

Abstract
The canonical model of “small cell lung cancer” (SCLC) depicts tumors arising from dual inactivation of TP53 and RB1. However, many genomic studies have persistently identified tumors with no RB1 mutations. Here, we examined RB1 protein expression and function in SCLC. RB1 expression was examined by immunohistochemical analysis of 62 human SCLC tumors. These studies showed that ~14% of SCLC tumors expressed abundant RB1 protein, which is associated with neuroendocrine (NE) gene expression and is enriched in YAP1 expression, but no other lineage proteins that stratify SCLC. SCLC cells and xenograft tumors with RB1 protein expression were sensitive to growth inhibition by the CDK4/6 inhibitor palbociclib, and this inhibition was shown to be dependent on RB1 expression by CRISPR knockout. Furthermore, a patient with biopsy-validated wt RB1 SCLC who received the CDK4/6 inhibitor abemaciclib demonstrated a dramatic decrease in mutant TP53 ctDNA allelic fraction from 62.1% to 0.4% and decreased tumor mass on CT scans. Importantly, immunohistochemistry of the diagnostic biopsy specimen showed RB1 positivity. Finally, we identified a transcriptomics-based RB1 loss-of-function signature that discriminates between SCLC cells with or without RB1 protein expression and validated it in the patient who was responsive to abemaciclib, suggesting its potential use to predict CDK4/6 inhibitor response in SCLC patients. Our study demonstrates that RB1 protein is an actionable target in a subgroup of SCLC, a cancer that exhibits no currently targetable mutations.
INTRODUCTION
The genomic characterization of tumors has often led to the discovery of new therapeutic targets. Therapeutic successes in “non-small cell lung cancer” (NSCLC) have targeted both high-frequency alterations, such as osimertinib treatment for mutant EGFR, which occurs in ~10% of NSCLC cases, and low-frequency alterations, such as alectinib treatment for ALK-fusions, which occur in <1% of NSCLC cases (1). Kinase inhibition is superior to chemotherapy in the front-line setting (2–4). Compared to NSCLC, there are fewer genomic studies on small cell lung cancer (SCLC) due to the low surgical resection rate and increasing reliance on needle biopsies and cytology for diagnosis. Nevertheless, data from many whole exome (WES) (5–8), targeted exome (9–11), and whole genome sequencing (WGS) (12) studies in SCLC suggest a model in which SCLC results from the dual inactivation of TP53 and RB1. The requirement for dual TP53/RB1 inactivation in SCLC tumorigenesis is also supported by knockout mouse models of this cancer (13). Beyond these two genes, no single mutated gene or signaling pathway alteration is highly enriched in human SCLC, although mouse models indicate that many genes, including MYC, can regulate SCLC tumorigenesis (14, 15). Thus, in the absence of actionable mutations, chemotherapy remains the primary treatment for SCLC. While immune checkpoint blockade (ICB) has been added recently to the standard-of-care (SOC) for SCLC, ICB demonstrates much less clinical benefit in SCLC than in NSCLC (16).
Transcriptomic analyses have led to the stratification of SCLC into four subgroups, which may have therapeutic implications. These subgroups are based on the expression of three clearly defined lineage-specific transcription factors (ASCL1, NEUROD1, and POU2F3) (17) and an enigmatic fourth group based on the expression of YAP1 in the Hippo pathway (18), the neuroendocrine (NE) transcription factor ATOH1 (19), or by an inflamed signature (20). It has been proposed that individual subgroups may show greater sensitivity toward PARP inhibitors (ASCL1, POU2F3), Aurora kinase inhibitors (NEUROD1), BCL2 inhibitors (ASCL1), cisplatin (POU2F3) and ICB agents (inflamed) (20). This approach could fulfill the compelling need to identify new therapeutic strategies for SCLC in the absence of recurrent actionable gene mutations.
Here, we adopted a different therapeutic strategy based on our belief that the canonical genomic model of SCLC ignores data from many genomic studies that persistently identify tumors with wt RB1 mutation status, ranging from 26–61%, which opens the possibility that some SCLC tumors can express functional RB1 protein. In support of this idea, we previously demonstrated RB1 protein expression in both SCLC tumors and cell lines (21). Importantly, we also demonstrated that RB1 protein expression in SCLC cell lines correlates with their sensitivity to CDK4/6 inhibitors (18). We now report that analyses of 62 SCLC tumors by immunohistochemistry (IHC) demonstrated that ~14% expressed RB1 protein, and RB1 expression was largely accompanied by neuroendocrine gene expression, which is a characteristic feature of SCLC. We further showed that ~50% of SCLC cell lines annotated as wt RB1 express functional RB1 protein, defined by sensitivity to the CDK4/6 inhibitor palbociclib, and that RB1 expression is required for palbociclib sensitivity. Finally, we identified a transcriptomics-based RB1 loss-of-function (LOF) signature that could correctly determine RB1 functional status in SCLC cell lines, as well as in a wt RB1 SCLC patient who responded to the CDK4/6 inhibitor abemaciclib.
MATERIALS and METHODS
Cell lines-
All cell lines were purchased from ATCC (Manassas, VA), except for DMS454 (Sigma, St. Louis, MO), and were authenticated within the last three years. The cells were maintained in the media recommended by the supplier.
Western blotting-
Protein lysates (40 μg) were prepared in the presence of protease inhibitors and analyzed as described previously (18) using 4–20% Criterion gels (Bio-Rad, Hercules, CA, USA). Antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA) (YAP1, RB1, CDKN2A/B, NEUROD1, CCND1, CDK4, CDK6, pRB-s780), Santa Cruz (Dallas, TX, USA) (INSM1, CDK2, E2F1), Sigma (St. Louis, MO, USA) (Actin, POU2F3), and BD Biosciences (Franklin Lakes, NJ, USA) (ASCL) and are listed in Sup. Table 1. Bands on western blots were quantified using Image J.
RB1 CRISPR/Cas9 knockout-
RB1 guide RNA on a pLentiCRISPRv2 vector was purchased from GenScript (Piscataway, NJ, USA) and stably introduced into cells using lentiviral transduction by seeding 100,000 cells per well of a 6 well plate in a 5 mL total volume that included 50 µL polybrene, virus (MOI = 5), and appropriate ATCC media to volume. Plates were centrifuged at 750 × g for 15 min and placed in a 37°C, 5% CO2 incubator overnight so that the viral media remained on the cells for 24 h. The viral media and cells in suspension were removed from the well into a 15 mL conical tube, centrifuged at 1,000 rpm for 5 min, and the cell pellet was resuspended in 5 ml new media. Viral medium was removed from the adherent cells and replaced with fresh medium. Puromycin was added the next day at 0.5 µg/ml for about two weeks to generate stable cell lines. Clones of H841 and SW1271 were isolated using serial dilutions. The cells were counted using a hemocytometer and diluted to 1000 cells/ml. 200 µl DMEM/F12 + 0.5 µg/ml puromycin was added to all wells of 96 well plates. Cells (200 µL) were added to all wells of column 1, and two-fold dilutions were performed in a 96 well plate. The plates were evaluated for single cells and placed in a 37°C incubator. Single cells were then expanded to generate stable clonal cell lines. RB1 knockout was verified by western blotting in both mixed populations and clones against the empty viral vector control mixed populations and clones (Sup. Fig. 1).
In vitro growth assays-
Growth response was measured in complete medium by live cell imaging of cells by phase contrast using IncuCyte ZOOM technology (18). At least six replicate wells of cells were seeded at 5,000 cells/well (10,000 cells/well for DMS53) in 96 well plates in ATCC recommended medium. Palbociclib was added at 1 µM to the test plate, and H2O was used as the vehicle control for a final volume of 200 µL per well. The plates were analyzed for five days (10 days for DMS53).
Cell cycle analysis was performed by our Cancer Center Cytometry and Imaging Core using standard techniques as previously described (22). The experiments were performed in triplicate.
The effect of palbociclib on cell growth was also examined by colony formation assays. Briefly, cells were seeded into six-well plates (125,000 cells/well for all cell lines except DMS53, which was 250,000 cells/well) and treated with 10-fold serial dilutions of palbociclib (0.001–1.0 µM) on the day of seeding. The medium was not changed during the 5-day course of the experiment (10 d for DMS53). After treatment, colonies were stained with 0.1% crystal violet and imaged.
Xenograft studies-
For xenograft experiments, 5 × 106 H841 cells (vector control or RB1 KO) were inoculated subcutaneously into one hind limb of each nude mouse (six weeks old; Charles River, Wilmington, MA, USA). Once tumors were readily palpable (~100 mm3), mice were randomized into two arms: vehicle (control) and palbociclib treatment. Each experimental arm contained 7–10 mice. Palbociclib was administered by gavage five days a week (100 mg/kg, Mon-Fri). Tumor volumes (LxW2/2) were monitored 3x per week using a caliper for three weeks, at which time the mice were euthanized and tumors were removed. The tumor growth rate was calculated as the relative tumor volume [tumor volume on day x / mean tumor volume of control/treated on day 1]. Growth inhibition by palbociclib between the arms was analyzed on day 17 using a two-way ANOVA. Animal well-being was monitored based on weight loss. No adverse effects of palbociclib were noted. Xenograft experiments followed a protocol approved by the Case Western Reserve University IACUC.
Transcriptomic analyses-
Patient RNA sequencing (Tempus xE) was checked using FastQC, and TrimGalore was used for adapter and quality trimming. RNA-seq reads were mapped against hg38 using STAR aligner. Differential gene expression analysis was performed using DESeq2. To determine RB1 LOF, we used a signature described by Chen et al. (23) that was developed using CCLE and TCGA data across many cancers. The RB1 LOF signature contained 186 genes, including 118 down-regulated and 68 up-regulated genes due to RB1 loss (Sup. Table 2). The RB1 LOF signature scoring system was developed by using a nearest shrunken centroid approach based on the expression pattern of these genes (23). The R package ‘pamr’ was used to generate the score. The RB1 LOF signature scores between 0 (functional RB1) and 1 (complete RB1 loss). We used area under the ROC curve (AUC) analysis to assess the accuracy of the RB1 LOF classifier. We also used 186 differentially expressed genes to perform enrichment analysis using GSEA (gene set enrichment analysis) in wt RB1 SCLC cells with or without RB1 protein expression.
Treatment of wt RB1 SCLC patients with abemaciclib-
A proof of principal, single-arm study of abemaciclib in wt RB1 SCLC was approved by the University Hospitals of Cleveland IRB and initiated based on our strong preclinical data (NCT04010357). Written informed consent was obtained from patients and the studies were conducted in accordance with the Declaration of Helsinki. Patients were required to have histologically confirmed ES-SCLC that is wt RB1 by genomic testing in biopsy tissue or blood using circulating DNA. Other enrollment criteria included the presence of chemo-refractory disease defined as 1) platinum refractory: no response after 1–2 cycles of chemotherapy, or 2) early relapse: with initial response but relapse <90 days; with measurable disease per the “Response Evaluation Criteria in Solid Tumors” (RECIST), version 1.1. CDK4/6 inhibitors, including abemaciclib, are already FDA-approved for breast cancer, with a good safety and efficacy profile for which the dose is already defined. Our two patients received abemaciclib orally on a continuous schedule every 12 (+/−2) hours (Q12H) on days 1 to 28 of a 28-day cycle until disease progression or unacceptable toxicity developed. The primary endpoint for our patients was “overall response rate” (ORR) at the end of abemaciclib cycle 2 (8 weeks). The ORR was determined according to RECIST v1.1. Patients received a lead in the scan at week four to assess rapid progression. Response was evaluated every 8 weeks (2 cycles) with radiographic imaging to assess the response to treatment.
Patient 2 presented with extensive stage SCLC. At time of diagnosis, patient 2 had bulky intrathoracic disease as well as malignant lymphadenopathy in the chest, supraclavicular and cervical lymph nodes. Patient 2 had superior vena cava syndrome and a baseline LDH greater than 600. Biopsy of the right cervical lymph nodes showed small cell lung cancer histology as well as INSM1, synaptophysin, and chromogranin positivity by IHC. Patient 2 went on to receive two cycles of carboplatin/etoposide with durvalumab but had primary refractory disease with disease progression. Patient 2 was then treated with abemaciclib as second line therapy and relapse was identified by ctDNA analysis on day 233 of CDK4/6 inhibitor treatment.
IHC analyses-
We analyzed three TMAs containing cores from SCLC tumors, as described previously (18). The construction and analyses of TMAs and the retrospective database of SCLC patients was approved by the University Hospitals of Cleveland IRB. The tumor sites are listed in Sup. Table 3. The demographic data of this cohort are presented in Sup. Table 4. Patient tumors (listed by de-identified patient ID) were represented on TMAs by 1–8 cores taken from to 1–2 tissue blocks. All patient biopsies were obtained pretreatment. TMA slides were deparaffinized with xylene and transferred through graded ethanol to H2O. Antigen retrieval was performed by boiling the slides in a pressure cooker filled with a sodium citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked by a 10-minute incubation in 3% hydrogen peroxide solution. Slides were blocked using Background Sniper (Biocare) for 20 min. Tissues were incubated with primary antibodies for one hour at room temperature, as described in Sup. Table 1. The specificity of the primary antibodies was validated by IHC analysis of TMA-containing SCLC cell lines. The bound antibody was detected using HRP–labeled polymer secondary antibody from Biocare for 30 min. The slides were rinsed in TBS and visualized with a 10-minute incubation of DAB in buffered substrate in the dark. Finally, the slides were counterstained with hematoxylin for 30 s and mounted using Biomount. Staining was graded on a scale of 0–3+, individualized for each antibody, and modified H scores were calculated as (staining intensity × % positivity), giving a range of values from 0–300. Only nuclear staining was considered. Scoring was performed by three pathologists. The H scores presented in Table 1 and Sup. Table 3 represent the maximum scores obtained for any of the cores for a given tumor. Individual H-scores are listed for any image shown in the manuscript and are specific to the core shown. The RB1 H-score cut-off point of 20 was based on our own judgment coupled with a desire to not overestimate RB1 positivity.
TABLE 1.
Features and H scores of RB1 positive SCLC tumors
| PID | # Cores | Site | RB1 | INSM1 | ASCL1 | NEUROD1 | POU2F3 | YAP1 |
|---|---|---|---|---|---|---|---|---|
| 326 | 2 | node | 120 | 50 | 50 | na | na | 120 |
| 549 | 2 | lung | 180 | 190 | 180 | 0 | 0 | 0 |
| 541 | 2 | lung | 140 | 10 | 0 | 10 | ? | 160 |
| 559 | 2 | pleura | 120 | 190 | 190 | na | na | 0 |
| 246 | 4 | mediastinum | 180 | 0 | 90 | 240 | 0 | 0 |
| 243 | 2 | lung | 90 | 240 | 0 | 0 | 0 | 0 |
| 636 | 2 | pleura | 100 | 270 | 160 | 255 | 0 | 0 |
| 776 | 4 | brain | 225 | 120 | 40 | 0 | 15 | 6 |
| 471 | 6 | brain | 180 | 285 | 270 | 270 | 0 | 40 |
na = no tissue available
Genomic analyses-
RB1 gene mutation status for 52 SCLC cell lines was obtained from the Cancer Cell Line Encyclopedia (CCLE) dataset (Broad 2019) available on the cBioPortal website (https://www.cbioportal.org/). We maintain a database of all SCLC patients treated at our medical center, which now has > 950 patient entries. In addition to demographic and clinical data, we also collect targeted and whole-exome sequencing data as part of the standard of care. Targeted sequencing was performed on pretreatment biopsies using either Foundation Medicine or Tempus platforms as CLIA-approved commercial vendors. Germline mutations were filtered out using proprietary company algorithms. Our cohort of SCLC patients, with matching clinical and genomic data, has now reached 120 patients. The demographic characteristics of this cohort are shown in Sup. Table 5. Analysis of the genomic alterations present in this cohort included only those annotated as biologically relevant and did not include variants of unknown significance (VUS) (Sup. Table 6). Only genes with ≥ 4 alterations are shown in Fig. 1. A chi-square test was used to determine the association between co-mutations and RB1 mutation status.
Figure 1. RB1 mutation and expression in SCLC tumors.
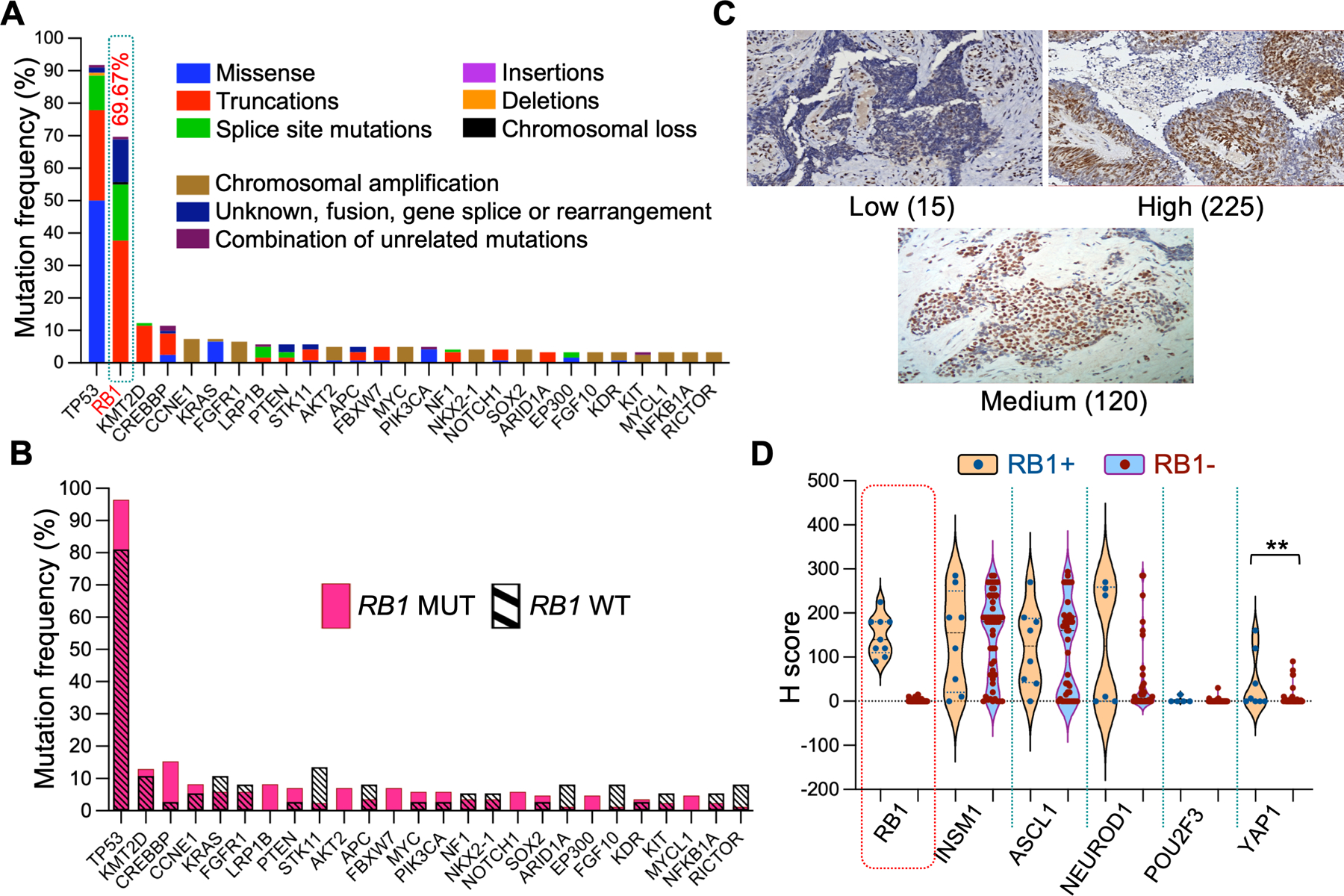
A. Gene mutation profile in SCLC tumors in the N=120 genomic cohort. Only genes that had ≥ 4 mutations are shown. Variants of unknown significance (VUS) were not considered. B. Same as in panel A except that the percentage of gene mutations between wt RB1 (magenta) and mutant RB1 (cross hatched) SCLC tumors is compared. TP53 (0.004) and CREBBP (0.045) exhibited a significantly lower % mutation rate in wt RB1; whereas STK11 (0.015), ARID1A (0.048), FGF10 (0.048), and RICTOR (0.048) exhibited a significantly higher mutation rate in wt RB1 (P values for individual genes in parentheses). Only genes that had ≥ 4 mutations were considered for analysis. C. Representative images of tumor RB1 staining (showing H scores). D. Violin plots of complete IHC profiles for the entire RB+ and RB- cohorts. Statistical differences in H score distribution between cohorts was only achieved for YAP1 and not for ASCL1 and NEUROD1. POU2F3 was not calculated.
Statistics-
The distribution of H scores in RB+ and RB- tumors was calculated using unpaired t-tests for each protein examined. Statistical analysis of growth inhibition by palbociclib was tested by unpaired or Mann Whitney T-tests at 48 h or the last time point (Day 5 or 10), respectively. At least 3–6 technical replicates were performed for each experimental arm. Experiments (western blotting, FACS, and colony formation assays) were repeated at least twice to ensure reproducibility. Statistical significance for all results is shown: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Data Availability-
Tumor and blood targeted DNA sequencing data are available in Supplementary Tables. Tumor RNA-seq data are deposited in Gene Expression Omnibus and are accessible using accession number GSE217646.
RESULTS
RB1 is expressed in a subgroup of human SCLC tumors and cell lines
The most recent analysis of 120 SCLC patients treated at our medical center by targeted exon sequencing produced a mutation profile similar to that reported previously, demonstrating high TP53 (N=110, 92%) and RB1 (N=85, 70%) mutation rates, then falling rapidly to much lower mutation rates (≤12%) among the remaining genes (Fig. 1A). Closer examination of the mutation distribution revealed a lack of alterations in cyclin D (CCND) or cyclin kinase inhibitor (CDKN) genes. When the wt and mutant RB1 cohorts were compared (Fig. 1B), TP53 and CREBBP demonstrated significantly lower alterations, whereas STK11, ARID1A, FGF10, and RICTOR demonstrated significantly higher alterations in the wt RB1 cohort. Overall, our results suggest that only a small number of SCLC tumors in our genomic cohort may have functional inactivation of the RB1 pathway or represent misclassification of tumors.
To directly address the question of RB1 expression in SCLC, we performed IHC staining on a second cohort of diagnostic tumor specimens (Sup. Fig. 2). To determine whether RB1 expression was specific to any of the four SCLC transcriptional subgroups, we performed IHC analysis for ASCL1, NEUROD1, POU2F3, YAP1, and an additional neuroendocrine gene, INSM1. This analysis involved 169 tumor tissue cores from 64 patients (Sup. Table 3). The results showed that nine out of 62 evaluable patient tumors (14.5%) expressed RB1 protein (Table 1, Fig. 1C). The RB1 positive cores originated from a variety of biopsy sites, both primary and metastatic, and were often associated with parallel expression of the neuroendocrine proteins INSM1, ASCL1, and NEUROD1 (Fig. 1D). YAP1 staining was present in both RB1 positive and negative tumors, although it was significantly higher in RB1 positive tumors. The staining results for our entire cohort were similar to those of previously reported studies, with 70.5% ASCL1 positive, 39% NEUROD1 positive, and 20.7% YAP1 positive (Sup. Table 3). However, only one tumor was POU2F3 positive (Sup. Fig. 3), and 41.9% of ASCL1 positive tumors were also NEUROD1 positive (Sup. Table 3). Taken together, these results indicate that RB1 protein expression is associated only with the YAP1 transcriptional subgroup.
The genomic mutation status of RB1 was known for eight IHC specimens, three wt RB1 and five mutant RB1 (Sup. Table 7). One wild-type RB1 tumor (PID 776) and one mutant RB1 tumor (PID 636) stained strongly positive for RB1. Interestingly, the RB1 positive, mutant RB1 tumor was annotated as an RB1-USP24 fusion, whereas the four RB1 negative, mutant RB1 tumors contained two nonsense mutations, a splice site mutation, and exon loss.
To further study the correlation between RB1 mutation status and RB1 protein expression, we used the Broad Cancer Cell Line Encyclopedia (CCLE, Broad 2019) database of 52 SCLC cell lines that annotated 13 (25%) as wild-type RB1 (Fig. 2A). We tested 11 wt RB1, two missense RB1 (H209, H1694), and five mutant RB1 SCLC cell lines for RB1 expression by western blotting. As shown in Fig. 2B, 7 of 11 SCLC cells annotated as wt RB1 expressed detectable protein (DMS114, DMS454, H211, SW1271, DMS53, H841, H1341), whereas none of the five cell lines with LOF RB1 mutations (H2171, H526, H1184, H2029, H2196) expressed RB1 protein. One cell line with a missense RB1 mutation (H209) expresses this protein. RB1 protein expression was notable in DMS454 cells because of its inconsistency, whereas RB1 expression in H841 cells was always present but often demonstrated a lower molecular weight than expected.
Figure 2. RB1 mutation and expression in SCLC cell lines.
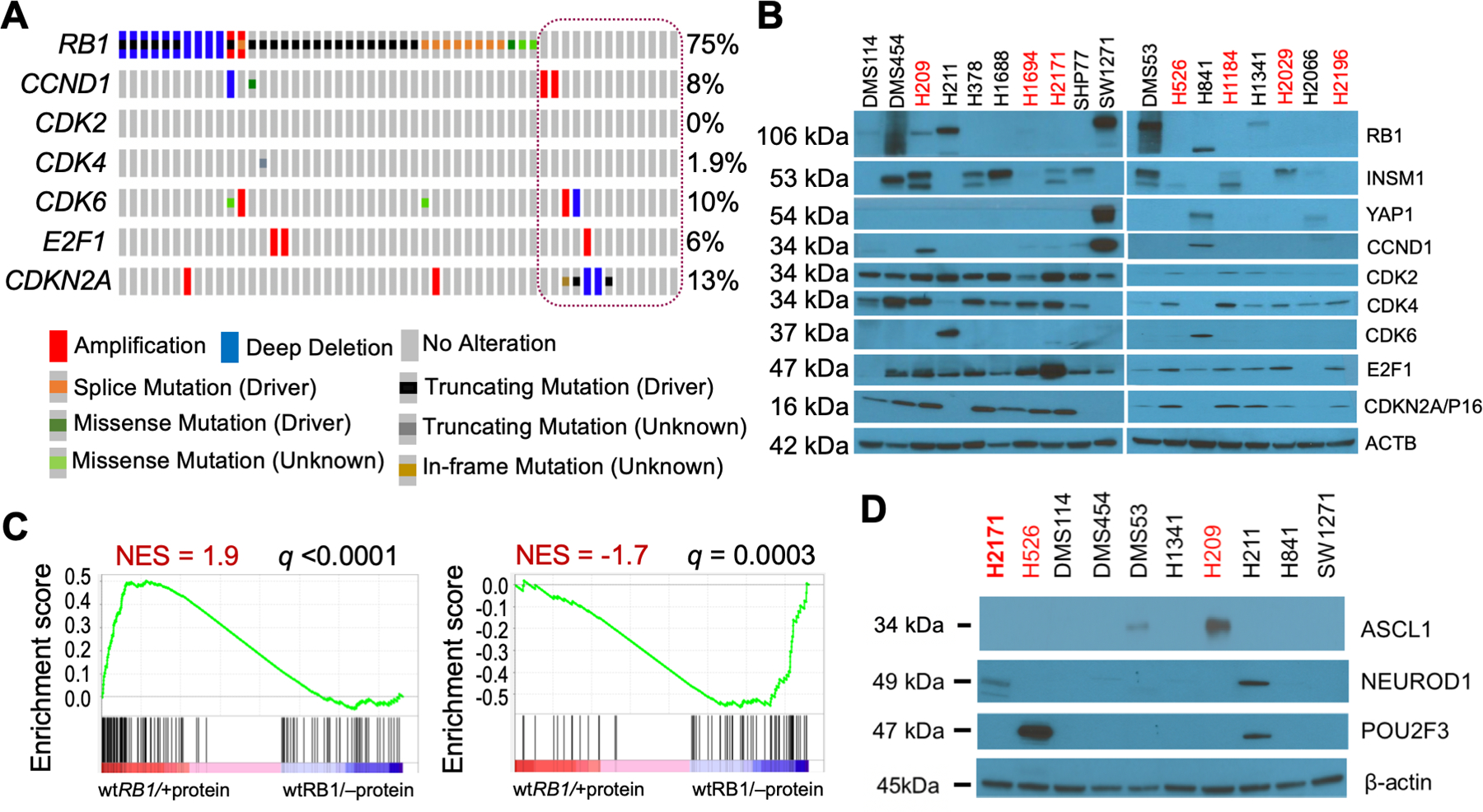
A. Oncoprint of mutations in select E2F pathway genes in SCLC cells. CCLE data (2019) for all 52 SCLC cells was visualized in cBioPortal using Oncoprint. The cells annotated as wt RB1 are circled. The genes queried were restricted to those examined by western blotting in Figure 2B. All the wt RB1 cells shown in Figure 2B are represented in the circle (column 1=DMS53, 2=DMS114, 3=DMS454, 4=H211, 5=H841, 6=H1092, 7=H1339, 8=H1341, 9=H2066, 10=H2286, 11=SBC5, 12=SHP77, 13=SW1271, L to R). B. Western blot analyses of wt RB1 (black text) and mutant RB1 (red text) cell lysates for expression of the proteins listed at right. Molecular size of bands is shown on the left. β-actin was used as a loading control. The specific mutations in the mutant RB1 cells are: H209 = missense p.C706F, H1694 = missense p.Q444L, H2171 = nonsense p.S567*, H526 = nonsense p.E31*, H1184 = nonsense p.E19*, H2029 = splice site, H2196 = splice site p.L738fs. C. GSEA analyses of wt RB1 SCLC cells with RB1 protein expression compared to wt RB1 SCLC cells lacking RB1 expression. GSEA analysis was performed using 186 RB1 LOF signature genes, including 118 downregulated and 68 upregulated genes. Downregulated genes due to RB1 loss are enriched in RB1 protein expressing cells (left panel), while upregulated genes due to RB1 loss are enriched in cells without RB1 protein (right panel). Cells used in wt RB1/+protein group: DMS114, DMS53, H1341, H211, H841, SW1271; and wt RB1/–protein group: H378, H1688, SHP77, H2066. D. Same as panel B.
Our results in SCLC tumors and cell lines demonstrated that the genomic mutation status of RB1 determined using WES may not correlate with RB1 protein expression and function. We used the RB1 western blotting results in Fig. 2B, together with matching RNA-seq data downloaded from the Cancer Cell Line Encyclopedia (CCLE), to screen RNA signatures that could accurately predict RB1 expression and function. We identified an RB1 LOF signature (23) that was developed using CCLE and TCGA data across many cancers. This RB1 LOF signature consists of 186 differentially expressed genes associated with RB1 loss, including 118 down-regulated and 68 up-regulated genes (Sup. Table 2). Our initial analysis indicated that the down-regulated genes of this signature showed enrichment in wt RB1 SCLC cells with RB1 protein expression (Fig. 2C, left panel), while the up-regulated genes of this signature were enriched in wt RB1 SCLC cells lacking RB1 protein expression (Fig. 2C, right panel), indicating that the signature can potentially discriminate between RB1 protein positive and negative wt RB1 cells. We later tested if this RB1 LOF signature could be used as a classifier to predict RB1 functionality based on CDK4/6 inhibition (see results described in the next section).
Previously, we showed that RB1 protein expression was positively correlated with YAP1 expression but negatively correlated with INSM1 expression in SCLC cells (18). Therefore, we investigated whether these associations were consistent in an expanded cohort of SCLC cell lines. As shown in Fig. 2B and Sup. Fig. 4, YAP1 expression was observed in only three of the seven consensus protein-positive wt RB1 cell lines (DMS114, H841, and SW1271), whereas INSM1 expression was absent in five of the seven (DMS114, H211, SW1271, H841, H1341). We then examined the expression of three genes (ASCL1, NEUROD1, POU2F3) used as transcriptional classifiers in SCLC in addition to YAP1 (17). As shown in Fig. 2D, only H211 cells demonstrated the expression of any additional classifier gene. H2171 and H526 cells were used as RB1 negative, but classifier-positive control cells. Because ASCL1, NEUROD1 and INSM1 are all considered NE genes, our data suggests that RB1 positive, wt RB1 SCLC cells are frequently NE negative, in contrast with human SCLC tumors from patients.
RB1 is functional within the E2F pathway
Our results show that over half of the SCLC cells annotated as wild-type RB1 and one of the two cell lines with missense RB1 mutations expressed detectable RB1 protein. Therefore, it was important to determine whether RB1 function in these cells was active or downregulated by endogenous cyclin/CDK overexpression or CDK inhibitor loss in the E2F pathway. Therefore, we examined the genomic mutation profile and protein expression of six additional members of the E2F pathway (Figs. 2A and 2B). Although the results of the seven wt RB1 SCLC cell lines (DMS114, DMS454, H211, SW1271, DMS53, H841, H1341) demonstrated that RB1 expression was associated with increased cyclin D (SW1271, H841) or CDK6 (H211, H841) expression and decreased CDK4 (H211, SW1271, DMS53, H841) and CDKN2A/p16 (H211, SW1271, H841) expression, none of these changes reflected matching genomic alterations in these cells, except for deletion of CDKN2A in SW1271 cells. Thus, most changes observed in E2F pathway proteins of wt RB1 SCLC cells are likely physiologic and not genetic.
To directly address RB1 functionality, we determined the sensitivity of wt RB1 SCLC cells to the CDK4/6 inhibitor palbociclib (PD0332991) because of the central role these two CDKs play in cell cycle progression. Initially, we examined the effect of 1.0 μM palbociclib on RB1 phosphorylation in wt RB1 SCLC cells using western blotting. The results, shown in Fig. 3A, demonstrated a sustained decrease in phospho-Ser780 RB1 in DMS53, H211, and SW1271 cells, but not in H1341 cells. H841 and DMS114 cells demonstrated parallel decreases in total RB1 and phospho-Ser780 RB1. The positive control NSCLC A549 cells demonstrated a rapid and transient decrease in phospho-Ser780 RB1, whereas the negative control H209 SCLC cells, in which RB1 was expressed but reportedly is non-functional (24), also displayed some loss of phospho-Ser780 RB1. FACS analyses showed an increased fraction of wt RB1 SCLC cells in the G1 phase of the cell cycle after drug treatment, except for DMS53 (Fig. 3B). Taken together, using palbociclib sensitivity as a tool, our results indicate that RB1 is part of a functional E2F pathway in DMS114, DMS53, H211, H841, and SW1271 SCLC cells.
Figure 3. RB1 is functional in wt RB1 SCLC cell lines.
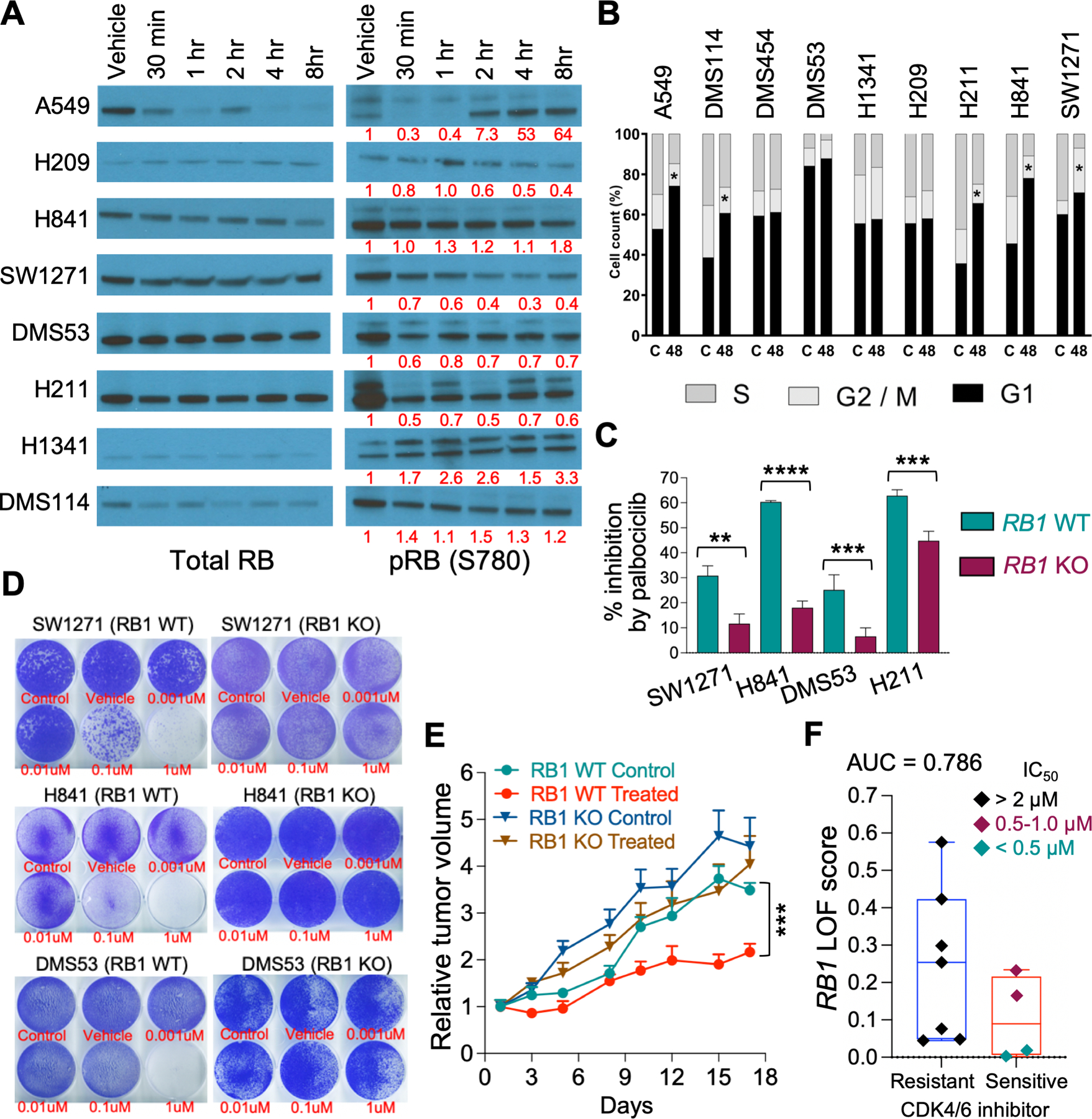
A. Western blot analyses of total RB1 and pSer780-RB1 expression in wt RB1 cell lysates after incubation with 1.0 µM palbociclib for the indicated times. β-actin was used as a loading control but is not shown. The bands for SCLC total RB1 were the correct molecular size and generally mirrored the β-actin loading control. Bands for RB1 were quantified by IMAGE J and the numbers under the pRB1 bands represent the pRB1/total RB1 ratio normalized to the vehicle control. B. FACS analyses of growth inhibition by 1.0 µM palbociclib after 48 hours. Results show the average of triplicates for the percentage of cells in each of the three cell cycles. C. Plot of the percent cell growth inhibition by 1.0 μM palbociclib in vector control (WT) and matching RB1 CRISPR knockout (KO) cells measured by real-time imaging after five (SW1271, H841, H211) or ten (DMS53) days. D. Images of growth inhibition by various concentrations of palbociclib, shown below wells in red text, in vector control (C) and matching RB1 CRISPR knockout (KO) WT RB1 cells, as determined by colony formation assays over five (SW1271 and H841) or ten (DMS53) days. E. Growth inhibition by 100 mg/kg palbociclib in xenograft tumors of H841 vector control (Control) and matching RB1 CRISPR knockout (KO) wt RB1 cells. Statistical differences were determined at Day 17. F. RB1 LOF signature scores between palbociclib resistant and sensitive cells. SCLC cell lines were treated with PD0332991 (palbociclib) and IC50 values calculated using linear regression analysis (18). Box plot showing RB1 LOF scores between palbociclib resistant (IC50 >2 μM: H1048, H1092, H1694, H209, H2171, H446, and SHP77) and sensitive (IC50 <1 μM: DMS114, H211, H841, and SW1271) groups. Area under the ROC curve analysis (AUC = 0.786) showed the accuracy of RB1 LOF classifier.
To confirm that RB1 protein expression was necessary for palbociclib sensitivity in wt RB1 SCLC cells, we used CRISPR to stably knock out RB1 expression in DMS53, H211, H841, and SW1271 cells. We isolated pools of RB1 knockout cells from all four cell lines and isolated individual clones from the H841 and SW1271 pools. The efficiency of the knockout was confirmed by western blotting for RB1 expression (Sup. Fig. 1). Interestingly, RB1 knockout had little effect on NE or YAP1 expression (Sup. Fig. 5). Initially, we studied the effect of RB1 knockout on palbociclib-induced growth inhibition by live-cell measurement of cell proliferation using the IncuCyte ZOOM. Palbociclib induced significant growth inhibition in the vector controls of all four cell lines, ranging from 30–60%, which was reduced in the matching RB1 knockout cell lines (Figure 3C). Interestingly, in all four cell lines, RB1 knockout had little effect on the basal growth rate compared to the matching vector controls (Sup. Fig. 6). We then performed colony formation assays to validate the IncuCyte ZOOM results (Fig. 3D). Once again, in all cell lines examined, the vector control cells displayed greater sensitivity to palbociclib growth inhibition than the matching RB1 knockout cells. H211 cells were not tested because they are loosely adherent. The effect of RB1 knockout on palbociclib sensitivity was further tested in xenograft tumor models of H841 cells. As shown in Fig. 3E, both control (RB1 WT control) and RB1 knockout (RB1 KO control) tumors demonstrated nearly equivalent rates of tumor growth. However, palbociclib treatment demonstrated significant growth inhibition only in tumors initiated from vector control H841 cells (RB1 WT treated). Taken together, these experiments clearly show that RB1 is part of a functional E2F pathway and is necessary for palbociclib sensitivity in SCLC cell lines expressing wild-type RB1 protein. Next, we used the RB1 LOF signature to determine if it could predict CDK4/6 inhibitor sensitivity. We treated 11 SCLC cell lines with the CDK4/6 inhibitor PD0332991 (palbociclib) and calculated IC50 values by linear regression analysis (described in ref. 18). Next, we grouped SCLC cells into resistant (IC50 >2 μM: H1048, H1092, H1694, H209, H2171, H446, and SHP77) and sensitive (IC50 <1 μM: DMS114, H211, H841, and SW1271) groups. Finally, we computed the RB1 LOF signature scores as described by Chen et al (23) for these cells and found that they correlated with CDK4/6 inhibitor sensitivity (Fig. 3F, AUC = 0.786).
RB1 expression is targetable in SCLC patients
Based on our preclinical research, we recently initiated a clinical trial and treated two patients with biopsy-validated wt RB1 SCLC and platinum-refractory disease with abemaciclib (LY2835219), a CDK4/6 inhibitor, as a single agent (NCT04010357, ClinicalTrials.gov). The first patient did not respond to abemaciclib; however, the second patient demonstrated dramatic and rapid shrinkage of the tumor within days of treatment, measured by either the allelic fraction of mutant TP53 in ctDNA (Fig. 4A) or CT scans of the lung and neck (Fig. 4B). Interestingly, patient 1, who did not respond, showed no RB1 expression on IHC. When we stained the diagnostic biopsy of patient 2 by IHC, the tumor was positive for RB1 expression and demonstrated the characteristic histological features of SCLC (Fig. 4C). Further IHC staining was positive for ASCL1, INSM1, synaptophysin, and Ki67, but negative for NEUROD1, POU2F3, YAP1, and chromogranin A (Fig. 4D and Sup. Fig. 7). Thus, similar to our previous IHC results, the RB1 positive tumor of patient 2 demonstrated robust expression of multiple NE genes. The genomic mutation profile of patient 2 contained no other biologically relevant oncogenes or tumor suppressors, except for a missense mutation in KEAP1 (Sup. Table 8).
Figure 4. Response of a wt RB1 SCLC patient to abemaciclib treatment.
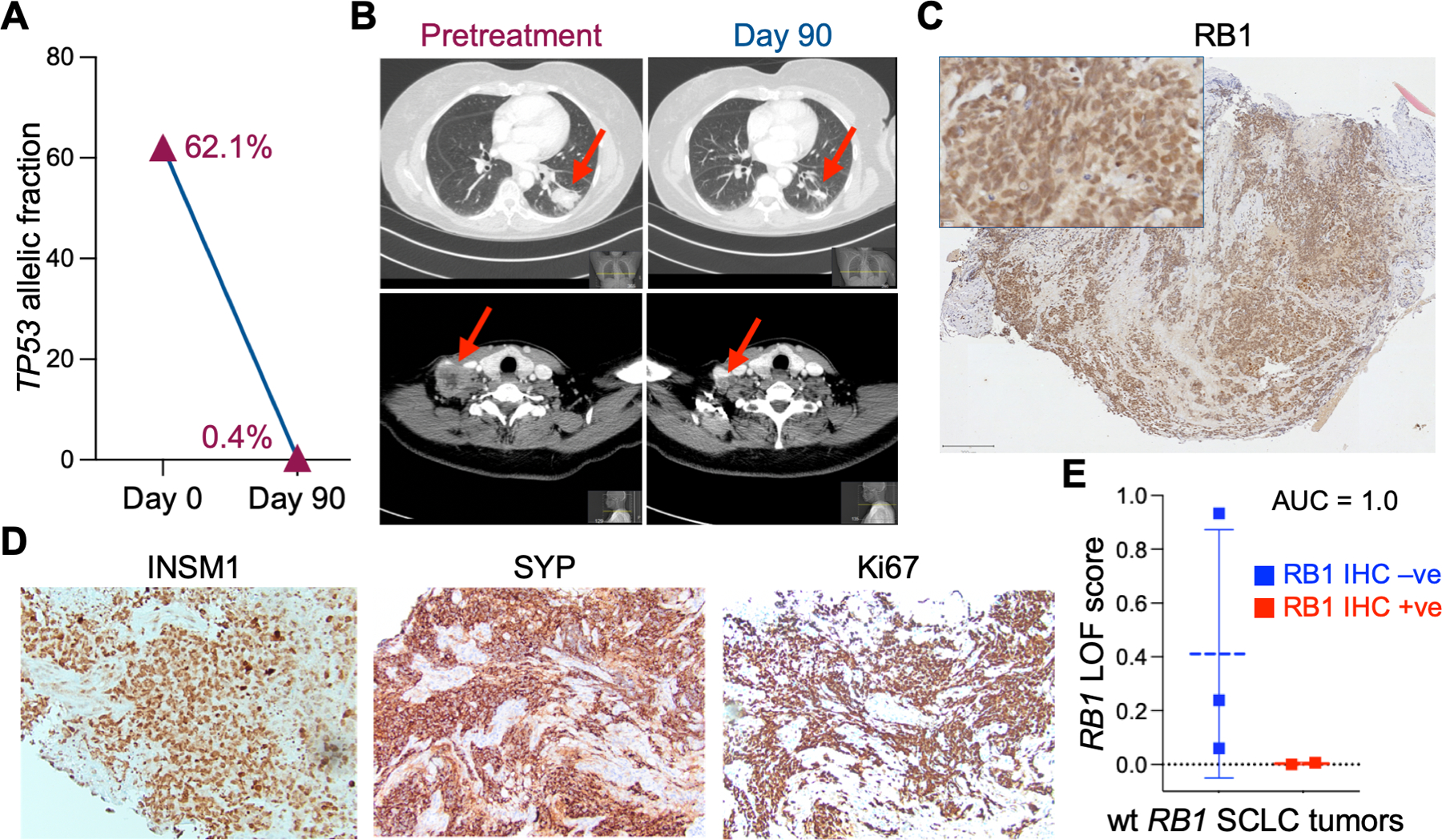
A. The allelic fraction of mutant TP53 in ctDNA was measured before (day 0) and during (day 92) abemaciclib treatment. B. CT scans from different axial planes of patient 2 taken before (Day 0) and during (Day 90) treatment with abemaciclib. Arrows indicate tumor in lung (top panels) and neck (bottom panels). C. RB1 IHC staining of a pretreatment biopsy of patient 2 tumor. Scale bar = 200 μM. D. Additional IHC stains of a second pretreatment biopsy of patient 2 tumor. E. RB1 LOF signature scores of wt RB1 SCLC patients with RB1 positive (+ive) IHC compared to wt RB1 patients with RB1 negative (-ve) IHC. Area under the ROC curve analysis (AUC = 1.0) showed high accuracy of the RB1 LOF classifier. Analysis of patient 2 data is included. Patient 1 had no RNA-seq data available. The four additional wt RB1 patients analyzed were not enrolled in the clinical trial.
We next determined how well the RB1 LOF signature score performed on human biopsy specimens. We calculated the RB1 LOF signature scores of 5 SCLC patients that had wt RB1 genomic status and matching RB1 IHC expression results. When we compared the RB1 LOF signature scores of RB1 IHC negative (-ve) with IHC positive (+ve, including patient 2), we found that the RB1 LOF signature scores were strongly correlated with RB1 protein expression (Fig. 4E, AUC = 1.0). Thus, although limited in cohort size, these results from human biopsies demonstrate the potential of the RB1 LOF signature score to predict the efficacy of CDK4/6 inhibitors in SCLC.
DISCUSSION
Although genomic analyses of SCLC have uniformly reported that almost all (>95%) SCLC tumors harbor missense mutations in TP53, the number of reported tumors harboring alterations in RB1 encompasses a much wider distribution. Although this broad range may be attributed, in part, to technical issues related to sequencing platforms, it does not diminish the idea that a cohort of SCLC harboring wt RB1 exists. Here, we validated the existence of this subgroup by demonstrating that ~14% of 62 SCLC patient tumors were positive for nuclear RB1 protein expression. Results from our SCLC cell line studies indicated that ~50% of cells annotated as wt RB1 expressed RB1 protein, indicating that ~12% of all SCLC cells in the CCLE demonstrated RB1 protein expression, which is in good agreement with our IHC results. Thus, given the limitations of the small cohorts analyzed and allowances for diagnostic variability, our data provide clear evidence for a small subgroup of SCLC with RB1 expression.
Our cell line studies demonstrated that most RB1-positive SCLC cells are sensitive to growth inhibition by the CDK4/6 inhibitor palbociclib, and that growth inhibition by palbociclib was dependent on the presence of RB1 protein. These results validate and extend our previous work showing that CDK4/6 sensitivity was largely restricted to wt RB1, and not mutant RB1, SCLC cells (18). Collectively, these two studies indicate that if RB1 is expressed, it is likely to exist in a functional and targetable E2F pathway. This idea is highlighted by the response of a SCLC patient with RB1 expression to the CDK4/6 inhibitor abemaciclib. Targeting the normal function of a pathway is appealing, especially in SCLC, where there are no actionable mutated targets.
Our previous (18) and current results in SCLC cells indicate that NE expression is largely absent in wt RB1 SCLC cells (Figs. 2B and 2D), which is in agreement with other studies (25). Lack of NE gene expression has always been viewed as a criterion to identify the so-called variant SCLC (26). Therefore, it was surprising that the majority of our RB1 positive tumors expressed the NE genes INSM1 and ASCL1 (Table 1, Fig. 1D). Although the detection of NE genes synaptophysin and chromogranin is typically used to support and improve SCLC diagnosis (27), INSM1 expression has recently been added as a diagnostic marker for SCLC (28). The other genes surveyed, ASCL1, NEUROD1, POU2F3 and YAP1, were all experimental and were only used to stratify SCLC into transcriptomic subgroups (17). However, our CDK4/6 responsive patient also exhibited strong INSM1, ASCL1, and synaptophysin expression (Fig. 4 and Sup. Fig. 7), indicating that RB1 positive SCLC is not restricted to a variant subgroup. Interestingly, at least in vitro, NE expression was not directly repressed by RB1, as CRISPR knockout of RB1 in the four wt RB1 KO SCLC cell lines did not induce any de novo upregulation of INSM1, ASCL1, or NEUROD1 expression (Sup. Fig. 5) and had only a small effect on pre-existing NE gene expression. Taken together, these results demonstrate that RB1 protein expression does not clearly correlate with a non-neuroendocrine phenotype in human tumors but may be associated with the YAP1 transcriptional subgroup of SCLC.
Since RB1 loss is regarded as a ‘default’ characteristic of SCLC, our results raise questions about its strict requirement in SCLC. Although the requirement for dual TP53/RB1 inactivation in SCLC tumorigenesis is supported by knockout mouse models of this cancer (13), it remains unclear whether functional knockout of the RB1 pathway, perhaps through endogenous cyclin amplification and/or CDK inhibitor loss, could achieve the same result. Although such genetic alterations are observed in some wild-type RB1 SCLC cell lines (Fig. 2A), they did not seem to abrogate palbociclib sensitivity (Fig. 3). Furthermore, although these alterations were present in our SCLC genomic cohort, their frequency was very low (<3/120 SCLC cases). Notably, our CDK4/6 responsive patient did not harbor any cyclin/CDK amplification or CDK inhibitor loss (Sup. Table 8). Thus, whether co-mutations in wt RB1 SCLC patients can alter sensitivity to CDK4/6 inhibitors awaits further patient recruitment.
CDK4/6 inhibitors are FDA-approved for use in estrogen hormone receptor (HR)-positive, HER2-negative advanced breast cancer. Unlike SCLC, these tumors rarely harbor RB1 loss, however they do demonstrate recurrent amplification of CCDN1 (~24%) or CDKN2A/p16 loss (<5%), making this cancer an ideal target for CDK4/6 inhibitors (29). Nevertheless, selection for these two gene alterations was not found to improve patient outcomes with CDK4/6 inhibitors (29, 30), suggesting that co-mutations in the E2F pathway do not alter sensitivity to CDK4/6 inhibitors. Thus, there remains an ongoing effort to identify predictive biomarkers for CDK4/6 inhibitor sensitivity for breast cancer beyond HR positivity, which itself has been challenged (31).
It is clear that not all SCLC patients with wt RB1 genomic status express RB1 protein, as shown by our cell line and human tumor results. This validates a previous assertion that gross chromosomal alterations beyond those detectable by WES can disrupt RB1 expression (12). This makes RB1 mutation status determined by targeted or WES a useful but imperfect predictor of RB1 expression and potential CDK4/6 inhibitor sensitivity. However, we showed that the RB1 LOF signature could better predict functional RB1 protein as the RB1 LOF signature score correlated well with CDK4/6 inhibitor sensitivity (Fig. 3F) and was further validated in human tumors (Fig. 4E). The CDK4/6 inhibitor trilaciclib has been shown in clinical trials to preserve immune cell function in SCLC patients receiving chemotherapy (32). Here, we provide data supporting the additional benefit of using these inhibitors in patients with wild-type RB1 tumors, making the translational potential of our findings immediate. Given the limitations of the small cohorts analyzed in our study, it is important to validate these findings in larger prospective clinical trials and to follow these patients to determine the durability of the CDK4/6 inhibitor response.
Supplementary Material
ACKNOWLEDGEMENTS
Funding was obtained from the National Institutes of Health grant R21 CA226322 (AD), a National Institutes of Health SCLC consortium grant U24 CA213274 (AD), and a National Institutes of Health grant P30 CA043703 to the Case Comprehensive Cancer Center to support its Integrated Genomics, Biostatistics & Bioinformatics, and Tissue Resources Cores. Additional support was provided by the Seidman Cancer Center of the University Hospitals of Cleveland Medical Center (AD).
Footnotes
Conflict-of-interest statement: “The authors declare that no conflicts of interest exist.”
REFERENCES
- 1.Majeed U, et al. Targeted therapy in advanced non-small cell lung cancer: current advances and future trends. J Hematol Oncol 2021,14:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Z, et al. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Can 2014,14:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas A, et al. Refining the treatment of NSCLC according to histological and molecular subtypes. Nat Rev Clin Oncol 2015,12:511–526. [DOI] [PubMed] [Google Scholar]
- 4.Tan W-L, et al. Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol 2016,17:e347–362. [DOI] [PubMed] [Google Scholar]
- 5.Rudin CM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Gen 2012,44:1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peifer M, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Gen 2012,44:1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umemura S, et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. J Thor Oncol 2014,9:1324–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwakawa R, et al. Expression and significance of genes frequently mutated in small cell lung cancers defined by whole exome/RNA sequencing. Carcinogenesis 2015,36:616–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross JS, et al. Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer. J Clin Pathol 2014,67:772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyoshi T, et al. Genomic profiling of large-cell neuroendocrine carcinoma of the lung. Clin Cancer Res 2017,23:757–765. [DOI] [PubMed] [Google Scholar]
- 11.Vollbrecht C, et al. Mutational analysis of pulmonary tumours with neuroendocrine features using targeted massive parallel sequencing: a comparison of a neglected tumour group. Br J Cancer 2015,113:1704–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.George J, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015,524:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meuwissen R, et al. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003,4:181–189. [DOI] [PubMed] [Google Scholar]
- 14.Gazdar AF, et al. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer 2017,17:725–737. [DOI] [PubMed] [Google Scholar]
- 15.Mollaoglu G, et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Can Cell 2017,31:270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iams WT, et al. Immunotherapeutic approaches for small-cell lung cancer. Nature Rev Clin Oncol 2020,17:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudin CM, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nature Rev Cancer 2019,19:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McColl K, et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 2017,8:73745–73756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simpson KL, et al. A biobank of small cell lung cancer CDX models elucidates inter- and intra-tumoral phenotypic heterogeneity. Nature Cancer 2020,1:437–451. [DOI] [PubMed] [Google Scholar]
- 20.Gay CM, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021,39:346–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dowlati A, et al. Clinical correlation of extensive-stage small cell lung cancer genomics. Annals Oncol 2016,27:642–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dabir S, et al. CD30 is a potential therapeutic target in malignant mesothelioma. Mol Cancer Ther 2015,14:740–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen WS, et al. Novel RB1-loss transcriptomic signature is associated with poor clinical outcomes across cancer types. Clin Can Res 2019,25:4290–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaye FJ, et al. A single amino acid substitution results in a retinoblastoma protein defective in phosphorylation and oncoprotein binding. Proc Natl Acad Science USA 1990,87:6922–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonkin D, et al. Are neuroendocrine negative small cell lung cancer and large cell neuroendocrine carcinoma with wt RB1 two faces of the same entity? Lung Cancer Manag 2019,8:LMT13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, et al. Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl Lung Cancer Res 2018,7:32–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thunnissen E, et al. The use of immunohistochemistry improves the diagnosis of small cell lung cancer and its differential diagnosis. An international reproducibility study in a demanding set of cases. J. Thorac Oncol 2017,12:334–346. [DOI] [PubMed] [Google Scholar]
- 28.Rooper LM, et al. INSM1 demonstrates superior performance to the individual and combined use of synaptophysin, chromogranin and CD56 for diagnosing neuroendocrine tumors of the thoracic cavity. Am J Surg Pathol 2017,41:1561–1569. [DOI] [PubMed] [Google Scholar]
- 29.Shah M, et al. CDK4/6 inhibitors: Game changers in the management of hormone receptor-positive advanced breast cancer? Oncology 2018,32:216–222. [PMC free article] [PubMed] [Google Scholar]
- 30.Finn RS, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomized phase 2 study. Lancet Oncol 2015,16:25–35. [DOI] [PubMed] [Google Scholar]
- 31.Cristofanilli M, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase-3 randomised controlled trial. Lancet Oncol 2016,17:425–439. [DOI] [PubMed] [Google Scholar]
- 32.Weiss JM, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Annals Oncol 2019,30:1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Tumor and blood targeted DNA sequencing data are available in Supplementary Tables. Tumor RNA-seq data are deposited in Gene Expression Omnibus and are accessible using accession number GSE217646.