

Abstract
Background and Aims
Allopolyploidization is a widespread phenomenon known to generate novel phenotypes by merging evolutionarily distinct parental genomes and regulatory networks in a single nucleus. The objective of this study was to investigate the transcriptional regulation associated with phenotypic novelty in coffee beans of the allotetraploid Coffea arabica.
Methods
A genome-wide comparative transcriptomic analysis was performed in C. arabica and its two diploid progenitors, C. canephora and C. eugenioides. Gene expression patterns and homeologue expression were studied on seeds at five different maturation stages. The involvement of homeologue expression bias (HEB) in specific traits was addressed both by functional enrichment analyses and by the study of gene expression in the caffeine and chlorogenic acid biosynthesis pathways.
Key Results
Expression-level dominance in C. arabica seed was observed for most of the genes differentially expressed between the species. Approximately a third of the genes analysed showed HEB. This proportion increased during seed maturation but the biases remained equally distributed between the sub-genomes. The relative expression levels of homeologues remained relatively constant during maturation and were correlated with those estimated in leaves of C. arabica and interspecific hybrids between C. canephora and C. eugenioides. Functional enrichment analyses performed on genes exhibiting HEB enabled the identification of processes potentially associated with physiological traits. The expression profiles of the genes involved in caffeine biosynthesis mirror the differences observed in the caffeine content of mature seeds of C. arabica and its parental species.
Conclusions
Neither of the two sub-genomes is globally preferentially expressed in C. arabica seeds, and homeologues appear to be co-regulated by shared trans-regulatory mechanisms. The observed HEBs are thought to be a legacy of gene expression differences inherited from diploid progenitor species. Pre-existing functional divergences between parental species appear to play an important role in controlling the phenotype of C. arabica seeds.
Keywords: Allopolyploidy, Coffea arabica, gene expression, homeologue expression bias, caffeine, additivity, expression-level dominance, phenotype, desiccation tolerance, seed
INTRODUCTION
Polyploidy is a common and recurrent feature in plants, and is recognized as a fundamental mechanism in their evolution and diversification (Jiao et al., 2011; Wu et al., 2020; Qiu et al., 2020). Whereas autopolyploids result from genome duplication within a species, allopolyploids derive from hybridization between two or more species, and are composed of divergent homeologous sub-genomes, each inherited from parental species. Compared with diploid progenitors, allopolyploids often show phenotypic innovations with potentially adaptive physiological and ecological advantages (Ramsey and Schemske, 2002; Chen, 2010). Over the past two decades, numerous studies have been conducted of the consequences of allopolyploidy at the genomic and transcriptomic level (reviewed in Van de Peer et al., 2017; Bottani et al., 2018; Nieto Feliner et al., 2020). For instance, the expression level of duplicated genes can deviate from parental additivity (i.e. typically considered to be the arithmetic average of the parental expression levels). Non-additive expression has been widely observed in diverse polyploids and includes at least three possible scenarios (Yoo et al., 2014): (1) the total gene expression level in a polyploid is similar to that of one of its parents (expression-level dominance); (2) total gene expression is lower or higher than in both parents (transgressive expression); and (3) the relative contribution of the parental copies (homeologues) to total gene expression is unequal (homeologue expression bias, HEB).
Although studies on the topic are relatively rare, plant metabolism can be strongly influenced by polyploidy. Several biochemical studies have shown changes in biosynthetic pathways in polyploids compared with in their parental species, for example phenolic compounds in Spartina anglica (Grignon-Dubois et al., 2020), flavonoids in Nicotiana tabacum (McCarthy et al., 2017) and organic acids in Ponkan mandarin (Tan et al., 2019). Other studies in which both metabolomic and transcriptomic changes have been examined also report variations in temporal gene expression for oil and flavonoid biosynthesis in upland cotton (Hovav et al., 2015) and higher vitamin E biosynthesis in hexaploid oat (Gutierrez-Gonzalez and Garvin, 2016), which may or may not be related to homeologue-specific contributions.
Coffee is one of the most important agricultural commodities. Of the more than 125 Coffea species identified (Davis et al., 2006), two, C. arabica L. and C. canephora P., are widely cultivated and account for almost all the world’s coffee production. All Coffea species are diploid, with the exception of C. arabica which is allotetraploid (2n = 4x = 44) and originates from interspecific hybridization between two diploid species, C. eugenioides and C. canephora, through a single allopolyploidization event (Lashermes et al., 1999, 2016). Homeologous genomes in C. arabica are designated Ea and Ca according to their parental origin. Coffea arabica and its parental species are perennial woody trees and display considerable variation in morphology, size and ecological adaptation. In contrast to C. arabica and C. eugenioides, which are found in highland environments, C. canephora is better adapted to warm and humid equatorial lowlands (Davis et al., 2006; DaMatta and Ramalho, 2006). Concerning the biochemical composition of the seeds, the concentrations of coffee-specific secondary metabolites vary significantly in C. arabica and its extant parental species, particularly chlorogenic acids (CGAs) and caffeine (Ky et al., 2001; Campa et al., 2005), two compounds that contribute to the bitterness of the coffee beverage. In C. arabica, the amount of CGA detected in its seeds (4.1 % dry matter) is close to that of C. eugenioides (5.2 %) and much lower than that detected in C. canephora (11.3 %), whereas the caffeine content in C. arabica seeds (around 1.2 % dry matter) is intermediate between those observed in C. eugenioides (0.5 %) and C. canephora (2.5 %). In addition to the accumulation of secondary metabolites, the seeds of C. arabica and its parental species vary considerably in desiccation tolerance (DT), which is acquired late in the seed maturation process (Dussert et al., 2000, 2018). Coffea arabica seeds display a relatively high level of DT, almost the same as that observed for C. eugenioides seeds, while C. canephora seeds display a low level of DT. Candidate genes and processes related to DT acquisition have recently been identified using interspecific comparative transcriptomics (Stavrinides et al., 2020).
Several recent studies explored the genomic and transcriptomic changes associated with the formation and diversification of the allotetraploid C. arabica species. Evidence was found for genome modifications that could have played a major role in the stabilization and survival of the ancestral allotetraploid and in its subsequent diversification (Lashermes et al., 2014, 2016). While the early phase of evolution mainly involved crossover exchanges between homeologous chromosomes, the later phase appears to have relied on more gradual duplicate gene evolution involving gene conversion and homeologue silencing. Gene expression patterns were also investigated in leaves of C. arabica. Compared with the expression profiles of its parental species, some genes show expression dominance, and their proportion seems to be modulated by the growing conditions of the trees (Bardil et al., 2011; Combes et al., 2013). However, neither of the two sub-genomes is preferentially expressed, and overall gene expression in C. arabica appears to be regulated by intertwined mechanisms (Combes et al., 2013). Based on the analysis of interspecific Coffea hybrids, Combes et al. (2015) reported that gene expression inheritance patterns and, in particular, expression level dominance are determined by regulatory divergences between parental alleles. Furthermore, differential contributions of homeologous genes in response to abiotic stress have been observed (Marraccini et al., 2011; Ferreira de Carvalho et al., 2013). However, although it is of considerable economic importance due to its involvement in cup quality (Joët and Dussert, 2018), the transcriptomic regulation associated with the biochemical characteristics of C. arabica bean remains largely unexplored.
The objective of this study was thus to evaluate the contribution of changes in gene expression to phenotypic novelty in coffee beans of the allopolyploid C. arabica. We performed a genome-wide comparative transcriptomic analysis in C. arabica and its two parental species. Gene expression patterns as well as homeologue expression were studied on seeds at five different maturation stages. The question of whether HEBs target genes involved in specific metabolic functions and regulatory processes was addressed both at the transcriptome-wide level through functional enrichment analyses and at the pathway level by focusing on the well-characterized caffeine and CGA biosynthetic pathways. The results provide novel insights into the patterns of gene expression associated with polyploidy in plants and the factors that govern shifts in gene expression. Among the possible mechanisms by which new phenotypes may arise, we highlight the effects of pre-existing functional divergences between parental species for key C. arabica traits such as caffeine content and the acquisition of DT during late seed maturation.
MATERIALS AND METHODS
Plant material
Seeds at five different development stages (ST3, ST4, ST5, ST6 and ST7; see Fig. 1) were collected as previously described (Stavrinides et al., 2020) from trees of the three species C. arabica, C. canephora and C. eugenioides grown in the International Coffea Collection (Saint-Pierre, Reunion Island). The developmental stages were selected based on marked anatomical and morphological traits of seeds and fruits that are shared across coffee species, as defined and described previously for C. arabica (Joët et al., 2009; Dussert et al., 2018). Briefly, the endosperm develops rapidly at stage 3, and growth ends during stage 4 when oil starts to accumulate. Stage 5 is the peak of reserve deposition and corresponds to endosperm hardening due to massive deposition of galactomannans in cell walls. Stage 6 coincides with fruit veraison and the end of the accumulation of reserves; finally, fruit and seed maturity are completed at stage 7 when the pericarp becomes red. In order to minimize the impact of genotypic effects, to facilitate interspecies comparisons and to optimize the recovery of plant material, especially in the early stages, seeds of each species were collected from different wild accessions and considered as biological replicates (C. arabica accessions AR36b-05, AR52-05, AR61-05 and AR38-05 for ST3 and ST4; AR28-06, AR02-06 and AR38b/05 for ST5, ST6 and ST7; C. canephora accessions BD 54 and BD 66 for ST3 and ST4; BD55, BD56 and DAF71 for ST5, ST6 and ST7; and C. eugenioides accessions DA71, DA78 and DA78c for all five stages).
Fig. 1.

Coffee seed development. Schematic representation of seed development stages in the upper panel (adapted from Dussert et al., 2018) and, in the lower panel, cross-sections of fruits at the different developmental stages studied.
RNA extraction and RNA sequencing
For all samples in the experimental design, a mix of >20 endosperms was ground to a fine powder, while frozen, in an analytical grinder (IKA A10, Staufen, Germany) and total RNA was extracted from 70 mg using the Qiagen RNeasy Lipid Tissue kit (Qiagen, Stanford, CA, USA). The quality and concentration of extracted RNAs were determined using the Agilent DNA 1000 (Agilent, Santa Clara, CA, USA). cDNA libraries were constructed using the TruSeq™ Stranded mRNA sample preparation kit (Illumina, USA) and sequenced on an Illumina HiSeq 2500 (single reads, 100 nt) at the MGX platform (Montpellier GenomiX, http://www.mgx.cnrs.fr/). After quality filtering using Cutadapt (quality score > Q30 and removal of reads shorter than 60 bp or longer than 140 bp), a total of 443, 320 and 411 million reads were retained for C. arabica, C. canephora and C. eugenioides, respectively (average of 29, 24 and 31 million reads per library for C. arabica, C. canephora and C. eugenioides, respectively). The entire dataset was deposited at the European Nucleotide Archive (ENA) under the project number PRJEB32533. In addition, previously available sequence datasets obtained from mature leaves were used for comparison. These RNA-seq data include samples of C. arabica (Lashermes et al., 2016; ENA project PRJEB5543) as well as samples of C. canephora (four accessions), C. eugenioides (four accessions) and from 11 F1 diploid interspecific hybrid plants resulting from crosses between C. canephora and C. eugenioides (Combes et al., 2015; ENA project PRJEB7565). The leaf data were processed as described above.
RNA-seq data processing
Owing to the low genetic divergence between the three Coffea species (average of 1.3 % gene sequence difference; Cenci et al., 2012), the trimmed reads of each library were all mapped to the C. canephora coding transcriptome DNA reference sequence (25574 CDS) (Denoeud et al., 2014) using BWA MEM (Li, 2013) with the default parameters. Reads were counted using IDXstats in SAMtools (Li et al., 2009). The aligned sequences of each seed library and leaf library were then analysed with the GATK toolkit (http://www.broadinstitute.org/gatk/) using the Unified Genotyper module with default parameters to identify SNPs (single nucleotide polymorphisms), and the Depth Of Coverage module to obtain information on depth coverage. To avoid artefacts due to reads from pseudogenes or repeat sequences, only CDS identified as single copy were used for subsequent analyses. Similarly, genes identified as having undergone DNA exchange between homeologous chromosomes in C. arabica (Lashermes et al., 2016) were excluded from the study. The biallelic SNPs detected in leaf and seed samples of the different accessions of both parental species, C. canephora and C. eugenioides, were compared. The SNPs for which the two groups of accessions of diploid parental species appeared homozygous for differences were retained. In this way, a list of 108 527 diagnostic SNPs (i.e. species specific) representing 12 860 CDS was produced.
Inheritance classification
Expression inheritance in C. arabica was determined at different seed development stages. Only genes with minimum cumulated read counts (depending on the number of replicates, >10 reads per replicate for at least one species) were considered (22 165 genes). In order to calculate ratios and perform statistical analyses, 0 mapped reads were changed to 1 (Marioni et al., 2008). Data were normalized with respect to library size with the DESeq2 package (using ‘varianceStabilizingTransformation’ and ‘VST’ as arguments; Love et al., 2014). Log-transformed expression values of parental diploid species and C. arabica were compared to examine changes in expression. As described by McManus et al. (2010), genes whose total expression in C. arabica deviated >1.25-fold from that of either parental species were considered to have non-conserved inheritance. Based on the magnitude and the direction of the changes, the genes were classified as displaying additivity (with E expression lower or higher than C expression), E or C expression level dominance (lower or higher than either parental species: down and up) and transgressivity (lower or higher than both parental species).
Estimation of homeologous gene expression
Using the established diagnostic SNP list, the reads of C. arabica were sorted into Ca or Ea homeologous bins using custom Perl scripts. Genes were discarded if >5 % of their reads showed inconsistencies in homeologous assignments (i.e. discrepancies between diagnostic SNPs for a given read, observed in <1 % of genes), if they showed high inter-replicate variation in their homeologue expression ratio or if they had <30 reads per replicate (cumulative numbers of C-specific and E-specific reads). Gene expression data for C. arabica homeologue replicates and parental species replicates were normalized using the DESeq2 package as previously described. The homeologous gene expression corresponding to homeologue-specific read counts (Ca or Ea) of the total read counts (Ca + Ea) was expressed as the percentage of the Ca homoelogue (% Ca) in the total gene expression of C. arabica. At each stage, data were treated as described and the successive stages were compared two by two. Based on the observation of highly significant correlation coefficients in the homeologous expression ratio dataset (Supplementary data Fig. S1; Pearson’s correlation between ST3 and ST4 equal to 0.97, P-value < 2.2 × 10–16; Pearson’s correlation between ST6 and ST7 equal to 0.98, P-value < 2.2 × 10–16), the data from two first (ST3–ST4) and two late stages (ST6–ST7) were combined by averaging homeologous gene expression estimations for further analysis.
Functional enrichment analysis
For MapMan analysis (Usadel et al., 2009), the lists of genes displaying Ca or Ea HEBs, the lists of genes displaying C up and E up expression dominance as well as the entire list of expressed genes at each developmental stage (background) were used for bin enrichment. Genes were assigned to a MapMan bin structure using Mercator (Lohse et al., 2014). A MapMan bin was classified as enriched for a given gene list if the number of genes belonging to that bin was statistically higher than expected from the background, using a two-tailed Fisher’s exact test, and adjusted for multiple testing using Benjamini–Hochberg correction.
RESULTS
Variation in gene expression dominance during seed development in C. arabica
Gene expression in C. arabica was compared with that of its two parental species at three stages of seed development for 22 165 genes. Based on the relative level of their expression in the three species, the genes were classified in eight categories: no difference, additive, transgressive up and down, E dominant up and down, and C dominant up and down (Table 1). The category of genes for which no significant difference in expression was found between the three species was by far the largest (i.e. ranging from 66.7 % to 70.5 % of genes) and varied significantly between maturation stages (P-value < 2.2 × 10–16). Among differentially expressed genes, the proportions of genes classified in both additive and transgressive categories were relatively stable whatever the stage considered (1.8–2.1, 3.9–4.2 and 2.5–2.8 %, for ST3–S4, ST5 and ST6–ST7, respectively). In contrast, the proportion of genes classified in E- and C-dominant categories involving dominance (i.e. gene expression level in C. arabica identical to one of the parental species) varied significantly between stages. The relative importance of the different categories involving dominance varied during the development of the seeds, but no particular trend was observed. In addition, the possibility that the variations observed in gene expression during seed development were the result of differences between the accessions used for each developmental stage was considered. Although unlikely, this scenario was possible in comparisons involving the ST3–ST4 stage. However, it can be completely ruled out in comparisons between stages ST5 and ST6–ST7 which involved the same combinations of accessions.
Table 1.
Gene expression levels in C. arabica compared with its parental species during seed maturation for 22 165 genes
| Expression pattern | ST3–ST4 | ST5 | ST6–ST7 | P-value | |
|---|---|---|---|---|---|
| No difference | 15 632 (70.5 %) | 15 067 (68.0 %) | 14 792 (66.7 %) | <2.2 × 10–16 | |
| Additive |
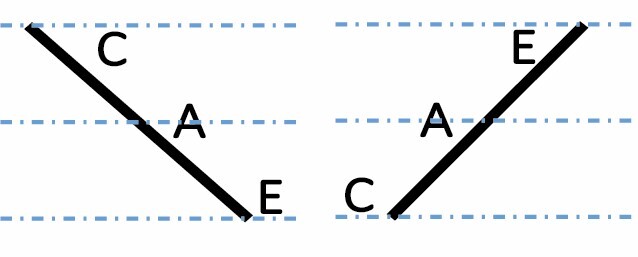
|
402 (1.8 %) | 441 (2.0 %) | 443 (2.1 %) | 0.2805 |
| Transgressive up |
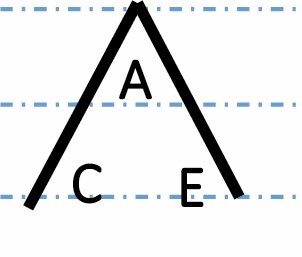
|
939 (4.2 %) | 854 (3.9 %) | 900 (4.1 %) | 0.1222 |
| Transgressive down |
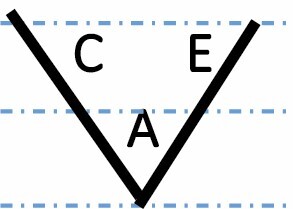
|
563 (2.6 %) | 635 (2.8 %) | 546 (2.5 %) | 0.0198 |
| E dominant up |
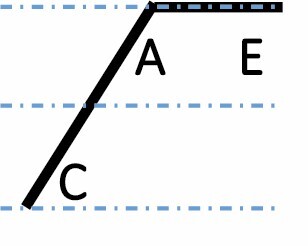
|
1048 (4.7 %) | 905 (4.1 %) | 921 (4.2 %) | 0.0012 |
| E dominant down |
|
1234 (5.6 %) | 1480 (6.7 %) | 1679 (7.5 %) | <2.2 × 10–16 |
| C dominant up |
|
1308 (5.9 %) | 1110 (5.0 %) | 1540 (6.9 %) | <2.2 × 10–16 |
| C dominant down |
|
1039 (4.7 %) | 1673 (7.5 %) | 1344 (6.1 %) | <2.2 × 10–16 |
A, C and E correspond to C. arabica, C. canephora and C. eugenioides, respectively. Differential gene expression between the three species is indicated by the dashed-dotted lines for each gene expression category (gene expression in C. arabica deviated >1.25-fold from that of either parental species). The proportions of each category were compared between maturation stages (prop.test, 0.01 as the significance threshold P-value).
Variations in homeologous gene expression patterns during C. arabica seed development
The relative expression level of homeologues was compared for 4604 genes expressed both during seed maturation and in leaves of C. arabica and interspecific hybrids between C. canephora and C. eugenioides. The genes were classified according to the percentage of Ca homeologue relative expression in ten categories ranging from 0–10 % to 90–100 % of the total gene expression (Fig. 2). While the average percentage of Ca homeologues was almost stable (50.2, 50.2 and 50.5 % in stages ST3–ST4, ST5 and ST6–ST7, respectively), the patterns of gene distribution differed significantly between the stages of seed maturation (χ 2 test = 272.07, d.f. = 318, P-value < 2.2 × 10–16). The proportion of genes exhibiting a HEB increased steadily during seed development (31.6 % for ST3–ST4, 36.6 % for ST5 and 42.0 % for ST6–ST7), and was always higher than the proportions observed in leaves in both C. arabica and the interspecific hybrids between C. canephora and C. eugenioides (pairwise prop.test between seed maturation stages and C. arabica or interspecific hybrids leaves, P-value < 2.2 × 10–16 in six tests). However, whatever the organ considered (seed or leaf) and the source of the plant material (C. arabica or interspecific hybrids between C. canephora and C. eugenioides), for genes showing HEB, equivalent proportions of genes were observed between genes preferentially expressed towards the Ca or Ea sub-genomes (prop.test P-value > 0.01 in the five tests).
Fig. 2.

Comparison of relative homeologous gene expression distributions for 4604 genes expressed both in C. arabica developing seeds (ST3–ST4 green, ST5 yellow, ST6–ST7 blue), and in leaves of C. arabica (light grey) and of Coffea hybrids (dark grey) (χ 2 test = 802, d.f. = 36, P-value < 2.2 × 10–16).
To complete comparisons between the seed and leaf datasets, the relative homeologous gene expression estimated at each seed maturation stage was compared with that observed in the leaves of C. arabica (Supplementary data Fig. S2). A highly significant correlation was observed at all seed stages (Pearson’s correlation, P-value < 2.2 × 10–16 in the three comparisons) with, however, a decrease in the correlation coefficient during seed maturation (0.60, 0.52 and 0.45 for ST3–ST4, ST5 and ST6–ST7, respectively). This decrease in the correlation coefficient is consistent with the increase in the proportion of HEB during seed development described above.
The relative expression of homologous genes was compared between successive stages of seed development (Fig. 3A). Highly significant correlation coefficients (Pearson’s correlation, P-value < 2.2 × 10–16) were observed: 0.7 and 0.9 for ST3–ST4 vs. ST5 and ST5 vs. ST6–ST7, respectively. As the cis-regulatory elements of the two sub-genomes are shared between the different stages of seed development, the modalities of action of the trans-regulatory factors can be assessed by comparing the genes whose total expression varies significantly between successive stages of seed development (ST3–ST4 vs. ST5 and ST5 vs. ST6–ST7). The relative expression level of homeologues was significantly correlated between stages (Pearson’s correlation, P-value < 2.2 × 10–16). with correlation coefficients of 0.77 and 0.93 for ST3–ST4 vs. ST5 and ST5 vs. ST6–ST7 respectively (Fig. 3B). The expression of homeologues thus appears to be mostly co-regulated during seed development.
Fig. 3.

(A) Comparison of relative homeologous gene expression between successive stages of maturation (Pearson’s correlation, P-value < 2.2 × 10–16, correlation coefficient of 0.70, 0.90 for 6269 genes between ST3–ST4 and ST5 and 7147 genes between ST5 and ST6–ST7, respectively). (B) For divergently expressed genes between successive maturation stages, comparison of homeologous gene expression ratios (Pearson’s correlation, P-value < 2.2 × 10–16, V0.77, 0.93 for 1004 genes between ST3–ST4 and ST5 and 277 genes between ST5 and ST6–ST7, respectively).
Relationship between homeologous gene expression and parental inheritance
The relationship between homeologous gene expression and patterns of gene expression relative to parental species (i.e. parental inheritance) was also investigated. At ST6–ST7 (the developmental stage with the highest proportion of genes exhibiting HEB), the distribution according to the relative expression level of homeologues of the genes included in the C- and E-dominant up and down categories was compared with that of all the genes studied (Fig. 4) (pairwise comparisons, χ 2 test, P-value < 2.2 × 10–16 for the four comparisons). The proportion of genes with HEB increased substantially among the genes classified in C- and E-dominant up and down categories. The expression of homeologues appeared to be biased towards Ea in the E-dominant up and C-dominant down categories and towards Ca in the C-dominant up and E-dominant down categories.
Fig. 4.

Comparison of homeologous gene expression distributions of C. arabica genes classified in C and E dominant up and down categories at ST6–ST7 with homeologous gene expression distribution of 8180 genes expressed at this seed maturation stage (χ 2 test = 741.72, d.f. = 9, P-value < 2.2 × 10–16 for E dominant up category, χ 2 test = 144.35, d.f. = 9, P-value < 2.2 × 10–16 for E dominant down category, χ 2 test = 584.49, d.f. = 9, P-value < 2.2 × 10–16 for C dominant up category and χ 2 test = 151.61, d.f. = 9, P-value < 2.2 × 10–16 for C dominant down category). The patterns of gene expression for the four categories considered are shown above the corresponding distribution.
Candidate genes were previously identified as positive and negative effectors of coffee seed desiccation tolerance (Clusters 1 and 2, respectively, described in Stavrinides et al., 2020). These two clusters contain genes displaying, respectively, E up and E down expression dominance during late developmental stages in C. arabica seeds. The distribution of genes according to the relative expression level of homeologues was also analysed among these candidate genes. The expression of DT-associated genes within mature C. arabica seeds mainly relied on that of the Ea sub-genome, while the expression levels observed for negative effectors of DT were mostly attributable to the Ca sub-genome (Supplementary data Fig. S3).
Metabolic functions and regulatory processes potentially affected by the divergent mobilization of the two sub-genomes
The question of the functional consequences of the homeologue transcriptional biases observed towards one parental sub-genome, i.e. the potential recruitment of biologically relevant transcriptional modules specific to a parental species, was addressed using MapMan enrichment analyses performed on groups of Ea- and Ca-biased genes at different developmental stages, as well as on genes displaying conserved bias throughout seed development (Supplementary data Table S1).
Enriched functions associated with genes displaying Ca bias throughout seed development included major functions related to the biosynthesis of amino acids (shikimate, glutamate and aspartate) and sugars (raffinose and trehalose), as well as photosynthesis-related processes (chlorophyll breakdown and interconversion, xanthophyll biosynthesis) and serine-type peptidase activities. Enriched functions associated with genes displaying Ca bias at specific stages pointed to phytohormones and the biosynthesis of auxin. It is worth noting that most of these functions were also detected to be significantly enriched when analyses were performed on the sub-groups of Ca-biased genes that display C up expression dominance at ST5 and ST6–ST7 (Supplementary data Table S1). Concerning phytohormones, enriched functions were related to auxin biosynthesis at ST5 (both indole-3-pyruvic acid and indole-3-acetamide biosynthetic pathways) as well as conjugation/degradation of jasmonate (ST5 and ST6–ST7). Significant enrichment was detected at ST5 for photosynthesis-related processes such as linear electron flow activity, photosystem II components, chlorophyll breakdown and interconversion, and carotenoid biosynthesis.
The main enriched functions associated with genes displaying Ea bias throughout seed development, each represented by several terms, concerned RNA processing and solute transport, while several other specific terms were associated with stress response, such as urease (involved in amino acid catabolism and nitrogen recycling), the plastid NADH dehydrogenase-like complex (involved in cyclic electron flow around photosystem I), PLD-α (phospholipase D; which catalyses the hydrolysis of phospholipids to phosphatidic acid, and is involved in stress signalling) as well as plasma membrane protein cold-responsive protein kinase which senses changes in membrane rigidity and permeability (Liu et al., 2017). Some of these functions, such as those related to urease and solute transport, were also observed to be significantly enriched when analyses were performed on the subgroups of Ea-biased genes that display E up expression dominance at ST3–ST4 and ST5 (Supplementary data Table S1).
Caffeine metabolism
A detailed comparison of gene expression among the three coffee species and a study of homeologue expression bias in C. arabica were conducted at the level of metabolic pathways focusing first on caffeine biosynthesis (Fig. 5). Caffeine is a purine alkaloid synthesized from xanthosine as initial substrate (Ashihara et al., 2008). Xanthosine is the main product of purine nucleotide catabolism and, in higher plants, predominantly derives from guanosine deamination through the activity of a specific guanosine deaminase (GSDA; Dahncke and Witte, 2013; Baccolini and Witte, 2019). Subsequently, the caffeine biosynthetic pathway is a four-step sequence involving one nucleosidase and three methylation reactions, for which the three specific genes and enzymes XMT, MXMT and DXMT N-methyl transferases (NMTs) have been characterized in either Arabica or Robusta coffee (Ogawa et al., 2001; Mizuno et al., 2003; McCarthy and McCarthy, 2007). In our transcriptome survey, the known caffeine biosynthetic genes (GSDA, XMT1, MXMT1 and DXMT1) exhibited similar expression patterns in developing seeds of the three species, i.e. a large expression peak was observed at ST4 at the beginning of the storage phase in the endosperm, followed by a sharp decrease in transcript levels at later stages (Fig. 5). At early stages, expression levels were significantly and recurrently higher in C. canephora than in C. eugenioides, with the largest differences (up to 4-fold at stage 4) observed for xanthosine methyltransferase (XMT1) and caffeine synthase (DXMT1). With the exception of MXMT1, overall transcript levels for C. arabica genes were intermediate between those of the parental species, and the differences in expression levels measured in the three species reflect the differences observed at the metabolite level for caffeine content in mature seeds. In addition, a marked homeologue expression bias was observed for GSDA, XMT1 and DXMT1 genes, with overall expression primarily associated with that of the Ca homeologue (Fig. 5). Indeed, the Ca homeologue of XMT1 was 20-fold more expressed than its Ea counterpart at early stages (ST3–ST4), whereas the Ca homeologue of the key caffeine synthase DXMT1 gene was 6-fold more expressed than the Ea homeologue at ST4, and up to 250-fold at ST3. For those two key caffeine biosynthetic enzymes, the difference in gene expression observed between the parental species was smaller than that noted between Ca and Ea homeologues, suggesting the quasi-silencing of the Ea homeologue and the specific expression of the Ca homeologue in C. arabica seeds.
Fig. 5.

Gene expression profiles for the caffeine biosynthetic pathway. Expression profiles for caffeine biosynthetic genes in C. arabica, C. canephora, and C. eugenioides (A), as well as allele-specific expression of Ca and Ea homeologue pairs in C. arabica (B). The x-axis represents seed developmental stages in chronological order and the y-axis represents the gene expression level as normalized read counts. DXMT1 (Cc01g00720), 3,7-dimethylxanthine methyltransferase (caffeine synthase); GSDA (Cc02g31500), guanosine deaminase; MXMT1 (Cc00g24720), 7-methylxanthine methyltransferase (theobromine synthase); NMN, N-methylnucleosidase; SAH, S-adenosyl-l-homocysteine; SAM, S-adenosyl-l-methionine; XMT1 (Cc09g06970), xanthosine methyltransferase.
Chlorogenic acid metabolism
A detailed pathway analysis of gene expression patterns and homeologue expression was also performed for phenylpropanoid genes involved in CGA biosynthesis (Fig. 6). The three first steps of 5-caffeoylquinic acid (5-CQA) biosynthesis involve the well-characterized enzymes of the ‘core phenylpropanoid pathway’, namely phenylalanine ammonia lyase (PAL), trans-cinnamate 4-hydroxylase (C4H) and 4-coumarate-CoA ligase (4CL). Subsequently, the main route involves two supplementary enzymatic steps: esterification of quinic acid on p-coumaroyl-CoA by C3′H (p-coumaroyl CoA 3-hydroxylase; Mahesh et al., 2007), and hydroxylation of p-coumaroyl quinate by HQT (hydroxycinnamoyl-CoA quinate hydroxycinnamoyl transferase; Niggeweg et al., 2004; Lallemand et al., 2012). An alternative route may require C3H, as recently described in the arabidopsis model plant (Barros et al., 2019), for direct hydroxylation of p-coumaric acid into caffeic acid, followed by 4CL and HQT enzymatic steps. Our study illustrates the tight co-ordination of gene expression of most of the key enzymes involved in the biosynthesis of CGA in early seed development, including PAL4, C4H, 4CL1, C3′H1 and C3H (Fig. 6), with the timing of expression patterns being very similar for all those genes in the three species. Potentially involved in 5-caffeoylquinic acid (5-FQA) biosynthesis, COMT1 and CCoAOMT1 genes also revealed very similar expression profiles. Finally, HQT, which is involved in both 5-CQA and 5-FQA biosynthesis as well as 3,5-diCQA synthesis (Lallemand et al., 2012), displayed a bimodal expression pattern with maximal expression at stages 3 and 6 and could be linked to CGA remobilization during the late maturation stage. Although the final concentration of CGA varies in a spectacular way in the mature seeds of the three coffee species, it is worth noting that the expression level of each of these CGA biosynthetic genes was quantitatively equivalent in the three species. The few differences observed were for PAL4, whose highest expression level was observed in seeds of C. canephora, and for 4CL1, whose expression was maximal in both C. canephora and C. arabica. In C. arabica, gene expression was balanced between the two parental homeologues for the vast majority of genes studied. Exceptions concerned PAL1, in which the Ca homeologue was twice as highly expressed as Ea, and HQT, with the Ea homeologue displaying up to 3-fold higher expression than its Ca counterpart. In addition, the parental origin of the dominant homeologue differed between PAL1 and HQT genes. The absence of any difference in gene expression between parental species and/or homeologues has also been observed for key genes involved in quinic acid synthesis and CGA remobilization, such as QDH (quinate dehydrogenase, Cc03g0247; Guo et al., 2014), F5H1 and CCR1 genes (ferulate 5-hydroxylase, Cc07g10360; and cinnamoyl-CoA reductase, Cc04g05040, respectively) (data not shown). At the level of gene and homeologue regulation, our results offer no clear molecular explanation for the differences observed in CGA biosynthesis between the three coffee species.
Fig. 6.

Gene expression profiles for phenylpropanoid genes and for the chlorogenic acid biosynthetic pathway. Expression profiles for putative biosynthetic genes in C. arabica, C. canephora and C. eugenioides (left panel), as well as allele-specific expression of Ca and Ea homeologue pairs in C. arabica (right panel). The x-axis represents seed developmental stages in chronological order and the y-axis represents the gene expression level as normalized read counts. C3H, 4-coumarate 3-hydroxylase; C3′H, p-coumaroyl CoA 3-hydroxylase; C4H, trans-cinnamate 4-hydroxylase; CCoAOMT, caffeoyl-CoA 3-O-methyltransferase; 4CL, 4-coumarate-CoA ligase; COMT, caffeic acid O-methyltransferase; CSE, caffeoyl shikimate esterase; HCT, hydroxycinnamoyl-CoA:shikimate/quinate hydroxycinnamoyl transferase; HQT, hydroxycinnamoyl-CoA quinate hydroxycinnamoyl transferase; PAL, phenylalanine ammonia lyase.
DISCUSSION
Allopolyploidization is a biological process that has played a major role in plant speciation and evolution. In particular, it is a widespread phenomenon known to generate novel phenotypes by merging evolutionarily distinct parental genomes and regulatory networks into a single nucleus (Nieto Feliner et al., 2020). The evolution and fate of duplicated gene loci in allopolyploid plants has become the subject of intensive study (Qiu et al., 2020). However, the mechanisms involved in the new gene expression patterns observed in allopolyploids as well as their effects on phenotype such as metabolic pathways are still poorly understood. In the present study, we considered C. arabica coffee beans as a model to study both the gene regulatory patterns associated with allopolyploidy and the gene expression profiles related to specific physiological/chemical traits.
For the majority of genes (around 70 %) expressed in the coffee bean, no significant difference in the level of total gene expression was observed between C. arabica and its two parental species, C. canephora and C. eugenioides. Among the genes whose expression levels differ in the three species, the majority were genes with non-additive regulation. In fact, most of the differentially expressed genes showed an expression level dominance characterized by a total gene expression level in C. arabica similar to that of one of its parents. However, in contrast to the observations made in leaves from interspecific hybrids between C. canephora and C. eugenioides (Combes et al., 2015), the cases of dominance in C. arabica seed were equally concerned with up- and downregulation, and their direction was balanced between the two parents. The absence of biased expression level dominance suggests balanced differences in the effects of the two parental trans-regulatory factors in C. arabica seeds. Furthermore, the non-additive gene regulation observed in C. arabica seeds appears to be developmentally sensitive. Indeed, an increase in dominance situations was observed during development and in particular at the later stages of maturation (i.e. comparison between stages ST5 and ST6–ST7) for which the same combination of tree accessions was compared, which excludes the possibility that the differences observed were the result of differences between accessions.
In allopolyploids, two homeologues of a gene can contribute equally or unequally to total gene expression, the latter case reflecting a homeologue expression bias (HEB). In C. arabica seed, about two-thirds of the genes analysed showed a balanced contribution of homeologues. This proportion of HEB is quite close to that previously estimated for C. arabica leaves (Combes et al., 2013). Moreover, whether in seeds or in leaves, the HEBs observed in C. arabica concern both the Ca and Ea sub-genomes, with neither sub-genome being preferentially expressed overall. Contrasting situations have been reported in other allopolyploid species. For example, in Brachypodium hybridum, similar proportions (approx. 60 %) of genes showed balanced homeologue expression in root and leaf tissues (Takahagi et al., 2018). In Tragopogon miscellus, the proportions of genes showing HEB were shown to vary between leaf and inflorescence, with a slight bias towards the sub-genome T. pratensis in both organs (Buggs et al., 2010; Boatwright et al., 2018; Shan et al., 2020). In hexaploid wheat, Triticum aestivum, balanced homeologue expression also varies between organs (approx. 63 % in the stigma to approx. 79 % in roots; Ramírez-González et al., 2018). Recent data (Khan et al., 2022) suggested that sub-genome biases are characteristic features of the Brassica napus seed throughout development, and that such bias might not be universal across the embryo, endosperm and seed coat of the developing seed.
It is interesting to note that in C. arabica seed, the proportion of genes exhibiting HEB increased from 32 % to 42.0 % during maturation. This shift in the frequency of genes exhibiting HEB during seed development has already been observed in other plants such as cotton and wheat (Hovav et al., 2015; Xiang et al., 2019). Furthermore, in a study targeting vitamin E biosynthesis genes in developing hexaploid oat seeds, the homeologues were predominantly expressed almost equally; however, expression bias was observed in some genes, some of which appeared to be key points in the regulation of vitamin E synthesis (Gutierrez-Gonzalez and Garvin, 2016).
Further analysis of the expression of homeologues provided information on how they are regulated in C. arabica seeds. In particular, strong conservation of the relative expression level of homeologues was observed for genes whose total expression level changes during seed maturation. Both homeologues appeared to be either over- or under-co-regulated by trans-regulatory factors and without bias towards either sub-genome. In addition, for the same set of genes, the relative expression levels of homeologues estimated in C. arabica seed were highly correlated with those estimated in leaves of C. arabica and interspecific hybrids between C. canephora and C. eugenioides, suggesting a strong parental legacy. In fact, gene expression in allopolyploids is thought to be due to many controlling factors acting separately or in concert. One of these factors is parental legacy, or the extent to which differences in gene expression between duplicate copies in an allopolyploid are the legacy of expression differences inherited from the progenitor diploid species (Yoo et al., 2013; Buggs et al., 2014). As already suggested based on observations in leaves (Combes et al., 2013), C. arabica homeologues are most probably regulated by intertwined mechanisms of cis- and trans-elements from both parents. In particular, the low sequence divergence between the two parental genomes is hypothesized to facilitate cross-talk between the parental copies of trans-elements.
The strong relationship we observed between expression level dominance and HEB in C. arabica seed is fully consistent with this interpretation. In agreement with previous studies that investigated both homeologous and total gene expression in allopolyploids (Yoo et al., 2013; Cox et al., 2014; Combes et al., 2015), the present data confirmed that biased expression level dominance toward one homeologue was mainly caused by up- or downregulation of the other homeologue. Since HEB and expression level dominance result from modulation of cis- and/or trans-regulatory elements, one can assume that these mechanisms are directly related to the evolutionary history of the parental species and to the evolution of cis- and/or trans-regulations of gene expression. In this context, the observed increase in the proportion of genes displaying HEB and in the divergence of the relative expression level of homeologues during C. arabica seed maturation is hypothesized to be the result of potentially adaptive divergence between the two parental species during the late stages of seed maturation.
We also investigated the functional consequences of transcriptional biases of observed homeologues towards a parental sub-genome, i.e. the potential recruitment of biologically relevant transcriptional modules specific to a parental species. Functional enrichment analysis performed on genes displaying either Ea or Ca bias was a way to identify processes potentially associated with physiological traits close to those observed in one parental species, such as seed desiccation tolerance in C. eugenioides. Indeed, enrichment analysis of Ea-biased genes pointed to key terms associated with stress response, including small heat shock proteins targeted to mitochondria (sHSP-M) and two key stress-related factors, PLDα and cold-responsive protein kinase (CRPK), associated with the fluidity of the plasma membrane. CRPK1 is a plasma membrane protein kinase that has been shown to sense changes in membrane rigidity and permeability during cold stress in arabidopsis (Liu et al., 2017). Phospholipase D activity is involved in the production of phosphatidic acid, a negatively charged phospholipid whose small head group influences membrane conformation, and PLDα1 has been recently identified as a key modulator of seed desiccation sensitivity in arabidopsis (Chen et al., 2018). Finally, sHSP-M has been recently shown to interact with a factor required for cytochrome c maturation, and to be involved in the fine-tuning of respiratory electron transfer chain activity during the development and germination of arabidopsis seed (Ma et al., 2019). Since membrane lipids have been reported to be a main site of damage in desiccation-sensitive coffee seeds and as the lack of co-ordinated repression of metabolism and mitochondrial respiration during drying is thought to play a predominant role in the oxidative burst that triggers lipid oxidation and membrane disruption (Dussert et al., 2006; Stavrinides et al., 2020), genes associated with these functions are interesting candidates to be functionally tested for their potential pivotal role in acquisition of partial desiccation tolerance in C. arabica and C. eugenioides seeds. Concerning Ca-biased genes, significant enrichment was detected for several terms associated with biosynthetic pathways of the phytohormone auxin, a key modulator of cell division and elongation as well as cellular metabolism during seed development and maturation (Cao et al., 2020; Winnicki, 2020; Verma et al., 2021). While many quantitative trait loci for seed size have been identified in various plants as being associated with genes involved in auxin biosynthesis, transport and signalling (Cao et al., 2020), the role of Ca auxin biosynthetic homeologues in the size of C. arabica seeds, which are bigger than the seeds of C. eugenioides, remains to be elucidated. Enriched functions associated with Ca-biased genes also revealed terms associated with energy metabolism, including mitochondrial oxidative phosphorylation, and many plastid-related processes such as chlorophyll metabolism, linear electron flow activity, photosystem II component and carotenoid biosynthesis. These functions, which are associated with the active cell metabolism encountered during early seed maturation, were also recently identified as being specifically repressed during late maturation of DT seeds of C. eugenioides and C. arabica, compared with desiccation-sensitive C. canephora seeds (Stavrinides et al., 2020). For these genes, the low expression levels observed during late maturation stages in C. arabica, intermediate between both extant parental progenitors, relies on that of the Ca sub-genome. A comparative transcriptomic study of seeds in C. arabica and parental species C. eugenioides and C. canephora has revealed a common transcriptional programme between the three species, but also, in some cases, divergences in gene regulation between C. canephora on the one hand and C. eugenioides and C. arabica on the other (Stavrinides et al., 2020).
With the aim of characterizing the allopolyploid phenotype in relation to parental divergence, parental and homeologous gene expression was also detailed for specific biosynthetic pathways. The purine alkaloid caffeine content in C. arabica seeds is intermediate between those observed in parental species, and additive inheritance for this chemical trait has been observed in interspecific hybrids (Barre et al., 1998). Our study is the first comprehensive transcriptional study of caffeine biosynthetic genes including total gene expression profiles throughout seed development in the three species, as well as the relative contributions of homeologue genes in C. arabica. Differences in caffeine biosynthetic gene expression between C. arabica and C. canephora have already been reported (Koshiro et al., 2006; Perrois et al., 2015). Our work not only confirms these differences but demonstrates that for three genes, namely GSDA, XMT and DXMT, the expression levels observed in C. arabica seeds are intermediate between those observed in the two parental species, the highest expression levels being observed in C. canephora seeds. These variations in gene expression mirror the difference observed in caffeine content of mature seeds of the three species, suggesting that tight transcriptional control of caffeine biosynthetic genes during early endosperm development plays a major role in regulating the accumulation of this metabolite. HEB was noted for those three genes, with the predominant expression of the Ca homeologue version. Concerning XMT and DXMT genes, we noted the quasi-silencing of Ea homeologues and specific recruitment of the Ca homeologues, with potentially major consequences for caffeine biosynthesis related to functional divergences of the encoded enzymes. Indeed, the two homeologous versions of DXMT have already been cloned and their recombinant proteins functionally characterized in Escherichia coli (Mizuno et al., 2003; Uefuji et al., 2003). Significant differences in their catalytic properties have been demonstrated, including their affinity for the substrate, theobromine, which has been shown to be 7-fold higher in the Ca version of DXMT (CCS1, AB086414, XM027206542), with its Km value being 157 µm (Mizuno et al., 2003), compared with the Ea-encoded enzyme (CaDXMT1, AB084125, XM027217280) whose Km value was around 1200 µm (Uefuji et al., 2003).
In contrast to the caffeine biosynthetic pathway, our results provide no clear molecular explanation for the differences in CGA biosynthesis observed between the three coffee species at the level of gene and homeologue regulation. Only faint HEB was detected for PAL1 and HQT. Such a discrepancy between the regulation of CGA-related gene expression and CGA accumulation suggests that major regulatory processes exist at the post-transcriptional level for this important seed chemical trait.
Conclusions
Neither sub-genome is preferentially expressed overall in C. arabica seeds, and total gene expression levels result from the expression of both homeologues with interoperability of parental regulatory networks. Furthermore, the HEB observed in one-third of the genes in C. arabica seeds appears to be a legacy of differences in gene expression inherited from the diploid progenitor species. Thus, we hypothesize that gene expression profiles in C. arabica depend on cis and/or trans functional divergences that occurred during the evolutionary history of its parental species. Functional enrichment analysis performed on genes exhibiting HEB enabled us to identify processes potentially associated with physiological traits. The expression profiles of the genes involved in caffeine biosynthesis appear to be consistent with the differences in the caffeine content of mature seeds of C. arabica and its parental species, suggesting that tight transcriptional control of caffeine biosynthetic genes during early endosperm development plays a major role in regulating the accumulation of this metabolite. We suggest that pre-existing transcriptional divergences between parental species play an important role in establishing phenotypic novelty in an allopolyploid species such as C. arabica.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Figure S1: comparison of homeologous gene expression between successive maturation stages. Figure S2: comparison of homeologous gene expression between leaves and seeds during maturation. Figure S3: comparison of homeologous gene expression distributions for candidate gene clusters related to desiccation tolerance and genes expressed at late seed maturation stages. Table S1: MAPMAN functional enrichment analysis of genes displaying homeologue expression bias during seed development.
ACKNOWLEDGEMENTS
The authors acknowledge the Coffea Biological Resources Center (BRC Coffea, maintained by IRD and CIRAD in Reunion Island) for providing the plant material used in this study. The authors would also like to thank Nathan Baschenis for providing photographs of the different stages of coffee seed development. This work was supported by funding from the Agropolis Fondation ‘GenomeHarvest’ project (ID 1504-006) through the French ‘Investissements d’avenir’ programme (Labex Agro:ANR-10-LABX-0001-01). The authors acknowledge the ISO 9001-certified IRD itrop HPC (member of the South Green Platform) at IRD Montpellier for providing HPC resources that contributed to the research results reported within this paper (https://bioinfo.ird.fr).
Contributor Information
Marie-Christine Combes, DIADE, Univ Montpellier, CIRAD, IRD, Montpellier, France.
Thierry Joët, DIADE, Univ Montpellier, CIRAD, IRD, Montpellier, France.
Anna K Stavrinides, DIADE, Univ Montpellier, CIRAD, IRD, Montpellier, France.
Philippe Lashermes, DIADE, Univ Montpellier, CIRAD, IRD, Montpellier, France.
LITERATURE CITED
- Ashihara H, Sano H, Crozier A. 2008. Caffeine and related purine alkaloids: biosynthesis, catabolism, function and genetic engineering. Phytochemistry 69: 841–856. doi: 10.1016/j.phytochem.2007.10.029. [DOI] [PubMed] [Google Scholar]
- Baccolini C, Witte C-P. 2019. AMP and GMP catabolism in Arabidopsis converge on xanthosine, which is degraded by a nucleoside hydrolase heterocomplex. The Plant Cell 31: 734–751. doi: 10.1105/tpc.18.00899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardil A, de Almeida JD, Combes MC, Lashermes P, Bertrand B. 2011. Genomic expression dominance in the natural allopolyploid Coffea arabica is massively affected by growth temperature. New Phytologist 192: 760–774. doi: 10.1111/j.1469-8137.2011.03833.x. [DOI] [PubMed] [Google Scholar]
- Barre P, Akaffou S, Louarn J, Charrier A, Hamon S, Noirot M. 1998. Inheritance of caffeine and heteroside contents in an interspecific cross between a cultivated coffee species Coffea liberica var dewevrei and a wild species caffeine-free C. pseudozanguebariae. Theoretical and Applied Genetics 96: 306–311. doi: 10.1007/s001220050741. [DOI] [Google Scholar]
- Barros J, Escamilla-Trevino L, Song L, et al. 2019. 4-Coumarate 3-hydroxylase in the lignin biosynthesis pathway is a cytosolic ascorbate peroxidase. Nature Communications 10: 1994. doi: 10.1038/s41467-019-10082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatwright JL, McIntyre LM, Morse AM, et al. 2018. A robust methodology for assessing differential homeolog contributions to the transcriptomes of allopolyploids. Genetics 210: 883–894. doi: 10.1534/genetics.118.301564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottani S, Zabet NR, Wendel JF, Veitia RA. 2018. Gene expression dominance in allopolyploids: hypotheses and models. Trends in Plant Science 23: 393–402. doi: 10.1016/j.tplants.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Buggs RJA, Chamala S, Wu W, et al. 2010. Characterization of duplicate gene evolution in the recent natural allopolyploid Tragopogon miscellus by next-generation sequencing and Sequenom iPLEX MassARRAY genotyping. Molecular Ecology 19: 132–146. doi: 10.1111/j.1365-294X.2009.04469.x. [DOI] [PubMed] [Google Scholar]
- Buggs RJA, Wendel JF, Doyle JJ, Soltis DE, Soltis PS, Coate JE. 2014. The legacy of diploid progenitors in allopolyploid gene expression patterns. Philosophical Transactions of the Royal Society B: Biological Sciences 369: 20130354. doi: 10.1098/rstb.2013.0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campa C, Doulbeau S, Dussert S, Hamon S, Noirot M. 2005. Qualitative relationship between caffeine and chlorogenic acid contents among wild Coffea species. Food Chemistry 93: 135–139. doi: 10.1016/j.foodchem.2004.10.015. [DOI] [Google Scholar]
- Cao J, Li G, Qu D, Li X, Wang Y. 2020. Into the seed: auxin controls seed development and grain yield. International Journal of Molecular Sciences 21: 1662. doi: 10.3390/ijms21051662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci A, Combes M-C, Lashermes P. 2012. Genome evolution in diploid and tetraploid Coffea species as revealed by comparative analysis of orthologous genome segments. Plant Molecular Biology 78: 135–145. doi: 10.1007/s11103-011-9852-3. [DOI] [PubMed] [Google Scholar]
- Chen ZJ. 2010. Molecular mechanisms of polyploidy and hybrid vigor. Trends in Plant Science 15: 57–71. doi: 10.1016/j.tplants.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Yu X, Zhang X, et al. 2018. Phospholipase Dα1-mediated phosphatidic acid change is a key determinant of desiccation-induced viability loss in seeds. Plant, Cell & Environment 41: 50–63. doi: 10.1111/pce.12925. [DOI] [PubMed] [Google Scholar]
- Combes M-C, Dereeper A, Severac D, Bertrand B, Lashermes P. 2013. Contribution of subgenomes to the transcriptome and their intertwined regulation in the allopolyploid Coffea arabica grown at contrasted temperatures. New Phytologist 200: 251–260. doi: 10.1111/nph.12371. [DOI] [PubMed] [Google Scholar]
- Combes M-C, Hueber Y, Dereeper A, Rialle S, Herrera J-C, Lashermes P. 2015. Regulatory divergence between parental alleles determines gene expression patterns in hybrids. Genome Biology and Evolution 7: 1110–1121. doi: 10.1093/gbe/evv057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MP, Dong T, Shen G, Dalvi Y, Scott DB, Ganley ARD. 2014. An interspecific fungal hybrid reveals cross-kingdom rules for allopolyploid gene expression patterns. PLoS Genetics 10: e1004180. doi: 10.1371/journal.pgen.1004180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahncke K, Witte C-P. 2013. Plant purine nucleoside catabolism employs a guanosine deaminase required for the generation of xanthosine in Arabidopsis. The Plant Cell 25: 4101–4109. doi: 10.1105/tpc.113.117184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaMatta FM, Ramalho JDC. 2006. Impacts of drought and temperature stress on coffee physiology and production: a review. Brazilian Journal of Plant Physiology 18: 55–81. doi: 10.1590/s1677-04202006000100006. [DOI] [Google Scholar]
- Davis AP, Govaerts R, Bridson DM, Stoffelen P. 2006. An annotated taxonomic conspectus of the genus Coffea (Rubiaceae). Botanical Journal of the Linnean Society 152: 465–512. doi: 10.1111/j.1095-8339.2006.00584.x. [DOI] [Google Scholar]
- Denoeud F, Carretero-Paulet L, Dereeper A, et al. 2014. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345: 1181–1184. doi: 10.1126/science.1255274. [DOI] [PubMed] [Google Scholar]
- Dussert S, Chabrillange N, Engelmann F, Anthony F, Louarn J, Hamon S. 2000. Relationship between seed desiccation sensitivity, seed water content at maturity and climatic characteristics of native environments of nine Coffea L. species. Seed Science Research 10: 293–300. doi: 10.1017/s0960258500000337. [DOI] [Google Scholar]
- Dussert S, Davey MW, Laffargue A, Doulbeau S, Swennen R, Etienne H. 2006. Oxidative stress, phospholipid loss and lipid hydrolysis during drying and storage of intermediate seeds. Physiologia Plantarum 127: 192–204. doi: 10.1111/j.1399-3054.2006.00666.x. [DOI] [Google Scholar]
- Dussert S, Serret J, Bastos-Siqueira A, et al. 2018. Integrative analysis of the late maturation programme and desiccation tolerance mechanisms in intermediate coffee seeds. Journal of Experimental Botany 69: 1583–1597. doi: 10.1093/jxb/erx492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira de Carvalho J, Poulain J, Da Silva C, et al. 2013. Transcriptome de novo assembly from next-generation sequencing and comparative analyses in the hexaploid salt marsh species Spartina maritima and Spartina alterniflora (Poaceae). Heredity 110: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignon-Dubois M, De Montaudouin X, Rezzonico B. 2020. Flavonoid pattern inheritance in the allopolyploid Spartina anglica – comparison with the parental species S. maritima and S. alterniflora. Phytochemistry 174: 112312. doi: 10.1016/j.phytochem.2020.112312. [DOI] [PubMed] [Google Scholar]
- Guo J, Carrington Y, Alber A, Ehlting J. 2014. Molecular characterization of quinate and shikimate metabolism in Populus trichocarpa. Journal of Biologicaa Chemistry 289: 23846–23858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Gonzalez JJ, Garvin DF. 2016. Subgenome-specific assembly of vitamin E biosynthesis genes and expression patterns during seed development provide insight into the evolution of oat genome. Plant Biotechnology Journal 14: 2147–2157. doi: 10.1111/pbi.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovav R, Faigenboim-Doron A, Kadmon N, et al. 2015. A transcriptome profile for developing seed of polyploid cotton. The Plant Genome 8: eplantgenome2014–eplantge.08.0041. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, et al. 2011. Ancestral polyploidy in seed plants and angiosperms. Nature 473: 97–100. doi: 10.1038/nature09916. [DOI] [PubMed] [Google Scholar]
- Joët T, Dussert S. 2018. Environmental and genetic effects on coffee seed biochemical composition and quality. In: Lashermes P, ed. Achieving sustainable cultivation of coffee. Cambridge: Burleigh Dodds Science Publishing, 49–68. [Google Scholar]
- Joët T, Laffargue A, Salmona J, et al. 2009. Metabolic pathways in tropical dicotyledonous albuminous seeds: Coffea arabica as a case study. New Phytologist 182: 146–162. doi: 10.1111/j.1469-8137.2008.02742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan D, Ziegler DJ, Kalichuk JL, et al. 2022. Gene expression profiling reveals transcription factor networks and subgenome bias during Brassica napus seed development. The Plant Journal 109: 477–489. [DOI] [PubMed] [Google Scholar]
- Koshiro Y, Zheng X-Q, Wang M-L, Nagai C, Ashihara H. 2006. Changes in content and biosynthetic activity of caffeine and trigonelline during growth and ripening of Coffea arabica and Coffea canephora fruits. Plant Science 171: 242–250. doi: 10.1016/j.plantsci.2006.03.017. [DOI] [Google Scholar]
- Ky C-L, Louarn J, Dussert S, Guyot B, Hamon S, Noirot M. 2001. Caffeine, trigonelline, chlorogenic acids and sucrose diversity in wild Coffea arabica L. and C. canephora P. accessions. Food Chemistry 75: 223–230. doi: 10.1016/s0308-8146(01)00204-7. [DOI] [Google Scholar]
- Lallemand LA, Zubieta C, Lee SG, et al. 2012. A structural basis for the biosynthesis of the major chlorogenic acids found in coffee. Plant Physiology 160: 249–260. doi: 10.1104/pp.112.202051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashermes P, Combes M-C, Robert J, et al. 1999. Molecular characterisation and origin of the Coffea arabica L. genome. Molecular and General Genetics 261: 259–266. doi: 10.1007/s004380050965. [DOI] [PubMed] [Google Scholar]
- Lashermes P, Combes M-C, Hueber Y, Severac D, Dereeper A. 2014. Genome rearrangements derived from homoeologous recombination following allopolyploidy speciation in coffee. The Plant Journal 78: 674–685. doi: 10.1111/tpj.12505. [DOI] [PubMed] [Google Scholar]
- Lashermes P, Hueber Y, Combes M-C, Severac D, Dereeper A. 2016. Inter-genomic DNA exchanges and homeologous gene silencing shaped the nascent allopolyploid coffee genome (Coffea arabica L.). G3: Genes|Genomes|Genetics 6: 2937–2948. doi: 10.1534/g3.116.030858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997. [Preprint]. [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Jia Y, Ding Y, et al. 2017. . Plasma membrane CRPK1-mediated phosphorylation of 14-3-3 proteins induces their nuclear import to fine-tune CBF signaling during cold response. Molecular Cell 66: 117–128. doi: 10.1016/j.molcel.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Lohse M, Nagel A, Herter T, et al. 2014. Mercator: a fast and simple web server for genome scale functional annotation of plant sequence data. Plant, Cell & Environment 37: 1250–1258. doi: 10.1111/pce.12231. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15: 550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Guan X, Li J, et al. 2019. Mitochondrial small heat shock protein mediates seed germination via thermal sensing. Proceedings of the National Academy of Sciences, USA 116: 4716–4721. doi: 10.1073/pnas.1815790116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahesh V, Million-Rousseau R, Ullmann P, et al. 2007. Functional characterization of two p-coumaroyl ester 3'-hydroxylase genes from coffee tree: evidence of a candidate for chlorogenic acid biosynthesis. Plant Molecular Biology 64: 145–159. [DOI] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. 2008. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Research 18: 1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraccini P, Freire LP, Alves GS, et al. 2011. RBCS1 expression in coffee: Coffea orthologs, Coffea arabica homeologs, and expression variability between genotypes and under drought stress. BMC Plant Biology 11: 85. doi: 10.1186/1471-2229-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy AA, McCarthy JG. 2007. The structure of two N-methyltransferases from the caffeine biosynthetic pathway. Plant Physiology 144: 879–889. doi: 10.1104/pp.106.094854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy EW, Berardi AE, Smith SD, Litt A. 2017. Related allopolyploids display distinct floral pigment profiles and transgressive pigments. American Journal of Botany 104: 92–101. doi: 10.3732/ajb.1600350. [DOI] [PubMed] [Google Scholar]
- McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ. 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Research 20: 816–825. doi: 10.1101/gr.102491.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K, Okuda A, Kato M, et al. 2003. Isolation of a new dual-functional caffeine synthase gene encoding an enzyme for the conversion of 7-methylxanthine to caffeine from coffee (Coffea arabica L.). FEBS Letters 534: 75–81. doi: 10.1016/s0014-5793(02)03781-x. [DOI] [PubMed] [Google Scholar]
- Nieto Feliner G, Casacuberta J, Wendel JF. 2020. Genomics of evolutionary novelty in hybrids and polyploids. Frontiers in Genetics 11: 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggeweg R, Michael AJ, Martin C. 2004. Engineering plants with increased levels of the antioxidant chlorogenic acid. Nature Biotechnology 22: 746–754. doi: 10.1038/nbt966. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Herai Y, Koizumi N, Kusano T, Sano H. 2001. 7-Methylxanthine methyltransferase of coffee plants. Gene isolation and enzymatic properties. Journal of Biological Chemistry 276: 8213–8218. doi: 10.1074/jbc.M009480200. [DOI] [PubMed] [Google Scholar]
- Perrois C, Strickler SR, Mathieu G, et al. 2015. Differential regulation of caffeine metabolism in Coffea arabica (Arabica) and Coffea canephora (Robusta). Planta 241: 179–191. doi: 10.1007/s00425-014-2170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu T, Liu Z, Liu B. 2020. The effects of hybridization and genome doubling in plant evolution via allopolyploidy. Molecular Biology Reports 47: 5549–5558. doi: 10.1007/s11033-020-05597-y. [DOI] [PubMed] [Google Scholar]
- Ramírez-González RH, Borrill P, Lang D, et al. 2018. The transcriptional landscape of polyploid wheat. Science 361: eaar6089. [DOI] [PubMed] [Google Scholar]
- Ramsey J, Schemske DW. 2002. Neopolyploidy in flowering plants. Annual Review of Ecology and Systematics 33: 589–639. doi: 10.1146/annurev.ecolsys.33.010802.150437. [DOI] [Google Scholar]
- Shan S, Boatwright JL, Liu X, et al. 2020. Transcriptome dynamics of the inflorescence in reciprocally formed allopolyploid Tragopogon miscellus (Asteraceae). Frontiers in Genetics 11: 888. doi: 10.3389/fgene.2020.00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrinides AK, Dussert S, Combes M-C, et al. 2020. Seed comparative genomics in three coffee species identify desiccation tolerance mechanisms in intermediate seeds. Journal of Experimental Botany 71: 1418–1433. doi: 10.1093/jxb/erz508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahagi K, Inoue K, Shimizu M, Uehara-Yamaguchi Y, Onda Y, Mochida K. 2018. Homoeolog-specific activation of genes for heat acclimation in the allopolyploid grass Brachypodium hybridum. GigaScience 7: giy020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan F-Q, Zhang M, Xie K-D, et al. 2019. Polyploidy remodels fruit metabolism by modifying carbon source utilization and metabolic flux in Ponkan mandarin (Citrus reticulata Blanco). Plant Science 289: 110276. [DOI] [PubMed] [Google Scholar]
- Uefuji H, Ogita S, Yamaguchi Y, Koizumi N, Sano H. 2003. Molecular cloning and functional characterization of three distinct N-methyltransferases involved in the caffeine biosynthetic pathway in coffee plants. Plant Physiology 132: 372–380. doi: 10.1104/pp.102.019679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel B, Poree F, Nagel A, Lohse M, Czedik-Eysenberg A, Stitt M. 2009. A guide to using MapMan to visualize and compare Omics data in plants: a case study in the crop species, Maize. Plant, Cell & Environment 32: 1211–1229. doi: 10.1111/j.1365-3040.2009.01978.x. [DOI] [PubMed] [Google Scholar]
- Van de Peer Y, Mizrachi E, Marchal K. 2017. The evolutionary significance of polyploidy. Nature Reviews. Genetics 18: 411–424. [DOI] [PubMed] [Google Scholar]
- Verma S, Attuluri VPS, Robert HS. 2021. An essential function for auxin in embryo development. Cold Spring Harbor Perspectives in Biology 13: a039966. doi: 10.1101/cshperspect.a039966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnicki K. 2020. The winner takes it all: auxin – the main player during plant embryogenesis. Cells 9: 606. doi: 10.3390/cells9030606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Han B, Jiao Y. 2020. Genetic contribution of paleopolyploidy to adaptive evolution in angiosperms. Molecular Plant 13: 59–71. doi: 10.1016/j.molp.2019.10.012. [DOI] [PubMed] [Google Scholar]
- Xiang D, Quilichini TD, Liu Z, et al. 2019. The transcriptional landscape of polyploid wheats and their diploid ancestors during embryogenesis and grain development. The Plant Cell 31: 2888–2911. doi: 10.1105/tpc.19.00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo M-J, Liu X, Pires C, Soltis PS, Soltis DE. 2014. Nonadditive gene expression in polyploids. Annual Review of Genetics 48: 485–517. [DOI] [PubMed] [Google Scholar]
- Yoo M-J, Szadkowski E, Wendel JF. 2013. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110: 171–180. doi: 10.1038/hdy.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.