

Abstract
Whole genome sequencing and assembly have revolutionized plant genetics and molecular biology over the last two decades. However, significant shortcomings in first and second generation technology resulted in imperfect reference genomes: numerous and large gaps of low quality or undeterminable sequence in areas of highly repetitive DNA along with limited chromosomal phasing restricted the ability of researchers to characterize regulatory non-coding elements and genic regions that underwent recent duplication events. Recently, advances in long-read sequencing have resulted in the first gapless, telomere-to-telomere (T2T) assemblies of plant genomes. This leap forward has the potential to increase the speed and confidence of genomics and molecular experimentation while reducing costs for the research community.
Keywords: Genomics, Gapless, Single-Molecule, Plants, Phasing
Graphical Abstract
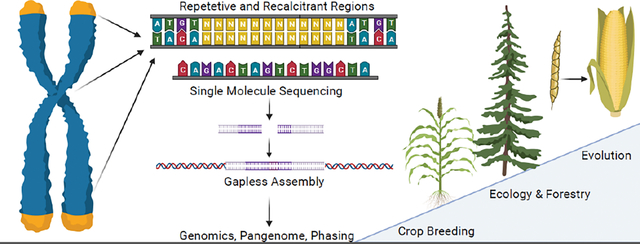
Introduction
Plant genomes represent a unique space within genome research. The breadth and depth of genetic architectural diversity among model and crop systems, even among strains of the same species, have constantly presented challenges for generating high quality assemblies without sequence gaps between telomeres. This concept of gapless genomes represents a compounding improvement for plant genomics that has recently been achieved through a combination of hybrid technologies that utilize short- and long-read single-molecule sequencing instruments to finally generate a telomere-to-telomere (T2T) reference for all chromosomes within simple or complex genomes (Figure 1). Telomeres, centromeres, and ribosomal repeats are common chromosomal features that have been recalcitrant regions for both human and plant genomes, but the fluid nature of plant genomes due to high levels of transposable elements (TE) and segmental duplications (SD) pose additional challenges. While there is an arms race between genome integrity and stability, plants develop novel advantageous genetic variation for species adaptation through TEs; however they have been difficult to characterize until recently. SDs are genomic repeats >1kb that are generally around 90% similar that often contain many variations of the same gene contributing to functional evolution [1–3]. Since these regions are very similar, they are often incorrectly assembled or collapsed into a single sequence, obscuring important functional variation. A recent gapless assembly of indica rice found 24% of the genome was composed of SDs and 150 duplicated genes showed tissue specific differential expression between the copies, suggesting sub- or neo-functionalization [4].
Figure 1. Gapless Genomes Resolve Recalcitrant Regions.
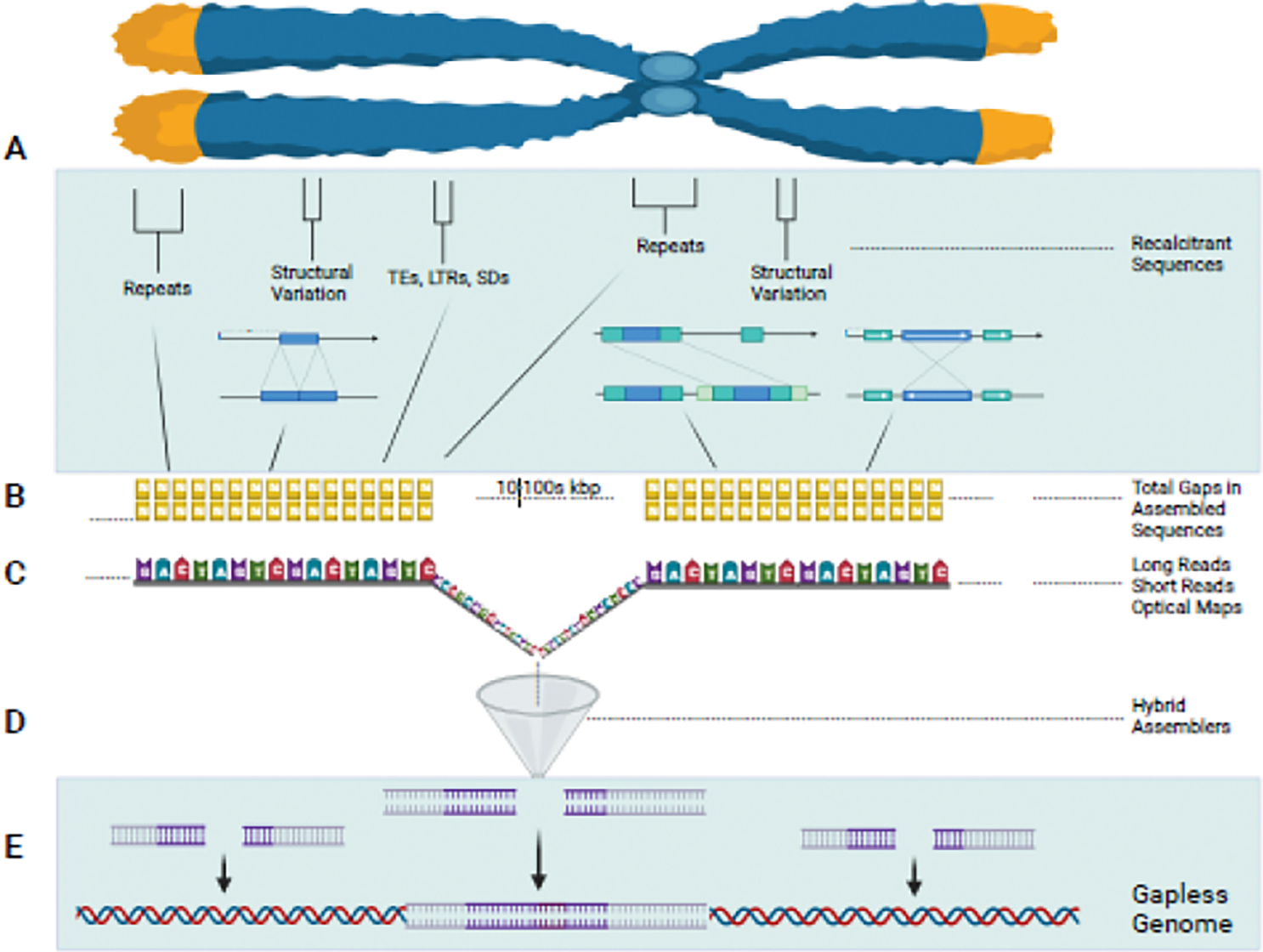
Telomere to telomere (T2T) chromosome assembly resolves all previously undeterminable sequences. A) Whole chromosomes are made up of many recalcitrant regions that are difficult for short-read sequencing approaches to resolve, such as TEs, highly repetitive regions, tandem duplications, ribosomal repeats, etc. B) These problematic regions result in un-callable sections that form gaps in the entire assembly, C) but single molecule technologies and hybrid approaches can create reads that completely cover these once unresolvable genomic sequences. D) These long-reads are then combined with hybrid assemblers to ultimately create the E) gapless genome. Created with BioRender.com
TEs often make up a substantial portion of plant genomes and are key components of regulatory architecture [5]. Long-read sequencing studies of many plant species frequently reveal notably more TE regions than observed in previous non-long-read studies because long-reads are able to sequence completely though the element. A PacBio-based resequencing of maize captured >100 thousand copies of intact TEs, and aided in defining a lineage-specific expansion of long terminal repeats (LTRs) in maize since its divergence from sorghum [6].
Ribosomal repeats resist coherent assembly via older sequencing methods. Cytogenetic studies in banana revealed clusters of 5S rDNA repeats on one arm of chr8 and in the centromeric regions of chr1 and chr3 [7], but these results were not seen in the v2 assembly, and in fact only seemed to represent 7.5 kb of the total sequence [8]. The banana genome was resequenced with long nanopore reads: 2.2 Mb of rDNA repeats were now included in the new T2T assembly, validating the original cytogenetic results [9] and further showing the power and potential of gapless genome strategies.
Regulatory element discovery can also be improved with T2T or near-gapless genome assemblies through the incorporation of epigenetic sequencing; long-read platforms can enable partial DNA methylation profiling, which can be combined with with improved genomic coverage to find cis-regulatory motifs by scanning hypomethylated regions across the assembly. This was done recently with long-read assemblies of 26 maize germplasms [10], and also shows how gapless genomes can augment pan-genome analyses.
Sequencing History
Short-read Sequencing
Next-Generation sequencing surpassed Sanger sequencing as the favored method for primary genome assembly in the late 2000s. Short-read methods were used in combination with Sanger to generate the first hybrid read type whole genome sequence of Vitis vinifera [11]. Pyrosequencing was rapidly eclipsed with the reversible terminator chemistry developed by Illumina Inc. (formerly Solexa) [12], which reduced sequencing costs, increased throughput and increased accuracy, despite initially providing contiguous reads of only 35bp, and heralded a new generation of genome science.
Next-Generation short-read sequencing strategies provide highly accurate sequence and high depth of coverage (>60x) for non-repetitive regions and can be unambiguously aligned into distinct contigs for assembly. As of publication, Illumina based technologies account for more than 90% of all sequencing done in the world, owing to its low cost and high capacity. Short-read approaches were initially designed for resequencing rather than de novo sequencing, which are still applied towards diversity and breeding panels as well as polyploid progenitors for the purpose of creating molecular breeding tools and evolutionary analysis [13,14]**. These methods have also been employed in the generation of large pan-genome projects that have sequenced 10s to 1000s [15–17] of individuals giving an unparalleled view into species diversity and the genomics that drive important phenotypes.
Long-read Sequencing
The subsequent, or third, generation of sequencing technologies tackled long DNA fragments (>1kb) by Pacific Biosciences (PacBio) SMRT cell technology [18] and Oxford Nanopore MinION [19]. Where short-reads use clonal amplification to overcome sequencing error and increase signal, these methods sequence a single-molecule at a time. Skipping amplification allows sequencing to continue far longer along the molecule than short-read methods, but leads to a reduction in accuracy. Thus, long-reads often require either higher coverage [20] or accurate short-reads to correct the long-reads [21] to create a highly contiguous assembly. Read length and accuracy improvements have continued from these initial platforms to generate reads of 10–25 kb through PacBio High Fidelity (HiFi - circular consensus sequencing) [22]* and >300 kb reads for Oxford Nanopore sequencing [23]. Continued improvement of HiFi sequencing has pushed read accuracy above 99% [22,24], and newer plant specific base calling models have pushed nanopore accuracy > 95%, further strengthening genome accuracy. Long-read incorporation has resolved significant issues plaguing larger, complex genomes, namely generating non-ambiguous reads and contigs that are long enough to span structural variants, repetitive regions, and TEs (Figure 2). For example, a gapless watermelon assembly discovered an additional 173 genes located in gap regions of the prior assembly, including two LRR-RLK genes, which play crucial roles in plant development [25]**. Long-read platforms also have the ability to partially sequence the epigenome and epitranscriptome [10,26].
Figure 2. Genome Assembly Improvement Over Sequencing Eras.
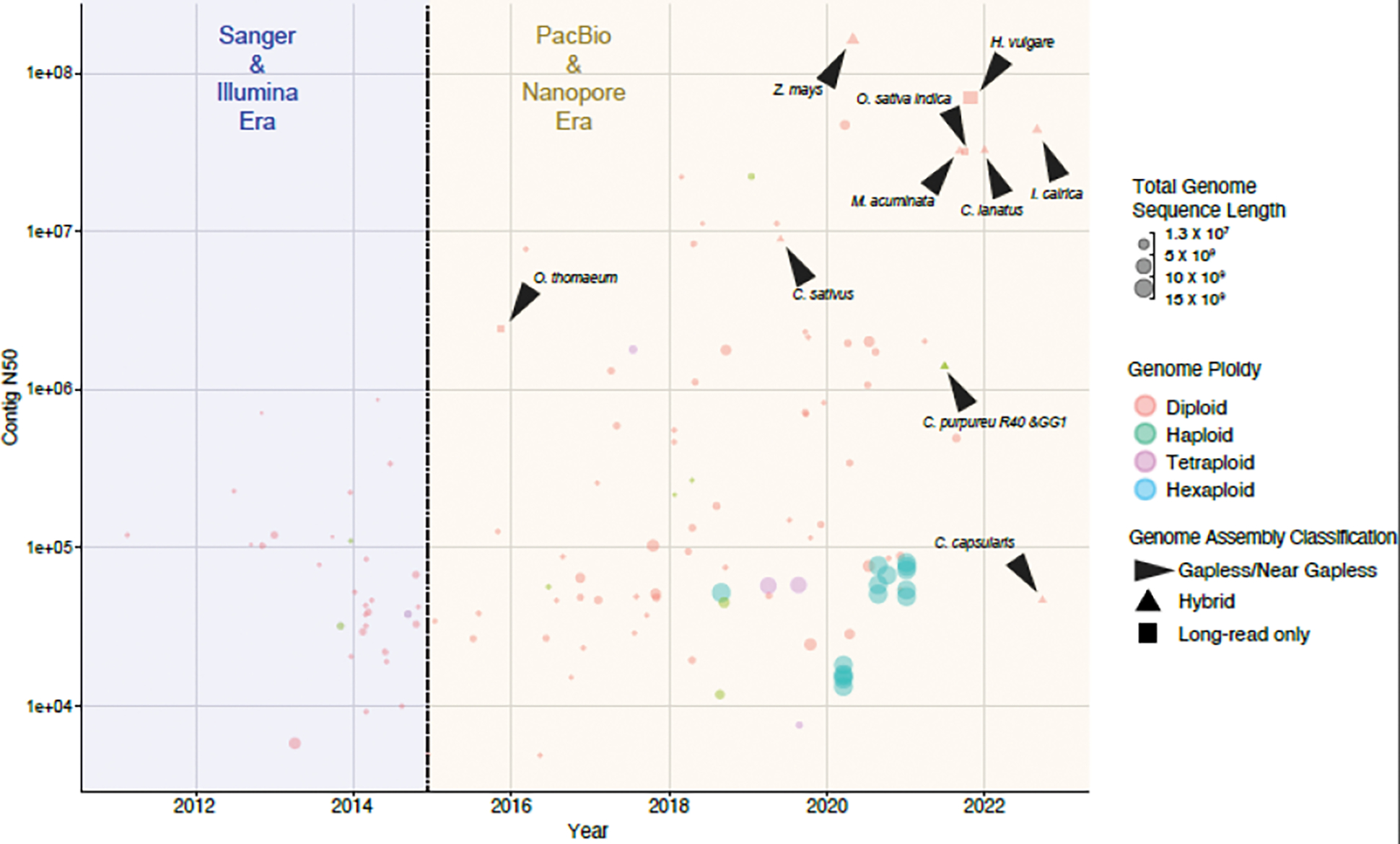
Contig N50 of plant genomes over the last 10 years and divided into two eras: pre- and post-single-molecule sequencing technologies (data taken from Ensembl Plants and NCBI). Genomes are colorized by ploidy (Orange = Diploid, Green = Haploid, Purple = Tetraploid, and Blue = Hexaploid). Gapless or near-gapless genomes are highlighted with black arrows and datapoint shapes indicate if genome assembly used hybrid (combination of short- and long-read) or long-read methods alone.
Optical Maps
Initially developed in the 1990’s, optical mapping used microscopy to visualize a restriction enzyme map over ranges from 0.2–1Mb. [27] Since then, genome quality has been bolstered by using optical maps to greatly reduce the total number of assembly contigs by expanding the effective length of the chromosome-wide pseudomolecule [28–30]. Even some long-read and most short-read hybrid assemblies that incorporate optical maps can see contig reduction by one or two orders of magnitude from sequencing-bases contig construction alone [31]. Zea mays is a paragon of this approach; optical mapping generated 63 contigs [32]* compared to the >100,000 contigs from the initial Sanger sequencing assembly a decade prior [33].
Hi-C
Hi-C is a chromatin conformation capture technique [34] that relies on crosslinking regions of chromatin prior to cleavage and ligation of sequencing compatible adapters. The resulting fragments are sequenced on a short-read sequencing instrument yielding unbiased genome wide chromatin interaction maps that reveal global patterns of interactions like translocation and inversion. Such long range information is essential to achieve phasing in polyploid plant samples. While long-reads alone can effectively phase diploid samples as seen in Z. mays [35]*, H. lupulus [36], and D. alata [37]**, phasing of polyploid samples has remained a major hurdle for crop science, since properly phased chromosomes can serve to identify regions of heterozygosity in hybrid breeding programs or within heavily heterozygous species, such as with obligate outcrossers. Phasing polyploids required developing new computational strategies such as those used to phase hexaploid bamboo [38]* and autotetraploid alfalfa [39]*. Hi-C methods are now being developed for long nanopore reads in place of short-reads; called Pore-C, it was recently used to assess chromatin interaction in Arabidopsis preserving methylation data in addition to long range DNA interactions [40]. These combined strategies revealed significant differences in haplotypes, which can contribute to plant phenotypes and ultimately the success or failure of breeding programs.
Assembly Technologies
Sequencing methods and longer DNA fragment extraction have accelerated the quality of assemblies significantly, but those advances rely on parallel improvements in assembly algorithms. Assemblers like ALLPATHS [41] use de Brujin graphs that integrate typical sequencing methods with longer range data via mate-pair libraries. However, they still rely on short-read sequencing technology and are not capable of effectively assembling long, noisy reads. Others, like Celera [42], were based on overlap layout consensus approaches and used Sanger sequencing to assemble early plant genomes like papaya [43]. Eventually, hybrid approaches employed both accurate short-reads and long noisy reads as seen in PBcR [44] and ALLPATHS-LG [45]. As sequencing throughput has increased the need for less computationally intense assembly methods has become critical. For perspective, the first long-read D. megolaster assembly was estimated to take 600,000 CPU hours using the Celera/PBcR assembler [46], contrasted to a CANU + early PacBio-based assembly of A. thaliana that required 925 CPU hours [47], while hexaploid wheat using MaSuRCA + PacBio took 470,000 CPU hours.
Extraction
While advances in sequencing technology have been crucial to genome assembly improvements, acquiring high quality plant DNA remains quite challenging. Unlike humans or mammals in general, plants have incredibly diverse metabolites that impact yield, quality, and performance of extracted DNA. While nanograms of DNA are required for short-read libraries, current single-molecule approaches often need more than 10 ug of DNA that is >10 kb in length [48]. Interestingly, as the capacity to sequence increasingly longer fragments of DNA emerged alongside the resurgence of optical mapping technologies, kits for high molecular weight DNA extraction are now being eschewed for older methods: CTAB based nuclei preparations were used for preparing DNA from the coast redwood [49] and gel-plus-lysis methods were applied for nanopore sequencing and optical mapping to generate a chromosome scale assembly of Sorghum [50]. Nevertheless, these methods remain difficult and time consuming, limiting the adoption of high throughput sequencing for diverse plant samples.
Applications and Examples
Strategies employed by T2T consortiums can be adapted to complex plant genomes to achieve truly contiguous, phased assemblies critical for annotating genes and non-coding regulatory elements (such as TEs and distal enhancers), genomic and marker-assisted breeding, transgenic and genome editing approaches, and GWAS. Trait discovery can be enhanced via gapless genomes through improved annotation of structural variants or highly duplicated gene families that confer environmental response phenotypes like disease resistance alleles [4,9,25,32]. Gapless genomes can also help in projection of conserved regions and gene models to different species [51], thus improving overall annotation quality and evolutionary understanding of priority traits; this applies toward more complex genomes, including highly repetitive and polyploid organisms. These genome assemblies are more stable and require less resequencing and version releases for the community, which lowers time cost for researchers that rely on genomics to underpin experiments and breeding programs. Additionally, current long-read platforms can detect certain forms of DNA methylation, conferring the benefit of partial epigenome sequencing as well [52]. A recent A. thaliana assembly showed distinct methylation patterns between pericentromeric and centromeric regions, and has applications towards dissecting important regulatory epigenomic sections in other plants and crop systems [53].
With continually decreasing costs, the ability to generate a high quality, phased reference genome for any organism lowers the boundaries for improving germplasm stock and broadening biotechnological approaches to more and varied types of crop systems that previously would go undervalued due to their complex genomes. This is the case for the watermelon gapless assembly that resulted in more confident gene identification and annotation and allowed the authors to more accurately quantify and locate EMS-induced SNPs within the Citrullus lanatus G42 lineage [25], providing a stronger understanding of mutagenic SNP saturation while mapping a male sterile trait. This T2T genome also yielded an example of accelerated functional gene identification in other cultivars, specifically by filling in the un-sequenced upstream region of Cla97C10G197910 gene (conveys rind hardness) within the 97103v2 cultivar gapped genome.
Summary and Concluding Remarks
These technologies have significant benefits, but each has limitations in achieving a fully contiguous T2T assembly. Short-reads are highly accurate, but fragment length curtails assembly and misses large amounts of the genome. Conversely, long-reads are able to sequence though complex regions of the genome and can provide unambiguous alignments to repetitive regions, but their overall lower accuracy and throughput can lead to missasemblies in addition to difficulties in isolating adequate high molecular weight DNA. While long-read technologies are generally thought to have very high costs, this is rapidly changing as methods and technologies are developed (Figure 3). These continuing improvements and cost reductions lower the entry barrier for researchers studying plants with complex genomes and ultimately removes barriers to apply conventional and molecular breeding approaches to a wider array of crop systems globally. The future of gapless genomes will continue to rely upon the iterative improvement of long-molecule sequencing, DNA isolation, assembly tools, and community cooperation for creating quality reference assemblies that inform pangenomes and are translatable across related species.
Figure 3. Cost and Output of Long-read Sequencing Over Time.
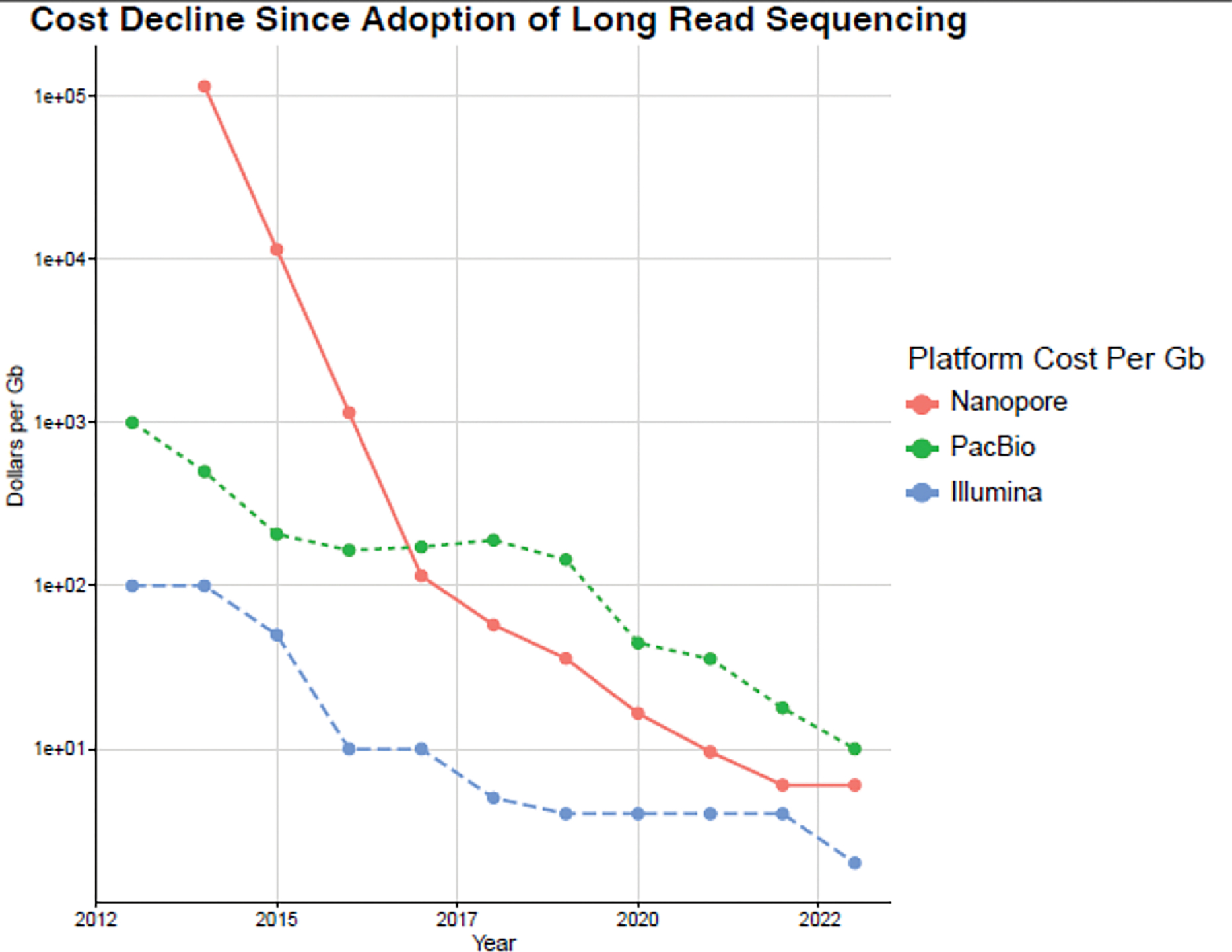
Dollars per Gb for Nanopore (red), PacBio (green), and short-read Illumina (blue) platforms from their first release to present day. Future costs for 2023 are using company estimates.
Highlights.
Sequencing technology has advanced and become inexpensive enough that gapless telomere-to-telomere (T2T) genome assemblies are possible for plant genomes.
Gapless genomes provide improved reference sequences compared to non-gapless constructions.
Gapless genomes improve and hasten researchers in molecular breeding and functional gene characterization.
Acknowledgements
The authors would like to acknowledge NCBI, Ensembl Plants, and Gramene for their data on plant genome assemblies that were used for figure generation. BioRender.com was used for the creation of the graphical abstract and Figure 1. KC, NG, DW are supported by the United States Department of Agriculture Agricultural Research Service (grant number 8062-21000-041-00D). SG is supported by the National Institutes of Health (grant number 5R50CA243890). WRM is The Davis Family Professor of Human Genetics and is also supported by the National Science Foundation (grant number IOS 1758800).
Footnotes
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
W. Richard McCombie is a founder and shareholder in Orion Genomics which works in plant genomics. Orion Genomics had no part in the preparation of this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Work Cited
- 1.Maron LG, Guimarães CT, Kirst M, Albert PS, Birchler JA, Bradbury PJ, Buckler ES, Coluccio AE, Danilova TV, Kudrna D, et al. : Aluminum tolerance in maize is associated with higher MATE1 gene copy number. Proc Natl Acad Sci U S A 2013, 110:5241–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu Z, Pu X, Gao R, Demurtas OC, Fleck SJ, Richter M, He C, Ji A, Sun W, Kong J, et al. : Tandem gene duplications drive divergent evolution of caffeine and crocin biosynthetic pathways in plants. BMC Biol 2020, 18:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panchy N, Lehti-Shiu M, Shiu S-H: Evolution of Gene Duplication in Plants. Plant Physiol 2016, 171:2294–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li K, Jiang W, Hui Y, Kong M, Feng L-Y, Gao L-Z, Li P, Lu S: Gapless indica rice genome reveals synergistic contributions of active transposable elements and segmental duplications to rice genome evolution. Mol Plant 2021, 14:1745–1756. [DOI] [PubMed] [Google Scholar]
- 5.Baduel P, Quadrana L: Jumpstarting evolution: How transposition can facilitate adaptation to rapid environmental changes. Curr Opin Plant Biol 2021, 61:102043. [DOI] [PubMed] [Google Scholar]
- 6.Jiao Y, Peluso P, Shi J, Liang T, Stitzer MC, Wang B, Campbell MS, Stein JC, Wei X, Chin C-S, et al. : Improved maize reference genome with single-molecule technologies. Nature 2017, 546:524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Čížková J, Hřibová E, Humplíková L, Christelová P, Suchánková P, Doležel J: Molecular analysis and genomic organization of major DNA satellites in banana (Musa spp.). PLoS One 2013, 8:e54808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin G, Baurens F-C, Droc G, Rouard M, Cenci A, Kilian A, Hastie A, Doležel J, Aury J-M, Alberti A, et al. : Improvement of the banana “Musa acuminata” reference sequence using NGS data and semi-automated bioinformatics methods. BMC Genomics 2016, 17:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belser C, Baurens F-C, Noel B, Martin G, Cruaud C, Istace B, Yahiaoui N, Labadie K, Hřibová E, Doležel J, et al. : Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing. Commun Biol 2021, 4:1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hufford MB, Seetharam AS, Woodhouse MR, Chougule KM, Ou S, Liu J, Ricci WA, Guo T, Olson A, Qiu Y, et al. : De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 2021, 373:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, Fitzgerald LM, Vezzulli S, Reid J, et al. : A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One 2007, 2:e1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. : Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456:53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu X, Liu Y, Luo H, Shang L, Leng C, Liu Z, Li Z, Lu X, Cai H, Hao H, et al. : Genomic footprints of sorghum domestication and breeding selection for multiple end uses. Mol Plant 2022, 15:537–551. By sequencing 445 diverse sorghum accessions, the authors generated a deep genotyping profile of over 23 million SNPs and 8 different haplotype models to explain the evolutionary and domestication diversity within the collection; the authors specifically evaluate the domestication genes SbTb1 and Sh1.
- 14. Gordon SP, Contreras-Moreira B, Levy JJ, Djamei A, Czedik-Eysenberg A, Tartaglio VS, Session A, Martin J, Cartwright A, Katz A, et al. : Gradual polyploid genome evolution revealed by pan-genomic analysis of Brachypodium hybridum and its diploid progenitors. Nat Commun 2020, 11:3670. The authors generated two chromosome-length assemblies for 2 Brachypodium species in addition to Illumina-based assemblies dozens more B. distachyon accessions. By comparing. Through multiple different comparative evolutionary methods (Kmer, structural variant, pan-genome analysis, plastome comparison, syntenic, etc), the authors described the history of Brachypodium pre- and post-polyploidization, gene loss/gain, and structural changes throughout different lineages.
- 15.Golicz AA, Batley J, Edwards D: Towards plant pangenomics. Plant Biotechnology Journal 2016, 14:1099–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao L, Gonda I, Sun H, Ma Q, Bao K, Tieman DM, Burzynski-Chang EA, Fish TL, Stromberg KA, Sacks GL, et al. : The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nature Genetics 2019, 51:1044–1051. [DOI] [PubMed] [Google Scholar]
- 17. Liu Y, Du H, Li P, Shen Y, Peng H, Liu S, Zhou G-A, Zhang H, Liu Z, Shi M, et al. : Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182:162–176.e13. 26 soybean varietals were genome sequenced and de novo assembled. This new pan-genome set was then compared to three existing reference genomes and characterized the core and dispensable gene sets as well as the genetic variation between lines in both core and dispensable gene models; specifically the authors looked at variation and core/dispensable makeup with genetic regions that recently underwent whole-genome duplication.
- 18.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. : Real-time DNA sequencing from single polymerase molecules. Science 2009, 323:133–138. [DOI] [PubMed] [Google Scholar]
- 19.Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M: Improved data analysis for the MinION nanopore sequencer. Nat Methods 2015, 12:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chin C-S, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O’Malley R, Figueroa-Balderas R, Morales-Cruz A, et al. : Phased diploid genome assembly with single-molecule real-time sequencing. Nature Methods 2016, 13:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, et al. : Mind the Gap: Upgrading Genomes with Pacific Biosciences RS Long-Read Sequencing Technology. PLoS ONE 2012, 7:e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hon T, Mars K, Young G, Tsai Y-C, Karalius JW, Landolin JM, Maurer N, Kudrna D, Hardigan MA, Steiner CC, et al. : Highly accurate long-read HiFi sequencing data for five complex genomes. Sci Data 2020, 7:399. 5 genomes (including maize) were subjected to HiFI sequencing and shown that median read accuracy was >99.8% for all samples; these genomes had >14 Kb library sizes and overall average quality values of Phred ~>30.
- 23.Vondrak T, Ávila Robledillo L, Novák P, Koblížková A, Neumann P, Macas J: Characterization of repeat arrays in ultra-long nanopore reads reveals frequent origin of satellite DNA from retrotransposon-derived tandem repeats. Plant J 2020, 101:484–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A, Olson ND, et al. : Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 2019, 37:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deng Y, Liu S, Zhang Y, Tan J, Li X, Chu X, Xu B, Tian Y, Sun Y, Li B, et al. : A telomere-to-telomere gap-free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding. Mol Plant 2022, 15:1268–1284. The authors create the first telomere-to-telomere reference genome assembly in watermelon. They compare this assembly to two gapped genomes of different accessions and show a higher resolution in SNP count as well as successfully sequencing previously unknown genetic regions that confer regulatory control over a gene conferring agricultural value to the commodity.
- 26.Lang D, Zhang S, Ren P, Liang F, Sun Z, Meng G, Tan Y, Li X, Lai Q, Han L, et al. : Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi reads of Pacific Biosciences Sequel II system and ultralong reads of Oxford Nanopore. Gigascience 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz DC, Li X, Hernandez LI, Ramnarain SP, Huff EJ, Wang Y-K: Ordered Restriction Maps of Saccharomyces cerevisiae Chromosomes Constructed by Optical Mapping. Science 1993, 262:110–114. [DOI] [PubMed] [Google Scholar]
- 28.Jing J, Reed J, Huang J, Hu X, Clarke V, Edington J, Housman D, Anantharaman TS, Huff EJ, Mishra B, et al. : Automated high resolution optical mapping using arrayed, fluid-fixed DNA molecules. Proc Natl Acad Sci U S A 1998, 95:8046–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu T, Wang L, Rimbert H, Rodriguez JC, Deal KR, De Oliveira R, Choulet F, Keeble-Gagnère G, Tibbits J, Rogers J, et al. : Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J 2021, 107:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mascher M, Wicker T, Jenkins J, Plott C, Lux T, Koh CS, Ens J, Gundlach H, Boston LB, Tulpová Z, et al. : Long-read sequence assembly: a technical evaluation in barley. Plant Cell 2021, 33:1888–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belser C, Istace B, Denis E, Dubarry M, Baurens F-C, Falentin C, Genete M, Berrabah W, Chèvre A-M, Delourme R, et al. : Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat Plants 2018, 4:879–887. [DOI] [PubMed] [Google Scholar]
- 32. Liu J, Seetharam AS, Chougule K, Ou S, Swentowsky KW, Gent JI, Llaca V, Woodhouse MR, Manchanda N, Presting GG, et al. : Gapless assembly of maize chromosomes using long-read technologies. Genome Biol 2020, 21:121. This study utilized a hybrid assembly approach with Nanopore and PacBio sequencing in addition to optical mapping to yield a high-resolution assembly of a maize inbred line B37-Ab10, including gapless assemblies of chromosome 3 and 9.
- 33.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. : The B73 maize genome: complexity, diversity, and dynamics. Science 2009, 326:1112–1115. [DOI] [PubMed] [Google Scholar]
- 34.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. : Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang B, Tseng E, Baybayan P, Eng K, Regulski M, Jiao Y, Wang L, Olson A, Chougule K, Van Buren P, et al. : Variant phasing and haplotypic expression from long-read sequencing in maize. Commun Biol 2020, 3:78. Long read sequencing was used to successfully phase the parental haplotypes within reciprocal maize hybrids (B73 and Ki11). These genomes were analyzed for variant content as well as allelic-specific expression and cis- and trans-regulatory effects by incorporating transcript reads into the analysis.
- 36.Padgitt-Cobb LK, Kingan SB, Wells J, Elser J, Kronmiller B, Moore D, Concepcion G, Peluso P, Rank D, Jaiswal P, et al. : A draft phased assembly of the diploid Cascade hop (Humulus lupulus) genome. The Plant Genome 2021, 14. [DOI] [PubMed] [Google Scholar]
- 37. Bredeson JV, Lyons JB, Oniyinde IO, Okereke NR, Kolade O, Nnabue I, Nwadili CO, Hřibová E, Parker M, Nwogha J, et al. : Chromosome evolution and the genetic basis of agronomically important traits in greater yam. Nat Commun 2022, 13:2001. A high-resolution reference genome for yam was created with long-read sequencing, HiC, and short read Illumina reads. The authors identified useful QTLs as well as variant characterization between other yam species in addition to evolutionary divergence that has occurred between lineages after a paleopolyploidization event.
- 38. Zheng Y, Yang D, Rong J, Chen L, Zhu Q, He T, Chen L, Ye J, Fan L, Gao Y, et al. : Allele-aware chromosome-scale assembly of the allopolyploid genome of hexaploid Ma bamboo (Dendrocalamus latiflorus Munro). J Integr Plant Biol 2022, 64:649–670. A complex long-read and HiC sequencing breakdown of bamboo showed haplotype information for the hexaploid, including sub-genome composition in addition to transcriptomic profiling in 8 different tissues. Unmapped repetitive regions were kept to <1% of the whole genome.
- 39. Chen H, Zeng Y, Yang Y, Huang L, Tang B, Zhang H, Hao F, Liu W, Li Y, Liu Y, et al. : Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat Commun 2020, 11:2494. A long-read and HiC reference genome construction of of alfalfa. The authors compare allelic sections across the chromosomes and show the TE expansion that has occurred across the genome in relation to whole genome duplication. The authors used this quality assembly as a means to confirm a novel CRISPR-CAS9 editing method, which they also developed for this paper.
- 40.Li Z, Long Y, Yu Y, Zhang F, Zhang H, Liu Z, Jia J, Mo W, Tian SZ, Zheng M, et al. : Pore-C simultaneously captures genome-wide multi-way chromatin interaction and associated DNA methylation status in Arabidopsis. Plant Biotechnol J 2022, 20:1009–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe DB: ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Research 2008, 18:810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denisov G, Walenz B, Halpern AL, Miller J, Axelrod N, Levy S, Sutton G: Consensus generation and variant detection by Celera Assembler. Bioinformatics 2008, 24:1035–1040. [DOI] [PubMed] [Google Scholar]
- 43.Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KLT, et al. : The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452:991–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, Richard McCombie W, Jarvis ED, et al. : Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature Biotechnology 2012, 30:693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. : High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A 2011, 108:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berlin K, Koren S, Chin C-S, Drake JP, Landolin JM, Phillippy AM: Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol 2015, 33:623–630. [DOI] [PubMed] [Google Scholar]
- 47.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM: Canu: scalable and accurate long-read assembly via adaptive -mer weighting and repeat separation. Genome Res 2017, 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li F-W, Harkess A: A guide to sequence your favorite plant genomes. Applications in Plant Sciences 2018, 6:e1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Workman R, Fedak R, Kilburn D, Hao S, Liu K, Timp W: High Molecular Weight DNA Extraction from Recalcitrant Plant Species for Third Generation Sequencing v1. [date unknown], doi: 10.17504/protocols.io.4vbgw2n. [DOI] [Google Scholar]
- 50.Deschamps S, Zhang Y, Llaca V, Ye L, May G, Lin H: A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. [date unknown], doi: 10.1101/327817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang F, Wang S, Wang H, Wang A, Xu D, Liu H, Yang B, Yuan L, Lei L, Chen R, et al. : A chromosome-level reference genome of a Convolvulaceae species Ipomoea cairica. G3 Genes|Genomes|Genetics 2022, 12:jkac187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ni P, Huang N, Nie F, Zhang J, Zhang Z, Wu B, Bai L, Liu W, Xiao C-L, Luo F, et al. : Genome-wide detection of cytosine methylations in plant from Nanopore data using deep learning. Nat Commun 2021, 12:5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang B, Yang X, Jia Y, Xu Y, Jia P, Dang N, Wang S, Xu T, Zhao X, Gao S, et al. : High-quality Arabidopsis thaliana Genome Assembly with Nanopore and HiFi Long Reads. Genomics Proteomics Bioinformatics 2022, 20:4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]