

Abstract

A Ru-catalyzed isomerization of Achmatowicz derivatives that opens unexplored routes to diversify the biogenic furanic platform is reported. The mechanistic insights of this formally redox-neutral intramolecular process were studied computationally and by deuterium labeling. The transformation proved to be a robust synthetic tool to achieve the synthesis of bioderived-monomers and a series of 4-keto-δ-valerolactones that further enabled the development of a flexible strategy for the synthesis of acetogenins. A concise and protective group-free asymmetric total synthesis of two natural products, namely, (S,S)-muricatacin and the (S,S)-L-factor, is also described.
Keywords: ruthenium catalysis, allylic alcohol isomerization, Achmatowicz rearrangement, biorefinery, bioactive lactones
Introduction
The biogenic furanic platform is considered to be a promising sustainable source of chemicals,1−3 for example, for the manufacture of resins and biofuels.3,4 Among other transformations that exploit this platform, the Achmatowicz rearrangement proved to be an efficient synthetic tool to produce structurally complex derivatives from simple furans and thus holds a unique place in the synthesis of many natural products and diverse organic syntheses.5−11 Its potential to serve as a key step in the production of biomass-derived C5 alcohols was recently reported by our group.12,13
The exploitation of the densely functionalized Achmatowicz dihydropyranones in organic synthesis has been reported in many instances.14,15 The hemiacetal and ketone groups received particular attention as numerous oxidations of the hemiacetal16−18 and reductions of the ketone group17−19 have been reported. To a lesser extent, the reduction of the olefin has also been described.12,20,21
The redox isomerization of allylic alcohols into saturated carbonyl compounds is among the most well-studied transition-metal-catalyzed processes.22,23 The development of efficient catalysts24−27 for this transformation showcased impressive advances; however, the majority of the reported examples have been devoted to the synthesis of simple α- and/or β-branched aldehydes and ketones. Despite their obvious utility,28 synthetic strategies that harness transition metal-catalyzed redox isomerization in the context of Achmatowicz derivatives have been scarcely described. Aiming at an expedient synthesis of carbohydrates, Wang et al. reported an Ir-catalyzed dynamic kinetic redox isomerization of Achmatowicz derivatives.29 This approach involves oxidation of the hemiacetal and consequent diastereoselective reduction of the ketone (Scheme 1A) and provided access to the key intermediates (1) in the synthesis of naturally occurring sugars. Later on, the same isomerization has been achieved by employing an enantioconvergent biocatalytic approach to yield γ-hydroxy-δ-lactones in an enantio- and diastereoselective fashion (Scheme 1B). The transformation has been shown to proceed also in vivo.30,31
Scheme 1. Redox Isomerization of Achmatowicz Derivatives and Downstream Applications.

To the best of our knowledge, transformations that utilize the reduction of the olefin instead of the ketone in a redox isomerization of Achmatowicz dihydropyranones still have not emerged. Herein, we present a new highly efficient Ru-catalyzed isomerization of a range of Achmatowicz derivatives to 4-keto-δ-valerolactones (2) by consecutive oxidation of the hemiacetal and reduction of the olefin. This strategy enabled the preparation of biorenewable monomers and a series of bioactive lactones (Scheme 1C).
Results and Discussion
We began our studies using 10 mol % [Ir(cod)MeO]2 as a catalyst. In the absence of a ligand, we observed full conversion of the starting material 3. The desired product 4 was formed in low yields alongside the formation of two major side products (Scheme 2), namely, 5, arising from the reduction of the ketone, and the oxidized product 6 (Table 1, entry 1). We were not able to identify other reaction products, and thus we anticipated that this was due to the occurrence of undesired polymerization reactions in which products could not be elucidated by NMR spectroscopy or detected by GC analysis.
Scheme 2. Redox Isomerization of 6-Hydroxy-2H-pyran-3(6H)-one 3.

Table 1. Catalytic Experimentsa,b.
| entry | catalyst | loading (mol %) | solvent | temp (°C) | ligand (mol %) | time (h) | 3 (%) | 4 (%) | 5 (%) | 6 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | [Ir(cod)MeO]2 | 10 | DCE | 60 | 1 | 18 | 3 | 26 | ||
| 2 | [Ir(cod)MeO]2 | 10 | DCE | rt | t-Bu-BiPy (10) | 1 | 84 | |||
| 3 | [Ir(cod)MeO]2 | 10 | DCE | 60 | BiPy (10) | 1 | 85 | |||
| 4 | [Ir(cod)MeO]2 | 10 | DCE | 60 | 4,4′-diMeO-BiPy (10) | 1 | 6 | 61 | ||
| 5 | [Ir(cod)MeO]2 | 10 | DCE | 60 | 1,10-Phen (10) | 1 | 53 | 27 | ||
| 6 | [RuCp*(MeCN)3]PF6 | 2 | THF | reflux | 4 | 70 | ||||
| 7 | [RuCp*(MeCN)3]PF6 | 2 | CH3CN | reflux | 4 | 99 | ||||
| 8 | [RuCp*(MeCN)3]PF6 | 1 | CH3CN | reflux | 6 | 99 | ||||
| 9 | [RuCp*(MeCN)3]PF6 | 0.5 | CH3CN | reflux | 6 | 89 | ||||
| 10 | [RuCp*(MeCN)3]PF6 | 1 | CH3CN | reflux | 17 | 87c,d | ||||
| 11 | [RuCp*(MeCN)3]PF6 | 1 | CH3CN | reflux | 18 | 85c,e |
Yields determined by GC analysis using dodecane as an internal standard.
0.5 mmol 1 in 5 mL of solvent.
Yield after chromatographic purification.
gram scale (3 = 1.14 g, 10 mmol).
gram scale (3 = 2.28 g, 20 mmol).
The use of t-Bu-BiPy and BiPy as ligands rendered product 4 in high yields of 84 and 85%, respectively (Table 1, entries 2 and 3). Noteworthy, the formation of the side products 5 and 6 was not detected. However, we still experienced a 16% unidentified loss of yield. The use of 4,4′-diMeO-BiPy or 1,10-phenanthroline ligands did not provide a positive effect (Table 1, entries 4 and 5). Unfortunately, our further attempts to solve that issue using [Ir(cod)MeO]2 as a catalyst failed under various reaction conditions (see Supporting Information 1, Table S1, entries 1–19).
Despite the fact that, in several instances, the [Ir(cod)MeO]2/BiPy catalyst provided 4 in very high yields (Table S1, entries 6, 7, and 12–14), the high catalyst loading and the unidentifiable loss of yield provoked us to continue our studies by screening a series of other catalysts. In this series, [Ir(cod)Cl]2, Pd(OAc)2, Pd2(dba)3, and [Ru(cod)Cl2]n were found to be ineffective under various conditions (Table S1, entries 20–26). Although, in some instances, the formation of 4 was observed in the presence of Ru(CO)(H)(Ph3P)3Cl or [Ru(p-cymene)Cl2]2, the yields have been far from satisfactory (Table S1, entries 27–37). The use of 2 mol % [RuCp*(MeCN)3]PF6 in THF rendered a more promising 70% yield of 4 (Table 1, entry 6). To our delight, 4 was formed in nearly quantitative analytical yields when the reaction was performed in refluxing MeCN (Table 1, entry 7). Noteworthy, this was the first experiment in which significant degradation of the starting material and/or the reaction products was not observed. The yield was reproducible in the presence of only 1 mol % catalyst (Table 1, entry 8). However, the decrease of the catalyst loading to 0.5 mol % rendered 4 in an 89% yield (Table 1, entry 9).
The 4-keto-δ-valerolactone (KVL) 4 has been previously reported from levulinic acid (LA) as a biorenewable monomer for the production of chemically recyclable and biodegradable polymer poly(4-ketovalerolactone) (PKVL).32 Such an approach utilizes 5-bromo levulinic acid (5-BrLA) as an intermediate, thus requiring extensive use of Br2. Furthermore, due to the availability of several activated positions, the regioselective bromination of LA proved to be troublesome and rendered a mixture of regioisomers and bis-brominated products.32
Stepping on the availability of 3 and our previous work on its upscale synthesis under flow conditions,12 we achieved the gram scale production of 4 in up to 87% of isolated yield (Table 1, entries 10 and 11). In contrast to the previous methods that rendered up to a 40% yield from LA, the overall yield of 4 from furfuryl alcohol was >80%. Noteworthy, our strategy is much more atom-efficient due to the lack of regioselectivity issues and extensive use of halogens.
To investigate the scope of the Ru-catalyzed isomerization (Scheme 3), we prepared a variety of alkyl-substituted Achmatowicz derivatives, namely, 7a–7l. In this series, we observed that the reaction rate was governed by the length of the substituent. The methyl- and ethyl-substituted lactones 8a and 8b were obtained in excellent yields in the 3 h reaction time. The n-pentyl 8c and phenethyl 8d derivatives were converted in only 30 and 35% isolated yields after 24 h, respectively, whereas the remaining starting material was predominantly preserved. The steric bulk of i-Pr 7e and cyclohexyl 7f derivatives was reflected in prolonged reaction times; however, the corresponding lactones 8e and 8f were obtained in excellent yields. The reaction tolerates a variety of other substituents including allyl (8g), spiro (8h–8j), and disubstituted derivatives (8k and 8l).
Scheme 3. Scope of the Ru-Catalyzed Isomerization,

Reactions were monitored by TLC.
Yields after chromatographic purification;
3 h;
18 h;
24 h;
48 h;
2 mol % cat.;
130 °C, Synthware pressure vessel.
We attempted to gain deeper understanding of the unusual effect of the length of the substituents by means of 2D 1H–1H NOESY NMR experiments. Unfortunately, the NMR data did not provide conclusive information on the proximity of those substituents to the reactive protons from the allylic system. However, the experimental data supported the involvement of steric effects.
The exposure of 7c to the less sterically bulky complex [RuCp(MeCN)3]PF6 rendered the corresponding lactone 8c in an improved 50% yield (Scheme 3), indicating that the steric bulk of the catalyst plays an important role; however, this role was not dominant. This finding led us to elevate the catalyst loading and the reaction temperature (2 mol %, 130 °C). To our delight, this resulted in a significant improvement, leading to the formation of 8c in an excellent yield of 90%. Due to decomposition at high temperatures, our attempt to increase the yield of the phenethyl derivative 8d was unsuccessful. On the other hand, the increased catalyst loading alone did not provide a pronounced effect. Furthermore, spiro lactone 8h, which was previously obtained in only 33%, was isolated in an 80% yield. Finally, we performed a gram-scale isomerization of Achmatowicz derivative 3 under the same conditions used for the [RuCp*(MeCN)3]PF6 catalyst (see Supporting Information 1, Section S2.3.1). Nevertheless, we did not observe any effect of the steric bulk of the catalyst over the reaction rate.
In all the cases, we observed slight decomposition of the products during purification on silica gel (for analytical yields, see Supporting Information 1, Section S2.3.2). Despite the hydrolysis of the lactones being the most obvious cause for the reduced yields, we were not able to identify the formation of the corresponding acids.
Next, we turned our attention to the synthesis of 5-hydroxyalkylbutan-4-olides, which are lactones widespread in nature that exhibit diverse biological properties, such as insect antifeedant activity33 and anti-tumor activity.34,35 Members of this family are found in microbial metabolite cultures of Erwinia quernica(36) and Streptomyces griseus.37 The short-chain homologues are flavor constituents in alcoholic beverages.38,39 Given this, it comes as no surprise that much attention has been given to their synthesis and exploitation as synthons in the preparation of complex natural products.40−44
We rationalized that a reduction of the ketone in derivatives 8a–8k will deliver simultaneous isomerization of the formed 4-hydroxy-δ-valerolactones to the more stable 5-hydroxyalkylbutan-4-olides, thus providing a concise flexible synthetic route to achieve the synthesis of these biologically significant lactones. To our delight upon simple treatment with NaBH4 in DCM in the presence of a catalytic amount of AcOH (Method A), 4 was directly converted in a nearly quantitative yield to lactone 10 without isolation of the intermediate hydroxylactone 9 (Scheme 4).
Scheme 4. Synthesis of 5-hydroxyalkylbutan-4-olides,

Method A: 2.0 equiv NaBH4, 2 drops of AcOH, DCM, rt, 24 h;
Method B: L-selectride, THF, −78 °C;
cat. HCl, MeOH, rt, 24 h.
In the presence of NaBH4, mono-substituted substrates 8a–8c were converted to the corresponding butyrolactones 12a–12c in good to high yields (Scheme 4). In the case of n-pentyl lactone 8c, the increased steric bulk of the substituent hampered the valerolactone to butyrolactone isomerization and we obtained an inseparable mixture of the intermediate hydroxyl lactone 11c and the desired product in a 65% yield. However, a simple treatment of this mixture with a catalytic amount of aq. HCl in MeOH rendered quantitative isomerization to 12c, which was isolated without further purification. Noteworthy, in all cases, we obtained an inseparable mixture of diastereoisomers in a near 1:1 ratio. The treatment of 8a–8c with bulkier L-selectride (Method B) delivered products 12a–12c as single syn-diastereoisomers in good yields. The L-selectride reduction reaction does not tolerate bulkier i-Pr and cyclohexyl substituents, and we observed the formation of complex mixtures of products. This observation is in accordance with steric effects governing the better diastereoselectivity achieved with L-selectride. However, upon treatment with NaBH4, the desired butyrolactones 12e and 12f were achieved in good yields and moderate diastereoselectivity (dr = 80:20 and 83:17, respectively). To our delight, the phenethyl 8d and allyl 8g substituted derivatives reacted smoothly in the presence of L-selectride. The corresponding lactones 12d and 12g were isolated as single syn-diastereoisomers in good yields. Noteworthy, one can foresee the latter as a precursor to higher acetogenin synthesis due to the presence of the terminal olefin prone to downstream modifications. Despite the fact that treatment with aq. HCl/MeOH was required, the spiro 8h–8j and disubstituted derivative 8k were converted in excellent yields to the corresponding products 12h–12k in the presence of NaBH4.
Finally, we wanted to demonstrate the utility of our synthetic strategy by its application to the protective-group-free total asymmetric synthesis of two important members of the acetogenin family, namely, L-factor and muricatacin. The L-factor is isolated from Streptomyces griseus and is considered an autoregulator of anthracycline biosynthesis.45 Muricatacin is isolated from the seeds of Annona muricata and has received attention in research due to its anti-proliferative activity and cytotoxicity against various human tumoral cell lines.46,47 It has been intensively exploited as a gateway synthon to higher acetogenin synthesis.48 To date, the synthetic approaches to achieve the synthesis of these chiral 5-hydroxyalkylbutan-4-olides mainly exploit nature’s chiral pool,49 namely, carbohydrates, which, in many instances, require laborious multistep synthesis and extensive use of protective groups.50−52 Catalytic asymmetric approaches have been rarely reported and are limited to the use of asymmetric dihydroxylation or epoxidation of synthetic unsaturated carboxylic acids.49,53,54
We initiated our asymmetric synthesis from furfuryl alcohols S-14c and S-14m that are readily available in their both enantiomeric forms from Noyori asymmetric hydrogenation of the corresponding ketones 13c and 13m (Scheme 5).55,56 Achmatowicz rearrangement using Oxone as an oxidant rendered S-7c and S-7m in high yields (75 and 80%, respectively). The valerolactones S-8c and S-8m obtained after isomerization with [RuCp(MeCN)3]PF6 were subjected to reduction by L-selectride, which proceeded in a stereoselective manner and delivered L-factor and muricatacin as single (+)-(4S,5S) diastereoisomers without racemization in 77 and 85% yields, respectively.
Scheme 5. Asymmetric Synthesis of (+)-L-factor and (+)-Muricatacin.

We also subjected the Achmatowicz derivative S-7m to isomerization in the presence of 1 mol % of the bulkier [RuCp*(MeCN)3]PF6 catalyst in MeCN under reflux. No formation of the desired lactone S-8m was observed, which is in accordance with our observation in that the isomerization reaction is highly influenced by the length of the alkyl substituents at C-6 in the Achmatowicz dihydropyranones.
A deuterium labeling study was undertaken to determine the mechanism of the Ru-catalyzed isomerization. To this end, we prepared D-3 isotopically labeled at the hemiacetal position. To obtain information on the molecularity of the process, we performed a crossover experiment between D-3 and 7b and analyzed the products by mass spectrometry and NMR spectroscopy (Scheme 6A). The deuterium was transferred intramolecularly to form product 3D-4, while deuterated product 8b was not observed. Noteworthy, by NMR experiments, we observed the incorporation of the deuterium solely at position 3, leading to the formation of product 3D-4 (Scheme 6B).
Scheme 6. Deuterium Labeling Studies.

There are two generally accepted reaction pathways for the intramolecular transition metal-catalyzed redox isomerization of allylic alcohols that do not require an isolated metal-hydride precatalyst or its in situ generation. Both mechanisms operate via an intramolecular 1,3-hydride shift57 with the involvement of either oxidative addition to the allylic C–H bond, forming a π-allyl metal-hydride complex58−60 (Scheme 7A) or the formation of metal alkoxide species57,61−63 (Scheme 7B). The latter has been proposed in several instances for Ru(II) catalysis.64,65 The hydride transfer (Scheme 7, A-II → A-III or B-II → B-III) in both previously established mechanisms should result in a regioselective deuterium incorporation at position 4, leading to compound 4D-4 (Scheme 6).
Scheme 7. Reported Mechanisms for the Transition Metal-Catalyzed Redox Isomerization of Allylic Alcohols.

In our case, the exclusive formation of product 3D-4 suggests the formation of a type B-II Ru π-olefin complex that undergoes 1,4-hydride addition relative to the ketone governed by electrophilic and/or thermodynamic reasons (Scheme 7).
We then performed an Ir-catalyzed isomerization of D-3 and observed a significant isotopic effect leading to a more sluggish reaction as compared to the non-deuterated substrate 3. Despite product 3D-4 being predominantly formed, the formation of 25% 4D-4 was also evident (Scheme 8). Therefore, we suggest that the occurrence of competitive mechanisms that involve enolization of the lactone and consequent side reactions might be a possible cause for the lower yields of the Ir-catalyzed redox isomerization.
Scheme 8. [Ir(cod)MeO]2/BiPy-Catalyzed Redox Isomerization of D-3.

The preference for the transposition of the hydrogen from the hemiacetal to the β-position of the enone was studied by means of density functional theory66 (DFT) calculations. The formation of a 16-electron complex is summarized in Figure 1, taking cation A as the starting point. The participation of such an intermediate is based on previous proposals67,68 on the complexation of allyl alcohols to [RuCp*(MeCN)3]PF6. Such a process can be achieved by a dissociative ligand exchange and the participation of a ruthenium-complexed product that assists in the deprotonation of the starting allyl alcohol (vide infra - Figure 4). The displacement of an acetonitrile ligand from A is a favorable process as the 16-electron ruthenium complex B becomes 11.0 kcal/mol more stable than the initial cation. A β-hydride elimination with an energy barrier of 14.5 kcal/mol results in the formation of 16-electron ruthenium complex D, which is slightly less stable (2.2 kcal/mol) than the previous tridentate complex B. The β-agostic intermediate C represents a mid-way geometry between the transition states involved in the β-hydride elimination. The more exigent basis set (B3LYP-cc-pVTZ, SMD) revealed intermediate C as a step in the uphill β-hydride elimination process, although the same intermediate was flanked by two more energetic transition states BC‡ and CD‡ at the gas phase B3LYP-cc-pVDZ. The intermediate nature of species C in the β-hydride elimination process is demonstrated by the elongation of the C–H bond of the allylic hydrogen (1.184 vs 1.108 Å in B) and the formation of the Ru–H bond (1.957 Å). This is also accompanied by the weakening of the C–H bond as demonstrated by the lower Wiberg index (0.89 in B and 0.72 in C) due to a stronger interaction by the metal center with the migrating hydrogen atom (WIRu-H = 0 in B and WIRu-H = 0.13 in C). The hydride elimination is accompanied by a growing interaction of the metal center with the carbon atom during its change in the oxidation state. Such an event is visible in the structure of the four-membered intermediate C as the interaction between the metal and the carbon atoms is described by a 0.16 WI even though they are distant by dRu-C = 2.419 Å. The β-hydride elimination process with the migration of the hydride to the metal becomes complete with D. This intermediate has the hydride placed 2.538 Å away from the newly established sp2 carbon (and a low 0.06 WI). The metal atom coordinates by η2 with the newly created C=O bond as demonstrated by the distances from the metal to each atom of the carbonyl group (dRu-O = 2.106 and dRu-C = 2.230 Å) and considerable Wiberg index (WIRu-O = 0.55 and dRu-C = 0.44).
Figure 1.
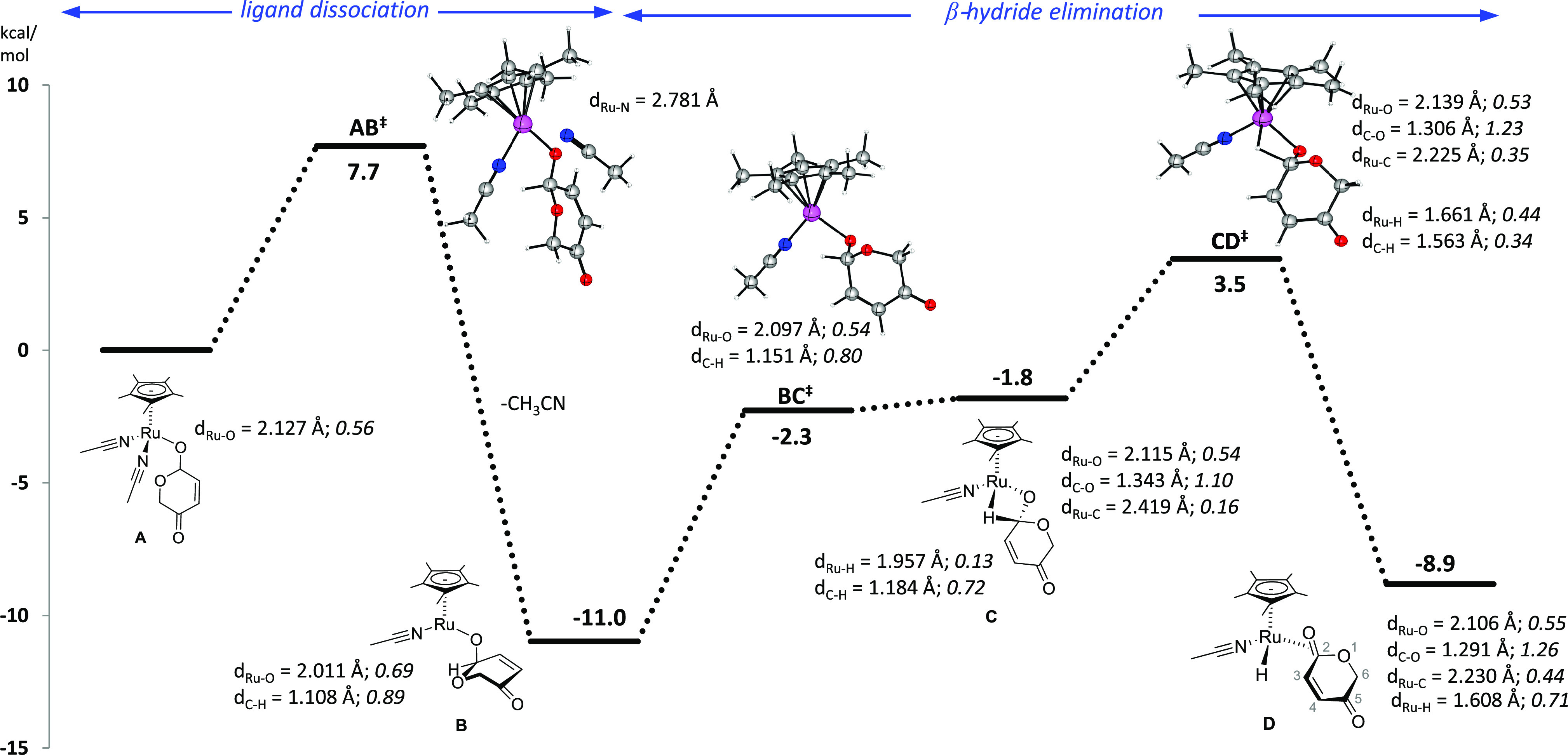
B3LYP/cc-pVTZ/SMD//B3LYP/cc-pVDZ relative free energies (kcal/mol) of stationary points along the β-hydride elimination pathway. Bond lengths (in Å) and Wiberg indexes (in italic) of relevant bonds are presented. The initial point (A) relates to the energy of the [RuCp*(MeCN)2alcohol]+ cation.
Figure 4.

B3LYP/cc-pVTZ/SMD//B3LYP/cc-pVDZ relative free energies (kcal/mol) of proton exchange from allyl alcohol and regeneration of the catalytic active species. Bond lengths (in Å) and Wiberg indexes (in italic) of relevant bonds are presented.
Other isomers of complex D that could form after decomplexation of the 5,6-dihydropyran-2,5-dione (DHPD) and further η2 coordination were optimized to identify more stable isomers (Figure 2). Indeed, the coordination of DHPD to the ruthenium center by the carbon–carbon π system leads to considerably more stable complexes (F1–F4) than the η2 coordination by the carbonyl π system (D and E) or by coordination to the oxygen lone pair (not shown). Although the natural charge of the metal-bonded hydrogen atom is kept the same in the D, E, and F isomers shown (qH = 0.11–0.18), the metal center becomes more negative when coordinated to the C=C bond (isomers D and E, qRu = −0.47 to −0.45) than when coordinated to the C=O bond (isomers F, qRu = −0.34 to −0.32). The stability of the complex upon coordination by the metal to the C=C bond was seen to be dependent on the orientation of DHPD to the pentamethylcyclopentadienyl unit. Geometries F2 and F4, both having co-planar DHPD and Cp* units, were determined to be 2.9–4.3 kcal/mol less stable than F1 and F3 in which the abovementioned rings are kept as far as possible.
Figure 2.

B3LYP/cc-pVTZ/SMD//B3LYP/cc-pVDZ relative free energies (kcal/mol) of stationary points for η2 complexes of RuCp*H(CH3CN) with DHPD. Bond lengths (in Å) and Wiberg indexes (in italic) of relevant bonds and selected atomic natural charges are presented.
Given the somewhat small differences in energies of the four isomers F1–F4, the reduction of DHPD by hydride delivery from the ruthenium complex was further studied (see the Supporting Information for further details). The two most energetically favorable paths are presented in Figure 3 in which conformer F1 is considered to undergo hydride delivery to the 4-position of DHPD and conformer F4 is to undergo hydride delivery to position 3. The energy barrier of the four-center transition state for the hydride delivery to the 3-position is 8.6 kcal/mol (G4) in contrast to the 13.7 kcal/mol required to reach intermediate G1. After complete delivery of the hydride to the α,β-unsaturated ketone, complex H4 is further stabilized in 1.6 kcal/mol upon bis-coordination of the ketone to the metal center in I4. The metal complex becomes even more stable as O-bound enolate J4 having a similar energy to F4. In contrast, the same type of coordination with the ester’s carbonyl group in I1 does not warrant any stabilization to complex H1. Moreover, the isomerization from the C-bound enolate of the ester moiety H1 to its O-bound enolate J1 is 13.1 kcal/mol higher in energy than η2 complex F1. An increase in the charge of the metal center during the hydride migration process indicates a change in the oxidation state of the metal as qRu changes from −0.46 in F4 to +0.04 in J4. The lack of stabilization in O-bound enolate J1 in comparison to its J4 congener is well in agreement with the observed isomerization of the isotopically labeled substrate.
Figure 3.

B3LYP/cc-pVTZ/SMD//B3LYP/cc-pVDZ relative free energies (kcal/mol) of stationary points along the pathways for the hydride delivery to the 3- and 4- positions of DHPD in black and gray, respectively. See the Supporting Information for further details and alternative pathways considered.
The release of the product occurs when another molecule of allyl alcohol is taken into the process (Figure 4). Such molecule coordinates to ruthenium by the hydroxyl group to form intermediate J′, which ultimately increases the proton lability and proton transfer to the oxygen atom of the O-bound enolate via transition state J′B′‡. Despite the energy demand for the approaching allyl alcohol to the metal complex, the proton transfer barrier is only 1.6 kcal/mol, leading to a more stable complex B′, which readily releases the enol of the final product, and complex B, which is ready to undergo β-hydride elimination and close the catalytic cycle.
Conclusions
In summary, we have presented a new practical intramolecular Ru-catalyzed redox isomerization of Achmatowicz derivatives, which allows easy access to a range of important lactones. In doing so, we provided a new high-yielding route to the biorenewable monomer 4-ketovalerolactone (KVL), thus paving the way for its larger use in the design of new polymers. Furthermore, we utilized our findings in the synthesis of several biologically significant lactones and developed a new concise protective-group-free asymmetric synthesis of two natural products, namely, muricatacin and L-factor. The selectivity of the process, namely, that concerning the preference for the delivery of the hydride to a specific position of the unsaturated heterocycle, was verified by isotopic labeling and rationalized by computational means.
Funding
This work received financial support from the
Bulgarian National
Science Fund (BNSF) under the National Scientific Program “VIHREN”
(grant no. KΠ-06- B-1) and from the PT national funds (FCT/MCTES, Fundação
para a Ciĉncia e Tecnologia and Ministério da Ciĉncia,
Tecnologia e Ensino Superior) through the project nos. UIDB/50006/2020,
UIDP/50006/2020, and PTDC/QUI-QOR/1131/2020. The project leading to
this application received funding from the European Union’s
Horizon 2020 research and innovation program under grant agreement
no. 951996.
B-1) and from the PT national funds (FCT/MCTES, Fundação
para a Ciĉncia e Tecnologia and Ministério da Ciĉncia,
Tecnologia e Ensino Superior) through the project nos. UIDB/50006/2020,
UIDP/50006/2020, and PTDC/QUI-QOR/1131/2020. The project leading to
this application received funding from the European Union’s
Horizon 2020 research and innovation program under grant agreement
no. 951996.
Acknowledgments
The authors acknowledge the National
Scientific
Program “VIHREN” (grant no. KΠ-06- B-1). N.R.C. thanks the FCT (Fundação
para a Ciĉncia e Tecnologia) for funding through the Scientific
Employment Stimulus (no. CEECINST/00026/2018). This work received
support from the PT national funds (FCT/MCTES, Fundação
para a Ciĉncia e Tecnologia and Ministério da Ciĉncia,
Tecnologia e Ensino Superior) through the project nos. UIDB/50006/2020,
UIDP/50006/2020, and PTDC/QUI-QOR/1131/2020. The CSC-IT Center for
Science Ltd., Finland, is acknowledged for the allocation of computational
resources. The authors are grateful to the INFRAMAT project (part
of the Bulgarian National Roadmap for research infrastructure, which
is supported by the Bulgarian Ministry of Education and Science) for
some of the research equipment that was used in this investigation.
Dr. Joao Ravasco is acknowledged for the HRMS analysis.
B-1). N.R.C. thanks the FCT (Fundação
para a Ciĉncia e Tecnologia) for funding through the Scientific
Employment Stimulus (no. CEECINST/00026/2018). This work received
support from the PT national funds (FCT/MCTES, Fundação
para a Ciĉncia e Tecnologia and Ministério da Ciĉncia,
Tecnologia e Ensino Superior) through the project nos. UIDB/50006/2020,
UIDP/50006/2020, and PTDC/QUI-QOR/1131/2020. The CSC-IT Center for
Science Ltd., Finland, is acknowledged for the allocation of computational
resources. The authors are grateful to the INFRAMAT project (part
of the Bulgarian National Roadmap for research infrastructure, which
is supported by the Bulgarian Ministry of Education and Science) for
some of the research equipment that was used in this investigation.
Dr. Joao Ravasco is acknowledged for the HRMS analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.2c04867.
Author Contributions
⊥ M.D. and A.F.-F. contributed equally. Conceptualization was done by S.P.S. Experimental methods were done by M.D., A.F.-F., M.A.R., E.V., M.K.M., and S.P.S. Computational methods were done by N.R.C. Funding acquisition was done by S.P.S. and N.R.C. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mariscal R.; Maireles-Torres P.; Ojeda M.; Sádaba I.; López Granados M. Furfural: a renewable and versatile platform molecule for the synthesis of chemicals and fuels. Energy Environ. Sci. 2016, 9, 1144–1189. 10.1039/C5EE02666K. [DOI] [Google Scholar]
- Li X.; Jia P.; Wang T. Furfural: a promising platform compound for sustainable production of C4 and C5 Chemicals. ACS Catal. 2016, 6, 7621–7640. 10.1021/acscatal.6b01838. [DOI] [Google Scholar]
- Lange J.-P.; van der Heide E.; van Buijtenen J.; Price R. Furfural – a promising platform for lignocellulosic biofuels. ChemSusChem 2012, 5, 150–166. 10.1002/cssc.201100648. [DOI] [PubMed] [Google Scholar]
- Yan K.; Wu G.; Lafleur T.; Jarvis C. Production, properties and catalytic hydrogenation of furfural to fuel additives and value-added chemicals. Renewable Sustainable Energy Rev. 2014, 38, 663–676. 10.1016/j.rser.2014.07.003. [DOI] [Google Scholar]
- Ma Y.; Vemula R.; Zhang Q.; Wu B.; O’Doherty G. A. Achmatowicz approach to the asymmetric synthesis of (+)- and (−)-monanchorin. Green Synth. Catal. 2022, 3, 156–161. 10.1016/j.gresc.2022.03.003. [DOI] [Google Scholar]
- Li N.; Zong M.-H. (Chemo)biocatalytic upgrading of biobased furanic platforms to chemicals, fuels, and materials: a comprehensive review. ACS Catal. 2022, 12, 10080–10114. 10.1021/acscatal.2c02912. [DOI] [Google Scholar]
- Bielski R.; Grynkiewicz G. Half a century with Achmatowicz rearrangement. Tetrahedron 2021, 85, 132058 10.1016/j.tet.2021.132058. [DOI] [Google Scholar]
- Ghosh A. K.; Brindisi M. Achmatowicz reaction and its application in the syntheses of bioactive molecules. RSC Adv. 2016, 6, 111564–111598. 10.1039/C6RA22611F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Tong R. Catalytic Environmentally friendly protocol for achmatowicz rearrangement. J. Org. Chem. 2016, 81, 4847–4855. 10.1021/acs.joc.6b00469. [DOI] [PubMed] [Google Scholar]
- Deska J.; Thiel D.; Gianolio E. The Achmatowicz rearrangement – oxidative ring expansion of furfuryl alcohols. Synthesis 2015, 47, 3435–3450. 10.1055/s-0035-1560345. [DOI] [Google Scholar]
- Achmatowicz O.; Bukowski P.; Szechner B.; Zwierzchowska Z.; Zamojski A. Synthesis of methyl 2,3-dideoxy-DL-alk-2-enopyranosides from furan compounds: A general approach to the total synthesis of monosaccharides. Tetrahedron 1971, 27, 1973–1996. 10.1016/S0040-4020(01)98229-8. [DOI] [Google Scholar]
- Simeonov S. P.; Ravutsov M. A.; Mihovilovic M. D. Biorefinery via achmatowicz rearrangement: synthesis of pentane-1,2,5-triol from furfuryl alcohol. ChemSusChem 2019, 12, 2748–2754. 10.1002/cssc.201900601. [DOI] [PubMed] [Google Scholar]
- Simeonov S. P.; Lazarova H. I.; Marinova M. K.; Popova M. D. Achmatowicz rearrangement enables hydrogenolysis-free gas-phase synthesis of pentane-1,2,5-triol from furfuryl alcohol. Green Chem. 2019, 21, 5657–5664. 10.1039/C9GC02888A. [DOI] [PubMed] [Google Scholar]
- Liang L.; Guo L.-D.; Tong R. Achmatowicz rearrangement-inspired development of green chemistry, organic methodology, and total synthesis of natural products. Acc. Chem. Res. 2022, 55, 2326–2340. 10.1021/acs.accounts.2c00358. [DOI] [PubMed] [Google Scholar]
- Kim S.; Oiler J.; Xing Y.; O’Doherty G. A. De novo asymmetric Achmatowicz approach to oligosaccharide natural products. Chem. Commun. 2022, 58, 12913–12926. 10.1039/D2CC05280F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong L.; Schill H.; Grange R. L.; Porzelle A.; Johns J. P.; Parsons P. G.; Gordon V. A.; Reddell P. W.; Williams C. M. Anticancer agents from the australian tropical rainforest: spiroacetals EBC-23, 24, 25, 72, 73, 75 and 76. Chem. – Eur. J. 2009, 15, 11307–11318. 10.1002/chem.200901525. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Talukdar A.; Zhang G.; Kedenburg J. P.; Wang P. G. A Divergent synthesis of uncommon sugars from furanaldehyde. Synlett 2005, 2005, 1547–1550. 10.1055/s-2005-869846. [DOI] [Google Scholar]
- Harris J. M.; Keränen M. D.; Nguyen H.; Young V. G.; O’Doherty G. A. Syntheses of four D- and L-hexoses via diastereoselective and enantioselective dihydroxylation reactions. Carbohydr. Res. 2000, 328, 17–36. 10.1016/S0008-6215(00)00031-8. [DOI] [PubMed] [Google Scholar]
- Croatt M. P.; Carreira E. M. Probing the role of the mycosamine C2′-OH on the activity of amphotericin B. Org. Lett. 2011, 13, 1390–1393. 10.1021/ol2000765. [DOI] [PubMed] [Google Scholar]
- Bartlett S.; Hodgson R.; Holland J. M.; Jones M.; Kilner C.; Nelson A.; Warriner S. Exploiting predisposition in the stereoselective synthesis of mono-, bi- and tetracyclic oxygen heterocycles: Equilibration between, and trapping of, alternative di- and tetraacetals. Org. Biomol. Chem. 2003, 1, 2393–2402. 10.1039/b303089j. [DOI] [PubMed] [Google Scholar]
- Georgiadis M. P.; Haroutounian S. A.; Couladouros E. A.; Apostolopoulos C. D.; Chondros K. P. Products from Furans. XVI . Novel synthetic routes to sympathomimetic amine analogues via 6-hydroxy-2H-pyran-3(6H)-ones. J. Heterocycl. Chem. 1991, 28, 697–703. 10.1002/jhet.5570280325. [DOI] [Google Scholar]
- Zhang X.-X.; Zhang Y.; Liao L.; Gao Y.; Su H. E. M.; Yu J.-S. Catalytic asymmetric isomerization of (homo)allylic alcohols: recent advances and challenges. ChemCatChem 2022, e202200126 10.1002/cctc.202200126. [DOI] [Google Scholar]
- Scalambra F.; Lorenzo-Luis P.; de los Rios I.; Romerosa A. Isomerization of allylic alcohols in water catalyzed by transition metal complexes. Coord. Chem. Rev. 2019, 393, 118–148. 10.1016/j.ccr.2019.04.012. [DOI] [Google Scholar]
- Liu T.-L.; Ng T. W.; Zhao Y. Rhodium-catalyzed enantioselective isomerization of secondary allylic alcohols. J. Am. Chem. Soc. 2017, 139, 3643–3646. 10.1021/jacs.7b01096. [DOI] [PubMed] [Google Scholar]
- Li H.; Mazet C. Iridium-catalyzed selective isomerization of primary allylic alcohols. Acc. Chem. Res. 2016, 49, 1232–1241. 10.1021/acs.accounts.6b00144. [DOI] [PubMed] [Google Scholar]
- Liu P. N.; Ju K. D.; Lau C. P. Highly efficient redox isomerization of allylic alcohols and transfer hydrogenation of ketones and aldehydes catalyzed by ruthenium complexes. Adv. Synth. Catal. 2011, 353, 275–280. 10.1002/adsc.201000667. [DOI] [Google Scholar]
- Cadierno V.; García-Garrido S. E.; Gimeno J.; Varela-Álvarez A.; Sordo J. A. Bis(allyl)–Ruthenium(IV) complexes as highly efficient catalysts for the redox isomerization of allylic alcohols into carbonyl compounds in organic and aqueous media: scope, limitations, and theoretical analysis of the mechanism. J. Am. Chem. Soc. 2006, 128, 1360–1370. 10.1021/ja054827a. [DOI] [PubMed] [Google Scholar]
- Georgiadis M. P.; Haroutounian S. A.; Apostolopoulos C. D. A convenient synthesis of 5-substituted 2-Pyrrolidinones via 2H-Pyran-3(6H)-ones. Synthesis 1991, 1991, 379–381. 10.1055/s-1991-26470. [DOI] [Google Scholar]
- Wang H.-Y.; Yang K.; Bennett S. R.; Guo S.-r.; Tang W. Iridium-catalyzed dynamic kinetic isomerization: expedient synthesis of carbohydrates from achmatowicz rearrangement products. Angew. Chem., Int. Ed. 2015, 54, 8756–8759. 10.1002/anie.201503151. [DOI] [PubMed] [Google Scholar]
- Liu Y.-C.; Merten C.; Deska J. Enantioconvergent biocatalytic redox isomerization. Angew. Chem., Int. Ed. 2018, 57, 12151–12156. 10.1002/anie.201804911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.-C.; Wu Z.-L.; Deska J. Coding synthetic chemistry strategies for furan valorization into bacterial designer cells. ChemSusChem 2022, e202201790 10.1002/cssc.202201790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Wang Y.; Hoye T. R. Poly(4-ketovalerolactone) from levulinic acid: synthesis and hydrolytic degradation. Macromolecules 2020, 53, 4952–4959. 10.1021/acs.macromol.0c00787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata A.; Hokimoto K.; Takemura T.; Katsuno T.; Yamamoto K. Plant constituents biologically active to insects. v. antifeedants for the larvae of the Yellow Butterfly, Eurema hecabe mandarina, in osmunda japonica. (1). Chem. Pharm. Bull. 1984, 32, 2815–2820. 10.1248/cpb.32.2815. [DOI] [PubMed] [Google Scholar]
- Cavé A.; Chaboche C.; Figadère B.; Harmange J. C.; Laurens A.; Peyrat J. F.; Pichon M.; Szlosek M.; Cotte-Lafitte J.; Quéro A. M. Study of the structure-activity relationships of the acetogenin of annonaceae, muricatacin and analogues. Eur. J. Med. Chem. 1997, 32, 617–623. 10.1016/S0223-5234(97)83287-4. [DOI] [Google Scholar]
- Rieser M. J.; Kozlowski J. F.; Wood K. V.; McLaughlin J. L. Muricatacin: A simple biologically active acetogenin derivative from the seeds of annona muricata (annonaceae). Tetrahedron Lett. 1991, 32, 1137–1140. 10.1016/S0040-4039(00)92027-6. [DOI] [Google Scholar]
- Wright A. E.; Schäfer M.; Midland S.; Munnecke D. E.; Sims J. J. Lateral root inducing compounds from the bacterium Erwinia quercina: Isolation, structure and synthesis. Tetrahedron Lett. 1989, 30, 5699–5702. 10.1016/S0040-4039(00)76174-0. [DOI] [Google Scholar]
- Gräfe U.; Reinhardt G.; Schade W.; Krebs D.; Eritt I.; Fleck W. F.; Heinrich E. Isolation and structure of novel autoregulators from Streptomyces Griseus. J. Antibiot. 1982, 35, 609–614. 10.7164/antibiotics.35.609. [DOI] [PubMed] [Google Scholar]
- Muller C. J.; Kepner R. E.; Webb A. D. Lactones in wines - a review. Am. J. Enol. Vitic. 1973, 24, 5. [Google Scholar]
- Muller C. J.; Maggiora L.; Kepner R. E.; Webb A. D. Identification of two isomers of 4,5-dihydroxyhexanoic acid gamma lactone in Californian and Spanish flor sherries. J. Agric. Food Chem. 1969, 17, 1373–1376. 10.1021/jf60166a013. [DOI] [Google Scholar]
- Hur J.; Jang J.; Sim J. A Review of the Pharmacological Activities and recent synthetic advances of γ-butyrolactones. Int. J. Mol. Sci. 2021, 22, 2769. 10.3390/ijms22052769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao B.; Fañanás-Mastral M.; Feringa B. L. Catalytic asymmetric synthesis of butenolides and butyrolactones. Chem. Rev. 2017, 117, 10502–10566. 10.1021/acs.chemrev.7b00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avedissian H.; Sinha S. C.; Yazbak A.; Sinha A.; Neogi P.; Sinha S. C.; Keinan E. Total synthesis of asimicin and bullatacin. J. Org. Chem. 2000, 65, 6035–6051. 10.1021/jo000500a. [DOI] [PubMed] [Google Scholar]
- Pearson W. H.; Hembre E. J. A practical synthesis of (−)-Swainsonine. J. Org. Chem. 1996, 61, 7217–7221. 10.1021/jo961101b. [DOI] [PubMed] [Google Scholar]
- Iwaki S.; Marumo S.; Saito T.; Yamada M.; Katagiri K. Synthesis and activity of optically active disparlure. J. Am. Chem. Soc. 1974, 96, 7842–7844. 10.1021/ja00832a055. [DOI] [Google Scholar]
- Gräfe U.; Eritt I. On the biological inactivity of 4, 5-dihydroxy-n-decanoic acid-4-lactones. J. Antibiot. 1983, 36, 1592–1593. 10.7164/antibiotics.36.1592. [DOI] [PubMed] [Google Scholar]
- Wang W.; Zhang R.; Wang J.; Tang J.; Wang M.; Kuang Y. Antitumour activity of muricatacin isomers and its derivatives in human colorectal carcinoma Cell HCT116. Anti-Cancer Agents Med. Chem. 2020, 20, 254–263. 10.2174/1871520619666191115111032. [DOI] [PubMed] [Google Scholar]
- Murcia M.; Navarro C.; Moreno A.; Csaky G. A. Naturally occurring δ-hydroxy-γ-lactones: muricatacins and related compounds. Curr. Org. Chem. 2010, 14, 15–47. 10.2174/138527210790226410. [DOI] [Google Scholar]
- Fernandes R. A.; Bhowmik A.; Choudhary P. Muricatacin, a gateway molecule to higher acetogenin synthesis. Chem. – Asian J. 2020, 15, 3660–3681. 10.1002/asia.202000955. [DOI] [PubMed] [Google Scholar]
- Fernandes R. A.; Gangani A. J.; Kumari A.; Kumar P. A Decade of muricatacin synthesis and beyond. Eur. J. Org. Chem. 2020, 2020, 6845–6858. 10.1002/ejoc.202000665. [DOI] [Google Scholar]
- Chaudhari D. A.; Ingle A. B.; Fernandes R. A. A concise synthesis of (4R,5R)-(−)-muricatacin and (4R,5R)-l-(−)-factor from d-glucono-δ-lactone. Tetrahedron: Asymmetry 2016, 27, 114–117. 10.1016/j.tetasy.2016.01.003. [DOI] [Google Scholar]
- Srećo B.; Benedeković G.; Popsavin M.; Hadžić P.; Kojić V.; Bogdanović G.; Divjaković V.; Popsavin V. Heteroannelated (+)-muricatacin mimics: synthesis, antiproliferative properties and structure–activity relationships. Tetrahedron 2011, 67, 9358–9367. 10.1016/j.tet.2011.09.132. [DOI] [Google Scholar]
- Ghosal P.; Kumar V.; Shaw A. K. A chiron approach to the total synthesis of cytotoxic (+)-muricatacin and (+)-5-epi-muricatacin from d-ribose. Carbohydr. Res. 2010, 345, 41–44. 10.1016/j.carres.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Dong H.-B.; Yang M.-Y.; Liu B.; Wang M.-A. Concise stereoselective total synthesis of (+)-muricatacin and (+)-epi-muricatacin. J. Asian Nat. Prod. Res. 2014, 16, 847–853. 10.1080/10286020.2014.916695. [DOI] [PubMed] [Google Scholar]
- Sabitha G.; Chandrashekhar G.; Vasudeva Reddy D.; Yadav S. J. Synthesis of (+)-(4S,5S)-Muricatacin via Pd-catalyzed stereospecific hydroxy substitution reaction of γ,δ-epoxy α,β-unsaturated ester with B(OH)3. Lett. Org. Chem. 2012, 9, 344–346. 10.2174/157017812801264656. [DOI] [Google Scholar]
- Ohkuma T.; Koizumi M.; Yoshida M.; Noyori R. General asymmetric hydrogenation of hetero-aromatic ketones. Org. Lett. 2000, 2, 1749–1751. 10.1021/ol0000814. [DOI] [PubMed] [Google Scholar]
- Fujii A.; Hashiguchi S.; Uematsu N.; Ikariya T.; Noyori R. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid–triethylamine mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. 10.1021/ja954126l. [DOI] [Google Scholar]
- Uma R.; Crévisy C.; Grée R. Transposition of allylic alcohols into carbonyl compounds mediated by transition metal complexes. Chem. Rev. 2003, 103, 27–52. 10.1021/cr0103165. [DOI] [PubMed] [Google Scholar]
- van der Drift R. C.; Bouwman E.; Drent E. Homogeneously catalysed isomerisation of allylic alcohols to carbonyl compounds. J. Organomet. Chem. 2002, 650, 1–24. 10.1016/S0022-328X(02)01150-6. [DOI] [Google Scholar]
- Hiroya K.; Kurihara Y.; Ogasawara K. Asymmetrization of meso 1,4-enediol ethers by isomerization with a chiral binap–RhI catalyst. Angew. Chem., Int. Ed. 1995, 34, 2287–2289. 10.1002/anie.199522871. [DOI] [Google Scholar]
- Bergens S. H.; Bosnich B. Homogeneous catalysis. Catalytic production of simple enols. J. Am. Chem. Soc. 1991, 113, 958–967. 10.1021/ja00003a032. [DOI] [Google Scholar]
- Kress S.; Johnson T.; Weisshar F.; Lautens M. Synthetic and mechanistic studies on the rhodium-catalyzed redox isomerization of cyclohexa-2,5-dienols. ACS Catal. 2016, 6, 747–750. 10.1021/acscatal.5b02387. [DOI] [Google Scholar]
- Batuecas M.; Esteruelas M. A.; García-Yebra C.; Oñate E. Redox isomerization of allylic alcohols catalyzed by osmium and ruthenium complexes containing a cyclopentadienyl ligand with a pendant amine or phosphoramidite group: X-ray structure of an η3-1-hydroxyallyl-metal-hydride intermediate. Organometallics 2010, 29, 2166–2175. 10.1021/om100126t. [DOI] [Google Scholar]
- Ahlsten N.; Martín-Matute B. Rhodium-catalysed coupling of allylic, homoallylic, and bishomoallylic alcohols with aldehydes and n-tosylimines: insights into the mechanism. Adv. Synth. Catal. 2009, 351, 2657–2666. 10.1002/adsc.200900448. [DOI] [Google Scholar]
- Cadierno V.; Crochet P.; Gimeno J. Ruthenium-catalyzed isomerizations of allylic and propargylic alcohols in aqueous and organic media: applications in synthesis. Synlett 2008, 2008, 1105–1124. 10.1055/s-2008-1072593. [DOI] [Google Scholar]
- van der Drift R. C.; Vailati M.; Bouwman E.; Drent E. Two reactions of allylic alcohols catalysed by ruthenium cyclopentadienyl complexes with didentate phosphine ligands: isomerisation and ether formation. J. Mol. Catal. A: Chem. 2000, 159, 163–177. 10.1016/S1381-1169(00)00221-1. [DOI] [Google Scholar]
- Parr R. G.; Weitao Y.. Density-Functional Theory of Atoms and Molecules. Oxford University Press: 1995, 1–333, 10.1093/oso/9780195092769.001.0001. [DOI] [Google Scholar]
- Trost B. M.; Kulawiec R. J. Chemoselectivity in the ruthenium-catalyzed redox isomerization of allyl alcohols. J. Am. Chem. Soc. 1993, 115, 2027–2036. 10.1021/ja00058a059. [DOI] [Google Scholar]
- Slugovc C.; Rüba E.; Schmid R.; Kirchner K. Improved efficiency of the ruthenium-catalyzed redox isomerization of allyl alcohols. Organometallics 1999, 18, 4230–4233. 10.1021/om990264y. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.