

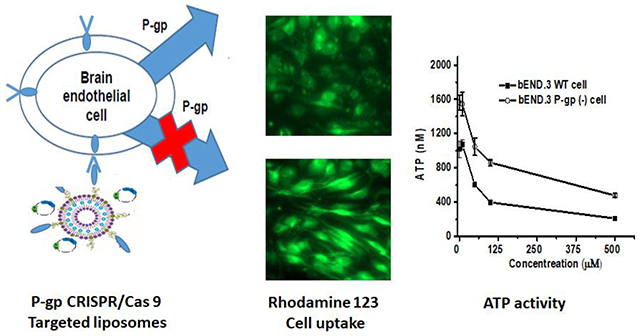
Abstract
This investigation aimed to use CRISPR-Cas9 gene-editing to knock down P-gp expression and then establish a feasible cell line to evaluate the potential pharmacoresistance of therapeutic agents mediated by efflux. A cationic liposome was prepared as a “smart bomb” by conjugating with a peptide-based targeting ligand (THRPPMWSPVWP, specifically binding to transferrin receptors at the blood-brain barrier (BBB), and then formed a nanocomplex with P-gp knockdown CRISPR/Cas9 plasmid. Higher uptakes of targeted and stable liposomes in bEND.3 cells were observed compared to non-peptide conjugated ones (p<0.05). The P-gp transporters were successfully knocked down by the cell-nontoxic CRISPR/Cas9 targeted liposomes and P-gp associated ATP activities were higher in the transfected cells (p<0.05). Functional studies of knocked down cells were evaluated by using prototypical P-gp substrates rhodamine 123 and doxorubicin. More accumulation of rhodamine 123 and higher cytotoxic sensitivity of doxorubicin was observed in the transfected cells as compared with those in the wild-type cells.
Keywords: liposome, brain targeting, transferrin receptor, CRISPR/Cas9, P-glycoprotein, pharmacoresistance
Graphical Abstract
INTRODUCTION
Treatment of brain diseases remains one of the biggest clinical challenges because of the difficulty of the adequate delivery of therapeutic agents to the brain (Geldenhuys et al., 2012, Chen and Liu, 2012, Krol, 2012). At the blood-brain barrier (BBB), brain cells are tightly packed by tight-junctional proteins and control the entry of compounds into the brain (Pardridge, 2007). The BBB restricts the penetration into the brain not only of large molecule drugs but also more than 98% of small molecule drugs (Tucker, 2011, Pardridge, 2005, Muldoon et al., 2007). Furthermore, physiological efflux transporters at the BBB, especially P-glycoprotein (P-gp), also called multidrug resistance protein 1 (MDR1; ABCB1), pump drugs from the brain endothelial membrane and cytosol compartment back into the blood for subsequent elimination (Bartels, 2011). Simultaneously responding to pathophysiological changes, efflux transporters overexpress at the BBB in neurological diseases including amyotrophic lateral sclerosis (ALS), epilepsy, brain cancer, spinal cord injury, Alzheimer’s disease, and Parkinson’s disease (Mahringer et al., 2011, Mahar Doan et al., 2002). P-gp efflux transporters at the BBB lead to the majority of drug failures to reach clinical approval due to low uptake of potential therapeutics into the brain and further induce pharmacoresistance of approved drugs in the later stage of diseases (Mahringer et al., 2011). To predict if therapeutic candidates are a substrate of P-gp, attempts have been made to develop evaluation models by knocking down endogenous P-gp in brain endothelial cells (Ke et al., 2019, Neuhaus et al., 2010, Ye et al., 2020).
Recently, advances in nuclease-based CRISPR/Cas9 systems, have allowed researchers to generate diverse genomic modifications in cultured cells and even in whole animals (Ding et al., 2016, Wang et al., 2016). Cas9 uses guide RNA (gRNA) to locate site-specific DNA sequences, directs Cas9 nuclease to specific sites, and therefore provides unique flexibility for genome editing (Ding et al., 2016). Superior to small interfering RNA (siRNA), the CRISPR/Cas9 system shows great promise for gene editing in terms of efficiency, simplicity, and versatility (Wang et al., 2016). However, CRISPR/Cas9 based therapeutics also face tremendous obstacles before entering the targeted cells, like other biological drugs and siRNA (Song, 2017, Mout et al., 2017). The development of feasible carriers and targeted delivery techniques for the delivery of CRISPR/Cas9 is highly desirable for improving the efficacy of CRISPR/Cas9 based gene editing tools (Mout et al., 2017).
Large and/or hydrophilic essential molecules such as hormones, transferrin, and insulin bind specific receptors and use transcytosis mechanisms across the BBB (Geldenhuys et al., 2012, Gabathuler, 2010). Conjugations of liposomes to ligands or antibodies, which recognize natural receptors expressed in brain endothelial cells at the BBB, are expected to deliver more drugs across the BBB via receptor-mediated transcytosis (Gabathuler, 2010, De Rosa et al., 2012). By providing a delivery vehicle for the therapeutics to the target cell, liposomes conjugated to the targeting ligand can increase the chance of binding to the cell surface receptor.
In this study, we designed a multifunctional liposome system for the delivery of CRISPR/Cas9. A transferrin-targeting peptide was conjugated on cationic liposomes. This targeted nanocarrier was used to specifically deliver P-gp CRISPR/Cas9 to the brain endothelial cells. The approach introduces a novel BBB-specific CRISPR/Cas9 delivery system to knock down the P-gp. The designed liposome-CRISPR/Cas9 system was expected to have a selective binding on the targeted cells and a significant gene silencing effect on the P-gp, resulting in the establishment of an in vitro BBB model cell line with low P-gp background.
MATERIALS AND METHODS
Materials
Mouse brain endothelial bEND.3 cells, Dulbecco’s Modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin plus streptomycin solution, and trypsin-EDTA were obtained from American Tissue Culture Collection (Rockville, MD, USA). Cholesterol, 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG 2000), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol) - 2000] (DSPE-PEG 2000 carboxylic acid), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (DSPE-NBD) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Transferrin receptor targeting peptide (THRPPMWSPVWP) was obtained from AnaSpec Corp. (Fremont, CA, USA). 2-morpholinoethanesulfonic acid (MES), N-ethylcarbodiimide (EDC), and N-hydroxysuccinimide (NHS) were purchased from Sigma Aldridge (St. Louis, MO, USA). ATPlite™ 1 step was purchased from PerkinElmer Inc (Branford, CT, USA). P-gp antibody and CRISPR/Cas9 KO plasmid targeting the sequence of MDR1 Exon5 GAGTACTACGACCACAAACC were from Santa Cruz Biotechnology, Inc (Dallas, TX, USA). Riluzole, rhodamine 123, doxorubicin, columns, and other chemical reagents were purchased from VWR (Radnor, PA, USA).
Liposome preparation
Liposomes composed of cholesterol, PEGylated lipid, and cationic lipids were prepared by using a hydration method (Yang et al., 2014a, Yang et al., 2012). Briefly, 2 mg of riluzole and lipid mixtures of cholesterol, 1,2-dioleoyl-3-trimethylammonium propane (DOTAP), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG 2000), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(poly ethyleneglycol)-2000] (DSPE-PEG 2000 carboxylic acid) with/without 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (DSPE-NBD) (molar ratio 6:5:0.6:0.3:0.1; the total amount of lipids was 12.7 mg) were dissolved in an organic solvent of chloroform and methanol (9:1, v/v). A dried thin lipid film was prepared in a round-bottom flask using a Buchi R-114 Rotavapor (Buchi Laboratories AG, Flawil, Switzerland) at a temperature of 50±2°C water bath. Subsequently, the residual solvent was completely removed by keeping the flask under vacuum for 2 hours. The dry lipid film was hydrated with 2 mL of 1x PBS solution. The liposome colloid thus was obtained by vortexing and then dispersing in an ultrasound water bath for 5 minutes. Final formulations were extruded in order through 0.4, 0.2, and 0.1 μm polycarbonate membrane filters (Coster Nucleopore, Cambridge, MA). Riluzole was encapsulated in the liposomes as an internal reference substance to evaluate the amounts of liposomes.
Liposome conjugation with transferrin peptide
Carboxylic acid groups on the prepared liposome were first activated by N-ethyl carbodiimide (EDC) and then reacted with N-hydroxysuccinimide (NHS) (Wei et al., 2015). Briefly, prepared liposomes were incubated with 3-dimethylaminopropyl carbodiimide (EDC) and N-hydroxysuccinimide (NHS) at room temperature for 15 minutes. The resulting surface of activated NHS-PEG-liposomes was first neutralized by 0.5 N NaOH to reach a pH of 8.5 and then reacted with different amounts of transferrin receptor binding-peptides (THRPPMWSPVWP) at 4 °C for 48 hours. The peptide-conjugated liposomes were purified by three methods. I) ultracentrifugation: the peptide-conjugated liposomes were precipitated using an XL-70 Ultracentrifuge (Beckman Coulter, Brea, CA, USA) with a 70.1 Ti rotor at 50,000 rpm for 1 hour at 4°C. II) size-exclusion column: Thermo Scientific™ Zeba™ Spin 0.5 mL Desalting Columns with a molecular weight cut-off (MWCO of 7KDa) were used to purify the liposomes. Samples were diluted in 25 mM Tris 500 mM NaCl at pH 7.5 and then desalted into the final buffer. All samples were processed according to the manufacturer’s recommended instructions. Or III) filter centrifugation: Vivaspin® 0.5mL ultrafiltration spin columns (MWCO of 3KDa) were used to purify the prepared liposomes. Samples were loaded in the insert column and centrifuged at 16,000g for 2 hours at 4°C.
Free reactants in the supernatant or filtrate and riluzole in the liposome were determined by a high-performance liquid chromatography (HPLC) method according to a previously validated method (Yang et al., 2018). Briefly, a gradient HPLC system with an auto-sampler and column oven was used (Agilent 1220 DAD; Santa Clara, CA, USA). Separation and quantitation were performed by using a ZORBAX C8 Column (5 μm, 4.6mm×250 mm) at 40 °C. The mobile phase, consisting of 0.05% formic acid in water and methanol (pH = 3.1, 70:30, v/v), was run at a constant flow rate of 1.0 mL/min. The sample injection volume was 10 μL and the detection wavelength was 264 nm.
Liposome characterization
Particle sizes and polydispersity index (PDI) of liposome nanoparticles were measured using a Delsa™ Nano C nanosizing system working at scattering angles of 165°C (Beckman Coulter, Brea, CA, USA) (Bai et al., 2009). Briefly, 20 μL of liposomal samples were dispensed into microsized cells for measurement with a 100 pinhole set to 20 mm for 70 continuous sample accumulation times. Each analysis was repeated three times to obtain the average particle size. The physicochemical stability of liposomes was assessed by determining the changes in particle size, PDI, and riluzole content upon storage of the formulations at 4°C for 1 month.
Cellular uptake of liposomes in brain endothelial cells
Mouse brain endothelial bEND.3 cells were grown in recommended Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% FBS, 2 mM L-glutamine, 100 μg/mL penicillin plus 100 μg/mL streptomycin, in a humidified 37°C incubator with 5% CO2. The bEND.3 cells were seeded on a 100 mm petri dish at a density of 2×106 cells/mL counted by a Cellometer® Auto T4 Cell Counter (Nexcelom Bioscience LLC, Lawrence, MA, USA). After 24 hours of incubation at 37°C and 5% CO2, cells were treated with targeted or non-targeted fluorescent liposomes for 48 hours. The cells were washed three times with 1x PBS and then imaged using an EVOS® FL cell imaging system (Life Technologies, Waltham, MA). After imaging, cells were scraped, suspended, and diluted to 1x106 cells/mL and then analyzed with an Acuri™ C6 flow cytometer (BD Biosciences, San Jose, CA, USA) and WinListTM 3D Flow Cytometry Analysis Software (Verity, Topsham, ME, USA) (Yang et al., 2017).
In vitro transfection of targeted CRISPR/Cas9 liposomes in brain endothelial cells
CRISPR/Cas9 plasmid was added to 150 μL RNase free water, followed by the addition of 150 μL of RNase-free water containing a series of targeted cationic liposomes with plasmids according to the different molar ratios of phosphate groups and cationic lipids as shown in Table I. The mixture was incubated for 15 minutes and then added to the cells. One day before the transfection, the bEND.3 cells were seeded in a 60 mm x 15 mm petri dish at a density of 2 x 105 cells/mL. 50 μL of formulations were added to the cells in the petri dish and mixed by gentle swirling of the dish. The petri dish was incubated at 37°C for 48 hours in a 5% CO2 incubator. The efficacy of liposomes on the inhibition of P-gp expression was tested by determining P-gp levels in brain endothelial bEND.3 cells according to previously published procedures (Bai et al., 2008). After treatments, proteins were isolated from the confluent bEND.3 cells using a cell lysis buffer. Total protein concentrations were measured with a Pierce BCA assay kit. 50 μg of proteins were electrophoresed and transferred to a polyvinylidene difluoride membrane. The membranes were treated with a primary P-gp antibody and then a secondary antibody. Signals for proteins were detected by Western Lightning Chemiluminescence reagents. The P-gp protein level was quantified from the densitometric intensity of each band using a UVP ChemiDoc-It® Imager (Upland, CA, USA). Results were normalized to total protein loading confirmed by BCA protein assay and expressed as a percentage of the band volume of treatment compared to that derived from the buffer control (Yang et al., 2017, Yang et al., 2014b, Chan et al., 2017).
Table 1.
Twelve formulation compositions including four amounts of CRISPR/Cas9 plasmid and three molar ratios of phosphate groups in plasmid and cationic lipids.
| Formulations | CRISPR/Cas9 plasmid amount | Phosphate group and cationic lipid molar ratio |
|---|---|---|
| F1 | 6 ng | 1:1 |
| F2 | 12 ng | 1:1 |
| F3 | 24 ng | 1:1 |
| F4 | 48 ng | 1:1 |
| F5 | 6 ng | 1:2 |
| F6 | 12 ng | 1:2 |
| F7 | 24 ng | 1:2 |
| F8 | 48 ng | 1:2 |
| F9 | 6 ng | 1:3 |
| F10 | 12 ng | 1:3 |
| F11 | 24 ng | 1:3 |
| F12 | 48 ng | 1:3 |
Cell cytotoxicity study in brain endothelial cells
The safety of CRISPR/Cas9 and liposome complexes F1 to F12 was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described (Yang et al., 2012). Briefly, brain endothelial bEND.3 cells were grown in flat-bottom, 96-well microtiter tissue culture plates. Immediately before the start of the experiment, the medium was removed from the wells and the cells were washed with 1x PBS. Subsequently, the cells were incubated with 50 μL of formulations or control samples for 24 hours. Each assay was performed on eight samples, and cell viability was calculated based on the absorbance from cells treated with samples and controls from cells treated with the medium.
ATP activity test in brain endothelial cells
An ATPase assay was performed as described previously (Dong et al., 2009). Wide-type and P-gp knockdown bEND.3 cells were seeded in an opaque 96-well plate at a density of 2x105 cells/well and incubated overnight. P-gp inhibitor, verapamil, was added to cells and incubated at 37°C for 2 hours. Following treatment, cells were washed twice with ice-cold 1x PBS and lysed with PBS containing 1% Triton X-100 at 37°C for 30 minutes. ATP in cell lysates was then measured using ATPlite™ 1 step according to the manufacturer’s instruction.
Rhodamine 123 uptakes in brain endothelial cells
The internalization of fluorescent rhodamine 123, a P-gp substrate, was evaluated in wild-type and CRISPR/Cas9 transfected brain endothelial bEND.3 cells as described previously (Yang et al., 2014b). Brain endothelial bEND.3 cells were seeded on a 6-well plate with 2 mL of cell media. After 24 hours of incubation at 37°C and 5% CO2, formulations including 0.2 mg/mL rhodamine 123 were added to the media. After 18 hours of incubation at 37°C, the media with treatment solutions were removed and the cells were washed with PBS three times. Cells were fixed in paraformaldehyde solution and permeabilized in Triton X-100. The final cell samples were imaged using an EVOS® FL cell imaging system (Life Technologies, Waltham, MA) (Yang et al., 2015, Yang et al., 2017). After imaging, cells were scraped, suspended, and diluted to 1x106 cells/mL and then analyzed with an Acuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA).
Doxorubicin effect in wild-type and P-gp knockdown brain endothelial cells
The effects of anticancer drug doxorubicin, a P-gp substrate, on wild-type and P-gp knockdown brain endothelial bEND.3 cells were evaluated by MTT assay as described previously (Yang et al., 2015). Wild-type and P-gp knockdown brain endothelial bEND.3 cells were grown in flat-bottom, 96-well microtiter tissue culture plates, respectively. When cells grew confluence, 50 μL of 1, 2, 4, 6, 8, 10, 20, and 30 μM of doxorubicin solutions and media control were added into each well for 24 hours. Increased cytotoxicity of doxorubicin was calculated based on the absorbance from cells treated with samples and controls from cells treated with the medium.
Statistical analysis
All data were expressed as the mean±SD. All sets of experiments were repeated at least 3 times. One-way ANOVA was used to compare the data. When the differences in the means were significant, post hoc pairwise comparisons were conducted using Newman-Keuls multiple comparisons (GraphPad Prism, version 3.03, GraphPad Software, San Diego, CA). Differences in p-values less than 0.05 were considered statistically significant.
RESULTS
Purification of targeted liposomes
Liposomes were prepared by a thin-film hydration method with four types of lipids, including cholesterol, cationic DOTAP lipid, PEG-2000 linked DSPE to reduce uptake of liposomes by the macrophage system, and DSPE-PEG 2000 carboxylic acid active lipid. Carboxylic acid groups in the prepared liposomes were activated by NHS and EDC and then reacted with amino groups in transferrin-binding peptides. Finally, the conjugated liposomes were purified. HPLC spectra demonstrated that all left reactants and reference substance riluzole were well separated with different retention times in the optimized system (Figure 1A). Of these, four peaks were related to NHS, EDC, peptide, and riluzole. The chromatographic profile for riluzole eluted at 7.347 min showed a single major peak that differs in retention times compared to other reactants, while three other peaks eluted before the main peak with retention times of 3.692, 4.628, and 6.197 min were related to NHS, EDC, and transferrin peptide, respectively. To remove unreacted chemicals, several methods were used to purify the liposomes and the final liposome products were quantitatively and qualitatively analyzed by HPLC assay. HPLC analysis indicated that better-purified targeted liposomes were obtained from the size-exclusion chromatography and spin filter columns compared to the ultracentrifugation method (Figures 1B, 1C, and 1D). After ultracentrifugation, the prepared liposomes were not precipitated and the reactants were still left in the collected liposomal layer (Figure 1B). After the purification process by the size-exclusion column or spin filter column, no peaks of NHS, EDC, and peptide were observed, suggesting both of these methods could remove all leftover reactants (Figures 1C and 1D). However, a much higher amount of liposomes indicated by a small riluzole peak area in HPLC spectra was adsorbed in the desalting size-exclusion column after elution than that from the filter column centrifugation. Overall, chromatograms of the final liposomes indicated that purified liposome product was better obtained by the filter column centrifugation.
Figure 1.

HPLC spectra of unpurified liposomes (A), liposomes after ultracentrifugation (B), liposomes after filter column centrifugation (C), and after desalting column size-exclusion liposomes (D) (I: NHS; II: EDC; III: peptide; and IV: riluzole).
Liposome characterization and stability
After the conjugation of peptides and liposomes was identified by the HPLC spectra, the size of liposomes was assessed before and after peptide conjugation. The particle size of the unconjugated liposomes was 164.7±3.1 nm. After the conjugation, the particle size of the targeted liposomes was 177.2±1.5 nm, slightly bigger than the plain liposomes (p<0.05). This significantly increased size indicated that peptide was conjugated on the liposomes. The size of the targeted liposomes was still less than 200 nm, which has less chance to activate the complement system in the liver and spleen and be quickly removed from the bloodstream (Kulkarni and Feng, 2013). The storage stability of liposome formulations at 4-8°C with a time course of one month was investigated. As shown in Figure 2, no significant alteration in the mean size diameters (Figure 2A), polydispersity index (PDI) (Figure 2B), and riluzole content (Figure 2C) under the testing condition was observed from either unconjugated or targeted liposomes. Altogether, targeted liposomes demonstrated consistent stability of particle size, size distribution, and drug content during one-month storage in the refrigerator.
Figure 2.

Changes in particle size (A), PDI (B), and riluzole content (C) of liposomes stored at 4°C for 1 month (I: unconjugated liposomes; II: targeted liposomes). Data represent the mean±SD, n=3. *Results are significantly different (p< 0.05).
Uptake study of liposomes in brain endothelial cells
In this study, a fluorescent probe lipid was adopted as an indicator to analyze the cellular uptake of both unconjugated liposomes and targeted liposomes. The cellular uptake of both liposomes was visualized in brain endothelial bEND.3 cells with a fluorescence microscope and quantified by a flow cytometer after treatments. The results showed that the cellular uptake of targeted liposomes was significantly higher than that of the unconjugated liposomes in bEND.3 cells (Figure 3). A peak shift to the right on the X-axis in a graphic representation of the flow cytometer results indicates an exponential increase of fluorescence in cells. Compared with unconjugated liposomes, targeted liposomes displayed more than 1.5-fold higher cellular internalization (Figures 3A and 3B). Similar results were obtained in the intracellular distribution of both liposomes as visualized in fluorescence microscope images (Figures 3C and 3D). Cells treated with targeted liposomes exhibited significantly higher fluorescence distribution in the bEND.3 cells than unconjugated liposomes. This suggested that the targeting effect of transferrin-binding peptide allowed targeted liposomes to interact with transferrin receptors on the brain endothelial cells, therefore significantly increasing the cellular uptake of the targeted liposomes.
Figure 3.

Representative stacked histograms were generated from a batch of flow cytometry analysis of quantified fluorescence intensity from unconjugated and targeted liposomes (A), quantified fluorescence intensity after repeats from flow cytometric analysis (B), and accumulation of fluorescence from unconjugated liposomes (C) as well as transferrin binding peptide-conjugated liposomes (D) in brain endothelial bEND.3 cells. Data represent the mean±SD, n=3. *Results are significantly different (p< 0.05).
Transfection efficiency of CRISPR/Cas9 liposomes in brain endothelial cells
After we successfully prepared and purified the targeted liposomes, the targeted liposomes with cationic lipids were used to form nanocomplexes with negatively charged CRISPR/Cas9 to assist in the transfection process. The expression of a fluorescent GFP tag was first used for analyzing the transfection efficiency of the plasmid. Twelve nanocomplexes of cationic liposomes with CRISPR/Cas9 plasmid were formulated with different molar ratios of negatively charged phosphate groups in CRISPR/Cas9 plasmid and cationic lipids. These twelve formulations included four amounts of CRISPR/Cas9 plasmid (6, 12, 24, and 48 ng), and 1:1; 1:2, and 1:3 of the molar ratios of phosphate groups in the plasmid to cationic lipids in the liposome, which are shown in Table 1. The transfection efficacy of nanocomplexes was assessed by the fluorescence intensity in the cells after the clonal expansion of the tagged flag marker. Highly expressed fluorescence quantified by the cell flow cytometer indicated that the cells were successfully transfected by the liposome-delivered CRISPR/Cas9 plasmid. The results showed that the transfection efficiency was in a linear relationship with the amounts of the plasmid from 6 to 48 ng related to formulations F5 to F8 and F9 to F12. Moreover, the transfection efficiency increased with the molar ratio increase of the plasmid of cationic liposomes in the nanocomplexes from 1:1 to 1:3 (Figure 4A). When the amount of plasmid was 48 ng, and the ratio of the plasmid to cationic liposomes was 1:3 in Formulation 12, the highest expressed fluorescence and transfection efficiency was obtained (Figure 4A). Consistent results were demonstrated in the images under a fluorescence microscope (Figure 4B). Higher fluorescence intensity was observed with the increase in the molar ratio of the plasmid to cationic liposomes in the nanocomplexes, and the increase in plasmid amount (Figure 4B). The best transfection was from the optimized formulation 12. Overall, the optimized formulation successfully delivered CRISPR/Cas9 into the cells, and then the flag GFP marker was significantly expressed in the transfected cells. Being consistent with the study results from other groups (Constantinescu et al., 2019, Sharma et al., 2012), we found that the optimized targeted liposomes successfully delivered more CRISPR/Cas9 into the cells, and the flag GFP marker was significantly expressed in the transfected cells with about 2-fold increase compared to unconjugated liposome control (data not shown).
Figure 4.
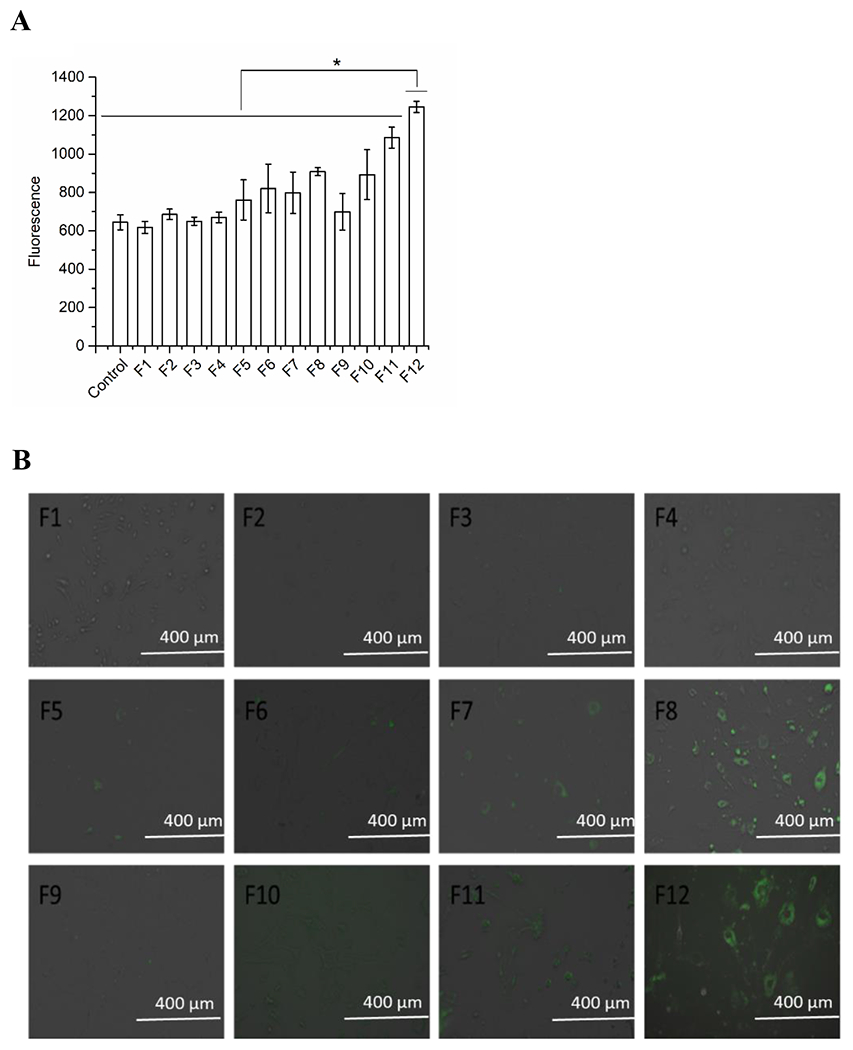
Fluorescence determination of CRISPR/Cas9 targeted liposomal formulations by cell flow cytometry (A) and under a fluorescence microscope (B) in brain endothelial bEND.3 cells. Data represent the mean±SD, n=3. *Results are significantly different (p< 0.05) (F1: 6 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F2: 12 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F3: 24 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F4: 48 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F5: 6 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F6: 12 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F7: 24 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F8: 48 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F9: 6 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F10: 12 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F11: 24 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F12: 48 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids).
Safety study of CRISPR/Cas9 liposomes
An ideal therapeutic nanocarrier should possess adequate delivery efficiency while maintaining low cytotoxicity. The cytotoxicity of various CRISPR/Cas9 targeted liposome formulations was evaluated by an MTT assay in bEND.3 cells. As presented in Figure 5, all formulations showed no apparent cytotoxicity even at a higher CRISPR/Cas9-lipid ratio (e.g., 1:3). These results demonstrated CRISPR/Cas9 targeted liposome formulations possess remarkably high compatibility when being used for CRISPR/Cas9 delivery, which could be used effectively to evaluate the level of P-gp knockdown in further studies without concerning cell viability. The changes in the P-gp expression and activity in the following studies could only be due to the CRISPR/Cas9 editing tool, not due to the cell cytotoxicity.
Figure 5.

Cytotoxicity of CRISPR/Cas9 targeted liposomal formulations in brain endothelial bEND.3 cells. Data represent the mean±SD, n=6-8 (F1: 6 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F2: 12 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F3: 24 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F4: 48 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F5: 6 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F6: 12 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F7: 24 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F8: 48 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F9: 6 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F10: 12 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F11: 24 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F12: 48 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids).
P-gp evaluation in brain endothelial cells
Based on high cellular uptake efficiency and low cytotoxicity, CRISPR/Cas9 liposomes seemed to possess a great potential to inhibit P-gp. Therefore, we further measured the in vitro transfection efficiency of CRISPR/Cas9 liposomes on knocking down P-gp by Western blotting assay in bEND.3 cells. In this study, cells treated by optimized nanocomplex formulations were lysed, and the expressions of P-gp were analyzed and quantified by Western blot. Cells without any treatments were taken as a control and the density of the P-gp band area was considered 100%. As indicated in Figure 6, the level of P-gp protein was significantly decreased when treated with an increasing amount of CRISPR/Cas9 plasmid. When the amount of plasmid was 48 ng (in Formulation 8), the downregulation effect on P-gp was 54.9±7.6%, which was significantly lower than other formulations containing 6 ng, 12 ng, or 24 ng of the plasmid in formulations 5, 6, or 7 with 100.7±6.3%, 78.3±13.5%, and 76.0±10.3% of the control, respectively. Similar results were obtained for formulation 12 with 48 ng of the plasmid (Figure 6). The downregulation effect on P-gp was 47.7±11.0% when formulation 12 was used, which was significantly lower than formulations containing less amount of plasmid. In addition, consistent with the results in the flow cytometer and fluorescence study (Figure 4), the P-gp efflux protein was significantly knocked down in cells that were treated by the nanocomplex formulations with the increased ratio of cationic liposomes to plasmid (Figure 6B). Overall, the optimized CRISPR/Cas9 targeted liposome showed significant efficiency in knocking down P-gp. The P-gp knockdown cells were further confirmed by ATP assay, fluorescent rhodamine 123 uptake study, and doxorubicin efficacy study.
Figure 6.

P-gp expression measured by Western blots in brain endothelial bEND.3 cells with CRISPR/Cas9 liposomes (the expression of P-gp in cells with buffer was noted as a baseline level). Data represent the mean±SD, n=3. *Results are significantly different (p< 0.05) (F1: 6 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F2: 12 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F3: 24 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F4: 48 ng CRISPR/Cas9 plasmid with 1:1 ratio of plasmid phosphate groups and cationic lipids; F5: 6 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F6: 12 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F7: 24 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F8: 48 ng CRISPR/Cas9 plasmid with 1:2 ratio of plasmid phosphate groups and cationic lipids; F9: 6 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F10: 12 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F11: 24 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids; F12: 48 ng CRISPR/Cas9 plasmid with 1:3 ratio of plasmid phosphate groups and cationic lipids).
ATP activity in wild-type and P-gp knockdown brain endothelial cells
To further investigate the P-gp levels in wild-type and P-gp knockdown bEND.3 cells, intracellular ATP levels in cells were measured after exposure to various concentrations of verapamil, a P-gp inhibitor. The results showed that in both wild-type and P-gp knockdown cells, with the concentrations of verapamil increasing, the intracellular ATP levels decreased (Figure 7). The ATP level in the wild-type cells was 1071.4± 58.2 nM when verapamil was used at 10 μM, and the ATP level decreased to 209.4±9.9 nM when verapamil was 500 μM. As the P-gp was knocked down by CRISPR/Cas9 targeted liposomes, there was a significant elevation in the ATP level, indicating a decreased level of P-gp, when verapamil was present. The ATP level was greatly increased in P-gp knockdown cells compared with the ATP level in wild-type cells. For example, when the concentration of verapamil was used as 100 μM, the ATP level was 396.4±11.4 nM in wild-type bEND.3 cells, while the ATP level increased to 863.0±37.5 nM in P-gp knockdown cells (Figure 7). The results suggested that the P-gp efflux transporter was successfully knocked down by the CRISPR/Cas9 cationic liposomes, which led to the increasing ATP levels in the P-gp knockdown bEND.3 cells.
Figure 7.

ATP activity in wild-type and P-gp knockdown brain endothelial bEND.3 cells treated with verapamil (WT: wild-type; P-gp (−): P-gp knockdown). Data represent the mean±SD, n=3.
Rhodamine 123 uptakes in wild-type and P-gp knockdown brain endothelial cells
CRISPR/Cas9 targeted cationic liposomes were used to treat bEND.3 cells to knock down the efflux transporter P-gp. Afterward, rhodamine 123, as a substrate of P-gp, was adopted as a fluorescent marker to analyze the cellular uptake of the marker in wild-type and P-gp knockdown bEND.3 cells. The cellular uptake was quantified by flow cytometry and visualized by a fluorescence microscope after treatments. Cells without the treatment of rhodamine 123 were used as a negative control. The flow cytometry results showed that cells with P-gp knockdown displayed significantly higher cellular internalization of rhodamine 123 when compared to cells without CRISPR/Cas9 treatment (Figures 8A and 8B). This indicated that CRISPR/Cas9 targeted liposomes successfully knocked down P-gp, the efflux pump. This led to a nearly two-fold increase of the substrate, rhodamine 123, uptake into bEND.3 cells. Similar results were obtained in the intracellular distribution of rhodamine 123 as visualized by a fluorescence microscope. Cells with P-gp knockdown exhibited significantly higher fluorescence distribution in the cytoplasm than wild-type cells did (Figures 8C and 8D).
Figure 8.

Quantified fluorescence intensity after repeats from flow cytometric analysis (A), and uptake of fluorescent rhodamine 123 in wild-type (B) and P-gp knockdown brain endothelial bEND.3 cells (C) (WT: wild-type cells; P-gp (−): P-gp knockdown cells). Data represent the mean±SD, n=3. *Results are significantly different (p< 0.05).
Doxorubicin effect in wild-type and P-gp knockdown brain endothelial cells
Doxorubicin, a chemotherapy drug, is implicated as a P-gp substrate. The P-gp efflux pump is one of the mechanisms that significantly contributes to doxorubicin chemotherapy resistance and failure. In this study, In vitro cytotoxic efficacy of doxorubicin with various concentrations was evaluated by a standard MTT assay in both wild-type and P-gp knockdown bEND.3 cells. As shown in Figure 9, the cytotoxic efficacy of doxorubicin was dose-dependent. The wild-type cell viability was 72.9±8.0% when doxorubicin was used at 6 μM, and the cell viability decreased to 50.2±11.4% when doxorubicin was 30 μM. As the P-gp was knocked down by CRISPR/Cas9 targeted liposomes, the cell viability was greatly decreased in P-gp knockdown cells compared with the cell viability in wild-type cells. Significant cytotoxic effect of doxorubicin started occurring at a concentration of 1 μM in P-gp knockdown cells with cell viability of 69.7±5.7%. At the same concentration of doxorubicin (1 μM), however, wild-type cells reached nearly 100% cell viability. When the concentration of doxorubicin was increased to 4 μM, the cell viability was 71.7±4.0% in wild-type bEND.3 cells, while the cell viability decreased to 45.9±11.2% in P-gp knockdown cells (Figure 9). The results indicated a higher sensitivity of doxorubicin was found in the P-gp knockdown bEND.3 cells, which resulted in significantly higher cytotoxic effects on bEND.3 cells.
Figure 9.

Cell viability of wild-type and P-gp knockdown brain endothelial bEND.3 cells treated with different concentrations of doxorubicin (WT: wild-type cells; P-gp (−): P-gp knockdown). Data represent the mean±SD, n=6-8.
DISCUSSION
Multiple drug resistance (MDR) is a term used to describe the broad-spectrum resistance to drug therapy in human diseases (Bar-Zeev et al., 2017, Gottesman et al., 2002). Transporter-mediated efflux of exogenous compounds is an important cellular defense mechanism and plays a central role in the barrier and excretory functions of tissues such as the intestinal mucosa, blood-brain barrier, renal proximal tubules, and liver. On the other side, drug resistance is the major cause of the failure of many therapeutic agents, including cancer chemotherapy and many other neuropharmaceuticals. Several members of the ATP-Binding Cassette (ABC) transporter superfamily including P-gp, also called multidrug resistance protein 1 (MDR1; ABCB1), breast cancer resistance protein (BCRP; ABCG2), and several members of the multidrug resistance-associated protein families are highly expressed in these barrier tissues and play an important role in drug disposition (Gottesman et al., 2002). P-gp has been shown to pump substrates drugs such as rhodamine 123, doxorubicin, and paclitaxel, out of brain cells through an ATP-dependent mechanism, which reduces the effective drug concentrations and consequently decreases the therapeutic activity.
At the BBB, brain endothelial cells are major components containing a monolayer with high transendothelial electrical resistance. They are the chief cell type responsible for BBB maintenance (Pardridge, 2005). As brain endothelial cells are derived from the BBB tissue, bEND.3 cells have been commonly used as an in vitro BBB model and are viable to evaluate the ability of drugs to cross the BBB (Yang et al., 2017). Several methods are available to determine the transportation mediated by the P-gp transporter in the brain endothelial cells, however, the discordance of the experimental condition leads to conflict reports of the P-gp substrate candidacy (He et al., 2009, Kido et al., 2002, McInerney et al., 2017, Miller et al., 2011). Over the years, knocking down endogenous P-gp in cells is commonly considered an easy approach to studying drug transport. A higher or lower degree of expression of P-gp was obtained by inhibitory RNA (Celius et al., 2004), clonal selection based on intracellular retention of fluorescent P-gp substrates (Gartzke and Fricker, 2014), and zinc-finger nucleases (ZFNs) (Gartzke et al., 2015). The CRISPR/Cas9 gene-editing technique introduces a double-strand break, which leads to the activation of DNA repair and small insertions or deletions at the target region. CRISPR/Cas9 gene-editing could be an attractive alternative approach for the inhibition of P-gp function because of its unique characteristics including specificity in gene targeting, low immunogenicity, and simplicity of design and production.
Although CRISPR/Cas9 holds exciting promise for different applications and utilities, its delivery challenges have hampered the use of CRISPR/Cas9 as a gene-editing agent. Such progress cannot be made until the gap for CRISPR/Cas9 delivery is bridged. Many researchers have developed novel nanomedicine carriers with the ultimate goal of these formulations being capable of safely and efficiently delivering CRISPR/Cas9 to cells in vitro and/or in vivo settings. These include liposomal/lipid-based nanoparticles (Yu et al., 2016), polymeric nanoparticles /polyplexes (Liu et al., 2018), and cell-penetrating peptides (Wang et al., 2018). Amongst these nanocarriers, liposomal/lipid-based ones have been the most researched, and Lipofectamine™, as a commercial transfection reagent, has been commonly used to complex CRISPR/Cas9 components to increase transfection efficiency and thus lead to DNA editing in cells (Cardarelli et al., 2016). Although the commercially available lipid-based transfection reagents were successfully used for the delivery of CRISPR/Cas9, there have been very few studies using targeted carriers to selectively deliver CRISPR/Cas9 to cells. Some researchers reported the potential of delivering nanocarrier-based CRISPR/Cas9 for a particular organ such as the lung or liver cells (Zhen et al., 2017). It is a significant concern as the nanosystem possibly knocks down P-gp beyond the targeted brain or cancer locations, e.g. in the liver, causing severe toxicity. Because the BBB is a complex and dynamic interface spontaneously responding to physiological changes, transporters can even be promoted by many brain diseases (Mahar Doan et al., 2002). This overexpression of P-gp efflux transporters, constituting the major mechanism of cell adaptation to disease and environmental stress, produces more drug export and resistance (Mahringer et al., 2011). Therefore, the expression of efflux transporters at the BBB leads to the majority of drug failures but possesses a potential target for P-gp knockdown. This also works for cancer cells, which leads to one of the best promising approaches using the P-gp knockdown strategy for the treatment of brain cancers. However, so far there has been no study on the targeted liposomes based delivery of CRISPR/Cas9 to the brain endothelial cells. We explored a targeted liposome conjugated with transferrin peptide as an alternative approach for CRISPR/Cas9 delivery. These targeted liposomes are expected to selectively deliver CRISPR/Cas9 to the cells and knockdown P-gp efflux pumps.
Studies have shown the transferrin receptors expressed in the brain endothelial bEND.3 cells (Chen et al., 2017, Lakkadwala and Singh, 2019). Essential nutrients such as iron use unique transferrin receptor-mediated transcytosis transport pathways across the BBB (Paterson and Webster, 2016). Mimicking those processes, the use of the transferrin receptor as a target for drug delivery remains in the context of ligand or antibody-based targeted nanocarriers. Researchers have described different kinds of nanocarriers that have been functionalized with transferrin receptor-targeting ligands (e.g., THRPPMWSPVWP), such as liposomes and polymer nanoparticles (Johnsen et al., 2019, Paterson and Webster, 2016). Drug delivery to the brain in vivo requires the nanoparticle-based drug delivery system (DDS) to be able to overcome two major barriers, the BBB and cell membrane, to reach the disease cells for treatment. These are crucial to enhance DDS transport across the BBB and penetrate disease cells. Ligand-conjugated liposomes have been widely studied for brain delivery and are considered a promising tool for brain disease therapy (de Wolf and Brett, 2000, Sharma et al., 2012). Studies reported that transferrin-linked liposomes significantly increased the cell internalization and cytotoxicity potentials of anticancer drugs when compared with control liposomes (non-modified) (Glaser et al., 2017, Gutman et al., 2000). The obtained results proved that surface modification was important to achieve higher cell uptake and the antiproliferative effect of anticancer drugs (Glaser et al., 2017, Gutman et al., 2000). However, the transcytosis efficiency must be carefully considered when designing a nanoformulation. Factors such as particle size, stability, ligand type with different binding affinity with receptors, and ligand density in the formulation impact the targeting ability of the nanoparticles (Bazak et al., 2015, Gutman et al., 2000).
Protein ligands and antibodies possess several disadvantages. Their poor stability, high immunogenicity, large molecular weight, and high cost in production limit the application of these ligands (de Wolf and Brett, 2000, Kholodenko et al., 2019, Bazak et al., 2015). Peptide-based ligands have gained increasing attention to avoid these problems. Peptides can be synthesized in quantity and at high purity. They are also relatively small in comparison with antibodies and proteins used for targeting receptors (Field et al., 2015, Ruoslahti, 2012, Delehanty et al., 2010). Transferrin receptor-targeting peptides have recently gained popularity due to their significant benefits, representing 38% of the explored strategies (Cheng et al., 2012). In addition, studies have reported that ligands with high molecular weight can prevent or reduce the modified nanoparticles transporting across the BBB, maintaining binding efficiency and high receptor specificity (Dixit et al., 2015). However, as the binding site of the peptide is relatively small by comparison with the protein, the affinity of the peptide for a receptor is likely to be lower than the affinity of an antibody or protein (Bien-Ly et al., 2014, Haqqani et al., 2018). It has been shown that high-affinity antibodies are less effective than medium-affinity antibodies for transcytosis across brain endothelium [35, 36], reducing antibody binding to the transferrin receptor can enhance its level of transcytosis (Yu et al., 2011). Hence, the lower affinity of peptides may be an advantage in targeting therapeutic agents in the brain. This may balance transferrin-conjugated nanocarriers used primarily for transcytosis to reach brain tissues rather than the endothelial cells themselves. Despite all efforts and decades of development, minimal clinical progression for actively targeting therapy has been made. Efforts are still highly needed to optimize active ligand-mediated systems.
In this study, four kinds of lipids, including cholesterol, DOTAP, DSPE PEG-2000, and DSPE PEG 2000 carboxylic acid, were used for the preparation of liposomes based on our previous studies of riluzole liposomes (Yang et al., 2018). The traditional cationic DOTAP lipids were added to the liposomes during the preparation. It was expected that the liposomes would form nanocomplexes via electrostatic interactions with a negative gene-editing tool for the delivery of CRISPR/cas9 plasmid. Numerous techniques have been developed to prepare peptide-conjugated liposomes using the reactivity of free amine groups of peptides. One of the most popular and commonly used methods is to covalently couple free carboxylic groups on the liposome surface to primary amines through activation of the carboxyl groups with EDC. The produced amine-reactive intermediate product can be easily displaced by nucleophilic attack from the primary amino groups in the peptides. Because the intermediate is unstable and hydrolyzed in aqueous solutions, NHS is added to produce a much more stable and soluble active intermediate. In this study, transferrin-targeted liposomes were prepared by a two-step coupling procedure: first, activating the free carboxyl group of the linker lipid incorporated in the liposomes with EDC and NHS, and then covalently conjugating the peptide to the lipids through the displacement of NHS groups by peptide amines. To promote the reactions of carboxylic acid groups on the liposomes, extra amounts of activation and protection agents and peptides were used. The removal of the unreacted peptide, EDC, and NHS was investigated and optimized by ultracentrifugation, desalting column size-exclusion chromatography, and spinning column filtration methods. Purified liposomes were obtained by the spinning column filtration as all leftover reactants were removed (Figure 1C). Non-targeted and targeted liposomes were evaluated for particle size and stability study. Overall, the stability of both non-targeted and targeted liposomes was found to be satisfactory (Figure 2).
As prepared liposomes with transferrin receptor binding peptides were highly internalized into brain endothelial bEND.3 cells (Figure 3), we investigated the potential of targeted liposomes to deliver CRISPR/Cas9 and knockdown P-gp. In this study, we formulated CRISPR/Cas9 plasmid to form nanocomplexes with the prepared targeted cationic liposomes. As shown in Figures 4 and 6, the transfection efficiency of different formulations displayed a nearly linear relationship with the ratio of cationic liposomes to CRISPR/Cas9 plasmid in the nanocomplexes. The difference in the transfection efficiency reflected the abilities of different CRISPR/Cas9 targeted liposomes to deliver CRISPR/Cas9 into bEND.3 cells were different. As the charge ratios and amount of CRISPR/Cas9 plasmids to cationic lipids are critical, increasing the ratio and amount of CRISPR/Cas9 in the cells corresponds to the increased transfection expression. Since P-gp efflux is an energy-dependent process, intracellular ATP levels were also investigated in the study. The results indicated that the ATP level was greatly elevated in P-gp knockdown cells compared with the ATP level in wild-type cells (Figure 7). The results, from another angle, also suggested that the P-gp efflux transporter was successfully knocked down by the CRISPR/Cas9 targeted liposomes, which led to the increasing ATP levels in the P-gp knockdown bEND.3 cells. Eventually, the uptake or cytotoxicity results from P-gp substrates, rhodamine 123, and anticancer drug doxorubicin (Figures 8 and 9), indicating that P-gp activities were significantly reduced in knockdown bEND.3 cells by CRISPR/Cas9 treatment. Although non-targeted nanoparticles could deliver CRISPR/Cas9 plasmid, it is crucial to select appropriate cell-specific moieties, carefully design effective and stable formulations, and improve efficiency profiles (Whitehead et al., 2009, Friberg and Nystrom, 2016). As brain cell-targeting and P-gp knockdown of developed liposomes were developed, we expect that the targeted liposomes incorporated with P-gp CRISPR/Cas9 knockdown plasmid could overcome issues of targeted delivery associated with many commercially available systems.
CONCLUSIONS
In this study, purified transferrin receptor binding peptide-conjugated liposomes were prepared and were able to be transported into brain endothelial cells. This new system could be used to successfully deliver CRISPR/Cas9 to inhibit the P-gp efflux pump. The uptake of the P-gp substrates, rhodamine 123 and doxorubicin, therefore, was significantly increased in the brain endothelial cell-constituted BBB model. We plan to continuously optimize the formulation of CRISPR/Cas9 brain-targeted liposomes, and the inhibitory efficacy of liposome-delivered CRISPR/Cas9 will be further evaluated by testing silenced P-gp levels in cells under disease-condition stimulations, which many times could overexpress P-gp. A new transferrin-knockdown brain endothelial cell model will be developed and less cellular uptake of designed liposomes will be expected in the cells as there is no receptor-mediated transport. We will further test the uptake of more reported P-gp substrates as well as non P-gp substrates to validate the cells as a predictive model. We also plan to use the developed cell model to test the uptake of compounds/therapeutic agents, especially to test some newly approved drugs to identify if they have potential pharmacoresistance in the treatment of brain diseases. Overall, multi-functioning liposomes containing targeted moiety and CRISPR/Cas9 could knock down P-gp in brain endothelial cells, making it an excellent tool for permeability studies without endogenous P-gp background and activity.
Highlights.
Transferrin receptor targeting peptides were conjugated on cationic liposomes
Peptide-conjugated liposome delivered CRISPR/Cas9 plasmid knocked down P-gp
P-gp associated ATP activities were high in the transfected cells
More accumulation of rhodamine 123 was observed in the transfected cells
Higher cytotoxic sensitivity of doxorubicin was observed in the transfected cells
ACKNOWLEDGMENTS
This study was supported by the Husson Faculty Research Fund and NIH 5R03EB028572 grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Yang, T. and Bai, S conceived of the presented idea, planned the experiments, supervised the findings of this work, and took the lead in writing the manuscript.
Curtis, S., Bai, A., Young, A., Derosier, D., and Ripley S. carried out the experiment.
All authors discussed the results and contributed to the final manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- BAI S, GUPTA V & AHSAN F 2009. Cationic liposomes as carriers for aerosolized formulations of an anionic drug: safety and efficacy study. Eur J Pharm Sci, 38, 165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAI S, YANG T, ABBRUSCATO TJ & AHSAN F 2008. Evaluation of human nasal RPMI 2650 cells grown at an air-liquid interface as a model for nasal drug transport studies. J Pharm Sci, 97, 1165–78. [DOI] [PubMed] [Google Scholar]
- BAR-ZEEV M, LIVNEY YD & ASSARAF YG 2017. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist Updat, 31, 15–30. [DOI] [PubMed] [Google Scholar]
- BARTELS AL 2011. Blood-brain barrier P-glycoprotein function in neurodegenerative disease. Curr Pharm Des, 17, 2771–7. [DOI] [PubMed] [Google Scholar]
- BAZAK R, HOURI M, EL ACHY S, KAMEL S & REFAAT T 2015. Cancer active targeting by nanoparticles: a comprehensive review of literature. J Cancer Res Clin Oncol, 141, 769–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIEN-LY N, YU YJ, BUMBACA D, ELSTROTT J, BOSWELL CA, ZHANG Y, LUK W, LU Y, DENNIS MS, WEIMER RM, CHUNG I & WATTS RJ 2014. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med, 211, 233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARDARELLI F, DIGIACOMO L, MARCHINI C, AMICI A, SALOMONE F, FIUME G, ROSSETTA A, GRATTON E, POZZI D & CARACCIOLO G 2016. The intracellular trafficking mechanism of Lipofectamine-based transfection reagents and its implication for gene delivery. Sci Rep, 6, 25879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CELIUS T, GARBERG P & LUNDGREN B 2004. Stable suppression of MDR1 gene expression and function by RNAi in Caco-2 cells. Biochem Biophys Res Commun, 324, 365–71. [DOI] [PubMed] [Google Scholar]
- CHAN GN, EVANS RA, BANKS DB, MESEV EV, MILLER DS & CANNON RE 2017. Selective induction of P-glycoprotein at the CNS barriers during symptomatic stage of an ALS animal model. Neurosci Lett, 639, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Y & LIU L 2012. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev, 64, 640–65. [DOI] [PubMed] [Google Scholar]
- CHEN Z, ZHAI M, XIE X, ZHANG Y, MA S, LI Z, YU F, ZHAO B, ZHANG M, YANG Y & MEI X 2017. Apoferritin Nanocage for Brain Targeted Doxorubicin Delivery. Mol Pharm, 14, 3087–3097. [DOI] [PubMed] [Google Scholar]
- CHENG Z, AL ZAKI A, HUI JZ, MUZYKANTOV VR & TSOURKAS A 2012. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science, 338, 903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONSTANTINESCU CA, FUIOR EV, REBLEANU D, DELEANU M, SIMION V, VOICU G, ESCRIOU V, MANDUTEANU I, SIMIONESCU M & CALIN M 2019. Targeted Transfection Using PEGylated Cationic Liposomes Directed Towards P-Selectin Increases siRNA Delivery into Activated Endothelial Cells. Pharmaceutics, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE ROSA G, SALZANO G, CARAGLIA M & ABBRUZZESE A 2012. Nanotechnologies: a strategy to overcome blood-brain barrier. Curr Drug Metab, 13, 61–9. [DOI] [PubMed] [Google Scholar]
- DE WOLF FA & BRETT GM 2000. Ligand-binding proteins: their potential for application in systems for controlled delivery and uptake of ligands. Pharmacol Rev, 52, 207–36. [PubMed] [Google Scholar]
- DELEHANTY JB, BOENEMAN K, BRADBURNE CE, ROBERTSON K, BONGARD JE & MEDINTZ IL 2010. Peptides for specific intracellular delivery and targeting of nanoparticles: implications for developing nanoparticle-mediated drug delivery. Ther Deliv, 1, 411–33. [DOI] [PubMed] [Google Scholar]
- DING Y, LI H, CHEN LL & XIE K 2016. Recent Advances in Genome Editing Using CRISPR/Cas9. Front Plant Sci, 7, 703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIXIT S, NOVAK T, MILLER K, ZHU Y, KENNEY ME & BROOME AM 2015. Transferrin receptor-targeted theranostic gold nanoparticles for photosensitizer delivery in brain tumors. Nanoscale, 7, 1782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONG X, MATTINGLY CA, TSENG MT, CHO MJ, LIU Y, ADAMS VR & MUMPER RJ 2009. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting P-glycoprotein and depleting ATP. Cancer Res, 69, 3918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIELD LD, DELEHANTY JB, CHEN Y & MEDINTZ IL 2015. Peptides for specifically targeting nanoparticles to cellular organelles: quo vadis? Acc Chem Res, 48, 1380–90. [DOI] [PubMed] [Google Scholar]
- FRIBERG S & NYSTROM AM 2016. NANOMEDICINE: will it offer possibilities to overcome multiple drug resistance in cancer? J Nanobiotechnology, 14, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GABATHULER R 2010. Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol Dis, 37, 48–57. [DOI] [PubMed] [Google Scholar]
- GARTZKE D, DELZER J, LAPLANCHE L, UCHIDA Y, HOSHI Y, TACHIKAWA M, TERASAKI T, SYDOR J & FRICKER G 2015. Genomic Knockout of Endogenous Canine P-Glycoprotein in Wild-Type, Human P-Glycoprotein and Human BCRP Transfected MDCKII Cell Lines by Zinc Finger Nucleases. Pharm Res, 32, 2060–71. [DOI] [PubMed] [Google Scholar]
- GARTZKE D & FRICKER G 2014. Establishment of optimized MDCK cell lines for reliable efflux transport studies. J Pharm Sci, 103, 1298–304. [DOI] [PubMed] [Google Scholar]
- GELDENHUYS WJ, ALLEN DD & BLOOMQUIST JR 2012. Novel models for assessing blood-brain barrier drug permeation. Expert Opin Drug Metab Toxicol, 8, 647–53. [DOI] [PubMed] [Google Scholar]
- GLASER T, HAN I, WU L & ZENG X 2017. Targeted Nanotechnology in Glioblastoma Multiforme. Front Pharmacol, 8, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOTTESMAN MM, FOJO T & BATES SE 2002. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer, 2, 48–58. [DOI] [PubMed] [Google Scholar]
- GUTMAN RL, PEACOCK G & LU DR 2000. Targeted drug delivery for brain cancer treatment. J Control Release, 65, 31–41. [DOI] [PubMed] [Google Scholar]
- HAQQANI AS, THOM G, BURRELL M, DELANEY CE, BRUNETTE E, BAUMANN E, SODJA C, JEZIERSKI A, WEBSTER C & STANIMIROVIC DB 2018. Intracellular sorting and transcytosis of the rat transferrin receptor antibody OX26 across the blood-brain barrier in vitro is dependent on its binding affinity. J Neurochem, 146, 735–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HE L, ZHAO C, YAN M, ZHANG LY & XIA YZ 2009. Inhibition of P-glycoprotein function by procyanidine on blood-brain barrier. Phytother Res, 23, 933–7. [DOI] [PubMed] [Google Scholar]
- JOHNSEN KB, BURKHART A, THOMSEN LB, ANDRESEN TL & MOOS T 2019. Targeting the transferrin receptor for brain drug delivery. Prog Neurobiol, 181, 101665. [DOI] [PubMed] [Google Scholar]
- KE XJ, CHENG YF, YU N & DI Q 2019. Effects of carbamazepine on the P-gp and CYP3A expression correlated with PXR or NF-kappaB activity in the bEnd.3 cells. Neurosci Lett, 690, 48–55. [DOI] [PubMed] [Google Scholar]
- KHOLODENKO RV, KALINOVSKY DV, DORONIN II, PONOMAREV ED & KHOLODENKO IV 2019. Antibody Fragments as Potential Biopharmaceuticals for Cancer Therapy: Success and Limitations. Curr Med Chem, 26, 396–426. [DOI] [PubMed] [Google Scholar]
- KIDO Y, TAMAI I, NAKANISHI T, KAGAMI T, HIROSAWA I, SAI Y & TSUJI A 2002. Evaluation of blood-brain barrier transporters by co-culture of brain capillary endothelial cells with astrocytes. Drug Metab Pharmacokinet, 17, 34–41. [DOI] [PubMed] [Google Scholar]
- KROL S 2012. Challenges in drug delivery to the brain: nature is against us. J Control Release, 164, 145–55. [DOI] [PubMed] [Google Scholar]
- KULKARNI SA & FENG SS 2013. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm Res, 30, 2512–22. [DOI] [PubMed] [Google Scholar]
- LAKKADWALA S & SINGH J 2019. Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Colloids Surf B Biointerfaces, 173, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU BY, HE XY, XU C, XU L, AI SL, CHENG SX & ZHUO RX 2018. A Dual-Targeting Delivery System for Effective Genome Editing and In Situ Detecting Related Protein Expression in Edited Cells. Biomacromolecules, 19, 2957–2968. [DOI] [PubMed] [Google Scholar]
- MAHAR DOAN KM, HUMPHREYS JE, WEBSTER LO, WRING SA, SHAMPINE LJ, SERABJIT-SINGH CJ, ADKISON KK & POLLI JW 2002. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther, 303, 1029–37. [DOI] [PubMed] [Google Scholar]
- MAHRINGER A, OTT M, REIMOLD I, REICHEL V & FRICKER G 2011. The ABC of the blood-brain barrier - regulation of drug efflux pumps. Curr Pharm Des, 17, 2762–70. [DOI] [PubMed] [Google Scholar]
- MCINERNEY MP, PAN Y, SHORT JL & NICOLAZZO JA 2017. Development and Validation of an In-Cell Western for Quantifying P-Glycoprotein Expression in Human Brain Microvascular Endothelial (hCMEC/D3) Cells. J Pharm Sci, 106, 2614–2624. [DOI] [PubMed] [Google Scholar]
- MILLER DW, HINTON M & CHEN F 2011. Evaluation of drug efflux transporter liabilities of darifenacin in cell culture models of the blood-brain and blood-ocular barriers. Neurourol Urodyn, 30, 1633–8. [DOI] [PubMed] [Google Scholar]
- MOUT R, RAY M, LEE YW, SCALETTI F & ROTELLO VM 2017. In Vivo Delivery of CRISPR/Cas9 for Therapeutic Gene Editing: Progress and Challenges. Bioconjug Chem, 28, 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULDOON LL, SOUSSAIN C, JAHNKE K, JOHANSON C, SIEGAL T, SMITH QR, HALL WA, HYNYNEN K, SENTER PD, PEEREBOOM DM & NEUWELT EA 2007. Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol, 25, 2295–305. [DOI] [PubMed] [Google Scholar]
- NEUHAUS W, STESSL M, STRIZSIK E, BENNANI-BAITI B, WIRTH M, TOEGEL S, MODHA M, WINKLER J, GABOR F, VIERNSTEIN H & NOE CR 2010. Blood-brain barrier cell line PBMEC/C1-2 possesses functionally active P-glycoprotein. Neurosci Lett, 469, 224–8. [DOI] [PubMed] [Google Scholar]
- PARDRIDGE WM 2005. Molecular biology of the blood-brain barrier. Mol Biotechnol, 30, 57–70. [DOI] [PubMed] [Google Scholar]
- PARDRIDGE WM 2007. Blood-brain barrier delivery. Drug Discov Today, 12, 54–61. [DOI] [PubMed] [Google Scholar]
- PATERSON J & WEBSTER CI 2016. Exploiting transferrin receptor for delivering drugs across the blood-brain barrier. Drug Discov Today Technol, 20, 49–52. [DOI] [PubMed] [Google Scholar]
- RUOSLAHTI E 2012. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater, 24, 3747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARMA G, MODGIL A, SUN C & SINGH J 2012. Grafting of cell-penetrating peptide to receptor-targeted liposomes improves their transfection efficiency and transport across blood-brain barrier model. J Pharm Sci, 101, 2468–78. [DOI] [PubMed] [Google Scholar]
- SONG M 2017. The CRISPR/Cas9 system: Their delivery, in vivo and ex vivo applications and clinical development by startups. Biotechnol Prog. [DOI] [PubMed] [Google Scholar]
- TUCKER IG 2011. Drug delivery to the brain via the blood-brain barrier: a review of the literature and some recent patent disclosures. Ther Deliv, 2, 311–27. [DOI] [PubMed] [Google Scholar]
- WANG H, LA RUSSA M & QI LS 2016. CRISPR/Cas9 in Genome Editing and Beyond. Annu Rev Biochem, 85, 227–64. [DOI] [PubMed] [Google Scholar]
- WANG HX, SONG Z, LAO YH, XU X, GONG J, CHENG D, CHAKRABORTY S, PARK JS, LI M, HUANG D, YIN L, CHENG J & LEONG KW 2018. Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide. Proc Natl Acad Sci U S A, 115, 4903–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEI M, GUO X, TU L, ZOU Q, LI Q, TANG C, CHEN B, XU Y & WU C 2015. Lactoferrin-modified PEGylated liposomes loaded with doxorubicin for targeting delivery to hepatocellular carcinoma. Int J Nanomedicine, 10, 5123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITEHEAD KA, LANGER R & ANDERSON DG 2009. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov, 8, 129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG T, BANTEGUI T, PIKE K, BLOOM R, PHIPPS R & BAI S 2014a. In vitro evaluation of optimized liposomes for delivery of small interfering RNA. J Liposome Res, 24, 270–9. [DOI] [PubMed] [Google Scholar]
- YANG T, BANTEGUI T, PIKE K, BLOOM R, PHIPPS R & BAI S 2014b. In vitro evaluation of optimized liposomes for delivery of small interfering RNA. J Liposome Res. [DOI] [PubMed] [Google Scholar]
- YANG T, FERRILL L, GALLANT L, MCGILLICUDDY S, FERNANDES T, SCHIELDS N & BAI S 2018. Verapamil and riluzole cocktail liposomes overcome pharmacoresistance by inhibiting P-glycoprotein in brain endothelial and astrocyte cells: A potent approach to treat amyotrophic lateral sclerosis. Eur J Pharm Sci, 120, 30–39. [DOI] [PubMed] [Google Scholar]
- YANG T, FOGARTY B, LAFORGE B, AZIZ S, PHAM T, LAI L & BAI S 2017. Delivery of Small Interfering RNA to Inhibit Vascular Endothelial Growth Factor in Zebrafish Using Natural Brain Endothelia Cell-Secreted Exosome Nanovesicles for the Treatment of Brain Cancer. AAPS J, 19, 475–486. [DOI] [PubMed] [Google Scholar]
- YANG T, MARTIN P, FOGARTY B, BROWN A, SCHURMAN K, PHIPPS R, YIN VP, LOCKMAN P & BAI S 2015. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res, 32, 2003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG T, NYIAWUNG D, SILBER A, HAO J, LAI L & BAI S 2012. Comparative studies on chitosan and polylactic-co-glycolic acid incorporated nanoparticles of low molecular weight heparin. AAPS PharmSciTech, 13, 1309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE D, HARDER A, FANG Z, WEINHEIMER M, LAPLANCHE L & MEZLER M 2020. Characterization and Validation of Canine P-Glycoprotein-Deficient MDCK II Cell Lines for Efflux Substrate Screening. Pharm Res, 37, 194. [DOI] [PubMed] [Google Scholar]
- YU X, LIANG X, XIE H, KUMAR S, RAVINDER N, POTTER J, DE MOLLERAT DU JEU X & CHESNUT JD 2016. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol Lett, 38, 919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU YJ, ZHANG Y, KENRICK M, HOYTE K, LUK W, LU Y, ATWAL J, ELLIOTT JM, PRABHU S, WATTS RJ & DENNIS MS 2011. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Transl Med, 3, 84ra44. [DOI] [PubMed] [Google Scholar]
- ZHEN S, TAKAHASHI Y, NARITA S, YANG YC & LI X 2017. Targeted delivery of CRISPR/Cas9 to prostate cancer by modified gRNA using a flexible aptamer-cationic liposome. Oncotarget, 8, 9375–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]