

Abstract
Macrophages are important players for the maintenance of tissue homeostasis1. Perivascular and leptomeningeal macrophages reside in close proximity to the central nervous system (CNS) parenchyma2, and their role in CNS physiology has not been well enough studied to date. Given their continuous interaction with the cerebrospinal fluid (CSF) and strategic positioning, we refer to these cells collectively as parenchymal border macrophages (PBMs). Here, we demonstrate that PBMs regulate CSF flow dynamics. We identify a subpopulation of PBMs expressing high levels of CD163 and Lyve1 (scavenger receptor proteins), located in close proximity to the brain arterial tree, and show that Lyve1+ PBMs regulate arterial motion that drives CSF flow. Pharmacological or genetic depletion of PBMs led to accumulation of extracellular matrix proteins, obstructing CSF access to perivascular spaces hence impairing CNS perfusion and clearance. Aging-associated alterations in PBMs and impairment of CSF dynamics were restored upon intracisternal injection of macrophage colony-stimulating growth factor (M-CSF). Human single-nuclei RNA sequencing data obtained from Alzheimer’s disease (AD) patients and healthy controls point to changes in phagocytosis/endocytosis and interferon-gamma (IFNγ) signaling on PBMs, pathways that are corroborated in a mouse AD model. Collectively, our results identify PBMs as novel cellular regulators of CSF flow dynamics, which could potentially be targeted pharmacologically to alleviate brain clearance deficits associated with aging and AD.
The meninges, a tripartite membranous covering of the brain parenchyma, are densely populated by immune cells whose derived cytokines have been shown to affect mouse behaviors3,4,5. Cytokines from the periphery and the meninges can be carried along in cerebrospinal fluid (CSF), which is driven by arterial pulsations and vasomotion to circulate throughout the brain parenchyma6–8. This parenchymal perfusion of CSF not only provides meningeal immune molecules a direct access to brain-cell signaling, but also performs “glymphatic clearance”9 by facilitating, via meningeal lymphatics, the removal and subsequent clearance of brain metabolites10,11. Antigens are transported along the same routes into the dura, where they are sampled by dural antigen-presenting cells (APCs) and presented to patrolling T cells to ensure immune surveillance of the central nervous system (CNS)12. Some of these brain-derived antigens subsequently also drain into the deep cervical lymph nodes (dCLNs) via meningeal lymphatic vessels10.
The CNS myeloid niche in the homeostatic brain is comprised of microglia and leptomeningeal and perivascular macrophages13 (we collectively term these two populations of border-macrophages as ‘parenchymal border macrophages’; PBMs). Unlike microglia, which are located within the brain parenchyma, PBMs reside in the subarachnoid and perivascular spaces along the vasculature, and are thus constantly in direct contact with the CSF14–16. Previous studies have suggested a detrimental role for such macrophages in chronic hypertension17, Alzheimer’s disease18,19, stroke, and experimental autoimmune encephalomyelitis (EAE)20. The functions of PBMs in brain homeostasis, however, are still largely unexplored.
Here, we show that PBMs regulate CSF flow dynamics, an integral aspect of brain physiology, under homeostatic conditions. We identify arterial-associated PBMs that display a transcriptomic profile of scavenger cells and control extracellular matrix (ECM) remodeling, affecting arterial motion, a driving force of CSF flow dynamics. Depletion of PBMs results in accumulation of ECM proteins and impairment of brain perfusion by the CSF. We demonstrate that normal aging is associated with PBM dysfunction, and we show here that treatment of aged mice with macrophage colony-stimulating factor (M-CSF) improves CSF dynamics.
Function and maintenance of PBMs
PBMs are found in leptomeningeal and perivascular spaces in the brain, at the vicinity of larger blood vessels, and can be distinguished from microglia by their location, and expression of the mannose receptor CD20616 (Fig. 1a; Extended Data Fig. 1a–c; Supplementary video 1). We were able to distinguish two subtypes of PBMs based on their expression of either Lyve1 or MHC-II (Fig. 1b, c). Using flow cytometry, PBMs can be distinguished using MHCII and CD38 as a substitute to Lyve1, as previously described21 (Extended Data Fig. 1d).
Fig. 1: PBMs sample CSF and regulate its flow dynamics.
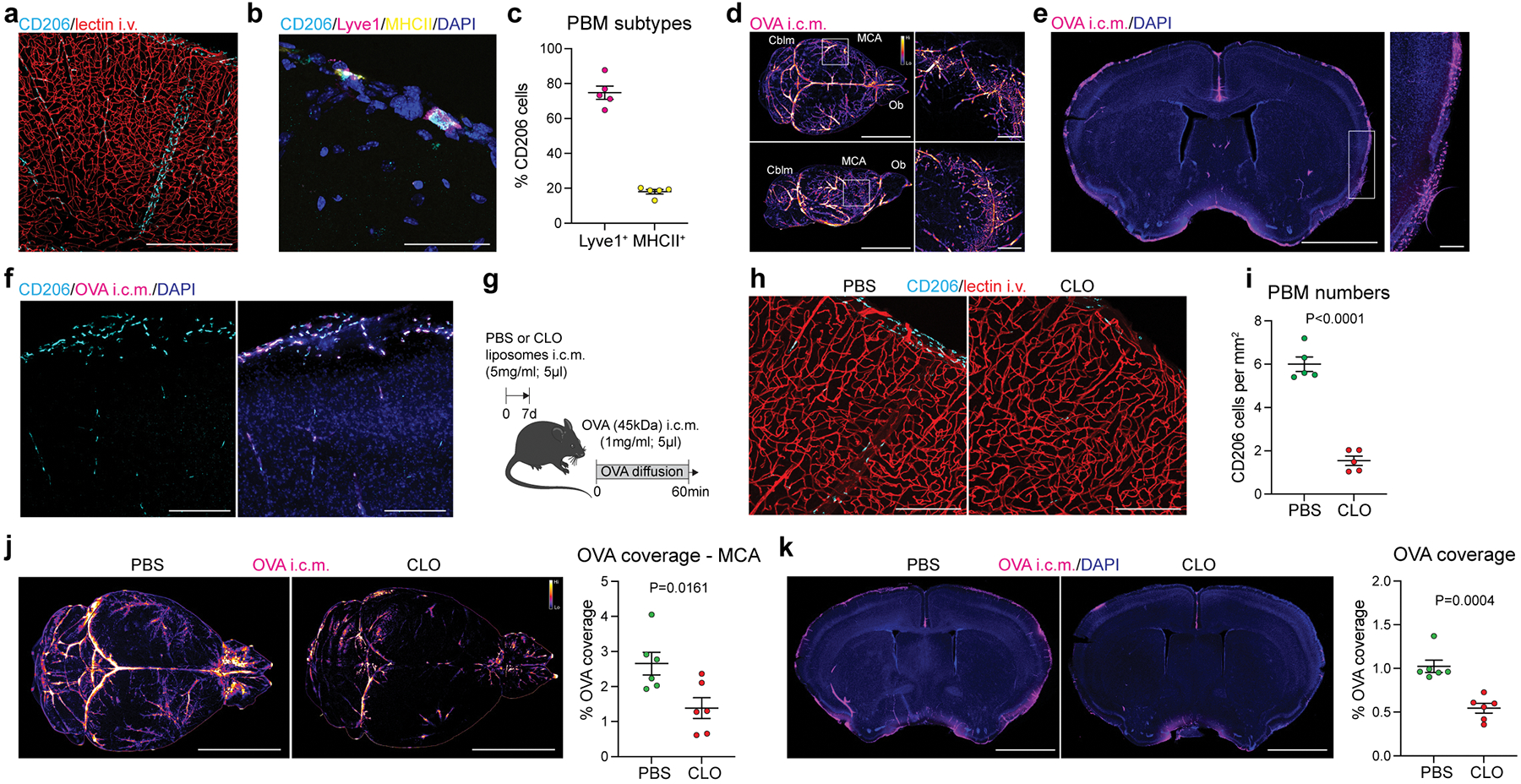
a, CD206+ PBMs are located in close association with i.v.-injected lectin+ blood vessels. Scale bar, 500μm. b, CD206+ PBMs can be separated into two major subtypes by their expression of Lyve1 or MHCII co-stained for DAPI. Scale bar, 50μm. c, Quantification of Lyve1+ versus MHCII+ PBMs (average of both perivascular space and leptomeninges). Data in c, n = 5 mice. d, WT mice received an i.c.m. injection of Alexa-647 conjugated ovalbumin (OVA). Mice were perfused one hour after OVA injection. Representative stereomicroscopy images showing whole brain OVA distribution from top (top raw) and side (bottom row) views. OVA is mostly distributed around the olfactory bulbs (Ob), middle cerebral artery (MCA) and cerebellum (Cblm). Scale bars, 5mm and 1mm (insets). e, OVA distribution in brain coronal sections (co-stained for DAPI) is largely found in leptomeninges and penetrating blood vessels. At higher magnification, OVA distribution appears also to be cellular (inset). Scale bars, 2mm and 200μm (inset). f, OVA+ cells are expressing PBM marker CD206. Scale bar, 100μm. g, One week after PBM depletion using i.c.m. clodronate liposomes (CLO), mice received an i.c.m. injection of OVA and were perfused one hour later. h, Representative images of brain sections showing CD206 and i.v.-injected lectin staining. Scale bar: 200μm. i, Quantification of CD206+ cells; n = 5 mice/group; two-tailed unpaired Welch’s t-test. j, Representative images showing OVA distribution in whole brains in PBM-depleted and control mice, and corresponding quantification. Scale bar, 5mm. k, OVA coverage in brain coronal sections, and corresponding quantification. Scale bar, 2mm. For j and k: n = 6 mice/group; two-tailed unpaired Welch’s t-test. All data are presented as mean values +/− SEM.
Perivascular spaces are filled with CSF and constitute an interface between the blood and the CNS parenchyma22. CSF flows along the perivascular space, can cross astrocytic endfeet and flow into the brain (a process termed glymphatic) 9,23. We studied CSF flow dynamics by injecting fluorescent tracers into the mouse cisterna magna (i.c.m.) and then assessing tracer diffusion into the brain. Fluorescent ovalbumin (OVA) was injected i.c.m., and the mice were perfused after one hour. The whole brain was extracted, fixed, and imaged by light sheet microscopy (Extended Data Fig. 1e; Supplementary video 2) or by stereomicroscopy (Fig. 1d; Extended Data Fig. 1f). Using these methods, we observed that OVA was located mostly in the regions of the olfactory bulbs (Ob), cerebellum (Cblm), and middle cerebral artery (MCA) (Fig. 1d). Although most of the tracer accumulated at the perivascular space, 28.9% of tracer was sampled by cells (Fig. 1d; Extended Data Fig. 1f).
Tracer penetration into the brain parenchyma is reportedly greater for small fluorescent tracers24. However, we found that tracers, independently of their size, accumulated in both perivascular and leptomeningeal spaces in CD206+ macrophages (or PBMs; Fig. 1e, f; Extended Data Fig. 1g). We also observed tracer uptake by PBMs when tracers were infused into the striatum (intra-striatal (i.s.); Extended Data Fig. 1h), suggesting that PBMs sample CSF content on its way into and out of the brain. Indeed, we can observe double positive PBMs two hours after the tracers were co-injected in both striatum and CSF, although CSF influx was reduced due to concomitant intrastriatal injection, as previously described23 (Extended Data Fig. 1i).
Given the close association of PBMs with CSF, we hypothesized that CSF flow dynamics may be partially controlled by PBMs. To test this hypothesis, we injected i.c.m. clodronate (CLO) encapsulating liposomes to deplete PBMs (~75% depletion was achieved when brain tissue was examined one week later; Fig. 1g–i). We confirmed that both number and morphology of microglial cells were not affected by i.c.m. CLO liposome administration (Extended Data Fig. 2a). Using IHC and single-cell RNA-sequencing (scRNA-seq), we also demonstrated that PBMs were the major population of brain border-associated cells phagocytosing i.c.m. injected DiI-labeled liposomes, whereas microglia and other stromal cells did not sample the tracer (Extended Data Fig. 2b–e) and hence could not be directly affected by the liposomes. Notably, we observed two major PBM subtypes based on their gene expression of Lyve1 and MHCII, as it was observed by both staining and flow cytometry (Extended Data Fig. 2f–i). More specifically, we found that CD206+Lyve1+MHCIIlow/neg PBMs were highly phagocytic/endocytic cells, expressing high levels of scavenger receptors such as Cd163, Mrc1, Lyve1, Msr1 and Siglec1. Interestingly, these cells also upregulate genes involved in interferon gamma pathway such as Irf8, Ifitm2 and Ifitm6 (Extended Data Fig. 2h, i). GO pathway analyses highlighted important roles of Lyve1+ PBMs in metabolic processes and chemotaxis (Extended Data Fig. 2i). On the other hand, we found that CD206+Lyve1−MHCII+ PBMs upregulate pathways involving immune response, response to viruses, cytokine production, cell-cell adhesion and antigen presentation (Extended Data Fig. 2h, i), suggesting that the two PBM subtypes have different functions.
To assess the role of PBMs in CSF dynamics, fluorescent OVA was injected i.c.m. one week after PBM depletion. After allowing the tracer to diffuse freely for one hour, mice were perfused and their whole brains extracted, fixed and imaged by stereomicroscopy (Fig. 1j). In PBM-depleted mice the OVA coverage was significantly reduced (Fig. 1j). Brains were then sectioned to evaluate OVA coverage in coronal sections, a method commonly used to evaluate CSF influx9,25 (Fig. 1k). OVA coverage of brain slices was also found to be significantly reduced in PBM-depleted mice (Fig. 1k). Three days after CLO treatment CSF flow was impaired to a lesser extent than one week after, which correlates with a milder (nevertheless, significant) PBM depletion at this time point (Extended Data Fig. 2j–m).
Our group recently showed that CSF flow was impaired after dural lymphatic ablation25. We found that at three days post CLO treatment, the number of CD206+ dural macrophages located at the vicinity of the superior sagittal sinus was reduced (Extended Data Fig. 2n–o), but there was no effect of CLO treatment after one week on either dural lymphatic vessels or dural CD206+ macrophages, which indicates that the observed impairment of CSF flow cannot be attributed to dural lymphatic ablation in this study (Extended Data Fig. 2p–s). We also did not find any effect of CLO treatment on choroid plexus CD206+ macrophages (Extended Data Fig. 2t). However, we found that a population of Iba1+ cells were absent in deep cervical lymph nodes (dCLNs) after CLO treatment (Extended Data Fig. 2u), presumably sinus subcapsular macrophages that were depleted once CLO liposomes drained to dCLNs.
We repeated the CSF flow experiment using 4-kDa dextran conjugated with fluorescein isothiocyanate (FITC-dextran; FITCdex) or 3-kDa Texas Red, which are more diffusive owing to their small molecular weight, and—as with OVA—influx of small tracers was found to be impaired after PBM depletion (Extended Data Fig. 3a, b). Furthermore, we observed a significant accumulation of tracers (both OVA and FITCdex) in the brain parenchyma one hour after intra-striatal (i.s.) injection in PBM-depleted mice, suggesting that both influx and efflux of CSF are impaired after PBM depletion (Extended Data Fig. 3c, d), or that CSF influx/efflux are interdependent, as previously described (for example, intrastriatal injection impairs influx of CSF tracers23). To assess whether the impairment of CSF flow dynamics could affect CSF protein content itself, we performed a proteomic analysis of CSF sampled from PBM-depleted and control mice (Extended Data Fig. 3e). Interestingly, we observed an accumulation of synapse-related proteins in PBM-depleted mice, such as NRXN1, PTPRS, NRCAM and CDH2 (Extended Data Fig. 3f–h). We also observed an accumulation of Clusterin (CLU), Apolipoprotein E (APOE) and Amyloid Precursor Protein (APP), proteins that have been associated with Alzheimer’s disease (AD), in the CSF of PBM-depleted mice (Extended Data Fig. 3i–k).
To better evaluate the dynamics of CSF flow in vivo, we used magnetic resonance imaging (MRI)26,27 (Fig. 2a) and demonstrated a strongly reduced diffusion of the contrast agent (Dotarem; 0.754 kDa) over time in PBM-depleted as compared to control-treated mice (Fig. 2b, c; Supplementary videos 3 and 4), without any noticeable effect of PBM depletion on ventricular size (Extended Data Fig. 3l–n). We also noticed an increased intracranial pressure one week after PBM depletion, which was normalized three weeks after depletion (Extended Data Fig. 3o), likely because cells started to repopulate the niche17,28. Interestingly, we observed a strong reduction of the tracer diffusion at the vicinity of the MCA by MRI (Extended Data Fig. 3p–r). The OVA coverage of dCLNs was reduced after PBM depletion using immunohistochemistry (Extended Data Fig. 3s–v). However, we did not find any difference in OVA drainage to dCLNs by in vivo imaging (Extended Data Fig. 3w–y). This is likely because the rapid efflux of OVA from the dCLNs in PBM-depleted mice as was evident from live imaging of drainage (Extended Data Fig. 3y, z), probably as a result of sinus subcapsular macrophage depletion by drained CLO liposomes (Extended Data Fig. 2u).
Fig. 2: In vivo evidence of PBMs regulating CSF flow dynamics.
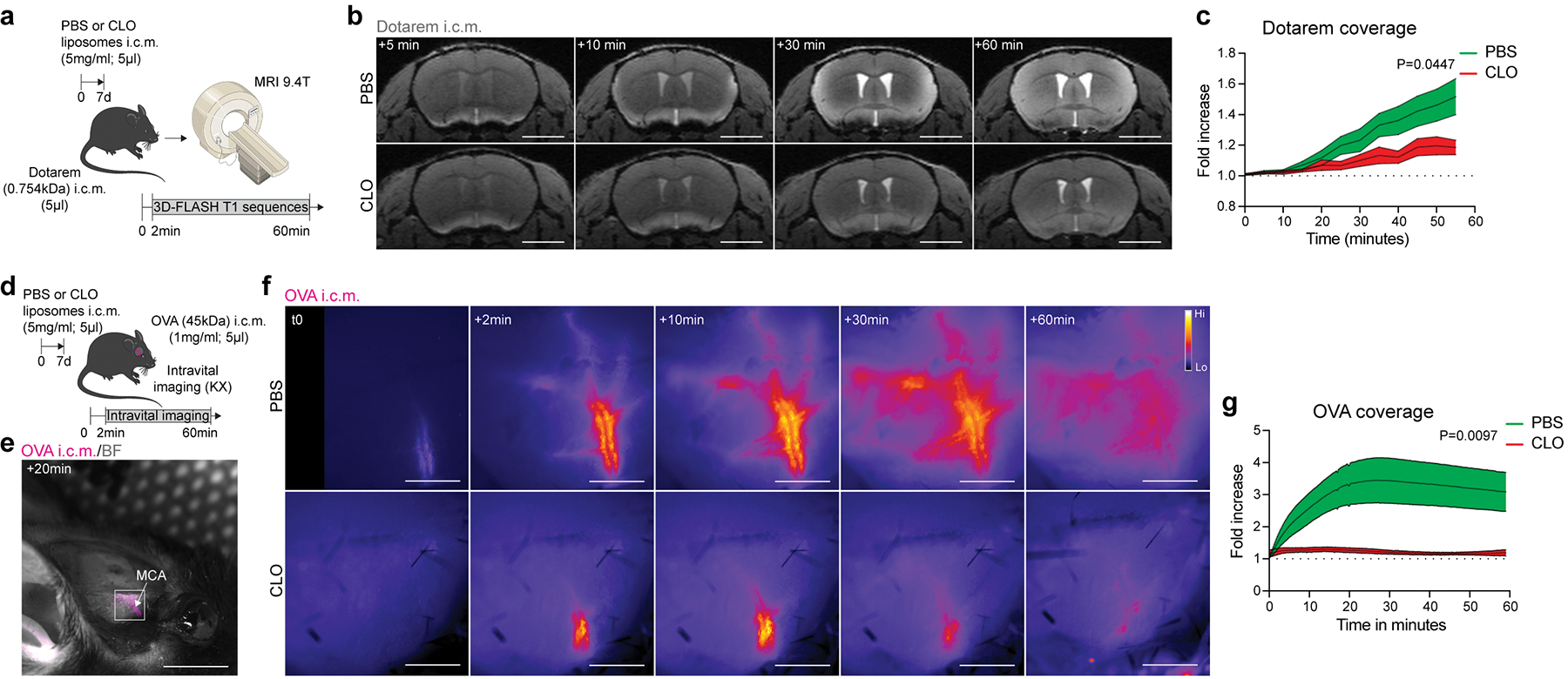
a, One week after PBM depletion, mice received an i.c.m. injection of Dotarem and were placed in prone position into the MRI device for dynamic imaging. b, Representative brain coronal images showing Dotarem distribution over an hour. Scale bar, 3mm. c, Quantification of Dotarem signal fold increase over time, n = 5 mice treated with PBS, and 7 mice treated with CLO; Repeated measures 2-way ANOVA with Geisser-Greenhouse correction. d, One week after CLO or PBS containing liposome injection, the right temporal bone was exposed and mice received an i.c.m. injection of OVA. Mice were immediately imaged on their side by stereomicroscopy. e, Representative in vivo OVA imaging at low magnification 20 minutes after i.c.m. injection. Scale bar, 5mm. f, Representative images showing OVA distribution over an hour at the proximal part of the MCA. Scale bar, 1mm. g, Quantification of OVA signal fold increase over time. n = 6 mice/group; Repeated measures 2-way ANOVA with Geisser-Greenhouse correction. All data are presented as mean values +/− SEM.
To better understand how PBM depletion affects CSF flow dynamics, we developed a new in vivo approach that allowed us to monitor fluorescent tracer movement over time. CSF tracers rapidly diffuse at the distal part of the MCA perivascular space6,27 and given our observed reduced tracer coverage around MCA by MRI (Extended Data Fig. 3p–r), we decided to visualize fluorescent macromolecule movement through the intact lateral parietal bone upon retraction of the right temporalis muscle (Fig. 2d, e). Immediately after i.c.m. injection of fluorescent OVA, mice were turned onto their side for in vivo stereomicroscopic imaging of the proximal part of the MCA through the intact skull (Fig. 2d; Extended Data Fig. 4a–c). Using this method, we observed in vivo that OVA was rapidly localized at the MCA perivascular space (Extended Data Fig. 4d) and sampled by perivascular cells (Extended Data Fig. 4e). We validated this method by exposing mice to different anesthetics known to either enhance (ketamine/xylazine cocktail) or inhibit (isoflurane) movement of CSF tracers29,30 (Extended Data Fig. 4f–j). Using this approach, we observed that OVA coverage over time was strongly reduced in PBM-depleted mice at the MCA level, in accordance with ex vivo results showing global reduction of tracer coverage (Fig. 2f, g; Supplementary videos 5 and 6).
Collectively, these data provide evidence that PBMs are strategically located at the interface between blood and brain parenchyma, they sample CSF, and regulate its flow dynamics.
PBMs regulate CSF flow dynamics through extracellular matrix remodeling and arterial motion.
Upon PBM depletion, OVA was barely able to reach the perivascular space of penetrating vessels (Extended Data Fig. 4k). This raised questions about the morphology of the perivascular space after PBM depletion. Aquaporin-4 (AQP4), a water channel present in the astrocytic end feet forming the glia limitans, i.e., the outer layer of the perivascular space, was proposed as a mediator of CSF influx23. Using immunohistochemistry, we were unable to detect any effect of PBM depletion on AQP4 coverage or AQP4 polarization (Extended Data Fig. 4l–n). By measuring the diameter of the perivascular space using the vascular marker CD31 in combination with AQP4 (Extended Data Fig. 4o), we found that the space between the two markers (arguably representing the perivascular space) was substantially smaller in PBM-depleted mice than in control mice (Extended Data Fig. 4o–q). Using i.c.m. injected fluorescent microbeads6 we were able to assess perivascular space in vivo (Extended Fig. 4r–t), but this method did not allow assessment of the perivascular space in PBM-depleted mice, because very few beads could be detected (Extended Data Fig. 4s, t). To circumvent in vivo imaging, we perfused the mice with PBS without PFA to preserve the perivascular space and bead accumulation was assessed at the vicinity of the MCA6 (Extended Data Fig. 4u, v). First, we compared the perivascular space measurement in PBS-treated mice using both methods and found a slight decrease in ex vivo compared to in vivo measurements (Extended Data Fig. 4u–w). Using this method, we found that the bead coverage was strongly reduced in PBM-depleted mice (Extended Data Fig. 4x). Although the perivascular space and MCA diameter did not change (Extended Data Fig. 4y), we observed that the space filled by the beads was reduced in PBM-depleted mice (Extended Data Fig. 4z).
The changes in CSF flow dynamics were accompanied by a mild behavioral phenotype (Extended Data Fig. 5). PBM-depleted mice froze more on the first day of the cued-fear conditioning test (Extended Data Fig. 5a). However, PBM-depleted mice did not show any deficits in anxiety (elevated plus maze; Extended Data Fig. 5b), general motor activity (Open field test; Extended Data Fig. 5c), response to stress (forced swim test; Extended Data Fig. 5d) or social recognition (3-chamber test; Extended Data Fig. 5e). We also measured vital signs such as respiratory rate, heart rate and arterial pulsations and diameter, and did not find any difference between groups (Extended Data Fig. 5f).
To understand whether stromal cells (i.e., endothelial cells and mural cells such as pericytes, vascular smooth muscle cells and fibroblasts) are affected by PBM depletion, we sorted CD45−CD13+CD31− (mural) and CD45−CD13−CD31+ (endothelial) cells and performed scRNA-seq one week after PBM depletion (Extended Data Fig. 6a). We found that fibroblasts (and pericytes, to a lesser extent) are the main producers of extracellular matrix (ECM), and that ECM-related genes (Lgals3, Col1a2, Tgfb2, Lum, Col11a1, Col8a2) were upregulated in PBM-depleted mice (Extended Data Fig. 6b, c). We also observed transcriptional changes in pericytes, mainly related to angiogenesis and DNA methylation (Extended Data Fig. 6c), as well as in capillaries (Extended Data Fig. 6d). However, we could not observe differences in terms of mural cell (Extended Data Fig. 6e), endothelial cell (Extended Data Fig. 6f), or vascular smooth muscle cell (VSMC) coverage (Extended Data Fig. 6g).
Macrophages have been proposed to regulate extracellular matrix (ECM) degradation in other organs via production of matrix metalloproteinases (MMPs) such as MMP2 and MMP9, which degrade the ECM secreted by other cell types such as fibroblasts and VSMCs26. We hypothesized that PBM depletion could lead to changes in MMP activity resulting in ECM accumulation that might interfere with CSF flow dynamics. Collagen type 4 and laminin, both ECM proteins highly expressed in the brain parenchyma, were indeed found to accumulate around CD31+ vessels in PBM-depleted mice, as compared to vehicle-treated controls (Fig. 3a–c; Extended Data Fig. 6h–l). When analyzed separately, we also observed an accumulation of ECM proteins at the vicinity of αSMA+ large surface/penetrating blood vessels (Extended Data Fig. 6m–n). Furthermore, we observed a significantly reduced MMP activity in the soluble brain fraction of PBM-depleted mice using fluorescence spectrometry (Fig. 3d, e), suggesting that the functional perivascular space is regulated by PBMs through ECM remodeling.
Fig. 3: PBMs regulate CSF flow dynamics through ECM remodeling and arterial motion.
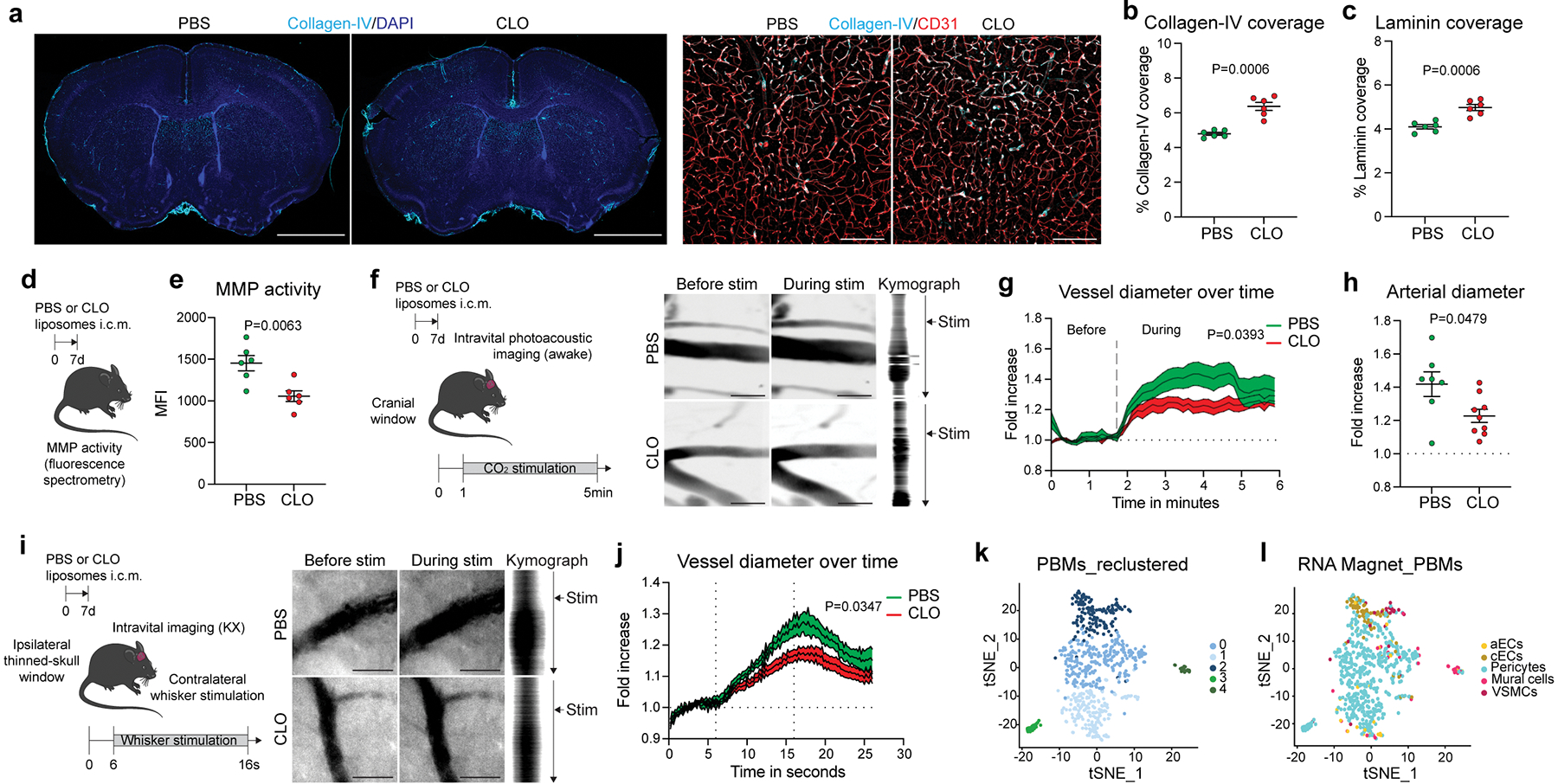
a, Representative images of brain coronal sections showing Collagen-IV and DAPI staining. High magnification images showing Collagen-IV association with CD31+ blood vessels. Scale bars, 2mm and 200μm. b, Quantification of Collagen-IV coverage. c, Quantification of Laminin coverage. For b and c, n = 6 mice/group; two-tailed unpaired Welch’s t-test. d, One week after CLO or PBS treatment, brains were extracted and incubated with MMP substrate. e, Quantification of MMP activity. n = 6 mice/group; two-tailed unpaired Welch’s t-test. f, One week after CLO or PBS treatment, mice were imaged using photoacoustic microscopy. Representative images showing vessels before (left) and during (middle) 10% CO2 challenge and corresponding kymographs (right). Scale bar: 30μm. g, Quantification of vessel diameter fold increase over time. n = 7 mice treated with PBS, and 9 mice treated with CLO; Repeated measures 2-way ANOVA with Geisser-Greenhouse correction. h, Quantification of vessel diameter fold increase during CO2 challenge. n = 7 mice treated with PBS, and 9 mice treated with CLO; two-tailed unpaired Welch’s t-test. i, One week after CLO or PBS liposome injection, mice were imaged by stereomicroscopy. Representative images showing vessels before (left) and during (middle) whisker stimulation and corresponding kymographs (right). Scale bar: 30μm. j, Quantification of vessel diameter fold increase over time. n = 6 mice treated with PBS, and 8 mice treated with CLO; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. k, Single-cell RNA sequencing demonstrating 5 PBM clusters. l, The RNA Magnet algorithm determined that PBM cluster 2 interacts preferentially with vascular smooth muscle cells (VSMCs) and capillary endothelial cells (cEC), while PBM clusters 0 and 1 preferentially interact with pericytes. All data are presented as mean values +/− SEM.
Arterial pulsation6,7 and vasomotion8 are known to be the major drivers of CSF flow dynamics. It was proposed that ECM deposition at the aorta level might affect its stiffness31. To test whether arterial motion might also be affected by PBM depletion, we created a small cranial window to observe vessel reactivity in vivo by photoacoustic microscopy (Fig. 3f). Head-restrained awake mice were placed on a moving platform and were able to move freely during the image acquisition. At one minute after the start of the imaging session, mice received 10% carbon dioxide (CO2) in medical air through a nose cone to induce vessel dilation. Using this approach, we were able to record vessel diameter before and during the CO2 challenge (Fig. 3f). Vasodilatory response of arteries from the PBM-depleted mice was impaired compared to control mice (Fig. 3g, h; Supplementary videos 7 and 8). To confirm these findings, we assessed neurovascular coupling by recording vascular dilation through a thinned-skull cranial window at the barrel cortex level during contralateral stimulation of the mouse whiskers32 (Fig. 3i). Vessel dilation upon whisker stimulation was indeed impaired in PBM-depleted mice, as compared to control counterparts (Fig. 3j; Supplementary videos 9 and 10). Systemic application of dobutamine, a β1 adrenergic agonist previously shown to enhance arterial pulsations7, rescued impaired CSF dynamics in PBM-depleted mice, as determined by i.c.m. injection of OVA and assessment of its distribution through the perivascular spaces as well as its influx into the parenchyma (Extended Data Fig. 6o–q).
To better understand the biology of PBMs, we performed scRNA-seq from mouse brain (Fig. 3k, i; Extended Data Fig. 7a). From the 14 different cell types obtained on the basis of their gene signatures, we isolated PBMs (based on their expression of canonical markers including Mrc1 and Ms4a7) and obtained 5 PBM clusters (Fig. 3k). To delineate crosstalk between PBMs and other cell types we used the RNA Magnet algorithm, which predicts paired physical and signaling interactions between cell types33. Notably, we found that cluster 2, exhibiting high expression of scavenger markers such as Lyve1 and Cd163, was interacting specifically with VSMCs, known to be located at the arterial level (Fig. 3l). Spatial proximity between PBMs and VSMCs were confirmed by electron microscopy, suggesting possible interactions, although these two cell types are separated by the basal lamina (Extended Data Fig. 7b). These results confirmed recent findings describing important interactions between VSMCs and PBMs to allow their migration at the perivascular space2. Looking at differentially expressed genes between PBM clusters, we found that cluster 2 could be identified as professional scavenger cells (Cd163, Cd38, Lyve1, Msr1, Cd36) when compared to the other clusters, which expressed genes related to antigen presentation (H2-Ab1, Cd74, Cd83, Cd14, Nlrp3) (Extended Data Fig. 7c–e). Immunostaining confirmed that Lyve1+ PBMs were highly concentrated at the vicinity of αSMA+ brain arteries/arterioles, while MHCII+ PBMs were mostly localized to αSMA− brain blood vessels (Extended Data Fig. 7f–g). We then took advantage of Lyve1 expression by PBMs to genetically target these cells using Lyve1Cre::Csf1rfl/fl mice34 (Cre+; Extended Data Fig. 7h). Mice at 3 months of age showed ~50% depletion of PBMs compared to their littermate controls not expressing Cre (Cre–; Extended Data Fig. 7i). We confirmed by flow cytometry that CD38+ PBMs were depleted by this genetic ablation, without affecting microglial cells or MHCII+ PBMs (Extended Data Fig. 7j–n). Supporting our studies using pharmacological ablation, ECM protein levels (Extended Data Fig. 7o, p) and CSF influx were also affected by genetic ablation of Lyve1+ PBMs (Extended Data Fig. 7q–v). However, we did not find changes in vessel coverage, MMP activity or ICP (Extended Data Fig. 7w–y), and CSF flow impairment was found to be more subtle in these mice (Extended Data Fig. 7q–v), likely because PBM depletion in these mice is to a lesser extent than after CLO treatment, or because a lifelong reduction in CSF flow resulted in adaptation and emergence of alternative pathways to regulate ICP.
In summary, these observations suggest that PBMs regulate CSF dynamics along the perivascular space and its efflux and influx of the brain parenchyma. The mechanism underlying PBM regulation of CSF dynamics is based on their ability to regulate ECM remodeling, affecting arterial stiffness.
PBMs in aging and Alzheimer’s disease
CSF flow is impaired in old mice and this impairment could be ameliorated in part by enhancement of meningeal lymphatic vessels25. We hypothesized that PBMs participate in the age-related deterioration of CSF dynamics. We first confirmed that CSF flow was indeed globally impaired in old mice by MRI, but also at the MCA level using fluorescent tracers, and in brain coronal sections using different molecular weight fluorescent tracers (Fig. 4a–c; Extended Data Fig. 8a–f). Comparison of the PBMs in young adult (3-month-old) and old (24-month-old) mice yielded no difference in overall CD206+ cell numbers, but aged mice exhibited a significant reduction in Lyve1+ cells and an increase in MHCII+ cells, in agreement with previous reports21 (Fig. 4d–g; Extended Data Fig. 8g–k). CD38+ PBMs in young mice were the major cell type that phagocytosed the pHrodo particles (Extended Data Fig. 8l–o). The change of PBM phenotype observed in aged mice appeared to be associated with impaired pHrodo bioparticle phagocytosis, (Extended Data Fig. 8p–r), reduction of the functional perivascular space filled by the beads (Extended Data Fig. 8s–y), and accumulation of ECM proteins (Fig. 4h–j; Extended Data Fig. 8z).
Fig 4. PBMs in aged mice.
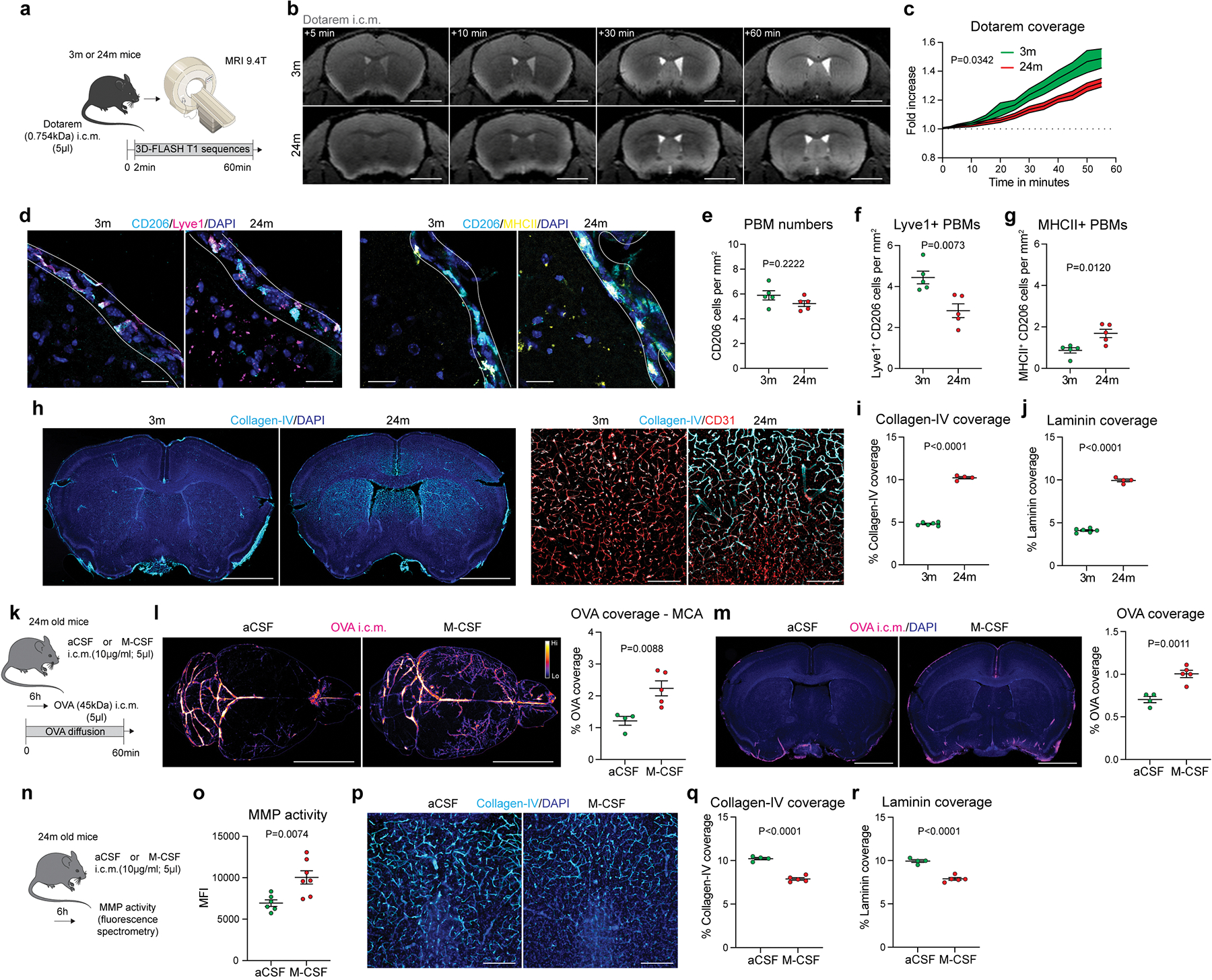
a, 3-month-old (3m) and 24-month-old (24m) mice received Dotarem i.c.m. and were placed into the MRI device. b, Representative images showing Dotarem distribution over an hour. Scale bar, 3mm. c, Quantification of Dotarem signal fold increase over time, n = 5 3m mice, and 6 24m mice; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. d, Representative images showing CD206, Lyve1, MHCII and DAPI staining. Scale bar, 20μm. e, Quantification of CD206+ PBMs. f, Quantification of Lyve1+CD206+ PBMs. g, Quantification of MHCII+CD206+ PBMs. For e-g: n = 5 mice/group; two-tailed unpaired Welch’s t-test. h, Representative images brain coronal sections showing Collagen-IV and DAPI staining. High magnification images showing Collagen-IV association with CD31+ blood vessels. Scale bars, 2mm and 200μm. i, Quantification of Collagen-IV coverage. j, Quantification of Laminin coverage. For i and j: n = 6 3m mice, and 4 24m mice; two-tailed unpaired Welch’s t-test. k, Six hours after i.c.m. injection of M-CSF or artificial CSF (aCSF), mice received OVA i.c.m. and were perfused one hour later. l, Representative images and quantification of OVA distribution in whole brains and m, brain coronal sections. n = 4 mice treated with aCSF, and 5 mice treated with M-CSF; two-tailed unpaired Welch’s t-test. n, Six hours after M-CSF treatment, brains were extracted and incubated with MMP substrate. o, Quantification of MMP activity. n = 6 mice treated with aCSF, and 7 mice treated with M-CSF; two-tailed unpaired Welch’s t-test. p, High magnification brain coronal sections images showing Collagen-IV and DAPI staining. Scale bar, 200μm. q, Quantification of Collagen-IV coverage. r, Quantification of Laminin coverage. For q and r: n = 4 mice treated with aCSF, and 5 mice treated with M-CSF; two-tailed unpaired Welch’s t-test. All data are presented as mean values +/− SEM.
Macrophage colony stimulating factor (M-CSF) has been previously shown to improve pathophysiology of Alzheimer’s disease (AD), supposedly through boosting phagocytosis of amyloid beta (Aβ) by blood-derived monocytes35. We hypothesized that such acute activation of PBMs in old mice might enhance their ECM degradation capacity and improve CSF dynamics. Using scRNA-seq, we confirmed that brain cells expressing Csf1r were mainly PBMs, as well as microglial cells and monocytes, although Csf1 was mostly expressed by microglial and mural cells (Extended Data Fig. 9a). A single i.c.m. injection of M-CSF (or artificial CSF as a control) was performed in old mice and CSF dynamics was evaluated after i.c.m. injection of OVA tracer six hours after M-CSF treatment (Fig. 4k). At one hour after OVA injection, brains were harvested and analyzed by stereomicroscopy, and OVA distribution at the MCA level in M-CSF treated old mice was found to be significantly enhanced compared to control old mice (Fig. 4l). The same phenotypes were also found in brain coronal sections (Fig. 4m). Importantly, acute M-CSF treatment in aged mice significantly increased MMP activity (Fig. 4n, o) and decreased ECM proteins deposition (Fig. 4p–r; Extended Data Fig. 9b, c). However, subacute M-CSF treatment (24h) in aged mice was ineffective to enhance CSF flow, although MMP activity was still significantly increased, but to a lesser extent than at six hours, suggesting that M-CSF treatment acutely drives MMP activity resulting in a short-term improvement in CSF flow (Extended Data Fig. 9d–g).
Our scRNA-seq results revealed that VSMC-associated PBMs can be differentiated from other PBMs by their expression of scavenger receptors (Extended Data Fig. 7c–e). Pathway analysis confirmed that cluster 2 showed upregulated genes characteristic of receptor-mediated endocytosis and phagocytosis (Extended Data Fig. 7e). This analysis also showed that cluster 2 was linked to a cellular response to Aβ. Furthermore, it was shown that depletion of perivascular macrophages worsens outcome in a mouse model of cerebral amyloid angiopathy (CAA)36, suggesting that PBMs are processing brain-derived Aβ. Finally, our CSF proteomics data indicate that PBM depletion leads to an accumulation of AD-associated risk factors such as CLU, APOE and APP (Extended Data Fig. 3i–k). We then hypothesized that PBMs might be involved in Aβ clearance in a mouse AD model. To test this hypothesis, 2-month-old 5xFAD mice each received a single i.c.m. injection of CLO liposomes or, as a control, PBS liposomes (Fig. 5a). We first found that CSF flow was impaired in PBM-depleted 5xFAD mice compared to their 5xFAD control littermates (Extended Data Fig. 10a, b). When their Aβ plaque loads were evaluated one month later, we found that PBM-depleted 5xFAD mice exhibited significantly increased plaque load compared to their 5xFAD control littermates (Fig. 5b), specifically in the brain cortex and amygdala (Extended Data Fig. 10c). To better understand the role of PBMs in AD pathophysiology, we performed scRNA-seq on brains from 6 to 7-month-old 5xFAD and their WT littermates (Fig. 5c; Extended Data Fig. 10d). We reclustered all macrophages and identified PBMs using the gene marker Mrc1 (Fig. 5d; Extended Data Fig. 10e). We also identified a damage-associated microglia (DAM) cluster specifically in 5xFAD mice, as previously described37 (Extended Data Fig. 10f). PBMs from 5xFAD mice exhibited altered phagocytosis/endocytosis and response to interferon gamma (IFNγ) pathways (Fig. 5e; Extended Data Fig. 10g). We confirmed using the RNA magnet algorithm that PBMs interact with mural cells, notably VSMCs and fibroblasts (FLCs) (Extended Data Fig. 10h). Interestingly, a human dataset that we had previously used to assess microglial function in AD38 also contained a small population of PBMs (Fig. 5f). PBM populations from control and familial AD patients had differed substantially, with 445 upregulated and 249 downregulated genes, respectively (Fig. 5g; Extended Data Fig. 10i). Among the most noticeably dysregulated gene signatures in human PBMs from familial AD patients were those involved in phagocytosis/endocytosis (CD163 expression) and IFNγ signaling (Fig. 5h; Extended Data Fig. 10j), recapitulating the findings from the AD mouse model. Interestingly, both IFNγ receptor genes Ifngr1 and Ifngr2 were more highly expressed in brain immune cells (i.e., microglia and PBMs) than stromal cells in our mouse single-cell dataset, suggesting an important interaction between PBMs and IFNγ (Extended Data Fig. 10k). To test the possibility that excess IFNγ in CSF may cause CSF dynamics dysfunction, we injected (i.c.m.) young adult WT mice with IFNγ and detected impaired CSF flow when compared to PBS injected mice (Extended Data. Fig. 10l, m).
Fig. 5: PBMs in human Alzheimer’s disease and 5xFAD mouse model of amyloidosis.
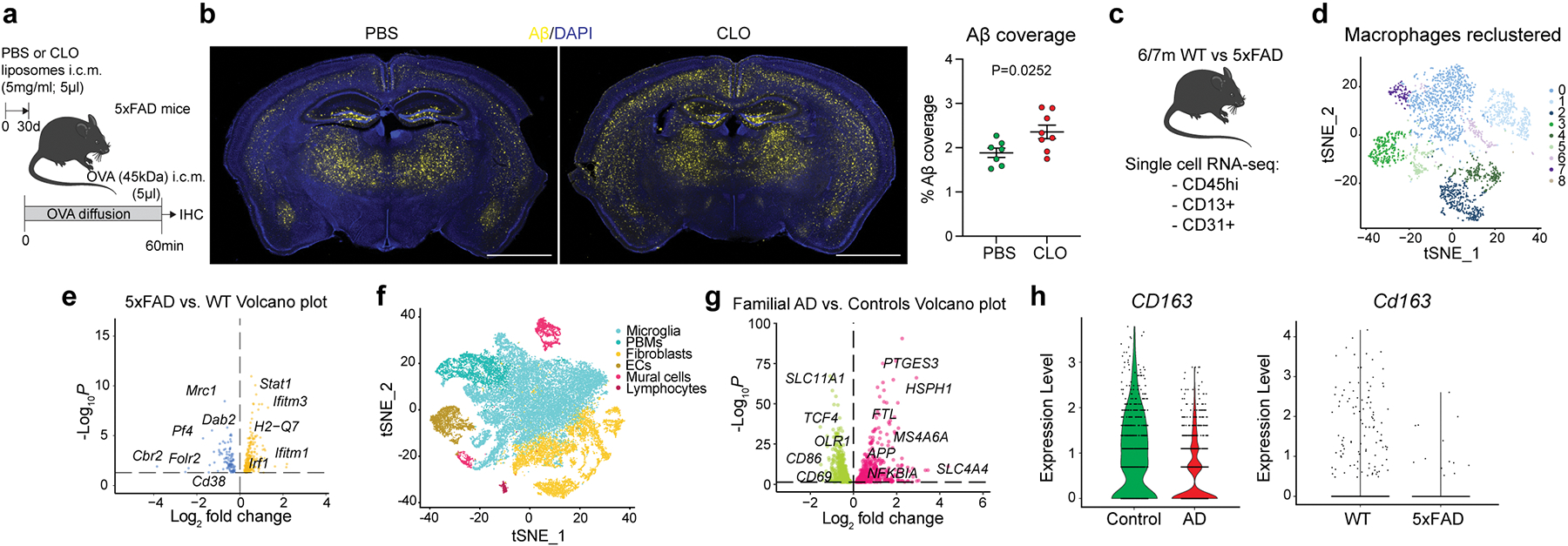
a, Experimental schematic: 5xFAD mice received an i.c.m. injection of PBS or CLO liposomes. One month later, mice received an i.c.m. injection of OVA and brains were harvested one hour later. b, Representative brain coronal section images showing amyloid-β (Aβ) plaque load and corresponding quantification. Scale bar, 2mm; n = 7 mice treated with PBS, and 8 mice treated with CLO; two-tailed unpaired Welch’s t-test. c, Single-cell RNA sequencing of the brain cortex from 6/7-month-old 5xFAD mice and their WT littermates. n = 8 mice/group. d, Macrophage populations were isolated from the 5xFAD mice single-cell RNA sequencing. Nine clusters could be identified. e, Volcano plot corresponding to down- and up-regulated genes in PBMs comparing 5xFAD mice vs. their WT littermates. F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P values. f, Human single-nuclei RNA sequencing from familial AD and control patients. Six cell populations were used in this dataset: microglia, fibroblasts, endothelial cells, mural cells, lymphocytes and PBMs. g, Volcano plot corresponding to down- and up-regulated genes in PBMs comparing patients suffering from familial AD vs. non-AD patients. F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P values. h, Violin plots corresponding to CD163 gene expression from both human single-nuclei (left) and mouse single-cell sequencing (right). All data are presented as mean values +/− SEM.
Collectively, these findings reveal that PBMs are critical players in CSF flow dynamics in aging and AD (Extended Data Fig. 10n), and should be further explored as potential new therapeutic targets for AD and other age-associated diseases characterized by protein aggregation and CSF dysfunction.
Discussion
The results of this study demonstrate that perivascular and leptomeningeal macrophages express similar markers and are located around the CNS parenchyma, constantly interacting with CSF. Given their location, function, and marker expression, we suggest referring to them as a single functional population, namely parenchymal border macrophages (PBMs).
PBMs are composed of two major subtypes: Lyve1+MHCIIlo/neg and Lyve1lo/negMHCII+. While PBMs and microglia are both derived from early erythro-myeloid progenitors in the yolk sac39, a recent study suggested that Lyve1+ macrophages predominantly originated from embryonic-derived progenitors and are maintained locally in the peritoneal mesothelium34. Our data demonstrate that Lyve1+MHCIIlo/neg PBMs regulate arterial motion and ECM remodeling (along large vessels as well as capillaries). Indeed, depletion or dysfunction of PBMs results in impaired arterial motion, accumulation of ECM and impairment of CSF flow. Previous studies using clodronate-liposome for depletion of perivascular macrophages did not observe major changes in cerebral blood flow17,19. The apparent discrepancy may be a result of the use of cranial windows and dura mater removal used in these studies, which might have masked the effects we observe with thinned skull preparations. Spontaneous low-frequency oscillations of arterioles in brain parenchyma have been proposed as the driving force of CSF/ISF clearance8. Interestingly, this paravascular clearance was found to be impaired in the context of cerebral amyloid angiopathy (CAA). Moreover, depletion of perivascular macrophages in CAA exacerbates the disease36, and perivascular macrophages can produce excessive reactive oxygen species leading to neurovascular dysfunction in the context of hypertension and AD17,19. Together with our data, it is plausible that PBMs are at the interface between spontaneous arterial oscillations, ECM remodeling, neuronal activity and CSF flow.
Notably, aged mice exhibit an increase in Lyve1lo/negMHCII+ PBMs, increased ECM deposition, and impaired CSF dynamics. We showed here that treatment of aged mice with M-CSF increased MMP activity, reduced ECM accumulation and acutely restored impaired CSF flow. PBM depletion also resulted in increased accumulation of parenchymal plaques in 5xFAD mouse model of amyloidosis. PBMs from familial AD patients exhibited altered expression profiles compared to non-AD patients, including dysregulated pathways involving phagocytosis/endocytosis and IFNγ signaling on PBMs. Interestingly, similar pathways were among the most differentially regulated in PBMs of mice from wild type vs. 5xFAD mice. Stimulation of PBMs in vivo in young mice by acute injection of IFNγ resulted in CSF flow impairment. Given the pleiotropic nature of IFNγ, however, its effect on CSF dynamics may not be solely as a result of its signaling on PBMs, although expression of both IFNγ receptors is higher in PBMs than other cell types in the brain. Future studies should focus on studying in further depth the role of IFNγ in CSF flow dynamics and developing intervention therapies for AD and other age-associated disorders based on IFNγ signaling or alternative modulations of PBMs.
Macrophages have previously been associated with cardiovascular diseases such as chronic hypertension17,40, and neurovascular coupling in both AD and homeostasis41. While our appreciation of the role played by tissue-resident macrophages in tissue homeostasis has been increasing in recent years, the exact functions performed by the PBM population are still largely unknown. In this study we unraveled an unexpected role for PBMs in CSF flow dynamics and demonstrated their potential therapeutic capacities in aging and in AD. These findings may lead to the development of novel therapeutic approaches for diseases associated with CSF dysfunction and with protein accumulation and aggregation, such as AD, Parkinson’s disease, and many others.
Methods:
Mice
Mice were bred in house or obtained from the Jackson Laboratory or provided by the National Institutes of Health/National Institute on Aging (24-month-old mice). Mice were housed in 12-hour light-dark cycle in a temperature- and humidity-controlled environment with water and food ad libitum. Mice were housed at least one week at the animal facility before any experimentation. Both males and females were used in this study. The following mouse strains were used in this study: C57BL/6J (WT; JAX 000664), C57BL/6-Tg (UbcGFP; JAX 004353), hemizygous B6SJL-Tg (APPSwFlLon, PSEN1*M146L*L286V)6799Vas/Mmjax (5xFAD mice; JAX 008730), Lyve1cre (B6;129P2-Lyve1tm1.1(EGFP/cre)Cys/J; JAX 012601), Csf1rfl/fl (B6.Cg-Csf1rtm1.2Jwp/J; JAX 021212), Aldh1l1Cre/ERT2(B6; JAX 031008). Sample sizes were chosen on the basis of standard power calculations (with α = 0.05 and power of 0.8) performed for similar experiments that were previously published. Animals from different cages, but within the same experimental group, were selected to assure randomization. Mice from the same cage received different treatments (for example, in a cage of 5 mice, 2 mice received PBS-loaded liposomes and 3 mice received CLO-loaded liposomes). Treatment was given in a blind manner and could be identified by a corresponding ear tag (treatment A: left ear was tagged; treatment B: right ear was tagged). All experiments were approved by the Institutional Animal Care and Use Committee of the University of Virginia and the Institutional Animal Care and Use Committee of the Washington University in Saint-Louis.
Intra-cisterna magna (i.c.m.) injections
Mice were anesthetized using an intraperitoneal injection of a cocktail of ketamine (100mg/kg) and xylazine (10mg/kg) (KX) diluted in 0.9% Na (saline) solution. The fur of the neck was shaved and cleaned with 70% iodine. Then, mice were placed in a stereotaxic frame to maintain the head fixed, and an ophthalmic solution was applied to prevent drying eyes. The skin from the neck was longitudinally incised and muscles were retracted using hooks to expose the cisterna magna. The solutions, diluted in artificial cerebrospinal fluid (aCSF), were injected using a 33-gauge Hamilton syringe (1 to 5μl; 2.5μl/min). The syringe was left in place for one minute after injection to prevent backflow. For survival surgeries, the skin was sutured and mice were kept on a heating pad until fully awake. Mice received a subcutaneous injection of Ketoprofen (2.5 mg/kg) at the end of the surgery.
To deplete parenchymal border macrophages (PBMs), mice received an i.c.m. injection of clodronate-loaded liposomes (CLO; 5μl; 5mg/ml; Fisher Scientific; CLD-8901). The control group consisted of mice that received an i.c.m. injection of PBS-loaded liposomes (PBS). The effect of PBM depletion was mostly studied one week after liposome injection 1) to avoid any side effect from an inflammatory reaction due to the depletion, and 2) at one week, we observed a robust depletion (80-to-85% depletion), as previously described28. We chose to wait for a month instead of a week to evaluate the mid-to-long term effect of PBM depletion on plaque accumulation in the 5xFAD Alzheimer’s disease mouse model.
To evaluate CSF flow dynamics in different contexts, Alexa647-conjugated Ovalbumin (OVA; 45kDa; 5μl; 1mg/ml diluted in artificial CSF; Thermo Fisher Scientific, O34784), Fluorescein Dextran (FITC-Dex, 4kDa; 5μl; 5mg/ml diluted in artificial CSF; Sigma-Aldrich, 46944), Texas Red Dextran (3kDa; 5μl; 1mg/ml diluted in artificial CSF; Fisher Scientific, D3328), fluorescent beads (0.1μm thick; 5μl; 1:5 dilution in artificial CSF; Life Technologies, F8888) or Dotarem (Gadolinium-based Molecular Resonance Imaging (MRI) contrast agent; 0.754kDa; 5μl; 0.5mmol/ml; Guerbet) were injected i.c.m. and mice were perfused one hour later.
To evaluate the role of PBMs on CSF flow in old mice, mice (24-month-old) received an i.c.m. injection of 5μl artificial CSF or M-CSF (10μg/ml diluted in artificial CSF; Sigma-Aldrich, M9170). Six- or 24 hours later, mice received an i.c.m. injection of OVA to evaluate CSF flow.
To evaluate the effect of interferon gamma (IFNγ) on CSF flow, young adult mice (2-to-3-month-old) received an i.c.m. injection of 1μl artificial CSF or recombinant IFNγ (20μg/ml diluted in artificial CSF; Fisher Scientific, 485-MI-100/CF). Three hours later, mice received an i.c.m. injection of 5μl OVA to evaluate CSF flow.
To evaluate PBM phagocytosis, mice received an i.c.m. injection of pHrodo particles (1mg/ml in artificial CSF; Deep Red E. coli bioparticles, Life Technologies, P35360). These particles will emit 647-wavelengh fluorescent only after being phagocytosed by cells (pH-dependent). The phagocytic activity was measured by pHrodo coverage by immunohistochemistry, as well as quantification of pHrodo+ cells by flow cytometry.
Intrastriatal injections
Anesthetized mice (KX) were shaved on the top of the head and placed in a stereotaxic frame. After skin incision, a small craniectomy was made using a drill, and the different solutions were injected using a glass capillary (1μl; 0.2μl/min) (coordinates from the Bregma: AP+1.5mm; ML−1.5mm; DV+2.5mm). The glass capillary was left in place for five additional minutes to prevent backflow. Mice were then sutured and placed on a heating pad until further experiments.
Dobutamine injection
For one experiment, mice received an intraperitoneal injection of dobutamine (40μg/kg diluted in saline; Sigma Aldrich, D0676), a beta-1 adrenergic agonist, or saline as a control, prior to CSF flow evaluation.
Proteomic analysis of cerebrospinal fluid
Cerebrospinal fluid collection
Mice were anesthetized using KX cocktail. The fur of the neck was shaved and cleaned with 70% iodine. Then, mice were placed in a stereotaxic frame to maintain the head fixed, and an ophthalmic solution was applied to prevent drying eyes. The skin from the neck was longitudinally incised and muscles were retracted using hooks to expose the cisterna magna. A glass capillary was inserted into the cisterna magna to collect the cerebrospinal fluid (CSF), which were transferred in 1.5ml Eppendorf tubes for further analyses.
Peptide preparation.
Cerebrospinal fluid (CSF; 7–10 μL) samples from mice were dried in a speedvac and solubilized with 30 μl of SDS buffer (4% (wt/vol), 100 mM Tris-HCl pH 8.0, 0.2% DCA). The protein disulfide bonds were reduced using 100 mM DTT with heating to 95 °C for 10 min. Peptides were prepared as previously described using a modification of the filter-aided sample preparation method (eFASP)42. The samples were mixed with 200 μl of 100 mM Tris-HCL buffer, pH 8.5 containing 8 M urea and 0.2% DCA (UA buffer). The samples were transferred to the top chamber of a 30,000 MWCO cutoff filtration unit (Millipore, part# MRCF0R030) and spun in a microcentrifuge at 14,000 rcf for 10 min. An additional 200 μl of UA buffer was added and the filter unit was spun at 14,000 rcf for 15 to 20 min. The cysteine residues were alkylated using 100 μl of 50 mM Iodoacetamide (Pierce, Ref. No. A39271) in UA buffer. Iodoacetamide in UA buffer was added to the top chamber of the filtration unit.
The samples were gyrated at 550 rpm for 30 min in the dark at RT using a Thermomixer (Eppendorf). The filter was spun at 14,000 rcf for 15 min and the flow through discarded. Unreacted iodoacetamide was washed through the filter with two sequential additions of 200 μl of 100 mM Tris-HCl buffer, pH 8.5 containing 8 M urea and 0.2% DCA, and centrifugation at 14,000 rcf for 15 to 20 min after each buffer addition. The flow through was discarded after each buffer exchange- centrifugation cycle. The urea buffer was exchanged with digestion buffer (DB), 50 mM ammonium bicarbonate buffer, pH 8, containing 0.2% DCA. Two sequential additions of DB (200 μL) with centrifugation after each addition to the top chamber was performed. The top filter units were transferred to a new collection tube and 100 μL DB containing 1 mAU of LysC (Wako Chemicals, cat. no. 129–02541) was added and samples were incubated at 37 °C for 2 h. Trypsin (1 μg) (Promega, Cat. No. V5113) was added and samples were incubated overnight at 37 °C. The filters were spun at 14,000 rcf for 15 min to recover the peptides in the lower chamber. The filter was washed with 50 μl of 100 mM ABC buffer and the wash was combined with the peptides. Residual detergent was removed by ethyl acetate extraction42. After extraction, the peptides were dried in a Speed-Vac concentrator (Thermo Scientific, Savant DNA 120 Speedvac Concentrator) for 15 min. The dried peptides were dissolved in 1% (vol/vol) TFA and desalted using stage tips (C18) as previously described (Rappsilber et al, nprot.2007.261). The peptides were eluted with 60 μl of 60% (vol/vol) MeCN in 0.1% (vol/vol) FA and dried in a Speed-Vac (Thermo Scientific, Model No. Savant DNA 120 concentrator). The peptides were dissolved in 20 μl of 1% (vol/vol) MeCN in water. An aliquot (10%) was removed for quantification using the Pierce Quantitative Fluorometric Peptide Assay kit (Thermo Scientific, Cat. No. 23290).
The remaining peptides were transferred to autosampler vials (Sun-Sri, Cat. No. 200046), dried and stored at −80 °C.
UPLC-Orbitrap Mass Spectrometry
The peptides were analyzed using ultraperformance HPLC Orbitrap mass spectrometry with the modifications described below. The samples in 1% (vol/vol) FA (1%) were loaded in 2.5 μl onto a 75 μm i.d. × 50 cm Acclaim® PepMap 100 C18 RSLC column (Thermo-Fisher Scientific) on an EASY nanoLC (Thermo Fisher Scientific) at a constant pressure of 700 bar at 100% A (0.1%FA). Prior to sample loading the column was equilibrated to 100%A for a total of 11 μl at 700 bar pressure. Peptide chromatography was initiated with mobile phase A (1% FA) containing 2%B (100%ACN, 1%FA) for 5 min, then increased to 20% B over 100 min, to 32% B over 20 min, to 95% B over 1 min and held at 95% B for 19 min, with a flow rate of 250 nl/min. The data was acquired in data-dependent acquisition (DDA) mode. The full-scan mass spectra were acquired with the Orbitrap mass analyzer with a scan range of m/z = 325 to 1500 and a mass resolving power set to 70,000. Ten data-dependent high-energy collisional dissociations were performed with a mass resolving power set to 17,500, a fixed lower value of m/z 110, an isolation width of 2 Da, and a normalized collision energy setting of 27. The maximum injection time was 60 ms for parent-ion analysis and production analysis. The target ions that were selected for MS/MS were dynamically excluded for 30 sec. The automatic gain control (AGC) was set at a target value of 1e6 ions for full MS scans and 1e5 ions for MS2. Peptide ions with charge states of unassigned or one were excluded for HCD acquisition
Identification of Proteins
MS raw data were converted to peak lists using Proteome Discoverer (version 2.1.0.81, Thermo-Fischer Scientific). MS/MS spectra with charges greater than or equal to two were analyzed using Mascot search engine (Matrix Science, London, UK; version 2.7.0). Mascot was set up to search against a custom non-redundant UniProt database of mouse (version March 2021, 16,997 entries), assuming the digestion enzyme was trypsin with a maximum of 4 missed cleavages allowed. The searches were performed with a fragment ion mass tolerance of 0.02 Da and a parent ion tolerance of 20 ppm. Carbamidomethylation of cysteine was specified in Mascot as a fixed modification. Deamidation of asparagine, deamidation of glutamine, formation of pyro-glutamic acid from N-terminal glutamine, acetylation of protein N-terminus and oxidation of methionine were specified as variable modifications. Peptides and proteins were filtered at 1% false-discovery rate (FDR) by searching against a reversed protein sequence database.
Intracranial Pressure measurements
Mice were anesthetized using KX cocktail and placed in a stereotaxic frame. After the cisterna magna being exposed, a glass capillary containing a pressure sensor was inserted into the cisterna magna. A baseline measure was done in saline solution. The capillary was let in place into the cisterna magna for 3 minutes. Intracranial pressure was recorded using the FISO Evolution Software (v. 2.2.0.0). Respiratory rates were also measured using this method (breaths/min).
Western Blots on isolated brain blood vessels
Brains were homogenized 2-mL tissue grinder in a total of 1.5 mL of microvessel isolation buffer (MIB; 15 mM HEPES, 147 mM NaCl, 4 mM KCl, 3 mM CaCl2, and 12 mM MgCl2), containing a cocktail of protease inhibitors (cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail, Sigma-Aldrich) and transferred in a 15-mL conical tube, and centrifuged at 1000g for 10 min at 4 °C. The supernatant was removed, and the pellet was resuspended in 5 mL of MIB containing 18% dextran (from Leuconostoc mesenteroides, M.W. 60 000–90 000; Sigma-Aldrich, St. Louis, MO) and spun at 4000g for 20 min at 4 °C. The resulting supernatant was discarded, and the pellet was resuspended in 1 mL of MIB. The homogenate was then filtered through a 20-μm nylon filter (Pluristrainer® 20 μm, Fisher Scientific). Microvessels were retained on the filter, whereas the parenchymal fraction was contained in the filtrate. The vascular fraction was then rinsed from the filter with PBS containing 1% BSA and pelleted at 4000g 10 min. The pellet was re-suspended in 1,5ml PBS and transferred in an Eppendorf tube and pelleted again at 4000g 10 min.
Isolated vessels were lysed in RIPA buffer (Bioworld) containing protease inhibitors and their concentration determined by the bicinchoninic acid assay kit (PIERCE). Thirty μg of protein was separated by PAGE on 7.5% Mini-PROTEAN® TGX™ Precast Protein Gels (BioRad) and transferred onto nitrocellulose membranes. Ponceau S (Sigma-Aldrich) was used to confirm loading of equal amounts of protein and to monitor the transfer procedure. After blocking with blocking buffer [TBS (50-mM Tris, 150-mM NaCl, pH 7.6) containing 0.1% Tween 20 and 5% milk], the membranes were probed overnight (4 °C) with a primary antibody (Collagen IV antibody 134001, BioRad, 1:500) diluted in blocking buffer. Membranes were rinsed in TBS containing 0.1% Tween 20 and incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (donkey anti-goat IgG (Abcam ab97120, 1:10000) diluted in blocking buffer.
Behavioral tests
Behavior tests were conducted at least one hour after the dark cycle started. Behavior tests were conducted in a sequence of ‘Elevated Plus Maze > Open Field Test > Three-chamber social interaction test > Forced Swim Test (cohort #1) or Cued Fear Conditioning test (cohort #2). Every behavior test was conducted with 50-dB white noise and at least 2 days apart to prevent stress. Experimental and social target mice were handled for three days before starting the first experiment. Before any behavior tests, cages were located in a dark room with 50-dB white noise for 30 minutes.
Three-chamber social interaction test
The size of the three-chambered apparatus was 40 cm W × 20 cm H × 26 cm D, with a center chamber of 12 cm W and side chambers of 14 cm W. The illuminance was kept at 50 lux. In the first session, the mouse could freely move around the whole three-chambered apparatus with two small containers in the left or right corner for 10 min (Session #1). The mouse was then gently confined in the center chamber while a novel ‘Object’ (O) and a wild-type stranger mouse, ‘Stranger 1 (aged-matched C57BL/6J strain)’(S1), was placed in one of the two plastic containers. The subject mouse was then allowed to freely explore all three chambers for 10 min (Session #2). In the third session, the subject mouse was again gently guided to the center chamber while the ‘Object’ was replaced with a wild-type ‘Stranger 2’ (S2) mouse. The subject mouse again freely explored all three chambers for 10 min (Session #3). Object/Stranger exploration was defined by the mouse’s nose being oriented toward the target and coming within 2 cm of it as measured by EthoVision XT 15 (Noldus).
Open field test
Mice were placed in an open field box (35 × 35 × 35 cm) and recorded with a video camera for 60 min. The center zone line was 9 cm apart from the edge. The testing room was illuminated at 0 lux. Mice movements were analyzed using EthoVision XT 15 program (Noldus).
Elevated plus-maze
The elevated plus-maze consisted of two open arms, two closed arms (for all arms, 35 cm × 7 cm), and a center zone, and was elevated to a height of 1 m above the floor. The illuminance of the closed arm was 80 lux, while the open arm was 120 lux. Mice were placed in the center zone and allowed to explore the space for 8 min. The data was analyzed using EthoVision XT 15 (Noldus).
Forced Swim Test
Mice were located in the ¾ tap water-filled 4L-beaker and recorded with a video camera on the top and side for 5 min. Water temperatures were kept at 20–21°C, and the room was 120-lux. Behavior was analyzed manually.
Cued-Fear conditioning test
On the first day, mice were located in the fear conditioning chamber (Ugo Basile). Before starting the experiment, 70% EtOH was sprayed once in the chamber. After 2 min of acclimation in white noise and the experimental condition, 1 kHz tone was applied for 20 sec accompanied by 0.7 mA electric shock in the last 2 sec. After 1 min waiting time, tone-to-waiting was repeated three times in total. After repetition, mice were taken out of the conditioning chamber and returned to their home cage. On the second day (24-hours after), the chamber was wiped with the vanilla-flavored oil, and the floor was exchanged with a gray-colored plastic plate. Mice were located in the chamber. After 2 min of acclimation to white noise, 1 kHz tone was applied for 1 min, followed by 2 mins of white noise. Mice were returned to the home cage. After seven days from the conditioning, the second day’s protocol was repeated. The data were analyzed using activity parameters of EthoVision XT 15 (Noldus). Freezing time was measured during the waiting time (for the first day), and during cue (for the second and eighth day).
Tissue collection and processing
Mice received a lethal intraperitoneal injection of euthasol (10% v/v in saline, 250μl), and transcardially perfused with PBS containing 10U/ml heparin. In some experiments, mice received an intravenous injection of lectin (30μl; Dylight 649 labeled Lycopersicon Esculentum; Fisher Scientific, DL-1178) five minutes prior to perfusion. After removal of the skin, muscles and mandibles, the head was drop-fixed in 4% paraformaldehyde (PFA) for 24h. Then, the skull caps (skull and attached dorsal dura mater) were detached and brains were kept in 4% PFA for an additional 24h (48h in total). When harvested, the deep cervical lymph nodes (dCLNs) were drop-fixed in 4% PFA for 12h. After post-fixation, the tissues were then cryoprotected with 30% sucrose solution and frozen in Tissue-Plus OCT compound (Thermo Fisher Scientific). Brains were sliced (100-μm thick sections) with a cryostat and kept in 24-well plates filled with PBS at 4°C. The dCLNs were sliced (30μm-thick sections) and collected on gelatin-coated slides. In one experiment, whole brains were post-fixed with 4%PFA, then washed with PBS, and were directly stained and imaged by stereomicroscopy.
Ex vivo Stereomicroscopy imaging
Mice received a lethal intraperitoneal injection of euthasol (10% v/v in saline, 250μl), and transcardially perfused with PBS containing 10U/ml heparin. After removal of the skin, muscles and mandibles, the head was drop-fixed in 4% paraformaldehyde (PFA) for 24h. Then, the skull caps (skull and attached dorsal dura mater) were detached and brains were kept in 4% PFA for an additional 24h (48h in total). The whole brains were then placed on a Petri dish and imaged by stereomicroscopy. For OVA measurements, whole brains were imaged using the following parameters: CY5 channel; zoom = 0.78; exposure time = 2s. Quantification of OVA coverage at the MCA level was done using the following parameters: CY5 channel; zoom = 5; exposure time = 500ms. For bead or pHrodo experiments, brains were placed on the side, and quantifications were done using the following parameters: GFP or CY5 channels, respectively; zoom = 5; exposure time = 250ms or 500ms, respectively.
Light sheet microscopy
For whole brain imaging, the vDISCO method was used to clear the brain. Briefly, mice received an i.c.m. injection of OVA (5μl). One hour later, mice received an intravenous injection of lectin (30μl), and were perfused five minutes later. Whole brain was post-fixed in 4%PFA for 24h, then permeabilized and cleared using vDISCO protocol with passive tissue immersion. Brains were immersed in ethyl cinnamate (Sigma-Aldrich, W243019) and placed in chambered cover-glass (Thermo Fisher, 155360) for light sheet imaging (LaVision BioTec).
Immunohistochemistry, imaging and quantifications
Tissues were rinsed in PBS and incubated in PBS 0.25% Triton with appropriate primary antibodies: rat anti-mouse CD206 (Bio-Rad MCA2235, 1:500), goat anti-mouse Iba1 (Abcam, ab5076, 1:500), Armenian hamster anti-mouse CD31 (Millipore Sigma, MAB1398Z, clone 2H8, 1:200), rabbit anti-mouse AQP4 (Millipore Sigma, A5971, 1:500), eFluor 660 conjugated anti-mouse Lyve1 (eBioscience, clone ALY7, 1:200), Alexa Fluor 594 conjugated anti-mouse I-A/I-E (MHCII; BioLegend, 107650, 1:200), rabbit anti-mouse Laminin (Abcam, ab7463, 1:500), rabbit anti-mouse Collagen-IV (EMD Millipore, AB756P, 1:500), FITC conjugated anti-mouse alpha SMA (Sigma Aldrich, F3777, 1:200), goat anti-mouse CD13 (R&D systems, AF2335, 1:200), rabbit anti-mouse amyloid beta (Cell Signaling Technology, 8243S, D54D2, 1:400). Sections were finally washed with a PBS solution containing 1:40000 4′,6-diamidino-2-phenylindole (DAPI) and mounted on Superfrost Plus slides (Fisher Scientific), and coverslipped with Aqua-Mount (Lerner) or ProLong Gold (Thermo Fisher). Slices were acquired using wide-field (Olympus, VS200-S6) or confocal (Leica, DMI-8 Stellaris) microscopes. Quantitative analyses of imaging measurements were performed using the FIJI package for ImageJ software. For brain tracer coverage measurements, six sections per mouse were used and manually thresholded to match observed signals. All six images were quantified as area of signal/total area (DAPI coverage) and averaged to get one value per mouse. For extracellular matrix coverage, whole brain sections, cortical regions, or lectin+ alpha SMA+ large vessels (two whole sections per mouse), were manually thresholded and quantified as area of signal/total area.
In vivo fluorescent tracer dynamics evaluation
Mice were anesthetized (KX), the head was shaved and mice were placed on a stereotaxic frame. A lateral incision was made between the right eye and the right ear. The temporalis muscle was gently separated from the temporal bone. The surface of the skull was cleaned with a cotton bud. After i.c.m. injection of the fluorescent tracer, mice were positioned on a heating pad on their side to expose the right temporal bone, under the stereomicroscope (Leica, M205 FA). The average time between the i.c.m. injection and the first image was 2 min. Mice were imaged over an hour (240 frames in total: 4 frames per min). At the end of the imaging session, mice were euthanized using euthasol and tissues were harvested for further analyses. For one experiment, mice were anesthetized with KX or isoflurane (induction at 4.5% and continuously exposed at 0.75–2%). The fold increase was measured by calculating the ratio between “x” value divided by the minimal value.
For in vivo deep cervical lymph node imaging, mice were anesthetized (KX), and the fur of the neck was shaved. After receiving an i.c.m. injection of OVA (5μl), mice were placed on supine position, the skin from the neck was incised and retracted using hooks to expose the deep cervical lymph nodes. Live imaging was done under the stereomicroscope, as previously described38 (average of 10min between the i.c.m. injection and the beginning of the imaging).
Magnetic Resonance Imaging
Right after i.c.m. injection of Dotarem under KX (Gadolinium-based MRI contrast agent; 0.754kDa; 5μl) (average time between i.c.m. injection and start of the MRI acquisitions: 2min), mice were placed in prone position on the magnetic resonance imaging device (9.4 Tesla MRI, Bruker Biospin). During the imaging session, 0.5–0.8% isoflurane was provided through a nose mask to prevent mice from waking up. Body temperature and respiratory rates were monitored during imaging. Isoflurane levels were adjusted with respiratory rates, and body temperature was controlled using a heating pad. Series of post-contrast T1 FLASH-3D weighted images were taken through the head with the following parameters: repetition time = 30 ms; echo time = 8 ms; number of echo images = 1; number of averages = 1; number of repetitions = 12; scan time = 272640 ms per sequence (4m54sec); flip angle = 20, FOV = 160 × 160 × 80 μm with a 128 × 128 × 64 matrix; spatial resolution = 125 × 125 × 125 μm (8 pixels per mm; voxel size = 0.125 mm3), number of slices = 64; receiving coil 4 elements RF ARR 400 1H M. The total acquisition time was of about 1 h per mouse (4m54sec × 12 sequences per mouse). To calculate the volume of ventricles, mice were placed on supine position on the molecular resonance imaging device (7 Tesla MRI, Bruker Biospin). T2-weighted sequences were taken through the mouse head using the following parameters: TR = 3000 ms, TE = 139 ms, FOV = 26 × 20.5 mm, slice thickness = 0.13 mm, number of slices = 160 and NEX = 3 (total acquisition = 16 min per mouse).
In vivo photoacoustic microscopy
Mice were anesthetized using a bolus of 4% isoflurane and medical air and maintained in 1–2% isoflurane during surgical procedure. After fixing the head in a stereotaxic frame, a longitudinal incision was made to expose the skull. A small cranial window was made to be able to image brain blood vessels. Mice were sutured and allowed to recover after surgery in a clean cage. On the same day, mice were anesthetized with 4% isoflurane and were transferred into the photoacoustic microscope, restrained using a nut and medical air was used for inhalation. The imaging session started 20 min after the mouse woke up. Thirty seconds after the acquisition started (average of 5,000 frames per mouse, frequency = 12Hz), mice received a mixture of 10% CO2 in medical air for 5 min. The vessel diameter fold increase was measured by calculating the ratio between the measured values and the average of the first thousand values (before CO2 challenge).
In vivo whisker stimulation
Mice were anesthetized using KX cocktail and placed in a stereotaxic frame. Importantly, body temperature was constantly monitored and adjusted using a heating pad. Whiskers on the right side of the mice were cut to let only about 0.5cm. A longitudinal incision was made to expose the skull, and a thinned-skull window was made at the left barrel cortex to be able to image the distal part of the middle cerebral artery (MCA). Mice were then placed under the stereomicroscope for imaging. Mechanical whisker stimulation was performed for 10 seconds, six seconds after the beginning of the imaging session. An average of 10 videos were made per mouse (5 with stimulations and 5 without stimulation), using the following parameters: brightfield channel; x16 zoom; exposure time:200ms (5 frames/second). Heart rates (beats/min) and basal arterial pulsations (pulsatile amplitude/vessel diameter) were measured using the videos without whisker stimulations.
Flow cytometry
Mice received a lethal intraperitoneal injection of euthasol (10% v/v in saline, 250μl), and transcardially perfused with PBS containing 10U/ml heparin. Lateral choroid plexuses were removed from half- or whole brains. Brains were then digested for 40 min at 37C with 1.4U/ml of Collagenase-VIII and 35U/ml of DNAse I in DMEM. The cell pellets were washed and resuspended in FACS buffer and stained with the following antibodies (1:200 dilution; eBioscience): rat anti-mouse Ly6C (BioLegend, 128033, BV510), rat anti-mouse XCR1 (BioLegend, 148216, BV421), rat anti-mouse CD24 (BioLegend, 101819, Pacific Blue), rat anti-mouse Ly6G (BD Biosciences, 741587, BUV661), rat anti-mouse CD45 (BD Biosciences, 746947, BV750), rat anti-mouse CD19 (BD Biosciences, 751213, BUV615), rat anti-mouse CD11b (BD Biosciences, 741242, BUV563), rat anti-mouse TCRb (BD Biosciences, 748405, BUV805), rat anti-mouse CD4 (BD Biosciences, 563790, BUV395), rat anti-mouse F4/80 (BioLegend, 123133, BV605), rat anti-mouse CD64 (BD Biosciences, 139308, PerCP-Cy5.5), rat anti-mouse CD206 (BioLegend, 141710, AF488), rat anti-mouse CD38 (BioLegend, 102716, AF647 or Biolegend, 102719, Pacific Blue), rat anti-mouse MHCII (BioLegend, 107641, BV650). Cell viability was determined using DAPI staining. Fluorescence data were acquired using Cytek Aurora spectral flow cytometer (Cytek) then analyzed using FlowJo Software (Tree Star, v5.0). For one experiment, mice received an i.c.m. injection of pHrodo particles (Life Technologies, P35360), that were detected by A647.
Electron microscopy
Mice received a lethal dose of i.p. euthasol and were transcardially perfused with warm Ringer’s solution followed by perfusion with warm fixative containing 2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 M cacodylate buffer (2mM CaCl2 at pH 7.4). Brains were transferred to a fixative solution and allowed to fix overnight at 4°C. Brains were rinsed in cacodylate buffer 3 times for 10 minutes and fixed in 1% osmium tetroxide/1.5% potassium ferrocyanide in cacodylate buffer for an hour. Brains were then washed in ultrapure water 3 times for 10 minutes and stained in 1% thiocarbohydrazide solution for one hour, followed by one hour staining in aqueous 2% osmium tetroxide. Brains were rinsed in ultrapure water 3 times for 10 minutes and stained overnight in 1% uranyl acetate at 4°C. The brains were then washed in ultrapure water 3 times for 10 minutes and stained with 20 mM lead aspartate at 60°C for 30 minutes. After staining was complete, samples were washed in ultrapure water, dehydrated in a graded acetone series (50%, 70%, 90%, 100% x3) for 10 minutes in each step, and infiltrated with microwave assistance (Pelco BioWave Pro, Redding, CA) into Durcupan resin. Samples were flat embedded and cured in an oven at 60°C for 48 hours. Post resin curing, 70nm-thick sections from brain cortex were prepared on copper grids, post-stained with uranyl acetate and Reynold’s lead and imaged on a Scanning Electron Microscope (Zeiss Merlin FE-SEM) using the followed parameters: voltage = 5.00kV; probe current = 3.0nA, WD = 6.9mm.
MMP activity assay
The left hemisphere of the brain was homogenized in 6 volumes of PBS using a mini bead beater (Sigma Aldrich) and 2.3 mm diameter zirconia/silica beads (Biospec). The homogenate was then centrifuged in a microcentrifuge (Eppendorf) at 14,000 rcf. for 20 min at 4°C. One hundred μg of protein from the supernatant (representing the soluble fraction of the brain) was incubated with 25 μM of the quenched fluorescent MMP substrate BML-P128–0001 (Enzo Life Sciences) in PBS and incubated at 37°C for 15 min. Afterwards, the fluorescence of the cleaved fluorescent product was measured using a BioTek Synergy H1 plate reader (Ex/Em= 340nm/440nm).
RNA sequencing
Mus musculus Dil Liposome Leptomeningeal scRNA-seq
Sample preparation.
Fifteen mice (3-month-old males; C57BL6/J) received an i.c.m. injection of 5μl (23mg/ml) of Dil-liposomes (Liposoma, I-005). Mice were perfused with heparinized PBS and tissues were harvested the next day. Subdural meninges were gently collected from the brains and were dissected in FACS buffer and digested in a digestion solution containing 1:50 Collagenase-VIII – 1:500 DNase – 1:50 FBS in FACS buffer for 15 minutes. Tissues were then mashed through 70μm strainers in 50ml tubes containing FACS buffer and 10% FBS to stop enzymatic digestion. After centrifugation, the supernatant was removed and cells were resuspended in FACS buffer and transferred to a V-bottom plate. Cells were sorted and only live Dil-positive cells were used for sequencing.
Single-Cell Data Preprocessing.
Reads were aligned to the mm10 genome using the Cellranger software pipeline (version 3.0.2) provided by 10x genomics. The resulting filtered gene by cell matrix of UMI counts was read into R using the read10xCounts function from the Droplet Utils package. Filtering was applied in order to remove low quality cells by excluding cells expressing fewer than 200 or greater than 5,000 unique genes, having fewer than 1,000 or greater than 30,000 UMI counts, as well as cells with greater than 20% mitochondrial gene expression. Expression values for the remaining cells were then normalized using the scran and scater packages. The resulting log2 values were transformed to the natural log scale for compatibility with the Seurat (v3) pipeline43–45.
Dimensionality Reduction and Clustering.
The filtered and normalized matrix was used as input to the Seurat pipeline and cells were scaled across each gene before the selection of the top 2,000 most highly variable genes using variance stabilizing transformation. Principal Components Analysis was conducted and an elbow plot was used to select the first ten principle components for tSNE analysis and clustering. Shared Nearest Neighbor (SNN) clustering optimized with the Louvain algorithm, as implemented by the Seurat FindClusters function was performed before manual annotation of clusters based on expression of canonical gene markers. Macrophages were then subset out, rescaled and clustered as above with the first twenty-one PCAs and a resolution of 0.3
Mus musculus CLO-depletion scRNA-seq
Sample preparation.
To deplete parenchymal border macrophages (PBMs), mice received an i.c.m. injection of clodronate-loaded liposomes (CLO; 5ul; 5mg/ml; Fisher Scientific; CLD-8901). The control group consisted of mice that received an i.c.m. injection of PBS-loaded liposomes (PBS). One week later, mice received a lethal intraperitoneal injection of euthasol (10% v/v in saline, 250ml), and transcardially perfused with PBS containing 10U/ml heparin. Lateral choroid plexuses were gently removed, then whole brains were digested, myelin was removed, and brain were stained with CD13 (to stain for mural cells), CD31 (endothelial cells), and CD45 (immune cells). Stromal cells (i.e., endothelial+mural) were then sorted and prepared for single cell sequencing.
Single-Cell Data Preprocessing.
Reads were aligned to the mm10 genome using the Cellranger software pipeline (version 6.0.0) provided by 10x genomics. The resulting filtered gene by cell matrix of UMI counts was read into R using the read10xCounts function from the Droplet Utils package. Filtering was applied in order to remove low quality cells by excluding cells expressing fewer than 200 or greater than 6,000 unique genes, having fewer than 1,000 or greater than 30,000 UMI counts, as well as cells with greater than 10% mitochondrial gene expression. Expression values for the remaining cells were then normalized using the scran and scater packages. The resulting log2 values were transformed to the natural log scale for compatibility with the Seurat (v3) pipeline43–45.
Dimensionality Reduction and Clustering.
The filtered and normalized matrix was used as input to the Seurat pipeline and cells were scaled across each gene before the selection of the top 2,000 most highly variable genes using variance stabilizing transformation. Principal Components Analysis was conducted and an elbow plot was used to select the first thirty principle components for tSNE analysis and clustering. Shared Nearest Neighbor (SNN) clustering optimized with the Louvain algorithm, as implemented by the Seurat FindClusters function was performed before manual annotation of clusters based on expression of canonical gene markers.
Mus musculus Brain scRNA-seq
Sample preparation.
Four mice (4-month-old males; Aldh1l1Cre-ERT2/RosaCAG-tdTomato (C57BL6/J background)) received 100μl of tamoxifen solution (20mg/ml) on three consecutive days to induce expression of tdTomato in astrocytes. A sham surgery of ligation of the lymphatic vessels afferent to the dCLNs consisting on skin incision and sternocleidomastoid muscle retractation was made. Afterwards, the skin was sutured and mice were allowed to recover on a heating pad until fully awake. Five weeks post-surgery, mice were perfused with heparinized PBS. Brains were gently dissected using scissors and incubated in digestion solution 3 times 20 minutes each (brains were also mechanically grinded using descending diameter plastic pipettes between incubations). Tissues were then mashed through 70μm strainers in 50ml tubes containing FACS buffer and 10% FBS to stop enzymatic digestion. Myelin was removed by transferring samples into 3ml FACS buffer containing 22% BSA and centrifuged (1000 g; 9 accelerations; 2 decelerations; 10 min at 4°C). The remaining supernatant and the myelin layer were carefully removed, the pellet resuspended in FACS buffer and transferred to a V-bottom plate. Fifty μl of FcBlock solution (1:50) were added to the wells and after 20min, antibodies (CD13 (BD Biosciences, 558744), CD31 (Biolegend, 102516) and CD45 (eBioscience, 550994); 1:200) were added (total volume = 200μl). Cells were enriched in CD11b and negative for Ly6G and sorted in four different categories: CD45+ CD13− CD31− (immune), CD45− CD13+ CD31− (mural), CD45− CD13− CD31+ (endothelial) and CD45-dTomato+ astrocytes. Overall, the recovered yield of astrocytes was low and the maximum number of sorted astrocytes was used for sequencing. For the remaining cell populations, equal cell numbers were pooled and used for subsequent analyses.
Single-Cell Data Preprocessing.
Reads were aligned to the mm10 genome using the Cellranger software pipeline (version 6.0.0) provided by 10x genomics. The resulting filtered gene by cell matrix of UMI counts was read into R using the read10xCounts function from the Droplet Utils package. Filtering was applied in order to remove low quality cells by excluding cells expressing fewer than 200 or greater than 7,000 unique genes, having fewer than 1,000 or greater than 50,000 UMI counts, as well as cells with greater than 15% mitochondrial gene expression. Expression values for the remaining cells were then normalized using the scran and scater packages. The resulting log2 values were transformed to the natural log scale for compatibility with the Seurat (v3) pipeline43–45.
Dimensionality Reduction and Clustering.
The filtered and normalized matrix was used as input to the Seurat pipeline and cells were scaled across each gene before the selection of the top 2,000 most highly variable genes using variance stabilizing transformation. Principal Components Analysis was conducted and an elbow plot was used to select the first thirty principle components for tSNE analysis and clustering. Shared Nearest Neighbor (SNN) clustering optimized with the Louvain algorithm, as implemented by the Seurat FindClusters function was performed before manual annotation of clusters based on expression of canonical gene markers.
Cell-Cell Interaction Analysis.
To evaluate potential cell-cell ligand-receptor interactions in an unbiased way, the RNAMagnet package 33 was utilized with Membrane, ECM, as well as Both, ligand receptor pairs queried and all vascular network cell types (arterial, capillary, and venous endothelial cells, pericytes, and vascular smooth muscle cells) included as anchors for RNAMagentAnchors. Signaling interactions were also investigated with RNAMagnetSignaling and the top signaling pair molecules were examined for both Endothelial cells and Mural cells with each other cell type present in the dataset. Macrophages were then subset out, rescaled and clustered as above with the first seven PCAs and a resolution of 0.3.
Mus musculus 5xFAD scRNA-seq
Sample preparation.
Mice received a lethal intraperitoneal injection of euthasol (10% v/v in saline, 250ml), and transcardially perfused with PBS containing 10U/ml heparin. Cortex were gently dissected in PBS, digested and stained after myelin removal with CD13 (to stain for mural cells), CD31 (endothelial cells), and CD45 (immune cells). Stromal cells (i.e., endothelial+mural) and immune cells were then sorted and prepared for single cell sequencing.
Single-Cell Data Preprocessing.
Reads were aligned to the mm10 genome using the Cellranger software pipeline (version 6.0.0) provided by 10x genomics. The resulting filtered gene by cell matrix of UMI counts was read into R using the read10xCounts function from the Droplet Utils package. Filtering was applied in order to remove low quality cells by excluding cells expressing fewer than 200 or greater than 7,500 unique genes, having fewer than 1,000 or greater than 40,000 UMI counts, as well as cells with greater than 25% mitochondrial gene expression. Expression values for the remaining cells were then normalized using the scran and scater packages. The resulting log2 values were transformed to the natural log scale for compatibility with the Seurat (v3) pipeline43–45.
Dimensionality Reduction and Clustering.
The filtered and normalized matrix was used as input to the Seurat pipeline and cells were scaled across each gene before the selection of the top 2,000 most highly variable genes using variance stabilizing transformation. Principal Components Analysis was conducted and an elbow plot was used to select the first twenty-two principle components for tSNE analysis and clustering. Shared Nearest Neighbor (SNN) clustering optimized with the Louvain algorithm, as implemented by the Seurat FindClusters function was performed before manual annotation of clusters based on expression of canonical gene markers.
Cell-Cell Interaction Analysis.
To evaluate potential cell-cell ligand-receptor interactions in an unbiased way, the RNAMagnet package33 was utilized with Membrane, ECM, as well as Both, ligand receptor pairs queried and all vascular, stromal, and mural cell types (arterial, capillary, and venous endothelial cells, ependymal cells, fibroblasts, pericytes, and vascular smooth muscle cells) included as anchors for RNAMagentAnchors. Macrophages were then subset out, rescaled and clustered as above with the first fourteen PCAs and a resolution of 0.6.
Homo sapiens snRNA-seq
Sample Preparation.
The Neuropathology Core of the Knight-Alzheimer’s Disease Research Center (Knight-ADRC) and the Dominantly Inherited Alzheimer Network (DIAN) provided the parietal lobe tissue of postmortem brains for each sample. These samples were obtained with informed consent for research use and were approved by the review board of Washington University in St. Louis. AD neuropathological changes were assessed according to the criteria of the National Institute on Aging-Alzheimer’s Association (NIA-AA). From the 60 frozen human parietal lobes, approximately 500 mg of tissue were cut and weighed on dry ice using sterile disposable scalpels. The parietal tissue was homogenized in ice-cold homogenization buffer (0.25 M sucrose, 150 mM KCl, 5 mM MgCl2, 20 mM tricine-KOH pH 7.8, 0.15 mM spermine, 0.5 mM spermidine, EDTA-free protease inhibitor, and recombinant RNase inhibitors) with a dounce homogenizer. Homogenates were centrifuged for 5 min at 500 × g, at 4°C, to pellet the nuclear fraction. The nuclear fraction was mixed with an equal volume of 50% iodixanol and added on top of a 35% iodixanol solution for 30 min at 10,000 × g, at 4°C. After myelin removal, the nuclei were collected at the 30%–35% iodixanol interface. Nuclei were resuspended in nuclei wash and resuspension buffer (1% BSA and recombinant RNase inhibitors in PBS) and pelleted for 5 min at 500 × g and 4°C. Nuclei were passed through a 40 μM cell strainer to remove cell debris and large clumps. Nuclei concentration was manually determined using DAPI counterstaining and hemocytometer. Nuclei concentration was adjusted to 1,200 nuclei per μL and processed immediately following the 10x Genomics® Single Cell Protocol instructions. We generated snRNA-seq libraries using the 10x Chromium single cell reagent Kit v3, for 10,000 cells per sample and sequenced 50,000 reads per cell from 31 frozen human parietal lobes.
Single-Cell Data Preprocessing.
We prepared a pre-mRNA reference according to the steps detailed by 10x Genomics based on the GRCH38 (3.0.0) reference and reads were aligned to the using the Cellranger software pipeline (version 3.0.2). The resulting filtered gene by cell matrices of UMI counts for each sample were read into R using the read10xCounts function from the Droplet Utils package. Filtering was applied in order to remove low quality cells by excluding cells expressing fewer than 500 or greater than 10,000 unique genes, having fewer than 2,000 or greater than 100,000 UMI counts, as well as cells with greater than 25% mitochondrial gene expression. Samples were then randomly assigned to one of five cohorts and individually processed to screen for the possible presence of perivascular macrophages. Expression values for the remaining cells in each cohort were merged by gene symbol into one dataframe and normalized using the scran and scater packages. The resulting log2 values were transformed to the natural log scale for compatibility with the Seurat (v3) pipeline45. Each was then scaled across each gene before the selection of the top 2,000 most highly variable genes using variance stabilizing transformation. Principal Components Analysis was conducted and an elbow plot was used to select principle components for tSNE analysis and clustering. Shared Nearest Neighbor (SNN) clustering optimized with the Louvain algorithm, as implemented by the Seurat FindClusters function was performed before manual annotation of clusters based on expression of canonical gene markers. After removal of cells identified as neurons, oligodendrocytes, oligodendrocyte precursor cells, and astrocytes, the remaining cells were split by original sample identity.
Integration, Dimensionality Reduction and Clustering.
Reference samples were chosen as those with more than 500 cells per sample and were prepped for integration utilizing the SCT normalization provided by Seurat with functions SelectIntegrationFeatures and PrepSCTIntegration. Sample integration was then performed with FindIntegrationAnchors specifying a k.filter = 100 and the reference samples determined above, and IntegrateData. Principal Components Analysis was conducted and an elbow plot was used to select the first thirty principle components for tSNE analysis and clustering. Shared Nearest Neighbor (SNN) clustering optimized with the Louvain algorithm, as implemented by the Seurat FindClusters function was performed before manual annotation of clusters based on expression of canonical gene markers.
Differential Expression.
For analysis of differentially expressed genes between conditions, each cluster was filtered to include genes that had at least 5 transcripts in at least 5 cells, then the top 2000 highly variable genes were determined and included for further analysis using the SingleCellExperiment modelGeneVar and getTopHVGs functions. After filtering, observational weights for each gene were calculated using the ZINB-WaVE zinbFit and zinbwave functions46. These were then included in the edgeR model, which was created with the glmFit function, by using the glmWeightedF function47. Results were then filtered using a Benjamini-Hochberg adjusted p-value threshold of less than 0.05 as statistically significant.
Pathway Enrichment.
Over representation enrichment analysis with Fisher’s Exact test was used to determine significantly enriched Gene Ontology (GO) terms (adj. p < 0.05) for the sets of significantly differentially expressed genes. For each gene set, genes were separated into up- and down-regulated and separately the enrichGO function from the clusterProfiler package was used with a gene set size set between 10 and 500 genes and p-values adjusted using the Benjamini-Hochberg correction.
Extended Data
Extended Data Fig 1. PBMs are distinct from microglia and sample CSF and ISF.
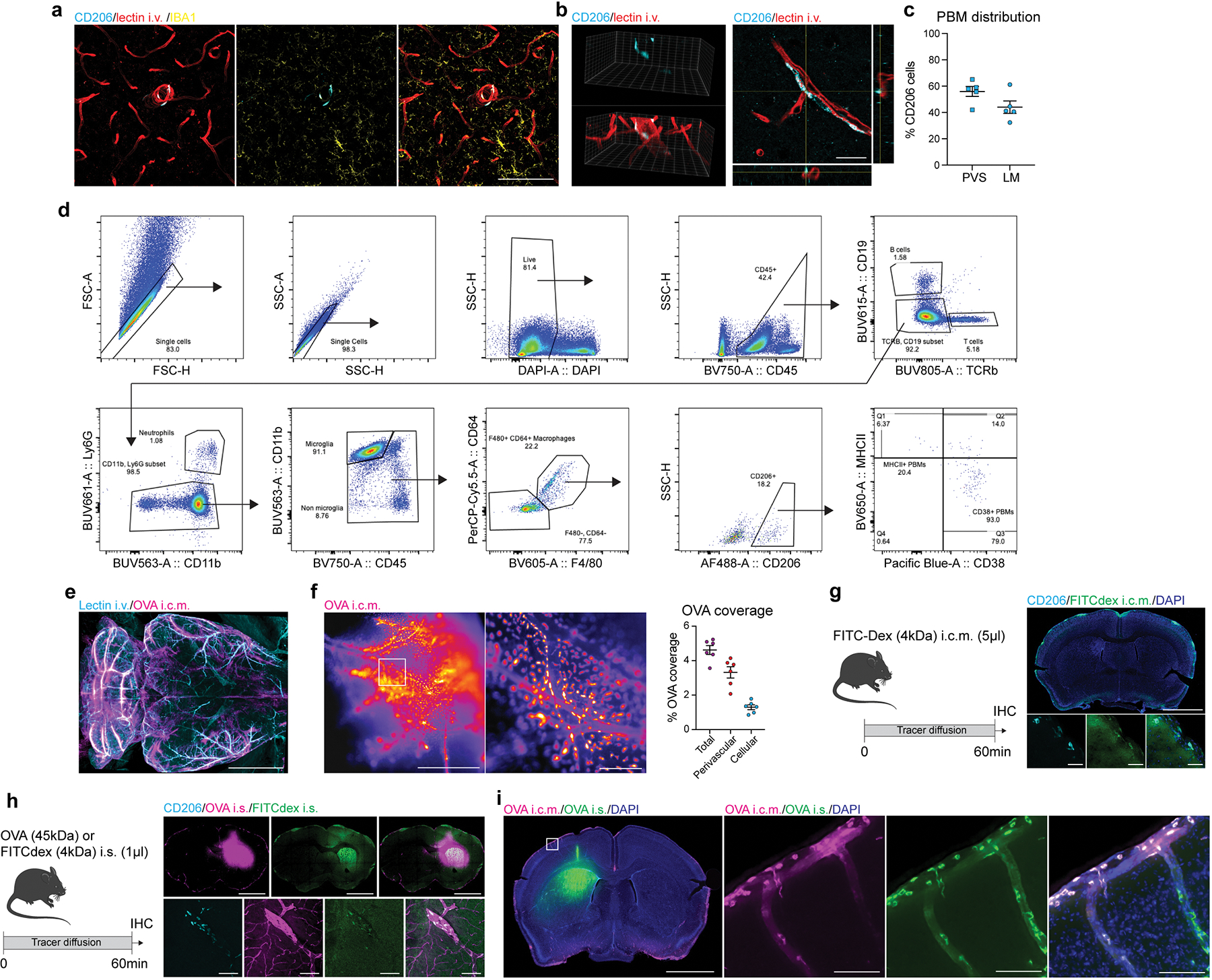
a, CD206+ PBMs (cyan) are easily distinguishable from IBA1+ microglia (yellow) and are located at the vicinity of i.v. lectin+ large blood vessels (red). Scale bar, 100μm. b, PBM are located outside of the brain vasculature, in the perivascular space. c, Quantification of whole brain sections showing spatial distribution of PBMs through both perivascular space (PVS) and leptomeninges (LM). Scale bar, 20μm. n = 5 mice. d, Gating strategy for PBM detection. PBMs were defined as DAPI−CD45+TCRb−CD19−CD11b+CD64hiF4/80hiCD206+ cells. PBMs can be divided in subtypes using MHCII and CD38. e, WT mice received an i.c.m. injection of Alexa-647 conjugated ovalbumin (OVA; 45kDa; 1mg/ml; 5μl). One hour after OVA injection, mice received an i.v. injection of Alexa-594 conjugated lectin (30μl) and were perfused five minutes later. Maximum projection image obtained by light sheet microscopy from a cleared mouse brain showing brain OVA (magenta) distribution at the vicinity of i.v.-injected lectin+ blood vessels (cyan). Scale bar, 1mm. f, WT mice received an i.c.m. injection of Alexa-647 conjugated ovalbumin (OVA; 45kDa; 1mg/ml; 5μl). Mice were perfused one hour after OVA injection. Representative stereomicroscopy images showing whole brain OVA distribution from the distal part of the middle cerebral artery (MCA), and quantification of both perivascular and cellular OVA distribution. Scale bars, 1mm and 200μm (inset). n = 6 mice. g, Experimental schematic: WT mice received an i.c.m. injection of FITC Dextran (FITCDex; 4kDa; 10mg/ml; 5μl) and brain were harvested one hour later. Brain coronal sections were stained for anti-CD206 (cyan) and DAPI. Scale bars, 2mm and 50μm (insets). h, Experimental schematic: WT mice received an intrastriatal (i.s.) injection of a cocktail containing 0.5μl of FITC-Dex (10mg/ml; green) and 0.5μl of OVA (1mg/ml; magenta) and brains were harvested one hour later. Brains were then stained for anti-CD206 (cyan). Scale bars, 2mm and 50μm (insets). i, Mice received an i.s. injection of A488-OVA (green) and an i.c.m. injection of A647-OVA (magenta) one hour later. Mice were perfused one hour later (two hours after the i.s. injection). Some cells sampled both i.s. and i.c.m. OVAs. Scale bars, 2mm and 100μm (inset). All data are presented as mean values +/− SEM.
Extended Data Fig 2. Effect of PBM depletion.
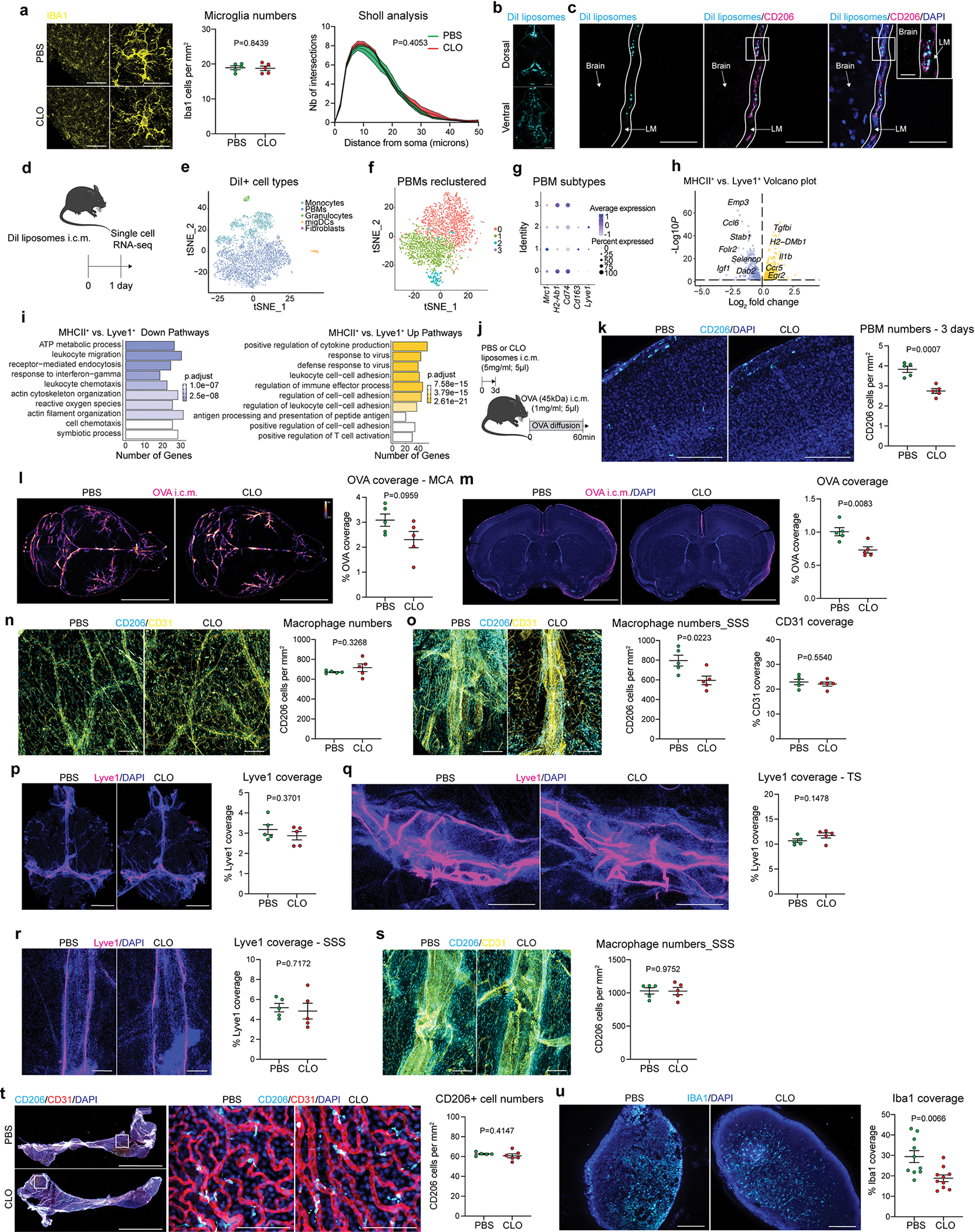
a, WT mice received an i.c.m. injection of clodronate-loaded liposomes (CLO) or PBS-loaded liposomes (PBS). Microglial cells were identified using anti-IBA1 staining (yellow). Cells that were not in the leptomeninges and CD206− were used for quantifications of cell numbers and Sholl analysis. Scale bars, 100μm and 20μm. n = 5 mice/group, two-tailed unpaired Welch’s t-test; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. b, WT mice received an i.c.m. injection of DiI-liposomes (5mg/ml; 5μl) and mice were perfused 24 hours later. Representative images showing DiI-liposome coverage in whole brains. Scale bar: 2mm. c, Representative images showing Dil-liposome (cyan) uptake by CD206+ PBMs (magenta) in leptomeninges (LM) co-stained for DAPI. Scale bars: 50μm and 10μm (inset). d, Experimental schematic: Twenty-four hours after i.c.m. injection of DiI-liposomes, leptomeninges were harvested, DiI-positive cells were sorted and single-cell RNA sequencing was performed. e, tSNE plot showing Dil-positive cells: monocytes, PBMs, granulocytes, migratory dendritic cells (migDCs), fibroblasts and NK/T cells. f, Single-cell RNA sequencing demonstrating 4 PBM clusters. g, Dot plots showing Mrc1, H2-Ab1, Cd74, Cd163 and Lyve1 gene expression in the 4 PBM subtypes. h, Volcano plot corresponding to down- and up-regulated genes comparing MHCIIhi vs. Lyve1hi PBMs. F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P values. i, GO Pathway analysis showing up- and down-regulated pathways comparing MHCIIhi vs. Lyve1hi PBMs. Over-representation test. j. Experimental schematic: WT mice received an i.c.m. injection of CLO or PBS liposomes. OVA was injected i.c.m. three days later and mice were perfused one hour later. k. Representative images showing CD206+ PBMs (cyan) on brain coronal section co-stained for DAPI, and corresponding quantification. Scale bar, 200μm. n = 5 mice/group; two-tailed unpaired Welch’s t-test. l, Representative images and quantification of OVA distribution in whole brains one hour after OVA injection. Scale bar, 5mm. m, Representative images and quantification of OVA coverage in brain coronal sections. Scale bar, 2mm. For l and m: n = 5 mice/group; two-tailed unpaired Welch’s t-test. n, Representative images from non-superior sagittal sinus region of the dura mater three days after CLO treatment co-stained for anti-CD206 (cyan) and anti-CD31 (yellow) and corresponding quantifications. o, Representative images from the superior sagittal sinus region from the dura mater three days after CLO treatment co-stained for anti-CD206 (cyan) and anti-CD31 (yellow), and corresponding quantifications. For n and o: Scale bars, 200μm; n = 5 mice/group; two-tailed unpaired Welch’s t-test. p-u, WT mice received an i.c.m. injection of CLO or PBS liposomes. Tissues were harvested one week later. p, Representative images of total Lyve1 (magenta) coverage in the dura mater co-stained for DAPI and corresponding quantification. Scale bar, 3mm. q, High magnification images showing Lyve1 staining at the transverse sinus level co-stained for DAPI and corresponding quantification. Scale bar, 500μm. r, High magnification images showing Lyve1 staining at the superior sagittal sinus level co-stained for DAPI and corresponding quantification. Scale bar, 200μm. s, High magnification images showing CD206 staining (cyan) co-stained for anti-CD31 (yellow) and corresponding quantification. Scale bar, 500μm. For p-s: n = 5 mice/group; two-tailed unpaired Welch’s t-test. t, Representative images from lateral choroid plexuses whole mounts co-stained for anti-CD206 (cyan), anti-CD31 (red) and DAPI with corresponding high magnifications and quantification. Scale bars, 2mm and 200μm (insets). n = 5 mice treated with PBS, and 6 mice treated with CLO; two-tailed unpaired Welch’s t-test. u, Representatives images from deep cervical lymph nodes co-stained for anti-IBA1 (cyan) and DAPI and corresponding quantification. Scale bar, 200μm. n = 10 mice/group; two-tailed unpaired Welch’s t-test. All data are presented as mean values +/− SEM.
Extended Data Fig 3. CSF flow after PBM depletion.
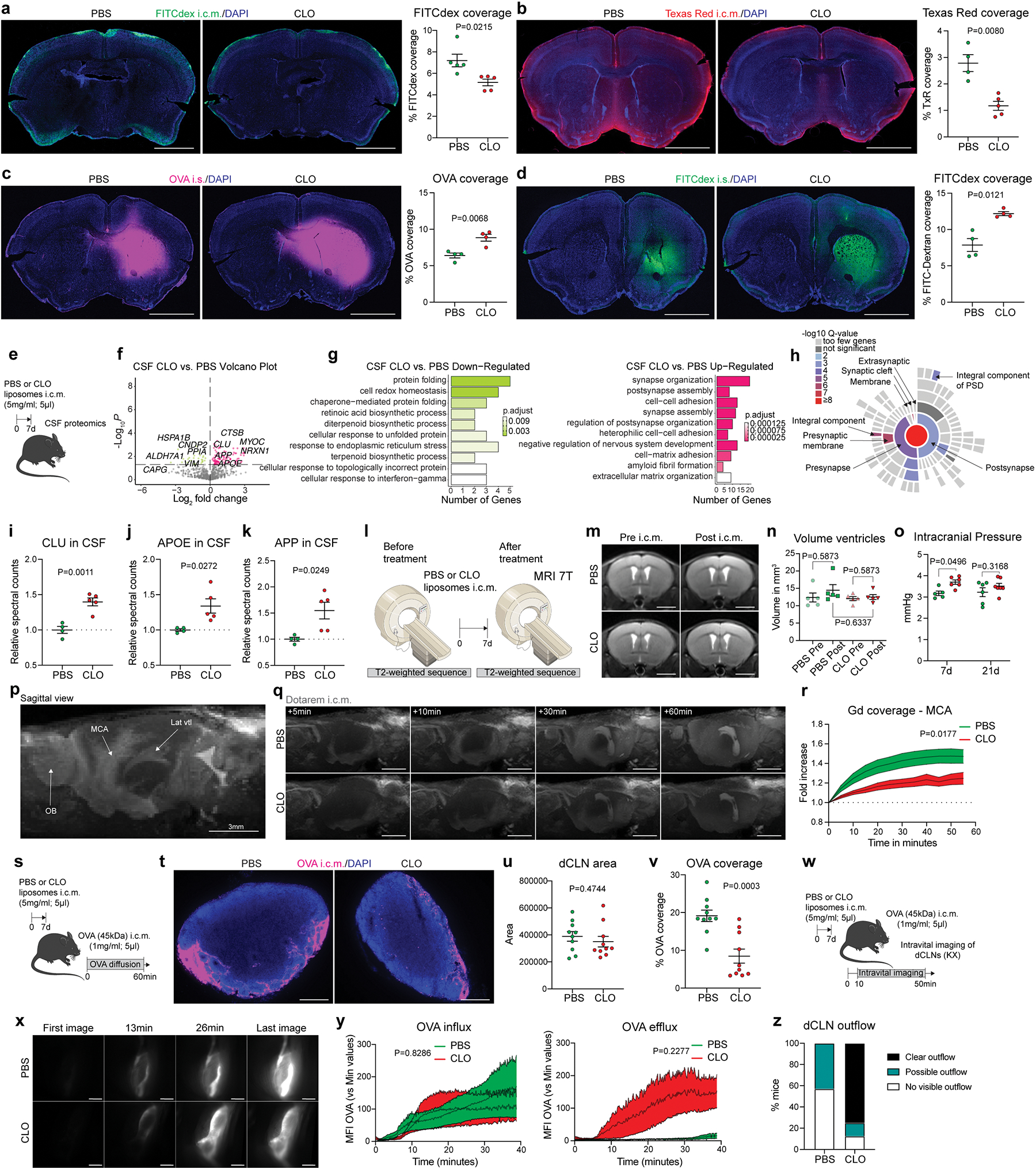
a, One week after PBM depletion, mice received an i.c.m. injection of FITC-Dextran (FITCDex; 4kDa; 5μl), brains were harvested one hour later and FITCDex (green) coverage was measured on coronal sections co-stained for DAPI. Representative images and corresponding quantifications are shown. Scale bar, 2mm. n = 5 mice/group; two-tailed unpaired Welch’s t-test. b, One week after PBM depletion, mice received an i.c.m. injection of Texas Red (3kDa; 5μl), brains were harvested one hour later and Texas Red (red) coverage was measured on coronal sections co-stained for DAPI. Representative images and corresponding quantifications are shown. Scale bar, 2mm. n = 4 mice treated with PBS, and 5 mice treated with CLO; two-tailed unpaired Welch’s t-test. c, One week after PBM depletion, mice received an intrastriatal injection of OVA (45kDa; 1μl) and brains were harvested one hour later. Representative images and corresponding quantifications are shown. Scale bar, 2mm. n = 4 mice treated with PBS, and 5 mice treated with CLO; two-tailed unpaired Welch’s t-test. d, One week after PBM depletion, mice received an intrastriatal (i.s.) injection of FITC-Dextran (FITCdex; 4kDa; 1μl) and brains were harvested one hour later. Representative images and corresponding quantifications are shown. Scale bar, 2mm. n = 4 mice/group; Two-tailed unpaired Welch’s t-test. e, One week after CLO or PBS liposome injection, mice were anesthetized, and a glass capillary was inserted i.c.m. to collect CSF for proteomic analyses. f, Volcano plot corresponding to down- and up-regulated proteins in CSF comparing PBM-depleted and control mice. F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P values. g, Corresponding GO Pathway analysis showing down- and up-regulated pathways in PBM-depleted and control mice. Over-representation test. h, Sunburst plot representing the location of the upregulated CSF-derived neuronal/synaptic-related proteins after PBM depletion. i-k, Quantification of relative spectral counts for i, Clusterin (CLU); j, Apolipoprotein E (APOE) and k, Amyloid Precursor Peptide (APP). For e-k: n = 4 mice treated with PBS, and 5 mice treated with CLO; two-tailed unpaired Welch’s t-test. l, MRI based T2-weighted anatomical sequences were performed before and one week after PBM depletion. m, Representative T2 images showing lateral ventricles (in hypersignal) before and after PBM depletion. Scale bar, 2mm. n, Quantification of ventricle volume in mm3. n = 5 mice/group; one-way ANOVA with Tukey multiple comparisons test. o, Intracranial pressure was measured one- (7d) and three (21d) weeks after PBM depletion. n = 5 mice treated with PBS, 7 mice treated with CLO at 7d; 6 mice treated with PBS, and 7 mice treated with CLO at 21d; two-way ANOVA with Sidak’s multiple comparisons test. p, Sagittal view of a T1-FLASH 3D image showing Dotarem (0.754kDa; 5μl) accumulation in different brain compartments, including the olfactory bulbs (OB), the lateral ventricles (Lat vtl) and the middle cerebral artery (MCA). Scale bar, 3mm. q, Representative T1-FLASH 3D images showing Dotarem distribution at the MCA level over time. Scale bar, 3mm. r, Quantification of Dotarem signal fold increase over an hour. n = 5 mice treated with PBS, and 7mice treated with CLO; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. s, One week after CLO or PBS liposome injection, mice received an i.c.m. injection of OVA, and deep cervical lymph nodes (dCLNs) were harvested one hour later. t, Representative images showing OVA coverage on dCLN sections. Scale bar, 200μm. u, Quantification of dCLN area. v, Quantification of OVA coverage. For u and v: n = 10 mice/group; two-tailed unpaired Welch’s t-test. w, One week after CLO or PBS liposome injection, mice received an i.c.m. injection of OVA and then placed in supine position under the stereomicroscope for dynamic imaging of OVA diffusion in the exposed lymph nodes. x, Representative images showing OVA coverage in dCLNs over time. Scale bar, 500μm. y, Quantifications of both OVA influx (left) and efflux (right) over time. n = 3 mice treated with PBS, and 7 mice treated with CLO; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. z, Proportion of mice that showed OVA outflow from dCLNs. All data are presented as mean values +/− SEM.
Extended Data Fig 4. In vivo recording of tracer diffusion at the middle cerebral artery (MCA) level and evaluation of the perivascular space.
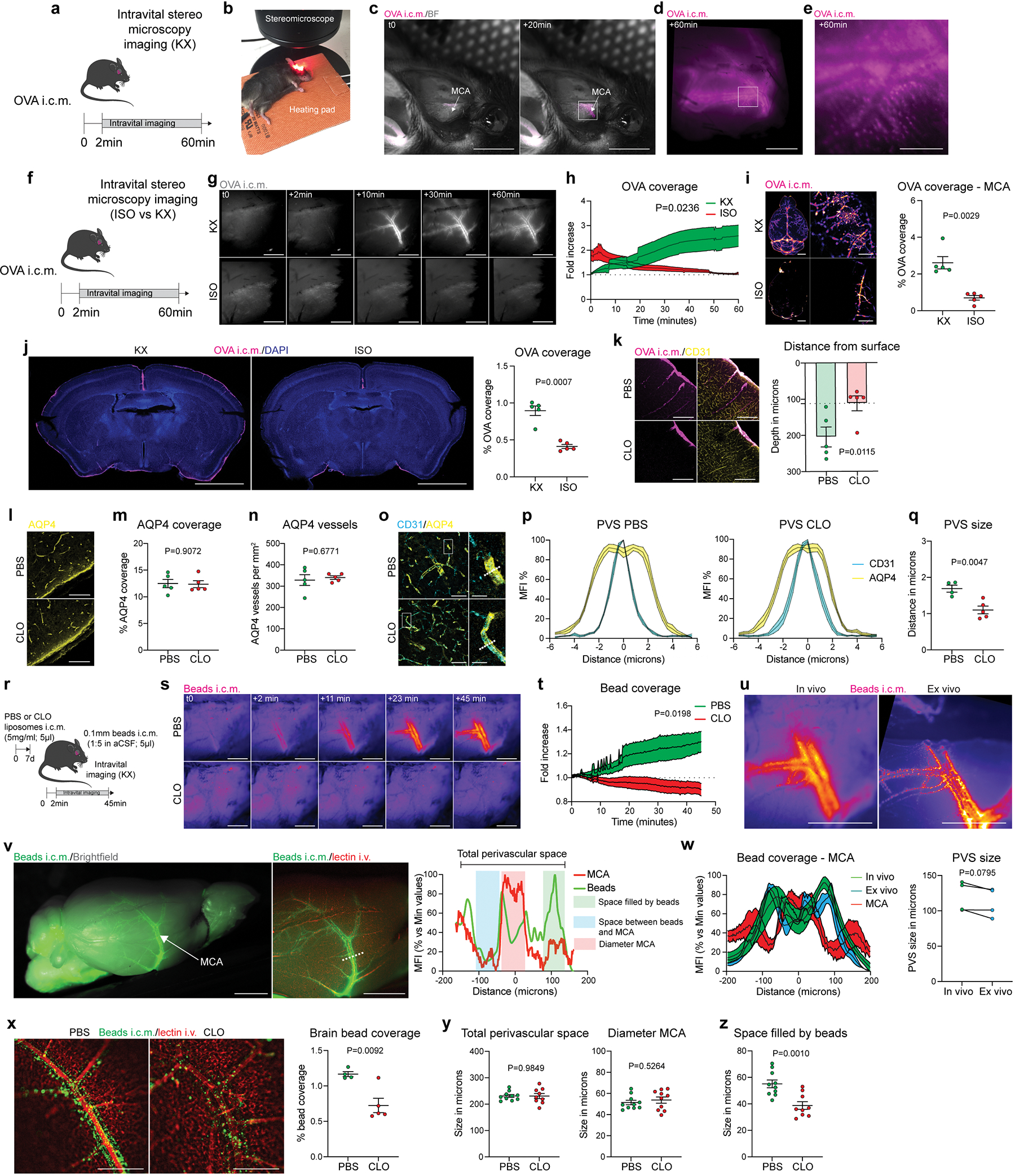
A, Experimental schematic: One week after CLO or PBS liposome injection, mice were placed in a stereotaxic frame, the top and the right side of the head were shaved, the skin was incised, and the right temporalis muscle was gently removed. b, After cleaning the area with a cotton bud, mice received an i.c.m. injection of OVA and were immediately placed on their side under the stereomicroscope. c, Example of OVA distribution at the middle cerebral artery level (MCA) before and 20min after injection. Scale bar, 5mm. d, Inset of c. Higher magnification image showing OVA distribution one hour after injection. The tracer is located around the vessel, at the perivascular level. Scale bar, 1mm. e, High magnification from d showing that OVA can be sampled by perivascular cells. Scale bar, 100μm. f, Experimental schematic: mice were anesthetized with either KX or isoflurane (4% induction, 1–2% during dynamic imaging) and received an i.c.m. injection of OVA. Mice were maintained with the same anesthesia regime during the dynamic imaging. g, Representative images showing OVA distribution over time. Scale bar, 1mm. h, Quantification of OVA coverage over time. n = 5 mice/group; two-way ANOVA mixed-effects analysis (30 last min only). i and j, Mice were then perfused and whole brains were imaged by stereomicroscopy, then the brains were sliced and analyses were made on brain coronal sections. i, Representative images showing OVA distribution and quantification in whole brains and a zoom on the middle cerebral artery. Scale bars, 2mm (left panels) and 1mm (right panels). j, Representative images showing OVA coverage on brain coronal sections. Scale bar, 2mm. For i and j, n = 5 mice/group; two-tailed unpaired Welch’s t-test. k, One week after PBM depletion and one hour after i.c.m. injection of OVA, mice were perfused, brains were extracted, and OVA distribution was analyzed on coronal sections stained for DAPI, and corresponding quantification of OVA depth distribution from the brain surface. Scale bars, 100μm and 50μm (insets). n = 5 mice/group; two-tailed unpaired Welch’s t-test. l, Representative images showing anti-aquaporin 4 (AQP4) staining. Scale bar, 50μm. m, Quantification of AQP4 coverage. n, Quantification of AQP4+ blood vessels. For m and n, n = 5 mice/group; two-tailed unpaired Welch’s t-test. o, Brain sections were co-stained for anti-AQP4 (yellow) and anti-CD31 (cyan), and perpendicular lines to blood vessels were used to measure the perivascular space. Scale bars, 50μm and 10μm (insets). p, Representation of the perivascular space (PVS) in PBS (left) and CLO (right) treated mice. q, Quantification of perivascular space. n = 4 mice treated with PBS, and 5 mice treated with CLO; two-tailed unpaired Welch’s t-test. r, One week after PBM depletion, mice received an i.c.m. injection of fluorescent beads (0.1μm thick; 5μl) and then were immediately placed on their side under the stereomicroscope for dynamic bead imaging at the MCA level. s, Representative images showing bead distribution over an hour at the proximal part of the MCA. Scale bar, 1mm. t, Quantification of bead coverage at the MCA level over time. n = 4 mice treated with PBS, and 7 mice treated with CLO; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. u, Representative images showing beads located at the MCA perivascular space in vivo (left) and ex vivo after perfusion with PBS and post-fixation in 4% PFA (right) from the same mouse. Scale bar, 1mm. v, Representative images from extracted whole brain showing bead repartition at low (left image) and higher magnification (right image) and corresponding plot profile. The beads (green line) are located outside of the MCA (red line), at the perivascular level. Scale bar, 2mm. w, Measure of the perivascular space (PVS) between in vivo and ex vivo from the same mice. n = 4 mice; Two-tailed paired t-test. x, Representative images showing ex vivo bead repartition at the MCA level in PBM-depleted mice and PBS-treated control mice, and corresponding quantification of bead coverage. Scale bar, 500μm; n = 4 mice treated with PBS, and 5 mice treated with CLO; two-tailed unpaired Welch’s t-test. y, Quantifications of total perivascular space (space between the two sides of the MCA where beads were found to accumulate) and MCA diameter (identified by the i.v. lectin injection). z, Quantification of the functional space where beads were found to be accumulated. For y and z: n = 10 mice treated with PBS, and 9 mice treated with CLO; two-tailed unpaired Welch’s t-test. All data are presented as mean values +/− SEM.
Extended Data Fig. 5. PBM depletion has mild effect on mouse behavior and no effect on vital signs.
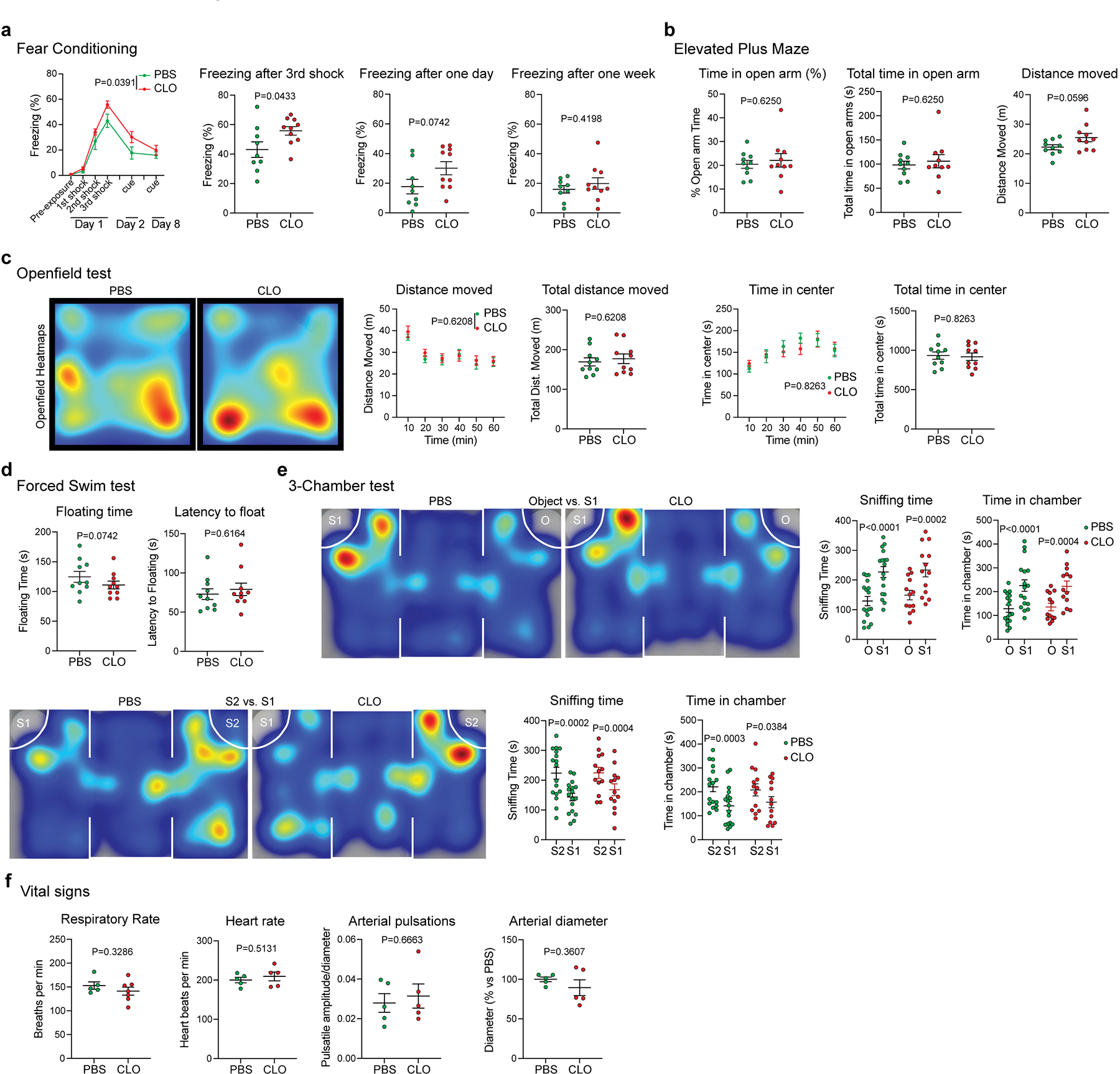
One week after PBM depletion, mice underwent a battery of different behavioral tests. a, Cued fear conditioning: quantification of the percentage of time spent freezing during the three shocks the first day, and exposure to conditioned clues at one- and seven days after fear conditioning. b, Elevated plus maze: quantifications of the percentage of time spent in the open arm (left), the total time spent in the open arm (middle) and the total distance moved (right). c, Open field test: quantification of the distance moved over an hour, the total distance moved, the time spent in the center of the box over an hour and the total time spent in the center of the box. d, Forced swim test: quantification of the total floating time (left) and the latency to float (right). For a-d: n = 10 mice/group; two-tailed unpaired Welch’s t-test. e, Three-chamber test: mice were first exposed to a mouse (S1) or an object (O), and then to a previously-exposed mouse (S1) or a new mouse (S2). Quantifications of the total sniffing time and the total time spent in the chamber for the two tests. n = 17 mice treated with PBS, and 13 mice treated with CLO; two-tailed unpaired Welch’s t-test. f, One week after PBM depletion, respiratory rate, heart rate, arterial pulsation and diameter were monitored. n = 5 mice/group; two-tailed unpaired Welch’s t-test. All data are presented as mean values +/− SEM.
Extended Data Fig 6. PBMs and extracellular matrix remodeling.
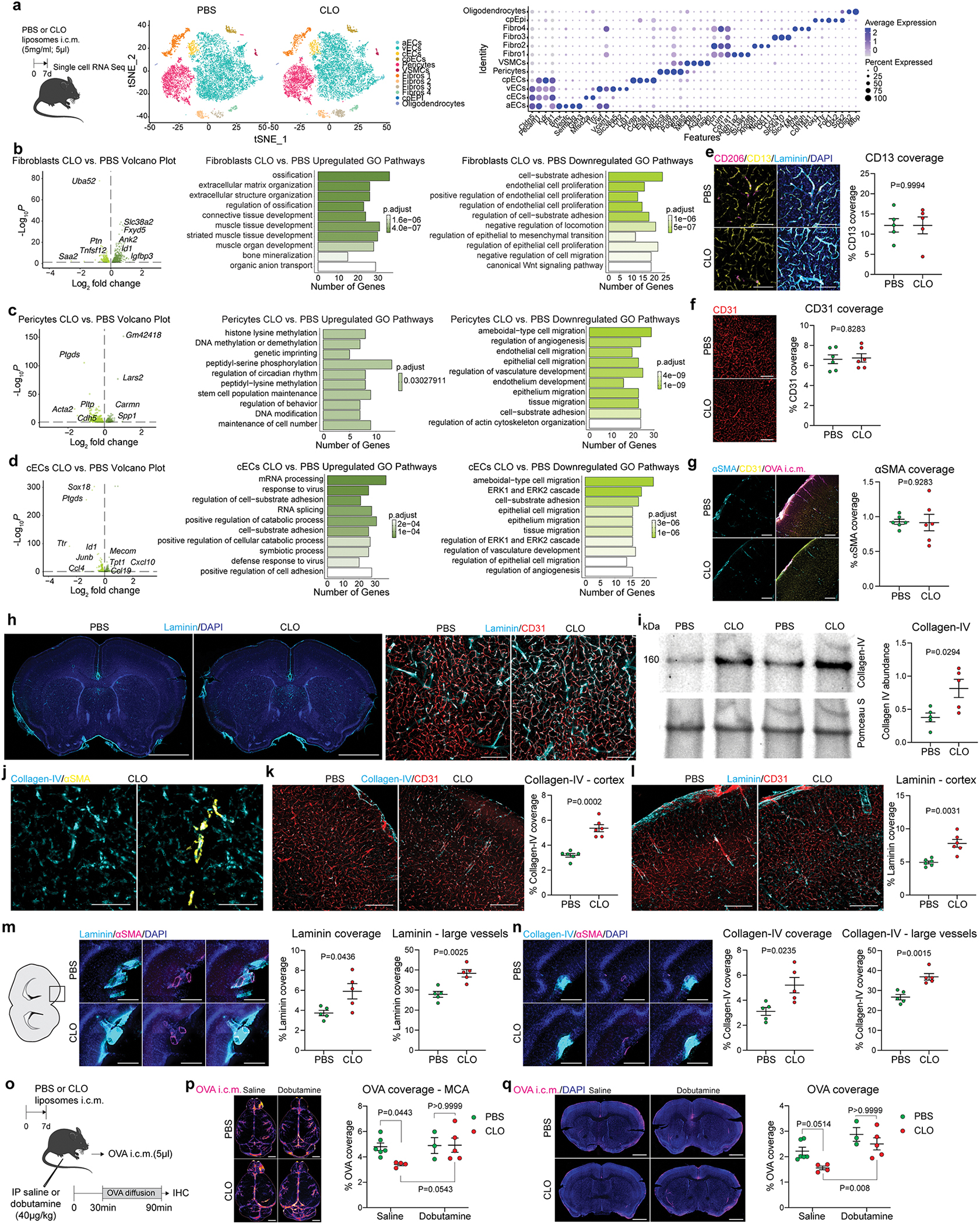
a, One week after CLO or PBS liposome injection, mice were perfused, brains were extracted, lateral choroid plexuses were removed, CD45-CD13+ and CD45-CD31+ cells were sorted and used for single-cell RNA sequencing. Nine different cell types were identified based on canonical markers. b, Volcano plot corresponding to up- and down-regulated genes comparing fibroblasts in PBM-depleted mice and PBS-treated control mice, and corresponding GO Pathway analyses showing up- (left) and down-regulated (right) pathways. c, Volcano plot corresponding to up- and down-regulated genes comparing pericytes in PBM-depleted mice and PBS-treated control mice, and corresponding GO Pathway analyses showing up- (left) and down-regulated (right) pathways. d, Volcano plot corresponding to up- and down-regulated genes comparing capillary endothelial cells (cECs) in PBM-depleted mice and PBS-treated control mice, and corresponding GO Pathway analyses showing up- (left) and down-regulated (right) pathways. For b-d, Volcano plots: F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P values; GO-pathways analyses: over-representation test. e, Representative images showing cortical brain sections stained for anti-CD13 (mural cells, yellow) co-stained for anti-CD206 (magenta), anti-Laminin (cyan) and DAPI, and corresponding quantification. Scale bar, 100μm; n = 5 mice/group; two-tailed unpaired Welch’s t-test. f, Representative images showing cortical brain sections stained for anti-CD31 (endothelial cells) and corresponding quantification. Scale bar, 200μm; n = 6 mice/group; two-tailed unpaired Welch’s t-test. g, Representative images showing cortical brain sections of mice that were i.c.m. injected with OVA (magenta), stained for anti-αSMA (vascular smooth muscle cells, cyan) and co-stained for anti-CD31 (yellow), and corresponding quantification. Scale bar, 200μm; n = 6 mice/group; two-tailed unpaired Welch’s t-test. h, One week after CLO or PBS liposome injection, brain coronal sections were stained for anti-Laminin (cyan) and DAPI (blue). Scale bar, 2mm. k, High magnification images showing Laminin (cyan) in association with CD31+ blood vessels (red). Scale bar, 200μm. i, Representative Western blot images of Collagen-IV (160kDa) and Ponceau S from isolated brain blood vessels one week after PBM depletion, and corresponding quantification. n = 5 mice/group; two-tailed unpaired Welch’s t-test. j, Representative images showing Collagen-IV (cyan) deposition at both αSMA+ (arteries/arterioles; yellow) and αSMA− blood vessels. Scale bar, 200μm. k and l, Representative images from mouse cortex showing accumulation of k: Collagen-IV; and l: Laminin (cyan), co-stained for anti-CD31 (red), and respective quantifications. Scale bars, 200μm; n = 6 mice/group; two-tailed unpaired Welch’s t-test. m, High magnification images showing Laminin (cyan) in association with αSMA+ surface and penetrating large blood vessels (magenta), and corresponding quantifications. n, High magnification images showing Collagen-IV (cyan) in association with αSMA+ surface and penetrating large blood vessels (magenta), and corresponding quantifications. For m and n: Scale bar, 200μm; n = 5 mice/group; two-tailed unpaired Welch’s t-test. o, Experimental schematic: mice received an i.p. injection of dobutamine (40μg/kg) or saline 30min prior to receiving an i.c.m. injection of OVA (1mg/ml; 5μl). Mice were perfused one hour later. p, Representative images showing OVA distribution in whole brains and corresponding quantifications. q, Representative images showing OVA coverage on brain coronal sections and corresponding quantifications. For p and q, Scale bars, 2mm; n = 6 mice treated with PBS, 4 mice treated with CLO in saline group; 3 mice treated with PBS, and 5 mice treated with CLO in dobutamine group; two-way ANOVA with multiple comparisons. All data are presented as mean values +/− SEM.
Extended Data Fig 7. Lyve1+ PBMs drive CSF flow dynamics.
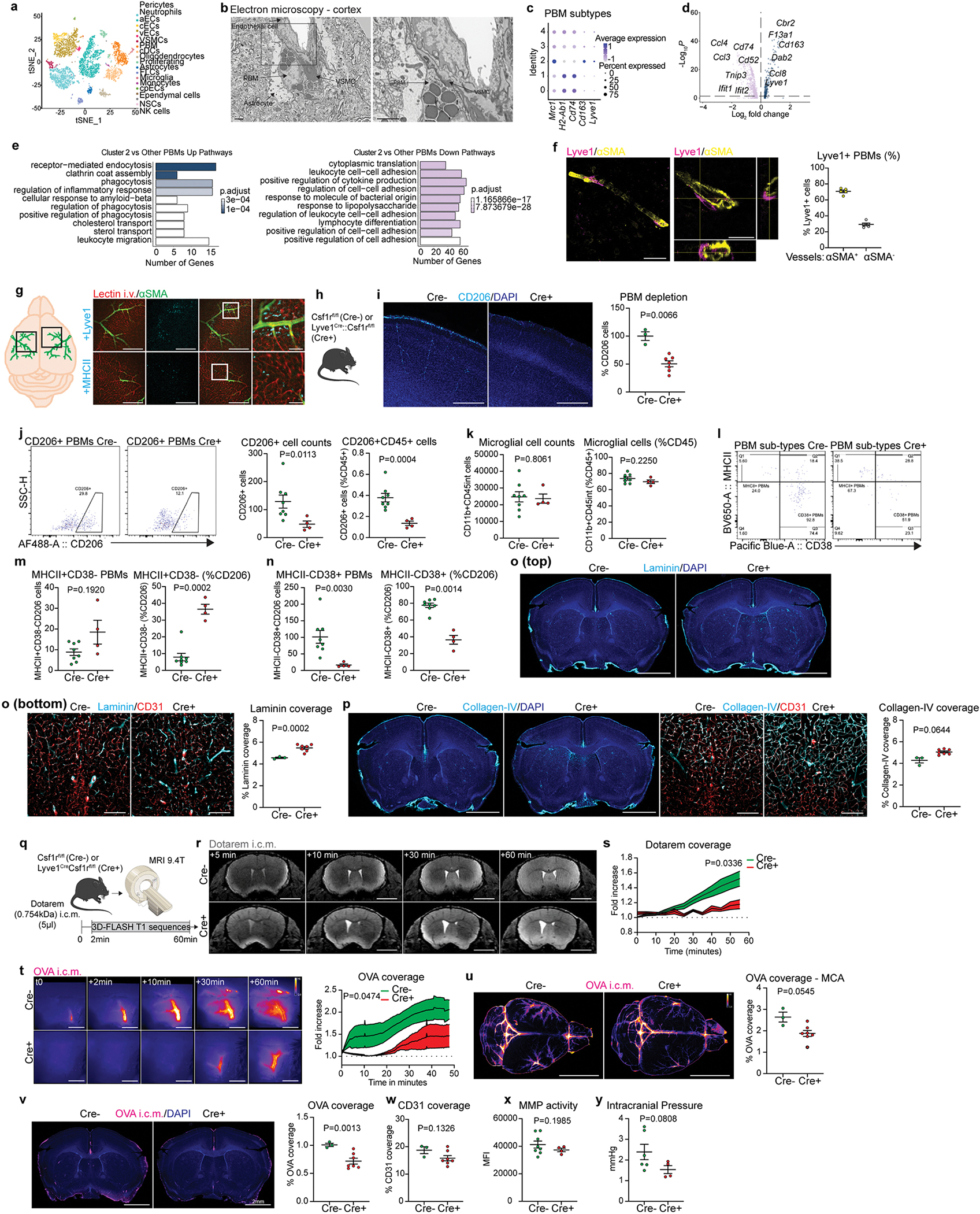
a, Single-cell RNA sequencing of the whole brain was performed. Eighteen cell types were identified based on canonical markers. b, Scanning electron microscopy image from a mouse cortex suggesting interactions between a PBM and a vascular smooth muscle cell (VSMC). Scale bars, 2μm. c, Dot plot for Mrc1 (CD206), H2-Ab1 (MHCII), Cd74, Cd163 and Lyve1 expression in each PBM cluster. d and e, Volcano plot and GO pathway analysis showing up- and down-regulated pathways in PBM cluster 2 versus other PBM clusters. Volcano plots: F-test with adjusted degrees of freedom based on weights calculated per gene with a zero-inflation model and Benjamini-Hochberg adjusted P values; GO-pathways analyses: over-representation test. f, Representative images suggesting interactions between Lyve1+ PBMs (magenta) and αSMA+ (yellow) VSMC. Scale bar, 200μm and 50μm (inset). g, Quantification of Lyve1+ cells associated or not with αSMA+ blood vessels. n = 5 mice. h, Mice received an i.v. injection of lectin and were perfused few minutes later. Whole brains were extracted, post-fixed with 4% PFA, and stained for anti-αSMA (green) and anti-Lyve1 (top panels) or anti-MHCII (bottom panels) (cyan). Scale bars: 1mm and 200μm (insets). i, Characterization of PBM depletion in Lyve1Cre::Csf1rfl/fl mice (Cre+) in brain coronal sections using CD206 staining (co-stained for DAPI) versus control littermates not expressing Cre (Csf1rfl/fl; Cre−). Scale bar, 500μm; n = 7 Cre− mice, and 3 Cre+ mice; two-tailed unpaired Welch’s t-test. j, Flow cytometry panels showing CD206+ cells in Csf1rfl/fl mice (Left; Cre−) and Lyve1Cre::Csf1rfl/fl mice (Right; Cre+), and quantifications of CD206+ PBM cell numbers (left), and frequency of CD206+ PBMs from total CD45+ cells (right). k, Quantification of CD11b+CD45int microglial cell numbers (left), and frequency of CD11b+CD45int microglial cells from total CD45+ cells (right). l, Flow cytometry panels showing CD38+ and/or MHCII+ CD206+ PBMs in Csf1rfl mice (Left; Cre−) and Lyve1Cre::Csf1rfl mice (Right; Cre+). m, Quantification of MHCII+CD38− PBMs (left), and frequency of MHCII+CD38− PBMs from total CD206+ cells (right). n, Quantification of MHCII-CD38+ PBMs (left), and frequency of MHCII-CD38+ PBMs from total CD206+ cells (right). For i-n: n = 8 Cre− mice, and 4 Cre+ mice; two-tailed unpaired Welch’s t-test. o, (Top) Representative images of coronal brains sections from Lyve1Cre::Csf1rfl/fl (Cre+) and Csf1rfl/fl (Cre−) mice co-stained for anti-Laminin (cyan) and DAPI. (Bottom) High magnification images showing Laminin location at the vicinity of CD31+ blood vessels (red), and corresponding quantification. Scale bars, 2mm (top) and 200μm (bottom). p, (Left) Representative images of coronal brains sections from Lyve1Cre::Csf1rfl/fl (Cre+) and Csf1rfl/fl (Cre−) mice co-stained for anti-Collagen-IV (cyan, right panels) and DAPI. (Right) High magnification images showing Collagen-IV location at the vicinity of CD31+ blood vessels (red), and corresponding quantification. Scale bars, 2mm (left) and 200μm (Right). q, Three-month-old Cre+ and Cre− mice received an i.c.m. injection of Dotarem (0.754kDa; 5μl) and were immediately placed in prone position into the MRI device for dynamic imaging. r, Representative brain coronal images showing Dotarem distribution over an hour. Scale bar, 3mm. s, Quantification of Dotarem signal fold increase over time, n = 8 Cre− mice, and 5 Cre+ mice; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. t, In vivo imaging of OVA coverage at the MCA level in Lyve1Cre::Csf1rfl/fl (Cre+) and Csf1rfl/fl (Cre−) mice, with corresponding representative images and quantification. Scale bar, 1mm; n = 7 Cre− mice, and 5 Cre+ mice; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. u, Representative images showing OVA distribution in whole brains in Lyve1Cre::Csf1rfl/fl (Cre+) and Csf1rfl/fl (Cre−) mice. Scale bar, 5mm. v, OVA coverage in coronal sections of Lyve1Cre::Csf1rfl/fl (Cre+) and Csf1rfl/fl (Cre−) mice. Scale bar, 2mm. For u and v: n = 7 Cre− mice, and 3 Cre+ mice; two-tailed unpaired Welch’s t-test. x, Quantification of CD31 coverage in Lyve1Cre::Csf1rfl/fl (Cre+) and Csf1rfl/fl (Cre−) mouse brain sections. n = 7 Cre− mice, and 3 Cre+ mice; two-tailed unpaired Welch’s t-test. y, Quantification of MMP activity from Lyve1Cre::Csf1rfl/fl (Cre+) and Csf1rfl/fl (Cre−) mice measured by fluorescence spectrometry after 15 min of incubation. n = 8 Cre− mice, and 4 Cre+ mice; two-tailed unpaired Welch’s t-test. z, Quantification of intracranial pressure from Lyve1Cre::Csf1rfl/fl (Cre+) and Csf1rfl/fl (Cre−) mice. n = 6 Cre− mice, and 4 Cre+ mice; two-tailed unpaired Welch’s t-test. All data are presented as mean values +/− SEM.
Extended Data Fig 8. PBMs in normal aging.
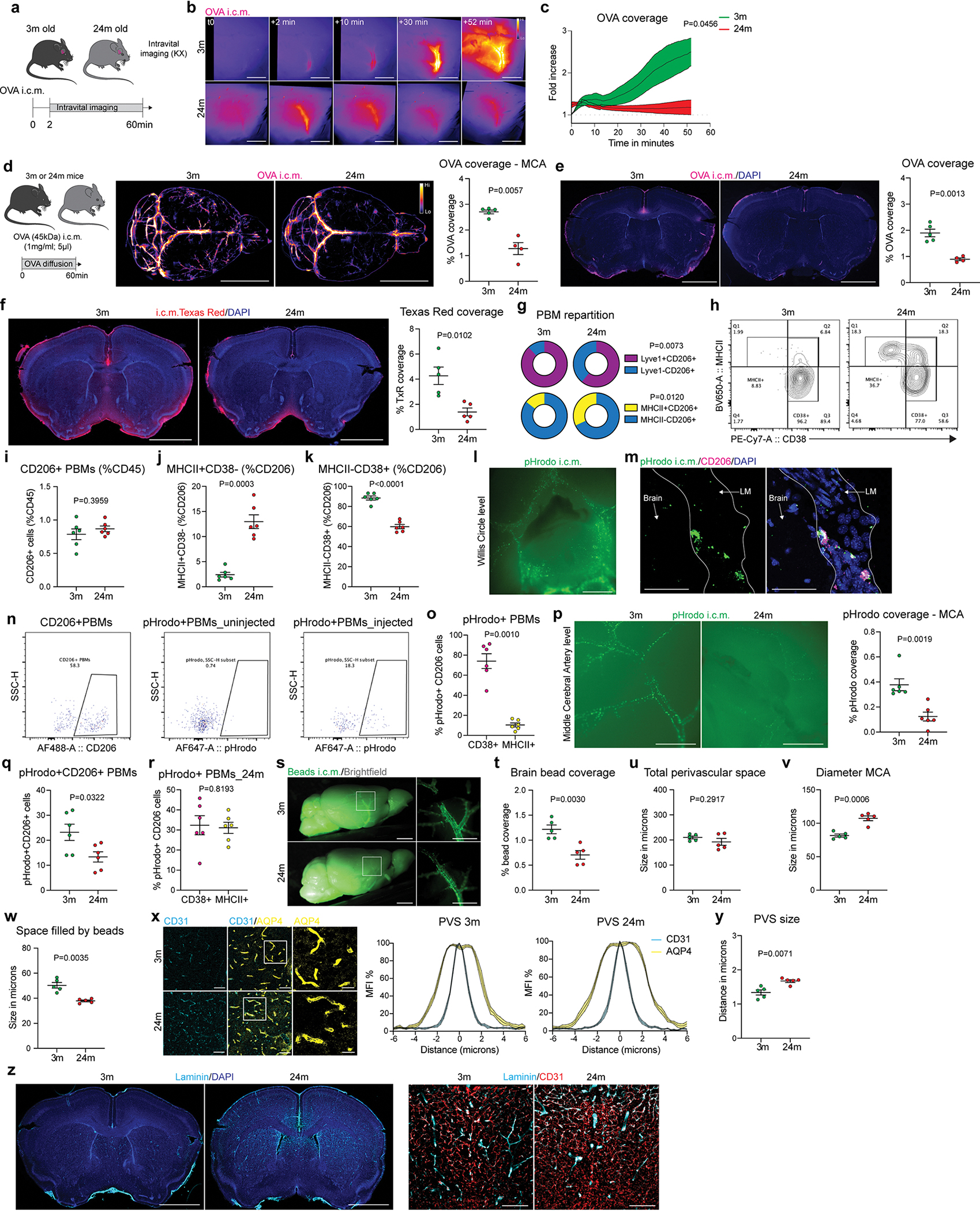
a, Experimental schematic: young adult (3-month-old) and aged (24-month-old) mice received an i.c.m. injection of OVA (45kDa; 1mg/ml; 5μl). Immediately after the injection, mice were placed on their side under the stereomicroscope for dynamic imaging. b, Representative images showing OVA distribution over time. Scale bar, 1mm. c, Quantification of OVA signal fold increase over time in 3m and 24m old mice. n = 5 mice/group; repeated measures 2-way ANOVA with Geisser-Greenhouse correction. d and e, 3m and 24m old mice received an i.c.m. injection of OVA. Mice were perfused one hour later. d, Representative images showing OVA distribution in whole brains in 3m and 24m old mice, and corresponding quantification. Scale bar, 5mm. e, OVA coverage in coronal sections in 3m and 24m old mice, and corresponding quantification. Scale bar, 2mm. For d and e: n = 5 3m mice, and 4 24m mice; two-tailed unpaired Welch’s t-test. f, 3m and 24m old mice received an i.c.m. injection of Texas Red (3kDa; 1mg/ml; 5μl), brains were harvested one hour later. Representative images of Texas Red coverage (red) and corresponding quantification. Scale bar, 2mm. n = 5 mice/group; two-tailed unpaired Welch’s t-test. g, Pie chart representation of the quantification of Lyve1+MHCII− versus Lyve1-MHCII+ PBMs by immunostaining. n = 5 mice/group; two-tailed unpaired Welch’s t-test. h, Representative flow cytometry plots from 3m and 24m old mice showing PBM subtypes, characterized by their expression of CD38 and MHCII. i-k, Quantification of i: CD206+ PBMs; j: MHCII+CD38− PBMs and k: MHCII-CD38+ PBMs. For i-k, n = 6 mice/group; two-tailed unpaired Welch’s t-test. l, 3m and 24m old mice received an i.c.m. injection of pHrodo particles (1μm; 5μl), which became fluorescent only after being phagocytosed. Scale bar, 2mm. m, Representative confocal image showing pHrodo particles being phagocytosed by CD206+ PBMs. Scale bar, 50μm. n, pHrodo-positive PBMs can also be detected by flow cytometry. o, Quantification of pHrodo+MHCII-CD38+ versus pHrodo+MHCII+CD38− PBMs in 3m old mice. n = 6 mice; paired t-test. p, Representative images of 3m and 24m old mice showing pHrodo particle repartition at the MCA level, and corresponding quantification. Scale bar, 2mm. q, Quantification of pHrodo+CD206+ PBMs in 3m and 24m old mice. For p and q: n = 6 mice/group; two-tailed unpaired Welch’s t-test. r, Quantification of pHrodo+MHCII-CD38+ versus pHrodo+MHCII+CD38− PBMs in 24m old mice. n = 6 mice; paired t-test. s, 3m and 24m old mice received an i.c.m. injection of fluorescent beads (0.1μm thick; 5μl). Mice were perfused one hour later. Representative image from extracted whole brain showing bead repartition at low (left images) and higher magnification (insets, right images). Scale bars, 2mm and 1mm (insets). t, Quantification of brain bead coverage. u, Quantifications of total perivascular space (space between the two sides of the MCA where beads were found to accumulate) v, Quantification of MCA diameter (identified by the i.v. lectin injection). w, Quantification of the functional space where beads were found to be accumulated. For t-w: n = 5 mice/group; two-tailed unpaired Welch’s t-test. x, Brain coronal sections from 3m and 24m old mice were co-stained for anti-AQP4 (yellow) and anti-CD31 (cyan) to measure the perivascular space size. Scale bars, 50μm and 10μm (insets), and representation of the perivascular space (PVS) diameter in 3m (middle) and 24m (right) old mice. y, Quantification of perivascular space diameter. n = 5 mice/group; two-tailed unpaired Welch’s t-test. z, (Left) Brain coronal sections were stained for anti-Laminin (cyan) and DAPI (blue). Scale bar, 2mm. (Right) High magnification images showing Laminin (cyan) in association with CD31+ blood vessels (red). Scale bar, 200μm. All data are presented as mean values +/− SEM.
Extended Data Fig 9. M-CSF treatment in old mice.
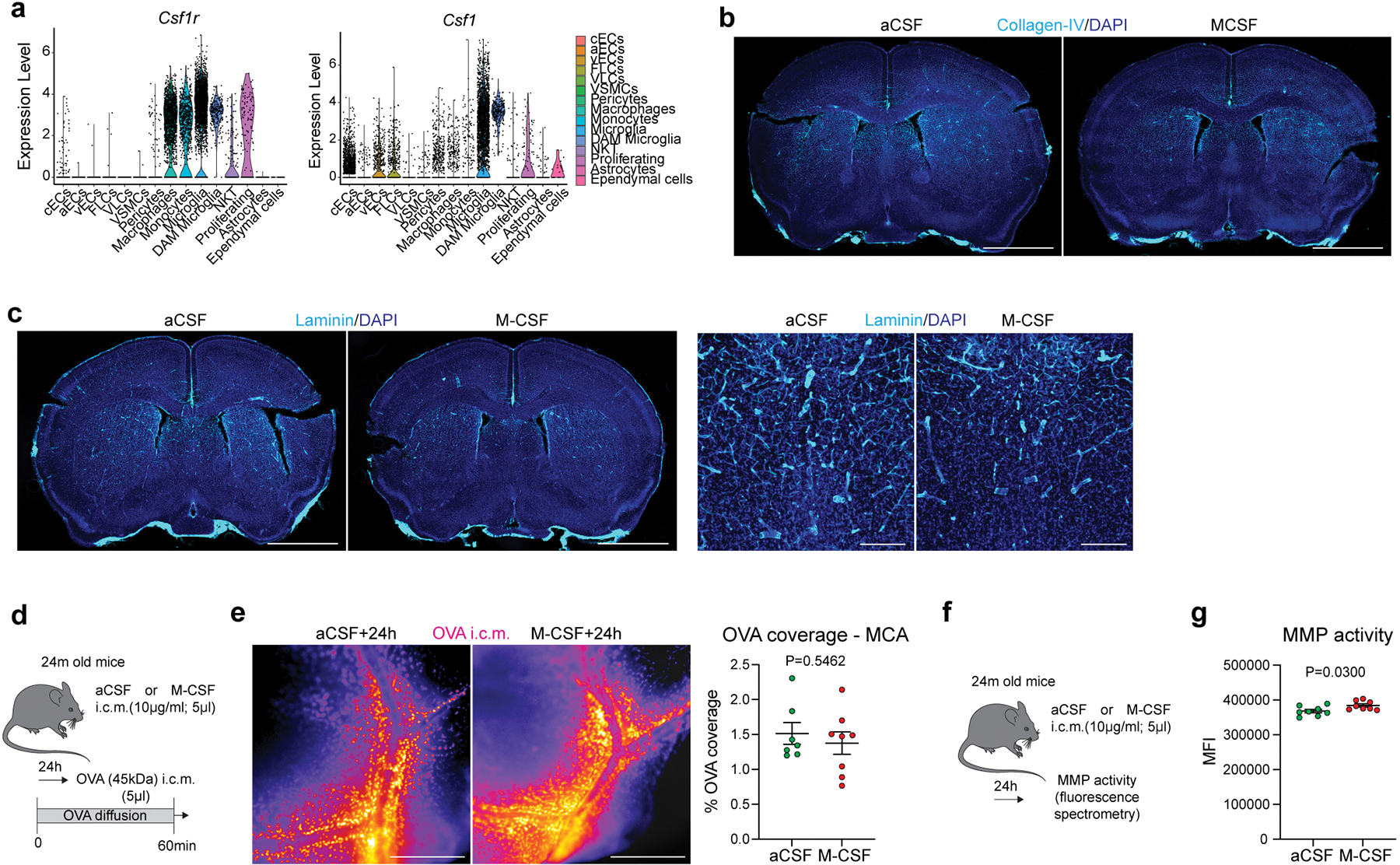
a, Violin plots showing expression of Csf1r mostly by PBMs, monocytes and microglia, as well as expression of Csf1, expressed mostly by endothelial, mural and microglial cells, from the mouse 5xFAD single-cell RNA sequencing dataset. b and c, Brain coronal sections of aged mice six hours after i.c.m. injection of artificial CSF (aCSF) or M-CSF (10μg/ml; 5μl) were stained for b: anti-Collagen-IV (left panels) or c: anti-Laminin (right panels) and co-stained for DAPI, Scale bars, 2mm and 200μm. d, Experimental schematic: 24m old mice received an i.c.m. injection of M-CSF (or aCSF as a control), and mice received an i.c.m. injection of OVA to assess CSF flow 24 hours later. e, Representative images showing OVA coverage at the MCA level, and corresponding quantification. Scale bar, 500μm. n = 7 mice treated with aCSF, and 8 mice treated with M-CSF; two-tailed unpaired Welch’s t-test. f, Experimental schematic: 24m old mice received an i.c.m. injection of M-CSF (or aCSF as a control), and MMP activity was assessed by fluorescence spectrometry 24 hours later. g, Quantification of MMP activity. n = 8 mice/group; two-tailed unpaired Welch’s t-test. All data are presented as mean values +/− SEM.
Extended Data Fig 10. PBMs in an Alzheimer’s disease mouse model and in AD patients.
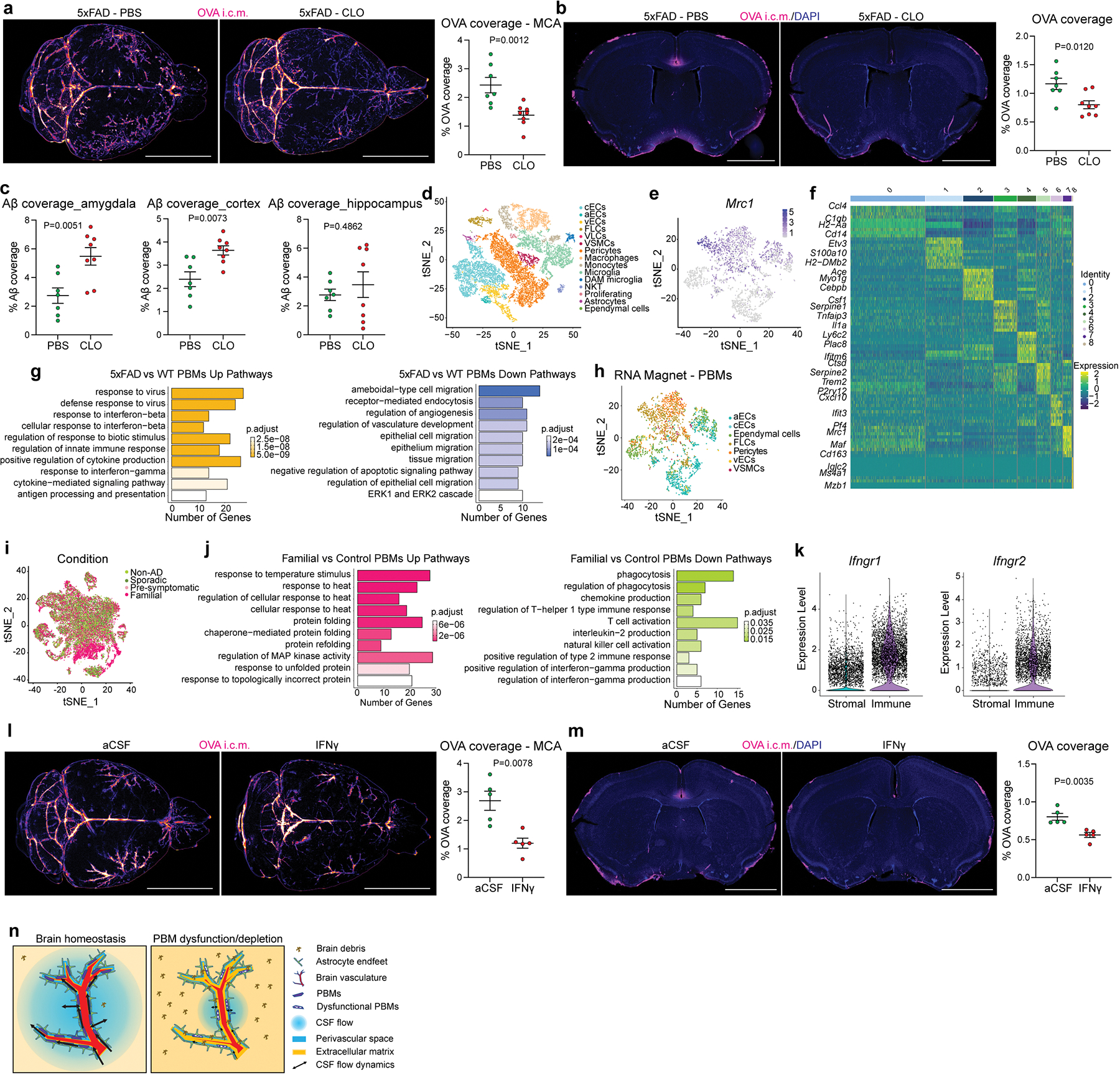
a and b, One month after CLO or PBS liposome injection, 5xFAD mice received an i.c.m. injection of OVA, and brains were analyzed one hour later. a, Representative images and quantification of OVA distribution in whole brains. Scale bar, 2mm. b, Representative images and quantification of OVA coverage on brain coronal sections. Scale bar, 2mm. For a and b, n = 7 5XFAD mice treated with PBS, and 8 5XFAD mice treated with CLO; two-tailed unpaired Welch’s t-test. c, Quantification of Aβ coverage in amygdala, cortex and hippocampus. n = 7 5XFAD mice treated with PBS, and 8 5XFAD mice treated with CLO; two-tailed unpaired Welch’s t-test. d, tSNE plot showing 35 different clusters on the 5xFAD mouse single-cell RNA sequencing dataset, based on expression of CD13, CD31 and CD45. e, Mrc1 expression in macrophage cluster allows PBM identification. f, Heatmap showing top 10 positively differentially expressed genes per cluster by adjusted p-value. g, GO Pathway analysis showing up- and down-regulated pathways in 5xFAD mice compared to their WT littermates. Over-representation test. h, The RNA Magnet algorithm determined that PBMs interact preferentially with vascular smooth muscle cells (VSMCs), pericytes and fibroblast-like cells (FLCs). i, Single-nuclei RNA sequencing on familial, pre-symptomatic, sporadic and non-AD patients. j, GO Pathway analysis showing up- and down-regulated pathways in familial AD patients compared to controls. Over-representation test. k, Gene expression levels of Ifngr1 and Ifngr2 from immune versus stromal cells from the mouse 5xFAD single-cell RNA sequencing dataset. l and m, Wild-type mice received an i.c.m. injection of artificial CSF (aCSF) or interferon gamma (IFNγ, 20ng/ml; 1μl). The same mice received an i.c.m. injection of OVA (1mg/ml; 5μl) 3 hours later and brains were harvested one hour later. l, Representative images and quantification of OVA distribution in whole brains. m, Representative images showing OVA coverage on brain coronal sections and corresponding quantification. For l and m, n = 5 mice/group; two-tailed unpaired Welch’s t-test. n, Proposed model that recapitulates the findings. All data are presented as mean values +/− SEM.
Supplementary Material
Acknowledgements.
We thank S. Smith for editing the manuscript. We thank S. Blackburn, N. Al-Hamadani, X. Wang, and E. Griffin for animal care, and S. Brophy for lab management. This work was supported by grants from the National Institutes of Health/National Institute on Aging (AG034113, AG057496, AG078106), the Cure Alzheimer’s Fund and the Ludwig Foundation to J. K.; AG057777 and AG067764 to O.H.; and AG062734 to C.M.K. O.H. is an Archer Foundation Research Scientist. We thank all the members of the Kipnis laboratory for their valuable comments during numerous discussions of this work. We thank all the members of the Washington University Center for Cellular Imaging core (WUCCI) for their valuable contribution of electron microscopy imaging. We thank the University of Virginia Flow Cytometry Core and from the Sequencing Core for their help with cell sorting and sequencing. We thank all the members of the Washington University Small Animal MR Imaging Facility and the University of Virginia Molecular Imaging Core Facility for their help in Magnetic Resonance Imaging. We acknowledge the expert technical assistance of Dr. Yiling Mi, Petra Erdmann-Gilmore, Alan Davis and Rose Connors for the CSF proteomic experiment performed at the Washington University Proteomics Shared Resource (WU-PSR), R Reid Townsend MD/PhD., Director and, Drs. Robert Sprung and Tim Zhang, Co-directors. The WU-PSR is supported in part by the WU Institute of Clinical and Translational Sciences (NCATS UL1 TR000448), the Mass Spectrometry Research Resource (NIGMS P41 GM103422; R24GM136766) and the Siteman Comprehensive Cancer Center Support Grant (NCI P30 CA091842). We acknowledge the staff of the Neuropathology Cores and other personnel of the Charles F. and Joanne Knight Alzheimer Disease Research Center (P30 AG066444, P01AG026276, P01AG03991). Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer’s Network (DIAN, UF1AG032438) funded by the National Institute on Aging (NIA), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI). This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the Knight ADRC and DIAN (see full list of DIAN consortium members at the end of the manuscript) research and support staff at each of the participating sites for their contributions to this study.
Footnotes
Competing interests.
J.K. is a scientific advisor and collaborator with PureTech. J.K. and A.D. are holding provisional patent applications related to the findings described herein. R.J.B co-founded and is on the scientific advisory board of C2N Diagnostics. C2N Diagnostics has licensed certain anti-tau antibodies to AbbVie for therapeutic development. He receives research support from Biogen, Eisai, and the DIAN-TU Pharma Consortium. He is also an advisor to Amgen and Hoffman La-Roche.
Data sharing:
Code Availability Statement:
Custom code used to analyze the mRNA sequencing data is freely available at https://doi.org/10.5281/zenodo.7047054
Statistical analyses and reproducibility
All data are presented as mean values +/− SEM. All the experiments (except single-cell and single-nuclei RNA sequencing data) have been repeated independently at least two times (biological replicates). Statistical significance was determined using Two-tailed unpaired Welch’s t-test (non-parametric) when comparing two independent groups, or by paired t-test when comparing values from the same group. For comparisons of multiple factors, one- or 2-way ANOVA with appropriate multiple-comparisons tests were used. Statistical analyses were performed using Prism 9.0 (GraphPad software). Exact P values were all provided in figures. We provide all raw data and the statistical analyses in supplementary Prism file. All experiments were done blinded and groups were revealed only after all the analyses were performed.
Dominantly Inherited Alzheimer Network Consortium
Full Names and Credentials
Richard Perrin2,6, Celeste M. Karch5,6, Randall J. Bateman6, Jared Brosch7, Jill Buck7, Marty Farlow7, Bernardino Ghetti7, Jasmeer Chhatwal8, Sarah Adams9, Nicolas Barthelemy9, Tammie Benzinger9, Susan Brandon9, Virginia Buckles9, Lisa Cash9, Charlie Chen9, Jasmin Chua9, Carlos Cruchaga9, Darcy Denner9, Aylin Dincer9, Tamara Donahue9, Anne Fagan9, Becca Feldman9, Shaney Flores9, Erin Franklin9, Nelly Joseph-Mathurin9, Alyssa Gonzalez9, Brian Gordon9, Julia Gray9, Emily Gremminger9, Alex Groves9, Jason Hassenstab9, Cortaiga Hellm9, Elizabeth Herries9, Laura Hoechst-Swisher9, David Holtzman9, Russ Hornbeck9, Gina Jerome9, Sarah Keefe9, Deb Koudelis9, Yan Li9, Jacob Marsh9, Rita Martinez9, Kwasi Mawuenyega9, Austin McCullough9, Eric McDade9, John Morris9, Joanne Norton9, Kristine Shady9, Wendy Sigurdson9, Jennifer Smith9, Peter Wang9, Qing Wang9, Chengjie Xiong9, Jinbin Xu9, Xiong Xu9, Ricardo Allegri10, Patricio Chrem Mendez10, Noelia Egido10, Aki Araki11, Takeshi Ikeuchi11, Kenji Ishii11, 27, Kensaku Kasuga11, Jacob Bechara12, William Brooks12, Peter Schofield12, Sarah Berman13, Sarah Goldberg13, Snezana Ikonomovic13, William Klunk13, Oscar Lopez13, James Mountz13, Neelesh Nadkarni13, Riddhi Patira13, Lori Smith13,Beth Snitz13, Sarah Thompson13, Elise Weamer13, Courtney Bodge14, Stephen Salloway14, Kathleen Carter15, Duc Duong15, Erik Johnson15, Allan Levey15, Lingyan Ping15, Nicholas T Seyfried15, Colleen Fitzpatrick16, Helena Chui17, John Ringman17, Gregory S Day18, Neill Graff-Radford18, Morgan Graham18, Sochenda Stephens18, Chrismary De La Cruz19, Jill Goldman19, Arlene Mejia19, Katie Neimeyer19, James Noble19, Anna Diffenbacher20, Yakushev Igor20, Johannes Levin20, Jonathan Vöglein20, Jane Douglas21, Nick Fox21, Miguel Grilo21, Cath Mummery21, Antoinette O’Connor21, Bianca Esposito22, Alison Goate22, Alan Renton22, Hisako Fujii23, Michio Senda23, Hiroyuki Shimada23, Samantha Gardener24, Ralph Martins24, Hamid Sohrabi24, Kevin Taddei24, Susanne Gräber-Sultan25, Lisa Häsler25, Anna Hofmann25, Mathias Jucker25, Stephan Käser25, Elke Kuder-Buletta25, Christoph Laske25, Oliver Preische25, Christian Haass26, Estrella Morenas-Rodriguez26, Brigitte Nuscher26, Ryoko Ihara27, Akemi Nagamatsu27, Yoshiki Niimi27, Clifford Jack28, Robert Koeppe29, Neal Scott Mason30, Colin Masters31, Ulricke Obermüller32.
Affiliations:
9Washington University in St. Louis School of Medicine, United States
10Institute of Neurological Research Fleni, Buenos Aires, Argentina
11Niigata University, Japan
12Neuroscience Research Australia
13University of Pittsburgh, United States
14Brown University-Butler Hospital, Rhode Island, United States
15Emory University School of Medicine, Atlanta, United States
16Brigham and Women’s Hospital – Massachusetts General Hospital, Boston, United States
17University of Southern California, Los Angeles, United States
18Mayo Clinic Jacksonville, Florida, United States
19Columbia University, New York, United States
20German Center for Neurodegenerative Diseases (DZNE) Munich, Germany
21University College London, United Kingdom
22Icahn School of Medicine at Mount Sinai, New York, United States
23Osaka City University, Japan
24Edith Cowan University, Perth, Australia
25German Center for Neurodegenerative Diseases (DZNE) Tubingen, Germany
26Ludwig-Maximilian’s University, Munich, Germany
27Tokyo University, Japan
28Mayo Clinic, Rochester, United States
29University of Michigan, Ann Arbor, United States
30University of Pittsburgh Medical Center, United States
31University of Melbourne, Australia
32Hertie Institute for Clinical Brain Research, Tubingen, Germany
Additional information
Extended data is linked to the online version of the paper at www.nature.com/nature.
Supplementary information is linked to the online version of the paper at www.nature.com/nature.
Reprints and permissions information is available at http://www.nature.com/reprints.
Data Availability Statement:
Mouse single-cell mRNA sequencing is available at the Gene Expression Omnibus under the accession number GSE188285. The human single-nucleus data from the Knight ADRC is publicly available by request from the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS) with accession number NG00108.v1
(https://www.niagads.org/datasets/ng00108). To access the data from the DIAN brain bank, special request must be made using this URL: https://dian.wustl.edu/our-research/for-investigators/.
The mice and MRI illustrations are available for a free access from Servier Medical Art (SMART) website (https://smart.servier.com). The brain cartoon and the summary illustration were created by the authors.
References
- 1.Ginhoux F & Guilliams M Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 44, 439–449 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Masuda T et al. Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature 604, 740–748 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Alves de Lima K et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol 21, 1421–1429 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filiano AJ et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 535, 425–429 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konsman JP, Parnet P & Dantzer R Cytokine-induced sickness behaviour: mechanisms and implications. Trends in Neurosciences 25, 154–159 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Mestre H et al. Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat Commun 9, 4878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iliff JJ et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 33, 18190–18199 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Veluw SJ et al. Vasomotion as a Driving Force for Paravascular Clearance in the Awake Mouse Brain. Neuron 105, 549–561.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iliff JJ et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4, 147ra111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louveau A et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X et al. Meningeal lymphatic vessels mediate neurotropic viral drainage from the central nervous system. Nat Neurosci 25, 577–587 (2022). [DOI] [PubMed] [Google Scholar]
- 12.Rustenhoven J et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 184, 1000–1016.e27 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kierdorf K, Masuda T, Jordão MJC & Prinz M Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci 20, 547–562 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Faraco G, Park L, Anrather J & Iadecola C Brain perivascular macrophages: characterization and functional roles in health and disease. J Mol Med (Berl) 95, 1143–1152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Hove H et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22, 1021–1035 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Goldmann T et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faraco G et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J Clin Invest 126, 4674–4689 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thanopoulou K, Fragkouli A, Stylianopoulou F & Georgopoulos S Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc Natl Acad Sci U S A 107, 20816–20821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park L et al. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ Res 121, 258–269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jordão MJC et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Mrdjen D et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48, 380–395.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Wardlaw JM et al. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol 16, 137–153 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Mestre H et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife 7, e40070 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L et al. Evaluating glymphatic pathway function utilizing clinically relevant intrathecal infusion of CSF tracer. J Transl Med 11, 107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Da Mesquita S et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature 560, 185–191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahn JH et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 572, 62–66 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Mestre H et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 367, eaax7171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polfliet MM et al. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J Neuroimmunol 116, 188–195 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Hablitz LM et al. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci Adv 5, eaav5447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gakuba C et al. General Anesthesia Inhibits the Activity of the ‘Glymphatic System’. Theranostics 8, 710–722 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim HY et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 49, 326–341.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Chow BW et al. Caveolae in CNS arterioles mediate neurovascular coupling. Nature 579, 106–110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baccin C et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol 22, 38–48 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang N et al. LYVE1+ macrophages of murine peritoneal mesothelium promote omentum-independent ovarian tumor growth. J Exp Med 218, e20210924 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boissonneault V et al. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer’s disease. Brain 132, 1078–1092 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Hawkes CA & McLaurin J Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A 106, 1261–1266 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Da Mesquita S et al. Meningeal lymphatics affect microglia responses and anti-Aβ immunotherapy. Nature 593, 255–260 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Utz SG et al. Early Fate Defines Microglia and Non-parenchymal Brain Macrophage Development. Cell 181, 557–573.e18 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Pires PW et al. Improvement in Middle Cerebral Artery Structure and Endothelial Function in Stroke-Prone Spontaneously Hypertensive Rats after Macrophage Depletion. Microcirculation 20, 650–661 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Császár E et al. Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. Journal of Experimental Medicine 219, e20211071 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Method References:
- 42.Erde J, Loo RRO & Loo JA Improving Proteome Coverage and Sample Recovery with Enhanced FASP (eFASP) for Quantitative Proteomic Experiments. Methods Mol Biol 1550, 11–18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lun ATL, McCarthy DJ & Marioni JC A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res 5, 2122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCarthy DJ, Campbell KR, Lun ATL & Wills QF Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van den Berge K et al. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol 19, 24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong G, Zhang W, Li H, Shen X & Guo Z Separate enrichment analysis of pathways for up- and downregulated genes. J R Soc Interface 11, 20130950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mouse single-cell mRNA sequencing is available at the Gene Expression Omnibus under the accession number GSE188285. The human single-nucleus data from the Knight ADRC is publicly available by request from the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (NIAGADS) with accession number NG00108.v1
(https://www.niagads.org/datasets/ng00108). To access the data from the DIAN brain bank, special request must be made using this URL: https://dian.wustl.edu/our-research/for-investigators/.
The mice and MRI illustrations are available for a free access from Servier Medical Art (SMART) website (https://smart.servier.com). The brain cartoon and the summary illustration were created by the authors.