

Abstract
High‐risk neuroblastoma patients have poor survival rates and require better therapeutic options. High expression of a multifunctional DNA and RNA‐binding protein, NONO, in neuroblastoma is associated with poor patient outcome; however, there is little understanding of the mechanism of NONO‐dependent oncogenic gene regulatory activity in neuroblastoma. Here, we used cell imaging, biochemical and genome‐wide molecular analysis to reveal complex NONO‐dependent regulation of gene expression. NONO forms RNA‐ and DNA‐tethered condensates throughout the nucleus and undergoes phase separation in vitro, modulated by nucleic acid binding. CLIP analyses show that NONO mainly binds to the 5′ end of pre‐mRNAs and modulates pre‐mRNA processing, dependent on its RNA‐binding activity. NONO regulates super‐enhancer‐associated genes, including HAND2 and GATA2. Abrogating NONO RNA binding, or phase separation activity, results in decreased expression of HAND2 and GATA2. Thus, future development of agents that target RNA‐binding activity of NONO may have therapeutic potential in this cancer context.
Keywords: DBHS, neuroblastoma, phase separation, RNA binding, super‐enhancer
Subject Categories: Molecular Biology of Disease, RNA Biology
NONO protein correlates with poor outcome in neuroblastoma. NONO binds 5′ ends of introns in oncogenic genes, including super‐enhancer‐regulated GATA2 and HAND2, enabling proper RNA processing, via RNA binding and phase separation activities.
Introduction
Neuroblastoma is derived from neural crest cells in the sympathetic nervous system and is the most common extracranial solid cancer in children (Munzer et al, 2008; Janoueix‐Lerosey et al, 2010). Whilst suitable treatments for low‐risk patients exist, high‐risk neuroblastoma patients have poor survival rates and a paucity of therapeutic options. High expression of the gene regulatory protein NONO (Non‐POU Domain Containing Octamer Binding) in neuroblastoma is associated with poor patient survival, suggesting this could be a potential therapeutic target (Liu et al, 2014). However, beyond knowledge of NONO binding to one long non‐coding RNA (lncRNA) in neuroblastoma, LncMycnUS, there is no further insight into NONO mechanistic activity in this cancer. NONO is a multifunctional protein with various roles in genome maintenance and gene regulation at the transcriptional and post‐transcriptional levels, including transcription initiation, elongation and termination, pre‐mRNA processing and splicing, and nuclear retention of RNA (Hennig et al, 2015; Knott et al, 2016; Hentze et al, 2018; Feng et al, 2020). NONO is a member of the highly conserved Drosophila behaviour/human splicing (DBHS) protein family, which also includes the splicing factor proline/glutamine‐rich (SFPQ) and paraspeckle protein component 1 (PSPC1) proteins. DBHS proteins have two N‐terminal RNA recognition motifs (RRM), a NonA/paraspeckle domain (NOPS), a coiled‐coil at the C‐terminus and N‐ and C‐terminal intrinsically disordered low complexity domains (LCDs). DBHS proteins form obligate dimers that can bind DNA, RNA, undergo oligomerisation, mediate additional protein–protein interactions and also undergo liquid–liquid phase separation. Combined, these different interactions suggest DBHS proteins act as “molecular scaffolds” to carry out their multipurpose activities in many facets of gene regulation.
Several studies have profiled the genome, and transcriptome‐wide DNA and RNA substrates bound by NONO in different biological contexts (Ma et al, 2016; Benegiamo et al, 2018; Xiao et al, 2019; Van Nostrand et al, 2020). Broadly, these studies have revealed widespread binding to diverse gene regulatory elements in chromatin as well as binding to mainly intronic elements of pre‐mRNAs. In some instances, this binding is linked to specific co‐regulatory networks such as NONO binding to ERK promoter targets in stem cells (Ma et al, 2016), or binding to pre‐mRNA of transcripts coding metabolic genes in liver hepatocytes (Benegiamo et al, 2018). Whilst NONO/ERK association at chromatin is required for mouse embryonic stem cell (mESC) pluripotency, there is little mechanistic insight explaining the consequences of NONO binding to RNA in different contexts. Other DBHS proteins have also been analysed by CLIP and ChIP (Takeuchi et al, 2018; Hosokawa et al, 2019; Iida et al, 2020; Stagsted et al, 2021); for instance, SFPQ binds to and enables processing of long introns in neurons, and prevents intron retention in ALS motor neurons (Luisier et al, 2018).
Given their extensive and diverse DNA and RNA targets, DBHS proteins are found throughout the nucleus as well as in specific sub‐nuclear sites, such as paraspeckles—where they bind and play an essential role stabilising the lncRNA NEAT1_2 scaffold (Knott et al, 2016)—and to sites of DNA damage (Krietsch et al, 2012; Li et al, 2014). Both paraspeckles and DNA damage foci are now classed as condensates built by the liquid–liquid phase separation properties of various component proteins (Fox et al, 2018; Spegg & Altmeyer, 2021). Liquid–liquid phase separation is a phenomenon explaining the dynamic association of molecules, including RNA‐binding proteins, into membrane‐less organelles, or condensates (Sabari et al, 2018; Alberti & Dormann, 2019; Zbinden et al, 2020). Recently, it was demonstrated that NONO undergoes phase separation at DNA damage foci (Fan et al, 2021); however, how phase separation by NONO plays a role in gene regulation is unknown.
Super‐enhancer (SE)‐regulated gene networks are defined by ChIP signatures and establish distinct cell lineage programs. Recently it was revealed that super‐enhancers are controlled by the formation of phase‐separated condensates composed of transcription factors and transcriptional cofactors (Boija et al, 2018; Guo et al, 2019). This regulation may be critical for neuroblastoma, as two main SE‐associated transcriptional networks control lineage identity, intra‐tumoral heterogeneity and cell‐type specific gene expression, including a mesenchymal cell state and a noradrenergic cell state (Boeva et al, 2017; van Groningen et al, 2017). The noradrenergic cells are further subdivided into three major SE‐driven epigenetic subtypes and their underlying master regulatory networks recapitulating three clinical groups in neuroblastoma tumours and cell lines (Gartlgruber et al, 2021). A small number of key transcription factors associated with SE were identified as members of the transcriptional core regulatory circuitry (CRC) that determine noradrenergic cell fate and growth, such as MYCN, PHOX2B, HAND2, GATA2 and GATA3 (van Groningen et al, 2017; Durbin et al, 2018; Gartlgruber et al, 2021).
In this study, we take a holistic look at NONO in neuroblastoma to determine the mechanisms linking NONO to poor outcome in this context. We combine cell imaging, biochemical analysis, and various RNA‐Seq analyses to reveal a complex picture of NONO‐dependent regulation of gene expression. We find NONO in numerous small non‐paraspeckle foci throughout the nucleus, tethered to these by RNA and DNA. Accordingly, NONO mutants that cannot bind RNA mis‐localise in larger spherical non‐functional puncta that more readily phase separate, confirming the critical role of NONO RNA binding in its function. Using PAR‐CLIP we show NONO binds to the 5′ ends of pre‐mRNA and influences pre‐mRNA processing. Notably, NONO‐bound transcripts are also more likely to be SE‐regulated, including HAND2 and GATA2. We find that the decreased expression of HAND2 and GATA2 after NONO depletion is likely mediated by inefficient pre‐mRNA processing at these loci.
Results
NONO puncta are condensates dependent on RNA and DNA
Given that high levels of NONO are correlated with poor patient outcome in neuroblastoma (Fig 1A; Liu et al, 2014), we set out to investigate the role of NONO in this biological context, to inform future rational design of therapeutics. At the cellular level, we have previously demonstrated that paraspeckles—as illustrated by Fluorescence In Situ Hybridisation (FISH) against NEAT1_2—are co‐localised with a subset of NONO immunofluorescence signal (Yamazaki et al, 2018; Naveed et al, 2021). When comparing the distribution of NEAT1_2 and NONO puncta between high‐risk neuroblastoma (KELLY and BE(2)‐C) and HeLa cell lines, we observed fewer paraspeckles in neuroblastoma cells (Fig 1B), as shown previously (Naveed et al, 2021). Instead of within paraspeckles, NONO is localised in numerous small puncta throughout the nucleus, in both neuroblastoma cell lines. We also checked if these small non‐paraspeckle NONO foci corresponded to “microspeckles,” individual non‐paraspeckle NEAT1_1 RNA foci; however, we did not observe co‐localisation of NONO with NEAT1_1 in KELLY and HeLa cells, consistent with previous observations (Li et al, 2017) (Appendix Fig S1A).
Figure 1. Both RNA and DNA are essential for distinct distribution of NONO puncta in neuroblastoma cell lines.
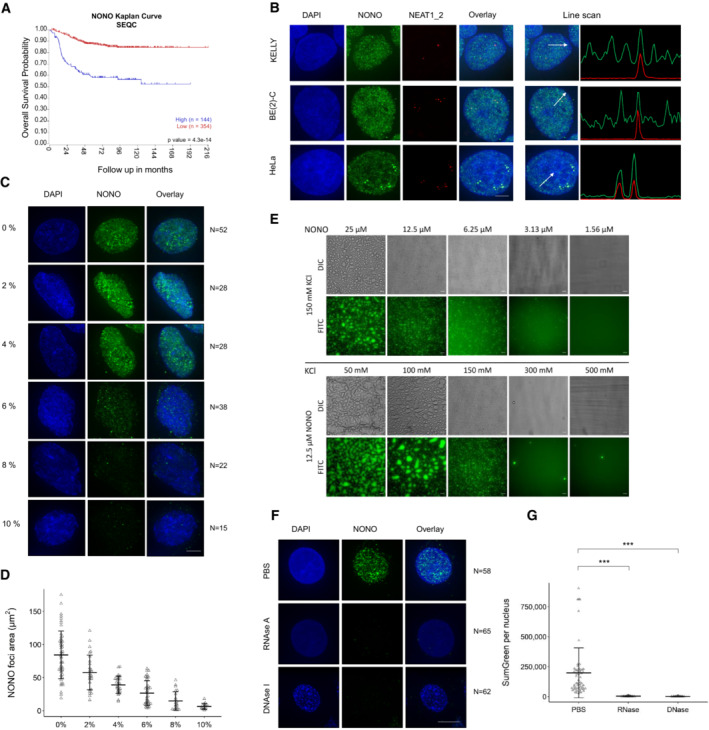
- The probability of overall survival is lower in neuroblastoma patients with high NONO expression based on Kaplan–Meier curve using the SEQC neuroblastoma dataset.
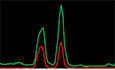
- Fluorescence micrograph images of representative cells stained for NONO and NEAT1_2 in KELLY and BE(2)‐C neuroblastoma and HeLa cells showing clear paraspeckle (as marked by NEAT1_2), and non‐paraspeckle NONO puncta. DAPI (blue) stain indicates cell nuclei, NONO immunofluorescence (green) and NEAT1_2 RNA FISH (red). Line scans correspond to the positions and directions of the arrows. Scale bar: 5 μm.
- Fluorescence micrograph images of representative cells stained for NONO in KELLY cells treated with 2, 4, 6, 8 or 10% 1,6 hexanediol showing dissolution of NONO puncta with increasing concentration. Scale bar: 5 μm.
- Dot plot of NONO foci area (μm2) per nucleus at different concentrations of 1,6 hexanediol as in (C). Bars are SD. The numbers of biological replicates are indicated in (C).
- Recombinant GFP‐NONO_WT can phase separate spontaneously. Its propensity to phase separate increases with increasing protein and decreasing KCl concentration. Scale bar: 20 μm.
- Fluorescence micrograph images of representative cells stained for NONO in KELLY cells treated with PBS, RNase A or DNase I, as indicated. Scale bar: 5 μm.
- Dot plot of summed green fluorescence per nucleus in (F). Bars are SD. The numbers of biological replicates are indicated in (F). Student's t‐test is used to compare the means. ***P < 0.001.
To address the nature of the small non‐paraspeckle NONO puncta, we incubated cells with 1,6‐hexanediol (a compound that disrupts liquid–liquid phase‐separated condensates) and observed dramatically reduced NONO signal intensity in KELLY (Fig 1C and D) and HeLa (Appendix Fig S1B and C) cells. Supporting a role for phase separation in the formation of NONO puncta, we also observed that recombinant full‐length GFP tagged NONO could form droplets in vitro, regulated by varying concentrations of either NONO protein, or KCl (Fig 1E). In addition, nuclease digestion using either RNase A or DNase I completely eradicated the nuclear NONO puncta signal in KELLY (Fig 1F and G) and HeLa (Appendix Fig S1D and E) cells. Together, these in vitro and in vivo observations suggest that the numerous non‐paraspeckle NONO puncta are dependent on both RNA (distinct from NEAT1_2) and DNA for their structural integrity. Further, the puncta may be condensates, as they are sensitive to 1,6‐hexanediol and NONO can form droplets in vitro.
RNA recognition motif 1 (RRM1) is essential for NONO to bind RNA targets
Given the importance of RNA in NONO puncta formation, we next addressed NONO RNA‐binding ability for its localisation and function. The canonical RRM1 is structurally characterised in NONO and is required for NONO binding RNA in vitro; however, its role in RNA binding has not been fully evaluated in different biological settings (Fox et al, 2005; Kuwahara et al, 2006; Passon et al, 2012; Knott et al, 2016, 2021). Thus, we compared the localisation of overexpressed YFP‐fused wild‐type NONO (YFP‐NONO_WT) with NONO lacking the RRM1 (YFP‐NONO_∆RRM1). Prior experiments showed NONO_∆RRM1 is structurally stable and can readily dimerise (Knott et al, 2021). We observed that, compared to YFP‐NONO_WT, YFP‐NONO_∆RRM1 no longer co‐localised with paraspeckles in KELLY (Fig 2A and B) and HeLa (Appendix Fig S2A–F) cells suggesting the loss of RRM1 abrogated the ability to bind NEAT1_2. The YFP‐NONO_∆RRM1 puncta were also more spherical, fewer in number, and larger, compared to YFP‐NONO_WT (Fig 2C–E). These NONO_∆RRM1 puncta were resistant to nuclease digestion, suggesting their formation is independent of RNA and DNA (Fig EV1A and B). Functionally, intact RRM1 in NONO was linked to KELLY cell proliferation, as measured by the percentage of BrdU‐positive cells increasing when exogenous YFP‐NONO_WT was overexpressed, but not YFP‐NONO_∆RRM1 (Fig 2F). We also mutated some additional residues in the NONO NOPS domain that may mediate RNA association, as identified in RBDMap RNA crosslinking studies (Castello et al, 2016). However, whilst these point mutants displayed a similar localisation to YFP‐NONO_∆RRM1, this is more likely the result of failure to dimerise than bind RNA, as, unlike YFP‐NONO_WT, or YFP‐NONO_∆RRM1, the YFP‐NONO point mutants no‐longer co‐immunoprecipitated endogenous SFPQ (Appendix Fig S2F).
Figure 2. RRM1 is an important region for NONO to bind RNA targets in KELLY cells.
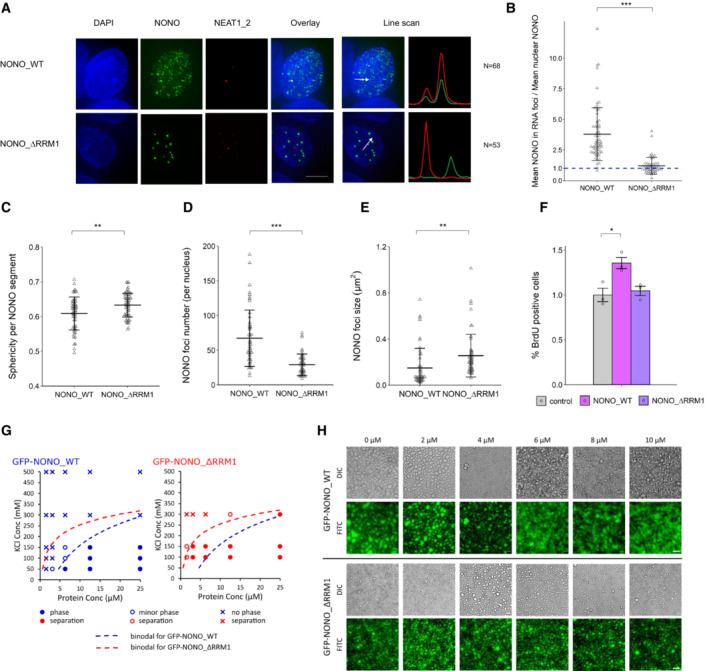
-
AFluorescence micrograph images of representative cells stained for NONO and NEAT1_2 after transfection with plasmids expressing YFP‐fused NONO_WT and NONO_∆RRM1 protein. DAPI (blue) stain indicates cell nuclei, YFP‐fused NONO (green) and NEAT1_2 RNA FISH (red). Scale bar: 5 μm.
-
BThe enrichment of mean NONO fluorescence detected within RNA FISH foci is quantitatively determined as a ratio relative to mean nuclear NONO fluorescence in (A). Bars are SD. The numbers of biological replicates are indicated in (A). Student's t‐test is used to compare the means. ***P < 0.001.
-
C–ESphericity per NONO segment, NONO foci number per nucleus and NONO foci size between YFP‐fused NONO_WT and NONO_∆RRM1 plasmids. Bars are SD. Biological replicates n = 60 in (C) and n = 50 in (D) and (E). Student's t‐test is used to compare the means. **P < 0.01, ***P < 0.001.
-
FPercentage of BrdU incorporation between control cells (YFP only transfection), cells with YFP‐fused NONO_WT and YFP‐NONO_∆RRM1 plasmids transfected. Bars are SEM. Biological replicates n = 3. Student's t‐test is used to compare the means. *P < 0.05.
-
GRecombinant GFP‐NONO_ΔRRM1 has a higher propensity to phase separate than GFP‐NONO_WT at the same KCl concentration. Blue and red colours denote GFP‐NONO_WT and GFP‐NONO‐ΔRRM1, respectively. Filled circles, open circles and crosses indicate distinct phase separation, minor phase separation and no phase separation, respectively. Dotted lines denote putative binodal line separating single‐ and two‐phase states.
-
HPS‐ASO against NEAT1 disrupts the phase separation of GFP‐NONO_WT but not GFP‐NONO_ΔRRM1 in a PS‐ASO concentration‐dependent manner. Scale bar: 40 μm.
Figure EV1. NONO_∆RRM1 puncta are resistant to nuclease digestion.
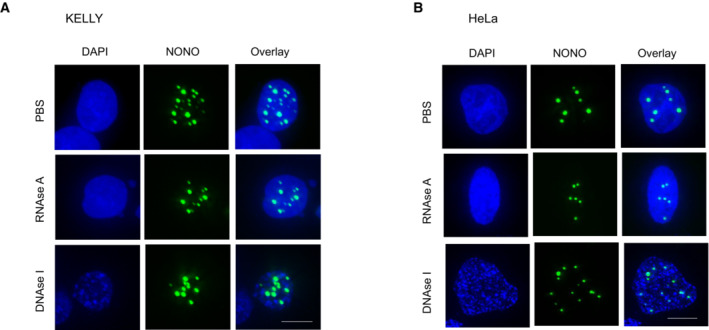
- Fluorescence micrograph images of representative cells stained for NONO in KELLY cells transfected with YFP‐NONO_∆RRM1 and then treated with PBS, RNase A or DNase I, as indicated.
- Fluorescence micrograph images of representative cells for NONO signal in HeLa cells transfected with YFP‐NONO_∆RRM1 and then treated with PBS, RNase A or DNase I.
Data information: Scale bar: 5 μm.
In vitro, recombinant GFP‐NONO_∆RRM1 could form droplets, with an increased propensity for droplet formation compared to GFP‐NONO_WT (Fig 2G). We next incubated recombinant NONO with a nucleic acid substrate, a 2′‐O‐methyl phosphorothioate antisense oligonucleotide (PS‐ASO) against NEAT1 (Vickers et al, 2019). PS‐ASOs with this chemistry are bound with high affinity by DBHS proteins, in an RRM1‐dependent manner (Knott et al, 2021). We observed that PS‐ASO addition to GFP‐NONO_WT solution caused NONO droplets to become small fibrils (Fig 2H, top). In contrast, adding PS‐ASO to GFP‐NONO_∆RRM1 had no effect on droplets, indicating NONO_∆RRM1 is impervious to the addition of nucleic acid NONO substrate (Fig 2H, bottom). Taken together, these results suggest RNA binding through RRM1 modulates the propensity for NONO to form droplets/condensates, thereby altering its sub‐nuclear distribution.
NONO binds predominantly within introns
We next assessed the transcriptome‐wide binding of NONO in KELLY and BE(2)‐C neuroblastoma cells. We performed PAR‐CLIP and found, similar to other DBHS protein CLIP experiments, the majority of NONO binding occurs within introns, even when normalising for the percentage of the transcriptome made up of introns (Fig 3A). NONO binding was biased towards the 5′ end of transcripts (Fig 3B), a pattern also previously observed in CLIP against NONO and other DBHS family proteins in other biological settings.
Figure 3. NONO preferentially binds 5′ introns and SE‐regulated target genes have more NONO binding.
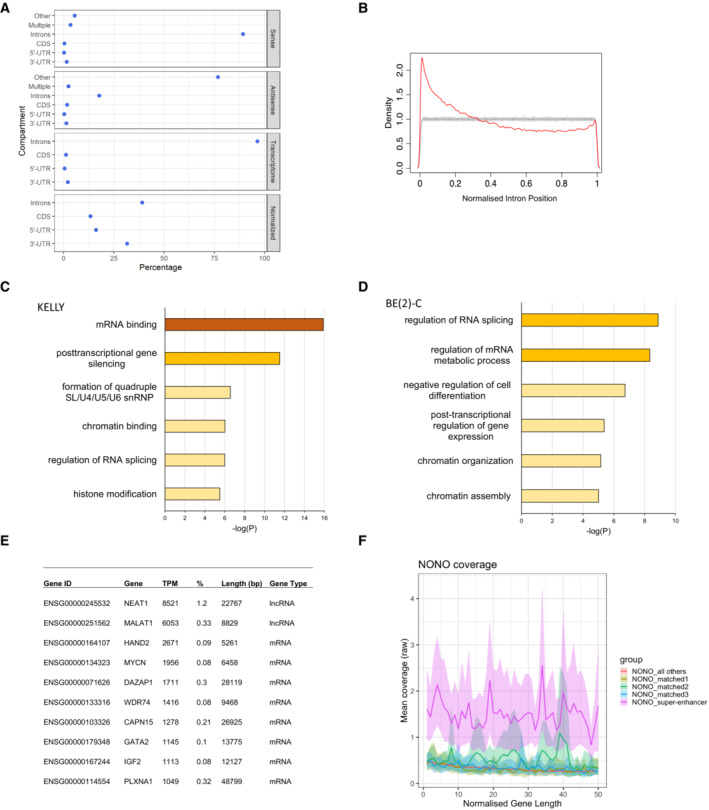
- NONO RNA‐binding sites are strongly biased towards introns, as determined by PAR‐CLIP and wavClusteR. Sense: percentage of annotated binding sites on sense strand. Antisense: percentage of annotated binding sites on antisense strand. Transcriptome: relative length of each annotation category within the transcriptome. Normalised: percentage of annotated binding sites corrected by total length of each annotation category in transcriptome.
- NONO binding sites within introns show preference for the 5′ end of genes as determined by PAR‐CLIP and PARpipe.
- Metascape gene ontology analysis of NONO‐bound target transcripts.
- Metascape gene ontology analysis of NONO‐bound target transcripts in BE(2)‐C cells.
- Summary of most highly NONO‐bound RNA targets by NONO TPM and % of all T‐to‐C reads.
- NONO PAR‐CLIP coverage profiles (metagene2) across SE‐regulated genes compared to expression and length‐matched controls. NONO binds to SE‐regulated target genes with greater coverage compared to matched controls.
(Jiang et al, 2017; Takayama et al, 2017; Benegiamo et al, 2018). To further define NONO targets, we summed aligned reads containing T‐to‐C transitions across genes (Hafner et al, 2010). Using TPM (transcripts‐per‐million; Wagner et al, 2012) and normalising for gene length, NONO targets (the top 20 percentile by TPM of all genes containing T‐to‐C read alignments, 1,831 genes) were enriched in several ontologies, including “mRNA binding” and “post‐transcriptional gene silencing” in KELLY and BE(2)‐C cells (Fig 3C and D).
Despite NEAT1 being less abundant in KELLY cells than other cell types, it was nevertheless the top NONO target RNA (Fig 3E), with by far the largest percentage of PAR‐CLIP reads of any RNA, reflecting the high affinity of NONO for NEAT1 (Naveed et al, 2021). Amongst the rest of the top 10 transcripts, HAND2 and GATA2 were notable as they encode super‐enhancer (SE) regulated transcription factors essential to mediate lineage control in neuroblastoma (Boeva et al, 2017; van Groningen et al, 2017). To determine if other SE‐regulated genes were also highly bound by NONO, we analysed publicly available H3K27 acetylation ChIP‐seq data from Chipumuro et al (2014) to identify the SE profile in KELLY cells, examining the overlap of NONO RNA binding for a subset of genes within SE regions from the H3K27ac ChIP‐seq analysis. We found that transcripts from genes within SE regions had substantially greater NONO RNA binding when compared with expression‐matched controls, suggesting a preferential RNA binding of NONO to SE‐regulated target gene transcripts (Fig 3F). We next sought to selectively inhibit SE‐associated foci, using the BET inhibitor JQ1 that prevents the SE‐assembling cofactor BRD4 from binding acetylated histones (Lovén et al, 2013). We observed that JQ1 treatment diminished NONO foci formation in KELLY (Fig EV2A and B) but not HeLa cells (Fig EV2C and D). Combined, these data suggest NONO associates with SE‐associated genes in neuroblastoma cells.
Figure EV2. JQ1 diminishes NONO foci formation in KELLY cells.
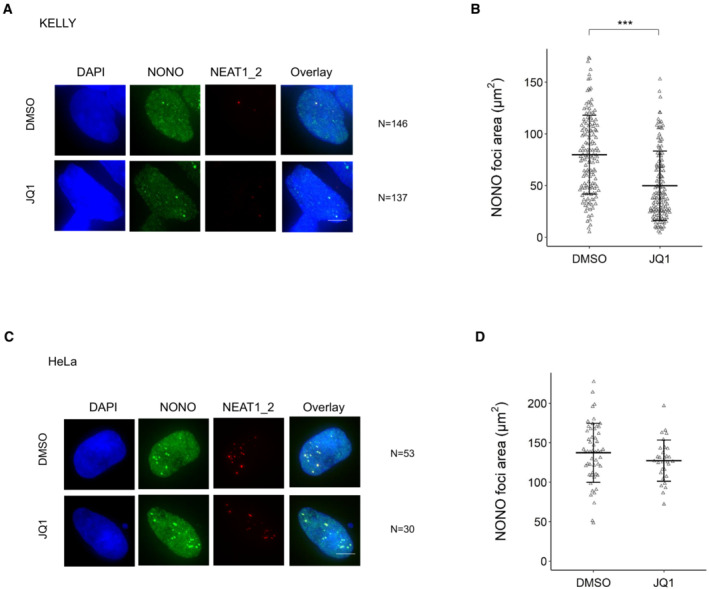
- Representative Fluorescence micrograph images of cells stained for NONO and NEAT1_2 in KELLY cells treated with BET inhibitor JQ1. DAPI (blue) stain indicates cell nuclei, NONO immunofluorescence (green) and NEAT1_2 RNA FISH (red). Scale bar: 5 μm.
- Dot plot of NONO foci area (μm2) per nucleus calculated using images of hundreds of cells, stained as in (A). Bars are SD. The numbers of biological replicates are indicated in (A). Student's t‐test is used to compare the means. ***P < 0.001.
- Representative Fluorescence micrograph images of cells stained for NONO and NEAT1_2 in HeLa cells treated with BET inhibitor JQ1. Scale bar: 5 μm.
- Dot plot of NONO foci area (μm2) per nucleus calculated using images of cells, stained as in (C). Bars are SD. The numbers of biological replicates are indicated in (C). Student's t‐test is used to compare the means.
To validate our PAR‐CLIP findings, we conducted RNA immunoprecipitation (RIP) against NONO followed by RNA quantification of mRNAs and pre‐mRNAs for MYCN, DAZAP1, GATA2 and HAND2, observing greater NONO binding to pre‐mRNAs (Fig 4A). We then used siRNA to knock down endogenous NONO and rescued with siRNA‐resistant YFP‐NONO_WT, or YFP‐NONO_∆RRM1 plasmids. When compared to controls, various target transcripts including total_NEAT1, DAZAP1, pre_HAND2 and pre_KCNQ2 were enriched with exogenous YFP‐NONO_WT, but not with YFP‐NONO_∆RRM1, confirming that binding to pre‐mRNA targets is lost in the mutant NONO (Fig 4B). The pre‐HAND2 and pre_KCNQ2 transcripts were also significantly different between the cells with YFP NONO_WT and YFP‐NONO_∆RRM1. To determine if any NONO foci corresponded to these target transcripts we used RNA FISH probes against NEAT1, or pre‐mRNAs of GATA2 and HAND2, in conjunction with immunofluorescence against NONO. Whilst each target transcript only had 1–2 foci per cell, likely representing nascent transcripts forming near gene loci, nevertheless, these foci were highly enriched in NONO (Fig 4C and D). Thus, we showed that some of the nuclear NONO foci represent NONO bound to GATA2 and HAND2, examples of SE‐regulated transcripts.
Figure 4. NONO binds more abundantly on pre‐mRNAs than mature counterparts in KELLY cells.
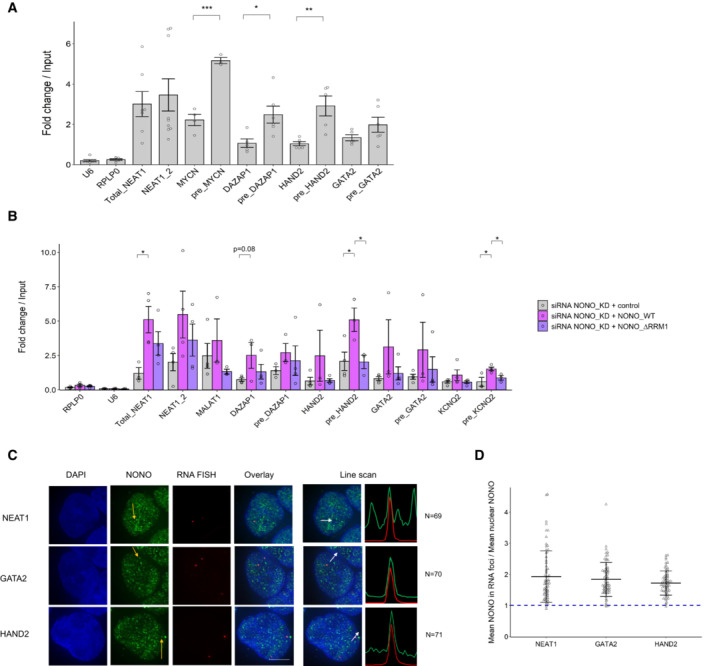
- Relative pre‐mRNA levels and their mature counterparts were measured by NONO RNA RIP followed by RT‐qPCR. RIP data with normal mouse serum IgG as controls are not displayed because all values are < 0.01. Bars are SEM. Biological replicates n ≥ 3. Student's t‐test is used to compare the means. *P < 0.05, **P < 0.01, ***P < 0.001.
- Relative pre‐mRNA levels and their mature counterparts were measured by NONO RNA RIP followed by RT‐qPCR in cells transfected sequentially with NONO KD siRNA and then siRNA‐resistant control (YFP only), YFP‐fused NONO_WT or NONO_∆RRM1 plasmids. Bars are SEM. Biological replicates n ≥ 3. Student's t‐test is used to compare the means. *P < 0.05.
- Fluorescence micrograph images of representative cells stained for NONO and NEAT1_2 (top), GATA2 (middle) and HAND2 (bottom). DAPI (blue) stain indicates cell nuclei, NONO immunofluorescence (green) and RNA FISH (red) for NEAT1_2, GATA2 and HAND2. Line scans correspond to the positions and directions of the arrows. Scale bar: 5 μm.
- In micrograph image quantitation analysis, the enrichment of mean NONO fluorescence detected within RNA FISH foci is determined as a ratio relative to mean nuclear NONO fluorescence in (C). Bars are SD. The numbers of biological replicates are indicated in (C).
NONO maintains proper RNA processing and splicing at the 5′ end of transcripts
To address the consequence of NONO knockdown (KD) on target gene expression, we used two siRNAs against NONO followed by RNA‐seq in KELLY cells. We first confirmed a significant reduction in NONO mRNA and protein levels after KD (Fig 5A) and then compared four controls with eight NONO KD samples (4 for each NONO siRNA) using RNA‐seq. In total, 3,594 genes were differentially expressed (DESeq2 P adj < 0.1), with no bias for up‐ or down‐regulation (Fig 5B) and no obvious difference according to gene length (Fig EV3A). Genes upregulated following NONO KD were enriched for ontologies relating broadly to the negative control of transcription, whereas genes downregulated were enriched in pathways relating to cholesterol synthesis and metabolism (Fig EV3B and C). Gene Set Enrichment Analysis (GSEA) also found the most highly enriched pathways amongst NONO KD samples (containing genes that increased in activity) were GO:0006342 “chromatin silencing” (GSEA P < 10−3, FDR < 10−3) and GO:0045814 “negative regulation of gene expression (epigenetic)” (GSEA P < 10−3, FDR < 10−3).
Figure 5. NONO KD results in altered RNA processing and splicing in KELLY cells.
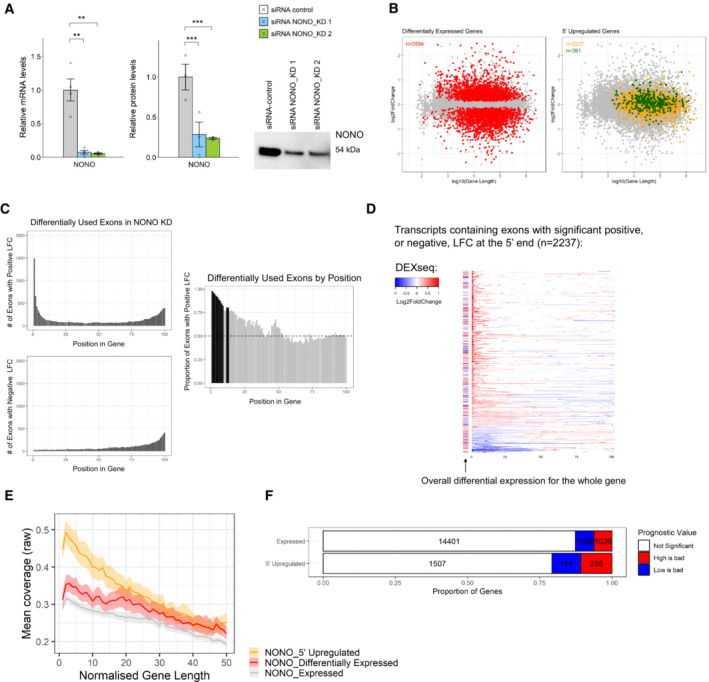
- Relative NONO mRNA and protein levels in KELLY cells treated with control or NONO KD siRNAs. Representative Western blot image for NONO protein. Bars are SEM. Biological replicates n ≥ 3. Student's t‐test is used to compare the means. **P < 0.01, ***P < 0.001.
- Differentially expressed genes do not demonstrate a length, or fold‐change bias (red). The upregulated genes at the 5′ end (orange), including those that are NONO bound (green), have a moderate bias towards longer genes.
- Top panel is a histogram of numbers of exons with significant positive differential usage events in NONO KD compared to control, ranked by gene position. Bottom panel is the same analysis, but for numbers of exons with negative differential usage events. Right panel shows the proportion of positive usage events, ranked by gene position. The dotted line at 0.5 indicates equal numbers of positive, and negative usage events. The black bars show where positive usage events are significantly occurring, over negative events (> 2 SD over the median).
- Individual transcripts with significant positive, or negative, differential usage exons at the 5′ end of the gene (bins 1–13 from Fig 5C). Each row represents a gene, split into 100 bins (exons and introns). Coloured bins represent significant events in differential usage. The bar to the left of each row indicates whether the gene as a whole is differentially expressed. Red indicates increased expression, and blue indicates decreased expression.
- NONO binding/coverage across genes showing 5′ upregulation (those with significant positive LFC events in the first 1–13 bins from Fig 5D) has a distinct 5′ bias when compared with genes which are differentially expressed, and compared to all expressed genes (metagene2).
- There is a higher proportion of neuroblastoma‐prognostic genes in the upregulated genes at the 5′ end compared with the other expressed genes based on the Kaplan–Meier tool using four neuroblastoma datasets (Cangelosi, Maris, SEQC and Versteeg).
Figure EV3. NONO KD induces significant changes in ATSS and A3 in KELLY cells.
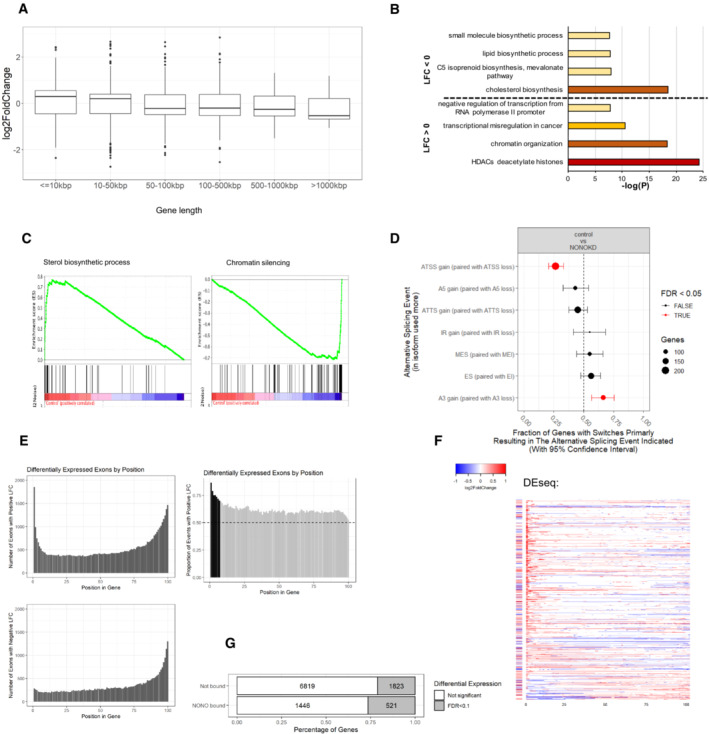
- Box plots showing log fold change for genes which were significantly differentially expressed (Padj < 0.1), separated by gene length. Biological replicates n = 4 in controls and n = 8 in NONO KD samples. Central band is median; boxes represent 1st and 3rd quartile (25th and 75th percentile, respectively) and whiskers 1.5× interquartile range. The numbers of genes are 948, 1,466, 534, 584, 48 and 14 for gene length ≤ 10, 10–50, 50–100, 100–500, 500–1,000 and > 1,000 kbp, respectively. (B) Summary of most highly enriched gene ontology categories produced by “Metascape” analysis of significantly upregulated (LFC > 0) and downregulated (LFC < 0) genes following NONO KD.
- Enrichment score profile plots produced by “GSEA” for the “sterol biosynthetic process” and “chromatin silencing” gene sets, respectively.
- NONO KD results in significant splicing events including ATSS and A3. The error bars represent 95% confidence intervals. Biological replicates n = 4 in controls and n = 8 in NONO KD samples.
- Top panel is a histogram of numbers of exons with significant positive differential expression events in NONO KD compared to control, ranked by gene position. Bottom panel is the same analysis, but for numbers of exons with negative differential expression events. Right panel shows the proportion of positive differential expression events, ranked by gene position. The dotted line at 0.5 indicates equal numbers of positive, and negative expression events. The black bars show where positive expression events are significantly occurring, over negative events (> 2 SD over the median).
- Individual transcripts with significant positive, or negative, differential expression exons at the 5′ end of the gene (bins 1–7 from Fig EV3E). Each row represents a gene, split into 100 bins (exons and introns). Coloured bins represent significant events in differential expression. The bar to the left of each row indicates whether the gene as a whole is differentially expressed. Red indicates increased expression, and blue indicates decreased expression.
- Proportions of differentially expressed genes between NONO‐bound and non‐bound target genes.
Given NONO's significant RNA‐binding activity, we next assessed the role of NONO in alternative splicing. To identify any overall changes, we used IsoformSwitchAnalyzR, which showed that NONO disruption induced a significant bias in the use of upstream “alternative transcription start sites” (ATSS) and downstream “alternative 3’ end acceptor sites” (A3) (Fig EV3D). To further focus on these splicing changes, we used DESeq2 to test changes in the absolute expression of each intron and exon between control and KD samples. In addition, we used DEXSeq to test changes in the expression of each intron and exon relative to the overall expression of the parent gene: “differential usage.” Thus, DEXSeq tests for changes that occur within a gene, controlling for expression changes of the gene as a whole. To look for a pattern of regulatory change with NONO KD, we divided each gene into 100 bins and looked at the position of each differentially expressed and differentially used exon/intron. Because this is standard RNA‐seq data, the number of reads for introns was insufficient to give clear results. However, we saw many thousands of exons, at different positions in transcripts, showing either significant positive, or negative LFC in NONO KD compared to control (Figs 5C and EV3E). We noticed a pattern towards greater usage, or expression, of exons in the 5′ parts of transcripts, for NONO KD (i.e. positive LFC), as opposed to negative LFC (compare Fig 5C, or Fig EV3E, top and bottom plots). To test if this pattern is significant, we calculated the proportion of positive LFC events for each bin position. We found, for bins at the 5′ end of transcripts that positive LFC events were significantly more likely than negative LFC events. Exons within the first 13 bins had a significantly greater proportion of upregulated usage events (Fig 5C, right) and exons within the first seven bins had a greater proportion of significantly upregulated expression changes (Fig EV3E, right). As only a subset of genes displayed this pattern, it is unlikely to be a technical artefact of RNA‐seq sample, or data, processing.
There were 2,237 genes containing exons that had significant expression and/or usage events (both positive LFC, and negative LFC) identified in Figs 5C and EV3E. Examining the full transcripts (exons and introns) of these 2,237 genes, again with their length normalised to 100 bins, there is a significant bias for 5′ upregulated usage and expression (Figs 5D and EV3F, red). However, despite this 5′ difference, there is no consistent change in the overall transcript expression and usage for these genes (Figs 5D and EV3F, row‐side colours, Fig 5B, yellow), even when overlapping with top NONO hits (Fig 5B, green). Thus, we observed that NONO KD induces upregulated usage and expression at the 5′‐most extent of genes, which is independent of overall expression changes. This suggests a potential deficiency in processing at the 5′ end of transcripts in the absence of NONO.
Combining the NONO KD RNA‐seq with NONO PAR‐CLIP, NONO‐bound RNAs were significantly more likely to be differentially expressed (χ2 = 76.9, P < 10−15, Fig EV3G). To test whether NONO RNA binding is implicated in the observed 5′ usage and expression changes, we looked at the average NONO PAR‐CLIP coverage across those genes that had significant exons with positive LFC in the 5′ ends. For this analysis we interrogated the subset of genes within the 2,237 shown in Fig 5D that had (bin 1–13) positive LFC only, creating a subset we called “NONO 5’ upregulated.” The 1,903 selected genes displayed a pronounced NONO binding bias at the 5′ end, compared to genes which were differentially expressed overall, yet showed approximately equivalent binding at the 3′ end (Fig 5E). Based on survival analysis, we also found that the 1,903 genes in this “NONO 5′ upregulated” class are more likely to be prognostic in neuroblastoma, including GATA2 and HAND2, compared with other expressed genes (Fig 5F). These results suggest that NONO RNA binding at the 5′ end may be involved in pre‐mRNA processing of transcripts with important roles in neuroblastoma.
Delayed RNA processing of GATA2 and HAND2 results in decreased expression
Given that GATA2 and HAND2 have been demonstrated to modulate differentiation and migration in neuroblastoma (Voth et al, 2009; Willett & Greene, 2011), we examined if their expression levels were regulated by NONO in KELLY cells. Indeed, GATA2 and HAND2 mRNA and protein levels were decreased after NONO KD (Fig 6A–C).
Figure 6. NONO KD reduces mRNA and protein expression of GATA2 and HAND2, which may result from inappropriate processing and splicing in KELLY cells.
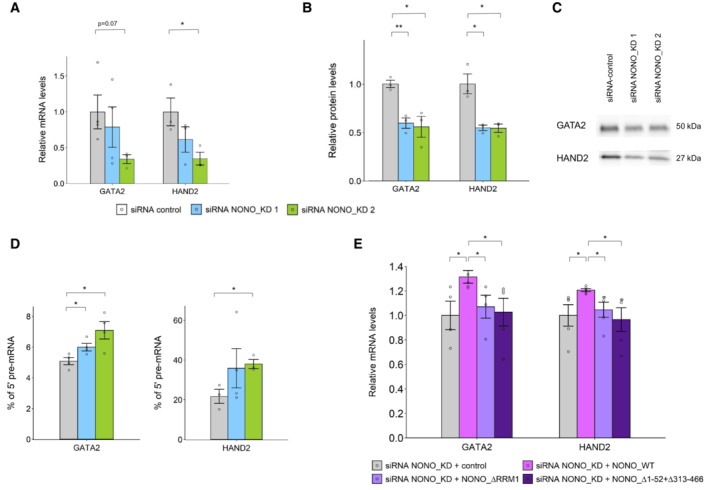
- Relative GATA2 and HAND2 mRNA levels via RT‐qPCR between control and NONO KD siRNAs. Bars are SEM. Biological replicates n ≥ 3.
- Western blot quantitation analysis of GATA2 and HAND2 protein levels between control and NONO KD siRNAs. Bars are SEM. Biological replicates n ≥ 3.
- Representative Western blot images for GATA2 and HAND2 proteins in (B).
- RT‐qPCR analysis using intron‐exon, and exon‐exon spanning primers, showing the percentage of GATA2 and HAND2 pre‐mRNAs at the 5′ end, relative to their mature transcripts. Results show NONO KD increases the relative proportion of GATA2 and HAND2 pre‐mRNA amplicon, over mRNA amplicon. Bars are SEM. Biological replicates n ≥ 3.
- Relative GATA2 and HAND2 mRNA levels in cells transfected sequentially with NONO KD siRNA and then siRNA‐resistant control (GFP only), GFP fused NONO_WT, NONO_∆RRM1 or NONO_∆1–52 + ∆313–466 plasmids. Bars are SEM. Biological replicates n ≥ 4.
Data information: Student's t‐test is used to compare the means. *P < 0.05, **P < 0.01.
We next determined the possible mechanisms underlying the decreased expression of GATA2 and HAND2. Although we could detect NONO binding to the GATA2 gene by ChIP‐qPCR, this chromatin binding did not alter when GATA2 transcription was reduced (Fig EV4A–C), suggesting the binding of NONO to DNA and nascent RNA were not linked. We also tested any NONO‐dependency for transcriptional elongation of GATA2 and HAND2, by ChIP‐qPCR of phosphorylated Serine 2 of RNA polymerase II, yet found no difference in signal with NONO KD (Fig EV4D), and no co‐localisation of NONO foci with PolII‐phospho‐Ser2 foci by immunofluorescence (Fig EV4E and F). In contrast, we found with RT‐qPCR using exon‐exon, and exon‐intron primer pairs, that NONO KD induced a higher relative expression of GATA2 and HAND2 pre‐mRNAs at the 5′ end when compared to their mature transcripts (Fig 6D).
Figure EV4. NONO displays some DNA binding to GATA2, which in turn is not sensitive to transcription levels, or linked to transcriptional elongation.
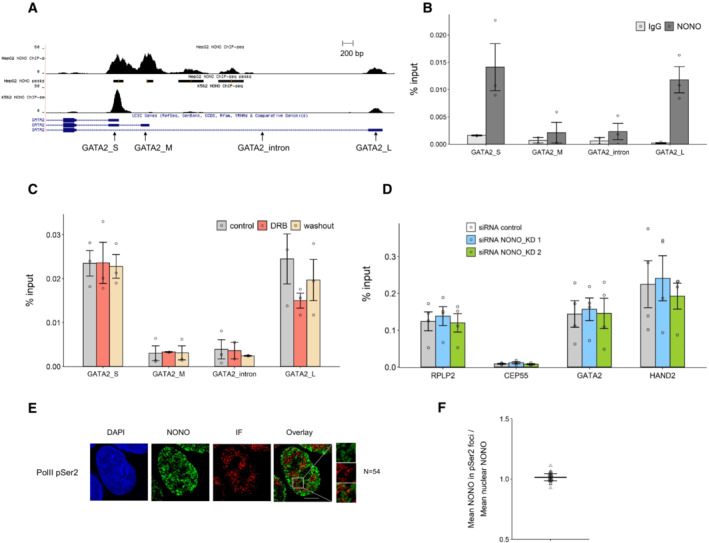
- NONO ChIP‐seq peaks for GATA2 in HepG2 and K562 cell lines shown as a Genome Browser image (Xiao et al, 2019). The primer pairs for GATA2 used in our ChIP‐qPCR are indicated.
- The enrichment of relative GATA2 chromatin fragments via ChIP‐qPCR against NONO and normal mouse serum IgG in KELLY cells. Bars are SEM. Biological replicates n = 3.
- NONO ChIP‐qPCR indicates the enrichment of relative GATA2 chromatin fragments in KELLY cells treated with control, DRB and DRB followed by a washout period. Bars are SEM. Biological replicates n = 3.
- The enrichment of relative GATA2 and HAND2 chromatin fragments after NONO KD via ChIP‐qPCR against RNA PolII phosphorated at Serine 2 in KELLY cells. Bars are SEM. Biological replicates n ≥ 3.
- Fluorescence micrograph images of representative cells stained for NONO and RNA PolII phosphorated at Serine 2. DAPI (blue) stain indicates cell nuclei, NONO immunofluorescence (green) and immunofluorescence for Pol II pSer2 (red). Scale bar: 5 μm.
- In micrograph image quantitation analysis, the enrichment of mean NONO fluorescence detected within immunofluorescence foci of Pol II pSer2 is determined as a ratio relative to mean nuclear NONO fluorescence in (E). The quantitation shows no enrichment of NONO in the foci. Bars are SD. Biological replicates n = 54.
To delineate which activities of NONO are responsible for maintaining robust GATA2 and HAND2 expression, we examined the ability of NONO mutants to rescue knockdown of endogenous NONO. Overexpression of NONO_WT increased GATA2 and HAND2 mRNA (Fig 6E) and protein levels (Appendix Fig S3A and B), following KD of endogenous NONO. However, NONO_∆RRM1 could not rescue expression to the same extent (Fig 6E), indicating RNA‐binding capacity is important. Finally, we looked at a NONO mutant lacking the N and C terminal low complexity regions (NONO_∆1–52 + ∆313–466). We previously found NONO_∆1–52 + ∆313–466 is globular, and forms crystals, not droplets, in vitro (Knott et al, 2021). Further, GFP‐NONO_∆1–52 + ∆313–466 exhibited a more diffuse signal in the nucleus than wildtype NONO (Fig EV5A and B). We therefore tested the ability of this construct to rescue GATA2 and HAND2 expression after NONO KD, but observed no rescue of their mRNA levels (Fig 6E). Taken together, these data indicate that NONO binds GATA2 and HAND2 pre‐mRNAs and enhances their RNA processing and splicing close to the 5′ end of transcripts. This NONO activity, driving optimal expression levels of these oncogenes, is dependent on RNA binding, as well as LCD‐mediated foci formation.
Figure EV5. NONO construct lacking the N‐ and C‐terminal low complexity domain is more diffuse in the nucleus than wildtype NONO.
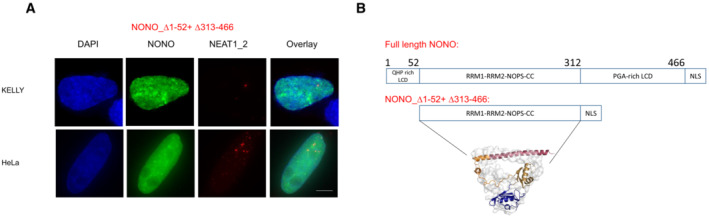
- Fluorescence micrograph images of representative cells stained for NONO and NEAT1_2 in KELLY and HeLa cells transfected with GFP fused NONO_∆1–52 + ∆313–466 plasmids. Scale bar: 5 μm.
- Construction of NONO_∆1–52 + ∆313–466 plasmids, indicating how the mutant lacking the N‐ and C‐terminal LCD is equivalent to a soluble structured dimer, previously solved by crystallography (PDB 3SDE).
Discussion
NONO acts as a molecular scaffold in gene regulation in many contexts. In this study, we identified NONO‐mediated enhancement of RNA processing at the 5′ end of important transcripts as a key molecular mechanism in neuroblastoma. NONO also forms numerous non‐paraspeckle nuclear foci, some of which co‐localise with the super‐enhancer‐regulated GATA2 and HAND2 transcription sites. In the absence of NONO, GATA2 and HAND2 protein levels decrease, with evidence for stalled 5′ RNA processing. Neither the NONO RNA‐binding mutant nor a NONO mutant lacking low complexity domains is able to rescue GATA2 and HAND2 levels. Therefore, we propose a model whereby NONO binds to, and coats the 5′ ends of GATA2 and HAND2 transcripts, forming gene‐body splicing‐associated condensates to enhance gene expression and support an oncogenic program. The genome‐wide PAR‐CLIP findings allow us to speculate that additional NONO foci may be sites of binding to other lncRNA and pre‐mRNA targets, particularly within the 5′ part of pre‐mRNAs regulated by super‐enhancers.
Here we show NONO forms numerous non‐paraspeckle foci in the nucleus, and that NONO readily forms droplets in vitro. NONO contains LCDs at the N‐ and C‐termini, with a central globular domain for RNA binding, dimerisation and coiled‐coil oligomerisation (Passon et al, 2012; Knott et al, 2015, 2016). In this study, we generated a NONO mutant that lacks both the N‐ and C‐terminal LCD. Recently, it was shown that the DBHS protein PSPC1 requires the C‐terminal LCDs to undergo phase separation (Li et al, 2021). We also showed that RNA binding, via RRM1, attenuates NONO localisation in vivo, and phase separation in vitro. A mutant with impaired RNA‐binding ability more readily phase separates in vitro, as well as forming large, nuclease‐resistant, spherical droplets in the nuclei of cells. In contrast, wildtype NONO binds RNA and forms small fibrils in vitro and much smaller, finer, irregular condensates inside the nucleus. The propensity of RNA to prevent aberrant, gross phase separation by NONO is similar to what is observed for FUS, where mutants lacking RNA‐binding capacity excessively phase separate, whilst the wild‐type protein is mostly diffuse (Maharana et al, 2018). However, NONO is different to FUS in that wildtype NONO is not diffuse, but instead forms many hundreds of smaller condensates, each likely representing a site of nascent transcription. For FUS, addition of small amounts of RNA promotes phase separation into droplets in vitro, whereas high RNA levels prevent droplet formation (Maharana et al, 2018). In contrast, addition of increasing concentration of nucleic acid in vitro causes NONO droplets to convert into small fibrils, a behaviour also observed for PSPC1 (Shao et al, 2022). Thus, distinct families of RNA‐binding proteins use their suite of multivalent interactions to respond differently to the presence of RNA in vitro and in vivo. Our work adds to the growing appreciation of how RNA‐binding domains influence the final material state and modulate the dynamics of condensates in general (Gotor et al, 2020; Wiedner & Giudice, 2021).
Paraspeckles are well‐known protein‐RNA condensates that form through microphase separation and are also not typically spherical. In the two main steps of paraspeckle formation, first, DBHS proteins, including NONO, bind and stabilise NEAT1 lncRNA. Secondly, FUS binds and carries out phase separation dependent on its prion‐like domain (Yamazaki et al, 2021). Whilst a role for NONO‐driven phase separation in paraspeckle formation is yet to be established, NONO phase separation is required for radiation‐induced DNA damage repair (Fan et al, 2021). In addition, NONO also promotes phase separation and activation of TAZ, the hippo pathway effector, to drive oncogenic transcription and tumorigenesis in glioblastoma, although a direct role for phase separation of NONO itself was not addressed in that study (Wei et al, 2021). Thus, NONO phase separation may be important for many of its regulatory activities in different contexts.
As well as binding to RNA, there is evidence from us and others, that NONO is bound to chromatin, albeit not through direct binding in many cases (Yang et al, 1997; Knott et al, 2016). In development, NONO and SPFQ are enriched at bivalent genes with high levels of poised RNA PolII, influencing lineage commitment (Yadav et al, 2014; Ma et al, 2016; Xiao et al, 2019; Van Nostrand et al, 2020). NONO is also responsible for recruitment of the 5‐hydroxymethylcytosine enzyme TET1 to chromatin in mESCs (Li et al, 2020). NONO RRM1 deletion did not abrogate its interaction with TET1, nor prevent NONO‐dependent recruitment of TET1 to chromatin in mESCs, therefore NONO RNA binding appears to be less important in regulation of pluripotency. This is in contrast to PSPC1, which recruits TET2 to chromatin, dependent on its RNA‐binding activity (Guallar et al, 2018). However, for neuroblastoma our data argue that chromatin‐associated NONO is not the main driver of the gene regulatory role in this context, but instead might have a permissive role, preparing and facilitating a rapid transcriptional response when stimulators are present. This interpretation is partly due to our ChIP evidence that NONO recruitment to the GATA2 promoter is insensitive to transcription inhibition. Further, NONO mutants lacking RNA binding were no longer recruited to normal NONO condensates, instead appearing in fewer, larger, rounder nuclear droplets that are non‐functional as they do not support enhanced cell proliferation in a similar manner to wildtype NONO.
Our sequencing and CLIP data suggest a subset of genes that display NONO‐dependent expression. These genes normally have high levels of NONO bound at the 5′ ends of their transcripts, principally in the introns. In the absence of NONO there is an increase in reads corresponding to the first ~ 13% of transcript length; however, this does not always correlate with overall differential expression. In the case of GATA2 and HAND2 we showed NONO loss leads to decreased protein production, with an increase in 5′ end of transcripts corresponding to the pre‐mRNA, implicating improper RNA processing in this downregulation. Thus, we propose a model in which a gene regulatory role for NONO in neuroblastoma hinges on binding to the pre‐mRNA of nascent SE‐regulated genes, promoting the formation of RNA‐processing condensates, allowing efficient splicing. Extensive prior literature supports a role for NONO in pre‐mRNA processing and splicing in different cell types and clinical samples (Yamamoto et al, 2018; Feng et al, 2020). Although NONO is not a crucial component in spliceosome assembly, it interacts with critical spliceosomal proteins (Zhang & Wu, 1996; Kameoka et al, 2004).
The primary mechanism of RNA processing enhanced by NONO condensates is still uncertain. Preventing intron retention is one explanation. However, using established bioinformatic pipelines we did not observe intron retention to be significantly altered with NONO KD. Given the 5′ usage bias, this may be due to an inability to differentiate first intron retention, from all‐intron retention. Others have shown NONO KD resulted in aberrant splicing, including intron retention, and altered gene expression in mouse developing retina (Yadav et al, 2014). Moreover, SFPQ, but not NONO, prevents intron retention in early neural differentiation (Luisier et al, 2018; Stagsted et al, 2021). Another factor to consider is the importance of NONO condensates for intron removal in co‐transcriptional splicing, as opposed to post‐transcriptional splicing. Whilst earlier findings support a role for NONO in co‐transcriptional splicing (through association both with nascent RNA and PolII CTD; Emili et al, 2002; Kameoka et al, 2004), new evidence of the importance of “nuclear anchoring” of partially processed, but fully transcribed, pre‐mRNA transcripts at the gene locus is emerging (Girard et al, 2012; Popp & Maquat, 2013; Quinn & Chang, 2016). In this context, a chromatin‐anchored nuclear pool of partially spliced, but polyadenylated RNA, may act in a regulatory manner as a reservoir for mature mRNA, upon splicing. Intriguingly, such a mechanism seems to be important in the neuronal gene regulation context (Yeom et al, 2021). Important future experiments would therefore include testing if NONO condensates act at the co‐ or post‐transcriptional level, by repeating NONO KD RNA‐seq, but with a polyA‐restricted library. If the 5′ usage bias is still apparent it suggests that pre‐mRNAs, already decorated with polyA tails, depend on NONO for proper splicing, supporting post‐transcriptional splicing.
How the formation of NONO RNA processing condensates impacts transcription initiation and elongation condensates remains open to debate. One possibility is that NONO condensates help release paused PolII (Core et al, 2008; Lee et al, 2008; Adelman & Lis, 2012), or prevent promoter‐proximal premature transcriptional termination (Ehrensberger et al, 2013; Kamieniarz‐Gdula & Proudfoot, 2019). Indeed, NONO co‐purifies with some subunits of the mediator complex (required for release of paused PolII; Huang et al, 2012). A signature of paused PolII is abundant 20–60 nt nascent RNA at the TSS (Rougvie & Lis, 1988; Rasmussen & Lis, 1993). Whilst we do see some genes with increased 5′ usage after NONO KD exhibiting such 20–60 nt fragments, we also observe more extensive 5′ transcript increases (over the first 13% of transcript length). Increased abundance of reads at the TSS may also be an increase in nascent transcription of NONO‐dependent genes to compensate for lower target protein levels. Additionally, NONO condensates may enhance PolII elongation, as SE's generally regulate transcriptional elongation (Henriques et al, 2018; Tang et al, 2020) and SFPQ influences elongation (Takeuchi et al, 2018; Hosokawa et al, 2019; Iida et al, 2020; Stagsted et al, 2021). NONO is also required in the recruitment of exonuclease XRN2 to facilitate pre‐mRNA 3′ processing and transcription termination (Kaneko et al, 2007). However, NONO KD leads to a significant upregulation only over the first 13% of the transcript, rather than a gradual decrease of transcript abundance across the entire gene, indicating 3′ exonuclease activity is unlikely to be playing a major role. Further, PolII‐Ser2 ChIP at the GATA2 locus is not altered by NONO KD. Thus, whilst NONO condensates may influence PolII release and elongation to some extent, these are not the only mechanisms at play here.
NONO disruption leads to decreased expression of HAND2 and GATA2, transcription factors that are amongst a small group that maintain noradrenergic cell fate and survival in neuroblastoma (Chipumuro et al, 2014; Oldridge et al, 2015; Boeva et al, 2017; van Groningen et al, 2017; Durbin et al, 2018; Gartlgruber et al, 2021). High expression levels of these and other transcription factors work cooperatively in a defined core regulatory circuit creating a feed‐forward loop to mediate sympathetic neuron specification, proliferation and differentiation (Voth et al, 2009; Rohrer, 2011; Willett & Greene, 2011). Enhancing expression of such targets suggests NONO may be part of a master transcriptional and post‐transcriptional nexus that concertedly assemble a high density of transcription factors and coactivators to drive robust expression in neuroblastoma, which is in agreement with cancer cells that acquire SEs to drive expression of prominent oncogenes (Chapuy et al, 2013; Lovén et al, 2013; Hnisz et al, 2017). Thus, NONO joins the ranks of oncogenic neuroblastoma coactivators such as BRD4, which forms nuclear phase‐separated condensates at sites of SE‐driven transcription (Sabari et al, 2018). Interestingly, BRD4/bromodomain inhibitors impair growth and induce apoptosis in neuroblastoma (Puissant et al, 2013). We show here one such inhibitor also diminishes NONO foci, suggesting NONO regulation may also be part of the therapeutic mechanism of this inhibitor. Furthermore, small molecules that interfere with RNA‐binding activity of NONO may serve as novel inhibitors of NONO biological functions and impede tumour cell growth. More broadly, this study may provide a new avenue for developing pharmaceutical drugs to manipulate RNA‐binding capacity of key regulators in combating cancer and other diseases.
Materials and Methods
Cell culture and transfections
Two human neuroblastoma cell lines including KELLY (originally, Sigma‐Aldrich 92110411) and BE (2)‐C (Originally, the American Type Culture Collection [ATCC] CRL‐2268) as well as HeLa cell line (Originally, ATCC CCL‐2) were used in this study. KELLY cells were grown in Gibco RPMI 1640 (Thermo Fisher, 11835055) with 10% Serana foetal bovine serum (FBS) (Fisher Biotec, FBS‐AU‐015) and 100 U/ml penicillin–streptomycin (Pen‐Strep) (Thermo Fisher, 15140122), whereas BE(2)‐C and HeLa cell lines were grown in Gibco DMEM medium (Thermo Fisher, 11995073) with 10% FBS and 100 U/ml Pen‐Strep. All cells were cultured in a 37°C incubator supplied with 5% CO2. These adherent cells were trypsinised using Gibco TrypLE Express (Thermo Fisher, 12604021) for routine passaging.
All transfections were performed in a forward transfection manner, with KELLY cells grown in RPMI (4% FBS) and HeLa cells grown in DMEM (10% FBS) during transfection. Lipofectamine 3000 (Thermo Fisher, L3000015) and Lipofectamine RNAiMAX (Thermo Fisher, 13778150) were the transfection reagents used in this study. All transfection mixtures were made up in serum‐reduced Gibco Opti‐MEM (Thermo Fisher, 11058021), as per the manufacturer's instructions. The cells were collected 2 days after the final transfection unless otherwise stated. The siRNAs used in the study were silencer select negative control No. 1 siRNA (Thermo Fisher 4390844), Silencer Select siRNA NONO s9612 (Thermo Fisher 4392422) and Silencer Select siRNA NONO s9613 (Thermo Fisher, 4392420). Plasmid NONO containing siRNA resistance site corresponding to siRNA NONO s9612 was made by QuikChange site‐directed mutagenesis kit (Stratagene, 200518) with base‐pair substitutions and the template plasmids pEYFP‐C1‐NONO and pEGFP‐C1‐NONO (human). Using siRNA‐resistant pEYFP‐NONO and pEGFP‐NONO plasmids as templates, plasmids pEYFP‐NONO_∆RRM1 (aa67‐141 deletion), R256I and F257I as well as pEGFP‐NONO_∆RRM1 and NONO_∆1–52 + 313–466 were made by Q5 site‐directed mutagenesis kit (New England BioLabs, E0554S) with base‐pair substitutions according to the manufacturer's instructions.
For siRNA‐only transfections, 25 nM of siRNAs and Lipofectamine RNAiMAX were used. For pEYFP‐ and pEGFP‐ controls and NONO plasmids‐only transfections, Lipofectamine 3000 was used, with 1.25 μg transfected into 6‐wells for Western blots and 200 ng (HeLa cells) or 400 ng (KELLY cells) transfected into 12‐wells for microscopic analysis. Sequential transfections for NONO RIP experiments were performed in 6‐well plates using 10 nM of siRNAs and Lipofectamine RNAiMAX to reduce the endogenous NONO followed by 1.25 μg of plasmid DNA and Lipofectamine 3000 on the next day. Co‐transfections of siRNAs and plasmids for BrdU assays were conducted in 12‐well plates using Lipofectamine RNAiMAX with 25 nM of siRNAs and 1 μg of plasmid DNAs.
RNA extraction and real‐time RT‐qPCR
Cells were lysed with NucleoZOL (Macherey‐Nagel, 740404) and RNA extractions were conducted according to the manufacturer's instructions. Lysed samples were heated at 55°C and vortexed at 1,000 rpm for 10 min. GlycoBlue Coprecipitant (Thermo Fisher, AM9516) was added prior to isopropanol precipitation to aid pellet visualisation.
RNA samples were reverse transcribed to cDNA using the QuantiTect reverse transcription kit (Qiagen, 205314). Real‐time qPCRs were performed in the Rotor‐Gene Q real‐time PCR cycler (Qiagen). A PCR reaction consisted of SensiMix SYBR No‐ROX (Bioline, QT650‐20), 250 nM of forward and reverse primers (Integrated DNA Technologies, Appendix Table S1), molecular‐grade H2O, and cDNA. Ribosomal protein P0 (RPLP0), U6 spliceosomal RNA (U6) and β‐actin were used as reference genes and relative mRNA expression level was presented using method. To assess differential processing at the 5′ end of the transcripts, primer pairs over adjacent exon‐intron junction (HAND2_2 and GATA2_2) were used to measure levels of pre‐mRNAs, and primers pairs across exon‐exon junction (HAND2_3) or located only in the exons (GATA2_3) were used to analyse the mature spliced transcripts. The level of pre‐mRNA processing was then normalised over the expression of each mature spliced transcript. In ChIP‐qPCR experiments, primer pairs of HAND2 and GATA2_4 were used to analyse the enrichment of chromatin fragments against RNA polymerase II phosphorated at Serine 2 with RPLP2 and CEP55 as positive and negative controls, respectively. Primer pairs of GATA2_S, GATA2_M and GATA2_L were used to analyse the enrichment of chromatin fragments against NONO with GATA2_intron (equivalent to pre_GATA2) as a negative control.
Protein extraction and Western blot
Cells were lysed with RIPA lysis buffer (150 mM NaCl, 25 mM Tris pH 7.5, 1% sodium deoxycholate, 0.1% SDS, 1% IGEPAL CA‐630). Protein samples were mixed with SDS gel‐loading buffer and heated to 95°C for 10 min. Samples and Precision Plus Protein™ All Blue Prestained Protein Standards (Bio‐Rad, 1610373) were loaded onto Mini‐PROTEAN TGX Pre‐Cast Gels (Bio‐Rad, 4561086). Gels were run in Tris/Glycine Buffer (Bio‐Rad, 1610771) at 200 V. Trans‐Blot Turbo Mini Nitrocellulose Transfer packs (Bio‐Rad, 1704158) were used for membrane transfer. Primary antibodies used were NONO (mouse monoclonal, in‐house made), GATA2 (CG2‐96) (mouse monoclonal, Santa Cruz, sc‐267), HAND2 (A‐12) (mouse monoclonal, Santa Cruz, sc‐398167) and SFPQ (mouse monoclonal, Merck, P2860). Primary antibodies of NONO and SFPQ were diluted 1:1,000 in 5% milk PBST, whilst GATA2 and HAND2 were used at 1:500 dilution in PBST. The secondary (horseradish‐peroxidase conjugated) antibodies including goat anti‐mouse IgG H&L HRP (Abcam, ab97023) and goat anti‐rabbit IgG H&L HRP (Abcam, ab97051) were diluted 1:10,000 in 5% milk PBST for NONO and SFPQ and 1:5,000 for GATA2 and HAND2. Luminata Crescendo Western HRP substrate (Merck, WBLUR0100) was added and blot images were acquired by Bio‐Rad Chemidoc. Bio‐Rad Imagelab Version 5.2 was used to quantify total protein levels and the intensity of the protein chemiluminescent bands. The relative intensity of chemiluminescent bands was normalised to the amount of total protein in each sample lane, and the sizes of chemiluminescent bands were determined in relation to the standard ladder bands.
GFP‐trap
Cells (2.5 × 106) were rinsed twice with ice‐cold PBS and lysed in 150 μl RIPA buffer (25 mM Tris pH 7.5, 150 mM NaCl, 1% IGEPAL CA‐630, 1% Sodium deoxycholate, 0.1% SDS) with 1× protease inhibitor cocktail (Merck, 04693132001). The cell lysates were centrifuged at 20,000 g for 15 min at 4°C and resuspended in TN buffer (25 mM Tris pH 7.5, 150 mM NaCl, 0.5% IGEPAL CA‐630). After taking pre‐GFP‐trap aliquots, the lysates were incubated with 10 μl GFP‐Trap Magnetic Agarose beads (ChromoTek, gtma‐20) for 3 h at 4°C, followed by washing three times with TN buffer. The samples were mixed with SDS buffer for Western blot.
Immunofluorescence and RNA Fluorescence In Situ Hybridisation
Cells were grown on and fixed onto coverslips (Schott, G405‐15) using 4% paraformaldehyde in PBS. Immunofluorescence started with permeabilisation in freshly made 1% Triton X‐100 diluted in PBS for 10 min. Epitope detection was conducted with a primary monoclonal mouse antibody against NONO at 1:500 dilution in PBST and a polyclonal rabbit antibody against RNA Pol II phosphorated at Serine 2 (Abcam, ab5095) at 1:500 dilution. After three times of PBST washes, coverslips were incubated with secondary FITC anti‐mouse antibody for NONO (Jackson Laboratories, 115‐095‐072) and TRITC anti‐rabbit antibody for Pol II pSer2 (Jackson Laboratories, 711‐026‐152) at 1:500 dilution in PBST. Coverslips were then counter‐stained with DAPI, and images were acquired. For dual immunofluorescence, the cells were incubated with primary antibodies raised in different species together.
For dual immunofluorescence and RNA FISH, after immunofluorescence with NONO, coverslips were then hybridised overnight with FISH probes at 37°C according to the manufacturer's instructions. The RNA FISH probes used in this study included human NEAT1 middle segment with Quasar R 570 Dye (NEAT1_2, Stellaris, SMF‐2037‐1), human NEAT1 5′ segment with Quasar R 570 Dye (NEAT1_1, Stellaris, SMF‐2036‐1) and custom‐made human GATA2 and HAND2 segments with Quasar R 570 Dye (Stellaris).
For the treatment with 1,6‐hexanediol, 1,6‐hexanediol (Merck, 240117‐50G) dissolved in the culture medium was added to cells at room temperature for 5 min. Then, the cells were fixed with 4% paraformaldehyde in PBS, followed by immunofluorescence procedure. The cells with low levels of overexpression were imaged.
For the treatment with JQ1, JQ1 (MedChem Express, HY‐13030) was dissolved in DMSO at 1 mM and further diluted with growth medium to a final concentration of 1 μM for 24 h incubation before imaging.
Coverslips with cells grown on were rinsed in PBS and reaction buffer (20 mM Tris pH 7.5, 5 mM MgCl2, 0.5 mM EGTA, 1× protease inhibitor cocktail). Cells were permeabilised with 0.1% Triton X‐100 diluted in reaction buffer for 5 min at room temperature, followed by rinsing subsequently in reaction buffer and PBS. Coverslips were then incubated with RNase A (Merck, R4642) or DNase I (Worthington, LS006333) to achieve a final concentration of 100 μg/ml in nuclease buffer (5 mM MgCl2 in PBS) for 20 min at room temperature. After nuclease digestion, cells were fixed with 4% paraformaldehyde in PBS and continued with immunofluorescence procedure.
BrdU cell replication assay
KELLY cells were plated onto coverslips in 12‐well plates and transfected with plasmids for 4 days. Two hours prior to fixation, medium was replaced with the culture medium containing 10 nM 5‐Bromo‐2′‐deoxyuridine (BrdU) (Merck, B5002‐100MG). Coverslips were then fixed, and cells were permeabilised in 1% Triton X‐100 diluted in PBS for 10 min at room temperature. After washing in PBS, coverslips were incubated in 1 M HCl for 30 min, rinsed twice in PBS and then incubated in 0.1 M sodium borate for 30 min, followed by blocking in 5% goat serum in PBST. Immunofluorescent staining was carried out using Anti‐BrdU antibody (Abcam, ab8955) at 1:500 dilution in PBST and then anti‐mouse Cy5‐conjugated secondary antibody (Jackson Laboratories, 115‐175‐072) at 1:300 dilution in PBST.
Expression and purification of recombinant proteins in vitro
The pEGFP‐NONO_WT or pEGFP‐NONO_ΔRRM1 plasmids were transformed into competent Rosetta 2(DE3)pLysS Escherichia coli cells (Novagen, 71400) and plated on LB agar plates with selection for kanamycin and chloramphenicol. When the optical density of the culture reaches 0.6–0.8, the flasks were cooled on ice for 15 min before expression was induced with 0.5 mM isopropyl β‐D‐1‐thiogalactopyranoside. After incubation for 16 h, the cells were harvested by centrifugation at 4,000 g. A single pellet from 500 ml of expression culture was gently resuspended on ice in 50 ml binding buffer (50 mM Tris, pH 7.5, 1 M KCl, 300 mM L‐arginine, 25 mM imidazole, 5% glycerol) supplemented with 5 μl Benzonase nuclease (Sigma, E1014‐25KU), 1 mM phenylmethanesulfonyl fluoride, 1× cOmplete™ protease inhibitor cocktail (Roche, 11873580001) and 1 mM MgCl2. The cells were lysed with an Emulsiflex C5 high‐pressure homogeniser (Avestin). The lysate was clarified by centrifugation at 24,000 g and then filtered through a 0.22 μM syringe filter. The supernatant was applied to a 5‐ml His‐Trap HF column charged with NiCl2 (GE Healthcare, 17‐5248‐02). GFP‐NONO_WT or GFP‐NONO_ΔRRM1 was eluted at room temperature with an imidazole gradient (25–500 mM) using the binding buffer and elution buffer (50 mM Tris, pH 7.5, 1 M KCl, 300 mM L‐arginine, 500 mM imidazole, 5% glycerol) over 10 column volumes. The peak fractions were pooled and loaded onto a Superdex 200 HiLoad 16/60 preparative‐grade column (GE Healthcare, 17‐1069‐01) in 5 ml injections and developed at room temperature with the size exclusion buffer (20 mM Tris, pH 7.5, 1 M KCl, 50 mM L‐Proline, 300 mM L‐arginine, 0.5 mM EDTA, 5% glycerol). Relevant peak fractions were pooled and concentrated with Amicon Ultra Centrifugal Filter Units 30 kDa MWCO (Merck Millipore, UFC903024) to the required concentration.
In vitro phase separation assay
Exogenously expressed NONO_WT and NONO_∆RRM1, both tagged at the N‐terminus with monomeric eGFP, were concentrated to 250 μM in storage buffer (20 mM Tris, pH 7.5, 500 mM KCl, 50 mM L‐proline, 300 mM L‐arginine, 0.5 mM EDTA, 5% glycerol). Four two‐fold serial dilutions of the proteins were made in storage buffer to obtain proteins with concentration, 125, 62.5, 31.3 and 15.6 μM. Proteins at these five concentrations were diluted 1 in 10 in appropriate dilution buffers to 25, 12.5, 6.25, 3.13 and 1.56 μM. The composition of the dilution buffer was 20 mM Tris, pH 7.5, 50 mM L‐proline, 0.5 mM EDTA, 5% glycerol and with the KCl concentration adjusted so that the final KCl concentrations after dilution were 50, 100, 150, 300 and 500 mM as required. Twenty microliter of each of the diluted proteins were pipetted into wells of a 384‐well flat‐bottomed non‐protein binding microplate (Grenier, #781906). The wells were sealed with clear film and incubated at room temperature for about 30 min. After incubation, DIC and FITC images of the wells were acquired with the Nikon Eclipse Ti2‐E inverted microscope. To observe the effect of 2′‐O‐methyl phosphorothioate antisense oligonucleotides (PS‐ASO) against NEAT1 on the phase separation of GFP‐NONO_WT and GFP‐NONO_ΔRRM1, the phase separation assays were repeated by diluting 1 in 10 the protein at 250 μM in appropriate dilution buffers to reach a final concentration of 100 mM KCL, 30 mM L‐Arg and 0, 2, 4, 6, 8 and 10 μM PS‐ASO after dilution, keeping other buffer component concentrations the same as the storage buffer. The sequence of the PS‐ASO was AUGAGUUUAGAACUCAAACUUUAUU.
Microscopy and image analysis
All images were acquired on a Deltavision Elite microscope (GE) using a 60× for BrdU assays or 100× objectives for others. For subsequent counting and quantitative analysis of fluorescent intensities, the Nikon NiS Elements software (Version 4.3, Nikon, Tokyo, Japan) was used. Acquisition parameters were kept consistent and intensity thresholds were set the same for samples within each experiment. Cells that had incorporated BrdU into their DNA during replication (labelled by Cy5) were measured and expressed relative to the total number of nuclei, as measured by DAPI staining. MeanGreen of NONO within each RNA FISH foci or each immunofluorescent foci of other proteins was calculated as a ratio relative to nuclear MeanGreen of NONO and then averaged for each cell. The ratio close to 1 indicates that NONO MeanGreen within specific foci is similar to nuclear NONO MeanGreen, implying no NONO enrichment and thus no co‐localisation between NONO signal and other molecules. Sphericity analysis was performed in ImageJ/Fiji with additional plugins (Ollion et al, 2013; Legland et al, 2016; preprint: Haase et al, 2020a, 2020b). Briefly, images were processed with CLIJ2 and the CLIJx‐Assistant, NONO puncta were segmented using MorpholibJ's Marker‐controlled Watershed, and sphericity was calculated from the resulting objects in the 3D ImageJ Suite. All image processing and segmentation parameters were standardised between experimental groups.
RNA immunoprecipitation (RIP)
KELLY cells (2.5 × 106) were rinsed twice with ice‐cold PBS, UV crosslinked at 400 mJ/cm2 on ice for 10 min in PBS, and then resuspended in 100 μl RIPA buffer (25 mM Tris pH 7.5, 150 mM NaCl, 1% IGEPAL CA‐630, 1% Sodium deoxycholate, 0.1% SDS, 1× protease inhibitor cocktail (Merck, 04693132001) and 2 U SUPERase‐In RNase inhibitor (Thermo Fisher, AM2696)). Cells were sonicated using S220 Focused‐ultrasonicator (Covaris, MA, USA) and cell lysates were centrifuged at 20,000 g for 15 min at 4°C. The supernatants were diluted in 300 μl TN buffer with RNase inhibitor (25 mM Tris pH 7.5, 150 mM NaCl, 0.5% IGEPAL CA‐630) and pre‐cleared with 15 μl Dynabeads Protein G (Thermo Fisher, 10004D) for 3 h at 4°C. After taking pre‐RIP aliquots, the pre‐cleared supernatants were divided into two parts equally and incubated with 15 μl Dynabeads pre‐bound with antibodies for 1.5 μg NONO or normal mouse IgG (Santa Cruz, sc‐2025) overnight at 4°C. The bead complexes were washed twice with RIPA buffer and twice with TN buffer. Then, the samples were incubated with TN buffer with 0.5% SDS and proteinase K (Thermo Fisher, EO0491) at 55°C for 30 min, followed by RNA extraction. After reverse transcription, qPCR was used to amplify immunoprecipitated transcripts and data were presented as fold change relative to the input.
Chromatin immunoprecipitation
KELLY cells (2 × 107) were fixed in 10 ml PBS with 1% formaldehyde for 20 min at room temperature. Crosslinking was quenched by the addition of glycine to a final concentration of 0.125 M for 5 min. Cells were centrifuged at 1,800 g for 5 min and resuspended in lysis buffer (50 mM Hepes pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 10% glycerol, 0.5% IGEPAL CA‐630, 0.25% Triton X‐100) with 1× protease inhibitor cocktail, followed by rinsing twice in wash buffer (10 mM Tris pH 8.0, 200 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA). The cell pellets were resuspended in nuclei lysis buffer (50 mM Tris pH 8.0, 10 mM EDTA pH 8.0, 1% SDS) with 1× protease inhibitor cocktail and sonicated with S220 Focused‐ultrasonicator to achieve 200–500 bp DNA fragments. After centrifuging at 10,000 g for 10 min at 4°C, the supernatants were mixed with ChIP dilution buffer (50 mM Tris pH 8.0, 167 mM NaCl, 1.1% Triton X‐100, 0.11% sodium deoxycholate) and RIPA‐150 buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA pH 8.0, 0.1% SDS, 1% Triton X‐100, 0.1% sodium deoxycholate). The samples were pre‐cleared with 20 μl Dynabeads, and pre‐ChIP aliquots were taken. The pre‐cleared supernatants were divided into two parts and incubated with 40 μl Dynabeads pre‐bound with antibodies for 3.5 μg NONO or RNA polymerase II phosphorated at Serine 2 (Abcam, ab5095) and normal mouse IgG overnight at 4°C. The bead complexes were sequentially washed twice each with RIPA‐150 buffer, RIPA‐500 buffer (50 mM Tris pH 8.0, 500 mM NaCl, 1 mM EDTA pH 8.0, 0.1% SDS, 1% Triton X‐100, 0.1% sodium deoxycholate), RIPA‐LiCl buffer (50 mM Tris pH 8.0, 1 mM EDTA pH 8.0, 1% IGEPAL CA‐630, 0.1% sodium deoxycholate, 500 mM LiCl) and TE/10 buffer (10 mM Tris pH 8.0, 0.1 mM EDTA pH 8.0). The complexes were eluted by proteinase K in proteinase K digestion buffer (20 mM Hepes pH 7.5, 1 mM EDTA pH 8.0, 0.5% SDS) for 15 min at 50°C, followed by adding 3 μl 5 M NaCl and 1 μl 30 mg/ml RNase A for reverse crosslinking overnight at 65°C. The samples were further digested by proteinase K for 1 h at 50°C. The DNAs were then purified by SparQ Pure Mag beads (Quantabio, 95196‐005) according to the manufacturer's instructions and diluted in TE/10 buffer. qPCR was used to amplify immunoprecipitated chromatin fragments and data were presented as the per cent input.
KELLY cells were treated with 100 μM 5,6‐Dichlorobenzimidazole 1‐β‐D‐ribofuranoside (DRB, Merck, D1916‐10MG) for 3 h at 37°C. Cells in washout group were treated with DRB for 3 h at 37°C, followed by replacing with the normal culture medium for 2 h at 37°C. Cells from control, DRB and washout groups were harvested for ChIP‐qPCR.
RNA sequencing
KELLY cells were plated in 6‐well plates and transfected with control siRNA and NONO KD siRNAs at a final concentration of 10 nM using Lipofectamine RNAiMAX for 72 h. Cells (2.5 × 106) were extracted for RNA samples as outlined above. RNA samples were sent to the Australian Genome Research Facility (AGRF) for sequencing. RNA quality was confirmed using a Bioanalyser (Perkin Elmar, MA, USA) prior to RNA‐seq. Whole transcriptome libraries were prepared with the TruSeq stranded total RNA library prep kit (Illumina, CA, USA), and ribosomal RNA was depleted with the Ribo‐Zero‐Gold kit (Illumina, CA, USA). Sequencing was performed using a HiSeq 2000 (Illumina, CA, USA) to generate 50 bp single‐end reads, resulting in an average 17–19 million reads per lane per sample. Reads from two lanes were pooled for each sample to generate 34–38 million reads for each sample.
Phosphoactivatable ribonucleoside‐enhanced crosslinking and immunoprecipitation
KELLY cells were grown on 20 × 14 cm dishes and treated for 14–16 h with 100 μM of 4‐Thiouridine (Merck, T4509). Cells (1.6 × 108) were rinsed with ice‐cold PBS, irradiated at 0.15 J/cm2 with 365 nm UV light, scraped and resuspended in NP40 lysis buffer (50 mM Hepes pH7.5, 1.5 M KCl, 2 mM EDTA, 0.5% NP‐40, 0.5 mM DTT, 1 × protease inhibitor cocktail). Then, cells were lysed and treated with 1 U/μl RNase T1 (Thermo Fisher Scientific, EN0541) for 5 min at room temperature to ensure that only RNA molecules bound by proteins were left. The lysates were pre‐cleared with 20 μl Dynabeads for 30 min at 4°C and then incubated with 100 μl Dynabeads conjugated with 50 μg NONO antibody for 2 h at 4°C. Samples were rinsed three times with NP40 lysis buffer and treated with 0.5 U/μl calf intestinal alkaline phosphatase (New England BioLabs, M0290) in dephosphorylation buffer (50 mM Tris pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT) for 10 min at 37°C. The bead complexes were rinsed twice in phosphatase wash buffer (50 mM Tris pH 7.5, 20 mM EGTA, 0.5% NP40) and twice in polynucleotide kinase buffer (PNK, 50 mM Tris pH 7.5, 50 mM NaCl, 10 mM MgCl2). The samples were end‐labelled with radioactive γ‐32P‐ATP (Perkin Elmar, NEG002250UC) to a final concentration of 0.5 μCi/μl and 0.8 U/μl T4 polynucleotide kinase (New England BioLabs, M0201S) for 30 min at 37°C. Then, the bead complexes were rinsed five times in PNK buffer and resuspended in 2× SDS buffer. After SDS–PAGE electrophoresis, gels were visualised on films and gel bands containing the target crosslinked protein‐RNA complexes cut out. Gel bands were treated to electro‐elution and the eluted complexes in SDS buffer were further incubated with 1% SDS and proteinase K for 30 min at 55°C. RNA extraction was then carried out using the miRNeasy Micro Kit (Qiagen, 217084) and RNA samples were sequenced.
Bioinformatics
Raw sequencing files were quality‐checked using FastQC (version 0.11.5) with all files passing. All subsequent analysis was performed using the gencodev37lift37 transcript model.
Par‐CLIP
PARpipe (https://github.com/ohlerlab/PARpipe) was used with the default parameters to process PAR‐CLIP datasets. In brief, after pre‐processing, the pipeline uses PARalyzer (version 1.5) to identify reads containing T‐to‐C transitions (which are indicative of successful RNA‐protein crosslinking) and aggregates these reads into clusters to define RNA‐protein binding sites. Alignment files from PARpipe were analysed using wavClusteR (version 2.24.0) to determine and annotate binding sites. TPM was calculated as described (Wagner et al, 2012) and used to define NONO‐bound genes as those in the top 20th percentile of genes featuring reads with T‐to‐C transitions. Gene ontology analysis was performed for NONO‐bound genes using Metascape (http://metascape.org; Tripathi et al, 2015), with expressed genes as the background. PAR‐CLIP coverage plots were generated using metagene2 (version 1.6.1).
Gene‐level RNA‐seq
Transcript quantification was performed using salmon (version 1.4.0), summarised to gene‐level counts and imported into R using tximport (1.18.0). Differential expression analysis was performed using DESeq2 (1.30.1) and the default parameters (α = 0.1). In the NONO KD siRNA experiment, four control samples were tested against eight NONO KD samples. Samples from the two NONO KD siRNAs were grouped in an attempt to correct for off‐target effects of the individual siRNAs. Gene ontology analysis was performed for differentially expressed genes using Metascape (http://metascape.org; Tripathi et al, 2015), separating genes by positive or negative log fold change (LFC). Gene Set Enrichment Analysis (GSEA, version 3.0) was also performed using the count data generated above and tested against the Biological Process gene sets maintained at MSigDB (https://software.broadinstitute.org/gsea/msigdb/).
Splicing analysis
IsoformSwitchAnalyzeR (version 1.12.0) was run using output from salmon to look for overall splicing changes. To extend this, DEXSeq (version 1.36.0) was used to extract all exons from the gencodev37lift37 gtf, and custom scripts were used to extract all introns. DESeq2 and DEXSeq were then run to determine differential expression and differential usage, respectively, at the level of exons and introns (separately). Then, for each gene, the maximum length transcript was divided into 100 bins. The position of each significant differentially expressed and differentially used exon and intron was then overlapped with these 100 bins. The number and direction of change of significant events within each bin was summed. Bins for which the proportion of positive fold change events was 2 standard deviations greater than the median proportion were deemed as significant. Heatmaps were created for all genes which showed significant events within the first 13 bins (as defined by significant difference in the proportion of positive and negative events). Heatmaps show the log2 fold change of significant events as a function of their relative position within the gene.
Super‐enhancer detection from ChIP‐seq
Publicly available KELLY (Chipumuro et al, 2014) cell line ChIP‐seq data for the H3K27ac histone mark (GSM1532401) and associated input (GSM1532403) were downloaded from GEO. ChIP‐seq reads were aligned using Bowtie 2 (Langmead & Salzberg, 2012) against the hg19 (GRCh37) reference genome with the parameters ‐k 2 ‐q. Aligned reads were filtered, sorted and indexed using SAMtools (Li et al, 2009) using a minimum mapping quality score of 30. ChIP‐seq peak calling was performed using MACS2 (Zhang et al, 2008) with the parameters: ‐‐keep‐dup auto –p 1 e‐9 ‐B. Super‐enhancer calling was performed using ROSE (Lovén et al, 2013; Whyte et al, 2013) with the parameters: ‐s 12500 ‐t 1000. Promoter‐associated enhancer exclusion in ROSE was changed to a 1,000 bp window (±500 bp) to allow the detection of MYCN‐associated super‐enhancer regions. Annotation of associated gene TSS for super‐enhancer regions was done using the org.Hs.eg.db and TxDb.Hsapiens.UCSC.hg19.knownGene packages in R. All genes whose TSS fell within 600 kbp flanking of detected super‐enhancer start and end sites were included as associated genes.
Survival analysis
The Kaplan–Meier tool from R2 genomics (https://hgserver1.amc.nl/cgi‐bin/r2/main.cgi?option=kaplan_main) was used to identify neuroblastoma datasets which showed MYCN as having a prognostic value for overall survival using the “scan” method and default settings. Using these four datasets (Cangelosi, Maris, SEQC and Versteeg) the tool was then used to identify all genes which were of prognostic value, using the “median” method and default settings. The median method here was used as a more conservative means of identifying prognostic genes of otherwise unproven clinical relevance. Any gene whose expression was significantly associated with prognosis in at least 3 of the 4 datasets was then taken as showing prognostic value in neuroblastoma.
Statistical analyses
All graphs show experiments from at least three biological replicates (n ≥ 3), and these numbers were clearly stated in figures and figure legends. For cell imaging‐related experiments, a two‐tailed unpaired Student's t‐test was conducted assuming unequal variance; graphs with error bars show means with standard deviations. For other experiments, a two‐tailed unpaired Student's t‐test was conducted assuming equal variance; graphs with error bars show means with standard errors. All graphs were generated in R.
Author contributions
Song Zhang: Conceptualization; formal analysis; investigation; writing—original draft; writing—review and editing. Jack AL Cooper: Formal analysis; writing—original draft; writing—review and editing. Yee Seng Chong: Formal analysis; writing—original draft; writing—review and editing. Alina Naveed: Investigation; writing—review and editing. Chelsea Mayoh: Methodology; writing—review and editing. Nisitha Jayatillek: Methodology; writing—review and editing. Tao Liu: Methodology; writing—review and editing. Sebastian Amos: Methodology; writing—review and editing. Simon Kobelke: Investigation; methodology. Andrew C Marshall: Methodology; writing—review and editing. Oliver Meers: Methodology; writing—review and editing. Yu Suk Choi: Methodology; writing—review and editing. Charles S Bond: Conceptualization; methodology; writing—review and editing. Archa H Fox: Conceptualization; formal analysis; supervision; funding acquisition; investigation; writing—original draft; writing—review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
PDF+
Acknowledgements
We thank other members of the Fox lab for helpful discussions about the manuscript. This work was supported by a research grant APP1147496 from the National Health and Medical Research Council of Australia to AF and CB and FT180100204 from the Australian Research Council to AF. Open access publishing facilitated by The University of Western Australia, as part of the Wiley – The University of Western Australia agreement via the Council of Australian University Librarians.
EMBO reports (2023) 24: e54977
Data availability
The datasets produced in this study are available in the following databases:
PAR‐CLIP seq: GEO database GSE114376, Secure reviewer token: ohchqogczxuztqz (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114376).
RNA‐Seq: GEO database GSE114376, Secure reviewer token: ohchqogczxuztqz (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114376).
References
- Adelman K, Lis JT (2012) Promoter‐proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Dormann D (2019) Liquid‐liquid phase separation in disease. Annu Rev Genet 53: 171–194 [DOI] [PubMed] [Google Scholar]
- Benegiamo G, Mure LS, Erikson G, Le HD, Moriggi E, Brown SA, Panda S (2018) The RNA‐binding protein NONO coordinates hepatic adaptation to feeding. Cell Metab 27: 404–418.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V, Louis‐Brennetot C, Peltier A, Durand S, Pierre‐Eugène C, Raynal V, Etchevers HC, Thomas S, Lermine A, Daudigeos‐Dubus E et al (2017) Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet 49: 1408–1413 [DOI] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall'Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM et al (2018) Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell 175: 1842–1855.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Frese CK, Horos R, Alleaume AM, Foehr S, Curk T, Krijgsveld J, Hentze MW (2016) Comprehensive identification of RNA‐binding domains in human cells. Mol Cell 63: 696–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MGM, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG et al (2013) Discovery and characterization of super‐enhancer‐associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24: 777–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A et al (2014) CDK7 inhibition suppresses super‐enhancer‐linked oncogenic transcription in MYCN‐driven cancer. Cell 159: 1126–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert‐Leahey N, He S, Krill‐Burger JM, Root DE, Vazquez F et al (2018) Selective gene dependencies in MYCN‐amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet 50: 1240–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrensberger AH, Kelly GP, Svejstrup JQ (2013) Mechanistic interpretation of promoter‐proximal peaks and RNAPII density maps. Cell 154: 713–715 [DOI] [PubMed] [Google Scholar]
- Emili A, Shales M, McCracken S, Xie W, Tucker PW, Kobayashi R, Blencowe BJ, Ingles CJ (2002) Splicing and transcription‐associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. RNA 8: 1102–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan XJ, Wang YL, Zhao WW, Bai SM, Ma Y, Yin XK, Feng LL, Feng WX, Wang YN, Liu Q et al (2021) NONO phase separation enhances DNA damage repair by accelerating nuclear EGFR‐induced DNA‐PK activation. Am J Cancer Res 11: 2838–2852 [PMC free article] [PubMed] [Google Scholar]
- Feng P, Li L, Deng T, Liu Y, Ling N, Qiu S, Zhang L, Peng B, Xiong W, Cao L et al (2020) NONO and tumorigenesis: more than splicing. J Cell Mol Med 24: 4368–4376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, Bond CS, Lamond AI (2005) P54nrb forms a heterodimer with PSP1 that localizes to paraspeckles in an RNA‐dependent manner. Mol Biol Cell 16: 5304–5315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, Nakagawa S, Hirose T, Bond CS (2018) Paraspeckles: where long noncoding RNA meets phase separation. Trends Biochem Sci 43: 124–135 [DOI] [PubMed] [Google Scholar]
- Gartlgruber M, Sharma AK, Quintero A, Dreidax D, Jansky S, Park Y‐G, Kreth S, Meder J, Doncevic D, Saary P et al (2021) Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat Cancer 2: 114–128 [DOI] [PubMed] [Google Scholar]
- Girard C, Will CL, Peng J, Makarov EM, Kastner B, Lemm I, Urlaub H, Hartmuth K, Lührmann R (2012) Post‐transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat Commun 3: 994 [DOI] [PubMed] [Google Scholar]
- Gotor NL, Armaos A, Calloni G, Torrent Burgas M, Vabulas RM, De Groot NS, Tartaglia GG (2020) RNA‐binding and prion domains: The Yin and Yang of phase separation. Nucleic Acids Res 48: 9491–9504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, Broekmans M, Haneveld F, Nowakowska NE, Bras J et al (2017) Neuroblastoma is composed of two super‐enhancer‐associated differentiation states. Nat Genet 49: 1261–1266 [DOI] [PubMed] [Google Scholar]
- Guallar D, Bi X, Pardavila JA, Huang X, Saenz C, Shi X, Zhou H, Faiola F, Ding J, Haruehanroengra P et al (2018) RNA‐dependent chromatin targeting of TET2 for endogenous retrovirus control in pluripotent stem cells. Nat Genet 50: 443–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall'Agnese A, Hannett NM, Spille J‐H, Afeyan LK, Zamudio AV, Shrinivas K et al (2019) Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572: 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase R, Jain A, Rigaud S, Vorkel D, Rajasekhar P, Suckert T, Lambert TJ, Nunez‐Iglesias J, Poole DP, Tomancak P et al (2020a) Interactive design of GPU‐accelerated image data flow graphs and cross‐platform deployment using multi‐lingual code generation. bioRxiv 10.1101/2020.11.19.386565 [PREPRINT] [DOI]
- Haase R, Royer LA, Steinbach P, Schmidt D, Dibrov A, Schmidt U, Weigert M, Maghelli N, Tomancak P, Jug F et al (2020b) CLIJ: GPU‐accelerated image processing for everyone. Nat Methods 17: 5–6 [DOI] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp AC, Munschauer M et al (2010) PAR‐CliP‐‐a method to identify transcriptome‐wide the binding sites of RNA binding proteins. J Vis Exp: 2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, Knott GJ, Iyer KS, Ho D, Newcombe EA et al (2015) Prion‐like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J Cell Biol 210: 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, Adelman K (2018) Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 32: 26–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, Preiss T (2018) A brave new world of RNA‐binding proteins. Nat Rev Mol Cell Biol 19: 327–341 [DOI] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA (2017) A phase separation model for transcriptional control. Cell 169: 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa M, Takeuchi A, Tanihata J, Iida K, Takeda S, Hagiwara M (2019) Loss of RNA‐binding protein Sfpq causes long‐gene Transcriptopathy in skeletal muscle and severe muscle mass reduction with metabolic myopathy. iScience 13: 229–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Li W, Yao X, Lin Q‐J, Yin J‐W, Liang Y, Heiner M, Tian B, Hui J, Wang G (2012) Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol Cell 45: 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida K, Hagiwara M, Takeuchi A (2020) Multilateral bioinformatics analyses reveal the function‐oriented target specificities and recognition of the RNA‐binding protein SFPQ. iScience 23: 101325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janoueix‐Lerosey I, Schleiermacher G, Delattre O (2010) Molecular pathogenesis of peripheral neuroblastic tumors. Oncogene 29: 1566–1579 [DOI] [PubMed] [Google Scholar]
- Jiang L, Shao C, Wu Q‐J, Chen G, Zhou J, Yang B, Li H, Gou L‐T, Zhang Y, Wang Y et al (2017) NEAT1 scaffolds RNA‐binding proteins and the microprocessor to globally enhance pri‐miRNA processing. Nat Struct Mol Biol 24: 816–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameoka S, Duque P, Konarska MM (2004) p54(nrb) associates with the 5′ splice site within large transcription/splicing complexes. EMBO J 23: 1782–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamieniarz‐Gdula K, Proudfoot NJ (2019) Transcriptional control by premature termination: a forgotten mechanism. Trends Genet 35: 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Rozenblatt‐Rosen O, Meyerson M, Manley JL (2007) The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre‐mRNA 3′ processing and transcription termination. Genes Dev 21: 1779–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, Lee M, Passon DM, Fox AH, Bond CS (2015) Caenorhabditis elegans NONO‐1: insights into DBHS protein structure, architecture, and function. Protein Sci 24: 2033–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, Bond CS, Fox AH (2016) The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res 44: 3989–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, Chong YS, Passon DM, Liang XH, Deplazes E, Conte MR, Marshall AC, Lee M, Fox AH, Bond CS (2021) Structural basis of dimerization and nucleic acid binding of human DBHS proteins NONO and PSPC1. Nucleic Acids Res 50: 522–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krietsch J, Caron M‐C, Gagné J‐P, Ethier C, Vignard J, Vincent M, Rouleau M, Hendzel MJ, Poirier GG, Masson J‐Y (2012) PARP activation regulates the RNA‐binding protein NONO in the DNA damage response to DNA double‐strand breaks. Nucleic Acids Res 40: 10287–10301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara S, Ikei A, Taguchi Y, Tabuchi Y, Fujimoto N, Obinata M, Uesugi S, Kurihara Y (2006) PSPC1, NONO, and SFPQ are expressed in mouse Sertoli cells and may function as coregulators of androgen receptor‐mediated Transcription1. Biol Reprod 75: 352–359 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Li X, Hechmer A, Eisen M, Biggin M, Venters BJ, Jiang C, Li J, Pugh B, Gilmour D (2008) NELF and GAGA factor are linked to promoter‐proximal pausing at many genes in drosophila. Mol Cell Biol 28: 3290–3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legland D, Arganda‐Carreras I, Andrey P (2016) MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32: 3532–3534 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li Z, Shu FJ, Xiong H, Phillips AC, Dynan WS (2014) Double‐strand break repair deficiency in NONO knockout murine embryonic fibroblasts and compensation by spontaneous upregulation of the PSPC1 paralog. Nucleic Acids Res 42: 9771–9780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Harvey AR, Hodgetts SI, Fox AH (2017) Functional dissection of NEAT1 using genome editing reveals substantial localization of the NEAT1_1 isoform outside paraspeckles. RNA 23: 872–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Karwacki‐Neisius V, Ma C, Tan L, Shi Y, Wu F, Shi YG (2020) Nono deficiency compromises TET1 chromatin association and impedes neuronal differentiation of mouse embryonic stem cells. Nucleic Acids Res 48: 4827–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Cui P, Sun Q, Du Z, Chen Z, Li Z, Liu C, Cao Y, Yang Z, Liu R et al (2021) PSPC1 regulates CHK1 phosphorylation through phase separation and participates in mouse oocyte maturation. Acta Biochim Biophys Sin 53: 1527–1537 [DOI] [PubMed] [Google Scholar]
- Liu PY, Erriquez D, Marshall GM, Tee AE, Polly P, Wong M, Liu B, Bell JL, Zhang XD, Milazzo G et al (2014) Effects of a novel long noncoding RNA, lncUSMycN, on N‐Myc expression and neuroblastoma progression. J Natl Cancer Inst 106: dju113 [DOI] [PubMed] [Google Scholar]
- Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA (2013) Selective inhibition of tumor oncogenes by disruption of super‐enhancers. Cell 153: 320–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luisier R, Tyzack GE, Hall CE, Mitchell JS, Devine H, Taha DM, Malik B, Meyer I, Greensmith L, Newcombe J et al (2018) Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat Commun 9: 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Karwacki‐Neisius V, Tang H, Li W, Shi Z, Hu H, Xu W, Wang Z, Kong L, Lv R et al (2016) Nono, a bivalent domain factor, regulates Erk signaling and mouse embryonic stem cell pluripotency. Cell Rep 17: 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillén‐Boixet J, Franzmann TM et al (2018) RNA buffers the phase separation behavior of prion‐like RNA binding proteins. Science 360: 918–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzer C, Menegaux F, Lacour B, Valteau‐Couanet D, Michon J, Coze C, Bergeron C, Auvrignon A, Bernard F, Thomas C et al (2008) Birth‐related characteristics, congenital malformation, maternal reproductive history and neuroblastoma: The ESCALE study (SFCE). Int J Cancer 122: 2315–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveed A, Cooper JA, Li R, Hubbard A, Chen J, Liu T, Wilton SD, Fletcher S, Fox AH (2021) NEAT1 polyA‐modulating antisense oligonucleotides reveal opposing functions for both long non‐coding RNA isoforms in neuroblastoma. Cell Mol Life Sci 78: 2213–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldridge DA, Wood AC, Weichert‐Leahey N, Crimmins I, Sussman R, Winter C, McDaniel LD, Diamond M, Hart LS, Zhu S et al (2015) Genetic predisposition to neuroblastoma mediated by a LMO1 super‐enhancer polymorphism. Nature 528: 418–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollion J, Cochennec J, Loll F, Escudé C, Boudier T (2013) TANGO: a generic tool for high‐throughput 3D image analysis for studying nuclear organization. Bioinformatics 29: 1840–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passon DM, Lee M, Rackham O, Stanley WA, Sadowska A, Filipovska A, Fox AH, Bond CS (2012) Structure of the heterodimer of human NONO and paraspeckle protein component 1 and analysis of its role in subnuclear body formation. Proc Natl Acad Sci USA 109: 4846–4850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW‐L, Maquat LE (2013) Organizing principles of mammalian nonsense‐mediated mRNA decay. Annu Rev Genet 47: 139–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P et al (2013) Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov 3: 308–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, Chang HY (2016) Unique features of long non‐coding RNA biogenesis and function. Nat Rev Genet 17: 47–62 [DOI] [PubMed] [Google Scholar]
- Rasmussen EB, Lis JT (1993) In vivo transcriptional pausing and cap formation on three drosophila heat shock genes. Proc Natl Acad Sci USA 90: 7923–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer H (2011) Transcriptional control of differentiation and neurogenesis in autonomic ganglia. Eur J Neurosci 34: 1563–1573 [DOI] [PubMed] [Google Scholar]
- Rougvie AE, Lis JT (1988) The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell 54: 795–804 [DOI] [PubMed] [Google Scholar]
- Sabari BR, Dall'Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC et al (2018) Coactivator condensation at super‐enhancers links phase separation and gene control. Science 361: eaar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Bi X, Pan Y, Gao B, Wu J, Yin Y, Liu Z, Peng M, Zhang W, Jiang X et al (2022) Phase separation of RNA‐binding protein promotes polymerase binding and transcription. Nat Chem Biol 18: 70–80 [DOI] [PubMed] [Google Scholar]
- Spegg V, Altmeyer M (2021) Biomolecular condensates at sites of DNA damage: more than just a phase. DNA Repair 106: 103179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagsted LVW, O'Leary ET, Ebbesen KK, Hansen TB (2021) The RNA‐binding protein SFPQ preserves long‐intron splicing and regulates circRNA biogenesis in mammals. Elife 10: e63088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama KI, Suzuki T, Fujimura T, Yamada Y, Takahashi S, Homma Y, Suzuki Y, Inoue S (2017) Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA‐binding protein PSF. Proc Natl Acad Sci USA 114: 10461–10466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Iida K, Tsubota T, Hosokawa M, Denawa M, Brown JB, Ninomiya K, Ito M, Kimura H, Abe T et al (2018) Loss of Sfpq causes long‐gene transcriptopathy in the brain. Cell Rep 23: 1326–1341 [DOI] [PubMed] [Google Scholar]
- Tang F, Yang Z, Tan Y, Li Y (2020) Super‐enhancer function and its application in cancer targeted therapy. NPJ Precis Oncol 4: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez‐Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC et al (2015) Meta‐ and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18: 723–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen J‐Y, Cody NAL, Dominguez D et al (2020) A large‐scale binding and functional map of human RNA‐binding proteins. Nature 583: 711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers TA, Rahdar M, Prakash TP, Crooke ST (2019) Kinetic and subcellular analysis of PS‐ASO/protein interactions with P54nrb and RNase H1. Nucleic Acids Res 47: 10865–10880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth H, Oberthuer A, Simon T, Kahlert Y, Berthold F, Fischer M (2009) Co‐regulated expression of HAND2 and DEIN by a bidirectional promoter with asymmetrical activity in neuroblastoma. BMC Mol Biol 10: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GP, Kin K, Lynch VJ (2012) Measurement of mRNA abundance using RNA‐seq data: RPKM measure is inconsistent among samples. Theory Biosci 131: 281–285 [DOI] [PubMed] [Google Scholar]
- Wei Y, Luo H, Yee PP, Zhang L, Liu Z, Zheng H, Zhang L, Anderson B, Tang M, Huang S et al (2021) Paraspeckle protein NONO promotes TAZ phase separation in the nucleus to drive the oncogenic transcriptional program. Adv Sci (Weinh) 8: e2102653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA (2013) Master transcription factors and mediator establish super‐enhancers at key cell identity genes. Cell 153: 307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedner HJ, Giudice J (2021) It's not just a phase: function and characteristics of RNA‐binding proteins in phase separation. Nat Struct Mol Biol 28: 465–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett RT, Greene LA (2011) Gata2 is required for migration and differentiation of retinorecipient neurons in the superior colliculus. J Neurosci 31: 4444–4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, Chen JY, Liang Z, Luo D, Chen G, Lu ZJ, Chen Y, Zhou B, Li H, Du X et al (2019) Pervasive chromatin‐RNA binding protein interactions enable RNA‐based regulation of transcription. Cell 178: 107–121.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav SP, Hao H, Yang HJ, Kautzmann MA, Brooks M, Nellissery J, Klocke B, Seifert M, Swaroop A (2014) The transcription‐splicing protein NonO/p54nrb and three NonO‐interacting proteins bind to distal enhancer region and augment rhodopsin expression. Hum Mol Genet 23: 2132–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R, Osawa T, Sasaki Y, Yamamoto S, Anai M, Izumi K, Matsumura Y, Sakai J, Aburatani H, Mizokami A et al (2018) Overexpression of p54(nrb)/NONO induces differential EPHA6 splicing and contributes to castration‐resistant prostate cancer growth. Oncotarget 9: 10510–10524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G, Hirose T (2018) Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol Cell 70: 1038–1053.e7 [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Yamamoto T, Yoshino H, Souquere S, Nakagawa S, Pierron G, Hirose T (2021) Paraspeckles are constructed as block copolymer micelles. EMBO J 40: e107270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YS, Yang MC, Tucker PW, Capra JD (1997) NonO enhances the association of many DNA‐binding proteins to their targets. Nucleic Acids Res 25: 2284–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom KH, Pan Z, Lin CH, Lim HY, Xiao W, Xing Y, Black DL (2021) Tracking pre‐mRNA maturation across subcellular compartments identifies developmental gene regulation through intron retention and nuclear anchoring. Genome Res 31: 1106–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbinden A, Pérez‐Berlanga M, De Rossi P, Polymenidou M (2020) Phase separation and neurodegenerative diseases: a disturbance in the force. Dev Cell 55: 45–68 [DOI] [PubMed] [Google Scholar]
- Zhang WJ, Wu JY (1996) Functional properties of p54, a novel SR protein active in constitutive and alternative splicing. Mol Cell Biol 16: 5400–5408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]