

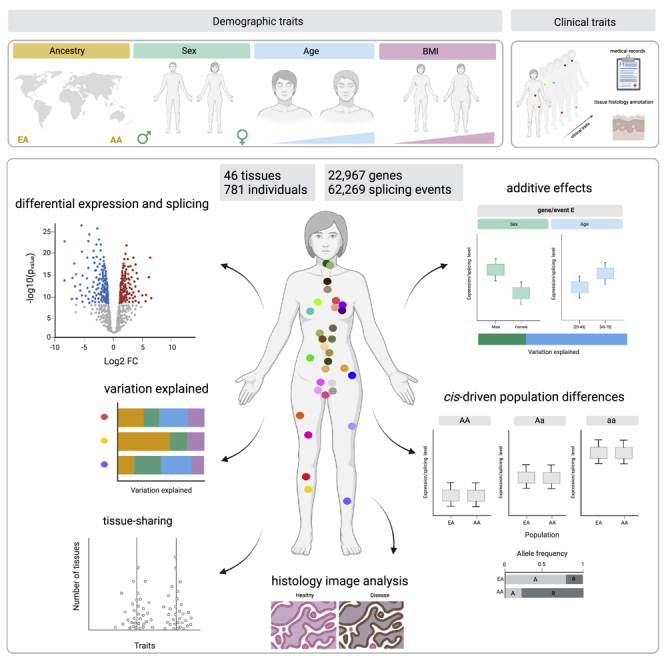
Summary
Understanding the consequences of individual transcriptome variation is fundamental to deciphering human biology and disease. We implement a statistical framework to quantify the contributions of 21 individual traits as drivers of gene expression and alternative splicing variation across 46 human tissues and 781 individuals from the Genotype-Tissue Expression project. We demonstrate that ancestry, sex, age, and BMI make additive and tissue-specific contributions to expression variability, whereas interactions are rare. Variation in splicing is dominated by ancestry and is under genetic control in most tissues, with ribosomal proteins showing a strong enrichment of tissue-shared splicing events. Our analyses reveal a systemic contribution of types 1 and 2 diabetes to tissue transcriptome variation with the strongest signal in the nerve, where histopathology image analysis identifies novel genes related to diabetic neuropathy. Our multi-tissue and multi-trait approach provides an extensive characterization of the main drivers of human transcriptome variation in health and disease.
Keywords: transcriptome, ancestry, sex, age, BMI, diabetes, alternative splicing, tissue, gene expression, human traits
Graphical abstract
Highlights
-
•
Ancestry, sex, age, and BMI make tissue-specific contributions to expression variation
-
•
Contributions to expression variation are mostly additive, and interactions are rare
-
•
Ribosomal proteins have widespread splicing differences between populations
-
•
Systemic analysis of diabetes reveals genes associated with neuropathy in the tibial nerve
García-Pérez et al. perform a multi-tissue analysis of the association between demographic and clinical traits and human transcriptome variation. Ancestry, sex, age, and BMI and certain diseases make large tissue-specific contributions to gene expression variation, whereas alternative splicing variation is mostly driven by ancestry and under genetic control.
Introduction
Over the past two decades, transcriptome analyses have revolutionized our understanding of a myriad of biological processes, allowing us to connect molecular changes to phenotypic traits. Analyses of gene and alternative splicing patterns across tissues,1,2 developmental time points,3,4,5 different physiological and pathological conditions,6,7,8 and species9,10,11,12,13,14 have provided insights into transcriptional and post-transcriptional regulatory mechanisms that underlie organismal phenotypes. A large-scale transcriptomic analysis in humans showed that gene expression, rather than alternative splicing, is key to defining tissue phenotypes, whereas both expression and alternative splicing contribute to interindividual variation.1 Further studies, focused on specific tissues, have shown that demographic traits such as ancestry, sex, age, and body mass index (BMI) are strongly associated with gene expression variation. Expression differences between populations are widespread, particularly in response to immune challenges.15,16 Sex expression differences are ubiquitous and can also be associated with the genetic regulation of gene expression,17 whereas changes in expression with age are mostly tissue specific and often correlated with mitochondrial activity.18 Alternative splicing (AS) also drives transcriptional heterogeneity by generating different exon combinations from the same gene. Several studies have identified AS events that vary with age,19,20 sex,21 and ancestry22 and have provided important insights into how splicing contributes to phenotypic variation.1,2
Despite the important contributions of these studies, analyses of transcriptome variation have mostly been restricted to single traits and a few tissues.17,18,19,20,21,23,24 Consequently, while demographic traits, such as ancestry, sex, and age, are simultaneously associated with human transcriptome variation, the nature of their joint effects across tissues remains largely unknown. Only studies in whole blood have started to address the synergic contribution of sex and age to differential gene expression variation upon immune stimulation, with sex associations being shared more across conditions than age associations.25 Similarly, it has been shown that gene expression varies between males and females during immune cell aging.26 However, beyond immune cell types, little is known about how different traits simultaneously interplay to define tissue, organ, and individual phenotypes.
Studies identifying gene and AS differences between healthy and diseased individuals have shed light on disease mechanisms by pinpointing the specific genes and pathways involved in disease progression and severity.27 However, most transcriptome analyses in the context of human disease have investigated their associations in well-known affected tissues (e.g., diabetes in pancreas28,29 or adipose tissues30,31), usually neglecting their systemic effects due to tissue sample collection limitations. The extensive medical history available for the Genotype-Tissue Expression (GTEx) cohort32 overcomes this drawback, allowing the study of the influence of different diseases on a multi-tissue scale. Similarly, the pathology annotation of GTEx samples’ images allows us to connect transcriptomic changes to disease-associated differences in tissue architecture.33
Here, we take advantage of GTEx data to systematically analyze the associations between multiple demographic and clinical traits and between gene expression and AS variation across human tissues. We identify differentially expressed genes and differentially spliced events across tissues, focusing on additive and interaction effects. We highlight commonalities and differences between tissues and traits and between expression and splicing. Overall, our multi-tissue and multi-trait approach provides an extensive characterization of the main drivers of human transcriptome variation, improving our understanding of how phenotypic variation emerges in health and disease.
Results
Demographic traits make different contributions to tissue transcriptional variation
We used the GTEx release v.8 data to simultaneously quantify gene expression changes with four demographic traits, genetic ancestry,2 sex, age, and BMI, across 46 different tissues from 781 individuals (Figure S1A). We considered a total of 22,967 genes (Figure S1B) and identified differentially expressed genes (DEGs) while controlling for known sources of technical variation and unobserved confounders such as cell-type composition (STAR Methods). Age had the largest number of DEGs followed by sex, ancestry, and BMI, with variations across tissues (Figure 1A). Skin, breast, and adipose tissues had the largest number of DEGs for ancestry, sex, and BMI, respectively, as previously observed.1 Interestingly, the arteries have the largest number of DEGs with age, which may relate to the observation of widespread aging changes in the cardiovascular system.34 These general patterns persist when controlling for sample size, although expression changes in the uterus and ovary with age become more apparent35 (Figure 1C). To assess replication, we compared our findings with four independent studies (STAR Methods) and found significant overlaps (one-tailed Fisher’s exact test, false discovery rate [FDR] <0.05) with all demographic traits in the expected tissues (Table S1A). We observed consistent replication of age-DEGs in blood reported in Pellegrino-Coppola et al.,36 which explicitly corrected for differences in cell-type abundances, suggesting that our differential expression analysis correctly controls for tissue composition.
Figure 1.

Contributions of demographic traits to gene expression variation
(A) Number of DEGs per tissue and demographic trait. Heatmap cell colors are normalized to the maximum value per trait. Tissues are sorted by sample size, and tissue labels correspond to tissue names described in Figure S1.
(B) Proportion of total tissue expression variation explained by each demographic trait. Top bars show the number of tissues for which each demographic trait explains the largest proportion of variation.
(C) Mean gene expression variation explained by each demographic trait in each tissue. This decreases with sample size since larger numbers of samples provide power to detect smaller contributions.
(D) Examples of genes with a large proportion of expression variation explained by a demographic trait.
Next, we assessed whether demographic traits made similar contributions to expression variation across tissues or their individual contributions varied independently by tissue. We used a hierarchical partitioning approach to quantify the contribution of each trait to gene expression variation while controlling for collinearity effects (STAR Methods). We found that, while one trait explains most of the variation in some tissues (e.g., sex in pituitary or age in aorta artery), the four demographic traits have comparable contributions in others (e.g., skeletal muscle or adipose subcutaneous) (Figure 1B). Ancestry was the principal contributor to expression variation in most tissues, followed by age, sex, and BMI (Figure 1B).
In general, the variation explained by demographic traits per gene is low (Figures 1C and S1D), consistent with previous observations.25 Age is associated with more genes but explains a lower proportion of their variation compared with sex, which is associated with fewer genes but makes larger contributions, consistent with observations in whole blood.25 The contribution of ancestry is similar to that of sex, and the contribution of BMI, in tissues where it contributes, is generally higher than that of age. We identified 3,196 genes for which a single trait explains more than 10% of their interindividual tissue differences (Figure 1D; Table S1B). Among these, some are known to be implicated in trait-related phenotypes, such as ACKR1, the malaria receptor gene, whose lower expression in individuals of African ancestry is associated with higher resistance to malaria infection.37
Overall, we observe that the associations of each demographic trait on interindividual variation in gene expression is largely tissue dependent.
Gene expression differences are restricted to one or a few tissues
We next sought to explore similarities between the contributions of each demographic trait across tissues. Most genes (90%) are differentially expressed (DE) in between one and five tissues (Figure 2A; Table S1C).17,18 This pattern cannot be explained by tissue-restricted expression because only 430 genes are exclusively expressed in the tissues where they are DE (Tables S1D and S1E) and the tissue where a gene is DE is often not the tissue with the highest expression of that gene (Figure S2A). Genes that are DE in many tissues for a given trait might be important drivers of phenotypic differences for that trait. We found 443 highly tissue-shared DEGs (DEGs in 10 or more tissues; Table S1F). Among these, ancestry-DEGs are enriched in glutathione-related metabolic processes (Figure 2B; Table S1G), mostly driven by glutathione genes clustered in a highly polymorphic locus associated with cancer risk.38,39,40 Most highly tissue-shared sex-DEGs are X-chromosome-inactivation escapees17,41 and Y-linked genes (Figure 2A), whereas highly tissue-shared age-DEGs are enriched in p53 pathway genes (Figure 2B; Table S1H), which are involved in aging and cancer.42 Genes DE with BMI in more than three tissues (159) include genes involved in body weight and food intake regulation, such as LEP or AKAP143,44 (Figure 2A).
Figure 2.

Tissue sharing of DEGs and contribution of genetic variants to expression differences between human populations
(A) Distribution of the number of tissues in which a gene is DE with each demographic trait. Labeled ancestry- and age-DEGs correspond to highly tissue-shared genes enriched in glutathione-related metabolic processes and p53 pathways, respectively. For sex-DEGs, black points correspond to highly shared X-chromosome-inactivation (XCI) escapees17 and Y genes. As examples, the well-known XCI escapee XIST41 and the ubiquitously transcribed Y gene UTY are labeled. The top three most tissue-shared BMI-DEGs are labeled. Bottom bars show the proportions of tissue-specific DEGs and DEGs in a low (2–5), moderate (6–9), or high (≥10) number of tissues with each demographic trait.
(B) Median tissue expression values for a highly tissue-shared ancestry (top) and age (bottom) DEG (EA, European American; AA, African American).
(C) Percentage of cis-driven of the total ancestry eGenes DE across tissues.
(D) Example of a cis-driven DEG in sun-exposed skin. Left: bar plot showing the allele frequency of the eQTL variant in each population. Right: PWP2 violin plots of gene expression levels stratified by population and individual genotype.
(E) cis-driven DEGs are associated with eQTLs with larger Fst values (Wilcoxon signed-rank test, p = 1.9e−10). Violin plots show the distribution of tissue median Fst values for cis-driven (left) and cis-independent (right) DEGs.
(F) cis-driven DEGs are more tissue shared (Wilcoxon signed-rank test, p = 3.9e−07). Violin plots show the distribution of median tissue-sharing values for cis-driven (left) and cis-independent (right) DEGs.
(G) cis-eQTLs explain a larger amount of gene expression variation than ancestry (Wilcoxon signed-rank test, p = 2.8e−14). Violin plots show the distribution of tissue median gene expression variation explained by eQTLs in cis-driven and cis-independent DEGs (left) and by ancestry in cis-independent DEGs (right).
Genetic effects underlie a large proportion of tissue gene expression differences between populations
Expression differences between human populations are partially driven by cis-regulatory variants (cis-eQTLs) with different allele frequencies between populations.15,16,45,46 Consistent with this, we observe that ancestry-DEGs are significantly enriched in eGenes (genes with at least a cis-eQTL)2 (two-tailed Fisher’s exact, FDR <0.05) (Table S2A). The contribution of cis-eQTLs to expression variation across populations has been explored only in immune cell types15,16 or cell lines.47,48 Here, we estimated the proportion of ancestry differences in expression variation attributable to cis-genetic effects across healthy tissues. We found that on average, 63% of the expression differences between populations in eGenes can be explained by cis-eQTLs (cis-driven DEGs) (Figures 2C, 2D, and S2B; STAR Methods). The proportion of cis-driven DEGs does not correlate with sample size (Spearman’s ρ = 0.15, p = 0.2914), suggesting that our ability to discriminate between cis-driven and cis-independent ancestry-DEGs does not depend on the number of eQTLs discovered. As expected, cis-driven DEGs have eQTLs with larger fixation indexes (Fsts), which measure the degree of differentiation between two populations49 (Figures 2E and S2C), and are DE in more tissues than cis-independent DEGs (Figures 2F and S2D), consistent with the observation that cis-eQTLs are often shared across tissues.2 Furthermore, on average, cis-driven ancestry effects explained a larger proportion of expression variation (∼22%) than genetic (∼11%) and ancestry effects (∼6%) in cis-independent DEGs (Figures 2G and S2E). In addition, although the numbers of both cis-driven and cis-independent DEGs are correlated with sample size (Spearman’s ρ 0.92 and 0.87, p 2.2e−16 and 3.8e−15, respectively), the number of cis-independent DEGs is much more variable across tissues than the number of cis-driven DEGs (Figure 2B). Overall, cis-driven genetic effects underlie a substantial fraction of ancestry differences and explain more expression variation than cis-independent effects, which have more subtle and tissue-specific influences and likely reflect a combination of developmental, environmental, and trans-genetic factors.
Additive contributions are widespread and tissue specific, whereas interactions are rare
Our study offers the opportunity to characterize the combined associations of demographic traits on specific genes across tissues. Unlike previous studies,25,26 by including several traits in the same model, we can explicitly assess whether the joint contributions of multiple traits are independent (additive) or dependent (interaction) and how these vary across tissues. First, we identified 7,458 DEGs with additive contributions for multiple traits across tissues (Figures S3A, S3B, and S3D). As expected, the expression variation explained per gene is larger for genes with additive contributions than for DEGs with one trait (Figure S3C). Most DEGs with two traits are restricted to a few tissues (e.g., 56% and 70% of ancestry-sex-DEGs and age-BMI-DEGs occur in not-sun-exposed skin and subcutaneous adipose tissue, respectively, consistent with their larger number of DEGs in those tissues) (Table S3A). Consequently, few genes (204) show the same additive contributions in more than two tissues. DEGs between populations and between sexes are the most tissue shared (Figures S3E and S3F), consistent with sex- and ancestry-DEGs being more tissue shared (Figure 2A).
We next explored whether particular combinations of demographic traits were more likely to be associated with the same genes. In 9 of the 21 tissues with a significantly larger than expected number of genes with additive contributions (two-tailed Fisher’s exact test, FDR <0.05) (STAR Methods), sex and age simultaneously affected gene expression, followed by sex and BMI (Figure 3A; Table S3B). Notably, in most cases, these additive contributions are driven by expression changes with specific directionalities (chi-square test, FDR <0.05) (Figure 3B; Table S3B), e.g., upregulation in males and older individuals in the tibial artery (Figure 3C) or upregulation in females and high BMI in the subcutaneous adipose tissue (Figure 3D). Importantly, these results are not confounded by differences in age or BMI between sexes (Figure S3G; STAR Methods). Such additive contributions might be especially relevant in genes whose expression levels are associated with disease risk, because healthy individuals in specific demographic groups may be at higher risk independent of their genetic background. Such is the case of CDKN2A, which has higher expression levels in males and older individuals (Figure 3C). CDKN2A is abundantly expressed in atherosclerotic lesions, particularly in cell types involved in atherogenesis,50 and positively correlates with CD68 (macrophages) and TNF (proinflammatory cytokine),51 both related to atherosclerosis.52
Figure 3.

Additive contributions of demographic traits are common and interactions rare
(A) Bar plot indicating the number of tissues with at least 20 DEGs with two demographic traits, of those how many have a significant overlap, and of those how many have a significant bias in the direction of change.
(B) Proportion of DEGs with sex and age (left) or sex and BMI (right) in each tissue that fall in each color-coded category.
(C and D) Left: examples of two tissues with more DEGs with two traits than expected that also have a bias in the direction of change. The scatterplots show the log2 fold change associated with each demographic trait, and each point represents a gene. Red, genes in categories with larger observed versus expected ratios. Labeled genes are among the ones with larger fold changes with both traits. Right: violin plots of expression levels for example genes, stratified by age range or sex. Bars at the bottom indicate the proportion of expression variation explained by each demographic trait.
(E) Comparison of age fold changes calculated separately for males and females in genes with a significant interaction between sex and age (Wilcoxon signed-rank test, p <2.2e−16).
(F) Example of a gene with a significant interaction between sex and age: its expression increases with age in males but it decreases with age in females. Expression levels are stratified by sex and age range.
We also tested whether the association of one demographic trait with gene expression variation could depend on another demographic trait (STAR Methods). We found 235 genes with a significant interaction between two demographic traits in 11 tissues (Table S3C). Most interactions (91%, 216 genes) occur in breast with sex and age and are driven by aging expression changes in females related to mammary gland development (Figures 3E and 3F; Table S3D).
Overall, these results show that demographic traits have tissue-specific additive contributions, whereas interactions are rare and highlight the importance of analyzing multiple individual traits simultaneously to assess the nature of their joint contributions.
Tissue distribution of alternative splicing events
Alternative processing of mRNAs contributes to transcriptional heterogeneity by generating transcripts with different exon combinations from the same gene. Such transcriptional heterogeneity has been shown to be important for development,53 disease,8 and evolutionary innovation.54 To improve our understanding of alternative mRNA processing variation across tissues and individuals, hereafter summarized as AS, we quantified AS based on the “percentage spliced-in” (PSI)55 for seven types of AS events (Figure 4A). We identified a total of 62,269 AS events (Figure S1B) (STAR Methods). The number of AS events per tissue was highly variable, but the distribution of type of event across tissues was similar: exon skipping and mutually exclusive exons were the most and least abundant, respectively (Figure S1B). In addition, alternative first and last exons are more tissue specific, consistent with previous observations,56 while retained intron events are more shared across tissues (Figure 4B).
Figure 4.

Contribution of demographic traits to AS variation
(A) Schematic illustration of the different types of splicing events. For each type of splicing event, we present the spliced-in and spliced-out versions of the splicing event. In black is the exonic/intronic sequence that is included in the spliced-in isoform and for which PSI values are calculated.
(B) Cumulative distribution of the number of tissues in which splicing events are AS.
(C) Functional characterization of AS events.
(D) Proportion of AS events associated with a switch between a non-coding and a coding isoform per type of event. Boxplots show the distribution of the proportion of AS events per tissue.
(E) Number of DSEs per tissue and demographic trait. Heatmap cell colors are normalized to maximum value per column.
(F) Proportion of the total tissue AS variation explained by each demographic trait. Top bars are the numbers of tissues for which each demographic trait explains the largest proportion.
(G) Examples of the potential functional consequences of DSEs. Shown are schematic representations of the PFAM domain and the transcript structure of isoforms that either include or exclude the splicing event and that contribute to the DSEs. For each event, PSI values are represented as boxplots with samples stratified by population or age range. Violin plots show the PSI distribution. Points correspond to individual PSI values. The number of individuals in each group is shown within the plot. Bars at the bottom indicate the proportion of alternative splicing variation explained by each demographic trait.
(H) Comparison of the relative contribution of each demographic trait to the total tissue expression and splicing variation explained. For each trait, the average value across tissues is plotted. The error bars correspond to the standard deviation. For each demographic trait, we considered only tissues with at least five DEGs and five DSEs.
We assessed whether AS events are associated with coding or non-coding isoform switches. Nearly half of the AS events were associated with a switch between two protein-coding isoforms (Figure 4C; Table S4A; STAR Methods). In 28% of those, the alternative usage of exonic/intronic sequences overlapped with a known protein-coding domain57 (STAR Methods), thus likely contributing to protein diversity. Most of the remaining AS events (40%) were associated with a switch between a non-coding and a protein-coding isoform. In these cases, inclusion of introns and alternative 5′ and 3′ events was more often associated with the non-coding isoform (binomial test, FDR <0.05; Figure 4D; Table S4B), suggesting that the inclusion rather than exclusion of additional bases is more associated with the loss of coding potential.
Ancestry explains most alternative splicing differences between individuals
We then explored the association between ancestry, sex, age, and BMI and AS variation by performing differential splicing analysis on the PSI value for each splicing event and correcting for known sources of technical variation and unobserved confounders such as cell-type composition (STAR Methods). We identified 16,197 differentially spliced events (DSEs) across tissues and demographic traits. In contrast to expression (Figure 1A), ancestry has the largest number of DSEs, followed by age, sex, and BMI (Figure 4E). DSEs affect a total of 6,909 genes (differentially spliced genes, DSGs; Figure S4A). Similar to expression, the largest number of DSEs with ancestry, sex, and BMI occur in not-sun-exposed skin, breast, and subcutaneous adipose tissue, respectively. However, the hypothalamus has the largest number of age-DSEs, closely followed by the arteries. The general patterns of differential splicing remain when controlling for the number of samples, but the aging signal in the brain becomes more apparent (Figure S4B). Splicing differences in the brain persist even if correcting for neuron abundance58 (Figure S4C), suggesting that age may be associated with splicing patterns in some brain regions independent of neuronal decay with aging.59 Further analyses may be needed to confirm this observation.
We found more alternative last exons but fewer alternative first exons and alternative 3′ events differentially spliced (DS) between populations than expected (Table S4C; chi-square test, FDR <0.05; STAR Methods). We also identified tissue- and trait-specific biases for some event types (Table S4D; binomial test, FDR <0.05; STAR Methods), with the strongest bias being increased intron retention in tibial artery with age and increased intron retention in not-sun-exposed skin in Europeans. We further explored the functional consequences of differential splicing. There were 7,925 (46.37%) DSEs associated with a switch between protein-coding isoforms, 1,892 (23.87%) of which affect a known protein-coding domain57 (Figure 4G; Table S4E; STAR Methods). Consistent with previous findings,5,22 more genes change both their expression and their splicing pattern than expected by chance, particularly between populations (two-tailed Fisher’s exact test, FDR <0.05) (Table S4F). These genes are enriched in eGenes and in genes with cis-sQTLs (cis-driven DSEs; sGenes) (Table S4G).
Next, we quantified the independent contributions of demographic traits to AS variation across tissues and compared them with expression (STAR Methods). Ancestry is the major contributor to splicing variation in most tissues, with a few exceptions, such as the aorta or brain regions such as the hypothalamus or amygdala, where age has a larger contribution (Figures 4F, S4D, and S4E). The contributions of sex and BMI are of note in only a few tissues, such as breast and muscle or the adipose tissues, respectively. The amount of variation explained by each demographic trait is lower for splicing than for expression (Figure S4F). However, proportionally, ancestry explained a larger proportion of splicing than expression variation, whereas the opposite was true for age, sex, and BMI (Figure 4H). Comparing splicing event types, exon skipping, alternative 5′ and 3′, and intron retention events explain larger amounts of variation than the alternative usage of transcript initiation or termination sites (Table S4H). This suggests that changes at the post-transcriptional level (i.e., exon skipping) might make a larger contribution to the overall AS variation than changes at the transcriptional level (i.e., alternative first or last exon). DSEs (653) with a large proportion (>10%) of AS variation explained by a given demographic trait might be relevant for trait-related phenotypes (Table S4I). For example, we found ancestry-DSEs in the CYP3A5 gene (Figure S4G), for which AS has been previously shown to abolish its enzymatic activity, mostly in European populations.60
Together, these results indicate that ancestry significantly explains a larger proportion of interindividual splicing variation than the other demographic traits.
cis-regulatory variants explain most alternative splicing variation between human populations
Differences in allele frequency between populations underlie a large proportion of splicing differences between populations.22 Genes with ancestry-DSEs are enriched in sGenes (two-tailed Fisher’s exact test, FDR <0.05) (Table S5A) and, on average, 77% of population splicing differences can be explained by cis-sQTLs with some variation across tissues (Figures 5A and S5A; STAR Methods). As expected, cis-driven DSEs are associated with sQTLs with larger genetic distances between populations and are more tissue-shared than cis-independent DSEs (Figures 5B, 5C, S5B, and S5C). Similar to expression, cis-sQTLs associated with cis-driven DSEs explained a larger proportion of splicing variation (∼9%) than either sQTLs (∼5%) or ancestry (∼6) in cis-independent DSEs (Figures 5D and S5D). The proportion of cis-driven DSEs is negatively correlated with sample size (Spearman’s ρ = −0.53, p = 0.0001548), suggesting that we identify splicing differences between populations that are likely driven by sQTLs yet to be identified.
Figure 5.

Splicing patterns of ribosomal proteins vary across human populations
(A) Percentage of cis-driven DSEs across tissues.
(B) cis-driven DSEs are associated with sQTLs with larger Fst values (Wilcoxon signed-rank test, p = 1.563e−12). Violin plots show the distribution of tissue median Fst values for cis-driven (left) and cis-independent (right) DSEs.
(C) cis-driven DSEs are more tissue shared (Wilcoxon signed-rank test, p = 1.395e−06). Violin plots show the distribution of median tissue-sharing values for cis-driven (left) and cis-independent (right) DSEs.
(D) cis-sQTLs explain a larger amount of splicing variation than ancestry (Wilcoxon signed-rank test, p = 1.421e−13). Violin plots show the distribution of tissue median splicing variation explained by sQTLs in cis-driven and cis-independent DSEs (left) and by ancestry in cis-independent DSEs (right).
(E) Distribution of the number of tissues in which a splicing event is DS with each demographic trait. Ancestry-DSEs in ribosomal proteins are highlighted in black and labeled if shared in 10 or more tissues. Bottom bars show the proportions of tissue-specific DSEs and DSEs in a low (2–5), moderate (6–9), or high (≥10) number of tissues with each demographic trait.
(F) Functional enrichment of genes with highly shared ancestry-DSEs.
(G) Bar plot shows the proportion and number of ancestry-DSEs in ribosomal proteins in two or more tissues that have the same or different directionality.
(H) Example of a genetic variant associated with the splicing pattern of a ribosomal protein not previously reported as an sGene. Bar plot shows the allele frequencies in each population. Violin plots show the PSI distribution stratified by population and genotype. Points correspond to individual PSI values (EA, European American; AA, African American).
(I) Examples of two highly tissue-shared ancestry-DSEs on ribosomal proteins that affect a protein-coding domain.
Alternative splicing differences between human populations in ribosomal proteins are widespread across tissues and under genetic control
We next sought to characterize the tissue-sharing pattern of DSEs. Only ancestry had highly shared DSEs (Figure 5E), and genes with highly shared ancestry-DSEs were strongly enriched in translation pathways, driven by ribosomal proteins (10 of 23 genes) (Figure 5F; Table S5B). This is consistent with previous observations that translation-related genes had the largest interindividual variation in AS.1 At the tissue level, ancestry-DSGs are also enriched in ribosomal proteins (Figure S6A). To explore further this functional enrichment, we used an independent dataset of DSGs between individuals of European and African ancestry in monocytes22 and found the same pathways enriched (Table S5C). Similarly, we found all DSGs between populations in monocytes also DS in at least one tissue (Figure S6A). Furthermore, isoform expression changes across tissues are highly concordant with those observed in monocytes,15 particularly for isoforms underlying highly shared splicing differences (Figure S6B). We do not observe this extensive variability in ribosomal proteins at the gene expression level, and neither was it previously observed in monocytes15 (Figure S6A).
The DSEs between populations in ribosomal proteins not only were tissue shared but also showed the same directionality across tissues (Figures 5G and S6C), which would be consistent with their splicing patterns being under genetic control.2 Consistent with this hypothesis, ∼90% of the population DSEs in ribosomal proteins previously reported as sGenes2 are cis-driven (Figure S6C; STAR Methods). For the remaining ancestry-DSEs in ribosomal proteins, we investigated if their AS patterns were significantly associated with nearby genetic variants (STAR Methods), given that sQTLs tend to be located in close proximity.61,62 Most splicing events (82%) have at least one significantly associated variant, and the majority of them are cis-driven (74%) (Figures 5H and S6C), which suggests that ancestry-differential splicing in ribosomal proteins is under genetic control. Reassuringly, five of the ribosomal proteins we find associated with genetic variants that were not reported as sGenes in the GTEx v.8 main paper2 were reported as sGenes using an isoform-based approach61 (Table S5D).
Finally, we explored the functional consequences of splicing differences between populations in ribosomal proteins and found that in 24% of the events associated with a switch between protein-coding isoforms, the alternative usage of exonic/intronic sequences overlaps a known protein-coding domain57 (Figure S5C). Two of these events are highly shared across tissues: an alternative 5′ splice site in the ribosomal protein RPLP2 that overlaps a 60s acidic ribosomal protein domain and an alternatively spliced exon in RPL10 (Figures 5I and S6D) that overlaps a ribosomal protein L16p/L10e domain. The 60s acidic ribosomal protein domain is an important component of the ribosomal stalk, a conserved structure involved in the recruitment of translation elongation factors,63 which affects the translation of some specific mRNAs.64
Our results suggest that there are widespread splicing differences in ribosomal proteins between populations, likely due to genetic control, and raises the possibility that some aspects of the translational machinery may consistently vary across human populations.
Clinical traits contribute to tissue expression variability
Many diseases are associated with differences in transcription, altering both expression6 and splicing.8 We leveraged the donors’ medical history and histopathological annotations to investigate transcriptomic changes associated with clinical traits and selected 17 disease-related phenotypes for analysis based on the number of affected donors and DEGs in the tissue of origin (Figure 6A; Tables S6A and S6B; STAR Methods). Hashimoto’s thyroiditis, pneumonia, and atherosclerosis have among the largest numbers of DEGs and DSGs in thyroid, lung, and tibial artery, respectively (Figures 6B and S7A). Except for Hashimoto’s thyroiditis, genes DE are not more likely to be also DS than expected by chance (two-tailed Fisher’s exact test, FDR <0.05) (Figure S7A; Table S6C), suggesting independent contributions. As expected, the contribution of clinical traits to expression and splicing variation is highly variable, depending on the tissue and the disease (Figures 6C and S7B). Notably, in some tissues, the contribution of clinical traits to expression variation is larger than that of the demographic traits (e.g., Hashimoto’s thyroiditis) (Figure S7B). Conversely, ancestry remains the principal driver of splicing variation (in 24 of 25 tissues) (Figure S7B). Among the genes (633) with a large proportion (>10%) of their interindividual transcriptomic differences explained by a clinical trait, some are well-known disease-related genes, such as INS in type 1 diabetes65 or FBLN2 in atherosclerosis66 (Figure S7C). Others, still uncharacterized, might also play important roles in disease phenotypes, such as CD37, related to immunity,67 in Hashimoto’s thyroiditis, or KYAT3, a regulator of KYNA, which is a biomarker for diabetes,68 in type 2 diabetes (Tables S6D and S6E). Nearly 10% of disease-DSEs overlap known protein domains, including an exon more frequently excluded in type 2 diabetic individuals in the NAGLU gene, which is associated with glucose metabolism and neuropathy69 (Figure S7D).
Figure 6.

Types 1 and 2 diabetes alter the transcriptome of multiple tissues, especially of the tibial nerve
(A) Clinical traits and affected tissues.
(B) Number of DEGs per tissue and clinical trait. Bars are colored according to the tissues.
(C) Proportion of the total tissue expression variation explained by each clinical trait.
(D) DEGs with type 1 or 2 diabetes in three or more tissues. The y axis corresponds to the fold change between healthy and diseased samples per tissue. In bold are known disease-related genes.70,71,72,73,74,75
(E) Left: the overlap between DEGs with types 1 and 2 diabetes in the tibial nerve. Right: DEGs with both diabetes have the same directionality. Genes driving functional enrichments (Table S6H) are labeled in the plot.
(F) On the left, tissue images from a healthy and a diabetic individual. Note the larger diameter of the fascicles (circles) and smaller interstitial spaces in the diabetic donor, consistent with previous observations.76 The right shows the ROC curves of the top and a median performing classifier.
(G) Top: DEGs with age and clinical traits show a biased directionality. Our observations are not confounded by age differences between healthy and diseased individuals (Figure S7F). Bars indicate the proportion (and number) of DEGs in each of the four possible directionalities. Bottom: scatterplot shows the fold change associated with age (x axis) versus the fold change between healthy and type 2 diabetes (y axis) in the tibial nerve. Labeled genes (24) have been previously associated with type 2 diabetes susceptibility in the tibial nerve through transcriptome-wide association studies.77
(H) LPL expression changes with age and type 2 diabetes. Gene expression levels are represented as boxplots with samples stratified by both traits. Bars at the bottom indicate the proportions of expression variation explained by age and type 2 diabetes.
Type 1 and type 2 diabetes are associated with transcriptome variation in multiple tissues, particularly the tibial nerve
Both type 1 and type 2 diabetes have systemic associations with transcriptome variation.65,78,79 Yet, previous work studying transcriptome changes in diabetes has focused on specific tissues80 or cell types.81 Here, we took advantage of the GTEx multi-tissue data to assess how types 1 and 2 diabetes are associated with changes in the transcriptome of multiple tissues (Figure S7E). We first assessed if types 1 and 2 diabetes’ association was similar across tissues and found 78 and 309 DEGs in two or more tissues with either type 1 or type 2 diabetes, respectively (Table S6F). The difference in the number of DEGs is likely due to decreased statistical power related to the lower number of individuals with type 1 diabetes. Seven of 14 DEGs in more than three tissues have been previously linked to diabetes, but the others are novel, like LDOC1, a regulator of NF-κB, which is involved in diabetic pathogenesis82,83 (Figure 6D). Conversely, only six events are DS with type 1 or 2 in more than one tissue (Figure S7F). In both the pancreas and the tibial nerve, we found a larger than expected overlap of DEGs with both type 1 and type 2 diabetes (two-tailed Fisher’s exact test, FDR <0.05; Figure S7E), with a significant bias in the nerve toward both diabetes changing expression in the same direction (chi-square test, FDR <0.05) (Figure 6E; Table S6G). Comparing the DE signal across tissues, we found that the tibial nerve is the most affected tissue in both types of diabetes. Pancreas had fewer DEGs than nerve, likely because pancreatic islets, central to the etiology of both types of diabetes,84,85 represent only ∼3% of the tissue, and thus, the whole pancreas is not representative of pancreatic islets.29,86 The observation that there are many genes associated with diabetes in other tissues may reflect the consequences of long-term exposure to hyperglycemia across tissues. The DEGs in the tibial nerve significantly overlap with those reported as dysregulated in the sciatic nerve of diabetic mice (two-tailed Fisher’s exact test, p = 1.195e−06).87 Functional enrichment analysis (Table S6H; STAR Methods) revealed that upregulated genes in the tibial nerve are enriched in immune receptor activity, whereas downregulated genes are enriched in ion channel activity. Our findings are consistent with the high incidence of diabetic neuropathy in diabetic patients, with ∼50% expected to develop this complication over time.88 Diabetic neuropathy is a type of nerve damage due to continuous high blood sugar levels that result in thicker nerve fascicles.76 To further validate our results, we used the GTEx tibial nerve samples’ histology image data to train a support vector machine to classify diabetic individuals (STAR Methods), and we obtained a maximum and median area under the receiver characteristic operator curve (AUC) of 81% and 75%, respectively (Figure 6F). We wondered whether the probability of being classified as diabetic could be a proxy for disease severity. We found 328 genes (Table S6I) whose expression significantly correlated with the probability of being diabetic (STAR Methods). These genes were highly enriched in neuropathy-related terms (Table S6J) and included genes previously linked with diabetic neuropathy pathogenesis, e.g., SYNDIG189 (Figure S6G), but also novel genes that could be important players in neuropathy progression, such as ARHGEF16, previously associated with diabetes but not diabetic neuropathy.90
Taken together, our findings expand previous work76,87,91 and suggest that, despite their different etiologies, types 1 and 2 diabetes are more strongly associated with transcriptome changes in tibial nerve than in other tissues. These changes are consistent with the high prevalence of diabetic neuropathy in patients88 due to hyperglycemia and provide novel gene candidates associated with diabetic neuropathy.
Clinical and demographic traits jointly contribute to gene expression variation
Demographic traits, such as sex, age, ancestry, and BMI, often influence complex disease risk, prevalence, and progression.92,93,94,95 Thus, we set out to investigate the interplay between demographic and clinical traits. We found 5,790 DEGs with at least one demographic and one clinical trait (Table S6K). These additive contributions are enriched in a tissue- and trait-specific fashion, e.g., larger than expected DEGs with sex and gynecomastia in the breast (two-tailed Fisher’s exact test, FDR <0.05). As we previously noted, many of these additive contributions are driven by expression changes with specific directionalities (chi-square test, FDR <0.05) (Table S6K): genes upregulated in individuals with Hashimoto’s thyroiditis and older individuals, consistent with the hypothyroidism caused by the disease96 and the subclinical hypothyroidism caused by age.97,98 Notably, most DEGs with age and both type 1 and type 2 diabetes in the tibial nerve are upregulated either in older and diabetic individuals or in younger and healthy individuals (Figure 6G). Among these genes is LPL, involved in the regeneration of the myelin sheath, which is deteriorated in diabetic neuropathy99 (Figure 6H). Importantly, this finding is not confounded by the greater incidence of type 2 diabetes in aged individuals (Figure S7H; Table S6K; STAR Methods), suggesting that diabetes affects the tibial nerve in a way similar to biological aging. These results indicate that some disease-related genes also have interindividual expression variation associated with non-disease traits and highlight the importance of characterizing their synergistic contribution to better understand disease mechanisms.
Demographic and clinical traits influence tissue cellular composition
The GTEx samples are heterogeneous bulk tissue samples that comprise diverse cell types. To identify changes in cell-type composition with demographic and clinical traits, we used enrichment scores for seven different cell types, benchmarked in Kim-Hellmuth et al.58 We found significant changes (FDR <0.05, STAR Methods) for six cell types across 18 tissues (Figure S7I) and replicated previously reported differences with sex.17 We found increased abundances of adipocytes in the liver with BMI100 and decreased adipocyte abundances in the subcutaneous adipose tissue with age.101,102 We observed lower enrichment scores for neurons in older individuals across brain regions, consistent with a decline in neuronal functions with aging.103,104 Changes in epithelial cells were associated with three demographic traits in different tissues: they decreased in colon transverse and prostate with age and they were more abundant in the sun-exposed skin of African American individuals and in the female breast. Epithelial cells are widespread throughout the body and perform different functions,105,106,107 some specific to particular body sites (e.g., the glandular epithelium secretes enzymes, hormones, and fluids,108 whereas the epithelial lining of internal organs absorbs nutrients109), which might explain the different patterns of change in the different tissues. We replicated previously reported changes with clinical traits58 but we did not find significant changes with either type 1 or type 2 diabetes, likely due to the limited number of cell types analyzed (Figure S7J). We further leveraged the histopathological annotations and found increased abundances of macrophages in lung samples with fibrosis (Figure S7J) (two-tailed Fisher’s exact test, p = 3.04e−07) and reduced spermatogenesis in older testis samples (two-tailed Fisher’s exact test, p = 9.8e−05). Together, these results suggest that both demographic and clinical traits are associated with differences in cell-type composition.33
Discussion
Understanding interindividual transcriptome variation is fundamental to deciphering human biology and disease. Our work quantifies the joint associations of different demographic and clinical traits on the transcriptome and identifies the most important drivers for each tissue. We show that demographic traits contribute to transcriptome variation additively, with few examples of interactions. Previous work focused on age and sex identified important aging differences between males and females in immune cells26 but did not address whether these were due to additive contributions or interactions. Our results suggest that age and sex make additive contributions to gene expression variation across tissues, often with biased directionalities. In addition, we also observe a larger contribution of aging in female reproductive tissues (i.e., breast, uterus, and ovary) than in those of males (i.e., testis and prostate). These changes may be related to female hormonal changes, as the age span of GTEx samples is centered around menopause age.110,111 However, larger sample sizes might be needed to identify more subtle interactions and to explore further interactions between demographic and clinical traits.
We show that many expression and splicing ancestry differences are under genetic control, and the proportion is larger for splicing (77%) than for expression (63%) (Figures 2C and 5A). This is likely explained by limitations in AS analysis due to the inherently noisier nature of splicing,112 which favors the detection of splicing differences with larger effect sizes, such as those driven by cis genetic effects (Figures 2F and 5D). In addition, we observed widespread splicing but not expression differences in ribosomal proteins between European Americans and African Americans across tissues that are mainly under genetic control. Notably, these results raise the intriguing possibility that specific aspects of the ribosome machinery differ between individuals of different genetic ancestries across tissues. Further analyses might be needed to address whether these differences arose as a product of positive selection simultaneously targeting ribosomal protein genes.113 Importantly, a recent publication reported expression—rather than splicing—differences in ribosomal proteins between populations using single-cell data,16 which highlights the power of bulk multi-tissue data analysis to distinguish between expression and splicing variation compared with single-cell analysis.
Although machine learning methods have been previously used on GTEx histology images to identify image quantitative trait loci,114,115 classify GTEx tissues,116 and make pathology annotations,117 our analysis on the diabetic nerve is the first to link genes with histology image features related to the association between diabetes and changes in tissue architecture. Importantly, this analysis successfully identified novel candidates associated with diabetic neuropathy that could be of interest given the limited treatment options.88 Furthermore, we observe that diabetic neuropathy and aging make additive contributions to gene expression variation in the same direction, consistent with aging and disease pathology often sharing common molecular mechanisms.118,119 Our result highlights the richness of the GTEx dataset to explore the relationship between transcriptome, histology, and disease.
Collectively, our results provide a comprehensive catalog of gene expression and AS differences across many different human tissues and traits. They offer insight into the traits that drive human transcriptome variation; can help unveil the role of transcriptome variation in complex traits, disease risk, and disease progression; and are a resource to be further exploited by the scientific community.
Limitations of the study
Despite our multi-tissue and multi-trait approach, there are certain inherent limitations in our study, such as the limited representation of human populations (only European Americans and African Americans) or the biased age span toward older individuals. Our findings highlight the urgent need to include individuals with diverse ancestries as well as developmental and pediatric samples in transcriptomic analyses, since both demographic traits are important drivers of transcriptome variation.120 We also have reduced statistical power to detect transcriptomic changes associated with certain clinical traits due to both reduced sample sizes and analysis of bulk tissue transcriptomes rather than specific cell types, such as pancreatic islet cells for type 1 diabetes.29 These limitations might explain why we find the same genes associated with type 1 and type 2 diabetes only in the tibial nerve and not in other tissues affected by long-term hyperglycemia, such as the heart or arteries.121,122 Alternatively, disease treatment is known to mitigate diabetic complications121,123,124 and could thus have an effect on the number of genes we observe DE in specific tissues. Hence, collecting information about donor prescribed drugs would be desirable in future studies. Our ability to detect changes in cell-type composition is limited by the analysis of bulk tissue transcriptomes (Figures S7I and S7J). Future organ single-cell atlases that include more donors and disease conditions will shed light on how demographic and clinical traits influence the transcriptomes at single-cell resolution.125
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marta Melé (marta.mele@bsc.es).
Materials availability
No materials were generated in the study.
Experimental model and subject details
GTEx subjects
All human donors were deceased, with informed consent obtained via next-of-kin consent for the collection and banking of deidentified tissue samples for scientific research. The research protocol was reviewed by Chesapeake Research Review Inc., Roswell Park Cancer Institute’s Office of Research Subject Protection, and the institutional review board of the University of Pennsylvania. There were 838 donors (557 biological sex male, 281 biological sex female). Donors ranged in age from 20-70, with most enrolled donors being older individuals. For more details on donor characteristics and sample collection, see the GTEx v8 main paper (DataS2).2
Method details
Biospecimen collection
The biospecimen collection is described in detail in the GTEx v8 main paper.2
Molecular analyte extraction and QC
Molecular analyte extraction and QC DNA and RNA extraction and sequencing details are provided in the GTEx v8 main paper.2
Quantification and statistical analysis
GTEx data
The GTEx version 8 dataset consists of 17,382 RNA-seq samples from 948 post-mortem donors and 54 tissues, with genotype data for 838 donors from whole genome sequencing available in a phased analysis freeze. The GTEx biospecimen collection, molecular phenotype data production, and quality control are described in detail in the GTEx v8 main paper.2 Here, we analyzed data from the 46 tissue sources with at least 100 RNA-seq samples. We only included samples (n=13,684) from donors (n=781) with available metadata for the covariates included in our differential expression and splicing analysis, as well as demographic trait information for the donors’ genetic inferred ancestry2 (we only included European American and African American donors), sex, age, and body mass index (BMI).
Gene and alternative splicing event quantification
Gene and transcript quantifications were based on the GENCODE 26 release annotation (https://www.gencodegenes.org/releases/26.html). We downloaded gene counts and TPM quantifications from the GTEx portal (https://gtexportal.org/home/datasets). We selected genes with the protein-coding and lincRNA biotype on the GTEX GENCODE v26 GTF. For the expression analysis, we considered expressed genes per tissue (TPM ≥ 1 and ≥ 10 reads (unnormalized) in ≥ 20% of tissue samples, excluding genes in the pseudoautosomal region (PAR). In total, we analyzed 22,967 genes (18,185 protein-coding and 4,782 lincRNA) across tissues (Figure S1B). For the splicing analysis, we downloaded transcripts TPM quantifications from the GTEx portal (https://gtexportal.org/home/datasets) and we used SUPPA2127 (https://github.com/comprna/SUPPA/) to calculate percentages of splicing inclusion (PSI) for 7 different types of splicing events: skipped exon (SE), mutually exclusive exons (MX), alternative 3 prime (A3), alternative 5 prime (A5), retained intron (RI), alternative first exon (AF), and alternative last exon (AL). Specifically, we used SUPPA2 to first generate the dictionary of splicing events from the GENCODE v26 annotation and then computed their PSI values for each sample and splicing event. Each splicing event is defined by a set of isoforms: those that include the exonic/intronic sequence (spliced-in isoform) and those that either exclude or include alternative exonic/intronic sequence (spliced-out isoform) (Figure 4A). We used the following criteria to select the alternatively spliced events (ASEs) in each tissue: events in protein-coding and lincRNA genes expressed in each tissue; events quantified in all tissue samples (no NAs); we excluded events with low complexity (fewer than 10 PSI unique values) or insufficient variability (near zero variance); we kept events from expressed isoforms (TPM ≥ 0.5 in ≥ 20% of tissue samples for both the most abundant spliced-in and spliced-out isoforms) and with a quantifiable contribution of the traits of interest (see hierarchical partition analysis). In total, we analyzed 62,269 AS events (18,491 SE, 1,203 MX, 7,128 A5, 8,296 A3, 4,191 RI, 18,111 AF and 4,849 AL) across tissues (Figure S1B). To investigate the potential functional consequences of ASEs, we first identified the isoforms that contribute to each splicing event. From the set of isoforms that contribute to each splicing event, we selected the two most abundant isoforms per tissue that include (spliced-in) and exclude (spliced-out) the splicing event. Depending on the biotype of these two isoforms (spliced-in and spliced out), ASEs can then be associated with a switch between protein-coding isoforms, a switch between a protein-coding and a non-coding isoform, or a switch between non-coding isoforms. For those events with a switch between protein coding isoforms, we implemented a pipeline to identify ASEss that disrupt PFAM domains (see identification of ASEs that disrupt known protein-coding domains) (https://github.com/Mele-Lab/2022_GTExTranscriptome_fromSplicingEventsToProteinDomains).
Differential gene expression analysis with demographic traits
To identify differentially expressed genes (DEGs) we used linear-regression models following the voom-limma pipeline.128,139 We ran the analyses separately for each of 46 tissue sources. We adjusted our differential expression analysis for technical covariates routinely included in previous GTEx publications.41,140 These covariates are related to parameters of donor death, ischemic time, RNA integrity number (RIN), and sequencing quality control metrics. To control for unknown sources of variation we explored the expression variance captured by the PEER factors2 and explained by known sample and donor covariates, as well as by the xCell enrichment scores.58 We also investigated the effect of including progressively increasing numbers of PEER factors in our model in the identification of DEGs. As previously noted, we found that the first PEER factor was mostly correlated with cell type heterogeneity (see Figure S4A from58), and the second PEER factor was mostly correlated with the sequencing batch (see Figure S8A from141). We also noted that, conversely to eQTL discovery, the effect of including additional PEER factors on the DEG discovery was variable across tissues and led to reduced power to detect expression differences. Thus, to control for unknown sources of variation mainly related to differences in tissue composition and sequencing batch, we included the first two PEER factors. For each tissue, we compared log-cpm gene expression values and evaluated the statistical significance of the demographic traits of interest: ancestry, sex, age, and BMI. Ancestry and sex were treated as categorical variables and age and BMI as continuous variables. We corrected all analyses for multiple testing using false discovery rate (FDR) through the Benjamini–Hochberg method and considered genes differentially expressed at an adjusted p-value below 0.05.
To investigate interactions between demographic traits, we expanded the linear models in each tissue adding interaction terms between categorical variables or between categorical and continuous variables. For an interaction to be tested in a tissue we required that (1) we had previously found DEGs with both demographic traits involved in the interaction term, and (2) we had sufficient sample size (n=20 samples in each group). To determine the number of samples in each combination we categorized the continuous variables: age in two groups (younger (donors younger than 45 years old), and older (45 years old or older donors)) and BMI in three groups (normal (BMI < 25), overweight (25≤ BMI < 30) and obese (BMI ≥30)) (Table S3C).
Differential splicing analysis with demographic traits
To perform differential splicing analysis, we used a method that allowed both a direct comparison with the differential gene expression analysis and a subsequent quantification of the alternative splicing variation explained by each trait (see hierarchical partition analysis). Thus, we implemented an approach as similar as possible to the one used to identify differentially expressed genes using generalized linear models but modeling Percentage of Spliced In (PSI) values using fractional regression. We chose fractional regression over the more popular beta regression because fractional regression is suited to work with bounded values that can assume the extremes, as is the case for PSI values (e.g. 0 and 1). Specifically, we used the R glm function from the R package stats126 setting family= ‘quasibinomial ('logit')’ as a parameter. For each splicing event within each tissue, we fitted logit transformed PSI values with the same model used in differential expression analysis and evaluated the statistical significance of the demographic traits of interest: ancestry, sex, age, and BMI.
To calculate robust standard errors for our coefficients we used the vcovHC function from the R package sandwich with type = "HC0".142 We corrected all analyses for multiple testing using false discovery rate (FDR) through the Benjamini-Hochberg method implemented in the R package stats.126 For all analyses we considered events differentially spliced at an adjusted p-value (FDR) below 0.05. Notably, we found significant overlaps (see DEGs and DSEs replication with independent datasets) with previously reported differentially spliced events between human populations22 (Table S1A).
Downsampling analysis for expression and splicing analysis with demographic traits
Different tissues have different sample sizes and demographic trait distributions (Figure S1B). To assess the influence of sample size in the detection of DEGs and DSEs, we run the differential expression and splicing analysis 10 times per tissue, randomly downsampling each tissue to 100 samples.
DEGs and DSEs replication with independent datasets
To validate and assess the replicability of our DEGs and DSGs with each of the four demographic traits, we compared our results to several biologically related studies that used independent transcriptome datasets (Table S1A). Regarding ancestry, we downloaded and parsed Table S1D from Quach et al.15 to obtain a list of DEGs between Africans and Europeans in resting (non-stimulated) human primary monocytes. Similarly, we downloaded and parsed Table S4A from Rotival et al.22 to obtain a list of DSGs between Africans and Europeans in resting (non-stimulated) human primary monocytes. In relation to sex, we downloaded and parsed additional file 1 from Jansen et al.143 to obtain a list of sex-biased genes in human peripheral blood. In relation to age, we downloaded and parsed Table S4 from Pellegrino-Coppola et al.36 to obtain a list of age associated genes from their extended model in human whole blood. Lastly, regarding BMI, we downloaded and parsed Tables S1 and S2 from van der Kolk et al.24 to obtain a list of DEGs between heavier and leaner co-twins in human adipose tissue and skeletal muscle, respectively. We performed a one-sided Fisher’s exact test to test if the number of DEGs or DSGs we identified per tissue significantly overlapped with the genes identified in previous studies. We corrected for multiple testing across tissues using the Benjamini-Hochberg method and determined significance at FDR < 0.05. For DEGs with demographic traits, if information about the fold change was available in the corresponding study, we considered a gene overlapped if it was DE in both studies and in the same direction. For sex-DEGs, we performed the analysis separately for genes located on autosomal or sex chromosomes. For all demographic traits, we found that our DEGs or DSGs significantly overlapped with the genes reported in the independent studies, and the tissues with the most significant overlap were the closest to the tissues used in the independent studies (Table S1A).
Hierarchical partition analysis
In observational data, regressors are usually correlated and it is not straightforward to decompose the explained variability in a model into its components from the individual regressors. To calculate the independent relative contribution of the different traits to the response variable, either gene expression or alternative splicing, we used a hierarchical partitioning approach. Specifically, hierarchical partitioning decomposes the model R2 through incremental partitioning where all possible orders of variables are used, and then obtains the average independent percentual contribution of each trait. We applied hierarchical partitioning on the residual values after regressing out the contribution of the covariates considered to be batch effects and previously included in the differential expression and splicing models (i.e., a linear model for differential expression, a fractional regression for differential splicing analysis). To do hierarchical partitioning on gene expression residuals, we used the hier.part R package.129 To do hierarchical partitioning on splicing residuals, we modified slightly the hier.part R package. Specifically, we used fractional regression using the standard glm function of R with the quasibinomial family and logit link functions and modified the code underlying the hier.part method so it would decompose the global R2, rather than the deviance, to have the same measure used in linear models for differential expression. In summary, we established for each gene and splicing event a linear or generalized linear regression model, respectively, that included the demographic traits and estimated their respective contribution to the residual variance once the batch effects are regressed out.
Identification of cis-driven ancestry-DEGs and ancestry-DSEs
We wanted to know if the expression and splicing differences between populations were due to cis genetic effects. To investigate so, we focused on ancestry-DEGs or genes with ancestry-DSEs previously reported as eGenes or sGenes (genes with significant gene-variant (eQTL) and splice-variants (sQTL) associations). To determine if the population differences are solely driven by genetic regulatory variants (cis-eQTLs and cis-sQTLs respectively), this is, if ancestry differences are due to cis-effects, we modeled the residual gene expression and the residual PSI values of the alternative splicing events (after regressing out batch effects) controlling for their associated genetic regulatory variants. We considered only eGenes and sGenes associated with at least one independent cis-e/sQTL.2
To elucidate if population differences are cis-driven or cis-independent we applied an anova F test to contrast a reduced (H0) versus a full model (H1).
The H0 is formulated as follows:
The H1 is formulated as follows:
To test the ancestry effect we applied an analysis of variance implemented in stats R package through anova function to contrast the hypothesis:
For a given gene/splicing event, the acceptance of H0 (FDR≥0.05) allows us to conclude that the ancestry effect (expression or splicing differences between populations) is fully driven by cis genetic effects. Conversely, the rejection of the null hypothesis (H0) (FDR<0.05) allows us to conclude that ancestry effect is not solely driven by cis genetic effects.
At the gene level, we fitted gene-specific linear models adding as covariates the genotype of the associated independent cis-eQTLs.2 At the splicing level, we fitted an event-specific quasibinomial logit generalized linear model adding the genotype of independent cis-sQTLs.2 Specifically, for each splicing event, we included all cis-sQTLs associated with the gene, since cis-sQTL mapping was performed testing at the gene level.2 This gene-level aggregation of cis-sQTL could result in the inclusion of a linearly dependent set of cis-sQTL. To reduce this dependency we computed the pairwise correlation matrix among the cis-sQTLs using the findCorrelation function in the caret R package144 and excluded highly correlated cis-sQTLs (correlation > 0.9). We also excluded cis-e/sQTLs with no variance and samples with missing genotypes. Finally, we only kept cis-e/sQTLs with at least 3 genotyped donors of each ancestry.
Fst values and tissue sharing of cis-driven ancestry-DEGs and ancestry-DSEs
We used the vcftools130 command “--weir-fst-pop” to compute the fixation indexes (Fst values)145 between individuals of European and African ancestry for all genotyped variants with a minimum allele frequency (MAF) of 0.01 in the latest GTEx release.2 Then, for every eGene and sGene in every tissue, we calculated the average Fst value of the associated cis-e/sQTLs. We used a one-tailed Mann Whitney U test to compare the Fst values associated with cis-driven and cis-independent ancestry-DEGs/DSEs. We corrected the associated P-values for multiple testing across tissues using the Benjamini-Hochberg method. We also used used a one-tailed Mann Whitney U test to compare the tissue sharing of cis-driven and cis-independent ancestry-DEGs/DSEs. We corrected the associated P-values for multiple testing across tissues using the Benjamini-Hochberg method (Figures S2 and S5).
Tissue enrichment of differentially expressed genes with additive effects
We used a two-tailed Fisher’s exact to test if genes are differentially expressed with two demographic traits more often than expected if they were independent. We ran the analysis in each tissue and for each pairwise combination of demographic traits and corrected p-values for multiple testing using the Benjamini-Hochberg method across tissues and pairwise combinations. We determined significance at an FDR < 0.05 (Table S3B). Then, we explored if there was a bias in the directionality of the additive effects. To do so, we focused on genes with additive effects in each tissue and with each pairwise combination of demographic traits. We used a Chi-square goodness of fit to test if the observed sample distribution (number of DEGs upregulated with both traits, number of DEGs downregulated with both traits, and number of DEGs either upregulated with one trait and downregulated with the other, and viceversa) corresponded to the expected probability distribution (based on the total number of genes upregulated and downregulated with each trait separately). We ran the analysis in tissues with at least 20 DEGs with a given pairwise combination of demographic traits. We corrected p-values for multiple testing using the Benjamini-Hochberg method across tissues and pairwise combinations. We determined significance at an FDR < 0.05 (Table S3B).
Enrichment of ancestry-DEGs and ancestry-DSGs in eGenes and sGenes
To investigate whether DEGs and DSGs between African Americans and Europeans Americans are overrepresented in eGenes and sGenes, we downloaded the latest analyses2 from the GTEx portal (https://gtexportal.org/home/datasets/GTEx_Analysis_v8_eQTL.tar and https://gtexportal.org/home/datasets/GTEx_Analysis_v8_sQTL.tar) and, as indicated in the portal, to obtain the list of eGenes and sGenes per tissue, we selected the rows with q-value < 0.05. Then, for each tissue, we computed a one-tailed Fisher’s Exact test to test if there were more ancestry-DEGs/DSGs that were also e/sGenes, respectively, than expected if they were independent. We determined a significant enrichment after correcting for multiple testing using the Benjamini-Hochberg method across tissues at an FDR < 0.05 (Tables S2A and S5A).
Inclusion exclusion of exonic/intronic sequence bias
For those ASEs associated with a switch between a protein-coding and a non-coding isoform, we tested if the inclusion of exonic/intronic sequence is more often associated with a non-coding biotype. To test this, for each splicing event, we considered the biotype (i.e coding or non-coding) of the most abundant spliced-in isoform, and the biotype of the most abundant spliced-out isoform (Figure 4A). We focused on those ASEs associated with a switch between a protein-coding and a non-coding isoform (Table S4A). Then, we investigated if the inclusion of the exonic/intronic sequence in the spliced-in isoform is more often associated with a non-coding biotype than with a coding biotype. To do so, in each tissue and for each type of splicing event, we counted the number of ASEs for which the spliced-in isoform was non-coding and the spliced-out was coding (NC-PC), and the number of ASEs for which the spliced-out isoform was non-coding and the spliced-in coding (PC-NC). We used a binomial test to test if the observed proportions significantly deviate from an equiprobable distribution (p=0.5). Since the same isoforms can contribute to different ASEs, to prevent redundancies in the statistical testing, we randomly selected one ASEs per spliced-in-spliced-out isoform combination. We corrected for multiple testing using the Benjamini-Hochberg method across tissues and types of splicing events and determined significance at an FDR < 0.05 (Table S4B).
Enrichment of DSEs in particular types of ASEs
We used a Chi-square test to investigate if there is a statistically significant difference between the expected frequencies and the observed frequencies of ASEs DS and not DS for each type of splicing event (Figure 4A). We ran the analyses separately for ASEs with each demographic trait and tissue. Since the same isoforms can contribute to different ASEs, to prevent redundancies in the statistical testing, we randomly selected one ASEs per spliced-in-spliced-out isoform combination. We determined whether the number of observed DSEs is significantly different than expected after correcting for multiple testing across tissues using the Benjamini-Hochberg method at an FDR < 0.05 (Table S4C).
Inclusion or exclusion bias in particular types of DSEs
We investigated if there was a bias towards positive or negative betas for the different types of DSEs with each demographic trait. The sign of the beta parameter indicates whether the exonic/intronic sequence included in the spliced-in isoform is more or less included with respect to the reference level: positive betas indicate more inclusion in African Americans, females, older individuals, or in individuals with higher BMI. Specifically, we used the binomial test to test if the observed proportions of positive and negative betas for the DSEs significantly deviate from an equiprobable distribution (p=0.5). We run one test for each type of splicing event (Table S4D). To prevent redundancies in the statistical testing, if two or more events of the same type were DS in a gene, we selected the beta of the DSE with the lowest adjusted p-value. We only ran the analysis in tissues with ≥ 10 DSEs of a given type with a demographic trait. We corrected for multiple testing using the Benjamini-Hochberg method across tissues and types of splicing events at an FDR < 0.05.
Overlap between DEGs and DSGs
We used a two-tailed Fisher’s exact test to investigate if genes DE are more likely to be DS, by testing if they overlap more often than expected if they were independent. We restricted the analysis to genes with ASEs, and hence tested for differential splicing, in each tissue. We ran the analyses separately for DEGs and DSGs with each demographic trait (Table S4F). We corrected for multiple testing using the Benjamini-Hochberg method across tissues and determined a larger than expected number of DEGs and DGSs at an FDR < 0.05.
Comparison of the AS variation explained by the different types of AS events
We computed the AS variation explained per DSE as described in section hierarchical partition analysis. We used a Kruskal–Wallis test (kruskal.test R function from the R package stats146) to compare the AS variation explained in the different types of splicing events. If the associated p-value was < 0.05, we further used the function pairwise.wilcox.test from the R package stats, which performs multiple testing correction, to compute all pairwise Mann–Whitney U-tests between the different types of splicing events. To quantify the differences between pairwise combinations of splicing events, we computed the effect sizes (Glass rank biserial correlation coefficient for Mann–Whitney U-test) for each pairwise combination of event types using the wilcoxonRG function from the R package rcompanion.147 We ran the analysis separately per tissue and demographic trait, considering only tissues with ≥ 3 trait-DSEs of each type of splicing event (Table S4H).
Assessing replication of differential expression and splicing patterns of ribosomal proteins
We downloaded and parsed Table S1D from Quach et al.15 to obtain a list of DEGs between Africans and Europeans in resting (non-stimulated) human primary monocytes. Similarly, we downloaded and parsed Table S4A from Rotival et al.22 to obtain a list of DSEs between Africans and Europeans in resting (non-stimulated) human primary monocytes. From those lists, we retrieved the ribosomal proteins DE and the ASEs in ribosomal proteins DS. To further validate our findings, we sought to investigate if the expression differences between populations of the ribosomal protein isoforms that participate in DSEs were correlated between GTEx tissues and monocytes. Monocyte isoforms expression matrices were provided by the authors and log2 FPKM values were transformed to TPM values to match GTEx quantifications. Then, we computed the differences in expression between populations (effect sizes) for the isoforms contributing to DSEs in ribosomal proteins for each GTEx tissue and in monocytes. Then, we tested whether the effect sizes of a particular GTEx tissue were correlated with that of monocytes. We corrected for multiple testing using the Benjamini-Hochberg method across tissues (FDR < 0.05).
Identification of cis-genetic variants associated with DSEs in ribosomal proteins
To identify candidate genetic regulatory variants of the splicing patterns of DSEs in ribosomal proteins, we focused on genotyped variants with MAF ≥ 0.01 within 1Kb of the splicing event. Then, in each tissue and for each DSE in ribosomal proteins, we fitted a generalized linear model using fractional regression as explained before, controlling for technical covariates and demographic traits as well as nearby genetic variants:
We used an anova test implemented in stats R package through anova function to determine if at least one genetic variant per DSE was significantly associated with the splicing patterns (FDR < 0.05). Then, we used the approach described in section identification of cis-driven ancestry-DEGs and ancestry-DSEs to determine if the splicing differences observed between populations are cis-driven.
Read coverage for highly tissue-shared DSEs in RPLP2 and RPL10
We downloaded the available RNAseq bam files for the samples from the SkinSunExposedLowerleg and SkinNotSunExposedSuprapubic tissues, which are part of the GTEx protected data stored in dbGap (accession number phs000424.v8.p2). We used deeptools131 to generate normalized coverage tracks (counts per million (CPM)) in 50 base-pairs windows considering uniquely mapped reads. We used the R package Gviz132 to plot the average read coverage per population (Figure S6D).
Identification of ASEs that disrupt known protein-coding domains
To further characterize the functional consequences of ASEs associated with a switch between protein-coding isoforms, we used an in-house developed pipeline that maps splicing events to protein-coding domains. Running the pipeline requires: (1) a gene annotation file (GTF file), (2) a list of transcripts IDs (isoforms), (3) a genome fasta file, and (4) a database of known protein-coding domains from the PFAM database.57 The computational method is implemented in Nextflow133 and publicly available at https://github.com/Mele-Lab/2022_GTExTranscriptome_fromSplicingEventsToProteinDomains. In brief, the pipeline extracts each isoform’s coding DNA sequence (CDS) from the GTF file and translates it into its corresponding amino acid sequence. Then, using the latter, it queries the PFAM database and selects high-confidence amino acid alignments to identify protein-coding domains in each isoform. We considered high-confidence protein-coding domain alignments those with: sequence alignment E-value < 1e-5, domain E-value < 0.01, domain score > 10, accuracy (hmmscan metrics) ≥ 0.8 and partiality ≥ 0.9 (where partiality is an in-house metric that represents the proportion of the sequence domain aligned).
To link ASEs to changes in protein-coding domains, the amino acid coordinates of the protein-coding domains are translated to genomic coordinates. Then, we evaluate if the exonic/intronic sequences that define an splicing event overlap with the genomic coordinates of the protein-coding domains. We determine that a splicing event affects a protein-coding domain if the exonic/intronic coordinates that define the splicing event overlap with at least one protein-coding domain in either one or both the spliced-in and spliced-out isoforms.
Selection of clinical traits
The clinical trait annotation was obtained either from the donors’ medical history, which are part of the GTEx protected data stored in dbGap (accession number phs000424.v8.p2) and obtained either from the donors’ medical record or information provided by the donors’ next-of-kin,148 or from histopathological annotations obtained through pathologist review, publicly accessible from the GTEx portal (https://gtexportal.org/home/histologyPage). Whereas the donors’ medical history annotations correspond to each individual (and thus all the tissues obtained from the same donor have the same annotation), the histopathological annotations are specific to tissue samples. Notably, annotations for some clinical traits were only available for a subset of donors and samples (Tables S6A and S6B). We only considered the clinical traits with at least five affected samples per tissue. We manually curated the histopathological annotations and, in some instances, combined phenotypically related annotations. Specifically, we labeled artery samples that suffered from atherosis, atherosclerosis, calcification, Mönckeberg's arteriosclerosis, and/or sclerosis with the wider term atherosclerosis, as more than one third (40.24%) of the tibial artery samples were annotated with at least two of these phenotypes. We also modified the spermatogenesis annotation to distinguish samples with normal and low levels of spermatogenesis based on the pathologists’ comments. We excluded clinical traits directly related to the donor’s cause of death, as these were strongly correlated with the Hardy scale index, and we also excluded donors annotated as type 1 diabetics that expressed insulin.149 Altogether, we analyzed expression and splicing changes associated with 48 different clinical traits (Table S6B).
Differential expression, differential splicing, and hierarchical partition analysis with clinical traits
To identify DEGs with clinical traits we used the same approach described in the section Differential gene expression analysis, but further including the clinical traits as covariates in the linear models. First, we ran one model per tissue and clinical trait (Table S6A) and selected those clinical traits with at least five DEGs in the affected tissues (n = 17 clinical traits) (Table S6B). For the histopathological annotations, the affected tissue was the tissue where we obtained the pathology annotation. For medical history annotations, we defined the affected tissue as the known tissue of origin according to the literature, e.g., the pancreas in type 1 diabetes.149 In cases with more than one known affected tissue, i.e. brain regions, we selected the tissue with the highest sample size, e.g., the brain cortex in multiple sclerosis (Table S6B). Then, we ran one model per tissue including all clinical traits selected in the previous step that had at least five DEGs in that tissue (Figures 6A and S7A). Note that the number of tissue samples is limited to the number of samples with available annotation for all clinical traits, and thus the number of samples analyzed might be smaller than previous analysis that considered only demographic traits. To test interactions we require at least 20 samples per group, but only type 2 diabetes in three tissues fulfilled this requirement. We only found 2 genes with a significant interaction between type 2 diabetes and ancestry (SMN1) and between type 2 diabetes and sex (STK26) in the adipose subcutaneous. To quantify the contribution of demographic and clinical traits to gene expression variation, we used hierarchical partition analysis as described in section hierarchical partition analysis expanding the model to include the clinical traits annotated in that tissue. To identify DSEs with clinical traits, we used the same approach as described in section Differential splicing analysis expanding the models to include clinical traits per tissue with at least 15 affected samples.
Tissue enrichment of genes with additive effects between clinical traits
We used two-tailed Fisher’s exact tests to investigate if two clinical traits affect the same genes per tissue more often than expected if they were independent. We ran the analysis per tissue and for each pairwise combination of clinical traits. We corrected for multiple testing using the Benjamini-Hochberg method in all tests and determined significance at an FDR < 0.05. Then, we used a Chi-square goodness of fit to investigate the directionality of the additive effects by comparing the observed and expected sample distributions as explained in tissue enrichment of differentially expressed genes with additive effects. We ran the analysis in tissues with at least 20 DEGs with additive effects. We corrected p-values for multiple testing using the Benjamini-Hochberg method across traits. We determined significance at an FDR < 0.05 (Table S6G).
Replication of tibial nerve DEGs with diabetes in mice
We downloaded and parsed Tables S1, S2, and S3 from Gu et al.87 to obtain a list of DEGs with type 1 and type 2 diabetes in the sciatic nerve of mice. We then compared this gene list with DEGs with type 1 and type 2 diabetes in the tibial nerve. We used a two-tailed Fisher’s exact test to test if the number of DEGs significantly overlapped between the two studies. We used as background the genes expressed in the human tibial nerve.
Prediction of diabetic status using tibial nerve histology images
The histology images are publicly available in the GTEx Histological Image Viewer (https://brd.nci.nih.gov/brd/image-search/searchhome). We used PyHist134 to transform tibial nerve whole slide images with diabetic annotation (n=971) from Aperio format (.svs) to png. We then segmented the images into tiles of 512x512 pixels (n=1399) with Otsu thresholding150 to keep the tiles with at least 75% of tissue content. We excluded 4 images that, by visual inspection, corresponded to mis-annotated tissues. We used the function computeFeatures.haralick() from the Bioconductor package EBImage135 to extract 13 Haralick features151 from RGB pixel values on three scales: 1, 10, and 100-pixel sliding windows, as described in.114 Using these features we trained a support vector machine136 with a linear kernel. We split the data into training (75% of the data) and testing sets (25% of the data) and ran 100 permutations. To identify genes associated with diabetic neuropathy progression, we used the probability of being classified as diabetic as a proxy for disease severity. Using only diabetic individuals, and for every data split, we computed for each gene the Pearson correlation between the probabilities of being diabetic obtained from the classifier and the residual gene expression values (after regressing out the effects of the covariates and demographic traits), and selected those genes that had a significant correlation (FDR < 0.05) in 90 % of the permutations (Table S6I).
Tissue enrichment of genes with additive effects between demographic and clinical traits
We used a two-tailed Fisher’s exact test to check if genes are differentially expressed with a clinical and a demographic trait more often than expected by chance. We ran the analysis per tissue and for each pair combination of demographic and clinical traits. We corrected p-values for multiple testing using the Benjamini-Hochberg method across tissues and pairs and determined significance at an FDR < 0.05. Then, we used a Chi-square test to investigate biases in the directionality of the additive effects as explained in tissue enrichment of differentially expressed genes with additive effects.
Matching demographic distributions for bias validation
To confirm that the biases in directionality we observe are not due to demographic differences between healthy and diseased populations, we ran differential expression analysis, as previously described, in subsamples of the data where the distribution of a given demographic trait in healthy individuals was matched to the distribution in diseased individuals. We used the function matchit from the R package MatchIt137 using the “optimal” method.152 For every clinical and demographic pair with a significant bias in the directionality of genes with additive effects, we subsampled with the maximum possible ratio of healthy to disease individuals, and ran the analysis explained in tissue enrichment of genes with additive effects between demographic and clinical traits (Figure S7F and Table S6K).
Differences in cell-type abundances with demographic and clinical traits
We downloaded cell-type abundance estimates for seven cell types (adipocytes, epithelial cells, hepatocytes, keratinocytes, myocytes, neurons, and neutrophils) described in more detail in Kim-Hellmuth et al.58 from the GTEx portal (https://gtexportal.org/home/datasets/GTEx_Analysis_v8_xCell_scores_7_celltypes.txt.gz). In each tissue, we only investigated changes in abundances for robustly estimated cell types (median xCell score > 0.1).58 To detect changes in abundances associated with demographic traits, we followed the same approach as described in Oliva et al 17,58. In brief, in each tissue, we fit a linear regression model as follows:
After correcting for multiple testing using the Benjamini-Hochberg method across all cell type-tissue-trait combinations we determined significance for each demographic trait at FDR < 0.05.
To investigate changes in tissue composition associated with clinical traits, we used the same approach expanding the models to include as covariates the corresponding clinical traits per affected tissue. We determined significance for each clinical trait at FDR < 0.05.
Functional enrichment analysis
We used the clusterProfiler R package138 for the different overrepresentation enrichment analyses (ORA) conducted throughout the paper, considering different databases (GO ontology, Reactome and DisGenet). We used the Benjamini-Hochberg method for multiple testing correction and report as significant gene sets with an FDR < 0.05. For each ORA, we carefully selected suitable background gene lists, rather than the default universe of all annotated genes. To investigate biological pathways associated with highly tissue-shared genes we used as input the list of highly tissue-shared ancestry or age-DEGs, and as background the list of all ancestry or age-DEGs. To investigate biological pathways associated with genes with highly tissue-shared ancestry-DSEs, we used as input the list of genes with highly tissue-shared ancestry-DSEs, and as background the list of all genes with at least one ancestry-DSE. To investigate biological pathways associated with genes with significant interactions between sex and age in breast, we used as input the latter genes and as background the list of genes expressed in breast. To explore functional pathways associated with DEGs with type 1 and type 2 diabetes, we used as input either the upregulated or downregulated DEGs and used as background genes expressed in the tibial nerve. To investigate the functional enrichments of genes whose expression correlates with the probability of our classifier to assign an image sample as diabetic we used as input all genes with a significant Pearson correlation (FDR<0.05) in all data permutations and as background, expressed genes in the tibial nerve.
Acknowledgments
This study was funded by the HumTranscriptom project with reference PID2019-107937GA-I00. R.G.-P. was supported by a Juan de la Cierva fellowship (FJC2020-044119-I) funded by MCIN/AEI/10.13039/501100011033 and “European Union NextGenerationEU/PRTR.” J.M.R. was supported by a predoctoral fellowship from “la Caixa” Foundation (ID 100010434) with code LCF/BQ/DR22/11950022. A.R.-C. was supported by a Formación Personal Investigador (FPI) fellowship (PRE2019-090193) funded by MCIN/AEI. R.C.-G. was supported by an FPI fellowship (PRE2020-092510) funded by MCIN/AEI. M.M. was supported by a Ramon y Cajal fellowship (RYC-2017-22249). Figures 4A and S1A and the graphical abstract were created with BioRender.com. We thank the donors and their families for their generous gifts of organ donation for transplantation and tissue donations for the GTEx research project and the GTEx consortium members.
Author contributions
M.M. conceived the study; M.M. and R.G.-P. designed and supervised all analyses; M.C. and F.R. assisted in statistical analysis design and implementation; S.C-.G. supervised M.B.; R.G.-P., J.M.R., R.C.-G., A.R.-C., W.O., O.S., M.B., P.J.R., M.C., and P.G.F. analyzed the data; R.G.-P. led data analysis related to demographic traits (Figures1–5); J.M.R. led data analysis related to clinical traits (Figure 6); M.M. and R.G.-P. wrote the manuscript with input from all co-authors; K.G.A. led the GTEx portal efforts; R.G., F.A., P.G.F., and K.G.A. advised in data analysis, provided helpful insight, and contributed to manuscript editing.
Declaration of interests
The authors declare no competing interests.
Published: December 30, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100244.
Supplemental information
(A) Replication of genes differentially expressed and differentially spliced with demographic traits using independent datasets. (B) Top 50 genes with a large proportion of their gene expression variation explained by a demographic trait. (C) Tissue sharing of genes differentially expressed with each demographic trait. (D) Differential gene expression is independent of gene expression breadth. (E) Genes expressed only in the tissues where they are differentially expressed. (F) Genes differentially expressed in 10 or more tissues. (G) Genes differentially expressed between populations in 10 or more tissues are enriched in the glutathione-related pathways. (H) Genes differentially expressed with age in 10 or more tissues are enriched in the p-53 signaling pathway.
(A) Genes differentially expressed with two demographic traits per tissue. (B) Tissue enrichment and biased expression in genes with additive contributions. (C) Genes with interactions between demographic traits. (D) Genes with a variable contribution of aging between males and females are enriched in mammary gland development.
(A) Functional characterization of alternatively spliced events per tissue and type of splicing event. (B) Non-coding isoforms are biased toward the inclusion of exonic/intronic sequence in intron retention, alternative 5′ and alternative 3′ splicing events, in alternatively spliced events associated with a switch between a non-coding and a protein-coding isoform. (C) There are more alternative last and fewer alternative 5′ and 3′ events than expected differentially spliced between populations. (D) Bias toward the inclusion or exclusion of exonic/intronic sequence is specific to the type of splicing events, tissues, and demographic traits. (E) Functional characterization of differentially spliced events. (F) Enrichment of DSGs in DEGs. (G) Genes differentially expressed and spliced between populations are enriched in eGenes and sGenes. (H) Comparison of the alternative splicing variation explained in each type of splicing event by demographic traits. (I) Top 50 splicing events with a large proportion of their alternative splicing variation explained by a demographic trait.
(A) Ancestry-DSGs are enriched in sGenes. (B) Functional enrichment of genes with highly tissue-shared differentially spliced events between populations. (C) Functional enrichment of genes differentially spliced between populations in human primary monocytes. (D) Ribosomal proteins with events differentially spliced between populations reported as sGenes in different studies.
(A) Number of DEGs per clinical trait and tissue (one model per clinical trait and tissue). (B) Number of samples per clinical trait and reason to include/exclude clinical traits with at least five positive samples. (C) Genes differentially expressed and spliced with clinical traits per tissue (one model per tissue). (D) Top genes (up to 50) with a large proportion of their gene expression variation explained by a clinical trait. (E) Top splicing events (up to 50) with a large proportion of their alternative splicing variation explained by a clinical trait. (F) Genes differentially expressed in more than one tissue per clinical trait (types 1 and 2 diabetes). (G) Genes differentially expressed with two clinical traits and tissue enrichment in genes with additive contributions. (H) Genes differentially expressed with both diabetes in the tibial nerve are enriched in immune receptor activity and ion channel activity (GO:MF). (I) Genes whose residual expression in the tibial nerve significantly correlates with the SVM-assigned probability of being classified as a diabetic individual. (J) Genes whose residual expression in the tibial nerve significantly correlates with the SVM-assigned probability of being classified as a diabetic individual are enriched in neuropathy-related diseases (GO:DisGeNET). (K) Genes differentially expressed with a clinical and a demographic trait per tissue and tissue enrichment analysis in genes with additive contributions, including results after matching trait distributions between healthy and diseased individuals.
Data and code availability
All GTEx protected data are available at the accession number dbGaP: phs000424.v8. Access to the raw sequence data is now provided through AnVIL: https://gtexportal.org/home/protectedDataAccess. Public-access data, including QTL summary statistics and expression levels, are available on the GTEx Portal: https://www.gtexportal.org, as well as in the UCSC and Ensembl browsers.
Analysis scripts are available at github: https://github.com/Mele-Lab/2022_GTExTranscriptome and all results tables derived from the analyses conducted in this paper are deposited at zenodo: https://doi.org/10.5281/zenodo.6797627.
References
- 1.Melé M., Ferreira P.G., Reverter F., DeLuca D.S., Monlong J., Sammeth M., Young T.R., Goldmann J.M., Pervouchine D.D., Sullivan T.J., et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardoso-Moreira M., Halbert J., Valloton D., Velten B., Chen C., Shao Y., Liechti A., Ascenção K., Rummel C., Ovchinnikova S., et al. Gene expression across mammalian organ development. Nature. 2019;571:505–509. doi: 10.1038/s41586-019-1338-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He P., Williams B.A., Trout D., Marinov G.K., Amrhein H., Berghella L., Goh S.-T., Plajzer-Frick I., Afzal V., Pennacchio L.A., et al. The changing mouse embryo transcriptome at whole tissue and single-cell resolution. Nature. 2020;583:760–767. doi: 10.1038/s41586-020-2536-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazin P.V., Khaitovich P., Cardoso-Moreira M., Kaessmann H. Alternative splicing during mammalian organ development. Nat. Genet. 2021;53:925–934. doi: 10.1038/s41588-021-00851-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee T.I., Young R.A. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K.C., Hua Y., Gueroussov S., Najafabadi H.S., Hughes T.R., et al. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806. doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scotti M.M., Swanson M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016;17:19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brawand D., Soumillon M., Necsulea A., Julien P., Csárdi G., Harrigan P., Weier M., Liechti A., Aximu-Petri A., Kircher M., et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- 10.Barbosa-Morais N.L., Irimia M., Pan Q., Xiong H.Y., Gueroussov S., Lee L.J., Slobodeniuc V., Kutter C., Watt S., Colak R., et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–1593. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- 11.Merkin J., Russell C., Chen P., Burge C.B. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. 2012;338:1593–1599. doi: 10.1126/science.1228186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braunschweig U., Barbosa-Morais N.L., Pan Q., Nachman E.N., Alipanahi B., Gonatopoulos-Pournatzis T., Frey B., Irimia M., Blencowe B.J. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014;24:1774–1786. doi: 10.1101/gr.177790.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarropoulos I., Marin R., Cardoso-Moreira M., Kaessmann H. Developmental dynamics of lncRNAs across mammalian organs and species. Nature. 2019;571:510–514. doi: 10.1038/s41586-019-1341-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z.-Y., Leushkin E., Liechti A., Ovchinnikova S., Mößinger K., Brüning T., Rummel C., Grützner F., Cardoso-Moreira M., Janich P., et al. Transcriptome and translatome co-evolution in mammals. Nature. 2020;588:642–647. doi: 10.1038/s41586-020-2899-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quach H., Rotival M., Pothlichet J., Loh Y.-H.E., Dannemann M., Zidane N., Laval G., Patin E., Harmant C., Lopez M., et al. Genetic adaptation and neandertal admixture shaped the immune system of human populations. Cell. 2016;167:643–656.e17. doi: 10.1016/j.cell.2016.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Randolph H.E., Fiege J.K., Thielen B.K., Mickelson C.K., Shiratori M., Barroso-Batista J., Langlois R.A., Barreiro L.B. Genetic ancestry effects on the response to viral infection are pervasive but cell type specific. Science. 2021;374:1127–1133. doi: 10.1126/science.abg0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliva M., Muñoz-Aguirre M., Kim-Hellmuth S., Wucher V., Gewirtz A.D.H., Cotter D.J., Parsana P., Kasela S., Balliu B., Viñuela A., et al. The impact of sex on gene expression across human tissues. Science. 2020;369:eaba3066. doi: 10.1126/science.aba3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glass D., Viñuela A., Davies M.N., Ramasamy A., Parts L., Knowles D., Brown A.A., Hedman A.K., Small K.S., Buil A., et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013;14:R75. doi: 10.1186/gb-2013-14-7-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viñuela A., Brown A.A., Buil A., Tsai P.-C., Davies M.N., Bell J.T., Dermitzakis E.T., Spector T.D., Small K.S. Age-dependent changes in mean and variance of gene expression across tissues in a twin cohort. Hum. Mol. Genet. 2018;27:732–741. doi: 10.1093/hmg/ddx424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balliu B., Durrant M., Goede O.d., Abell N., Li X., Liu B., Gloudemans M.J., Cook N.L., Smith K.S., Knowles D.A., et al. Genetic regulation of gene expression and splicing during a 10-year period of human aging. Genome Biol. 2019;20:230. doi: 10.1186/s13059-019-1840-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trabzuni D., Ramasamy A., Imran S., Walker R., Smith C., Weale M.E., Hardy J., Ryten M., North American Brain Expression Consortium Widespread sex differences in gene expression and splicing in the adult human brain. Nat. Commun. 2013;4:2771. doi: 10.1038/ncomms3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rotival M., Quach H., Quintana-Murci L. Defining the genetic and evolutionary architecture of alternative splicing in response to infection. Nat. Commun. 2019;10:1671. doi: 10.1038/s41467-019-09689-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muniandy M., Heinonen S., Yki-Järvinen H., Hakkarainen A., Lundbom J., Lundbom N., Kaprio J., Rissanen A., Ollikainen M., Pietiläinen K.H. Gene expression profile of subcutaneous adipose tissue in BMI-discordant monozygotic twin pairs unravels molecular and clinical changes associated with sub-types of obesity. Int. J. Obes. 2017;41:1176–1184. doi: 10.1038/ijo.2017.95. [DOI] [PubMed] [Google Scholar]
- 24.van der Kolk B.W., Saari S., Lovric A., Arif M., Alvarez M., Ko A., Miao Z., Sahebekhtiari N., Muniandy M., Heinonen S., et al. Molecular pathways behind acquired obesity: adipose tissue and skeletal muscle multiomics in monozygotic twin pairs discordant for BMI. Cell Rep. Med. 2021;2:100226. doi: 10.1016/j.xcrm.2021.100226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piasecka B., Duffy D., Urrutia A., Quach H., Patin E., Posseme C., Bergstedt J., Charbit B., Rouilly V., MacPherson C.R., et al. Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc. Natl. Acad. Sci. USA. 2018;115:E488–E497. doi: 10.1073/pnas.1714765115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Márquez E.J., Chung C.-H., Marches R., Rossi R.J., Nehar-Belaid D., Eroglu A., Mellert D.J., Kuchel G.A., Banchereau J., Ucar D. Sexual-dimorphism in Human Immune System Aging. Nat Commun. 2022;11:751. doi: 10.1101/755702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skol A.D., Jung S.C., Sokovic A.M., Chen S., Fazal S., Sosina O., Borkar P.P., Lin A., Sverdlov M., Cao D., et al. Integration of genomics and transcriptomics predicts diabetic retinopathy susceptibility genes. Elife. 2020;9:e59980. doi: 10.7554/eLife.59980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yip L., Fuhlbrigge R., Alkhataybeh R., Fathman C.G. Gene expression analysis of the pre-diabetic pancreas to identify pathogenic mechanisms and biomarkers of type 1 diabetes. Front. Endocrinol. 2020;11:609271. doi: 10.3389/fendo.2020.609271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alonso L., Piron A., Morán I., Guindo-Martínez M., Bonàs-Guarch S., Atla G., Miguel-Escalada I., Royo R., Puiggròs M., Garcia-Hurtado X., et al. TIGER: the gene expression regulatory variation landscape of human pancreatic islets. Cell Rep. 2021;37:109807. doi: 10.1016/j.celrep.2021.109807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carruthers N.J., Strieder-Barboza C., Caruso J.A., Flesher C.G., Baker N.A., Kerk S.A., Ky A., Ehlers A.P., Varban O.A., Lyssiotis C.A., et al. The human type 2 diabetes-specific visceral adipose tissue proteome and transcriptome in obesity. Sci. Rep. 2021;11:17394. doi: 10.1038/s41598-021-96995-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samaras K., Botelho N.K., Chisholm D.J., Lord R.V. Subcutaneous and visceral adipose tissue gene expression of serum adipokines that predict type 2 diabetes. Obesity. 2010;18:884–889. doi: 10.1038/oby.2009.443. [DOI] [PubMed] [Google Scholar]
- 32.Keen J.C., Moore H.M. The genotype-tissue expression (GTEx) project: linking clinical data with molecular analysis to advance personalized medicine. J. Pers. Med. 2015;5:22–29. doi: 10.3390/jpm5010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breschi A., Muñoz-Aguirre M., Wucher V., Davis C.A., Garrido-Martín D., Djebali S., Gillis J., Pervouchine D.D., Vlasova A., Dobin A., et al. A limited set of transcriptional programs define major cell types. Genome Res. 2020;30:1047–1059. doi: 10.1101/gr.263186.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakatta E.G. The reality of getting old. Nat. Rev. Cardiol. 2018;15:499–500. doi: 10.1038/s41569-018-0068-y. [DOI] [PubMed] [Google Scholar]
- 35.Nelson S.M., Telfer E.E., Anderson R.A. The ageing ovary and uterus: new biological insights. Hum. Reprod. Update. 2013;19:67–83. doi: 10.1093/humupd/dms043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pellegrino-Coppola D., Claringbould A., Stutvoet M., BIOS Consortium. Boomsma D.I., Ikram M.A., Slagboom P.E., Westra H.-J., Franke L. Correction for both common and rare cell types in blood is important to identify genes that correlate with age. BMC Genom. 2021;22:184. doi: 10.1186/s12864-020-07344-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tournamille C., Colin Y., Cartron J.P., Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy–negative individuals. Nat. Genet. 1995;10:224–228. doi: 10.1038/ng0695-224. [DOI] [PubMed] [Google Scholar]
- 38.Zhao Y., Marotta M., Eichler E.E., Eng C., Tanaka H. Linkage disequilibrium between two high-frequency deletion polymorphisms: implications for association studies involving the glutathione-S transferase (GST) genes. PLoS Genet. 2009;5:e1000472. doi: 10.1371/journal.pgen.1000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Josephy P.D. Genetic variations in human glutathione transferase enzymes: significance for pharmacology and toxicology. Hum. Genom. Proteomics. 2010;2010:876940. doi: 10.4061/2010/876940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Traverso N., Ricciarelli R., Nitti M., Marengo B., Furfaro A.L., Pronzato M.A., Marinari U.M., Domenicotti C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013;2013:972913. doi: 10.1155/2013/972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tukiainen T., Villani A.-C., Yen A., Rivas M.A., Marshall J.L., Satija R., Aguirre M., Gauthier L., Fleharty M., Kirby A., et al. Landscape of X chromosome inactivation across human tissues. Nature. 2017;550:244–248. doi: 10.1038/nature24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rufini A., Tucci P., Celardo I., Melino G. Senescence and aging: the critical roles of p53. Oncogene. 2013;32:5129–5143. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 43.Obradovic M., Sudar-Milovanovic E., Soskic S., Essack M., Arya S., Stewart A.J., Gojobori T., Isenovic E.R. Leptin and obesity: role and clinical implication. Front. Endocrinol. 2021;12:585887. doi: 10.3389/fendo.2021.585887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ji L., Zhao Y., He L., Zhao J., Gao T., Liu F., Qi B., Kang F., Wang G., Zhao Y., et al. AKAP1 deficiency attenuates diet-induced obesity and insulin resistance by promoting fatty acid oxidation and thermogenesis in Brown adipocytes. Adv. Sci. 2021;8:2002794. doi: 10.1002/advs.202002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Neill M.B., Quach H., Pothlichet J., Aquino Y., Bisiaux A., Zidane N., Deschamps M., Libri V., Hasan M., Zhang S.-Y., et al. Single-cell and bulk RNA-sequencing reveal differences in monocyte susceptibility to influenza A virus infection between Africans and Europeans. Front. Immunol. 2021;12:768189. doi: 10.3389/fimmu.2021.768189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nédélec Y., Sanz J., Baharian G., Szpiech Z.A., Pacis A., Dumaine A., Grenier J.-C., Freiman A., Sams A.J., Hebert S., et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell. 2016;167:657–669.e21. doi: 10.1016/j.cell.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 47.Martin A.R., Costa H.A., Lappalainen T., Henn B.M., Kidd J.M., Yee M.-C., Grubert F., Cann H.M., Snyder M., Montgomery S.B., Bustamante C.D. Transcriptome sequencing from diverse human populations reveals differentiated regulatory architecture. PLoS Genet. 2014;10:e1004549. doi: 10.1371/journal.pgen.1004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A.C., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holsinger K.E., Weir B.S. Genetics in geographically structured populations: defining, estimating and interpreting FST. Nat. Rev. Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hannou S.A., Wouters K., Paumelle R., Staels B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: what have we learned from GWASs? Trends Endocrinol. Metab. 2015;26:176–184. doi: 10.1016/j.tem.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 51.Holdt L.M., Sass K., Gäbel G., Bergert H., Thiery J., Teupser D. Expression of Chr9p21 genes CDKN2B (p15INK4b), CDKN2A (p16INK4a, p14ARF) and MTAP in human atherosclerotic plaque. Atherosclerosis. 2011;214:264–270. doi: 10.1016/j.atherosclerosis.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 52.Hansson G.K., Libby P. The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 53.Baralle F.E., Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017;18:437–451. doi: 10.1038/nrm.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verta J.-P., Jacobs A. The role of alternative splicing in adaptation and evolution. Trends Ecol. Evol. 2022;37:299–308. doi: 10.1016/j.tree.2021.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Schafer S., Miao K., Benson C.C., Heinig M., Cook S.A., Hubner N. Alternative splicing signatures in RNA-seq data: percent spliced in (PSI) Curr. Protoc. Hum. Genet. 2015;87:11.16.1–11.16.14. doi: 10.1002/0471142905.hg1116s87. [DOI] [PubMed] [Google Scholar]
- 56.Reyes A., Huber W. Alternative start and termination sites of transcription drive most transcript isoform differences across human tissues. Nucleic Acids Res. 2018;46:582–592. doi: 10.1093/nar/gkx1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj S., Richardson L.J., et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021;49:D412–D419. doi: 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim-Hellmuth S., Aguet F., Oliva M., Muñoz-Aguirre M., Kasela S., Wucher V., Castel S.E., Hamel A.R., Viñuela A., Roberts A.L., et al. Cell type-specific genetic regulation of gene expression across human tissues. Science. 2020;369:eaaz8528. doi: 10.1126/science.aaz8528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raj T., Li Y.I., Wong G., Humphrey J., Wang M., Ramdhani S., Wang Y.-C., Ng B., Gupta I., Haroutunian V., et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018;50:1584–1592. doi: 10.1038/s41588-018-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuehl P., Zhang J., Lin Y., Lamba J., Assem M., Schuetz J., Watkins P.B., Daly A., Wrighton S.A., Hall S.D., et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001;27:383–391. doi: 10.1038/86882. [DOI] [PubMed] [Google Scholar]
- 61.Garrido-Martín D., Borsari B., Calvo M., Reverter F., Guigó R. Identification and analysis of splicing quantitative trait loci across multiple tissues in the human genome. Nat. Commun. 2021;12:727. doi: 10.1038/s41467-020-20578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao K., Lu Z.-X., Park J.W., Zhou Q., Xing Y. GLiMMPS: robust statistical model for regulatory variation of alternative splicing using RNA-seq data. Genome Biol. 2013;14:R74. doi: 10.1186/gb-2013-14-7-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bargis-Surgey P., Lavergne J.-P., Gonzalo P., Vard C., Filhol-Cochet O., Reboud J.-P. Interaction of elongation factor eEF-2 with ribosomal P proteins. Eur. J. Biochem. 1999;262:606–611. doi: 10.1046/j.1432-1327.1999.00434.x. [DOI] [PubMed] [Google Scholar]
- 64.Remacha M., Jimenez-Diaz A., Santos C., Briones E., Zambrano R., Rodriguez Gabriel M.A., Guarinos E., Ballesta J.P. Proteins P1, P2, and P0, components of the eukaryotic ribosome stalk. New structural and functional aspects. Biochem. Cell. Biol. 1995;73:959–968. doi: 10.1139/o95-103. [DOI] [PubMed] [Google Scholar]
- 65.Atkinson M.A., Eisenbarth G.S., Michels A.W. Type 1 diabetes. Lancet. 2014;383:69–82. doi: 10.1016/s0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu L., Liu Y., Liu C., Zhang Z., Du Y., Zhao H. Analysis of gene expression profile identifies potential biomarkers for atherosclerosis. Mol. Med. Rep. 2016;14:3052–3058. doi: 10.3892/mmr.2016.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gartlan K.H., Wee J.L., Demaria M.C., Nastovska R., Chang T.M., Jones E.L., Apostolopoulos V., Pietersz G.A., Hickey M.J., van Spriel A.B., Wright M.D. Tetraspanin CD37 contributes to the initiation of cellular immunity by promoting dendritic cell migration. Eur. J. Immunol. 2013;43:1208–1219. doi: 10.1002/eji.201242730. [DOI] [PubMed] [Google Scholar]
- 68.Zhen D., Liu J., Zhang X.D., Song Z. Kynurenic acid acts as a signaling molecule regulating energy expenditure and is closely associated with metabolic diseases. Front. Endocrinol. 2022;13:847611. doi: 10.3389/fendo.2022.847611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tétreault M., Gonzalez M., Dicaire M.-J., Allard P., Gehring K., Leblanc D., Leclerc N., Schondorf R., Mathieu J., Zuchner S., Brais B. Adult-onset painful axonal polyneuropathy caused by a dominant NAGLU mutation. Brain. 2015;138:1477–1483. doi: 10.1093/brain/awv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Watada H., Fujitani Y. Minireview: autophagy in pancreatic β-cells and its implication in diabetes. Mol. Endocrinol. 2015;29:338–348. doi: 10.1210/me.2014-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MacDonald M.J., Longacre M.J., Langberg E.-C., Tibell A., Kendrick M.A., Fukao T., Ostenson C.-G. Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia. 2009;52:1087–1091. doi: 10.1007/s00125-009-1319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mulder H. Transcribing β-cell mitochondria in health and disease. Mol. Metab. 2017;6:1040–1051. doi: 10.1016/j.molmet.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Manu M.S., Rachana K.S., Advirao G.M. Altered expression of IRS2 and GRB2 in demyelination of peripheral neurons: implications in diabetic neuropathy. Neuropeptides. 2017;62:71–79. doi: 10.1016/j.npep.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 74.Wang C., Calcutt M.W., Ferguson J.F. Knock-out of DHTKD1 alters mitochondrial respiration and function, and may represent a novel pathway in cardiometabolic disease risk. Front. Endocrinol. 2021;12:710698. doi: 10.3389/fendo.2021.710698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prashanth G., Vastrad B., Tengli A., Vastrad C., Kotturshetti I. Investigation of candidate genes and mechanisms underlying obesity associated type 2 diabetes mellitus using bioinformatics analysis and screening of small drug molecules. BMC Endocr. Disord. 2021;21:80. doi: 10.1186/s12902-021-00718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh K., Gupta K., Kaur S. High resolution ultrasonography of the tibial nerve in diabetic peripheral neuropathy. J. Ultrason. 2017;17:246–252. doi: 10.15557/JoU.2017.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cao C., Wang J., Kwok D., Cui F., Zhang Z., Zhao D., Li M.J., Zou Q. webTWAS: a resource for disease candidate susceptibility genes identified by transcriptome-wide association study. Nucleic Acids Res. 2022;50:D1123–D1130. doi: 10.1093/nar/gkab957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eid S., Sas K.M., Abcouwer S.F., Feldman E.L., Gardner T.W., Pennathur S., Fort P.E. New insights into the mechanisms of diabetic complications: role of lipids and lipid metabolism. Diabetologia. 2019;62:1539–1549. doi: 10.1007/s00125-019-4959-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Solis-Herrera C., Triplitt C., Cersosimo E., DeFronzo R.A. In: Pathogenesis of Type 2 Diabetes Mellitus. Endotext K.R.F., Anawalt B., Boyce A., G. Chrousos W.W. de H., Dhatariya K., Dungan K., Hershman J.M., J. Hofland S.K., et al., editors. MDText.com, Inc.; 2021. [Google Scholar]
- 80.Fadista J., Vikman P., Laakso E.O., Mollet I.G., Esguerra J.L., Taneera J., Storm P., Osmark P., Ladenvall C., Prasad R.B., et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA. 2014;111:13924–13929. doi: 10.1073/pnas.1402665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bury J.J., Chambers A., Heath P.R., Ince P.G., Shaw P.J., Matthews F.E., Brayne C., Simpson J.E., Wharton S.B., Cognitive Function and Ageing Study Type 2 diabetes mellitus-associated transcriptome alterations in cortical neurones and associated neurovascular unit cells in the ageing brain. Acta Neuropathol. Commun. 2021;9:5. doi: 10.1186/s40478-020-01109-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao S., Wang Q., Li Z., Ma X., Wu L., Ji H., Qin G. LDOC1 inhibits proliferation and promotes apoptosis by repressing NF-κB activation in papillary thyroid carcinoma. J. Exp. Clin. Cancer Res. 2015;34:146. doi: 10.1186/s13046-015-0265-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patel S., Santani D. Role of NF-κB in the pathogenesis of diabetes and its associated complications. Pharmacol. Rep. 2009;61:595–603. doi: 10.1016/s1734-1140(09)70111-2. [DOI] [PubMed] [Google Scholar]
- 84.Eizirik D.L., Pasquali L., Cnop M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat. Rev. Endocrinol. 2020;16:349–362. doi: 10.1038/s41574-020-0355-7. [DOI] [PubMed] [Google Scholar]
- 85.Krentz N.A.J., Gloyn A.L. Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat. Rev. Endocrinol. 2020;16:202–212. doi: 10.1038/s41574-020-0325-0. [DOI] [PubMed] [Google Scholar]
- 86.Viñuela A., Varshney A., van de Bunt M., Prasad R.B., Asplund O., Bennett A., Boehnke M., Brown A.A., Erdos M.R., Fadista J., et al. Genetic variant effects on gene expression in human pancreatic islets and their implications for T2D. Nat. Commun. 2020;11:4912. doi: 10.1038/s41467-020-18581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gu Y., Qiu Z.-L., Liu D.-Z., Sun G.-L., Guan Y.-C., Hei Z.-Q., Li X. Differential gene expression profiling of the sciatic nerve in type 1 and type 2 diabetic mice. Biomed. Rep. 2018;9:291–304. doi: 10.3892/br.2018.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Feldman E.L., Callaghan B.C., Pop-Busui R., Zochodne D.W., Wright D.E., Bennett D.L., Bril V., Russell J.W., Viswanathan V. Diabetic neuropathy. Nat. Rev. Dis. Primers. 2019;5 doi: 10.1038/s41572-019-0092-1. 41–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo K., Elzinga S., Eid S., Figueroa-Romero C., Hinder L.M., Pacut C., Feldman E.L., Hur J. Genome-wide DNA methylation profiling of human diabetic peripheral neuropathy in subjects with type 2 diabetes mellitus. Epigenetics. 2019;14:766–779. doi: 10.1080/15592294.2019.1615352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kirchner H., Sinha I., Naslund E., Zierath J. Altered DNA methylation of glycolytic and lipogenic genes in liver of obese and type 2 diabetic patients. Exp. Clin. Endocrinol. Diabetes. 2015;122 doi: 10.1055/s-0035-1547613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ray P.R., Khan J., Wangzhou A., Tavares-Ferreira D., Akopian A.N., Dussor G., Price T.J. Transcriptome analysis of the human tibial nerve identifies sexually dimorphic expression of genes involved in pain, inflammation, and neuro-immunity. Front. Mol. Neurosci. 2019;12:37. doi: 10.3389/fnmol.2019.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Niccoli T., Partridge L. Ageing as a risk factor for disease. Curr. Biol. 2012;22:R741–R752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 93.Field A.E., Coakley E.H., Must A., Spadano J.L., Laird N., Dietz W.H., Rimm E., Colditz G.A. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch. Intern. Med. 2001;161:1581–1586. doi: 10.1001/archinte.161.13.1581. [DOI] [PubMed] [Google Scholar]
- 94.Goldman N., Weinstein M., Cornman J., Singer B., Seeman T., Goldman N., Chang M.-C. Sex differentials in biological risk factors for chronic disease: estimates from population-based surveys. J. Womens Health. 2004;13:393–403. doi: 10.1089/154099904323087088. [DOI] [PubMed] [Google Scholar]
- 95.Kittles R.A., Weiss K.M. Race, ancestry, and genes: implications for defining disease risk. Annu. Rev. Genom. Hum. Genet. 2003;4:33–67. doi: 10.1146/annurev.genom.4.070802.110356. [DOI] [PubMed] [Google Scholar]
- 96.Caturegli P., De Remigis A., Rose N.R. Hashimoto thyroiditis: clinical and diagnostic criteria. Autoimmun. Rev. 2014;13:391–397. doi: 10.1016/j.autrev.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 97.Bremner A.P., Feddema P., Leedman P.J., Brown S.J., Beilby J.P., Lim E.M., Wilson S.G., O’Leary P.C., Walsh J.P. Age-related changes in thyroid function: a longitudinal study of a community-based cohort. J. Clin. Endocrinol. Metab. 2012;97:1554–1562. doi: 10.1210/jc.2011-3020. [DOI] [PubMed] [Google Scholar]
- 98.Gesing A., Lewiński A., Karbownik-Lewińska M. The thyroid gland and the process of aging; what is new? Thyroid Res. 2012;5:16. doi: 10.1186/1756-6614-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rachana K.S., Manu M.S., Advirao G.M. Insulin-induced upregulation of lipoprotein lipase in Schwann cells during diabetic peripheral neuropathy. Diabetes Metab. Syndr. 2018;12:525–530. doi: 10.1016/j.dsx.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 100.Wree A., Kahraman A., Gerken G., Canbay A. Obesity affects the liver - the link between adipocytes and hepatocytes. Digestion. 2011;83:124–133. doi: 10.1159/000318741. [DOI] [PubMed] [Google Scholar]
- 101.Mancuso P., Bouchard B. The impact of aging on adipose function and adipokine synthesis. Front. Endocrinol. 2019;10:137. doi: 10.3389/fendo.2019.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tchkonia T., Morbeck D.E., Von Zglinicki T., Van Deursen J., Lustgarten J., Scrable H., Khosla S., Jensen M.D., Kirkland J.L. Fat tissue, aging, and cellular senescence. Aging Cell. 2010;9:667–684. doi: 10.1111/j.1474-9726.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Esiri M.M. Ageing and the brain. J. Pathol. 2007;211:181–187. doi: 10.1002/path.2089. [DOI] [PubMed] [Google Scholar]
- 104.Castelli V., Benedetti E., Antonosante A., Catanesi M., Pitari G., Ippoliti R., Cimini A., d’Angelo M. Neuronal cells rearrangement during aging and neurodegenerative disease: metabolism, oxidative stress and organelles dynamic. Front. Mol. Neurosci. 2019;12:132. doi: 10.3389/fnmol.2019.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shashikanth N., Yeruva S., Ong M.L.D.M., Odenwald M.A., Pavlyuk R., Turner J.R. Epithelial organization: the gut and beyond. Compr. Physiol. 2017;7:1497–1518. doi: 10.1002/cphy.c170003. [DOI] [PubMed] [Google Scholar]
- 106.Guillot C., Lecuit T. Mechanics of epithelial tissue homeostasis and morphogenesis. Science. 2013;340:1185–1189. doi: 10.1126/science.1235249. [DOI] [PubMed] [Google Scholar]
- 107.Larsen S.B., Cowley C.J., Fuchs E. Epithelial cells: liaisons of immunity. Curr. Opin. Immunol. 2020;62:45–53. doi: 10.1016/j.coi.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rehfeld A., Nylander M., Karnov K. Glandular epithelium and glands. Compendium of Histology. 2017:101–120. doi: 10.1007/978-3-319-41873-5_6. [DOI] [Google Scholar]
- 109.Johansson M.E.V., Sjövall H., Hansson G.C. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013;10:352–361. doi: 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Monteleone P., Mascagni G., Giannini A., Genazzani A.R., Simoncini T. Symptoms of menopause — global prevalence, physiology and implications. Nat. Rev. Endocrinol. 2018;14:199–215. doi: 10.1038/nrendo.2017.180. [DOI] [PubMed] [Google Scholar]
- 111.Arinkan S.A., Gunacti M. Factors influencing age at natural menopause. J. Obstet. Gynaecol. Res. 2021;47:913–920. doi: 10.1111/jog.14614. [DOI] [PubMed] [Google Scholar]
- 112.Pickrell J.K., Pai A.A., Gilad Y., Pritchard J.K. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 2010;6:e1001236. doi: 10.1371/journal.pgen.1001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pritchard J.K., Pickrell J.K., Coop G. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr. Biol. 2010;20:R208–R215. doi: 10.1016/j.cub.2009.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barry J.D., Fagny M., Paulson J.N., Aerts H.J.W.L., Platig J., Quackenbush J. Histopathological image QTL discovery of immune infiltration variants. iScience. 2018;5:80–89. doi: 10.1016/j.isci.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ash J.T., Darnell G., Munro D., Engelhardt B.E. Joint analysis of expression levels and histological images identifies genes associated with tissue morphology. Nat. Commun. 2021;12:1609–1612. doi: 10.1038/s41467-021-21727-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Badea L., Stănescu E. Identifying transcriptomic correlates of histology using deep learning. PLoS One. 2020;15:e0242858. doi: 10.1371/journal.pone.0242858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gallins P., Saghapour E., Zhou Y.-H. Exploring the limits of combined Image/’omics analysis for non-cancer histological phenotypes. Front. Genet. 2020;11:555886. doi: 10.3389/fgene.2020.555886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.MacNee W., Rabinovich R.A., Choudhury G. Ageing and the border between health and disease. Eur. Respir. J. 2014;44:1332–1352. doi: 10.1183/09031936.00134014. [DOI] [PubMed] [Google Scholar]
- 119.Li Z., Zhang Z., Ren Y., Wang Y., Fang J., Yue H., Ma S., Guan F. Aging and age-related diseases: from mechanisms to therapeutic strategies. Biogerontology. 2021;22:165–187. doi: 10.1007/s10522-021-09910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sirugo G., Williams S.M., Tishkoff S.A. The missing diversity in human genetic studies. Cell. 2019;177:1080. doi: 10.1016/j.cell.2019.04.032. [DOI] [PubMed] [Google Scholar]
- 121.Nathan D.M., DCCT/EDIC Research Group The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care. 2014;37:9–16. doi: 10.2337/dc13-2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zheng Y., Ley S.H., Hu F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018;14:88–98. doi: 10.1038/nrendo.2017.151. [DOI] [PubMed] [Google Scholar]
- 123.Call J.T., Cortés P., Harris D.M. A practical review of diabetes mellitus type 2 treatment in primary care. Rom. J. Intern. Med. 2022;60:14–23. doi: 10.2478/rjim-2021-0031. [DOI] [PubMed] [Google Scholar]
- 124.Rawshani A., Rawshani A., Franzén S., Eliasson B., Svensson A.-M., Miftaraj M., McGuire D.K., Sattar N., Rosengren A., Gudbjörnsdottir S. Mortality and cardiovascular disease in type 1 and type 2 diabetes. N. Engl. J. Med. 2017;376:1407–1418. doi: 10.1056/NEJMoa1608664. [DOI] [PubMed] [Google Scholar]
- 125.Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M., et al. The human cell atlas. Elife. 2017;6:e27041. doi: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.R Core Team . 2019. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 127.Trincado J.L., Entizne J.C., Hysenaj G., Singh B., Skalic M., Elliott D.J., Eyras E. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018;19:40. doi: 10.1186/s13059-018-1417-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nally R.M., Walsh C.J. Hierarchical partitioning public-domain software. Biodivers. Conserv. 2004;13:659–660. doi: 10.1023/b:bioc.0000009515.11717.0b. [DOI] [Google Scholar]
- 130.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mathe E., Davis S. 2018. Statistical Genomics: Methods and Protocols (Methods in Molecular Biology) [Google Scholar]
- 133.Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 134.Muñoz-Aguirre M., Ntasis V.F., Rojas S., Guigó R. PyHIST: a histological image segmentation tool. PLoS Comput. Biol. 2020;16:e1008349. doi: 10.1371/journal.pcbi.1008349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pau G., Fuchs F., Sklyar O., Boutros M., Huber W. EBImage—an R package for image processing with applications to cellular phenotypes. Bioinformatics. 2010;26:979–981. doi: 10.1093/bioinformatics/btq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Meyer D., Dimitriadou E., Hornik K., Weingessel A., Leisch F. Misc Functions of the Department of Statistics. TU Wien; 2021. Probability theory group (formerly: e1071) p. e1071. R package version 1.7-10/r494. [Google Scholar]
- 137.Ho D.E., Imai K., King G., Stuart E.A. MatchIt: nonparametric preprocessing for parametric causal inference. J. Stat. Soft. 2011;42 doi: 10.18637/jss.v042.i08. [DOI] [Google Scholar]
- 138.Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Law C.W., Chen Y., Shi W., Smyth G.K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ferreira P.G., Muñoz-Aguirre M., Reverter F., Sá Godinho C.P., Sousa A., Amadoz A., Sodaei R., Hidalgo M.R., Pervouchine D., Carbonell-Caballero J., et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat. Commun. 2018;9:490. doi: 10.1038/s41467-017-02772-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zeileis A. Object-oriented computation of sandwich estimators. J. Stat. Soft. 2006;16 doi: 10.18637/jss.v016.i09. [DOI] [Google Scholar]
- 143.Jansen R., Batista S., Brooks A.I., Tischfield J.A., Willemsen G., van Grootheest G., Hottenga J.-J., Milaneschi Y., Mbarek H., Madar V., et al. Sex differences in the human peripheral blood transcriptome. BMC Genom. 2014;15:33. doi: 10.1186/1471-2164-15-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kuhn M. 2020. Caret: Classification and Regression Training. R Package Version 6.0-86. [Google Scholar]
- 145.Weir B.S., Cockerham C.C. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 146.The R project for statistical computing. https://www.r-project.org/
- 147.Mangiafico S. 2022. Functions to Support Extension Education Program Evaluation [R Package Rcompanion Version 2.4.15] [Google Scholar]
- 148.Carithers L.J., Ardlie K., Barcus M., Branton P.A., Britton A., Buia S.A., Compton C.C., DeLuca D.S., Peter-Demchok J., Gelfand E.T., et al. A novel approach to high-quality postmortem tissue procurement: the GTEx project. Biopreserv. Biobank. 2015;13:311–319. doi: 10.1089/bio.2015.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Atkinson M.A., Eisenbarth G.S., Michels A.W. Type 1 diabetes. Lancet. 2014;383:69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979;9:62–66. doi: 10.1109/tsmc.1979.4310076. [DOI] [Google Scholar]
- 151.Haralick R.M., Shanmugam K., Dinstein I. Textural features for image classification. IEEE Trans. Syst. Man Cybern. 1973;3:610–621. doi: 10.1109/tsmc.1973.4309314. [DOI] [Google Scholar]
- 152.Hansen B.B., Klopfer S.O. Optimal full matching and related designs via network flows. J. Comput. Graph Stat. 2006;15:609–627. doi: 10.1198/106186006X137047. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Replication of genes differentially expressed and differentially spliced with demographic traits using independent datasets. (B) Top 50 genes with a large proportion of their gene expression variation explained by a demographic trait. (C) Tissue sharing of genes differentially expressed with each demographic trait. (D) Differential gene expression is independent of gene expression breadth. (E) Genes expressed only in the tissues where they are differentially expressed. (F) Genes differentially expressed in 10 or more tissues. (G) Genes differentially expressed between populations in 10 or more tissues are enriched in the glutathione-related pathways. (H) Genes differentially expressed with age in 10 or more tissues are enriched in the p-53 signaling pathway.
(A) Genes differentially expressed with two demographic traits per tissue. (B) Tissue enrichment and biased expression in genes with additive contributions. (C) Genes with interactions between demographic traits. (D) Genes with a variable contribution of aging between males and females are enriched in mammary gland development.
(A) Functional characterization of alternatively spliced events per tissue and type of splicing event. (B) Non-coding isoforms are biased toward the inclusion of exonic/intronic sequence in intron retention, alternative 5′ and alternative 3′ splicing events, in alternatively spliced events associated with a switch between a non-coding and a protein-coding isoform. (C) There are more alternative last and fewer alternative 5′ and 3′ events than expected differentially spliced between populations. (D) Bias toward the inclusion or exclusion of exonic/intronic sequence is specific to the type of splicing events, tissues, and demographic traits. (E) Functional characterization of differentially spliced events. (F) Enrichment of DSGs in DEGs. (G) Genes differentially expressed and spliced between populations are enriched in eGenes and sGenes. (H) Comparison of the alternative splicing variation explained in each type of splicing event by demographic traits. (I) Top 50 splicing events with a large proportion of their alternative splicing variation explained by a demographic trait.
(A) Ancestry-DSGs are enriched in sGenes. (B) Functional enrichment of genes with highly tissue-shared differentially spliced events between populations. (C) Functional enrichment of genes differentially spliced between populations in human primary monocytes. (D) Ribosomal proteins with events differentially spliced between populations reported as sGenes in different studies.
(A) Number of DEGs per clinical trait and tissue (one model per clinical trait and tissue). (B) Number of samples per clinical trait and reason to include/exclude clinical traits with at least five positive samples. (C) Genes differentially expressed and spliced with clinical traits per tissue (one model per tissue). (D) Top genes (up to 50) with a large proportion of their gene expression variation explained by a clinical trait. (E) Top splicing events (up to 50) with a large proportion of their alternative splicing variation explained by a clinical trait. (F) Genes differentially expressed in more than one tissue per clinical trait (types 1 and 2 diabetes). (G) Genes differentially expressed with two clinical traits and tissue enrichment in genes with additive contributions. (H) Genes differentially expressed with both diabetes in the tibial nerve are enriched in immune receptor activity and ion channel activity (GO:MF). (I) Genes whose residual expression in the tibial nerve significantly correlates with the SVM-assigned probability of being classified as a diabetic individual. (J) Genes whose residual expression in the tibial nerve significantly correlates with the SVM-assigned probability of being classified as a diabetic individual are enriched in neuropathy-related diseases (GO:DisGeNET). (K) Genes differentially expressed with a clinical and a demographic trait per tissue and tissue enrichment analysis in genes with additive contributions, including results after matching trait distributions between healthy and diseased individuals.
Data Availability Statement
All GTEx protected data are available at the accession number dbGaP: phs000424.v8. Access to the raw sequence data is now provided through AnVIL: https://gtexportal.org/home/protectedDataAccess. Public-access data, including QTL summary statistics and expression levels, are available on the GTEx Portal: https://www.gtexportal.org, as well as in the UCSC and Ensembl browsers.
Analysis scripts are available at github: https://github.com/Mele-Lab/2022_GTExTranscriptome and all results tables derived from the analyses conducted in this paper are deposited at zenodo: https://doi.org/10.5281/zenodo.6797627.