

Abstract
Genomic architecture appears to play crucial roles in health and a variety of diseases. How nuclear structures reorganize over different timescales is elusive, partly since the tools needed to probe and perturb them are not as advanced as needed by the field. To fill this gap, the National Institutes of Health Common Fund started a program in 2015, called the 4D Nucleome (4DN), with the goal of developing, and ultimately applying technologies, to interrogate the structure and function of nuclear organization in space and time.
eTOC Blurb:
In this Perspective, Roy et al. share the progress of the NIH Common Fund 4DNucleome Program, which aims to map the structure and function of genomic architecture in space and time by developing necessary tools and techniques and delivering these resources for the wide scientific community.
The 4D Nucleome (4DN) Program—setting the stage
One of the most remarkable feats of nature is packing two meters of genomic DNA into a tiny (<10 micrometer) nuclear space, achieving more than 200,000-fold linear reduction1. This phenomenon raises several questions: Is this a random process? If not, what are the rules of folding? How does folding relate to gene expression and regulation? Are there cell-type and cell-state specific folding patterns? Is this folding regulated and does it have multiple stable configurations or is it dynamic? And finally, is folding functionally relevant for health? Three-dimensional structure plays a role beyond the linear genome and its modifications2 and a concerted and multi-prong approach to understand the constraints, interactions and structure-function relationship of the genome packed into the nucleus is needed. This will allow to comprehensively understand the organization of the genome and its relationship to normal health and disease in both three-dimensional space and time (the fourth dimension in the 4DN).
Many people have been working on elucidating the basic scheme for multi-level packaging of linear DNA into the nucleus and have identified many important physical and chemical constraints3. More importantly, the relationship between genome folding and regulation of gene expression has been studied for at least the last two decades4. The research community has known for some time that the nucleus is partitioned into euchromatin and heterochromatin regions—and that actively transcribed genes are associated with the euchromatin, while repressed genes are most frequently associated with heterochromatin5–7. This is not static--rather, genomic regions move in and out of these regions as they are expressed under certain conditions and in certain cellular states8. For instance, during mitosis, the nuclear envelope breaks down and transcription of most genes ceases before a new daughter cell is produced. In addition, tools have been developed that enabled the identification of chromosome territories, domains, and the process for chromosome segregation9, paving the way for an organized research program such as the 4DN to better understand the spatial-temporal dynamics of the nucleus, such as longer-range interactions for example between promoter and enhancer regions. However, many unanswered questions remained. How do folding patterns facilitate or constrain gene transcription that may vary from cell to cell and over time? How do these long-range interactions between different segments of the genome occur despite folding and nuclear dynamics and how can we visualize or detect them? Central to the conceptualization of the 4DN program was the need to develop methods to detect these interactions and elucidate their significance in various cell types and in distinct cell states10.
A landmark sequencing method, termed chromatin conformation capture (3C), developed by Dekker et al.11, changed the way these interactions are detected. This method cross-links spatially close genomic loci to quantify 3D interactions between different genomic locations. By visualizing the data as a contact map based on linear genomic sequence, different long-range interactions and their frequencies can be resolved. Several variants and refinements of the method had been developed over the years leading up to the 4DN program, including Hi-C12, ChIA-PET13, and Micro-C14.
In parallel, there have been significant advances in light and electron imaging methods in the past decade. Fluorescence in-situ hybridization (FISH) has been a mainstay in visualizing chromatin conformations, and the combination of super-resolution microscopy and high affinity, stable, chromosome painting techniques15 have enabled measurement of genomic architecture from megabase to nucleotide resolution. Imaging advances have opened the possibility for more comprehensive mapping including the extension of FISH to 3D-FISH16, and the development of variants such as cryo-FISH17. Additionally, during phase 1 of the 4DN program (2015 – 2020), researchers at the intersection of genomic and imaging approaches developed cutting-edge techniques such as genome architecture mapping (GAM)18, and beginning to make an impact in cancer and neuroscience research19,20.
Establishing 4DN (2013 – 2015)
Following a series of discussions with panels of scientific experts and stakeholders, as well as input from the broad community, the NIH Common Fund established the 4DN program in 2015 to investigate the question of how, when a normal, healthy cell becomes diseased, does the chromatin architecture change and what can we learn about the relationship between structure and function? The 4DN program was conceived as two five-year phases, the first phase focusing on establishment of the tools and principles for spatiotemporal mapping of the architecture and the second phase emphasizing the relationship between spatial organization and function. It was clear from the beginning that to achieve these goals and generate resources for the community, a concerted team effort would be needed that could not be achieved by any individual laboratories or even through a series of individual projects. In a consortium of this proportion, investigators are tasked to adopt a community-centric focus rather than concentrating on their own individual research interests. This represents a shift in thinking as well as decision-making for themselves and their laboratories. While mapping the functional organization of the genome was deemed critical to fully understand disease pathways, it was equally important to systematically develop next generation tools and techniques as key deliverables for the broad scientific community to probe the nuclear architecture, requiring a synergistic and global effort. In addition, metrics and standards needed to be developed and adopted by a community of investigators, not just individuals. Thus, a broad community of practice needed to be established to fully achieve these objectives.
The following scientific areas were identified as priorities for the first phase of the program: 1) next-generation, high-resolution and high-throughput tools to explore the relationships between nuclear organization and the regulation of gene expression programs, down to single cell resolution; 2) computational tools to integrate existing data sets, and to manage, analyze, and visualize the datasets generated by a community effort; 3) predictive models of nuclear structure/function relationships in the context of various cellular states or transitions (differentiation, reprogramming, cell cycle, responses to external stimuli, disease development); 4) next-generation tools to explore nuclear dynamics through controlled perturbation of nuclear conformation; and finally 5) “pilot” maps (using in-development technologies) and “reference” maps (using robust and validated technologies) of the genomic architecture of the interphase nucleus for a variety of eukaryotic cells and tissues. The overall vision for the program was that these initiatives together would develop and deliver necessary technologies and tools and creating a space for a community of users to probe the structure and function of the nucleus.
The philosophy of, and Congressional mandate for, the NIH Common Fund is to identify cross-cutting areas of science that would benefit from a short-term (must not be more than 10 years), goal-driven research program that can catalyze and transform the chosen field. These programs are intended to change paradigms, develop or implement innovative tools and methods, and/or provide fundamental resources for research that can be used by the global biomedical science community. These programs most often involve a combination of highly coordinated research to develop large datasets or deploy innovative technology to further enhance the capabilities of the consortium, and pilot demonstration projects to road test the value of the data resources. 4DN Phase 1 consisted of four technology-driven funding initiatives aligned with the priorities stated above, an organizational hub was established to manage the consortium internally as well as coordinate with other national and international entities working on related topics and to engage in outreach efforts to inform the broad scientific community about the progress and resources generated by the consortium. Finally, given that the consortium was designed to generate multiple genomic and imaging data sets, a data coordination center was created to develop data and metadata standards along with data ingestion management, analysis, and visualization of data for public consumption in coordination with the organizational hub. 29 projects were funded, with a total budget of ~$128 million over the first 5 years21.
4DN Phase I (2015 – 2020)
While nucleomic research began before the NIH program started, the field blossomed since the 4DN was established in 2015. We acknowledge that covering the entire field of 4DN research is beyond the scope of this perspective and thus, we specifically outline some of the achievements of the first 5 years of the 4DN program and how they might benefit the goals of phase 2 and the wider research community.
Technology Development
Accelerating technology development was a key goal of the first phase of 4DN, with the focus on techniques that would be crucial in phase 2 of the program. As a result, the consortium produced an abundance of new genomic and imaging technologies as well as refinements and extensions of existing approaches (see Table 1). Below is a brief synopsis of a selection of these technologies.
Table 1.
Selection of technologies developed during the first phase of tchia-he 4DN Program and the number of citations found using the NIH iCite (https://icite.od.nih.gov) tool (Hutchins, Yuan, Anderson, & Santangelo, 2016)50.
| Technology | Reference | Citations |
|---|---|---|
| CUT&Tag | (Kaya-Okur, et al., 2019)26 | 489 |
| ChromEMT | (Ou, et al., 2017)31 | 418 |
| sn-Hi-C | (Flyamer, et al., 2017)22 | 403 |
| OptoDroplet | (Shin, et al., 2017)40 | 377 |
| DNA SPRITE | (Quinodoz, et al., 2018)24 | 376 |
| CRISPRrainbow | (Ma, et al., 2016)32 | 224 |
| PLAC-seq | (Fang, et al., 2016)56 | 167 |
| TSA-seq | (Chen, et al., 2018)57 | 155 |
| Micro-C (in mammalian cells) | (Krietenstein, et al., 2020)58; (Hsieh, et al., 2020)59 | 152; 155 |
| ChIA-Drop | (Zheng, et al., 2019)25 | 103 |
| RNA-DNA SPRITE | (Quinodoz, et al., 2021)60 | 67 |
| C-BERST | (Gao, et al., 2018)61 | 67 |
| NAD-seq | (Vertii, et al., 2019)62 | 41 |
| TrAC-loop | (Lai, et al., 2018)63 | 39 |
| High Resolution Repli-seq | (Zhao, Sasaki, & Gilbert, 2020)64 | 35 |
| Sci-Hi-C | (Ramani, et al., 2020)65 | 34 |
| pa-DamID | (van Schaik, Vos, Peric-Hupkes, Hn Celie, & van Steensel, 2020)66 | 30 |
| Liquid Chromatin Hi-C | (Belaghzal, et al., 2021)67 | 30 |
| immuno-GAM | (Winick-Ng, et al., 2021)20 | 25 |
| Multi-contact 3C | (Tavares-Cadete, Norouzi, Dekker, Liu, & Dekker, 2020)68 | 25 |
| iMARGI | (Wu, et al., 2019)27 | 15 |
| Hi-C 3.0 | (Lafontaine, Yang, Dekker, & Gibcus, 2021)69 | 7 |
Genomic Technologies
At the start of the 4DN, there were limited methods to study the dynamics of chromatin changes during development, largely due to the small sample sizes. To overcome this problem, Mirny and colleagues developed a Hi-C method protocol for single nuclei (snHi-C22) that provides >10-fold more contacts per cell than the previous methods23 and demonstrated that the chromatin topology is significantly reorganized during the oocyte-to-zygote transition in murine model system. During development, the chromatin is reorganized after fertilization to produce a totipotent zygote that has the capability to generate a new organism. Strikingly, it is distinct in paternal and maternal nuclei even within a single-cell zygote. They observed that topologically associating domains (TADs) and loops but not compartments are present in zygotic maternal chromatin. These observations suggest that these features are generated by different mechanisms.
An exciting advance is the ability to detect higher order interactions using a variety of methods; previous methods were limited to detection of pairwise complexes and could not distinguish models such as ternary complexes or alternative pairwise complexes. Guttman and colleagues developed split-pool recognition of interactions by tag extension (SPRITE), to detect genome-wide higher-order interactions within the nucleus24 using a split-pool barcoding strategy. Most methods to study genome-wide structure are based on proximity ligation, the probability of which has been experimentally quantified18, and can fail to detect some interactions. Guttman and colleagues demonstrated via SPRITE that long-range nuclear interactions include two major inter-chromosomal hubs and that these two inter-chromosomal hubs correspond to DNA organization around the nuclear bodies--nucleolus and nuclear speckles, respectively-- thereby providing a global model of 3D packaging of genome in the nucleus. Unlike Hi-C and ChIA-PET, which rely on pairwise proximity ligation and reflects population-level analysis, ChIADrop is a method developed by the 4DN to score for multiplex chromatin-interactions using droplet-based and barcode-linked sequencing at single cell level with single molecule precision25. Importantly, the protocol required a small number of cells (~5 × 103cells) and functionally characterized transcriptional multiplex interactions, providing evidence for transcriptional factories within the nucleus. Applying this technique to small populations of Drosophila cells, the researchers found that promoter-promoter interactions are not as extensive as previously thought, and that progressive sets of contacts are related to active promoters in the direction of transcription.
Cleavage Under Targets and Tagmentation (CUT&Tag), is based on the same general principles as ChIP-seq and uses an enzyme-tethering strategy to target chromatin-bound proteins to reveal information about their binding sites26. It offers several advantages over ChIP-seq including utilizing less starting material. Another major advantage of CUT&Tag is that the entire method, from live cells to sequencing-ready libraries, can be done in a single tube on the benchtop in a high-throughput fashion, taking less than one day thereby reducing costs, time and improving robustness. This chromatin profiling technique can efficiently reveal regulatory information in genomes and unlike RNA-seq, can also profile silenced regions and markers of closed chromatin such as H3K27me3, thereby aiding in cell fate determination.
RNA-chromatin interactions appear to be important for gene regulation and chromatin structure. Recently methods have been developed to discover chromatin-associated RNAs and to map their interactions to the genome such as iMARGI27, ChAR-seq28, GRID-seq18, and RADICL-seq28. iMARGI, GRID-seq and RADICL-seqIt use low input number of cells, while iMARGI can be executed in a few hours, thereby reducing both time and material costs. Mapping of chromatin-associated RNAs provides a rich map of genomic locations associated with chromosome silencing, nucleolus-chromosome interactions, and RNA-mediated epigenetic inheritance.
Imaging Technologies
At the start of the 4DN program it became apparent that, although there are approaches for quality control, interpretation, reproducibility and sharing of microscopy images, there is a lack of consistent approaches for generating quantitative images and that metadata schemas vary significantly. This has complicated image comparisons, data harmonization and reproduction when comparing imaging data from different microscopes or different labs. To address both these points, researchers in the consortium developed the Micro-Meta App, an intuitive, highly interoperable, open-source software tool that is designed to provide these features to facilitate the extraction and collection of relevant microscopy metadata29. Similarly, researchers worked closely with the Open Microscopy Environment (OME) community, to extend their community driven metadata models for microscopy30.
O’Shea and colleagues developed ChromEMT31, a technique that combines DNA staining and contrast enhancement (an intercalating DNA dye (DRAQ5) locally catalyzes deposition of polymerized diaminobenzidine (DAB) upon exposure to light, and the polymer is visualized with osmium tetroxide OsO4)) with improvements in multi-tilt electron tomography, for higher resolution imaging of chromatin structure. They demonstrated that chromatin is a disordered 5- to 24-nanometer-diameter curvilinear chain, which is packed together at different 3D concentration distributions in interphase and mitotic nuclei. They further showed that chromatin chains could have many different arrangements and bend at various lengths to achieve structural compaction and high packing densities.
Pederson and colleagues developed a technique to image multiple genomic loci in living cells termed CRISPRainbow32. This is a method for labeling DNA in living cells by combining Cas9d with engineered sgRNA scaffolds that bind sets of fluorescent proteins. They simultaneously imaged up to six chromosomal loci in individual live cells and showed significant differences in the dynamic properties of different chromosomal loci.
FISH has been the mainstay of nuclear imaging and the consortium has contributed numerous advances to the field. Cai and colleagues developed a combined CRISPR imaging and DNA seqFISH assay that enabled them to track multiple genomic loci in living cells and then identify them later33 that complements their development of seqFISH+ that can image mRNAs from 10,000 genes in single cells34. More recently, Bewersdorf and colleagues have reported significant improvements in DNA-PAINT using a two-color approach and improved probes and hybridization strategy35. The challenge of imaging low copy numbers was tackled by Raj and colleagues who developed clampFISH for high gain amplification of multiplexed RNA and DNA FISH experiments36. Using this technique, they were able to detect and characterize patterns of low-level rDNA, mRNA and lncRNA in HeLa cells using a low magnification microscope.
On the analysis side, there have been considerable efforts to develop single particle tracking to resolve spatiotemporal dynamics, including the development of a web interface for stroboscopic photo-activation particle tracking that minimizes motion blur and some tracking errors37. The consortium has also worked on correlating the results of genomic and imaging studies to resolve observed organizational differences and come up with integrated approaches38.
Perturbation Technologies
It has been recognized for several years now that phase transitions are driven by proteins with intrinsically disordered regions (IDRs) and emerged as a wide-spread mechanism for assembling membrane-less organelles such as nucleoli or liquid-like RNA/protein (RNP) bodies39. However, effective tools to study this phenomenon have been slow to develop. As part of their 4DN award, the Brangwynne lab developed an optogenetic method using light to artificially activate IDR-mediated phase transitions in living cells40. They used this “optoDroplet” system to study condensed phases driven by the IDRs of various ribonucleoproteins (RNPs), thereby elucidating not only physiological phase transitions but also establishing their potential link to pathological aggregates.
New Insights into Biology and Chromatin Architecture
In addition to demonstrating new technologies, the network published many papers advancing our understanding of how the chromatin folding affects biology. For instance, Hnisz et al.41, showed that while oncogenes are tucked away and repressed in chromosome structures called insulated neighborhoods in normal cells, they are exposed and activated via disruption of insulated neighborhood boundaries in cancer cells.
Imprinting is an epigenetic phenomenon in mammals which is reflected in expression of genes from a single parental allele. By measuring allele-specific expression in single cells via a novel in situ fluorescence hybridization technique, Raj, and colleagues42 showed that imprinting disorders can display remarkable heterogeneity at single cell level with respect to their molecular phenotypes. This observation suggests that such heterogeneity may contribute to the fundamental epigenetic mosaicism in human imprinting disorders.
In related studies, the Levine lab43 and the Blobel lab44 studied transcriptional bursting. Levine and colleagues showed that in drosophila embryos, strong enhancers produce more bursts compared to weak enhancers when linked to synthetic reporter genes. However, linked reporter genes exhibited coordinated bursting features when regulated by a common enhancer, thereby challenging established models of enhancer-promoter looping. Blobel and colleagues, using the β-globin as a model gene, showed that in mammalian cells β -globin transcriptional burst fraction and sizes increased during erythroid maturation. However, the β-globin enhancer primarily determines burst fraction and not the burst size. These studies have implications for how different promoters are used by the same enhancer in cis during development.
The Casellas lab showed that the oncogenic transcription factor c-Myc regulates chromatin decompaction and nuclear architecture during B-cell activation/differentiation45. They showed that histone acetylation, methylation and nucleosome remodeling are primed by preloaded enzymes, and upon activation result in decompaction of clusters of nanodomains in the nucleus. In addition, the number of contact domains and loops also doubled during activation. They showed that c-Myc regulates both nanodomain compaction as well as domain and loop formation in B cells. Similarly, the Murre lab46 showed that during T cell differentiation, non-coding transcripts regulate chromatin folding and compartmentalization by activating epigenetic marks across the loop domain, thereby regulating enhancer-promoter looping and potentially enhancing phase separation.
Integrated Cell Line Studies
A key decision made by the consortium early on was to analyze genomic interactions using imaging and genomics approaches in a select few cell systems and conditions. Five cell lines were provided to each 4DN member to use and provide pilot data for all studies (tier 1) and later, 11 additional cell lines were distributed within the network based on popular usage for specific projects (tier 2)47. To systematically evaluate various chromosome conformation capture assay experimental parameters, Dekker, Mirny and colleagues used four different cell types selected by the 4DN consortium: pluripotent H1 human embryonic stem cells (H1-hESCs), differentiated endoderm (DE) cells derived from H1-hESCs, fully differentiated human foreskin fibroblast (HFF) cells, and HeLa-S3 cells48. Likewise, Nicodemi, Pombo and colleagues compared Hi-C, GAM, and SPRITE in silico and found that bulk data faithfully reproduces the same reference 3D features as observed in the chosen cell line systems, however single cell data does not correlate as well because of significant variability49.
Measuring the Success of 4DN Phase I
The NIH Common Fund regularly assesses all of its programs using multiple metrics to understand their performance and impact. During the first five years (Phase 1), the Consortium published 746 papers that have been cited more than 13,000 times in the literature (see Figure 1b), and the average Relative Citation Ratio (RCR)50 for these publications significantly exceeds the benchmark measure of scientific influence for similar papers (see Figure 1b). However, as a relatively young program, a broader impact of the program in terms of adoption of methods, tools and technologies cannot be fully evaluated yet. For instance, are scientists in various disciplines using the generated information, tools, and methods in their research? What impact is the 4DN program having on integrating genomics and imaging research? Is cancer research or disease-focused research more broadly impacted by 4DN? Although a systematic analysis to address these questions is not possible yet, there are some early indications. For instance, the Optodroplet method is increasingly being used as a key tool to determine if components of eukaryotic transcription machinery, such as transcription factors and cofactors including BRD4, subunits of the Mediator complex, and RNA polymerase II, form liquid-liquid phase separated compartments to regulate gene expression51. In general, many of the methods discussed here have more than 100 citations, reflecting a general interest from the broad scientific community and a measure of how widely these methods are recognized.
Figure 1.
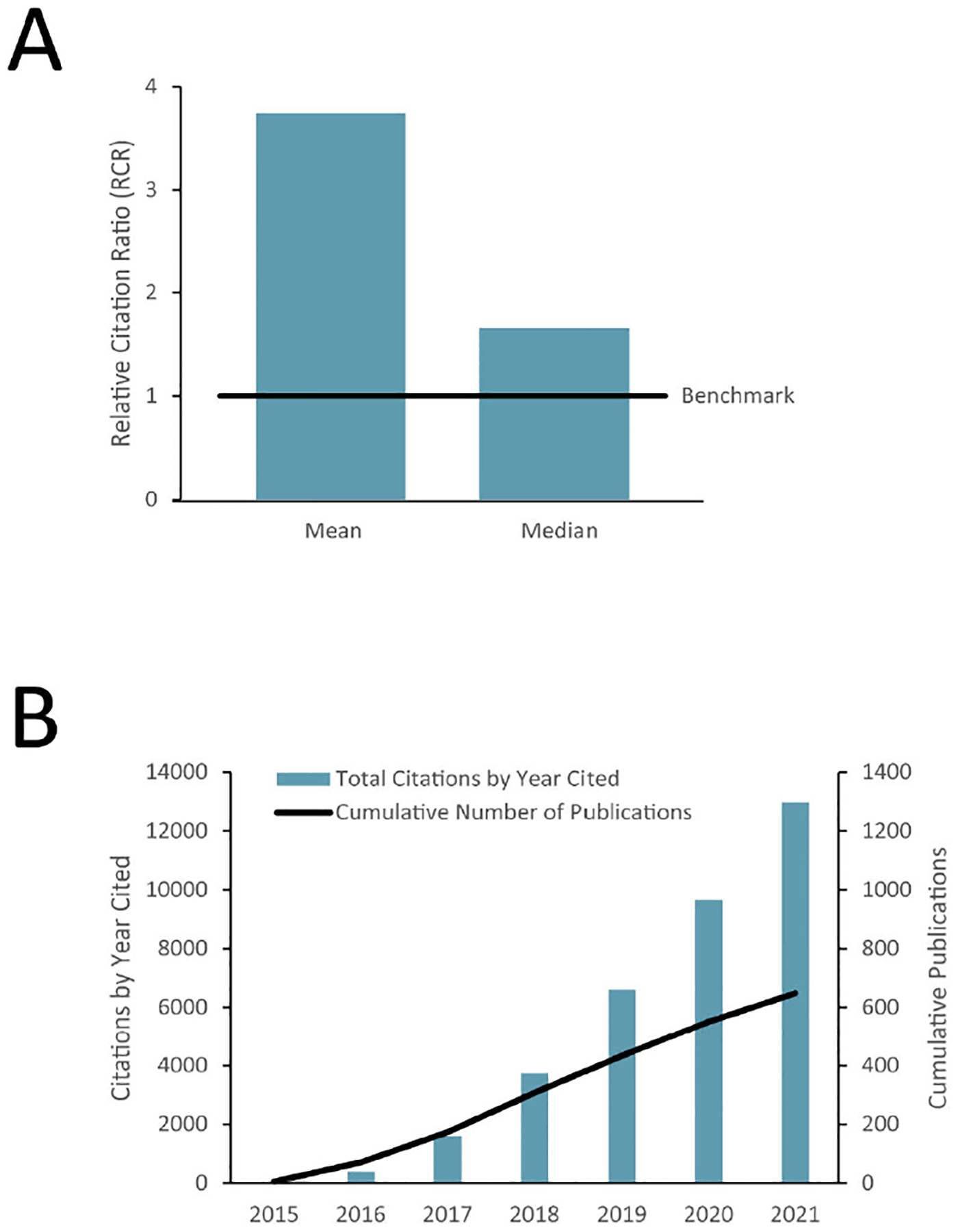
a) Median and mean Relative Citation Ratio (RCR) for 4DN publications and b) Cumulative count of 4DN publications and citation of these publications.
Altogether, the 4DN data portal now hosts results from more than 4,600 experiments, 110 TB of data and 36,000 files52. Complementing these outputs, the consortium has published 33 experimental protocols, 52 software packages and seven analysis pipelines. In addition to making resources available for download, they can also be explored interactively in 3D using an integrated genomic and imaging browser53. Datasets are downloaded more than 250 times each month, with more than 50% of the users coming from outside the U.S, indicating both the utility and demand for 4DN data. The Hi-C and micro-C datasets are the most in-demand, with significant interest in the multimodal datasets associated with the H1-hESC cell line. These types of outputs were expected, but the consortium also delivered several that were not initially planned, including the ingestion and sharing of data from other sources on the portal (nearly 20% of the data on the portal was generated by groups who are not funded as part of the consortium), development of a microscope calibration tool54 and establishment of a curated set of 16 publicly available cell lines47. The 4DN consortium also conducted joint meetings with both The American Society for Cell Biology and Keystone Symposia to have a greater outreach effort to the broader scientific community. Apart from joint scientific sessions, 4DN investigators held in person workshops to illustrate the utility of tools and techniques along with demonstration of the 4DN data portals.
4DN Phase II (2020 – 2025)
While the focus of phase 1 (2015–2020) was to develop technologies enabling mapping of the nucleus and to study various nuclear structures, phase 2 of the 4DN Program (2020–2025) is applying many of these technologies to understand the functional significance of genomic architecture and its role in human health and disease because it is now clear that the folding of the genome is functionally relevant for gene expression and health. Thus, the goals for phase 2 of the program are to address outstanding technological and conceptual challenges to studying nuclear organization and dynamics in mammalian cells and tissues; to provide deeper mechanistic insights of functional genome organization; and to elucidate the functional role of nuclear organization in development, health, and disease.
To achieve these goals, the consortium is moving from analyzing cell lines, which were necessary in most instances for methods development, to more physiologically relevant systems including tissues, primary cells, and animal models. For example, Pombo and colleagues developed immuno-GAM and demonstrated highly specific chromatin conformations in brain cells that are tightly related to gene regulation mechanisms and specialized functions20. These results suggest there are cell-type specialized 3D chromatin structures at multiple genomic scales that regulate patterns of gene expression in different cell types.
Further, Shen and colleagues have developed CRISPRview, a technique that integrates immunostaining, CRISPR interference, RNAscope, and image analysis to validate cell-type-specific cis-regulatory elements in heterogeneous populations of primary cells55. They set out to study cell-type-specific cis-regulatory chromatin interactions, open chromatin peaks, and transcriptomes for radial glia, intermediate progenitor cells, excitatory neurons, and inter-neurons isolated from mid-gestational samples of the human cortex. They show that chromatin interactions determine several layers of gene regulation, with transposable elements and disease-associated variants enriched at distal interacting regions in a cell-type-specific manner. These results provide further insights into chromatin architecture and cell-type-specific gene expression patterns in the developing human cortex.
Another direction coalescing from the collaborative environment of the 4DN, relates to development and refinement of techniques that focus on single cell architecture. One project in its early stages is focusing on single cell analysis of chromatin structure, epigenetics from neuronal cells, their precursors, and cardiac cells as they develop.
Conclusion
As part of its mission to study the three-dimensional organization of the nucleus in space and time, the 4DN consortium is constantly expanding a comprehensive set of durable outputs that can be utilized by the broad scientific community. These include genomics, imaging, and computational tools to study nuclear architecture in space and time as well as protocols, methods, reference maps and paradigm-shifting discoveries that have the potential to influence and strengthen work in nuclear spatiotemporal dynamics in the coming decades. It can be rather difficult to measure the success of a scientific program while it is ongoing because it might be confounding to disentangle the direct effects of the program versus other non-programmatic effects. In addition, scientific advances often take many years to achieve their ultimate impact on human health and disease. We primarily focused on three areas to provide a quantitative measure of success: (i) use of data, publications, and resources; (ii) new discoveries of paradigm shifting ideas; and (iii) new, high-value technologies. According to these measures, the initial indications of the impact of 4DN are beginning to be seen. However, we acknowledge that the wider impacts of the program on various scientific disciplines relevant for human health and disease as well as adoption of tools and technologies are yet to be evaluated and could still take another few years to be fully realized. Nevertheless, the success that the 4DN consortium has achieved so far is likely to lead to a lasting impact on the wider research community.
Acknowledgments:
We thank James M Anderson and Olivier Blondel for advice and critical reading of the manuscript. We also thank many members of the NIH 4DN working group for fruitful discussions and help with managing the 4DN program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
The authors declare no competing interests.
References
- 1.Holmes VF, and Cozzarelli NR (2000). Closing the ring: links between SMC proteins and chromosome partitioning, condensation, and supercoiling. Proc Natl Acad Sci U S A 97, 1322–1324. 10.1073/pnas.040576797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kragesteen BK, Spielmann M, Paliou C, Heinrich V, Schopflin R, Esposito A, Annunziatella C, Bianco S, Chiariello AM, Jerkovic I, et al. (2018). Dynamic 3D chromatin architecture contributes to enhancer specificity and limb morphogenesis. Nat Genet 50, 1463–1473. 10.1038/s41588-018-0221-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Annunziato AT (2008). DNA Packaging: Nucleosomes and Chromatin. Nature Education 1, 26. [Google Scholar]
- 4.Fraser P (2006). Transcriptional control thrown for a loop. Curr Opin Genet Dev 16, 490–495. 10.1016/j.gde.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Hildebrand EM, and Dekker J (2020). Mechanisms and Functions of Chromosome Compartmentalization. Trends Biochem Sci 45, 385–396. 10.1016/j.tibs.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sexton T, Schober H, Fraser P, and Gasser SM (2007). Gene regulation through nuclear organization. Nat Struct Mol Biol 14, 1049–1055. 10.1038/nsmb1324. [DOI] [PubMed] [Google Scholar]
- 7.Lamond AI, and Earnshaw WC (1998). Structure and function in the nucleus. Science 280, 547–553. 10.1126/science.280.5363.547. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Chen J, Muir LA, Ronquist S, Meixner W, Ljungman M, Ried T, Smale S, and Rajapakse I (2015). Functional organization of the human 4D Nucleome. Proc Natl Acad Sci U S A 112, 8002–8007. 10.1073/pnas.1505822112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parada L, and Misteli T (2002). Chromosome positioning in the interphase nucleus. Trends Cell Biol 12, 425–432. 10.1016/s0962-8924(02)02351-6. [DOI] [PubMed] [Google Scholar]
- 10.Jerkovic I, and Cavalli G (2021). Understanding 3D genome organization by multidisciplinary methods. Nat Rev Mol Cell Biol 22, 511–528. 10.1038/s41580-021-00362-w. [DOI] [PubMed] [Google Scholar]
- 11.Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing chromosome conformation. Science 295, 1306–1311. 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 12.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. (2009). An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462, 58–64. 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, and Rando OJ (2015). Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell 162, 108–119. 10.1016/j.cell.2015.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyle S, Rodesch MJ, Halvensleben HA, Jeddeloh JA, and Bickmore WA (2011). Fluorescence in situ hybridization with high-complexity repeat-free oligonucleotide probes generated by massively parallel synthesis. Chromosome Res 19, 901–909. 10.1007/s10577-011-9245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solovei I, Cavallo A, Schermelleh L, Jaunin F, Scasselati C, Cmarko D, Cremer C, Fakan S, and Cremer T (2002). Spatial preservation of nuclear chromatin architecture during three-dimensional fluorescence in situ hybridization (3D-FISH). Exp Cell Res 276, 10–23. 10.1006/excr.2002.5513. [DOI] [PubMed] [Google Scholar]
- 17.Branco MR, Branco T, Ramirez F, and Pombo A (2008). Changes in chromosome organization during PHA-activation of resting human lymphocytes measured by cryo-FISH. Chromosome Res 16, 413–426. 10.1007/s10577-008-1230-x. [DOI] [PubMed] [Google Scholar]
- 18.Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas LM, Branco MR, et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. 10.1038/nature21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yanchus C, Drucker KL, Kollmeyer TM, Tsai R, Winick-Ng W, Liang M, Malik A, Pawling J, De Lorenzo SB, Ali A, et al. (2022). A noncoding single-nucleotide polymorphism at 8q24 drives IDH1-mutant glioma formation. Science 378, 68–78. 10.1126/science.abj2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winick-Ng W, Kukalev A, Harabula I, Zea-Redondo L, Szabo D, Meijer M, Serebreni L, Zhang Y, Bianco S, Chiariello AM, et al. (2021). Cell-type specialization is encoded by specific chromatin topologies. Nature 599, 684–691. 10.1038/s41586-021-04081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.NIH (2022). NIH RePORT Search. https://reporter.nih.gov/search/hwod18pmFUKAd0yQdj4BUw/projects?sort_field=FiscalYear%26sort_order=asc%26shared=true.
- 22.Flyamer IM, Gassler J, Imakaev M, Brandao HB, Ulianov SV, Abdennur N, Razin SV, Mirny LA, and Tachibana-Konwalski K (2017). Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114. 10.1038/nature21711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, and Fraser P (2013). Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64. 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, et al. (2018). Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 174, 744–757 e724. 10.1016/j.cell.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng M, Tian SZ, Capurso D, Kim M, Maurya R, Lee B, Piecuch E, Gong L, Zhu JJ, Li Z, et al. (2019). Multiplex chromatin interactions with single-molecule precision. Nature 566, 558–562. 10.1038/s41586-019-0949-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930. 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu W, Yan Z, Nguyen TC, Bouman Chen Z, Chien S, and Zhong S (2019). Mapping RNA-chromatin interactions by sequencing with iMARGI. Nat Protoc 14, 3243–3272. 10.1038/s41596-019-0229-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell JC, Jukam D, Teran NA, Risca VI, Smith OK, Johnson WL, Skotheim JM, Greenleaf WJ, and Straight AF (2018). Chromatin-associated RNA sequencing (ChAR-seq) maps genome-wide RNA-to-DNA contacts. Elife 7. 10.7554/eLife.27024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rigano A, Ehmsen S, Ozturk SU, Ryan J, Balashov A, Hammer M, Kirli K, Boehm U, Brown CM, Bellve K, et al. (2021). Micro-Meta App: an interactive tool for collecting microscopy metadata based on community specifications. Nat Methods 18, 1489–1495. 10.1038/s41592-021-01315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammer M, Huisman M, Rigano A, Boehm U, Chambers JJ, Gaudreault N, North AJ, Pimentel JA, Sudar D, Bajcsy P, et al. (2021). Towards community-driven metadata standards for light microscopy: tiered specifications extending the OME model. Nat Methods 18, 1427–1440. 10.1038/s41592-021-01327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, and O’Shea CC (2017). ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357. 10.1126/science.aag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, and Pederson T (2016). Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol 34, 528–530. 10.1038/nbt.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takei Y, Shah S, Harvey S, Qi LS, and Cai L (2017). Multiplexed Dynamic Imaging of Genomic Loci by Combined CRISPR Imaging and DNA Sequential FISH. Biophys J 112, 1773–1776. 10.1016/j.bpj.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan GC, and Cai L (2019). Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239. 10.1038/s41586-019-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chung KKH, Zhang Z, Kidd P, Zhang Y, Williams ND, Rollins B, Yang Y, Lin C, Baddeley D, and Bewersdorf J (2022). Fluorogenic DNA-PAINT for faster, low-background super-resolution imaging. Nat Methods 19, 554–559. 10.1038/s41592-022-01464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rouhanifard SH, Mellis IA, Dunagin M, Bayatpour S, Jiang CL, Dardani I, Symmons O, Emert B, Torre E, Cote A, et al. (2018). ClampFISH detects individual nucleic acid molecules using click chemistry-based amplification. Nat Biotechnol. 10.1038/nbt.4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen AS, Woringer M, Grimm JB, Lavis LD, Tjian R, and Darzacq X (2018). Robust model-based analysis of single-particle tracking experiments with Spot-On. Elife 7. 10.7554/eLife.33125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nir G, Farabella I, Perez Estrada C, Ebeling CG, Beliveau BJ, Sasaki HM, Lee SD, Nguyen SC, McCole RB, Chattoraj S, et al. (2018). Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet 14, e1007872. 10.1371/journal.pgen.1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Julicher F, and Hyman AA (2009). Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732. 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- 40.Shin Y, Berry J, Pannucci N, Haataja MP, Toettcher JE, and Brangwynne CP (2017). Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 168, 159–171 e114. 10.1016/j.cell.2016.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. (2016). Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458. 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ginart P, Kalish JM, Jiang CL, Yu AC, Bartolomei MS, and Raj A (2016). Visualizing allele-specific expression in single cells reveals epigenetic mosaicism in an H19 loss-of-imprinting mutant. Genes Dev 30, 567–578. 10.1101/gad.275958.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fukaya T, Lim B, and Levine M (2016). Enhancer Control of Transcriptional Bursting. Cell 166, 358–368. 10.1016/j.cell.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartman CR, Hsu SC, Hsiung CC, Raj A, and Blobel GA (2016). Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Mol Cell 62, 237–247. 10.1016/j.molcel.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kieffer-Kwon KR, Nimura K, Rao SSP, Xu J, Jung S, Pekowska A, Dose M, Stevens E, Mathe E, Dong P, et al. (2017). Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Mol Cell 67, 566–578 e510. 10.1016/j.molcel.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP, et al. (2017). Non-coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 171, 103–119 e118. 10.1016/j.cell.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.4DN (2022). 4DN consortium recommended cell lines. https://www.4dnucleome.org/cell-lines.html.
- 48.Akgol Oksuz B, Yang L, Abraham S, Venev SV, Krietenstein N, Parsi KM, Ozadam H, Oomen ME, Nand A, Mao H, et al. (2021). Systematic evaluation of chromosome conformation capture assays. Nat Methods 18, 1046–1055. 10.1038/s41592-021-01248-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fiorillo L, Musella F, Conte M, Kempfer R, Chiariello AM, Bianco S, Kukalev A, Irastorza-Azcarate I, Esposito A, Abraham A, et al. (2021). Comparison of the Hi-C, GAM and SPRITE methods using polymer models of chromatin. Nat Methods 18, 482–490. 10.1038/s41592-021-01135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hutchins BI, Yuan X, Anderson JM, and Santangelo GM (2016). Relative Citation Ratio (RCR): A New Metric That Uses Citation Rates to Measure Influence at the Article Level. PLoS Biol 14, e1002541. 10.1371/journal.pbio.1002541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.4DN (2022). 4D Nucleome Data Portal. https://data.4dnucleome.org/.
- 53.4DN (2022). Nucleome Browser. http://vis.nucleome.org/entry/. [DOI] [PMC free article] [PubMed]
- 54.Huisman M (2019). Vision Beyond Optics: Standardization, Evaluation and Innovation for Fluorescence Microscopy in Life Sciences.
- 55.Song M, Pebworth MP, Yang X, Abnousi A, Fan C, Wen J, Rosen JD, Choudhary MNK, Cui X, Jones IR, et al. (2020). Cell-type-specific 3D epigenomes in the developing human cortex. Nature 587, 644–649. 10.1038/s41586-020-2825-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, and Ren B (2016). Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 26, 1345–1348. 10.1038/cr.2016.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, and Belmont AS (2018). Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol 217, 4025–4048. 10.1083/jcb.201807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krietenstein N, Abraham S, Venev SV, Abdennur N, Gibcus J, Hsieh TS, Parsi KM, Yang L, Maehr R, Mirny LA, et al. (2020). Ultrastructural Details of Mammalian Chromosome Architecture. Mol Cell 78, 554–565 e557. 10.1016/j.molcel.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsieh TS, Cattoglio C, Slobodyanyuk E, Hansen AS, Rando OJ, Tjian R, and Darzacq X (2020). Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding. Mol Cell 78, 539–553 e538. 10.1016/j.molcel.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinodoz SA, Jachowicz JW, Bhat P, Ollikainen N, Banerjee AK, Goronzy IN, Blanco MR, Chovanec P, Chow A, Markaki Y, et al. (2021). RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790 e5730. 10.1016/j.cell.2021.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao XD, Tu LC, Mir A, Rodriguez T, Ding Y, Leszyk J, Dekker J, Shaffer SA, Zhu LJ, Wolfe SA, and Sontheimer EJ (2018). C-BERST: defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat Methods 15, 433–436. 10.1038/s41592-018-0006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vertii A, Ou J, Yu J, Yan A, Pages H, Liu H, Zhu LJ, and Kaufman PD (2019). Two contrasting classes of nucleolus-associated domains in mouse fibroblast heterochromatin. Genome Res 29, 1235–1249. 10.1101/gr.247072.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lai B, Tang Q, Jin W, Hu G, Wangsa D, Cui K, Stanton BZ, Ren G, Ding Y, Zhao M, et al. (2018). Trac-looping measures genome structure and chromatin accessibility. Nat Methods 15, 741–747. 10.1038/s41592-018-0107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao PA, Sasaki T, and Gilbert DM (2020). High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol 21, 76. 10.1186/s13059-020-01983-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramani V, Deng X, Qiu R, Lee C, Disteche CM, Noble WS, Shendure J, and Duan Z (2020). Sci-Hi-C: A single-cell Hi-C method for mapping 3D genome organization in large number of single cells. Methods 170, 61–68. 10.1016/j.ymeth.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Schaik T, Vos M, Peric-Hupkes D, Hn Celie P, and van Steensel B (2020). Cell cycle dynamics of lamina-associated DNA. EMBO Rep 21, e50636. 10.15252/embr.202050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Belaghzal H, Borrman T, Stephens AD, Lafontaine DL, Venev SV, Weng Z, Marko JF, and Dekker J (2021). Liquid chromatin Hi-C characterizes compartment-dependent chromatin interaction dynamics. Nat Genet 53, 367–378. 10.1038/s41588-021-00784-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tavares-Cadete F, Norouzi D, Dekker B, Liu Y, and Dekker J (2020). Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nat Struct Mol Biol 27, 1105–1114. 10.1038/s41594-020-0506-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lafontaine DL, Yang L, Dekker J, and Gibcus JH (2021). Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Curr Protoc 1, e198. 10.1002/cpz1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]