

Summary
To assess the transcriptomic profile of disease-specific cell populations, fibroblasts from patients with primary open-angle glaucoma (POAG) were reprogrammed into induced pluripotent stem cells (iPSCs) before being differentiated into retinal organoids and compared with those from healthy individuals. We performed single-cell RNA sequencing of a total of 247,520 cells and identified cluster-specific molecular signatures. Comparing the gene expression profile between cases and controls, we identified novel genetic associations for this blinding disease. Expression quantitative trait mapping identified a total of 4,443 significant loci across all cell types, 312 of which are specific to the retinal ganglion cell subpopulations, which ultimately degenerate in POAG. Transcriptome-wide association analysis identified genes at loci previously associated with POAG, and analysis, conditional on disease status, implicated 97 statistically significant retinal ganglion cell-specific expression quantitative trait loci. This work highlights the power of large-scale iPSC studies to uncover context-specific profiles for a genetically complex disease.
Keywords: human induced pluripotent stem cells, retinal organoids, retinal ganglion cells, single-cell RNA sequencing, glaucoma, transcriptomics, eQTL
Graphical abstract
Highlights
-
•
Large-scale scRNA-seq study of human iPSC-derived retinal organoids
-
•
23 subpopulations spread across 247,520 cells
-
•
Identified 312 significant eQTLs specific to retinal ganglion cells
-
•
Identified eQTLs associated with POAG
Daniszewski et al. performed scRNA-seq on retinal organoids derived from 110 human iPSC lines. They identified 97 expression qualitative trait loci specific to the ganglion cell population and associated with POAG. The data are a valuable resource to improve our understanding of a genetically complex disease like glaucoma.
Introduction
Glaucoma is the leading cause of irreversible blindness worldwide and experts predict it will affect approximately 80 million people by 2040.1 The most common subtype—primary open-angle glaucoma (POAG)—is characterized by an open iridotrabecular meshwork angle and progressive degeneration of retinal ganglion cells (RGCs), which culminates in loss of visual field.2 Therapeutic options are currently limited; all are directed at lowering intraocular pressure (IOP), which has been shown to slow but not fully prevent or reverse visual loss.3 Elevated IOP was long considered a distinguishing feature of POAG; however, it is now clear that it is not a direct determinant of disease development.4 Patients with elevated IOP may not develop glaucomatous optic neuropathy, while those with IOP within the normal population range may sustain significant RGC loss.5, 6, 7
POAG has one of the highest heritabilities of all common and complex diseases,8,9 and much work has focused on dissecting its genetic architecture. The genetic etiology of POAG is varied: rare genetic variants, e.g., myocilin (MYOC)10 and optineurin (OPTN),11 cause disease with high penetrance, while common variants have smaller effect sizes. Genome-wide association analyses (GWAS) have identified over 100 independent loci that carry a common risk allele associated with an increased risk of open-angle glaucoma,12 although many of these are associated with variation in IOP.13, 14, 15, 16, 17, 18, 19, 20 Unlike rare variants that largely contribute to disease by changing protein coding, common variants predominantly act via changes in gene regulation.21 Mapping expression quantitative trait loci (eQTL) is one of the powerful approaches used to provide functional evidence of the mechanisms of the common genetic variants, allowing the allelic effect of a variant on disease risk to be linked to changes in gene expression. To avoid spurious associations, and to best understand the cellular effects of changes in gene expression, eQTL mapping needs to be conducted for cells that are pathophysiologically relevant to the disease. In the case of glaucoma, this includes trabecular meshwork cells and RGCs.
The molecular profiling of RGCs in normal and diseased tissue would improve our understanding of the mechanisms that bear a disease risk or contribute to glaucoma development. Unfortunately, it is impossible to obtain or molecularly profile RGCs from living donors in a non-invasive manner. To overcome this, somatic cells can be reprogrammed into patient-specific induced pluripotent stem cells (iPSCs),22,23 which can then be differentiated into RGCs.24,25 Over the years, multiple protocols have been developed to generate RGCs in vitro.26 Human retinal organoids show a stratified cellular organization closely resembling the developing human neural retina,27, 28, 29, 30, 31, 32 and thus it is now possible to generate organoid-derived RGCs for downstream applications, including disease modeling25,33,34 and cell transplantation.35 These constructs can also be subjected to single-cell RNA sequencing (scRNA-seq) to distinguish cell types based on their unique transcriptional signature and examine rare populations that would be missed using bulk RNA-seq.36, 37, 38 Here, we used scRNA-seq to characterize the transcriptomic profile of the organoid-derived retinal cells, in particular RGCs generated from a large cohort of the patient-derived iPSCs. We identified a number of cell-type- and disease-specific eQTLs. Using an additive linear model, a total of 4,443 eQTLs were found to be associated with 3,860 SNPs. By combining our data with recent GWAS results in a transcriptome-wide association study (TWAS), seven genes were identified to be significantly associated with glaucoma development.
Results
Large-scale generation of patient iPSCs, differentiation into retinal organoids, and scRNA-seq
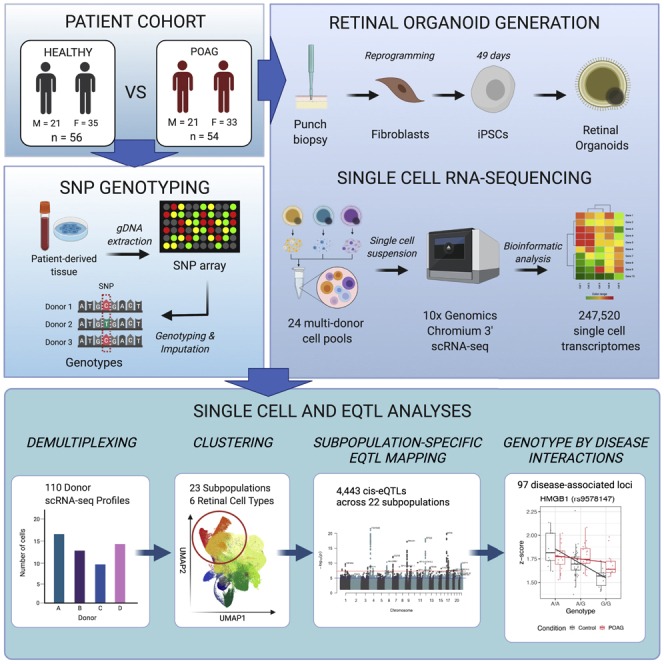
We recruited a large cohort of 183 individuals, which included healthy (n = 92, of whom 50 were female) and patients with advanced POAG (n = 91, of whom 50 were female) (Table S1). The mean ± SD age at biopsy for controls was 68.1 ± 8.2 years, and for case subjects it was 69.1 ± 14.4 years. Participants underwent skin biopsy, and fibroblasts were cultured and reprogrammed to iPSCs using episomal vectors as we described previously.39 Genotyping data were also generated from participants and, after quality control and imputation, yielded 7,691,208 autosomal SNPs at a minor allele frequency above 0.01.
iPSC lines were differentiated in batches (25 batches, 6–8 lines with equal numbers of control and POAG lines per batch) to neural retina for 28 days in adherent cultures. Retinal organoids were then excised, cultured in suspension for 7 days and plated onto Matrigel for an additional 2-week period to allow neuronal outgrowth from RGCs, and harvested for scRNA-seq (Figure 1A). This timeline was based on work by others, which described RGC emergence by day 35 of retinal organoid differentiation29 and RGC neurite extension following plating of dissociated organoids by day 40.30 Twenty-two lines did not differentiate to retinal organoids and were discarded (healthy, 11 lines, 1 of which was female; POAG, 11 lines, 5 of which were female) (Figure S1). Cells from the remaining 161 individual cell lines were harvested and divided into 25 batches for scRNA-seq, with each batch containing cells from 6 to 8 cell lines with a targeted capture of 2,000 cells per line.
Figure 1.

Generation of retinal organoids and identification and characterization of cell subpopulations
(A) Retinal organoids were generated from iPSCs over a period of 49 days. iPSCs were differentiated as a monolayer for the first 28 days, and then cultured in 3D as a suspension for 7 days. Resulting organoids were then plated onto Matrigel and grown until retinal ganglion cells started to project out of the organoid at 49 days. These were harvested for scRNA-seq.
(B) Uniform Manifold Approximation and Projection (UMAP) representation of cells grouped into 23 subpopulations, as identified by Louvain clustering.
(C) UMAP plot of the cell types and lineages, as determined by analysis of differentially expressed genes of individual subpopulations and trajectory analysis. RGC clusters form one branch of the trajectory. Other cell types—RPE, interneurons, photoreceptors, and lens—arise from another branch of the trajectory. The last main branch consists of differentiating RPC subpopulations. RPC, retinal progenitor cell; RGC, retinal ganglion cell; RPE, retinal pigmented epithelium.
(D) Heatmap of the mean expression of cell-type-specific gene markers across each subpopulation. Expression values have been scaled and converted to Z scores and genes have been grouped by cell type. AC, amacrine cell; BC, bipolar cell; HC, horizontal cell; MG, Müller glia; PR, photoreceptor; RPC, retinal progenitor cell; RPE, retinal pigment epithelium; RGC, retinal ganglion cell.
A total number of 330,569 cells were captured via scRNA-seq and sequenced to a mean read depth of 41,020 per cell (Table S2). Individual cells were traced back to their cell-line donor using a combination of transcriptome- and genotype-based methods (Table S3). Lines were removed based on the following criteria: failed genotype and virtual karyotyping quality control, monogenic POAG, non-European background, and low cell-capture numbers (Figure S1). Individual cells were removed based on scRNA-seq metrics as described in the STAR Methods. A total of 247,520 cells (healthy, 128,175; POAG, 119,345) from 110 iPSC lines (healthy, 56 of which 35 were female, mean ± SD age of samples 67.5 ± 7.8 years; POAG, 54 of which 33 were female, 71.8 ± 11.5 years) were retained for subsequent analyses.
Identification and characterization of 23 subpopulations from 247,520 cells
Clustering identified 23 subpopulations distributed across all cell lines and conditions (Figures 1B and 1C). Differential expression markers were used to classify the subpopulations to different retinal cell classes based on canonical markers40, 41, 42, 43 (Figure 1D; Tables S4 and S5). We compared the distribution of cell types between patients with POAG and healthy controls and observed no statistically significant differences between the groups after correction for multiple testing (Figure S2; Table S6). However, the proportions of subpopulations between individual cell lines differed (Pearson’s chi-square test, p < 2.2 × 10−16).
Retinal progenitor cells (RPCs) represented 77.4% of all cells and localized across 16 subpopulations. RPCs expressed PAX6 and SOX2 transcription factors, which are key regulators of neuronal fate,44,45 LHX2, required for maintenance of open chromatin during retinogenesis46 and gliogenesis,47 and the RPC markers VSX2 and ASCL1 (Figures 1D and S2B). Cell-cycle genes were not evenly distributed within progenitor subpopulations. The G2/M phase marker MKi67 was predominantly expressed by cells in RPC1, RPC2, and RPC5. The S phase marker PCNA was distributed more broadly; however, the majority of PCNA-positive cells were identified in RPC clusters 1, 2, 4, and 5 (Figure 1D).
RGCs were the second most predominant cell group, representing 17.0% of all cells across the cohort. Based on previous work, RGCs were classified by the expression of the following genes: POU4F2, ISL1, RBPMS, SNCG, GAP43, NEFL/M, ELAVL4, EOMES, and DCX.48, 49, 50 Three distinct RGC subpopulations (RGC1-3), arising from one subpopulation of RPCs (RPC9), were identified (Figures 1B–1D; Table S4). Pseudotime analysis confirmed the lineage development of RGC1-3 cell types from a single progenitor population (Figure S3), with POU4F2 and ISL1 showing increasing expression as cells differentiated further from a progenitor state (Figure 1D). The expression of both genes was required for RGC specification and differentiation.51, 52, 53 POU4F2 expression was generally higher in the RGC1 and RGC2 subpopulations than in RGC3, while ISL1 expression was higher in RGC1 and RGC3 compared with RGC2. The low levels of ATOH7 expression in RGC1 and RGC3, in conjunction with the fact that cells in these subpopulations expressed markers of differentiated RGCs, such as SNCG, RBPMS, GAP43, and NEFM (Figure 1D), suggests that these subpopulations represent more mature RGCs compared with those from RGC2. We also identified cells expressing markers for photoreceptors/bipolar cells (2.6%) and interneurons (1.7%; Table S4). Retinal pigmented epithelial (RPE) cells localized in one subpopulation and constituted 1.3% of all cells (Table S4). No Müller cells were identified. The various subpopulations are fully described in Data S1. These data are consistent with Sridhar et al.,42 who also found that RPCs and RGCs are the predominant populations of cells within early retinal organoids. Finally, to directly compare the similarity of our iPSC-derived retinal cell types with actual somatic cells, we used scPred—an unbiased gene-marker free cell classification method.54 Our cells were re-classified based on a previously described adult human retinal RNA-seq dataset,55 and a strong overlap between cell type classifications was observed (Figure S3C).
Trajectory analysis of subpopulations reveals disease-specific mechanisms in RGC lineages
We studied the ordering of the subpopulations across pseudotime by performing trajectory inference using the slingshot package,56 as described in STAR Methods. Trajectory inference revealed a complex, branching trajectory that consisted of 12 lineages (Figures 1C and 2A). Three of these lineages (6, 7, and 9) comprise RGC subpopulations branching off from RPC9 (Figures 2A and 2B). We examined these lineages in greater detail by studying gene expression patterns related to disease and pseudotime. Gene ontology analysis of the RGC lineages revealed an overrepresentation of genes involved in neurogenesis (Figure 2C). Further to this, there was a significant difference in the distribution of cells across pseudotime in the lineage terminating with RGC3 (lineage 7) based on disease status (Kolmogorov-Smirnov test: p = 0.028).
Figure 2.

Trajectory analysis of cell subpopulations and characterization of pseudotime-related disease pathways
(A) Global lineages were constructed using proliferative subpopulation, RPC1 as the origin, and subpopulations RGC1, photoreceptors, interneurons, RPE, and lens as the endpoints. This resulted in a branching trajectory, with RGC subpopulations—RGC1, RGC2, RGC3, and their progenitors—RPC9 and RPC13, segmenting off the main trajectory at RPC11.
(B) The branching trajectory consists of 12 lineages. Three of these lineages, 6, 7, and 9, belong to the RGC subpopulations.
(C) Pathways associated with the RGC lineage over pseudotime.
(D) Relationship between genotype and expression profiles of HMGB1 at SNP rs9578147, in subpopulations RGC1, RGC2, RGC3, and RPE.
We then used tradeSeq57 to investigate the nature of this lineage and determine if disease status affected gene expression patterns across the trajectory. We identified 1,471 genes that were differentially expressed between the conditions, across pseudotime (Benjamini-Hochberg FDR < 0.05) (Table S7). Disease ontology of these genes was performed using gene set enrichment analysis,58 and revealed association with four disease pathways—schizophrenia, psychotic disorders, disease of mental health, and cognitive disorder, as annotated by the Disease Ontology database.59 We also applied pseudotime to cis-eQTL mapping to determine if this had a significant interaction between genotype and POAG. This uncovered one new eGene—HMGB1—that had pseudotime as a significant interaction term (p = 1.76 × 10−7) at SNP rs9578147 (Figure 2D). Interestingly, HMGB1 is involved with nucleosome stabilization, and is released from injured cells and induces an inflammatory response.60 It has been shown to induce RGC death in NMDA-mediated retinal neurodegeneration,61,62 and is present in glaucomatous retina.63
The genetic control of gene expression is highly cell-type specific
We tested for cis-eQTL for each cell population independently and identified a total of 4,443 eQTL that surpassed the study-wide significance threshold of FDR < 0.05 (Benjamini-Hochberg procedure), and the eGene was expressed in at least 30% of tested donors (Table 1; for full results, see Table S8). The mean ± SD of eQTL identified per cell subpopulation was 202 ± 120 (Table 2), indicating consistent power to detect eQTL and a similarity of cell types in each population as expected. We assessed the overlap of eQTL between cell types and found that the majority of cis-eQTL are cell type specific (Figure 3A). A total of 647 out of 3,091 genes with an eQTL (eGenes) were detected in more than one cell type, and only 215 of these eGenes had an eQTL observed in two or more cell types (Figures 3B and 3C; Table S9). RPE and RPC14 did not have any overlapping eQTL with any other cell types, while RPC1 and RGC1 had the greatest number of overlapping eQTL (27) (Pearson’s correlation: r2 = 0.46, p = 0.02) (Figure 3B). As the majority of subpopulations are retinal progenitors, non-RPC subpopulations share more eQTL signals with RPC subpopulations than each other. Two genes had an eQTL in all subpopulations but RPC2—RPS26 and GSTT1 (Figure 3D). Only GSTT1 had overlapping eQTL in 16 subpopulations, which indicates that the variants associated with this eQTL are either in linkage disequilibrium with each other or are targeting the same causal variant. Approximately half of the eGenes detected in the RGC subpopulations (RGC1, 46.9%; RGC2, 58.9%; and RGC3, 51.1%) were exclusive to this cell type, and only seven of these eGenes (PPP1R17, RASD1, NXPH1, IGFBPL1, SAPCD2, KRTAP5-AS1, and TK1) were found in at least one RGC subpopulation (Figure 3D).
Table 1.
Breakdown of significant cis-eQTLs detected in the full cohort and by disease status-conditional tests
| Model | No. of eQTL | No. of eGenes | No. of eSNPs |
|---|---|---|---|
| Population | 4,484 | 3,102 | 3,892 |
| Disease | 4,443 | 3,091 | 3,860 |
| Control | 2,985 | 2,394 | 2,492 |
| POAG | 2,460 | 2,090 | 2,136 |
The relationship between genotype and expression was tested at loci within 1 MB of each gene, using four different models. Population and disease models were tested using Z scores of quantile-normalized mean expression of all donors, and the population model was used to test control and POAG donors separately. eQTL were significant based on the following criteria: FDR < 0.05, gene expressed in at least 30% of the donors.
Table 2.
Summary of lead cis-eQTL per subpopulation
| Subpopulation | No. of lead cis-eQTL | No. of eGenes | No. of eSNPs |
|---|---|---|---|
| RPC1 | 539 | 539 | 537 |
| RPC2 | 233 | 233 | 232 |
| RPC3 | 180 | 180 | 180 |
| RPC4 | 377 | 377 | 368 |
| RPC5 | 245 | 245 | 245 |
| RPC6 | 115 | 115 | 115 |
| RPC7 | 108 | 108 | 103 |
| RPC8 | 268 | 268 | 263 |
| RPC9 | 122 | 122 | 113 |
| RPC10 | 261 | 261 | 257 |
| RPC11 | 205 | 205 | 201 |
| RPC12 | 227 | 227 | 226 |
| RPC13 | 147 | 147 | 147 |
| RPC14 | 38 | 38 | 38 |
| RPC15 | 208 | 208 | 206 |
| RGC1 | 382 | 382 | 381 |
| RGC2 | 124 | 124 | 123 |
| RGC3 | 131 | 131 | 130 |
| Interneurons | 245 | 245 | 239 |
| Photoreceptors | 184 | 184 | 183 |
| RPE | 97 | 97 | 94 |
| Lens | 7 | 7 | 7 |
cis-eQTL from disease model were filtered by the following thresholds: FDR < 0.05; eGene expression > 30%. Lead cis-eQTL were selected based on SNP-gene interaction with lowest FDR value.
Figure 3.

Cell subpopulation-specific eQTLs—Summary of eQTLs specific to cell subpopulations
(A) Minimal overlap of genes with significant eQTLs (eGenes) show that they are predominantly subpopulation specific.
(B) For eGenes present in more than one subpopulation, we compared the allelic effect from the original lead eQTL in one subpopulation with the allelic effect after conditioning the eQTL with an eSNP for the same eGene from another subpopulation. The heatmap represents the pairwise correlations between allelic effects of each tested subpopulation. The scatterplots represent the allelic effects—measured as beta—in RGC1 conditioned against RGC2 and RGC1 conditioned against RPC1. The original beta values are plotted on the x axis, and the conditioned beta values are plotted on the y axis. The color of the points shows if the change in allelic effect was significant (red).
(C) Chromosomal map of significant loci in RGC subpopulations RGC1 (light orange), RGC2 (red), RGC3 (dark orange), and RPE (blue). Loci were labeled significant if FDR <5 × 10−8. Table S8 contains full details of significant loci.
(D) Relationships between genotype and Z score for examples of common eQTL (RPS26 [rs1131017] and GSTT1 [rs5760147]) and RGC-exclusive eQTL (PPP1R17 [rs2284227]) in comparison with corresponding loci within RPE subpopulation.
To identify eQTL that had alternative allelic effects under different disease statuses, we included an interaction term (SNP:disease status) in the original linear model for each eQTL identified by the first round of analysis. eQTL with interacting effects were determined to be significant based on a threshold of FDR < 0.05 of the interaction term. We tested 4,443 eQTL that had been mapped with the original linear model and, from these, we identified 1,399 eQTLs with evidence of significant interaction between the SNP allelic effect and POAG disease status. Ninety-seven of these eQTLs were specific to RGC lineage subpopulations and are of particular interest, as the data suggest that the allelic effect of the SNP differs due to disease (Figure 4A; Table S10). Interestingly, rs28368130 at chromosome 9p21, a locus that has been definitively associated with POAG,12,64 was found to influence CDKN2B expression in the RGC1 cell population with a disease status FDR p = 7.97 × 10−4. In further analysis, we identified that there was variation in the number of donors with a non-zero expression of the CDKN2B gene, supportive of previous work showing allele-specific methylation of this promoter region.65 We then tested if there was a relationship between the percentage of donors with non-zero expression and genotype classes for cases and controls and identified a significant association across all RGC subtypes (χ2, RGC1 p = 5.17 × 10−11; RGC2 p = 9.42 × 10−10, RGC3 p = 1.57 × 10−14) (Figure 4B).
Figure 4.

Disease-associated cis-eQTLs specific to cell subpopulations
SNP-by-disease interactions were tested for all SNPs within one MBP of each gene for each cell subpopulation.
(A) p values of significant eQTL (x axis) in RGC subpopulations RGC1, RGC2, and RGC3 were plotted against the p values of SNP-by-disease interactions (y axis). The dotted red line represents the p value threshold of 0.05 for the SNP-by-disease interactions. Points and labels in red represent interactions that are significant in only the RGC subpopulations.
(B) Expression profile of CDKN2B, based on the rs28368130 genotype for POAG cases and controls. Expression is displayed in Z scores.
(C) Boxplots displaying the Z scores of the following genes: IGFBPL1, SAR1A, and VAMP2 were grouped by disease and genotype. Regression lines for each condition represent the relationship between genotype and expression. Table S8 contains full details of significant loci.
IGFBPL1 regulates axonal growth in RGCs66 and was also found to have a statistically significant disease-state interaction eQTL in the RPC9, RGC1, and RGC2 subpopulations of cells (Figure 4C). Similarly, SAR1A, which is involved in transport between the ER to the Golgi apparatus and is associated with axonal growth,67 has an eQTL identified by rs4746023, in RGC1 cell types. In patients with POAG, carrying each additional copy of the A allele causes an increase by an average of 1.4 transcripts per cell, which is approximately 2 times higher than in healthy controls (Figure 4C).
Disease-specific differential expression of genes across cell types identifies altered transthyretin expression in POAG RGCs
We next sought to evaluate the relationship between disease status and regulation of the transcriptome and a cellular level, testing for differences in the expression levels of genes in each cell population. In total, after Bonferroni correction, we identified 3,118 genes whose expression was either up- or downregulated in POAG cases relative to the controls. We can be confident that these results are due to the genetic effects underlying POAG risk, as at all steps from iPSC generation, differentiation, cell capture, and library preparation, the cell lines were either managed in shared conditions or randomized with respect to disease status (STAR Methods). Furthermore, no firm environmental factors have been found to definitively predispose to POAG risk and are unlikely to account to a difference in gene expression in differentiated cells, given the epigenetic profile of fibroblast-derived iPSCs is reset during reprogramming.68,69 Focusing on the three RCG populations, we identified 144 genes differentially expressed between POAG cases and controls (Figure 5A; Tables S10 and S11). Consistent with our observations of cell-type-specific eQTL, 68.06% of genes were identified as differentially expressed in only one cell type, reinforcing the conclusions that the genetic effects of POAG are highly cell-type specific. Interestingly, TTR was found to be differentially expressed between POAG cases and healthy controls across all RGC subpopulations (Figure 5A). Coding variants in TTR are known to cause familial amyloidotic polyneuropathy, which is frequently associated with glaucoma.70,71
Figure 5.

Prioritization of glaucoma risk genes
(A and B) (A) Volcano plot displaying genes differentially expressed between POAG cases and healthy controls and (B) Manhattan plot displaying the transcriptome-wide association analysis from the scRNA-seq data.
TWAS identifies novel and refines known genetic associations for glaucoma
We leveraged these iPSC-derived retinal organoid single-cell eQTL data with our recently reported multitrait glaucoma GWAS summary statistics to prioritize glaucoma risk genes in a TWAS.12 In previous work, we combined GWAS for multiple genetically correlated traits, and thus used the glaucoma-specific effect size estimates and p values for SNPs across the genome.12 In the single-cell TWAS, we identified seven genes associated with POAG after Bonferroni correction (Figure 5B). Of the five genes identified in the RGC1 subpopulation, one is located at a locus (chromosome 17q21) recently associated with POAG.72 Here, we implicate KANSL1-AS1, which was also identified as a major eGene for RGCs (Figure 3B). KANSL1-AS1 was also found by TWAS to be associated with POAG in the RPC9 subpopulation. The TWAS results also helped to fine-map potential causal genes at known GWAS loci.73,74,75,76 Most of the identified genes map to loci that have been previously associated with POAG.72 In brief, the five TWAS genes on chromosome 2 are located near GWAS-reported gene BRE and share the same GWAS variant rs6741499 (or in strong LD),12 which is also associated with IOP.20 Of these, the top TWAS hit MPV17 encodes a mitochondrial inner membrane protein involved in the metabolism of reactive oxygen species75 and has been found to play an important role in the pathogenesis of RGC damage.76 The gene CTD-3074O7.5 on chromosome 11 from RGC2 subpopulation is near MALAT1, which is also associated with vertical cup-to-disc ratio.77 We then compared the TWAS results based on scRNA-seq data with bulk RNA-seq data. The bulk retinal transcriptome data were described previously.78 Of the three genes with available bulk TWAS results, only KANSL1-AS1 was significant after Bonferroni correction (Pbulk TWAS = 5.99 × 10−6).
Discussion
Here, we present a large-scale scRNA-seq analysis of iPSC-derived RGCs. We generated over 100 patient-specific iPSC lines and differentiated them into RGCs using retinal organoids. Following the capture of over 330,000 cells, we analyzed 258,071 cells from 110 individuals. We identified a total of 4,443 eQTL across all cell types using aggregated, single-cell-level expression data, including 312 eGenes specific to RGCs. We tested for shared allelic effects between eSNPs of the 647 eGenes common to more than one subpopulation and discovered that only 215 are due to shared eQTL. This strategy, as opposed to testing for eGene overlap, helps ensure that the estimated proportion of shared eQTL is not inflated due to occurrences where the same gene’s expression is associated with two independent eQTL in different cell types. POAG culminates in loss of RGCs, and in iPSC-derived RGCs we identified disease-associated loci. Analysis, conditional on disease status, implicated 97 statistically significant RGC eQTLs, and single-cell TWAS identified seven genes at loci previously associated with POAG.
Several recent studies have employed scRNA-seq to characterize transcriptomic changes during human retinal development using fetal tissue,50,79 retinal organoids,80, 81, 82,82, 83, 84 or both.42,43 Our results complement these findings and are in concordance with those of Sridhar et al.42 who also observed RPCs as the major cell type in early organoids followed by RGCs, photoreceptors, and interneurons. Similar observations were made by Lu et al.43 The absence of glial cells within the retinal organoids in our dataset is not surprising given that these are generally the last retinal cells to develop83 and emerged in older retinal organoids.42 Furthermore, we did not observe statistically significant differences in cellular composition of organoids derived from healthy controls and patients with POAG, which suggests a high level of consistency across differentiation batches.
In the TWAS framework, the gene expression data (association between SNPs and genes) were used to train prediction models to determine gene expression levels by genetic variants (genetically regulated gene expression, GReX).84,85 The prediction models were used to impute gene expression levels in the GWAS dataset based on the trained weights from multiple-SNP prediction models, which could be further used to evaluate the association between imputed gene expression levels and the GWAS phenotype (i.e., glaucoma), and to identify genes associated with disease traits. In this study, we performed the first glaucoma TWAS based on cell-type-specific expression profiling. The gene expression profiling between different tissues (e.g., bulk retinas versus subpopulations belonging to the RGC lineage) could be markedly different. In our TWAS analysis, the single-cell gene expression data from different subpopulations were used to train prediction models, and then to impute the gene expression levels in GWAS dataset based on summary statistics.86 The single-cell level resolution TWAS can provide new insights into the potential causal genes for glaucoma in specific cell types.
It is becoming widely recognized that the pharmaceutical pipeline for drug development has stalled, and that there is a pressing need for human models of disease to improve our molecular understanding of common, complex diseases and facilitate preclinical trials.87 We investigated the impact of genetic background and disease status on gene expression through eQTL mapping. Highlighting the power of large-scale iPSC studies to uncover disease-specific profiles, this work lays the foundation for context-specific drug screening and underscores the efficiency of using stem cell models for dissecting complex disease.
Limitations of the study
Certain limitations of our study should be noted. Due to the complexity of the retinal organoid culturing, we only analyzed organoids derived from 110 individuals. This would have limited our ability to identify rare and/or low-penetrant genetic variants associated with glaucoma. Although sufficient for RGC identification, the duration for differentiation of our retinal organoids (49 days) limited our ability to identify all retinal cell types. For example, “late born” retinal subtypes, e.g., rods or Müller glia were not present in our organoids. Nevertheless, recent work of Sridhar et al. demonstrated similar cellular composition between retinal organoids and fetal retina at equivalent stages of development.42
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-hOCT-4 | Santa Cruz | Cat# sc-5279; RRID: AB_628051 |
| AlexaFluor 488 goat anti-mouse IgG | Thermo Fisher Scientific | Cat# A11029; RRID: AB_2534088 |
| Mouse anti-hTRA-1-60 (PE-conjugated) | Thermo Fisher Scientific | Cat# MA-1-023-PE; RRID: AB_2536704 |
| Mouse anti-TRA-1-60 | Abcam | Cat# ab16288; RRID: AB_778563 |
| AlexaFluor 488 goat anti-mouse IgM | Thermo Fisher Scientific | Cat# A21042: RRID: AB_2535711 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM, high glucose | Life Technologies | Cat# 11965118 |
| Fetal Bovine Serum, qualified | Life Technologies | Cat# 26140079 |
| L-glutamine (200mM) | Life Technologies | Cat# 25030081 |
| Penicillin-Streptomycin (10,000 U/ml) | Life Technologies | Cat# 15140122 |
| TeSR™-E7™ | StemCell Technologies | Cat# 05914 |
| CellAdhere™ Dilution Buffer | StemCell Technologies | Cat# 07183 |
| Vitronectin™ | StemCell Technologies | Cat# 07180 |
| StemFlex™ | Life Technologies | Cat# A3349401 |
| DPBS, no calcium, no magnesium | Life Technologies | Cat# 14190250 |
| ReLeSR™ | StemCell Technologies | Cat# 05873 |
| DMEM/F-12 | Life Technologies | Cat# 11320082 |
| L-glutamine (200mM) | Life Technologies | Cat# 25030081 |
| MEM Non-Essential Amino Acids solution (100x) | Life Technologies | Cat# 11140050 |
| Corning® Matrigel® basement membrane matrix | VWR | Cat# 734-1100 |
| B-27 supplement (50x) | Life Technologies | Cat# 17504044 |
| FGF basic protein, human recombinant | Merck | Cat# GF003 |
| Neurobasal™ medium | Life Technologies | Cat# 21103049 |
| GlutaMAX™ supplement | Life Technologies | Cat# 35050061 |
| D-(+)-Glucose solution 45 % in H2O | Sigma | Cat# D8769 |
| N-2 supplement (100x) | Life Technologies | Cat# 17502048 |
| Bovine Serum Albumin | Sigma | Cat# A4503-50G |
| Critical commercial assays | ||
| MycoAlert mycoplasma detection kit | Lonza | Cat# LT07-318 |
| Human Dermal Fibroblasts Nucleofector™ Kit | Lonza | Cat# VVPD-1001 |
| LS columns | Miltenyi Biotec | Cat# 130-042-401 |
| MidiMACS Separator | Miltenyi Biotec | Cat# 130-42-301 |
| MACSQuant Analyzer 10 | Miltenyi Biotec | Cat# 130-096-343 |
| Anti-TRA-1-60 microbeads, human | Miltenyi Biotec | Cat# 130-100-832 |
| Papain Dissociation System | Worthington Biochemical Corporation | Cat# LK003153 |
| Countess II™ Fl Automated cell counter | Thermo Fisher Scientific | Cat# AMQAF1000 |
| QIAamp DNA Mini kit | Qiagen | Cat# 51306 |
| SimpliNano™ Spectrophotometer | GE Life Sciences | Cat# 29061711 |
| Chromium™ Single Cell 3’ Library, Gel Bead & Multiplex Kit, 16 rxns | 10X Genomics | Cat# PN-120233 |
| Dynabeads™ MyOne™ Silane | Thermo Fisher Scientific | Cat# 37002D |
| SPRIselect beads | Beckman Coulter | Cat# B23318 |
| NovaSeq 6000 S4 Reagent kit | Illumina | Cat# 20039236 |
| Deposited data | ||
| Single-cell RNA-seq | This manuscript (https://data.humancellatlas.org) | 77780d56-03c0-481f-aade-2038490cef9f |
| Experimental models: cell lines | ||
| Human iPSCs | This manuscript (https://data.humancellatlas.org) | 77780d56-03c0-481f-aade-2038490cef9f |
| Software and algorithms | ||
| NovaSeq Control Software v1.6 | Illumina | https://support.illumina.com/sequencing/sequencing_instruments/novaseq-6000/downloads.html |
| GenomeStudio v2.0.4 | Illumina | https://support.illumina.com/array/array_software/genomestudio/downloads.html |
| GenomeStudio PLINK Input Report Plug-in v2.1.4 | Illumina | |
| 10x Cellranger | 10X Genomics | https://www.10xgenomics.com/software/ |
| Minimac3 | Das et al., 201688 | https://github.com/Santy-8128/Minimac3 |
| Eagle v2.3 | Loh et al., 201689 | https://data.broadinstitute.org/alkesgroup/Eagle/ |
| ascend R package | Senabouth et al., 201990 | https://github.com/powellgenomicslab/ascend |
| demuxlet | Kang et al., 201891 | https://github.com/statgen/demuxlet |
| scrublet | Wolock et al., 201992 | https://github.com/AllonKleinLab/scrublet |
| Monocle 3 R package | Cao et al., 201993 | http://cole-trapnell-lab.github.io/monocle-release/monocle3 |
| Seurat 3.1 R package | Butler et al., 201894 and Stuart et al., 201995 | https://github.com/satijalab/seurat |
| UMAP | Becht et al., 201896 | https://github.com/lmcinnes/umap |
| Slingshot 1.4.0 R package | Street et al., 201856 | https://github.com/kstreet13/slingshot |
| tradeSeq 0.99.80 | Van den Berge et al., 202057 | https://statomics.github.io/tradeSeq/index.html |
| limma 3.42.2 R package | Ritchie et al., 201597 | https://bioconductor.org/packages/release/bioc/html/limma.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alex Hewitt: hewitt.alex@gmail.com.
Material availability
There are restrictions to the availability of the human iPSC lines generated in this study due to MTA.
Experimental model and subject details
Participant recruitment
All participants gave informed written consent.98 All experimental work was approved by the Human Research Ethics committees of the Royal Victorian Eye and Ear Hospital (11/1031H, 13/1151H-004), University of Melbourne (1545394), University of Tasmania (H0014124) and the University of Western Australia (RA/4/1/5255) as per the requirements of the National Health & Medical Research Council of Australia (NHMRC) and in accordance with the Declarations of Helsinki. We recruited a large cohort of patients with POAG and sex-, ethnically-matched individuals, through the Glaucoma Inheritance Study in Tasmania and the Australian and New Zealand Registry of Advanced Glaucoma, local ophthalmic clinics and adjunct studies (mean ± SD age: 69.1 ± 14.4 years at biopsy for case subjects; 68.1 ± 8.2 years at biopsy for controls). POAG patients required a clinical diagnosis of advanced normal tension glaucoma, as they are presumed to have RGCs with an increased susceptibility to degeneration compared to people with trabecular dysfunction or a very high intraocular pressure. As described previously, advanced glaucoma was defined by severe visual loss resulting from POAG.64,99,100 In the worst eye a vertical cup:disc ratio >0.95 and a best-corrected visual acuity worse than 6/60 due to POAG or on a reliable Humphrey Visual Field a mean deviation of ≤ -22dB; or at least 2 out of 4 central squares involved with a Pattern Standard Deviation of <0.5%. The maximum documented pre-treatment IOP, measured by Goldmann applanation tonometry, was recorded. To fulfil a standard clinical diagnosis of Normal Tension POAG the maximum recorded IOP was required to be < 22 mmHg.101 Clinical-exclusion criteria were signs of secondary or syndromic glaucoma. For each control participant, a complete ophthalmologic evaluation (incorporating automated visual field testing, fundus and optic disc imaging, corneal pachymetry) was performed. An ophthalmic history was obtained, with questions centered on age at diagnosis, family history, surgical intervention for glaucoma or cataract, macular degeneration, retinal detachment, and refractive surgery. Control subjects, with no known family history of glaucoma, and who had normal IOP, optic discs (optical coherence tomography retinal nerve fibre layer analysis within age-matched normal limits), and visual fields, were selected for analysis.
Method details
Fibroblast culture
Skin biopsies were obtained from non-sun exposed regions using a 3mm2 dermal punch. Fibroblasts were expanded, cultured, and banked in DMEM with high glucose, 10% fetal bovine serum (FBS), L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (all from Thermo Fisher Scientific, USA). All cell lines were mycoplasma-free. Fibroblasts at passage (p) 2 were used for reprogramming.
Generation, selection, and iPSC maintenance
A TECAN liquid-handling platform was used to maintain and passage iPSCs, as described in.38 iPSCs were generated by nucleofection with episomal vectors expressing OCT-4, SOX2, KLF4, L-MYC, LIN28 and shRNA against p53102 in feeder- and serum-free conditions using TeSR™-E7™ medium as described previously.39 The reprogrammed cells were maintained on the automated platform using TeSR™-E7™medium, with daily medium change. Pluripotent cells were selected by sorting with anti-human TRA-1-60 Microbeads.39 Cell number was determined; cells were subsequently plated onto vitronectin XF™-coated plates and in StemFlex™ medium. Subsequent culturing was performed on the automated platform using StemFlex™ medium, which was changed every 2-3 days. Passaging was performed weekly on the automated platform using ReLeSR™ onto vitronectin XF™-coated plates as described in 39. Pluripotency was assessed by expression of the markers OCT3/4 (sc-5279, Santa Cruz Biotechnology) and TRA-1-60 (MA1-023-PE, Thermo Fisher Scientific, USA) by immunocytochemistry. Virtual karyotyping was undertaken using Illumina Human Core Exome or UK Biobank Axiom™ Arrays, as described in.38 The iPSC lines FSA0001, FSA0002, FSA0005, FSA0006, IST2168, IST2607, MBE1006, MBE2900, MBE2906, MBE2909, TOB0199, TOB0224, TOB0412, TOB0421, TOB0431, TOB0435, TOB0474, WAB0450, WAB0069 were characterised in.38
Differentiation of iPSCs into retinal organoids
Retinal organoids were generated following29 with modifications. Briefly, on day 28 formed retinal organoids with surrounding tissue were excised using a 21G needle. They were maintained in suspension culture for 7 days in PRO medium (DMEM/F12, 1:1, L-glutamine, 1% non-essential amino acids, Penicillin-Streptomycin 10,000 U/ml) supplemented with B27 and FGF2 (10 ng/mL). On day 35, organoids were transferred to Matrigel-coated 24-well tissue culture plates and maintained for 14 days in NDM medium (Neurobasal, 1% MEM non-essential amino acids, 1% GlutaMAX, 1% glucose (45%), Penicillin-Streptomycin 10,000 U/ml) with 2% B27 and 1% N2 added fresh. Medium was changed every 2-3 days. Optic cups were dissociated with Papain Dissociation System following the manufacturer’s instructions. Briefly, cells were harvested with papain (20 U/mL) and DNase I (2,000 U/mL) for 30 min at 37°C. Subsequently, NDM medium was added at a 1:1 ratio and cells were gently triturated with a P1000 pipette followed by centrifugation (5 min, 300 g, 4°C). Cells were resuspended in 1 mL of 0.1% BSA in PBS solution. Subsequently, cells were counted and assessed for viability with Trypan Blue using a Countess II automated counter, then pooled at a concentration of 1000 cells/μL (1 × 106 cells/mL). Final cell viability estimates ranged between 79-99%. Cell lines were harvested in 25 batches (6-8 lines per batch) and multiplexed for scRNA-Seq with a targeted capture of 2,000 cells per line.
Quantification and statistical analysis
Transcriptome profiling of single cells from retinal organoids and cell-based quality control
Multi-donor single-cell suspensions were prepared for scRNA-seq using the Chromium Single Cell 3′ Library & Gel bead kit (10x Genomics; PN-120237). Each pool was loaded onto individual wells of 10x Genomics Single Cell A Chip along with the reverse transcription (RT) master mix to generate single-cell gel beads in emulsion (GEMs). Reverse transcription was performed using a C1000 Touch Thermal Cycler with a Deep Well Reaction Module (Bio-Rad) as follows: 45 min at 53°C; 5 min at 85°C; hold 4°C cDNA was recovered and purified with DynaBeads MyOne Silane Beads (Thermo Fisher Scientific; catalog no. 37002D). Subsequently, it was amplified as follows: for 3 min at 98 °C; 12× (for 15 s at 98°C; for 20 s at 67°C; for 60 s at 72 °C); for 60 s at 72°C; hold 4°C followed recommended cycle number based on targeted cell number. Amplified cDNA was purified with SPRIselect beads (Beckman Coulter; catalog no. B23318) and underwent quality control following manufacturer’s instructions. Sequencing libraries for each pool were labeled with unique sample indices (SI) and combined for sequencing across two 2 × 150 cycle flow cells on an Illumina NovaSeq 6000 (NovaSeq Control Software v1.6) using S4 Reagent kit (catalog no. 20039236). Raw base calls from the sequencer then underwent demultiplexing, quality control, mapping and quantification with the Cell Ranger Single Cell Software Suite 3.1.0 by 10x Genomics (https://www.10xgenomics.com/). Processed reads from the sequencer were mapped to the Homo sapiens reference hg19/GRCh37 from ENSEMBL (release 75), and the pipeline was run using the estimated cell count value of 20,000. scRNA-seq data from each pool underwent quality control separately in R using the ascend package.90 Cells were removed if they did not meet thresholds calculated from 3 Median Absolute Deviations (MAD) of the following statistics: total Unique Molecular Identifier (UMI) counts, number of detected genes, and fraction of mitochondrial and ribosomal transcripts to total expression.
SNP genotype analysis and imputation
DNA was extracted from cell pellets using QIAamp DNA Mini Kit (QIAGEN, 51306) following the manufacturer's instructions. DNA concentration was determined with SimpliNano spectrophotometer (GE Life Sciences), diluted to a final concentration of 10-15 ng/μL and genotyped on UK Biobank Axiom™ Arrays at the Ramaciotti Centre for Genomics, Sydney, Australia. Samples previously screened using Illumina arrays, were re-genotyped on UK Biobank Axiom™ Arrays. Genotype data were exported into PLINK data format using GenomeStudio PLINK Input Report Plug-in v2.1.4 and screened for SNP and individual call rates (<0.97), HWE failure (p < 10−6), and MAF (<0.01).103 Samples with excess autosomal heterozygosity or with sex-mismatch were excluded. In addition, a genetic relationship matrix from all the autosomal SNPs were generated using the GCTA tool and one of any pair of individuals with estimated relatedness larger than 0.125 were removed from the analysis.104 Individuals with non-European ancestry were excluded outside of an “acceptable” box of +/− 6SD from the European mean in PC1 and PC2 in an SMARTPCA analysis. The 1000G Phase 3 population was used to define the axes, and the samples were projected onto those axes (Figure S4). Imputation was performed on each autosomal chromosome using the Michigan Imputation Server with the Haplo-type Reference Consortium panel (HRC r1.1 2016) and run using Minimac3 and Eagle v2.3 88,89. Only SNPs with INFO >0.8 were retained.
Demultiplexing of cell pools into individual donors
demuxlet v1.0 91 and scrublet v0.20 92 were used to demultiplex cells from mixed-donor pools using transcriptome and genotype data. Each pool was demultiplexed separately. demuxlet was run with exon-only SNPs and the following arguments: “--field GP, --geno.error = 0.01, --min-mac 1, --min-callrate 0.5, alpha = 0.5, doublet-prior = 0.5”. Cells were initially assigned a putative donor based on the maximum likelihood of reads from scRNA-seq overlapping sets of unique variants (SNPs) mapped by genotyping. scrublet was then used to confirm if a cell was a neotypic doublet by building a simulation of doublets based on sampled transcriptome data and scoring the cell based on its neighbors in k-means nearest neighbor graph. A cell was designated a singlet if scrublet agrees, and if the posterior probability of it being a singlet in demuxlet is greater than 0.99. Unassigned donors, doublets and cells with ambiguous assignments were omitted from downstream analyses.
Aggregation, normalization and dimensionality reduction of scRNA-seq datasets
The unfiltered count matrices of all batches were combined into one dataset using the cellranger aggr pipeline. This pipeline equalized the read depth of all batches by downsampling reads from higher-depth libraries to match the lowest depth library.105 Cells that had been removed from single-batch analyses due to low quality, being labeled as a doublet, or with conflicting assignments, were also removed from the combined expression matrix. The SCTransform function from Seurat (v3.0.2) was applied to the filtered count matrix to perform cell-cell and batch normalization.106 The fraction of mitochondrial and ribosomal expression of total expression was regressed out as a part of this step, and the top 3000 most variable genes were used to calculate residuals. The residuals were then reduced to 30 principal components (PCs) using PC Analysis (PCA). These PCs were reduced further into two dimensions, for visualization and clustering via Uni-form Mani-fold Approximation Projection (UMAP).96
Identification and annotation of cell subpopulations
Graph-based clustering via the Louvain algorithm that was implemented in Seurat was used to identify cell subpopulations.107,108 First, cell-cell Euclidean distances calculated from PCs were projected onto a K-means nearest-neighbor graph (KNN). Next, the Louvain algorithm was implemented at resolutions between 0 and 1, at 10 equal intervals of 0.1. Finally, the movement of cells between subpopulations at these resolutions were visualized on to a clustree plot (Figure S2A) as implemented in the clustree R package.109 Regions of stability were identified from the plot, and the resolution where this region began - 0.4, was selected as the optimal resolution. Cells were divided into 22 subpopulations at this resolution. Cells that could not be assigned to a subpopulation - singletons, were assigned to a group designated ‘Cluster 0’. To annotate each subpopulation, the combined Likelihood Ratio Test (LRT) as described by110 was applied to normalized, log-transformed UMI counts. Disease-specific markers within subpopulations were identified using the Wilcoxon Rank Sum test.111 Genes were classified as markers if they were differentially expressed, based on the thresholds of average log2 fold change > |0.25| and Bonferroni-corrected p value < 0.01. Annotation of retinal cell types was performed using eye-field and retinal marker genes as described in Data S1.
Identification of differentiation lineages via pseudotime analysis
Differentiation lineages were identified using the slingshot R package.56 Singletons were excluded from the trajectory, and the UMAP matrix was used as input for the ‘slingshot’ function. RPC1 was selected as the root of the trajectories due to the expression of the proliferative marker MKI67, and the endpoints of trajectories were defined as the relatively mature cell subpopulations: RGC1, Photoreceptors, Interneurons, RPE and Lens. A Kolmogorov-Smirnov test was used to test for significant differences in the distribution of cells across pseudotime in RGC lineages 6, seven and 9. Pseudotime values for RGC3 were then averaged for use with cis-eQTL analysis. Trajectory-specific differential expression analysis was performed with the tradeSeq R package 57, using the raw counts of cells from RPC9, RGC1, RGC2 and RGC3. These cells were used to build a new lineage via slingshot, using RPC9 as the origin and RGC1 as the endpoint. The counts and resulting slingshot object were then used to run evaluateK with the number of knots ranging from 3 to 10. The optimal number of knots was determined to be 7. The fitGAM function was then run with this value, and lineages defined as condition. The associationTest function was used to identify markers specific to each condition sub-lineage, while the conditionTest function was used to identify markers that differed between the conditions.
Identification of cis-eQTL using transcriptome and genotype data
We investigated cis-eQTL associated with each subpopulation using the R package Matrix eQTL.112 To prepare scRNA-seq data for this stage of analysis, the normalized, corrected UMI count matrix was split by subpopulation. The counts from cells for each individual were aggregated by taking the mean of each gene and used to build a gene-donor count matrix. Genes that were expressed in less than 30% of the donor population were excluded, and remaining mean counts were log-transformed. These values were then quantile normalised using the `normalizeBetweenArrays` function from the limma R package.97 SNPs with a Minor Allele Frequency (MAF) of less than 10% were filtered from the genotype dataset. SNPs within one mega-base pairs of a gene were tested. To identify lead eQTL based on disease, Matrix eQTL was run with an additive linear model using sex, age, disease status and the top six genotype principal components as covariates. To identify eQTL that had alternative allelic effects under different disease statuses, we included an interaction term (SNP:disease status) in the original linear model for each eQTL identified by the first round of analysis. eQTL with interacting effects were determined to be significant based on a threshold of FDR <0.05 of the interaction term. The FDR was calculated using the Benjamini–Hochberg procedure for all gene-SNP interactions that pass the p value threshold of 0.05, as applied using MatrixEQTL.112
To identify eGenes with overlapping eQTL signals in more than one subpopulation, we performed multi-directional conditional analysis on pairs of subpopulations. If a gene had an eQTL in subpopulation A and subpopulation B, we tested whether the allelic effects of eSNPA and eSNPB were dependent on each other by including eSNPA as a fixed covariate in the linear model for subpopulation B, and eSNPB in the linear model for subpopulation A. eSNPs were independent if the association remained significant. To determine if eSNPs tag the same causal variant in both subpopulations or were in LD, we tested the change in allelic effect between this model and the original model for significance. If the change was not significant, then the eSNPs tag independent causal variants for the same gene in different cell subpopulations.
Transcriptome wide association study analysis
Transcriptome-wide association study analysis (TWAS) was performed using summary statistics generated by eQTL analysis. We customized our prediction models based on the online tutorial (available through https://github.com/hakyimlab/MetaXcan) to evaluate the association between predicted gene expression and glaucoma risk using GWAS summary statistics. The gene-expression prediction models were constructed from the single-cell eQTL data using the following models: “blup”, “lasso”, “top1” and “enet”. The GWAS summary statistics from a recent multitrait meta-analysis of glaucoma were used.12 S-PrediXcan was used to harmonize the GWAS summary statistics and to compute the gene-level association results using each subpopulation of single-cell expression data and glaucoma GWAS summary statistics.86 A total of 3,573 genes (across tested subpopulations) were tested for glaucoma. To correct for multiple testing, we adjusted the gene-level association p values using the Bonferroni correction method (0.05/(total number of tests across all subpopulations)). We then compared the transcriptome-wide association results based on scRNA-seq data to bulk RNA-seq data. The bulk retinal transcriptome data were described previously.78 Briefly, 406 controls and age-related macular degeneration cases that passed RNA-seq and genotyping quality control were modeled with mixed models, LASSO, or elastic net according to Gusev et al85 The effect sizes from these models were used as weights to calculate the gene-trait associations.
Acknowledgments
We thank Vikrant Singh, Anthony Cook, Alison Conquest, and Dominik Kaczorowski for providing technical support. This research was supported by National Health and Medical Research Council Practitioner Fellowships (A.W.H., J.E.C., and D.A.M.), Senior Research Fellowship (A.P., 1154389; S.M.), Career Development Fellowship (J.E.P., 1107599) and Investigator Fellowship (J.E.P., 117578), an Australian Research Council Future Fellowship (A.P., FT140100047), an International Postgraduate Research Scholarship & Research Training Program Scholarship (M.D.), and by grants from the National Health and Medical Research Council (1150144 and 1143163) and the Australian Research Council (180101405), the Joan and Peter Clemenger Foundation, the Goodridge Foundation, the Ophthalmic Research Institute of Australia, the BrightFocus Foundation, the Philip Neal bequest, Stem Cells Australia – the Australian Research Council Special Research Initiative in Stem Cell Science, the University of Melbourne, and Operational Infrastructure Support from the Victorian Government.
Author contributions
Conceptualization, J.E.P., A.P., and A.W.H.; methodology, M.D., A.S., X.H., S.M., J.E.P., A.P., and A.W.H.; investigation, M.D., H.H.L., X.H., G.E.L., D.H., L.R., P.S., J.E., C.-L.C., U.N., R.A.McC., V.G., S.Y.L., J.G.L., and L.G.; resources, H.H.L., E.S., S.L.G., L.C., L.K., D.A.M., J.E.C., J.E.P., A.P., and A.W.H.; data analysis, M.D., A.S., X.H., S.M., J.E.P., A.P., and A.W.H.; writing – original draft, M.D., A.S., X.H., S.M., J.E.P., A.P., and A.W.H.; writing – review & editing, M.D., A.S., X.H., G.E.L., D.H., D.A.M., J.E.C., S.M., J.E.P., A.P., and A.W.H.; supervision and project administration, J.E.P., A.P., and A.W.H.; funding acquisition, D.A.M., J.E.C., S.M., J.E.P., A.P., and A.W.H.
Declaration of interest
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure sex balance in the recruitment of human participants. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: June 8, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100142.
Contributor Information
Joseph E. Powell, Email: j.powell@garvan.org.au.
Alice Pébay, Email: apebay@unimelb.edu.au.
Alex W. Hewitt, Email: hewitt.alex@gmail.com.
Supplemental information
Summary of all cell lines generated for their project, and details regarding the removal of cell lines.
Single-cell libraries were generated using the Chromium Single Cell Gene Expression V3 workflow by 10x Genomics. Cells derived from 160 donors were pooled and sequenced in 25 batches with an expected cell capture value of 20,000. This table summarizes the quality of read mapping and quantification of each batch.
Differentially expressed genes were used as markers for each cluster to assist in cell classification. The expression of each cluster was contrasted against the expression of all other cells, and DE genes were identified using a likelihood-ratio test, as implemented via the “bimod” function in Seurat.
Differentially expressed genes from POAG cell lines in the RGC lineage were identified via tradeSeq’s “conditionTest” function. Genes were significant if FDR (Benjamini-Hochberg) < 0.05.
cis-eQTLs were mapped using an additive linear model as implemented via MatrixEQTL. Lead cis-eQTLs were selected based on significance (FDR < 0.05), expression in >30% of donors and lowest FDR value. Lead cis-eQTLs were then tested for significance of genotype-disease interactions.
eGenes identified in initial round of cis-eQTL mapping were selected for conditioning if they had an eQTL in more than one cell type and were associated with more than one eSNP. eSNPs were first tested for independence, followed by allelic change after conditioning.
Differential gene expression was conducted via Wilcoxon rank-sum test on RGC clusters, testing POAG cells versus control cells.
Data and code availability
-
•
Single-cell RNA-seq data have been deposited at the Human Cell Atlas (https://data.humancellatlas.org/explore/projects/77780d56-03c0-481f-aade-2038490cef9f). Accession number: 77780d56-03c0-481f-aade-2038490cef9f
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Tham Y.C., Li X., Wong T.Y., Quigley H.A., Aung T., Cheng C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040. Ophthalmology. 2014;121:2081–2090. doi: 10.1016/j.ophtha.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Quigley H.A. Open-angle glaucoma. N. Engl. J. Med. 1993;328:1097–1106. doi: 10.1056/nejm199304153281507. [DOI] [PubMed] [Google Scholar]
- 3.Garway-Heath D.F., Crabb D.P., Bunce C., Lascaratos G., Amalfitano F., Anand N., Azuara-Blanco A., Bourne R.R., Broadway D.C., Cunliffe I.A., et al. Latanoprost for open-angle glaucoma (UKGTS): a randomised, multicentre, placebo-controlled trial. Lancet. 2015;385:1295–1304. doi: 10.1016/s0140-6736(14)62111-5. [DOI] [PubMed] [Google Scholar]
- 4.Wilson M.R. The myth of "21". J. Glaucoma. 1997;6:75–77. doi: 10.1097/00061198-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Friedman D.S., Wilson M.R., Liebmann J.M., Fechtner R.D., Weinreb R.N. An evidence-based assessment of risk factors for the progression of ocular hypertension and glaucoma. Am. J. Ophthalmol. 2004;138:S19–S31. doi: 10.1016/j.ajo.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 6.Leske M.C. Open-angle glaucoma -- an epidemiologic overview. Ophthalmic Epidemiol. 2007;14:166–172. doi: 10.1080/09286580701501931. [DOI] [PubMed] [Google Scholar]
- 7.Sommer A. Ocular hypertension and normal-tension glaucoma: time for banishment and burial. Arch. Ophthalmol. 2011;129:785–787. doi: 10.1001/archophthalmol.2011.117. [DOI] [PubMed] [Google Scholar]
- 8.Polubriaginof F.C.G., Vanguri R., Quinnies K., Belbin G.M., Yahi A., Salmasian H., Lorberbaum T., Nwankwo V., Li L., Shervey M.M., et al. Disease heritability inferred from familial relationships reported in medical records. Cell. 2018;173:1692–1704.e11. doi: 10.1016/j.cell.2018.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K., Gaitsch H., Poon H., Cox N.J., Rzhetsky A. Classification of common human diseases derived from shared genetic and environmental determinants. Nat. Genet. 2017;49:1319–1325. doi: 10.1038/ng.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stone E.M., Fingert J.H., Alward W.L.M., Nguyen T.D., Polansky J.R., Sunden S.L.F., Nishimura D., Clark A.F., Nystuen A., Nichols B.E., et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 11.Rezaie T., Child A., Hitchings R., Brice G., Miller L., Coca-Prados M., Heon E., Krupin T., Ritch R., Kreutzer D., et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295:1077–1079. doi: 10.1126/science.1066901. [DOI] [PubMed] [Google Scholar]
- 12.Craig J.E., Han X., Qassim A., Hassall M., Cooke Bailey J.N., Kinzy T.G., Khawaja A.P., An J., Marshall H., Gharahkhani P., et al. Multitrait analysis of glaucoma identifies new risk loci and enables polygenic prediction of disease susceptibility and progression. Nat. Genet. 2020;52:160–166. doi: 10.1038/s41588-019-0556-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Koolwijk L.M.E., Ramdas W.D., Ikram M.K., Jansonius N.M., Pasutto F., Hysi P.G., Macgregor S., Janssen S.F., Hewitt A.W., Viswanathan A.C., et al. Common genetic determinants of intraocular pressure and primary open-angle glaucoma. PLoS Genet. 2012;8:e1002611. doi: 10.1371/journal.pgen.1002611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hysi P.G., Cheng C.Y., Springelkamp H., Macgregor S., Bailey J.N.C., Wojciechowski R., Vitart V., Nag A., Hewitt A.W., Hohn R., et al. Genome-wide analysis of multi-ancestry cohorts identifies new loci influencing intraocular pressure and susceptibility to glaucoma. Nat. Genet. 2014;46:1126–1130. doi: 10.1038/ng.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Springelkamp H., Iglesias A.I., Cuellar-Partida G., Amin N., Burdon K.P., van Leeuwen E.M., Gharahkhani P., Mishra A., van der Lee S.J., Hewitt A.W., et al. ARHGEF12 influences the risk of glaucoma by increasing intraocular pressure. Hum. Mol. Genet. 2015;24:2689–2699. doi: 10.1093/hmg/ddv027. [DOI] [PubMed] [Google Scholar]
- 16.MacGregor S., Ong J.S., An J., Han X., Zhou T., Siggs O.M., Law M.H., Souzeau E., Sharma S., Lynn D.J., et al. Genome-wide association study of intraocular pressure uncovers new pathways to glaucoma. Nat. Genet. 2018;50:1067–1071. doi: 10.1038/s41588-018-0176-y. [DOI] [PubMed] [Google Scholar]
- 17.Choquet H., Paylakhi S., Kneeland S.C., Thai K.K., Hoffmann T.J., Yin J., Kvale M.N., Banda Y., Tolman N.G., Williams P.A., et al. A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nat. Commun. 2018;9:2278. doi: 10.1038/s41467-018-04555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nag A., Venturini C., Small K.S., International Glaucoma Genetics C., Young T.L., Viswanathan A.C., Mackey D.A., Hysi P.G., Aung T., Hammond C., et al. A genome-wide association study of intra-ocular pressure suggests a novel association in the gene FAM125B in the TwinsUK cohort. Hum. Mol. Genet. 2014;23:3343–3348. doi: 10.1093/hmg/ddu050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khawaja A.P., Cooke Bailey J.N., Wareham N.J., Scott R.A., Simcoe M., Igo R.P., Jr., Song Y.E., Wojciechowski R., Cheng C.Y., Khaw P.T., et al. Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat. Genet. 2018;50:778–782. doi: 10.1038/s41588-018-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao X.R., Huang H., Nannini D.R., Fan F., Kim H. Genome-wide association analyses identify new loci influencing intraocular pressure. Hum. Mol. Genet. 2018;27:2205–2213. doi: 10.1093/hmg/ddy111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards S.L., Beesley J., French J.D., Dunning A.M. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R., et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 24.Gill K.P., Hung S.S.C., Sharov A., Lo C.Y., Needham K., Lidgerwood G.E., Jackson S., Crombie D.E., Nayagam B.A., Cook A.L., et al. Enriched retinal ganglion cells derived from human embryonic stem cells. Sci. Rep. 2016;6:30552. doi: 10.1038/srep30552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohlemacher S.K., Sridhar A., Xiao Y., Hochstetler A.E., Sarfarazi M., Cummins T.R., Meyer J.S. Stepwise differentiation of retinal ganglion cells from human pluripotent stem cells enables analysis of glaucomatous neurodegeneration. Stem Cell. 2016;34:1553–1562. doi: 10.1002/stem.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gill K.P., Hewitt A.W., Davidson K.C., Pebay A., Wong R.C.B. Methods of retinal ganglion cell differentiation from pluripotent stem cells. Transl Vis Sci. Technol. 2014;3:7. doi: 10.1167/tvst.3.3.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakano T., Ando S., Takata N., Kawada M., Muguruma K., Sekiguchi K., Saito K., Yonemura S., Eiraku M., Sasai Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell. 2012;10:771–785. doi: 10.1016/j.stem.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Zhong X., Gutierrez C., Xue T., Hampton C., Vergara M.N., Cao L.H., Peters A., Park T.S., Zambidis E.T., Meyer J.S., et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014;5:4047. doi: 10.1038/ncomms5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reichman S., Slembrouck A., Gagliardi G., Chaffiol A., Terray A., Nanteau C., Potey A., Belle M., Rabesandratana O., Duebel J., et al. Generation of storable retinal organoids and retinal pigmented epithelium from adherent human iPS cells in xeno-free and feeder-free conditions. Stem Cell. 2017;35:1176–1188. doi: 10.1002/stem.2586. [DOI] [PubMed] [Google Scholar]
- 30.Fligor C.M., Langer K.B., Sridhar A., Ren Y., Shields P.K., Edler M.C., Ohlemacher S.K., Sluch V.M., Zack D.J., Zhang C., et al. Three-Dimensional retinal organoids facilitate the investigation of retinal ganglion cell development, organization and neurite outgrowth from human pluripotent stem cells. Sci. Rep. 2018;8:14520. doi: 10.1038/s41598-018-32871-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Capowski E.E., Samimi K., Mayerl S.J., Phillips M.J., Pinilla I., Howden S.E., Saha J., Jansen A.D., Edwards K.L., Jager L.D., et al. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development. 2019;146:dev171686. doi: 10.1242/dev.171686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volkner M., Zschatzsch M., Rostovskaya M., Overall R.W., Busskamp V., Anastassiadis K., Karl M.O. Retinal organoids from pluripotent stem cells efficiently recapitulate retinogenesis. Stem Cell Rep. 2016;6:525–538. doi: 10.1016/j.stemcr.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arno G., Agrawal S.A., Eblimit A., Bellingham J., Xu M., Wang F., Chakarova C., Parfitt D.A., Lane A., Burgoyne T., et al. Mutations in REEP6 cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2016;99:1305–1315. doi: 10.1016/j.ajhg.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.A Tucker B., Solivan-Timpe F., Roos B.R., Anfinson K.R., Robin A.L., Wiley L.A., Mullins R.F., Fingert J.H. Duplication of TBK1 stimulates autophagy in iPSC-derived retinal cells from a patient with normal tension glaucoma. J. Stem Cell Res. Ther. 2014;04:161. doi: 10.4172/2157-7633.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li K., Zhong X., Yang S., Luo Z., Li K., Liu Y., Cai S., Gu H., Lu S., Zhang H., et al. HiPSC-derived retinal ganglion cells grow dendritic arbors and functional axons on a tissue-engineered scaffold. Acta Biomater. 2017;54:117–127. doi: 10.1016/j.actbio.2017.02.032. [DOI] [PubMed] [Google Scholar]
- 36.Wills Q.F., Livak K.J., Tipping A.J., Enver T., Goldson A.J., Sexton D.W., Holmes C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol. 2013;31:748–752. doi: 10.1038/nbt.2642. [DOI] [PubMed] [Google Scholar]
- 37.Kolodziejczyk A.A., Kim J.K., Tsang J.C., Ilicic T., Henriksson J., Natarajan K.N., Tuck A.C., Gao X., Buhler M., Liu P., et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell. 2015;17:471–485. doi: 10.1016/j.stem.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daniszewski M., Nguyen Q., Chy H.S., Singh V., Crombie D.E., Kulkarni T., Liang H.H., Sivakumaran P., Lidgerwood G.E., Hernandez D., et al. Single-cell profiling identifies key pathways expressed by iPSCs cultured in different commercial media. iScience. 2018;7:30–39. doi: 10.1016/j.isci.2018.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crombie D.E., Daniszewski M., Liang H.H., Kulkarni T., Li F., Lidgerwood G.E., Conquest A., Hernandez D., Hung S.S., Gill K.P., et al. Development of a modular automated system for maintenance and differentiation of adherent human pluripotent stem cells. SLAS Discov. 2017;22:1016–1025. doi: 10.1177/2472555217696797. [DOI] [PubMed] [Google Scholar]
- 40.Hoshino A., Ratnapriya R., Brooks M.J., Chaitankar V., Wilken M.S., Zhang C., Starostik M.R., Gieser L., La Torre A., Nishio M., et al. Molecular anatomy of the developing human retina. Dev. Cell. 2017;43:763–779. doi: 10.1016/j.devcel.2017.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark B.S., Stein-O'Brien G.L., Shiau F., Cannon G.H., Davis-Marcisak E., Sherman T., Santiago C.P., Hoang T.V., Rajaii F., James-Esposito R.E., et al. Single-cell RNA-seq analysis of retinal development identifies NFI factors as regulating mitotic exit and late-born cell specification. Neuron. 2019;102:1111–1126.e5. doi: 10.1016/j.neuron.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sridhar A., Hoshino A., Finkbeiner C.R., Chitsazan A., Dai L., Haugan A.K., Eschenbacher K.M., Jackson D.L., Trapnell C., Bermingham-McDonogh O., et al. Single-cell transcriptomic comparison of human fetal retina, hPSC-derived retinal organoids, and long-term retinal cultures. Cell Rep. 2020;30:1644–1659.e4. doi: 10.1016/j.celrep.2020.01.007. e1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu Y., Shiau F., Yi W., Lu S., Wu Q., Pearson J.D., Kallman A., Zhong S., Hoang T., Zuo Z., et al. Single-cell analysis of human retina identifies evolutionarily conserved and species-specific mechanisms controlling development. Dev. Cell. 2020;53:473–491.e9. doi: 10.1016/j.devcel.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ericson J., Rashbass P., Schedl A., Brenner-Morton S., Kawakami A., van Heyningen V., Jessell T.M., Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
- 45.Graham V., Khudyakov J., Ellis P., Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 46.Zibetti C., Liu S., Wan J., Qian J., Blackshaw S. Epigenomic profiling of retinal progenitors reveals LHX2 is required for developmental regulation of open chromatin. Commun Biol. 2019;2:142. doi: 10.1038/s42003-019-0375-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Melo J., Zibetti C., Clark B.S., Hwang W., Miranda-Angulo A.L., Qian J., Blackshaw S. Lhx2 is an essential factor for retinal gliogenesis and notch signaling. J. Neurosci. 2016;36:2391–2405. doi: 10.1523/JNEUROSCI.3145-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langer K.B., Ohlemacher S.K., Phillips M.J., Fligor C.M., Jiang P., Gamm D.M., Meyer J.S. Retinal ganglion cell diversity and subtype specification from human pluripotent stem cells. Stem Cell Rep. 2018;10:1282–1293. doi: 10.1016/j.stemcr.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rheaume B.A., Jereen A., Bolisetty M., Sajid M.S., Yang Y., Renna K., Sun L., Robson P., Trakhtenberg E.F. Single cell transcriptome profiling of retinal ganglion cells identifies cellular subtypes. Nat. Commun. 2018;9:2759. doi: 10.1038/s41467-018-05134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu Y., Wang X., Hu B., Mao Y., Chen Y., Yan L., Yong J., Dong J., Wei Y., Wang W., et al. Dissecting the transcriptome landscape of the human fetal neural retina and retinal pigment epithelium by single-cell RNA-seq analysis. PLoS Biol. 2019;17:e3000365. doi: 10.1371/journal.pbio.3000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mu X., Fu X., Beremand P.D., Thomas T.L., Klein W.H. Gene regulation logic in retinal ganglion cell development: isl1 defines a critical branch distinct from but overlapping with Pou4f2. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6942–6947. doi: 10.1073/pnas.0802627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan L., Deng M., Xie X., Gan L. ISL1 and BRN3B co-regulate the differentiation of murine retinal ganglion cells. Development. 2008;135:1981–1990. doi: 10.1242/dev.010751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu F., Kaczynski T.J., Sethuramanujam S., Li R., Jain V., Slaughter M., Mu X. Two transcription factors, Pou4f2 and Isl1, are sufficient to specify the retinal ganglion cell fate. Proc. Natl. Acad. Sci. U. S. A. 2015;112:E1559–E1568. doi: 10.1073/pnas.1421535112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alquicira-Hernandez J., Sathe A., Ji H.P., Nguyen Q., Powell J.E. scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 2019;20:264. doi: 10.1186/s13059-019-1862-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan W., Peng Y.R., van Zyl T., Regev A., Shekhar K., Juric D., Sanes J.R. Cell Atlas of the human fovea and peripheral retina. Sci. Rep. 2020;10:9802. doi: 10.1038/s41598-020-66092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Street K., Risso D., Fletcher R.B., Das D., Ngai J., Yosef N., Purdom E., Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018;19:477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van den Berge K., Roux de Bezieux H., Street K., Saelens W., Cannoodt R., Saeys Y., Dudoit S., Clement L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020;11:1201. doi: 10.1038/s41467-020-14766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 59.Schriml L.M., Arze C., Nadendla S., Chang Y.W.W., Mazaitis M., Felix V., Feng G., Kibbe W.A. Disease Ontology: a backbone for disease semantic integration. Nucleic Acids Res. 2012;40:D940–D946. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lotze M.T., Tracey K.J. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 61.Sakamoto K., Okuwaki T., Ushikubo H., Mori A., Nakahara T., Ishii K. Activation inhibitors of nuclear factor kappa B protect neurons against the NMDA-induced damage in the rat retina. J. Pharmacol. Sci. 2017;135:72–80. doi: 10.1016/j.jphs.2017.09.031. [DOI] [PubMed] [Google Scholar]
- 62.Sakamoto K., Mizuta A., Fujimura K., Kurauchi Y., Mori A., Nakahara T., Ishii K. High-mobility group Box-1 is involved in NMDA-induced retinal injury the in rat retina. Exp. Eye Res. 2015;137:63–70. doi: 10.1016/j.exer.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 63.Schallenberg M., Prokosch V., Thanos S. Regulation of retinal proteome by topical antiglaucomatous eye drops in an inherited glaucoma rat model. PLoS One. 2012;7:e33593. doi: 10.1371/journal.pone.0033593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burdon K.P., Macgregor S., Hewitt A.W., Sharma S., Chidlow G., Mills R.A., Danoy P., Casson R., Viswanathan A.C., Liu J.Z., et al. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat. Genet. 2011;43:574–578. doi: 10.1038/ng.824. [DOI] [PubMed] [Google Scholar]
- 65.Burdon K.P., Awadalla M.S., Mitchell P., Wang J.J., White A., Keane M.C., Souzeau E., Graham S.L., Goldberg I., Healey P.R., et al. DNA methylation at the 9p21 glaucoma susceptibility locus is associated with normal-tension glaucoma. Ophthalmic Genet. 2018;39:221–227. doi: 10.1080/13816810.2017.1413659. [DOI] [PubMed] [Google Scholar]
- 66.Guo C., Cho K.S., Li Y., Tchedre K., Antolik C., Ma J., Chew J., Utheim T.P., Huang X.A., Yu H., et al. IGFBPL1 regulates axon growth through IGF-1-mediated signaling cascades. Sci. Rep. 2018;8:2054. doi: 10.1038/s41598-018-20463-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aridor M., Fish K.N. Selective targeting of ER exit sites supports axon development. Traffic. 2009;10:1669–1684. doi: 10.1111/j.1600-0854.2009.00974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lister R., Pelizzola M., Kida Y.S., Hawkins R.D., Nery J.R., Hon G., Antosiewicz-Bourget J., O'Malley R., Castanon R., Klugman S., et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Polo J.M., Anderssen E., Walsh R.M., Schwarz B.A., Nefzger C.M., Lim S.M., Borkent M., Apostolou E., Alaei S., Cloutier J., et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reynolds M.M., Veverka K.K., Gertz M.A., Dispenzieri A., Zeldenrust S.R., Leung N., Pulido J.S. Ocular manifestations of familial transthyretin amyloidosis. Am. J. Ophthalmol. 2017;183:156–162. doi: 10.1016/j.ajo.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 71.Beirao J.M., Malheiro J., Lemos C., Beirao I., Costa P., Torres P. Ophthalmological manifestations in hereditary transthyretin (ATTR V30M) carriers: a review of 513 cases. Amyloid. 2015;22:117–122. doi: 10.3109/13506129.2015.1015678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gharahkhani P., Jorgenson E., Hysi P., Khawaja A.P., Pendergrass S., Han X., Ong J.S., Hewitt A.W., Segre A.V., Rouhana J.M., Hamel A.R., Igo R.P., Jr., Choquet H., Qassim A., Josyula N.S., Cooke Bailey J.N., Bonnemaijer P.W.M., Iglesias A., Siggs O.M., Young T.L., Vitart V., Thiadens A.A.H.J., Karjalainen J., Uebe S., Melles R.B., Nair K.S., Luben R., Simcoe M., Amersinghe N., Cree A.J., Hohn R., Poplawski A., Chen L.J., Rong S.S., Aung T., Vithana E.N., NEIGHBORHOOD consortium. ANZRAG consortium. Biobank Japan project. FinnGen study. UK Biobank Eye and Vision Consortium. GIGA study group. 23 and Me Research Team. Tamiya G., Shiga Y., Yamamoto M., Nakazawa T., Currant H., Birney E., Wang X., Auton A., Lupton M.K., Martin N.G., Ashaye A., Olawoye O., Williams S.E., Akafo S., Ramsay M., Hashimoto K., Kamatani Y., Akiyama M., Momozawa Y., Foster P.J., Khaw P.T., Morgan J.E., Strouthidis N.G., Kraft P., Kang J.H., Pang C.P., Pasutto F., Mitchell P., Lotery A.J., Palotie A., van Duijn C., Haines J.L., Hammond C., Pasquale L.R., Klaver C.C.W., Hauser M., Khor C.C., Mackey D.A., Kubo M., Cheng C.Y., Craig J.E., MacGregor S., Wiggs J.L. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat. Commun. 2021;12:1258. doi: 10.1038/s41467-020-20851-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wainberg M., Sinnott-Armstrong N., Mancuso N., Barbeira A.N., Knowles D.A., Golan D., Ermel R., Ruusalepp A., Quertermous T., Hao K., et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 2019;51:592–599. doi: 10.1038/s41588-019-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gusev A., Mancuso N., Won H., Kousi M., Finucane H.K., Reshef Y., Song L., Safi A., Ophoff R.A., O’Donovan M.C., et al. Schizophrenia Working Group of the Psychiatric Genomics Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet. 2018;50:538–548. doi: 10.1038/s41588-018-0092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spinazzola A., Viscomi C., Fernandez-Vizarra E., Carrara F., D'Adamo P., Calvo S., Marsano R.M., Donnini C., Weiher H., Strisciuglio P., et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 76.McMonnies C. Reactive oxygen species, oxidative stress, glaucoma and hyperbaric oxygen therapy. J Optom. 2018;11:3–9. doi: 10.1016/j.optom.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramdas W.D., van Koolwijk L.M.E., Ikram M.K., Jansonius N.M., de Jong P.T.V.M., Bergen A.A.B., Isaacs A., Amin N., Aulchenko Y.S., Wolfs R.C.W., et al. A genome-wide association study of optic disc parameters. PLoS Genet. 2010;6:e1000978. doi: 10.1371/journal.pgen.1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ratnapriya R., Sosina O.A., Starostik M.R., Kwicklis M., Kapphahn R.J., Fritsche L.G., Walton A., Arvanitis M., Gieser L., Pietraszkiewicz A., et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019;51:606–610. doi: 10.1038/s41588-019-0351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aldiri I., Xu B., Wang L., Chen X., Hiler D., Griffiths L., Valentine M., Shirinifard A., Thiagarajan S., Sablauer A., et al. The dynamic epigenetic landscape of the retina during development, reprogramming, and tumorigenesis. Neuron. 2017;94:550–568.e10. doi: 10.1016/j.neuron.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mao X., An Q., Xi H., Yang X.J., Zhang X., Yuan S., Wang J., Hu Y., Liu Q., Fan G. Single-cell RNA sequencing of hESC-derived 3D retinal organoids reveals novel genes regulating RPC commitment in early human retinogenesis. Stem Cell Rep. 2019;13:747–760. doi: 10.1016/j.stemcr.2019.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim S., Lowe A., Dharmat R., Lee S., Owen L.A., Wang J., Shakoor A., Li Y., Morgan D.J., Hejazi A.A., et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc. Natl. Acad. Sci. U. S. A. 2019;116:10824–10833. doi: 10.1073/pnas.1901572116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Collin J., Queen R., Zerti D., Dorgau B., Hussain R., Coxhead J., Cockell S., Lako M. Deconstructing retinal organoids: single cell RNA-seq reveals the cellular components of human pluripotent stem cell-derived retina. Stem Cell. 2019;37:593–598. doi: 10.1002/stem.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vecino E., Acera A. Development and programed cell death in the mammalian eye. Int. J. Dev. Biol. 2015;59:63–71. doi: 10.1387/ijdb.150070ev. [DOI] [PubMed] [Google Scholar]
- 84.Gamazon E.R., Wheeler H.E., Shah K.P., Mozaffari S.V., Carroll R.J., Eyler A.E., Nicolae D.L., Cox N.J., Consortium G.T., Nicolae D.L., Im H.K. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W.J.H., Jansen R., de Geus E.J.C., Boomsma D.I., Wright F.A., et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barbeira A.N., Dickinson S.P., Bonazzola R., Zheng J., Torres J.M., Torstenson E.S., Garcia T., Edwards T.L., Huckins L.M., Nicolae D.L., et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018;9:1825. doi: 10.1038/s41467-018-03621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cook D., Brown D., Alexander R., March R., Morgan P., Satterthwaite G., Pangalos M.N. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat. Rev. Drug Discov. 2014;13:419–431. doi: 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- 88.Das S., Forer L., Schonherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Loh P.R., Danecek P., Palamara P.F., Fuchsberger C., A Reshef Y., K Finucane H., Schoenherr S., Forer L., McCarthy S., Abecasis G.R., et al. Reference-based phasing using the Haplotype reference Consortium panel. Nat. Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Senabouth A., Lukowski S.W., Hernandez J.A., Andersen S.B., Mei X., Nguyen Q.H., Powell J.E. ascend: R package for analysis of single-cell RNA-seq data. GigaScience. 2019;8 doi: 10.1093/gigascience/giz087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M., et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wolock S.L., Lopez R., Klein A.M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cao J., Spielmann M., Qiu X., Huang X., Ibrahim D.M., Hill A.J., Zhang F., Mundlos S., Christiansen L., Steemers F.J., et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502. doi: 10.1038/s41586-019-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 97.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McCaughey T., Liang H.H., Chen C., Fenwick E., Rees G., Wong R.C.B., Vickers J.C., Summers M.J., MacGregor C., Craig J.E., et al. An interactive multimedia approach to improving informed consent for induced pluripotent stem cell research. Cell Stem Cell. 2016;18:307–308. doi: 10.1016/j.stem.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 99.Souzeau E., Goldberg I., Healey P.R., Mills R.A., Landers J., Graham S.L., Grigg J.R., Usher B., Straga T., Crawford A., et al. Australian and New Zealand Registry of advanced glaucoma: methodology and recruitment. Clin. Exp. Ophthalmol. 2012;40:569–575. doi: 10.1111/j.1442-9071.2011.02742.x. [DOI] [PubMed] [Google Scholar]
- 100.Wu J., Hewitt A.W., Green C.M., Ring M.A., McCartney P.J., Craig J.E., Mackey D.A. Disease severity of familial glaucoma compared with sporadic glaucoma. Arch. Ophthalmol. 2006;124:950–954. doi: 10.1001/archopht.124.7.950. [DOI] [PubMed] [Google Scholar]
- 101.Burdon K.P., Crawford A., Casson R.J., Hewitt A.W., Landers J., Danoy P., Mackey D.A., Mitchell P., Healey P.R., Craig J.E. Glaucoma risk alleles at CDKN2B-AS1 are associated with lower intraocular pressure, normal-tension glaucoma, and advanced glaucoma. Ophthalmology. 2012;119:1539–1545. doi: 10.1016/j.ophtha.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 102.Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K.i., et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 103.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir E.a., Amir el A.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Blondel V.D., Guillaume J.-L., Lambiotte R., Lefebvre E. Fast unfolding of communities in large networks. J. of Statistical Mechanics: Theory and Experiment. 2008:P10008. [Google Scholar]
- 109.Zappia L., Oshlack A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. GigaScience. 2018;7 doi: 10.1093/gigascience/giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McDavid A., Finak G., Chattopadyay P.K., Dominguez M., Lamoreaux L., Ma S.S., Roederer M., Gottardo R. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics. 2013;29:461–467. doi: 10.1093/bioinformatics/bts714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hollander M., Wolfe D.A., Chicken E. John Wiley & Sons; 2013. Nonparametric Statistical Methods. [Google Scholar]
- 112.Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of all cell lines generated for their project, and details regarding the removal of cell lines.
Single-cell libraries were generated using the Chromium Single Cell Gene Expression V3 workflow by 10x Genomics. Cells derived from 160 donors were pooled and sequenced in 25 batches with an expected cell capture value of 20,000. This table summarizes the quality of read mapping and quantification of each batch.
Differentially expressed genes were used as markers for each cluster to assist in cell classification. The expression of each cluster was contrasted against the expression of all other cells, and DE genes were identified using a likelihood-ratio test, as implemented via the “bimod” function in Seurat.
Differentially expressed genes from POAG cell lines in the RGC lineage were identified via tradeSeq’s “conditionTest” function. Genes were significant if FDR (Benjamini-Hochberg) < 0.05.
cis-eQTLs were mapped using an additive linear model as implemented via MatrixEQTL. Lead cis-eQTLs were selected based on significance (FDR < 0.05), expression in >30% of donors and lowest FDR value. Lead cis-eQTLs were then tested for significance of genotype-disease interactions.
eGenes identified in initial round of cis-eQTL mapping were selected for conditioning if they had an eQTL in more than one cell type and were associated with more than one eSNP. eSNPs were first tested for independence, followed by allelic change after conditioning.
Differential gene expression was conducted via Wilcoxon rank-sum test on RGC clusters, testing POAG cells versus control cells.
Data Availability Statement
-
•
Single-cell RNA-seq data have been deposited at the Human Cell Atlas (https://data.humancellatlas.org/explore/projects/77780d56-03c0-481f-aade-2038490cef9f). Accession number: 77780d56-03c0-481f-aade-2038490cef9f
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.