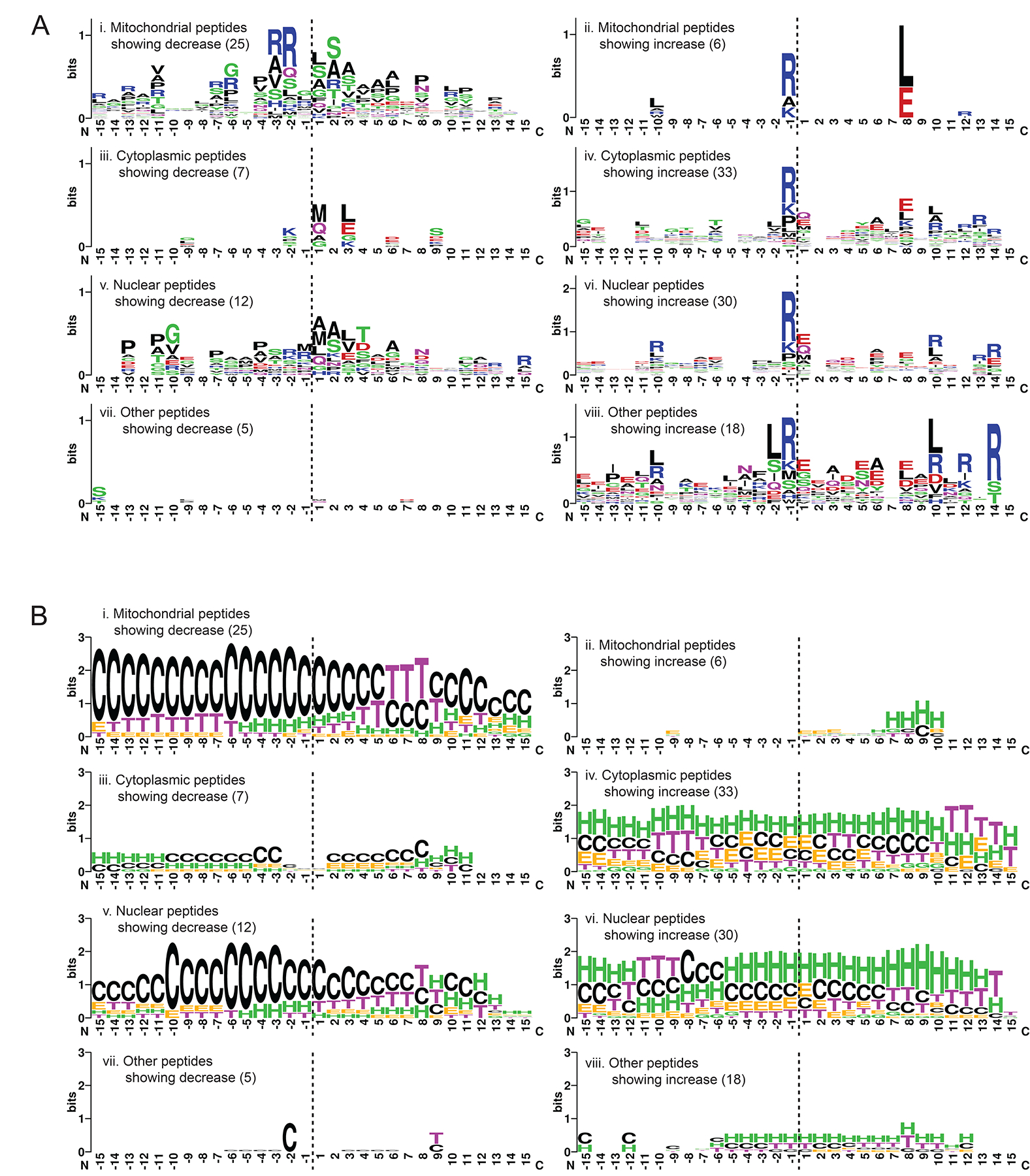
(A) Analysis of the 30-residue sequences representing the 15 residues flanking both sides of the the cleavage site (horizontal dashed line) generating the novel N-terminus. Sequences were categorized by the subcellular localization of their originating proteins. Depleted peptides (left panel) were analyzed separately from accumulated ones (right panel), with the number of sequences included per analysis shown in brackets. The results are presented in WebLogo format using default settings of the WebLogo online generator44. The residues N-terminal of the cleavage site are labeled on the x-axis from −15 to −1 and those C-terminal are labeled from 1 to 15. The y-axis is shown in bits (information unit) that corresponds to the entropy of amino acid variation with maximum entropy being log2 20 (amino acids) = 4.3 bits.
(B) Secondary structural analysis of the 30-residue sequences described in A. The required structural information was extracted from the Protein Data Bank (PDB) of the originating proteins with solved X-ray or NMR structures. Otherwise, the information was obtained using primary sequence-based structural predictions generated by AlphaFold39, either as existing models already generated and curated at the AlphaFold Protein Structure Database (URL: https://alphafold.ebi.ac.uk/) or as de novo predictions following the published AlphaFold protocol. The results are shown in the WebLogo format using the following settings: G = 310 helix, H = α-helix and I = Π-helix are colored green; E = extended β-strand and B = isolated β-bridge are colored orange; T = H-bond turn is colored purple; S = non-H-bond bend is colored blue; C = unstructured coil is colored black. The y-axis is shown in bits, with maximum entropy at log2 8 (secondary structures) = 3 bits.
Interestingly, for depleted peptides, Arg residues were favored at positions −2 and −3 (i.e. 2nd and 3rd position upstream of the proteolytic site) in mitochondrial peptides (Fig. 6A, panel i). For accumulated peptides, the Arg residue was strongly favored at position −1 across all subcellular locations (Fig. 6A, panels ii, iv, vi and viii). Arg was also favored to various degrees at positions 12 to 14, depending on the subcellular localization of the peptide. The ~14 residue periodicity correlates well with the average distance between any two catalytic serine residues within the ClpP chamber in extended conformation (~ 25 Å), indicating the possibility of tandem cleavage events on a fully extended polypeptide. Other preferred residues in accumulated peptides include Glu (and Gln in some cases) at positions 1 and 8, and to a lesser degree Ala at position 6 (Fig. 6A, panels ii, iv, vi and viii). Leu is also preferred at position 8 in mitochondrial peptides showing increase (Fig. 6A, panel ii). These alternate cleavage site sequence preferences may reflect the transient remodeling of the active site’s shape and surface potential in consequence of the conformational changes required for ClpP function.
Next, the secondary structure profiles of the categorized novel N-termini were analyzed. Secondary structure data were extracted from Protein Data Bank (PDB) coordinates of the peptides’ originating proteins or from AlphaFold structural models38,39 (see Methods). For depleted peptides, proteolysis occurred predominantly in inherently unstructured (i.e. coil) regions (Fig. 6B, panels i, iii, v and vii). This was especially evident for mitochondrial and nuclear peptides given their larger sample size (Fig. 6B, panels i and v). In contrast, proteolysis of accumulated peptides seems to occur at multiple types of secondary structures, although α-helices were preferred (Fig. 6B, panels ii, iv, vi and viii).
Taken together, the TR-27-induced, ClpP-dependent proteolysis appeared to be facilitated by specific sequence and structural motifs present in substrate proteins: (1) unstructured regions were predominantly cleaved without a strong dependence on any specific sequence motifs, although the presence of Arg at the second or third residues upstream of the proteolytic site was likely to promote cleavage of substrates; (2) α-helical segments tended to be cleaved immediately downstream of an Arg residue, and cleavage was promoted further by the presence of charged residues (both positive and negative) at specific sites downstream of the cleavage site. Importantly, it seems that novel N-termini that were generated in predominantly unstructured regions of the substrate proteins were physically accessible for subsequent proteolytic activities and were depleted accordingly. Similarly, novel N-termini that were generated in helical or other structured regions were more protected from further degradation, resulting in their accumulation.
DISCUSSION
Using X-ray crystallography and molecule binding studies, we elucidated the structural basis for the TR analogs’ enhanced affinity for ClpP relative to both the parent molecule ONC201 and ADEP. Improved binding affinity arose from better surface charge and shape complementarity between TR compounds and ClpP H-sites as well as differential distribution of hydrogen bonding patterns that tightened molecule sequestration. All five TR compounds showed a significant improvement in potency compared to the parent molecule ONC201 (Fig. 1 and Table 1). Importantly, these compounds targeted ClpP specifically, given that deletion of CLPP conferred strong resistance (Fig. 1 and Table 1).
Through HYTANE, we were able to monitor global proteomic changes that occurred in consequence of ClpP activation by TR compounds. Comparison of our data with those from the published proximity-dependent biotin identification (BioID) analyzing the proteomic impact of ONC201-induced ClpP activation8, revealed many proteins that were depleted in common between the two datasets (Table S3). Other proteins that were unique for each dataset but were constituents of the same protein complexes were also similarly depleted (Table S3). These include: subunits of the respiratory Complexes I (NDUF proteins), II (SDH proteins), III (UQCR proteins) and IV (COX proteins) and assembly factors (NDUFAF proteins) dedicated to the maturation of the Complex I α-subcomplex; proteins associated with the TCA cycle (OGDH, DLST, SUCLG1 and 2, MDH2, IDH3A and 3G); protein components of the 39S (MRPL proteins) and 28S (MRPS proteins) subunits of the mitochondrial ribosome; proteins involved in the transcription (TFAM) and translation (GFM1) of genes encoded by mitochondrial genome; and enzymes that facilitate specific steps in glutamate metabolism (GLUD1), β-oxidation of fatty acids of various lengths (HADH, ECHS1, ACAT1) and purine metabolism (HINT2) (Table S3). Hence several pathways that are considered to be hallmarks of cancer and cancer metabolism40,41 are being impacted upon ClpP dysregulation. Importantly, we verified the targeting of TFAM and Grp75 by compound activated ClpP. This clearly demonstrates the direct impact of these compounds on mitochondrial transcription and protein homeostasis.
While the BioID approach identified candidate substrates and interactors of activated ClpP8, our HYTANE approach enabled not only a similar substrate profiling, but also the characterization of proteolytic events and their structural underpinnings, which are important aspects that were not explored before in human cells. Furthermore, mapping of novel N-terminal peptides to their originating proteins allowed us to deconstruct both primary and secondary structural requirements for protein cleavage for TR-activated ClpP.
Importantly, novel N-termini that accumulated were found to originate from structured regions of targeted proteins that are less accessible. Conversely, N-termini that were depleted tended to be structurally flexible and, therefore, more accessible for proteolysis. Furthermore, the majority of accumulated N-termini were mostly generated via cleavage after an Arg residue while the depleted ones were generated without relying on any particular sequence motifs. Importantly, this single-Arg motif is distinct from the twin-Arg motif associated with the cleavage of mitochondrial targeting sequence during the maturation and translocation of mitochondrial proteins42,43.
Previously, Hofsetz and co-workers43 reported that N-terminal peptides generated by proteolytic cleavage immediately after an Arg residue in specific mitochondrial proteins were highly abundant in CLPP-expressing mice, echoing our own finding for novel N-termini that accumulated in TR-27-treated WT CLPP-expressing cells. Furthermore, the N-terminal peptides derived from 34 mitochondrial proteins identified by Hofsetz et al. as potential ClpXP substrates were also detected in our HYTANE experiment as showing significant changes in abundance in TR-27-treated WT cells; ten of which (AK2, CPT2, DNAJA3, HINT2, HSPA9, IVD, MRPL4, MRPL49, NDUFV2 and TOMM40) correspond to novel N-termini. These commonalities likely reflect the molecular features and mechanical aspects of the proteolytic process that remain unchanged in both ClpXP and the TR-27-activated ClpP.
Overall, our results provided the structural and mechanistic bases for the anticancer properties of the TR compounds and their potential use in the clinic either as a single agent or in combination with other targeted agents.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Walid A. Houry (walid.houry@utoronto.ca).
Materials availability
This study did not generate new unique reagents.
Data and code availability
X-ray diffraction data have been deposited at the RCSB Protein Data Bank, while the HYTANE mass spectrometry data have been deposited at the MassIVE repository. All data are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-HsClpP | Abcam | RRID:AB_124822 |
| Rabbit monoclonal anti-HsClpX | Abcam | RRID:AB_168338 |
| Rabbit monoclonal anti-Mcl1 | Cell Signaling Technology | RRID:AB_2799149 |
| Rabbit monoclonal anti-Caspase 8 | Cell Signaling Technology | RRID:AB_10545768 |
| Rabbit monoclonal anti-Caspase 9 | Cell Signaling Technology | RRID:AB_2068621 |
| Rabbit polyclonal anti-TFAM | Proteintech | RRID:AB_11182588 |
| Rabbit polyclonal anti-Grp75 | Proteintech | RRID:AB_2120458 |
| Goat monoclonal anti-Caspase 3 | R&D Biosystems | RRID:AB_354518 |
| Mouse monoclonal anti-GAPDH | Abcam | RRID:AB_8245 |
| HRP-conjugated goat anti-rabbit IgG | BioRad | RRID:AB_11125142 |
| HRP-conjugated goat anti-mouse IgG | BioRad | RRID:AB_11125547 |
| HRP-conjugated rabbit anti-goat IgG | Sigma-Aldrich | RRID:AB_258242 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium acetate, ACS Reagent, ≥ 99.0% | Sigma-Aldrich | Cat#241245 |
| PEG 4,000, powder | Sigma-Aldrich | Cat#8.17006 |
| TR-27 | reference19 | N/A |
| TR-57 | reference19 | N/A |
| TR-65 | reference19 | N/A |
| TR-107 | This paper | N/A |
| TR-133 | This paper | N/A |
| ADEP-14 | reference6 | N/A |
| Doxorubicin (hydrochloride) | Cayman Chemical Co. | Cat#15007 |
| Sulforhodamine B (SRB) | Sigma-Aldrich | Cat#230162 |
| jetPRIME Transfection Reagent | Polyplus Transfection | Cat#114–07 |
| jetPRIME Transfection Buffer | Polyplus Transfection | Cat#712–60 |
| Reprosil-Pur 120 C18-AQ, 1.9 μm | Dr. Maisch | Cat#r119.aq. |
| Casein-FITC | Sigma-Aldrich | Cat# C3777 |
| Untagged human mitochondrial ClpP | This paper | N/A |
| Yeast SUMO protease | reference52 | N/A |
| Critical Commercial Assays | ||
| PureLink Quick Plasmid Miniprep Kit | Thermo-Fisher Scientific | Cat#K210011 |
| Pierce BCA Protein Assay Kit | Thermo-Fisher Scientific | Cat#23225 |
| Deposited Data | ||
| Structure of human ClpP complex with ADEP-28 | Protein Data Bank reference6 | 6BBA |
| Structure of human ClpP complex with ONC201 | Protein Data Bank reference8 | 6DL7 |
| Structure of human ClpP complex with ZG111 | Protein Data Bank reference34 | 7VP9 |
| Structure of human ClpP Y118A mutant complex with D9 | Protein Data Bank reference7 | 6H23 |
| Structure of human ClpP complex with TR-27 | Protein Data Bank (This paper) | 7UVM |
| Structure of human ClpP complex with TR-57 | Protein Data Bank (This paper) | 7UVN |
| Structure of human ClpP complex with TR-65 | Protein Data Bank (This paper) | 7UVR |
| Structure of human ClpP complex with TR-107 | Protein Data Bank (This paper) | 7UVU |
| Structure of human ClpP complex with TR-133 | Protein Data Bank (This paper) | 7UW0 |
| HYTANE data | MassIVE repository (This paper) | MSV000089642 |
| Experimental Models: Cell Lines | ||
| HEK293 T-REx | Gift from A. Trifunovic | RRID:CVCL_U427 |
| HEK293 T-RExCLPP−/− | reference53 | N/A |
| HEK293 T-RExCLPX−/− | reference6 | N/A |
| MDA-MB-231 | Gift from L. Attisano | RRID:CVCL_0062 |
| MDA-MB-231 CLPP−/− | This paper | N/A |
| MDA-MB-231 CLPX−/− | This paper | N/A |
| Experimental Models: Organisms / Strains | ||
| Escherichia coli DH5α | Lab stock | NCBI:txid668369 |
| Escherichia coli BL21(DE3) ΔclpP::cat (SG1146) | reference54 | N/A |
| Oligonucleotides | ||
| CLPP Exon 1 pX330 F: CACCGAAGCCGACCGGGGCGTGCGG |
This paper | N/A |
| CLPP Exon 1 pX330 R: AAACCCGCACGCCCCGGTCGGCTTC |
This paper | N/A |
| CLPP Exon 1 ssODN: GCATGACGCCACCCGGGCCCCCCCTACCAATATTCATTATTATCACCTTCCGCACGCCCCGGTCGGCTTCCGTCCGATGGCGGAACTACAGCTTCCGGCG |
This paper | N/A |
| CLPX Exon1 pX330 F: CACCGCGGTGCTTGTACTTGCGGCG |
reference6 | N/A |
| CLPX Exon1 pX330 R: AAACCGCCGCAAGTACAAGCACCGC |
reference6 | N/A |
| CLPX Exon1 ssODN: GGCCTCGCGGAGATGCCCAGCTGCGGTGCTTGTACTTGCGGCGCGGCGTAGGTCCGGCTCATCACCTCCTCACTCGCCTCCGCGCAGAGA |
reference6 | N/A |
| Recombinant DNA | ||
| pX330 | reference48 | RRID:Addgene_42230 |
| pX330-CLPP KO | This paper | N/A |
| pX330-CLPX KO | reference6 | N/A |
| pETSUMO2-CLPP(-MTS) | reference6 | N/A |
| pETSUMO2-TFAM(-MTS) | This paper | N/A |
| Software and Algorithms | ||
| Proteome Discoverer 2.2 | Thermo-Fisher Scientific | RRID:SCR_014477 |
| WebLogo 2.8.2 | reference44 | RRID:SCR_010236 |
| AlphaFold | reference39 | N/A |
| NIS-Elements Basic Research Software | Nikon | RRID:SCR_002776 |
| XDS | reference55 | https://xds.mr.mpg.de/ |
| Phenix | reference46 | https://phenix-online.org/ |
| COOT | reference45 | http://www2.mrc-lmb.cam.ac.uk/Personal/pemsley/coot |
| PISA | reference47 | https://www.ebi.ac.uk/pdbe/pisa/ |
| The PyMOL Molecular Graphics System | Schrödinger, LLC | https://pymol.org/2/ |
| Other | ||
| BbsI | New England Biolabs | Cat#R0539S |
| FastAP | Thermo-Fisher Scientific | Cat#EF0651 |
| T4 Polynucleotide Kinase | New England Biolabs | Cat#M0201S |
| T4 DNA Ligase | New England Biolabs | Cat#M0202S |
| EnSpire 2300 Multilabel Reader | Perkin-Elmer | Cat#655077 |
| P-2000 Laser-Based Micropipette Puller | Sutter Instrument Co. | Cat#P-2000 |
| EkspertNanoLC 425 HPLC System | Eksigent Technologies | Cat#Ekspert425 |
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | Thermo-Fisher Scientific | Cat#IQLAAEGAAPF ADBMBHQ |
| Microcon-10kDa Ultracel YM-10 filter unit | Millipore | Cat#Z648078 |
| C18 Stage Tips | Supelco | Cat#66883-U |
| Eclipse 80i fluorescence microscope | Nikon | RRID:SCR_015572 |
| X-Cite Series 120Q excitation light source | Excelitas Technologies | https://www.excelitas.com/product/x-cite-120q |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial cell cultures
The strains of Escherichia coli used for DNA propagation and protein expression (see Key Resources Table for details) were grown in Luria-Bertani Broth (LB; 10 g/L bio-tryptone + 5 g/L yeast extract + 10 g/L NaCl) supplemented with the appropriate antibiotics, unless otherwise stated. For DNA propagation, cells were grown at 37 °C with shaking. For protein expression, cells were grown in pre-cultures for 16–18 hours at 37 °C with shaking. Cells in protein expression cultures were also grown at 37 °C with shaking until induction of protein expression with 1 mM IPTG. The cultures were then maintained at 37 °C to express the desired protein for 4 hours.
Mammalian cell cultures
All mammalian cell lines used in this study are female (see Key Resources Table for details). HEK293 T-REx cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 100 U/mL penicillin-streptomycin, unless stated otherwise. MDA-MB-231 cell lines were maintained in Dulbecco’s modified Eagle’s medium/Ham’s F-12 50/50 mix (DMEM/F-12) supplemented the same way. Cells were grown and maintained in standard tissue culture plates or tissue culture flasks with ventilated caps. For DNA transfection, cells were kept in the same media but without any antibiotics at least 24 hours prior to the procedure. All cells were passaged by standard trypsinization procedures at least three times before use in any experiments.
For long-term cryogenic storage, cells were first trypsinized and re-suspended in media, followed by gradual addition of 80% FBS + 20% DMSO to a final 1:1 (v:v) ratio. The cell stocks were then transferred into cryogenic storage vials and stored in foam storage racks at −80 °C for at least 24 hours before long-term storage in a liquid nitrogen storage tank. To recover cells from the frozen stocks, the frozen cells were thawed at 37 °C and re-suspended in media at 9x the frozen stock’s volume. The media was applied gradually to the cells to minimize osmotic shock. Cells were then collected by centrifugation and re-suspended in fresh media after which, they were transferred to tissue culture plates or ventilated tissue culture flasks for growth and maintenance as described.
METHOD DETAILS
All reagents used are listed in the Key Resources Table.
Synthesis of TR compounds
TR-27 and TR-65 syntheses are described in US 11,091,483. TR-57 synthesis is described in US 11,091,477. TR-107 and TR-133 syntheses are described in PCT/US2020/019944. NMR, HPLC and LC-MS were used to evaluate isolated compounds and to evaluate reaction mixtures. For LC-MS, a Symmetry C18, 5 μm, 4.6 × 50 mm column was used with water and MeCN as the two solvents. Typically, a linear gradient from time 0 (10% MeCN) to time 4.5 min (95% MeCN) was employed. The flow rate was 1.7 mL/min with absorbance monitored at 254 nm.
Synthesis of TR-27 and TR-65 (Figure S6). A 50 mL three necked flask was charged with compound 1 (1 g, 5.2 mmol), 1,2 dichloroethane (10 mL), and DIEA (665 mg, 5.2 mmol). The mixture was stirred for 15 minutes at 25 °C. Compound 2A (5.2 mmol) was added, followed by NaBH(OAc)3 (6.7 mmol). The mixture was stirred for 2 h at 25 °C. LC-MS analysis of the reaction mixture confirmed that the reaction was complete. The reaction was quenched with ice water (20 mL), extracted twice with dichloromethane 20 mL. The combined organic phase was washed with saturated aqueous NaHCO3, (2 × 25 mL) and concentrated in vacuo. Compound 3A was obtained as an off-white solid. The product was used for the next steps without further purification. Intermediate 3B was prepared in a similar fashion.
Compound 4 (6.1 g, 59.8 mmol) was dissolved in methanol (70 mL) and CH3I (12.7 g, 89.7 mmol) was added dropwise at 25 °C. The reaction mixture was heated to reflux for 30 minutes, and the solvent was removed under vacuum. The residue was suspended in MTBE (50 mL) and filtered. The resulting solid was dried under vacuum to afford compound 5 (5.8 gr yield 83%) as white solid. Compound 5 (230 mg, 2 mmol) and (4-chlorophenyl)methanamine (compound 6, 590 mg, 4.2 mmol) were dissolved in dioxane (5 mL). The reaction mixture was heated to reflux for 12 hours. Analysis of the reaction mixture by LC-MS confirmed that the reaction was complete. The solvent was removed in vacuo, and the residue was suspended with toluene for 12 hours. The suspension was filtered, and the filtered cake was dried under vacuum to afford compound 7.
A 10 mL three necked flask was charged with compound 3A (109 mg, 0.4 mmol), compound 7 (85 mg, 0.4 mmol), methanol (3 mL) and MeONa (65 mg, 1.2 mmol). The mixture was refluxed for 15 hours. Analysis of the reaction mixture by LC-MS confirmed that the reaction was complete. The reaction was cooled to room temperature. Half of the solvent was removed under vacuum, and water (2 mL) was added drop wise. A light tan colored solid precipitated, which was isolated by filtration and washed with water. The resulting solid was dried under vacuum to afford TR-27 (42 mg, yield 25%). TR-65 was prepared in a similar fashion using compound 3B.
TR-27, 7-[(4-chlorophenyl)methyl]-11-[(3-ethynylphenyl)-2,5,7,11-tetraazatricyclo[7.4.0.02,6]trideca-1(9),5-dien-8-one: Yield 30% (last step); 1HNMR (400 MHz, DMSO-d6) δ 2.51 (s, 2H), 2.62 (s, 2H), 3.02 (s, 2H), 3.61 (s, 2H), 3.7 (t, J = 8.8Hz, 2H), 3.94 (d, J = 9.2Hz, 2H), 4.19 (s, 1H), 4.88 (s, 2H), 7.3–7.42 (m, 8H); LC-MS: m/z = 430.8 (M+1).
TR-65, 3-((4-(4-Chlorobenzyl)-5-oxo-1,2,4,5,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-7(6H)-yl)methyl)benzonitrile: Yield 25% (last step); 1HNMR (400MHz, CDCl3) δ 2.49 (t, J = 5.6Hz, 2H), 2.68 (t, J = 5.6Hz, 2H), 3.26 (s, 2H), 3.68 (s, 2H), 3.91 (s, 4H), 5.0 (s, 2H), 7.25 (t, J = 5.6Hz, 2H), 7.42 (t, J = 8Hz, 3H), 7.55–7.57 (d, J = 8Hz, 2H), 7.66 (s, 1H); LC-MS: m/z = 432.2 (M+1).
Synthesis of TR-57 (Figure S7A). A mixture of compound 3B (8.55g. 20.2 mmol), sodium carbonate (1.76 g, 17 mmol) and ammonia solution (4.5 mL, 25%) in ethanol (70 ml) was heated at 70 °C for 5 h. The solution was concentrated, extracted with dichloromethane (2 × 200 mL) and washed with brine. The extracts were dried over Na2SO4 and evaporated under reduced pressure to give 7.88 g of compound 8 which was directly used for the next steps.
To a solution of compound 8 (1.7 g, 6.9 mmol) in 20 mL toluene was added 4-chlorobenzylisocyanate 9 (1.1 g, 7.5 mmol) and triethylamine (1.1g, 10.4 mmol). The solution was heated to 80 °C for 8 h. The reaction solution was cooled to rt and concentrated in vacuo forming a white solid. The mixture was filtered and the resulting solid dissolved in MeOH (20 ml). NaOMe (340 mg, 6.3 mmol) was added, and the mixture was refluxed overnight. Then ca 10–15 mL of methanol were removed, and the precipitate was filtered. The desired product, compound 10, was obtained as pale-yellow solid (0.8 g, yield 37%).
To a solution of compound 10 (200 mg, 0.49 mmol) in DMF (2 mL) was added potassium carbonate (150 mg) and methyl iodide (120 mg, 0.84 mmol). The mixture was heated at 100 °C for 12 h. The water was added, and the solution was extracted with EtOAc (5 ml × 3). The combined extracts were washed with brine 3 times. The final product, TR-57 was obtained by preparative TLC (40 mg, yield 20 %).
TR-57, 3-((3-(4-chlorobenzyl)-1-methyl-2,4-dioxo-1,2,3,4,7,8-hexahydropyrido[4,3-d]pyrimidin-6(5H)-yl)methyl)benzonitrile: Yield 20% (last step); 1HNMR (400MHz, CDCl3) δ 2.65 (s, 2H), 2.73 (s, 2H), 3.34 (s, 2H), 3.37 (s, 3H), 3.71 (s, 2H), 5.08 (s, 2H), 7.26 (t, J = 8Hz, 2H), 7.4–7.46 (m, 3H), 7.57–7.59 (d, J = 8Hz, 2H), 7.67 (s, 1H); LC-MS: m/z = 421.1 (M+1).
Synthesis of TR-107 and TR-133 (Figure S7B). A 10 mL three necked flask was charged with 3B (109 mg, 0.4 mmol), formamidine acetate (42 mg, 0.4 mmol), methanol (3 mL) and K2CO3 (130 mg, 1.2 mmol). The mixture was refluxed for 12 hours. Evaluation of the reaction mixture by LC-MS confirmed that the reaction was complete. The vessel was cooled to room temperature. Half of the solvent was removed under vacuum. Water (2 mL) was added drop wise while stirring resulting in an off-white-colored precipitant that was isolated by filtration and washed with water. The resulting solid was dried under vacuum to afford compound 11.
A 10 mL three necked flask was charged with compound 11 (107 mg, 0.4 mmol), 1-bromomethyl-4-chloro-benzene (82 mg, 0.4 mmol), acetonitrile (3 mL) and Cs2CO3 (390 mg, 1.2 mmol). The mixture was refluxed for 4 h. Evaluation of the reaction mixture by LC-MS confirmed that the reaction was complete. The reaction vessel was cooled down to room temperature. Half of the solvent was removed under vacuum, and water (2 mL) was added drop wise while stirring resulting in a white solid. The solid was isolated by filtration, washed with water, and dried under vacuum to afford TR-107. This procedure was used to prepare TR-133 using 1-bromomethyl-4-bromo-benzene as the alkylating agent.
TR-107, 3-([3-[(4-chlorophenyl)methyl]-4-oxo-3H,4H,5H,6H,7H,8H-pyrido[4,3-d]pyrimidin-6-yl]methylbenzonitrile: 1HNMR (400MHz, CDCl3) δ 3.06 (s, 2H), 3.42 (s, 2H), 3.92 (s, 2H), 4.35 (s, 2H), 5.03 (s, 2H), 7.24 (s, 2H), 7.33–7.35 (d, J=8Hz, 2H), 7.6 (t, J=8Hz, 1H), 7.72–7.81 (m, 3H), 8.14 (s, 1H); LC-MS: m/z = 390.9(M+1).
TR-133, 3-([3-[(4-bromophenyl)methyl]-4-oxo-3H,4H,5H,6H,7H,8H-pyrido[4,3-d]pyrimidin-6-yl]methyl)benzonitrile: 1HNMR (400MHz, CDCl3) δ 2.73–2.79 (m, 4H), 3.46 (s, 2H), 3.75 (s, 2H), 5.03 (s, 2H), 7.21–7.23 (d, 2H), 7.43–7.5 (m, 3H), 7.59 (t, J=8.8Hz, 2H), 7.7 (s, 1H), 8.06 (s, 1H); LC-MS: m/z = 434.1(M+2).
Casein-FITC degradation assays
Recombinant human ClpP was expressed and purified as previously described6. ClpP protease activity was assessed by measuring the degradation rate of bovine milk casein labeled with fluorescein isothiocyanate (casein-FITC) in the presence of TR-27, TR-57, TR-65, TR-107, TR-133, and ONC201. Reactions were conducted in buffer containing 20 mM HEPES, pH 8.0, 100 mM KCl, and 1% DMSO. Monomeric ClpP concentrations varied from 0–10 μM in the presence of 0–80 μM of the compounds. Reactions were preincubated for 10 min at 37 °C before initiating the reactions with addition of 2.5 μM casein-FITC in a 96-well black flat-bottom plate. Fluorescence (485 nm excitation, 535 nm emission) was monitored for 100 min at 1 min intervals on an EnSpire Multilabel Plate Reader (PerkinElmer). Kinetic analysis was then performed by determining the initial velocities from data collected over three independent experiments. Nonlinear regression analysis was performed on initial velocity data using the Hill equation:
V0 is the initial velocity, Vmax is the maximal velocity, h is the Hill coefficient, and the EC50 is the half maximal effective concentration to activate ClpP. Using mass conservation, it can be readily shown that the relationship between EC50 and Kd, app is:
Where n represents the number of bound ligands and [ClpP]T is the total ClpP concentration. The above equation was used in the fits for Fig. 2C and to derive Kd, app given in Table 1.
Surface plasmon resonance measurements
SPR measurements were performed on a Biacore X100 (Cytiva Life Sciences). Experiments were carried out at 25 °C in buffer containing 25 mM sodium phosphate, pH 7.5, 200 mM KCl, 0.05% Tween-20, and 0.004% DMSO. Human ClpP was immobilized to flowcell 2 of a CM5 chip (Cytiva Life Sciences) using the amine immobilization method in the Biacore X100 Control Software. A pulse of NHS/EDC activation solution was followed by a 420 sec pulse of 250 μg/mL human ClpP in 10 mM sodium acetate buffer, pH 5.0. A subsequent injection of ethanolamine blocked off any remaining active groups on the chip surface. 8300 RU of human ClpP was immobilized on the CM5 surface using this procedure. Flowcell 1, used as the reference, was unmodified. For binding experiments, a concentrated dilution series of each compound was made using DMSO as dilutant. The compounds were diluted into buffer containing no DMSO so that the final concentration of DMSO was 0.004%. Each concentration of compound was injected in triplicate. Data was analyzed using Biacore X100 Evaluation Software. A sensorgram of DMSO diluted into buffer (i.e., no compound) was subtracted from each sensorgram. Steady states were calculated from the last 15 sec before the end of injection. Binding curves were fit to a 1:1 Langmuir binding model.
Expression and Purification of TFAM
The expression of TFAM lacking the MTS in E. coli BL21(DE3) SG1146 was performed as follows. Cells carrying pETSUMO2-TFAM(-MTS), a modified pETSUMO plasmid (Invitrogen) for expressing His6-His6-SUMO-TFAM(-MTS), were plated on LB containing 30 μg/mL kanamycin and grown into colonies at 37 °C overnight. A pre-culture was prepared by inoculating a single colony in 20 mL of LB supplemented with 30 μg/mL kanamycin and grown at 37 °C overnight. The pre-culture was then used to inoculate 2 L of LB containing 30 μg/mL kanamycin, and cells were grown at 37 °C until an optical density at 600 nm of 0.5. 1 mM of IPTG was then added to induce protein expression at 37 °C for 4 hours with shaking. Cells were spun down at 4,500 rpm for 25 minutes. The pellets were frozen until purification.
The frozen pellet from the 2-L culture was resuspended in 17 mL of 10 mM imidazole lysis buffer (25 mM Tris-HCl, pH 7.5, 0.5 M NaCl, and 10 % glycerol). Cells were then lysed using the French Press and insoluble cell debris were subsequently removed by spinning down at 15,000 rpm for 35 minutes at 4 °C. 3.5 mL of Ni-NTA beads were equilibrated with 10 column volumes (CVs) of water and then 10 CVs of 10 mM imidazole lysis buffer. The cell lysate was incubated with the equilibrated beads for 1 h at 4 °C while rocking; the flowthrough was then discarded. The column was then washed with 5 CVs of 50 mM imidazole lysis buffer, 5 CVs of 100 mM imidazole lysis buffer, 5 CVs of 150 mM imidazole lysis buffer, 5 CVs of 200 mM imidazole lysis buffer, and 5 CVs of 400 mM imidazole lysis buffer. The fractions containing eluted TFAM were dialyzed for 15 h in dialysis buffer (25 mM TrisHCl, pH 7.5, 0.1 M NaCl, and 10 % glycerol). The dialysis buffer also contained SUMO protease to cleave the His6-His6-SUMO tag off TFAM(-MTS). After dialysis, the products were passed through a second column of Ni-NTA to remove the His6-His6-SUMO tags. The TFAM(-MTS)-containing fractions were analyzed with SDS-PAGE to determine the presence and purity of TFAM(-MTS). The purified protein was then concentrated using Amicon ultra-15 centrifugal filter units with a molecular weight cut-off of 10,000 Da. The concentrated TFAM(-MTS) was then flash frozen and stored at −80 °C.
In vitro degradation assays using TFAM
The buffer used in the in vitro degradation assay consisted of 25 mM HEPES, pH 7.5, 35 mM KCl, 25 mM MgCl2, 0.03 % Tween-20 (v:v), 10 % glycerol (v:v), 1 mM DTT, and 16 mM creatine phosphate. The degradation assay also contained 13.5 μM TFAM, 15.5 μM ClpP, and 20 μM TR-65. The reaction was carried out in an Eppendorf tube incubated at 37 °C and shaking at 300 rpm. At time points 0 h, 1 h, 2 h, 3 h, 4 h, and 5 h, samples of 9 μL were collected after tubes were spun down for 3 sec. A 9 μL sample was mixed with 3.5 μL of 4xSDS-PAGE sample buffer containing β-mercaptoethanol, then heated at 95 °C for 5 min. Samples were loaded onto a 0.75 mm 15 % SDS-PAGE gel for 60 min at 200 V and stained with Coomassie blue.
Sample preparation for Edman Degradation
The buffer used in the in vitro degradation assay consisted of 25 mM HEPES, pH 7.5, 35 mM KCl, 25 mM MgCl2, 0.03 % Tween-20 (v:v), 10 % glycerol (v:v), 1 mM DTT, and 16 mM creatine phosphate. The degradation assay also contained 65.9 μM TFAM, 43.7 μM ClpP, and 68 μM TR-65. The reaction was carried out in an Eppendorf tube incubated at 37 °C with shaking at 300 rpm. At time points 0 h, 2 h, and 4 h, samples of 100 μL were collected after tubes were spun down for 3 sec. A 100 μL sample was mixed with 33 μL of 4x SDS-PAGE sample buffer containing β-mercaptoethanol, then heated at 95 °C for 5 min. Samples were loaded on a 1.5 mm 15 % SDS-PAGE gel for 60 min at 200 V. The gel was transferred onto a 0.2 mm PVDF membrane. Blot was then ponceau S stained and the two desired bands were cut using a razor blade. The two bands were de-stained using PBS containing 5% Tween-20 and rinsed using deionized water. Bands were air-dried for 2 hours and then individually placed in Eppendorf tubes for shipping. The samples were sent to Creative Proteomics (https://www.creative-proteomics.com/) for N-terminal sequencing using Edman degradation.
Crystallization and structure determination of human ClpP-TR complexes
Human ClpP was concentrated to 10 mg/mL and mixed separately with each TR compound at a protein-ligand concentration ratio of 1:3. The mixtures were then incubated at 37 °C for 1 hr. Crystals of human ClpP-TR complexes were grown using the sitting drop vapor diffusion method by mixing 5 μL of protein-ligand mixture and 5 μL of the precipitant solution, composed of 0.1 M sodium acetate, pH 4.6–5.2 and 5% PEG 4,000. Crystals grew to maximum dimensions within a month at 4 °C. Prior to X-ray data collection, crystals were frozen in liquid nitrogen using a cryoprotectant composed of the precipitant solution supplemented with 20% glycerol. Diffraction data were collected at the CMCF-BM Line of the Canadian Light Source using synchrotron X-rays at 0.98 Å wavelength and were reduced using XDS. Structures of human ClpP-TR complexes were determined by molecular replacement using the structure of human ClpP without bound ligands (PDB id 6DL7) as search model. Initial structure refinement was performed in Phenix with simulated annealing and coordinate shaking to remove model bias. Subsequent model building and refinement steps were performed in COOT and Phenix, with individual coordinate, occupancy, and B-factor parameter optimization45,46. Data collection and refinement statistics are shown in Table S1. Molecular surface areas were calculated using the coordinates of refined models in the PISA server available at https://www.ebi.ac.uk/msd-srv/protint/cgi-bin/piserver47.
Mammalian cell culture maintenance
All mammalian cell lines used in this study are female (see Key Resources Table for details). HEK293 T-REx and HEK293 T-REx CLPP−/− were gifts from Professor Aleksandra Trifunovic (CECAD, Cologne, Germany). HEK293 T-REx CLPX−/− was generated in our previous study6. MDA-MB-231 was provided by Professor Liliana Attisano (University of Toronto, Toronto, Canada). Both MDA-MB-231 CLPP−/− and MDA-MB-231 CLPX−/− were generated from MDAMB-231 via CRISPR/Cas9 methodology (see below). All HEK293 T-REx cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 100 U/mL penicillin-streptomycin. Similarly, all MDA-MB-231 cell lines were maintained in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) supplemented in the same manner. Cells were grown in standard tissue culture plates or tissue culture flasks with ventilated caps, at 37 °C under a 5% CO2 atmosphere with moisture. Cells were passaged upon reaching 80–90% confluence. All cells were passaged at least three times before use in any experiments. For DNA transfection, cells were kept in their respective growth media without antibiotics 24 hr prior to the procedure.
For long-term cryogenic storage, cells were trypsinized and resuspended in their respective growth medium at 1 mL or smaller volume (per culture on a 100-mm plate). Next, an equal volume of 80% FBS + 20% DMSO was added gradually with periodic mixing to a final 1:1 (v:v) ratio. The cell stocks were stored into cryogenic vials and kept in foam boxes at −80 °C for at least 24 hr before transferring to a standard liquid nitrogen storage tank for long term storage. To reactivate the frozen cells, cell stocks were thawed at 37 °C and resuspended in growth medium at 9x the stock’s volume. Growth medium was added gradually with periodic mixing to minimize osmotic shock. Cells were collected by centrifugation, resuspended in fresh growth medium and seeded in a tissue culture plate or vented flask for growth and maintenance as described above.
Generation of clonal CLPP and CLPX null mutants
The plasmids required for knocking out CLPP and CLPX were generated using the SpCas9- and gRNA-expressing vector pX330 (pX330-U6-Chimeric_BB-CBh-hSpCas9) – a gift from Feng Zhang (Addgene plasmid #42230; http://n2t.net/addgene:42230; RRID:Addgene_42230). DNA oligonucleotides for expressing gRNA to target exon 1 of CLPP and CLPX, respectively, were designed following the protocol described by Cong et al. with minor changes48. The oligonucleotides are listed in the Key Resources Table. Briefly, pX330 was digested with the restriction enzyme BbsI (New England Biolabs). Next, each pair of DNA oligonucleotides were phosphorylated at the 5′-end with T4 polynucleotide kinase (New England Biolabs) and annealed by heating and slow-cooling (from 95 °C to 25 °C, at 5 °C/min) in a PCR thermocycler. The annealed oligonucleotides were then ligated into the BbsI-digested pX330 with T4 DNA ligase (New England Biolabs), and the completed pX330-CLPP KO and pX330-CLPX KO were transformed into competent DH5α, followed by selection on LB-agar plates containing 100 μg/mL ampicillin. The plasmids were propagated and extracted using the PureLink Quick Plasmid Miniprep Kit (Thermo-Fisher Scientific).
Null mutants of CLPP and CLPX were generated in MDA-MB-231 cells by CRISPR/Cas9 using the SpCas9- and gRNA-expressing plasmids pX330-ΔCLPP and pX330-ΔCLPX, respectively, via the method that we have previously established6. Briefly, MDA-MB-231 cells were cultured in a 6-well tissue culture plate until reaching 60–70% confluence. All antibiotics were excluded from the growth medium at least 24 hr prior to DNA transfection was performed. Next, cells were co-transfected with pX330-CLPP KO and the oligonucleotide CLPP Exon 1 ssODN, or pX330-CLPX KO and CLPX Exon 1 ssODN, using the jetPRIME transfection reagent and buffer (Polyplus Transfection). Cells were incubated in the presence of DNA-liposome complexes for up to 5 hr, at which time the growth medium (no antibiotics) was replaced to allow cells to recuperate and propagate. Expression of ClpP and ClpX was monitored by western blotting, and the transfection protocol was repeated up to three times to ensure CLPP or CLPX was knocked out in a sufficient number of cells.
Clonal populations of CLPP and CLPX null mutants were established by isolating single cells followed by their expansion. First, cells were trypsinized and resuspended at ~10 cells/mL and were seeded onto 96-well tissue culture plates at 150 μL per well (~1.5 cell per well). Wells containing exactly one cell were then identified. After sufficient cellular propagation, the clonal populations were passaged multiple times onto successively larger plates for expansion. Complete abolishment of CLPP and CLPX expression was confirmed for each clonal population by western blotting. All antibodies used are listed in Key Resources Table.
One-dose NCI-60 human tumor cell line screens for TR-57 and TR-107
The one-dose NCI-60 screens for TR-57 and TR-107 were performed by the Division of Cancer Treatment & Diagnosis (DCTD) at the National Cancer Institute (NIH, USA) following the protocol described at https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm. Briefly, each of the 60 cell lines in 9 cancer panels were seeded at 5,000 to 40,000 cells/well in 96-well tissue culture plates using RPMI 1640 media supplemented with 5% fetal bovine serum (FBS) and 2 mM L-glutamine. Cells were grown for 24 hours at 37 °C under an atmosphere of 5% CO2 and 100% relative moisture. Two sets of replicates were prepared: one for quantifying cell growth before drug treatment and one for after. For cultures that are to be drug-treated, TR-57 or TR-107 was applied at a final concentration of 10 μM and cells were incubated with or without drugs for 48 hours. To quantify cell growth, adherent cells were fixed by the addition of cold trichloroacetic acid (TCA) at 10% (v:v) final concentration and incubated at 4 °C for 60 minutes. Non-adherent cells were first settled by light centrifugation and fixed by gentle application of cold TCA at 16% final concentration. After which, the supernatant was discarded, and the plates were rinsed five times with water and air-dried. To stain the fixed cells, 0.4% (w:v) sulforhodamine B (SRB) solution (in 1% acetic acid) was applied at 100 μL per well, and plates were incubated at room temperature for 10 minutes. Unbound SRB was then removed, and the plates were rinsed five times with 1% acetic acid and then air-dried. Bound SRB was extracted with 10 mM trizma base at equal volume across all wells and the absorbance at 515 nm (A515) was measured with an automatic plate reader. The % cell growth for each cell line was calculated using the following equations:
if [A515(48 hr)]with drug ≥ A515(0 hr) or
if [A515(48 hr)]with drug < A515(0 hr)
Growth inhibition profiling
The growth inhibition of TR compounds was assessed using WT, CLPP−/− and CLPX−/− of HEK293 T-REx and MDA-MB-231. Cells were seeded onto 96-well plates at 1.5 × 103 cells via 50 μL per well and grown for 24 hr. Next, serial dilutions were prepared using the appropriate growth medium for each compound at twice the final concentration and were applied at equal volume (i.e., 50 μL per well) to designated wells of the 96-well plates, yielding a final volume at 100 μL per well with the compounds at the required concentration. Final DMSO concentration was kept at 0.25% (v:v) in all wells. Cells were incubated in the presence of compounds for 72 hr.
Adherent live cells were first fixed by adding trichloroacetic acid (TCA) (Sigma-Aldrich) at 10% (w:v), followed by incubation at 4 °C for approximately 2 hr. Fixed cells were then washed with ddH2O for 4–5 rounds and air-dried, followed by staining of fixed cellular material with 0.4% (w:v) sulforhodamine B (SRB) (Sigma-Aldrich) dissolved in 1% acetic acid (v:v) for 30 min. Stained cells were washed for 4–5 rounds with 1% acetic acid and were again air-dried. To extract the SRB stain retained by the fixed cellular material, 10 mM Tris base was applied at 150 μL per well followed by gentle agitation of the plates. The extracted SRB in each well was quantified by measuring absorbance at 510 nm using the EnSpire 2300 Multilabel Reader (Perkin-Elmer). A510 is linearly proportional to the amount of fixed cellular material in each well that in turn corresponds directly to cell viability.
Western Blotting
WT, CLPP−/− and CLPX−/− cells of HEK293 T-REx and MDA-MB-231 were grown as described above and harvested by standard trypsinization and centrifugation. For samples used in validation of HYTANE results, the required MDA-MB-231 WT and CLPP−/− were grown in the presence of 1 μM TR-27 or DMSO for 24 hours prior to harvesting. Alternatively, the same cell lines were grown in the presence of 2 μM doxorubicin hydrochloride (Cayman Chemical Co.) or DMSO for 72 hours for use as positive controls of apoptotic induction. For confirmation of reduction in expression of Grp75 and TFAM, cells were grown in the presence of 1 μM TR-27 or 6 μM ADEP-14 for 24 hours. Cell lysates were prepared by re-suspending cell pellets in Dulbecco’s PBS and sonication. Protein concentration of the lysates was measured and equalized by Dulbecco’s PBS addition. Proteins in the cell lysates were segregated by SDS-PAGE and electro-transferred to Immobilin-P PVDF membranes (EMD Millipore) for western blotting following the standard protocol. All blots were imaged by film. Primary antibodies for ClpP, ClpX and GAPDH were purchased from Abcam. Primary antibodies for Mcl1, Caspase-8 and Caspase-9 were purchased from Cell Signaling Technologies. The primary antibody for Caspase-3 was purchased from R&D Systems. Primary antibodies for TFAM and Grp75 were purchased from Proteintech. HRP-conjugated goat anti-rabbit IgG (for ClpP, ClpX, Mcl1, Caspase-8, Caspase-9, TFAM and Grp75) and HRP-conjugated goat anti-mouse IgG (for GAPDH) were purchased from BioRad. HRP-conjugated rabbit anti-goat IgG (for Caspase-3) was purchased from Sigma-Aldrich.
HYTANE sample preparation and processing
MDA-MB-231 WT and CLPP−/− cells were grown to approximately 70% confluence, at which point 1 μM (final concentration) of TR-27 or DMSO was applied to the designated cultures, followed by 24-hr incubation. Cells were then trypsinized, washed with Dulbecco’s phosphate-buffered saline (PBS) and cell pellets were flash-frozen and stored at −80 °C until use.
For HYTANE sample processing, cell pellets were lysed with 6 M guanidine hydrochloride containing 100 mM triethylammonium bicarbonate (TEAB) at pH 8.0. Lysates (kept on ice) were sonicated for 15 sec (5 sec on, 3 sec off for 3 cycles) at 30% amplitude on a sonicator with 1/8″ microtip. Protein concentration was determined with Pierce BCA Protein Assay Kit (Cat#: 23225, ThermoFisher Scientific). 0.2 mg was diluted in 6 M guanidine hydrochloride containing 100 mM TEAB (pH 8.0) to final protein concentration of 1 mg/mL. Samples were reduced with 5 μL 100 mM DTT at 56 °C for 1 hr. 5 μL of 300 mM acrylamide was then added and samples were incubated at room temperature for 1 hr to protect free sulfhydryls. An additional 5 μL of 100 mM DTT was subsequently added and samples were incubated at 25 °C for 10 min to quench unreacted acrylamide. The quenched sample was centrifuged at 15,000 × g through 10K MWCO filters to remove residual reagents. The filters were then washed with 6 M guanidine hydrochloride + 100 mM TEAB (pH 8.0). The sample was dissolved in 6 M guanidine hydrochloride + 100 mM TEAB (pH 8.0). For dimethyl labeling: (1) 20 μL of 4% (wt%) formaldehyde and 20 μL of 0.6 M NaCNBH3 for light label corresponding to “+TR-27” samples; (2) 20 μL of 4% (wt%) CD2O and 20 μL 0.6 M NaBH3CN for intermediate label corresponding to another ClpP activator (Drug 2) samples that will be described in a future study; (3) 20 μL of 4% (wt%) 13CD2O and 20 μL 0.6 M NaBD3CN, for heavy label corresponding to “+DMSO” control samples, was added and incubated for 2 hr at 37 °C. The filters were centrifuged at 15,000 × g to remove excess reagents, followed by two washes with 6 M guanidine hydrochloride containing 1 M glycine and 50 mM ammonium bicarbonate to quench the remaining residual reagents. Importantly, during the first wash the filter was incubated with the wash solution for 1 hr before it was centrifuged. Next, the filters were washed twice with 50 mM HEPES (pH 8.0). The retained peptides were dissolved in 100 μL of 50 mM HEPES (pH 8.0). 100 μL of 0.04 mg/mL trypsin was then applied and the peptides were digested at 37 °C for 12 hr. The trypsin-digested peptides were collected by centrifugation at 15,000 × g. Filters were washed with 100 μL ddH2O and this was pooled with the peptides eluted prior. To the eluate, 100 μL of ddH2O, 40 μL 0.6 M NaBH3CN (dissolved in 1-propanol) and 1 mL of 10 mg/mL hexadecanal (dissolved in 1-propanol) was added in sequence. This solution was incubated at 50 °C for 8 hr. 20 μL of freshly made NaBH3CN (dissolved in 1-propanol) was then added, and the solution was dried under vacuum at 50 °C to reduce its volume to less than 50 μL. Samples were redissolved in 2% acetonitrile containing 5% formic acid, followed by ultrasonication to disperse any precipitate. The samples were then centrifuged at 12,000 × g for 15 min to allow any precipitant to rise to the top. The supernatant was removed with a needle to a new Eppendorf tube. The collected supernatant was dried under vacuum and then cleaned with a C18 Stage Tip (Supelco). The eluted peptides were dried down and stored at −40 °C until mass spectrometry data acquisition.
Mass Spectrometry (MS) data acquisition for HYTANE samples
For data-dependent acquisition (DDA) LC-MS/MS, digested peptides were analyzed using a nano-HPLC (high-performance liquid chromatography) coupled to MS. One-tenth of each sample of eluted peptides was used for each duplex experiment (light-dimethylated + heavy-dimethylated samples). Coated nano-spray emitters (75 μm × 15 cm) were generated from fused silica capillary tubing, with 75 μm internal diameter, 365 μm outer diameter and 5–8 μm tip opening, using a P-2000 Laser-Based Micropipette Puller (Sutter Instrument Co.) with parameters set as heat: 280, FIL = 0, VEL = 18, DEL = 2000. Nano-spray emitters were packed with C18 reversed-phase material (Reprosil-Pur 120 C18-AQ, 1.9 μm) resuspended in methanol using a pressure injection cell. To begin the MS acquisition process, the prepared sample suspended in 5% formic acid was directly loaded at 400 nL/min for 20 min onto a Nano-spray emitter. Peptides were eluted from the column with an acetonitrile gradient generated by the EkspertNanoLC 425 HPLC System (Eksigent Technologies) and analyzed on the Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo-Fisher Scientific). The gradient was delivered at 200 nL/min from 2.5% acetonitrile + 0.1% formic acid to 35% acetonitrile + 0.1% formic acid using a 90-min linear gradient. This was followed by an 8-min gradient from 35% acetonitrile with 0.1% formic acid to 80% acetonitrile with 0.1% formic acid. Afterwards, an 8-min wash with 80% acetonitrile + 0.1% formic acid was performed followed by equilibration with 2.5% acetonitrile with 0.1% formic acid for 23 min. The complete DDA protocol lasted 150 min. The MS1 scan had an accumulation time of 50 ms within a mass range of 400–1500 Da, using the Orbitrap resolution of 120000, 60% RF lens, AGC target of 125% and 2400 V. This was followed by MS/MS scans for a total cycle time of 3 sec, with an accumulation time of 50 ms and 33% HCD collision energy per scan. Candidate ions that met the required charge state from 1–6 and an AGC target of 400% were isolated using the Orbitrap resolution of 15000. Previously analyzed candidate ions were dynamically excluded for 9 sec.
Raw HYTANE MS data processing via Proteome Discoverer
The collected raw MS data were processed using the Proteome Discoverer 2.2 (Thermo-Fisher Scientific). The default processing workflow PWF_Fusion_Dimethylation_Quan_ITHCD_Sequest_HT_Percolator.pdProcessingWF and the default consensus workflow CWF_Comprehensive_EnhancedAnnotation_LFQ_and_Precursor_Quant.pdConsensusWF were used, with the following modifications. In the processing workflow, protein database used was UP000005640 and PD_Contaminants_2015_5 (EMBL_EBI) in all “Sequest HT” nodes. The “Spectrum Files RC” node was replaced with “Spectrum Files”. There were 2 “Sequest HT” nodes per dimethyl label. In the first “Sequest HT” node, oxidized methionine (+15.995 Da) and the appropriate dimethyl label (+28.031 Da, +32.056 Da, +36.076 Da) on N-terminus was used as dynamic modifications. Propionamide (+71.037 Da) on cysteine was used as a static modification along with the appropriate dimethyl label for light, medium or heavy (+28.031 Da, +32.056 Da or +36.076 Da) on lysine. For the second “Sequest HT” node, oxidized methionine (+15.995 Da) was used as a dynamic modification. Pyroglutamate (−17.027 Da) on glutamine, pyrogluamate (−18.011 Da) on glutamic acid and acetyl (+42.011 Da) on the N-terminus was used as dynamic modifications on the peptide terminus. Propionamide (+71.037 Da) on cysteine was used as a static modification along with the appropriate dimethyl label for light, medium or heavy (+28.031 Da, +32.056 Da or +36.076 Da) on lysine. The N-terminal peptides were subsequently identified, annotated and their abundance quantified, and those that were deemed high confidence hits were selected for further analysis. Data have been deposited as a submission to the MassIVE repository (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and assigned the accession number MSV000089642.
Manual analysis of processed HYTANE MS data and profiling of N-terminomes
The processed HYTANE MS data were manually analyzed as follows (Fig. S4): (1) the abundance score of each identified N-terminal peptide was normalized by the mean abundance score of its corresponding data set to enable comparison across data sets; (2) for each peptide, the mean-normalized abundance scores for “WT + TR-27” (corresponds to TR-27-treated WT cells) were normalized against the corresponding scores for “WT + DMSO” (corresponds to DMSO-treated WT controls) in a bootstrapping manner (i.e. three “WT + TR-27” scores against three “WT + DMSO” for a total of 3×3=9 abundance ratios), and similarly for “CLPP−/− + TR-27” and “CLPP−/− + DMSO”, to obtain the peptide’s abundance ratios that describes any change induced by TR-27; (3) log2 was calculated for each abundance ratio derived such that a positive value corresponds to the peptide’s accumulation, a negative value to its depletion and zero represents no change; (4) the mean and standard deviation for each set of log2 of abundance ratios were calculated; (5) for each peptide, the WT abundance ratio was normalized by the CLPP−/− abundance ratio and the log2 of the quotient was calculated to obtain a score that describes any change in the peptide’s abundance that was induced by TR-27 in a ClpP-dependent manner; (6) for each peptide, t-test was performed to compare the WT and CLPP−/− abundance ratios and the corresponding p-value was calculated with p < 0.05 corresponds to a change in the peptide’s abundance considered to be induced by TR-27 in a ClpP-dependent manner; (7) for each peptide with p < 0.05, if its N-terminal amino acid residue, i.e. the residue that occurs immediately downstream of the proteolytic site, belongs to the first three residues of the peptide’s originating protein based on the annotated primary sequence from UniProt or other public protein databases or if it occurs within known signal peptides or pro-peptides that are cleaved off as part of the originating protein’s maturation process, then the peptide was considered a naturally occurring N-terminus; (8) a peptide with p < 0.05 that does not meet the criteria of a natural N-terminus as outlined was then considered a novel N-terminus.
Both natural and novel N-termini were profiled based on their sequence and local secondary structure. For sequence-based profiling, considering the N-terminal residue of the identified peptide as being immediately downstream of the peptide bond that was cleaved, the 15-residue sequences flanking each side of the cleavage site were identified based on the peptide’s originating protein. After sorting based on abundance change (i.e. accumulation vs. depletion) and subcellular localization (i.e. mitochondrion, cytoplasm, nucleus or others), the peptides were aligned manually by their cleavage sites and a sequence profile was constructed in the sequence logo format using the WebLogo online generator44. For structure-based profiling, the secondary structural information associated with each peptide was extracted from solved protein structures available from the Protein Data Bank or from predicted structural models by AlphaFold that was available or that we generated39,49. A secondary structure profile was then constructed with WebLogo using the DSSP (Dictionary of Secondary Structure of Proteins) secondary structural assignment50,51.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses and software that were used for specific experiments have been described in the relevant subsections under Method Details and in the Key Resources Table. Additional statistical parameters, such as standard deviations (SD) of quantitative data, are explained in the corresponding figure legends. In general, all quantitative data were collected from at least three independent replicates and are presented as means ± SD. All enzyme kinetics experiments and cytotoxicity assays on mammalian cells were repeated multiple times to ensure data reproducibility.
Supplementary Material
Data S1. Supplemental data table providing data and analysis of native and novel N-terminal peptides identified by HYTANE, related to Figures 5 and 6.
Highlights.
TR compounds have nM binding affinity for human mitochondrial ClpP protease.
TR compounds have great shape and charge complementary with their binding sites.
TR compounds target ClpP and exhibit antiproliferative activity on cancer cells.
N-terminome analysis reveals cellular changes caused by TR-induced ClpP activation.
ACKNOWLEDGMENTS
We thank Dr. Sadhna Phanse for help with analyzing the HYTANE data. We also thank the staff scientists of the Canadian Macromolecular Crystallography Facility at the Canadian Light Source for assistance with X-ray data collection. M. F. M. was supported by the Precision Medicine Initiative (PRiME) fellowship at the University of Toronto (internal fellowship number PRMF2019-007). M. M. B. was supported by an Ontario Graduate Scholarship and by Dorthy Sterling Dow Walsh Award. A.-C.G. is the Canada Research Chair in Functional Proteomics and the Lea Reichmann Chair in Cancer Proteomics. Part of the research described in this paper was performed using beamline CMCF-BM at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan. Proteomics work was performed at the Network Biology Collaborative Centre at the Lunenfeld-Tanenbaum Research Institute, a facility supported by Canada Foundation for Innovation funding, the Ontarian Government, and Genome Canada and Ontario Genomics (OGI-139). This work was supported by a grant from National Institutes of Health (GM R01 138520) to L. M. G., and by the Canadian Cancer Society Innovation Grant (Grant #706282) and Canadian Institutes of Health Research Project Grant (PJT-173345) to W. A. H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
E. J. I. is President of Madera Therapeutics, LLC. The company has commercial interests and intellectual property related to this work. The rest of the authors declare no competing interests.
REFERENCES
- 1.Voos W, Jaworek W, Wilkening A, and Bruderek M (2016). Protein quality control at the mitochondrion. Essays Biochem 60, 213–225. 10.1042/EBC20160009. [DOI] [PubMed] [Google Scholar]
- 2.Moehle EA, Shen K, and Dillin A (2019). Mitochondrial proteostasis in the context of cellular and organismal health and aging. J Biol Chem 294, 5396–5407. 10.1074/jbc.TM117.000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mabanglo MF, Bhandari V, and Houry WA (2022). Substrates and interactors of the ClpP protease in the mitochondria. Curr Opin Chem Biol 66, 102078. 10.1016/j.cbpa.2021.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Mabanglo MF, and Houry WA (2022). Recent structural insights into the mechanism of ClpP protease regulation by AAA+ chaperones and small molecules. J Biol Chem 298, 101781. 10.1016/j.jbc.2022.101781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang SG, Maurizi MR, Thompson M, Mueser T, and Ahvazi B (2004). Crystallography and mutagenesis point to an essential role for the N-terminus of human mitochondrial ClpP. J Struct Biol 148, 338–352. 10.1016/j.jsb.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Wong KS, Mabanglo MF, Seraphim TV, Mollica A, Mao YQ, Rizzolo K, Leung E, Moutaoufik MT, Hoell L, Phanse S, et al. (2018). Acyldepsipeptide Analogs Dysregulate Human Mitochondrial ClpP Protease Activity and Cause Apoptotic Cell Death. Cell Chem Biol 25, 1017–1030 e1019. 10.1016/j.chembiol.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Stahl M, Korotkov VS, Balogh D, Kick LM, Gersch M, Pahl A, Kielkowski P, Richter K, Schneider S, and Sieber SA (2018). Selective Activation of Human Caseinolytic Protease P (ClpP). Angew Chem Int Ed Engl 57, 14602–14607. 10.1002/anie.201808189. [DOI] [PubMed] [Google Scholar]
- 8.Ishizawa J, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y, Zhao R, St-Germain J, Heese LE, Egan G, et al. (2019). Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 35, 721–737 e729. 10.1016/j.ccell.2019.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang SG, Ortega J, Singh SK, Wang N, Huang NN, Steven AC, and Maurizi MR (2002). Functional proteolytic complexes of the human mitochondrial ATP-dependent protease, hClpXP. J Biol Chem 277, 21095–21102. 10.1074/jbc.M201642200. [DOI] [PubMed] [Google Scholar]
- 10.Kardon JR, Yien YY, Huston NC, Branco DS, Hildick-Smith GJ, Rhee KY, Paw BH, and Baker TA (2015). Mitochondrial ClpX Activates a Key Enzyme for Heme Biosynthesis and Erythropoiesis. Cell 161, 858–867. 10.1016/j.cell.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yien YY, Ducamp S, van der Vorm LN, Kardon JR, Manceau H, Kannengiesser C, Bergonia HA, Kafina MD, Karim Z, Gouya L, et al. (2017). Mutation in human CLPX elevates levels of delta-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc Natl Acad Sci U S A 114, E8045–E8052. 10.1073/pnas.1700632114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitman JC, Paw BH, and Chung J (2018). The role of ClpX in erythropoietic protoporphyria. Hematol Transfus Cell Ther 40, 182–188. 10.1016/j.htct.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammerling BC, and Gustafsson AB (2014). Mitochondrial quality control in the myocardium: cooperation between protein degradation and mitophagy. J Mol Cell Cardiol 75, 122–130. 10.1016/j.yjmcc.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rondelli CM, Perfetto M, Danoff A, Bergonia H, Gillis S, O’Neill L, Jackson L, Nicolas G, Puy H, West R, et al. (2021). The ubiquitous mitochondrial protein unfoldase CLPX regulates erythroid heme synthesis by control of iron utilization and heme synthesis enzyme activation and turnover. J Biol Chem 297, 100972. 10.1016/j.jbc.2021.100972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Furoukh N, Ianni A, Nolte H, Holper S, Kruger M, Wanrooij S, and Braun T (2015). ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. Biochim Biophys Acta 1853, 2580–2591. 10.1016/j.bbamcr.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 16.Cormio A, Musicco C, Gasparre G, Cormio G, Pesce V, Sardanelli AM, and Gadaleta MN (2017). Increase in proteins involved in mitochondrial fission, mitophagy, proteolysis and antioxidant response in type I endometrial cancer as an adaptive response to respiratory complex I deficiency. Biochem Biophys Res Commun 491, 85–90. 10.1016/j.bbrc.2017.07.047. [DOI] [PubMed] [Google Scholar]
- 17.Seo JH, Rivadeneira DB, Caino MC, Chae YC, Speicher DW, Tang HY, Vaira V, Bosari S, Palleschi A, Rampini P, et al. (2016). The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol 14, e1002507. 10.1371/journal.pbio.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishigaki R, Osaki M, Hiratsuka M, Toda T, Murakami K, Jeang KT, Ito H, Inoue T, and Oshimura M (2005). Proteomic identification of differentially-expressed genes in human gastric carcinomas. Proteomics 5, 3205–3213. 10.1002/pmic.200401307. [DOI] [PubMed] [Google Scholar]
- 19.Graves PR, Aponte-Collazo LJ, Fennell EMJ, Graves AC, Hale AE, Dicheva N, Herring LE, Gilbert TSK, East MP, McDonald IM, et al. (2019). Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem Biol 14, 1020–1029. 10.1021/acschembio.9b00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, et al. (2015). Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 27, 864–876. 10.1016/j.ccell.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hackl MW, Lakemeyer M, Dahmen M, Glaser M, Pahl A, Lorenz-Baath K, Menzel T, Sievers S, Bottcher T, Antes I, et al. (2015). Phenyl Esters Are Potent Inhibitors of Caseinolytic Protease P and Reveal a Stereogenic Switch for Deoligomerization. J Am Chem Soc 137, 8475–8483. 10.1021/jacs.5b03084. [DOI] [PubMed] [Google Scholar]
- 22.Jacques S, van der Sloot AM,C,CH, Coulombe-Huntington J, Tsao S, Tollis S, Bertomeu T, Culp EJ, Pallant D, Cook MA, et al. (2020). Imipridone Anticancer Compounds Ectopically Activate the ClpP Protease and Represent a New Scaffold for Antibiotic Development. Genetics 214, 1103–1120. 10.1534/genetics.119.302851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonner ER, Waszak SM, Grotzer MA, Mueller S, and Nazarian J (2021). Mechanisms of imipridones in targeting mitochondrial metabolism in cancer cells. Neuro-oncology 23, 542–556. 10.1093/neuonc/noaa283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang J, Zhang J, Luo B, Qiao W, Qiu Z, Song R, Dai Z, Sui J, Xu X, Ruan S, et al. (2022). Discovery of a Novel Series of Imipridone Compounds as Homo sapiens Caseinolytic Protease P Agonists with Potent Antitumor Activities In Vitro and In Vivo. Journal of medicinal chemistry 65, 7629–7655. 10.1021/acs.jmedchem.1c02071. [DOI] [PubMed] [Google Scholar]
- 25.Allen JE, Krigsfeld G, Patel L, Mayes PA, Dicker DT, Wu GS, and El-Deiry WS (2015). Identification of TRAIL-inducing compounds highlights small molecule ONC201/TIC10 as a unique anti-cancer agent that activates the TRAIL pathway. Mol Cancer 14, 99. 10.1186/s12943-015-0346-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prabhu VV, Morrow S, Rahman Kawakibi A, Zhou L, Ralff M, Ray J, Jhaveri A, Ferrarini I, Lee Y, Parker C, et al. (2020). ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia 22, 725–744. 10.1016/j.neo.2020.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Czuczi T, Muranyi J, Barany P, Mora I, Borbely A, Csala M, and Csampai A (2022). Synthesis and Antiproliferative Activity of Novel Imipridone-Ferrocene Hybrids with Triazole and Alkyne Linkers. Pharmaceuticals (Basel) 15. 10.3390/ph15040468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duchatel RJ, Mannan A, Woldu AS, Hawtrey T, Hindley PA, Douglas AM, Jackson ER, Findlay IJ, Germon ZP, Staudt D, et al. (2021). Preclinical and clinical evaluation of German-sourced ONC201 for the treatment of H3K27M-mutant diffuse intrinsic pontine glioma. Neuro-oncology advances 3, vdab169. 10.1093/noajnl/vdab169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen L, Shan Y, Weng Y, Sui Z, Zhang X, Liang Z, Zhang L, and Zhang Y (2016). Hydrophobic Tagging-Assisted N-Termini Enrichment for In-Depth N-Terminome Analysis. Anal Chem 88, 8390–8395. 10.1021/acs.analchem.6b02453. [DOI] [PubMed] [Google Scholar]
- 30.Shoemaker RH (2006). The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer 6, 813–823. nrc1951 [pii] 10.1038/nrc1951 [doi]. [DOI] [PubMed] [Google Scholar]
- 31.Binepal G, Mabanglo MF, Goodreid JD, Leung E, Barghash MM, Wong KS, Lin F, Cossette M, Bansagi J, Song B, et al. (2020). Development of Antibiotics That Dysregulate the Neisserial ClpP Protease. ACS Infect Dis 6, 3224–3236. 10.1021/acsinfecdis.0c00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aristotelous T, Hopkins AL, and Navratilova I (2015). Chapter Twenty-Three - Surface Plasmon Resonance Analysis of Seven-Transmembrane Receptors. In Methods in Enzymology, Shukla AK, ed. (Academic Press; ), pp. 499–525. 10.1016/bs.mie.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 33.Mabanglo MF, Leung E, Vahidi S, Seraphim TV, Eger BT, Bryson S, Bhandari V, Zhou JL, Mao YQ, Rizzolo K, et al. (2019). ClpP protease activation results from the reorganization of the electrostatic interaction networks at the entrance pores. Commun Biol 2, 410. 10.1038/s42003-019-0656-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang P, Zhang T, Wang X, Xiao H, Li H, Zhou LL, Yang T, Wei B, Zhu Z, Zhou L, et al. (2022). Aberrant human ClpP activation disturbs mitochondrial proteome homeostasis to suppress pancreatic ductal adenocarcinoma. Cell Chem Biol 29, 1396–1408.e1398. 10.1016/j.chembiol.2022.07.002. [DOI] [PubMed] [Google Scholar]
- 35.Bhandari V, Wong KS, Zhou JL, Mabanglo MF, Batey RA, and Houry WA (2018). The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem Biol 13, 1413–1425. 10.1021/acschembio.8b00124. [DOI] [PubMed] [Google Scholar]
- 36.Kleifeld O, Doucet A, Prudova A, auf dem Keller U, Gioia M, Kizhakkedathu JN, and Overall CM (2011). Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat Protoc 6, 1578–1611. 10.1038/nprot.2011.382. [DOI] [PubMed] [Google Scholar]
- 37.The UniProt C (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res 45, D158–D169. 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Zidek A, Bridgland A, Cowie A, Meyer C, Laydon A, et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596. 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell metabolism 23, 27–47. 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calvo SE, Julien O, Clauser KR, Shen H, Kamer KJ, Wells JA, and Mootha VK (2017). Comparative Analysis of Mitochondrial N-Termini from Mouse, Human, and Yeast. Mol Cell Proteomics 16, 512–523. 10.1074/mcp.M116.063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofsetz E, Demir F, Szczepanowska K, Kukat A, Kizhakkedathu JN, Trifunovic A, and Huesgen PF (2020). The Mouse Heart Mitochondria N Terminome Provides Insights into ClpXP-Mediated Proteolysis. Mol Cell Proteomics 19, 1330–1345. 10.1074/mcp.RA120.002082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 48.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50, D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kabsch W, and Sander C (1983). Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 51.Joosten RP, Joosten K, Cohen SX, Vriend G, and Perrakis A (2011). Automatic rebuilding and optimization of crystallographic structures in the Protein Data Bank. Bioinformatics 27, 3392–3398. 10.1093/bioinformatics/btr590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee C-D, Sun H-C, Hu S-M, Chiu C-F, Homhuan A, Liang S-M, Leng C-H, and Wang T-F (2008). An improved SUMO fusion protein system for effective production of native proteins. Protein Sci 17, 1241–1248. 10.1110/ps.035188.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, Konig T, Kukat A, and Trifunovic A (2016). Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep 17, 953–964. 10.15252/embr.201642077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kimber MS, Yu AY, Borg M, Leung E, Chan HS, and Houry WA (2010). Structural and theoretical studies indicate that the cylindrical protease ClpP samples extended and compact conformations. Structure (London, England : 1993) 18, 798–808. 10.1016/j.str.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 55.Kabsch W (2010). XDS. Acta Crystallogr D Biol Crystallogr 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental data table providing data and analysis of native and novel N-terminal peptides identified by HYTANE, related to Figures 5 and 6.
Data Availability Statement
X-ray diffraction data have been deposited at the RCSB Protein Data Bank, while the HYTANE mass spectrometry data have been deposited at the MassIVE repository. All data are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-HsClpP | Abcam | RRID:AB_124822 |
| Rabbit monoclonal anti-HsClpX | Abcam | RRID:AB_168338 |
| Rabbit monoclonal anti-Mcl1 | Cell Signaling Technology | RRID:AB_2799149 |
| Rabbit monoclonal anti-Caspase 8 | Cell Signaling Technology | RRID:AB_10545768 |
| Rabbit monoclonal anti-Caspase 9 | Cell Signaling Technology | RRID:AB_2068621 |
| Rabbit polyclonal anti-TFAM | Proteintech | RRID:AB_11182588 |
| Rabbit polyclonal anti-Grp75 | Proteintech | RRID:AB_2120458 |
| Goat monoclonal anti-Caspase 3 | R&D Biosystems | RRID:AB_354518 |
| Mouse monoclonal anti-GAPDH | Abcam | RRID:AB_8245 |
| HRP-conjugated goat anti-rabbit IgG | BioRad | RRID:AB_11125142 |
| HRP-conjugated goat anti-mouse IgG | BioRad | RRID:AB_11125547 |
| HRP-conjugated rabbit anti-goat IgG | Sigma-Aldrich | RRID:AB_258242 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium acetate, ACS Reagent, ≥ 99.0% | Sigma-Aldrich | Cat#241245 |
| PEG 4,000, powder | Sigma-Aldrich | Cat#8.17006 |
| TR-27 | reference19 | N/A |
| TR-57 | reference19 | N/A |
| TR-65 | reference19 | N/A |
| TR-107 | This paper | N/A |
| TR-133 | This paper | N/A |
| ADEP-14 | reference6 | N/A |
| Doxorubicin (hydrochloride) | Cayman Chemical Co. | Cat#15007 |
| Sulforhodamine B (SRB) | Sigma-Aldrich | Cat#230162 |
| jetPRIME Transfection Reagent | Polyplus Transfection | Cat#114–07 |
| jetPRIME Transfection Buffer | Polyplus Transfection | Cat#712–60 |
| Reprosil-Pur 120 C18-AQ, 1.9 μm | Dr. Maisch | Cat#r119.aq. |
| Casein-FITC | Sigma-Aldrich | Cat# C3777 |
| Untagged human mitochondrial ClpP | This paper | N/A |
| Yeast SUMO protease | reference52 | N/A |
| Critical Commercial Assays | ||
| PureLink Quick Plasmid Miniprep Kit | Thermo-Fisher Scientific | Cat#K210011 |
| Pierce BCA Protein Assay Kit | Thermo-Fisher Scientific | Cat#23225 |
| Deposited Data | ||
| Structure of human ClpP complex with ADEP-28 | Protein Data Bank reference6 | 6BBA |
| Structure of human ClpP complex with ONC201 | Protein Data Bank reference8 | 6DL7 |
| Structure of human ClpP complex with ZG111 | Protein Data Bank reference34 | 7VP9 |
| Structure of human ClpP Y118A mutant complex with D9 | Protein Data Bank reference7 | 6H23 |
| Structure of human ClpP complex with TR-27 | Protein Data Bank (This paper) | 7UVM |
| Structure of human ClpP complex with TR-57 | Protein Data Bank (This paper) | 7UVN |
| Structure of human ClpP complex with TR-65 | Protein Data Bank (This paper) | 7UVR |
| Structure of human ClpP complex with TR-107 | Protein Data Bank (This paper) | 7UVU |
| Structure of human ClpP complex with TR-133 | Protein Data Bank (This paper) | 7UW0 |
| HYTANE data | MassIVE repository (This paper) | MSV000089642 |
| Experimental Models: Cell Lines | ||
| HEK293 T-REx | Gift from A. Trifunovic | RRID:CVCL_U427 |
| HEK293 T-RExCLPP−/− | reference53 | N/A |
| HEK293 T-RExCLPX−/− | reference6 | N/A |
| MDA-MB-231 | Gift from L. Attisano | RRID:CVCL_0062 |
| MDA-MB-231 CLPP−/− | This paper | N/A |
| MDA-MB-231 CLPX−/− | This paper | N/A |
| Experimental Models: Organisms / Strains | ||
| Escherichia coli DH5α | Lab stock | NCBI:txid668369 |
| Escherichia coli BL21(DE3) ΔclpP::cat (SG1146) | reference54 | N/A |
| Oligonucleotides | ||
| CLPP Exon 1 pX330 F: CACCGAAGCCGACCGGGGCGTGCGG |
This paper | N/A |
| CLPP Exon 1 pX330 R: AAACCCGCACGCCCCGGTCGGCTTC |
This paper | N/A |
| CLPP Exon 1 ssODN: GCATGACGCCACCCGGGCCCCCCCTACCAATATTCATTATTATCACCTTCCGCACGCCCCGGTCGGCTTCCGTCCGATGGCGGAACTACAGCTTCCGGCG |
This paper | N/A |
| CLPX Exon1 pX330 F: CACCGCGGTGCTTGTACTTGCGGCG |
reference6 | N/A |
| CLPX Exon1 pX330 R: AAACCGCCGCAAGTACAAGCACCGC |
reference6 | N/A |
| CLPX Exon1 ssODN: GGCCTCGCGGAGATGCCCAGCTGCGGTGCTTGTACTTGCGGCGCGGCGTAGGTCCGGCTCATCACCTCCTCACTCGCCTCCGCGCAGAGA |
reference6 | N/A |
| Recombinant DNA | ||
| pX330 | reference48 | RRID:Addgene_42230 |
| pX330-CLPP KO | This paper | N/A |
| pX330-CLPX KO | reference6 | N/A |
| pETSUMO2-CLPP(-MTS) | reference6 | N/A |
| pETSUMO2-TFAM(-MTS) | This paper | N/A |
| Software and Algorithms | ||
| Proteome Discoverer 2.2 | Thermo-Fisher Scientific | RRID:SCR_014477 |
| WebLogo 2.8.2 | reference44 | RRID:SCR_010236 |
| AlphaFold | reference39 | N/A |
| NIS-Elements Basic Research Software | Nikon | RRID:SCR_002776 |
| XDS | reference55 | https://xds.mr.mpg.de/ |
| Phenix | reference46 | https://phenix-online.org/ |
| COOT | reference45 | http://www2.mrc-lmb.cam.ac.uk/Personal/pemsley/coot |
| PISA | reference47 | https://www.ebi.ac.uk/pdbe/pisa/ |
| The PyMOL Molecular Graphics System | Schrödinger, LLC | https://pymol.org/2/ |
| Other | ||
| BbsI | New England Biolabs | Cat#R0539S |
| FastAP | Thermo-Fisher Scientific | Cat#EF0651 |
| T4 Polynucleotide Kinase | New England Biolabs | Cat#M0201S |
| T4 DNA Ligase | New England Biolabs | Cat#M0202S |
| EnSpire 2300 Multilabel Reader | Perkin-Elmer | Cat#655077 |
| P-2000 Laser-Based Micropipette Puller | Sutter Instrument Co. | Cat#P-2000 |
| EkspertNanoLC 425 HPLC System | Eksigent Technologies | Cat#Ekspert425 |
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | Thermo-Fisher Scientific | Cat#IQLAAEGAAPF ADBMBHQ |
| Microcon-10kDa Ultracel YM-10 filter unit | Millipore | Cat#Z648078 |
| C18 Stage Tips | Supelco | Cat#66883-U |
| Eclipse 80i fluorescence microscope | Nikon | RRID:SCR_015572 |
| X-Cite Series 120Q excitation light source | Excelitas Technologies | https://www.excelitas.com/product/x-cite-120q |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.