

Abstract
LC-HRMS experiments detect thousands of compounds, with only a small fraction of them identified in most studies. Traditional data processing pipelines contain an alignment step to assemble the measurements of overlapping features across samples into a unified table. However, data sets acquired under nonidentical conditions are not amenable to this process, mostly due to significant alterations in chromatographic retention times. Alignment of features between disparately acquired LC-MS metabolomics data could aid collaborative compound identification efforts and enable meta-analyses of expanded data sets. Here, we describe metabCombiner, a new computational pipeline for matching known and unknown features in a pair of untargeted LC-MS data sets and concatenating their abundances into a combined table of intersecting feature measurements. metabCombiner groups features by mass-to-charge (m/z) values to generate a search space of possible feature pair alignments, fits a spline through a set of selected retention time ordered pairs, and ranks alignments by m/z, mapped retention time, and relative abundance similarity. We evaluated this workflow on a pair of plasma metabolomics data sets acquired with different gradient elution methods, achieving a mean absolute retention time prediction error of roughly 0.06 min and a weighted per-compound matching accuracy of approximately 90%. We further demonstrate the utility of this method by comprehensively mapping features in urine and muscle metabolomics data sets acquired from different laboratories. metabCombiner has the potential to bridge the gap between otherwise incompatible metabolomics data sets and is available as an R package at https://github.com/hhabra/metabCombiner and Bioconductor.
Graphical Abstract
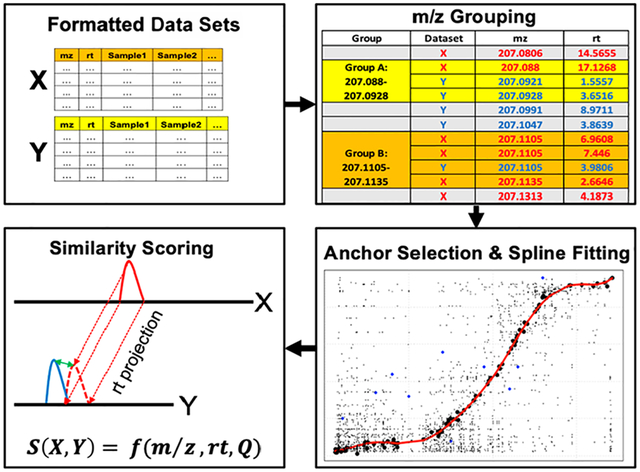
Untargeted metabolomics assays provide valuable information about the composition of organic samples and biochemical phenomena underlying diverse phenotypes. The most common method for profiling metabolites is liquid chromatography coupled to electrospray ionization mass spectrometry (LC-ESI-MS) due to its sensitivity, resolution, and versatility.1 Analysis and interpretation of metabolomics data are challenging due to the intricacy of mass spectral data and the difficulty of assigning unambiguous compound identities to detected features; in most studies, only a small portion is readily identified.2
A key step in LC-MS data processing pipelines is the alignment of mass spectral peaks represented as < mass-to-charge ratio (m/z) and retention time (RT) > features, across a set of experimental samples. The common goal is to maximize the discovery of shared constituent analytes and assemble their respective abundance measurements into a unified table of spectral features to be used for further downstream analyses. Dozens of algorithms have been developed for the alignment of LC-MS peaks,3 many of which have been implemented in popular open-source processing software, such as XCMS4 and MZMine2.5 Feature alignment methods should account for a drift in RTs resulting from distinct sample matrices and changes in chromatographic conditions between individual runs. In recent years, new approaches such as BatchCorr,6 MetMatch,7 and DIMEDR8 have emerged for alignment of metabolomics data acquired in separate experimental batches in order to account for significant interbatch alterations in compound RTs which may lead to misalignments, especially when long interval periods occur between analyses of experimental batches. Other methods, such as metaXCMS,9 facilitate meta-analysis of multiple processed data sets by aligning features nearest in m/z and RT distance among different sample groups.
These approaches are designed for aligning LC-MS features acquired under roughly identical settings within a single laboratory, which may harbor slight alterations in analytical conditions between runs. A greater challenge is presented when aligning features detected under varied experimental factors, such as differences in gradient methods, chromatographic columns, or mobile phase solvents. Such disparities typically result in significant deviations between chromatograms that cannot be corrected by conventional feature alignment approaches. Protocols for the untargeted analysis of biological mixtures have not been standardized across laboratories, creating “incompatible islands” of data sets where feature measurements cannot be directly and thoroughly compared.8 Multilaboratory comparison studies focused on determining the consistency of global LC-MS metabolomics measurements typically use identical assays,10 employ indirect quantitative approaches such as CCSWA,11 or limit their analyses to shared identified compounds12 due to the challenges of matching and comparing the measurements of unidentified features.
The primary hurdle in aligning disparate LC-MS data sets is correcting significant deviations in observed RTs for shared analytes. Numerous approaches have been published for translating RTs between similar, but nonidentical, chromatography methods. Retention indices, calculated from internal standards as a dimensionless, transferrable alternative to retention times, are traditionally applied to GC-MS data and have seen limited application to LC-MS.13–15 The analogous iRT is empirically derived using synthetic peptides of varying hydrophobicity for proteomics data.16 Abate-Patella et al. described a “retention projection” approach that uses experimentally measured retention factor (k) vs solvent composition (Φ) relationships.17,18 The PredRet tool contains a database of compounds whose RTs have been recorded by different chromatography systems, and the tool uses a spline-fitting approach to predict RTs in a new system with an overlap of measured compounds within this database.19 Recently, the CALLC tool coupled chromatographic systems mapping with Quantitative Structure Relationship Relationship (QSRR) modeling, a technique that applies machine learning approaches to predict metabolite retention times from their underlying chemical descriptors.20 While these approaches are designed to facilitate compound identification, they cannot be directly applied for the related but distinct goal of matching and aligning unknown spectral features. One tool for metabolomics feature matching between data sets is PAIR-UP MS,21 which utilizes the shared correlation structure of similar biological specimens, effectively bypassing RT comparisons. However, this method requires a large number of samples and shared known identified metabolites to work effectively. Various proteomics alignment approaches take advantage of MS2-based identifications, using shared peptide identities between runs to nonlinearly model RT shifts, followed by score-based matching.22–25 This solution is not always viable in metabolomics since LC-MS assays are often performed without comprehensive MS/MS analysis or alternative means of determining metabolite coverage overlaps. Mitra et al.26 proposed a quality control method for determining elution order distortions in LC-MS proteomics data generated by different laboratories, using m/z and abundance ranks to perform RT mapping and peak matching.26
Here, we present a novel computational pipeline, implemented in the metabCombiner R package, for matching features corresponding to shared known and unknown metabolites and concatenating their spectral measurements to form a combined feature table. metabCombiner takes a pair of conventionally processed metabolomics data sets as input, fits a nonlinear spline to map between RTs, and ranks all feature pair alignments (hereafter denoted as FPAs), assigning to each a similarity score based on differences in m/z, retention time (fitted vs observed), and relative abundance. metabCombiner has no sample size limit and does not require identified compounds, though prior knowledge may be incorporated for enhanced results. We evaluated the accuracy of RT mapping and feature matching of shared known compounds in a pair of plasma data sets generated with two different reversed-phase liquid chromatography (RPLC) protocols. We further demonstrated the method in two separate cross-data set alignment cases consisting of human urine and rat skeletal muscle, analyzed under varied conditions in separate metabolomics laboratories.
METHODS
Software Overview.
metabCombiner is a software package written in the R statistical language. The inputs for this package are two peak-picked and conventionally aligned feature tables. Rows represent individual features whose m/z, retention time, and per-sample abundance values are displayed in separate columns. The data sets must be acquired in the same ionization mode, with no prior scaling or normalization that may distort their ranked abundance order. The data sets used as inputs must be acquired from biologically similar specimens with a strong expected overlap in their metabolic composition. Finally, chromatographic protocols used to acquire the data sets must be similar enough that the elution order of compounds is largely comparable, if not identical. Postprocessing steps for removal or annotation of features of nonsample origin (e.g., blank sample features, noise, processing artifacts) and isotopologues are desirable but not required. Users may include recommended but optional, feature identifiers, as well as adduct, fragment, or formula labels for validation and parameter optimization purposes. Additional input data set columns may be included in the output report table as “extra” nonanalyzed columns.
metabCombiner workflow.
For a pair of data sets, one is designated as the projection (X) data set, whose retention times will be mapped to the chromatogram of its complement, denoted as the reference (Y) data set. We recommend that the data set with the shorter retention time range be designated as the reference as we generally observe smaller absolute prediction errors when mapping from a highly separated chromatogram to a less separated one than vice versa. The metabCombiner workflow is constructed around key observations for shared compounds in data sets that conform to the following assumptions. The first is that m/z deviations for identical compounds are generally small, rarely exceeding 5–10 mDa even if measured by different high-resolution mass analyzers, as long as proper instrument calibration was maintained throughout the analyses. The second is that while raw spectral abundances are generally not comparable between experiments, relative abundance (Q)–here calculated as ranked median or mean intensity quantile values between 0 and 1–can serve as an additional dimension for comparison besides m/z and rt. Finally, due to the requirement for biological sample similarity, a number of highly abundant common endogenous metabolites (e.g., creatinine in urine) are assumed to be present in both data sets, and these can be used to anchor a nonlinear mapping of retention times. The workflow is depicted in Figure 1, and the specific steps are described below.
Figure 1.
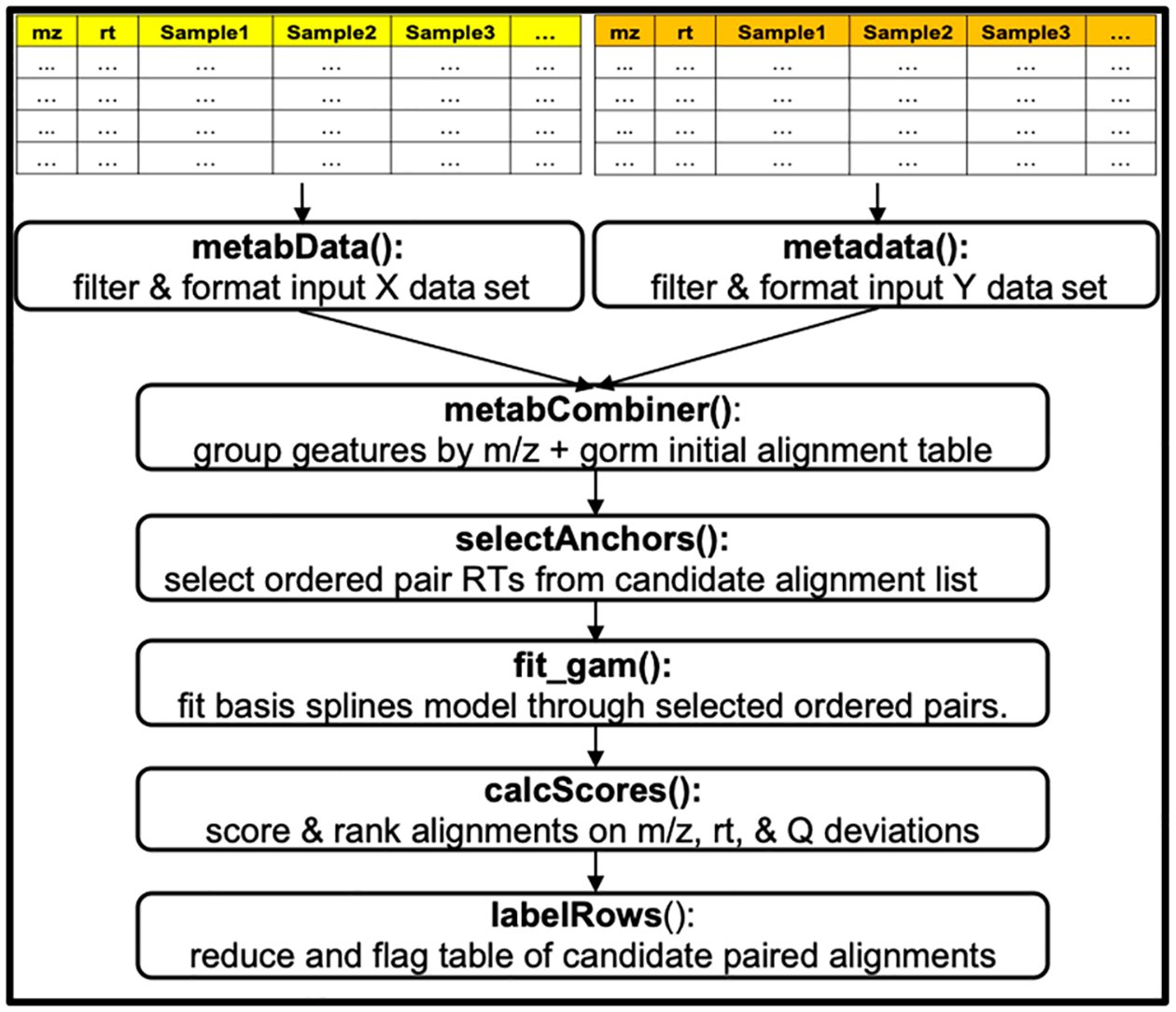
metabCombiner workflow with associated function names.
1. Data Preprocessing.
Each data set is separately processed and formatted, checking for all required and optional metadata. Subsequently, multiple filters are applied to reduce the input feature list. A retention time range filter can limit to features between a start and end retention time, eliminating the head and/or tail of the chromatogram which often contain features mostly of solvent origin. The second filter eliminates features that are missing in more than a certain percentage of the analyzed samples. Finally, pairs of features within a specified m/z and RT tolerance values are deemed duplicates, with one copy retained. Relative abundance quantile Q values are calculated for the remaining features in each data set.
2. Grouping by m/z.
Features from both input tables are pooled, sorted, and binned in the m/z dimension. Distinct feature groups form whenever the difference between consecutive m/z values is less than a user-specified binGap argument (by default 5 mDa). Each group contains m features from data set X and n features from data set Y (m > 0 and n > 0), with m * n total possible FPAs. Subsequent steps assess which FPAs correspond to shared metabolite entities.
3. RT Ordered Pair Selection.
A set of ordered pairs is required to anchor RT mapping. Ideally, confidently identified compounds would be useful for this purpose; however, we often do not observe a sufficient coverage of known metabolites to span the full retention time range of both chromatograms. Therefore, the ordered pairs are selected among all possible FPAs using the process illustrated in Figure 2A. First, the most abundant feature (i.e., with the largest Q value) from the X data set is selected and denoted as x1. The most abundant Y data set feature in the same m/z group containing x1 is selected as the corresponding y-ordinate, y1. Together, the RTs of x1 and y1 serve as the first anchor. All features within a small RT window (e.g., 0.03 min) of x1 and y1 in their respective data sets are excluded from consideration as potential anchors. This selection approach is iterated for the remaining features, until all feature pairs have been either included or excluded as anchors, providing an initial list of ordered pairs (x1,y1), (x2, y2), …, (xn,yn) which we denote as Set A. This process is repeated, now choosing the most abundant features of data set Y and their counterparts in data set X, deriving a second anchor list, Set B. The final anchor set is the intersection of sets A and B and is expected to provide a rough outline of the nonlinear smooth curve between two sets of chromatographic retention times through which a spline may be fit. This largely unsupervised process may select a number of inaccurate FPAs, which manifest as outliers from the outlined curve. Constraints can be placed on m/z and Q differences of anchoring X and Y features to increase the robustness of anchor selection. Users may also incorporate features with shared identities as anchors, which the program will select first as ordered pairs before performing the outlined process based on relative abundance. Incorporating prior knowledge improves and refines the RT mapping steps.
Figure 2.
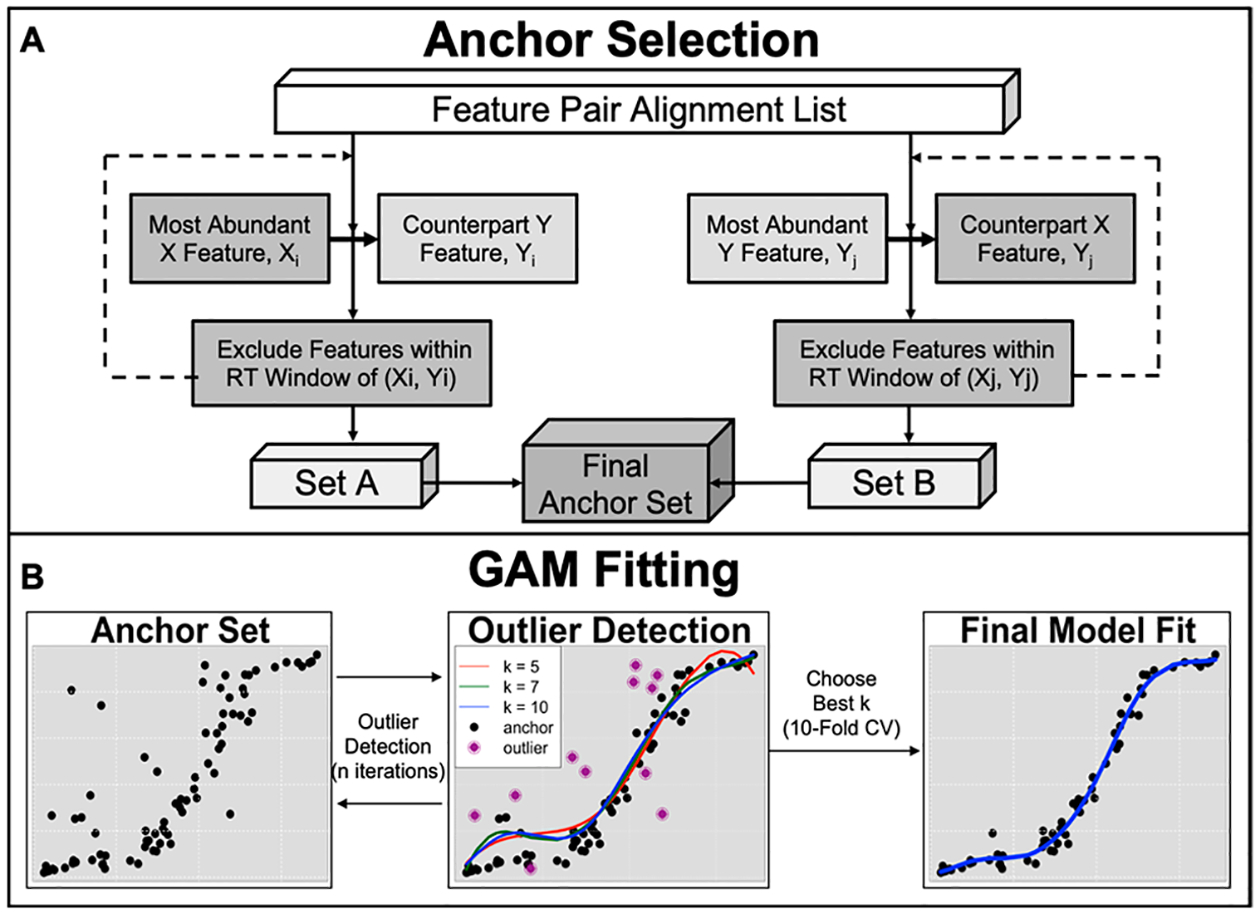
metabCombiner RT mapping procedure. In (A), RT ordered pairs are selected from shared abundant (or identified) features, generating two lists that are subsequently intersected to obtain a final set of anchors. Features within a close retention time window of anchors are excluded. In (B), multiple GAM fits determine outlier anchors before the best k parameter value is chosen through 10-fold CV, which generates the final mapping.
4. Spline-Fitting.
Basis splines, implemented in the mgcv R package, is the main method for RT mapping in this workflow.27,28 Basis splines are a type of generalized additive model (GAM), where a smooth curve is computed based on the sum of low-order polynomial basis functions joined at k control points. k determines the flexibility of the smooth curve and must be optimized from the underlying data. The RT mapping process is illustrated in Figure 2B. First, multiple GAMs with different values of k (e.g., 5, 7, 10, …) are fit to the ordered pairs computed in the anchor selection step, modeling Y-ordinates as a function of the X-ordinates, i.e.
In each individual model, we calculate for each ordered pair (rtx, rty) the absolute value of the fitted vs observed residual value. Anchors which have consistently high residuals (by default, defined as twice the mean model error in over half of the model fits) are excluded as outliers. This is repeated for a specified number of iterations, removing anchors that deviate significantly from the outlined curve. Anchors selected by matching identity are a key exception as they are never filtered, even if relatively high errors are observed. With the remaining points, the optimal k value is selected from among the provided options using 10-fold cross-validation, minimizing mean absolute deviation. The final model is computed using this k value, which then maps RTs between data sets.
5. Similarity Scoring.
Each feature may have a multitude of potential candidate matches in its counterpart data set. To determine the most plausible FPAs, we assign all pairs of grouped features Fx and Fy a similarity score between 0 and 1 according to the expression
where mzx, rtx, and Qx are the respective m/z, RT, and Q values of feature Fx; mzy, rty, and Qy are the respective m/z, RT, and Q values of feature Fy; and f denotes the computed RT mapping function, with prediction errors normalized by the range of the Y data set RT values. A, B, and C are positive weight parameters penalizing differences in features m/z, rt, and Q, respectively. Detailed guidelines for choosing effective weights are contained in S1. Briefly, the most effective ranges of values used in testing for A, B, and C are 50–120, 5–20, and 0–1, respectively. The choice of values should account for instrument mass accuracy, model fit, chromatographic range, and sample similarity. If the data sets contain a sufficiently representative set of shared identified compounds, the package method evaluateParams() finds the set of A, B, and C weight values that optimize an objective function maximizing the positive difference between scores of true FPAs of these known compounds from their respective misaligned pairings. A score of 1 implies perfect concordance of m/z, RT prediction, and relative abundance of a pair of complementary features from the input data sets, implying a likely aligned compound match, whereas scores closer to 0 may be disregarded as misaligned pairs. Each feature’s potential matches from the complementary data set are ranked in reverse score order, with best matches (rankX = 1 and rankY = 1) displayed first.
6. Row Reduction.
The final step is to reduce the report table by removing misaligned pairs. Ideally, every feature should be aligned with at most one feature from the counterpart data set; however, in some cases, multiple matches may need to be considered. The labelRows() package method facilitates this process by placing thresholds on score and pairwise ranking; then, lower-ranked FPAs are flagged for review if either the score or mz/rt values are within a small distance from the top-scoring FPA or rejected as removable rows otherwise. This process typically eliminates 80–90% of misalignments. Further details on FPA table reduction are contained in S2.
Evaluation Data Set.
Untargeted RPLC metabolomics data were acquired twice in the positive ionization mode for ten human plasma samples, five from a pooled plasma obtained from deidentified Red Cross (RC) blood donors and five from a pooled plasma purchased from a commercial supplier for the NIH Children’s Health Exposure Analysis Resource (CHEAR) consortium. Samples and process blanks were analyzed using the same instrumentation and column but with two different gradient elution methods: one with a total run time of 20 min, the other 30 min. Both data sets were processed with XCMS and reduced by isotopologue and negative sample control filtering. We identified 137 metabolites in common according to Metabolomics Standards Initiative (MSI) criteria 1 or 229 and annotated 532 in-source adducts, fragments, and multimers of these metabolites using a custom R script and the Binner annotation software,30 which are listed in Supplementary Sheet S1. For this pair of data sets, we evaluated the RT fitting and score-based matching of compounds, using identified features as a benchmark. First, known metabolites were partitioned into 50% training, 50% test sets. RT fitting was both semisupervised (with all training set compounds included as anchors) or unsupervised (rt fitting without prior knowledge), with mean absolute deviation (MAD) of the fit calculated for the test set compounds. To evaluate score-based matching, we used evaluateParams to guide A, B, and C weight value selection on the training set compounds. An “accurate match” is defined to be a best-scoring FPA (rankX = 1 and rankY = 1) between two identically annotated features with a score greater than 0.5. Feature matching accuracy is assessed “per-variant”–that is, weighing each adduct/fragment feature equally–and “per-compound”–summing the fractions of accurately matched adducts and fragments for each compound over the total number of test compounds. Different sample subsets (CHEAR, RC) analyzed in the 30 min analysis are designated as data set X, with the opposite set in the 20 min analysis designated as data set Y. Further analysis details for this pair of data sets are contained in S3.
Exploration Data Sets.
We used metabCombiner to align untargeted metabolomics data sets acquired at the University of Michigan Metabolomics Core with data acquired from other institutions. First, data from three pooled replicates of healthy human urine obtained from BioIVT (Westbury, NY) and NIST Standard Reference Material SRM3673 (together called ‘B3N3’) were aligned to that of 43 samples from interstitial cystitis patients (“IC43”) obtained from a study published by Blaženović et al.31 We replicated sample preparation and HILIC-positive chromatographic approaches from this study but with a shorter column, inducing major RT shifts. We use a QTOF mass spectrometer as opposed to an Orbitrap. MS files from B3N3 and IC43 (downloaded from Metabolomics Workbench32 Study ID ST001122) were both processed by MZMine2 using the ADAP pipeline,33 extracting 10624 and 22313 features, respectively. In the published study, hundreds of compounds were putatively identified at MSI levels 1 and 2, which are annotated in IC43. For B3N3, we assigned 123 compound identities, all at MSI level 2 using a simple MS/MS-based workflow using NIST MSPepSearch.34 More experimental and computational analysis details for this pair of data sets can be found in S4.
Second, muscle tissue from 10 sedentary and 10 exercised rats was analyzed in our facilities (“MiSE10”) as well as by the Broad Institute (“BrSE10”) in the negative ionization mode. Data were processed using XCMS and Progenesis QI (nonlinear dynamics), detecting 5335 and 8573 features, respectively. There were greater differences in the experimental methods used in this case, including column type (Waters H3 TSS modified C18 vs unmodified C18), mobile phase solvents (methanol vs acetonitrile), mass analyzer (QTOF vs Orbitrap), and m/z scan range (50–1000 vs 70–850). See S5 for more experimental and computational analysis details for this pair of data sets. Of the named identified compounds (200 in MiSE10 and 80 in BrSE10), there were only 14 (mostly nonpolar) overlapping identities, which are listed in Supplementary Sheet S2.
RESULTS
metabCombiner Output.
The main output of metabCombiner is a table containing FPAs organized into separate m/z groups in order of increasing m/z. The signal abundance values of each feature are concatenated to form a combined table. An example of the m/z group from the plasma data sets is shown in Figure 3A. It consists of three features from data set X (30 min analysis) and three features from data set Y (20 min analysis), all within the m/z range 426.3205–426.3223. Two features each from the two complementary data sets are unidentified isomers (X7710 and X7753, Y7385 and Y7434) with the third feature previously identified in both data sets as cholate [M + NH4]+. A pairwise top match (rankX = 1 and rankY = 1) is assigned between [X7710, Y7385] and [X7785, Y7434], respectively, with alignment scores very close to 1. Alternative possible alignments for this pair of compounds are displayed as separate rows which can be quickly dismissed as misalignments. The alignment score of cholate [M + NH4]+ with itself is lower due to a higher retention time prediction error and a slight difference in the relative abundance of this compound between assays; nonetheless, it is correctly assigned the top-scoring FPA with itself, and all other pairs score very poorly. Thus, three FPAs corresponding to three separate compounds remain in the final table, and six misalignments are eliminated. A visual inspection of the peaks shown in Figure 3B and 3C confirms this matching.
Figure 3.
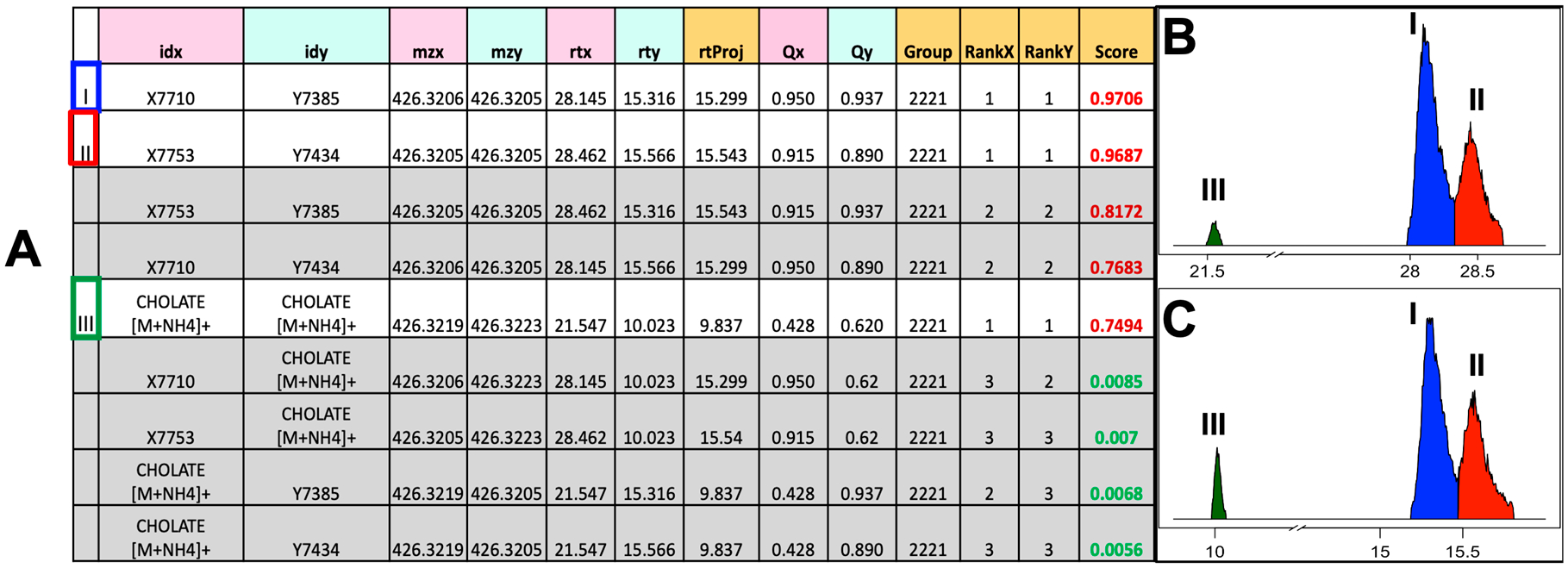
Example scored feature group output with matching peaks. (A) Columns with pink and turquoise headings are feature metadata provided by the input data sets. Orange headings–rtProj (model-predicted RT), Group number, Score, and Feature Ranks with respect to alternative matches–are generated by the program. Rows in gray contain mismatched features and should be discarded. Rows 1 and 2 are unidentified isomers; row 5 was previously identified in both data sets as cholate [M + NH4]+. (B) and (C) are the plotted chromatographic peaks corresponding to these features in data sets X and Y, respectively.
m/z Grouping.
The size of the initial table of FPAs is a function of input data set feature counts, their degree of m/z overlap, and the binGap parameter in the m/z grouping step. Table S1 reports the initial FPA count in the three test cases using different values for binGap. In the plasma data sets, the number of FPAs is comparable to the initial data set sizes and grows steadily with increased gap values. In the urine data analysis, the FPA space grows very rapidly due to a high density of features in the low (m/z = 100–400 Da) range in both data sets. On the other hand, we observe a smaller FPA list between the muscle data sets, likely due to differences in m/z ranges surveyed by the respective analyses. While the majority of matching known compounds displays small (<1 mDa) m/z differences, larger errors (>5 mDa) may be observed in some cases as a result of instrumental and preprocessing software factors. Thus, the value of binGap reflects a trade-off between compactness of the initial combined table and the ability to detect all true FPAs. The binGap value is set to 5 mDa by default and can be altered as necessary.
Retention Time Mapping.
The plot package method is useful for visualizing results of anchor selection and GAM-fitting. Plots for plasma, urine, and muscle data sets are displayed in Figure 4. In each case, we observe a moderate to high degree of fluctuation in the center of the chromatogram along the gradient slope, indicating that these regions are generally more difficult to model accurately. Moreover, there are differences in how well-represented each chromatographic region is in the three plots. In the first case, all regions are well-represented, whereas the third case contains a noticeable gap along the gradient. The second plot also has a relatively sparse representation of ordered pair anchors in the early chromatogram. The plot serves as a useful tool for tuning the parameters associated with model fitting as well as determining an appropriate RT penalty weight.
Figure 4.
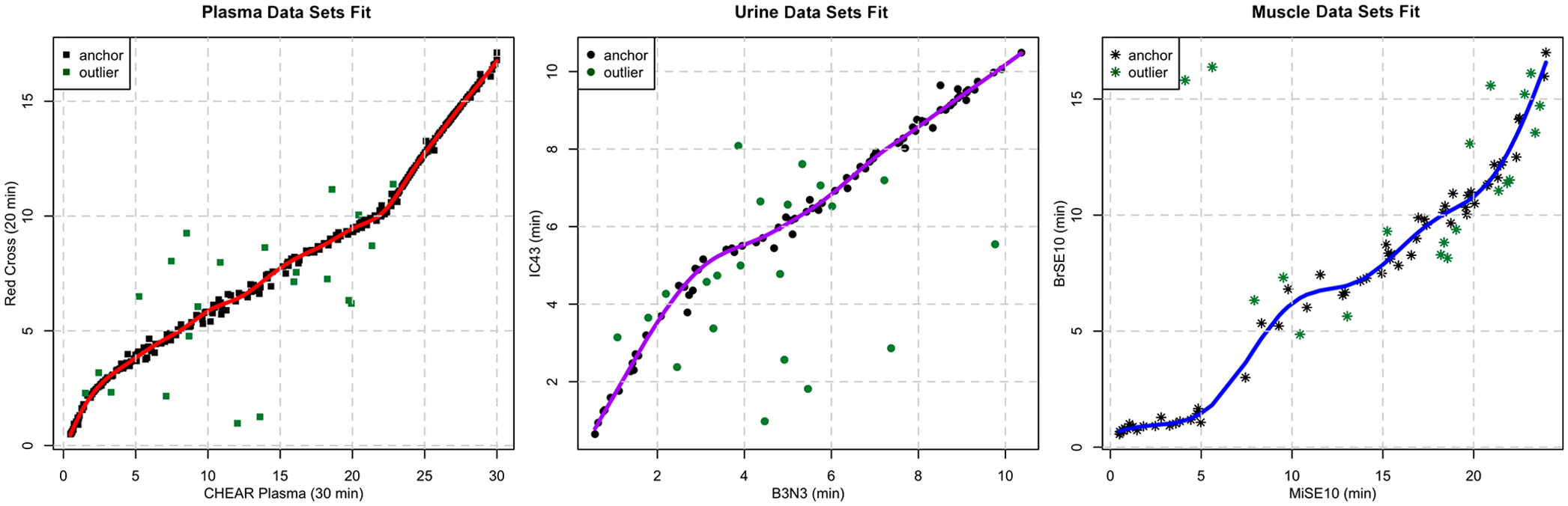
Images of the RT mapping curve for each of the three cases described in this study. Anchor points with high residuals in over half the GAM fits at a given iteration are marked as outliers (indicated in green), except for those with matching identities.
Evaluation with Plasma Data sets.
We analyzed CHEAR and Red Cross plasma aliquots together in our laboratory using two different RPLC protocols with 20 and 30 min total chromatography times. Of the 137 identified compounds common to both plasma data sets, all but three could be grouped by m/z using the default 5 mDa binGap value. The principal ions of caffeine, glutamylphenylalanine, and creatine deviated by 0.006, 0.0088, and 0.02 Da, respectively. We opted for a binGap value of 0.0075 to be used for all analyses of this data set pair, grouping all metabolite ions except for creatine. Therefore, 136 compounds were used for our analysis, with 68 each randomly partitioned into training and test sets. The choices of sample subset (CHEAR vs Red Cross) and whether or not to use known identity information affect the selection of anchors and the subsequent modeling and feature matching accuracy. The results of our evaluation are displayed in Table 1. The mean absolute error of each model is consistently around 0.06 min, with a slight advantage observed in semisupervised models in which training set compounds are selected as anchors. Prediction errors vs observed retention times for selected test set compounds are shown in Figure S1. In each model, more than 50 out of the 68 compounds could be predicted within 0.1 min. Polar and very nonpolar metabolite RTs are mostly well-predicted, whereas compounds of intermediate polarity were less predictable due to alterations in gradient slopes, with the highest retention time errors between 0.25 and 0.35 min. Notably, the inclusion of prior information provides a distinct advantage in predicting metabolite RTs in relatively sparse chromatographic regions.
Table 1.
Evaluation Resultsa
| mode | X data set | Y data set | RT MA.D. | accuracy (per variant) | weighted accuracy (per compd) |
|---|---|---|---|---|---|
| semisupervised (including identities) | 30 MIN CHEAR | 20 MIN Red Cross | 0.056 | 259/270 | 0.91 |
| unsupervised | 30 MIN CHEAR | 20 MIN Red Cross | 0.066 | 254/270 | 0.88 |
| semisupervised (including identities) | 30 MIN Red Cross | 20 MIN CHEAR | 0.054 | 254/270 | 0.88 |
| unsupervised | 30 MIN Red Cross | 20 MIN CHEAR | 0.07 | 250/270 | 0.86 |
Different subsets (CHEAR or Red Cross plasma) are used as data set X from the 30 min data set, with the opposite set from the 20 min data set serving as data set Y. Semisupervised model fits (inclusion of training set compounds) vs unsupervised (no prior information) in terms of mean absolute deviation (MAD) RT fit, feature matching accuracy per-feature and per-compound.
The fitted models were then used to evaluate scoring, using 270 annotated variants (adducts, in-source fragments, and multimers) of the test set metabolites as points of comparison. Score parameter arguments were chosen to be to be A = 100, B = 15, and C = 0.3, as guided by evaluateParams on training set compounds. Most compounds accurately achieve the highest alignment score for all of their variants, with weighted per-compound average scores between 0.85 and 0.9 in all analyses. Four shared compounds scored at or below the threshold 0.5 level, mostly due to penalization of high m/z differences. In cases for which the correct alignment is ranked below a misalignment, at least one feature may be more similar in m/z, Q, or RT fit. The feature(s) may arise from a structural isomer eluting within close proximity, while in other cases, a peak may be divided as a result of processing errors. No accurate FPA ranked poorer than third best for the respective compounds, and the scores of all but one compound variant were within 0.2 from the top-scoring alignment. Proceeding with table reduction, we set score, rankX, rankY, and delta score tolerance values at 0.5, 3, 2, and 0.2 to automatically reduce the set of 14024 FPAs by 6765; further inspection reduces an additional 400, reducing the table to less than 6900 FPAs.
Data Exploration with metabCombiner.
Urine Data Sets Analysis.
In a preliminary survey of the data sets, we observed relatively subpar mass accuracy, particularly for our B3N3 data set; therefore, we opted for a wider m/z grouping binGap tolerance value of 0.01, despite the substantial increase to 95898 rows in the initial FPA table. We conducted the metabCombiner analysis for this pair of data sets in two stages. First, we performed analysis without relying on named features and assessed the validity of these alignments. This was followed by a semisupervised analysis using consistently named compound identities to obtain an improved RT mapping and reduced table of FPAs.
The anchor selection step produced 66 ordered pairs for mapping between retention times in both data sets, using B3N3 as data set X and IC43 as data set Y. Scoring parameter values were set to A = 60, B = 8, and C = 0.3. Forty-one consistently named compounds between both data sets achieved the highest-scoring alignments among their respective groups. Three more top alignments were between features named as positional isomers, e.g., 4- and 3-hydroxypyridine, which we count among the consistent set. The computed GAM mapped retention time accurately for the majority of these compounds, with 28 and 33 fitted within 0.1 and 0.2 min (1–2%) of the observed retention time, respectively. One named compound, ornithine, eluted 0.67 min later than predicted as its elution order changed considerably between the two data sets. Seven identically named features did not score highly, mainly due to excessive RT fit deviations. In 14 cases, we found high-scoring alignments between features with mismatched identities; six of these could be resolved through manual review of MS/MS or correcting adduct annotations of these features. Many assigned features had no probable match in the counterpart data set (particularly among drug-related metabolites), and a few others could not be definitively assigned as a match due to low scores or the presence of conflicting feature(s). On the other hand, aligning B3N3 to the well-annotated IC43 provides moderate-to-high scoring alignments to 167 distinct features that were named in IC43 but not B3N3. These alignments provide a list of putative identities which can be subsequently verified through the use of authentic standards. The list of consistent, inconsistent, and putative compound identities is provided in Supplementary Sheet S3.
We proceeded with the semisupervised analysis aided by the 44 consistently named metabolites. With this adjustment, 98 ordered pairs were selected for anchoring the updated RT mapping. A visual of the two model fits is shown in Figure S2. The greatest differences in the model-predicted RTs are observed in the early to middle chromatographic regions. Score parameters, as guided by the evaluateParams method, were similar to those used before, with only B changed to 7. Table reduction proceeded with similar thresholds to the previous case, with alignment scores below 0.5 and ranks above 3 filtered; together with inspection of flagged rows, we reduced the table from 95898 to 3265 FPAs or roughly 3% of the original table size.
Muscle Data Sets Analysis.
Experimental variables varied more for this pair of data sets as compared to the previous analyses. The protocols used to acquire BrSE10 are optimized for the measuring metabolites of intermediate polarity (e.g., bile acids and free fatty acids), whereas MiSE10 is acquired with a more generalized metabolomics assay. This has several implications when aligning features in this pair of data sets. First, as a consequence of different column types (Waters HSS T3 C18, which has an embedded polar retention functionality, vs Waters BEH unmodified C18), polar compounds were difficult to map and differentiate properly as they eluted very rapidly in BrSE10 as compared to MiSE10. Second, differences in coverage of highly nonpolar metabolites caused major distortions in the model fit (see Figure S3). To correct this, we excised the late chromatographic portions where highly nonpolar compounds elute, setting a maximum retention time of 24 and 17 min for MiSE10 and BrSE10, respectively. These constraints remove 10–20% of the input features in each data set. Third, we observed that a number of fatty acids present at high abundances in BrSE10 were barely or not at all detectable in MiSE10 samples, likely due to differences in sample extraction protocols between the two assays; therefore, quantile Q differences are less reliable in some cases. Finally, while we did not encounter significant mass errors for shared compounds in preliminary analysis, the overall mean m/z for these two data sets differed by more than 200 Da (549.3 vs 316.1 in MiSE10 and BrSE10, respectively). With the binGap parameter kept to its default value of 5 mDa, this generated a small initial set of only 3247 possible FPAs, indicating a limited coverage overlap between the assays.
We optimized anchor selection and GAM-fitting using a grid search of parameter values, with the mean absolute RT deviation for 14 shared identified compounds serving as the error metric. The final model mapped five compounds accurately to within 0.1 min; two were predicted within 0.25 min; five had errors of between 0.4 and 0.6 min; two (cholate and glycocholate) could not be predicted by any of the models to within 1.25 min. Given these factors, scoring parameters were chosen as A = 100, B = 7, and C = 0.2. Of the shared known compounds, cholate (0.35) and glycocholate (0.39) score the lowest due to the higher RT fitting errors. Ten compounds accurately achieved the highest alignment score in all of their respective adduct forms; the remaining metabolites have one misaligned variant each, and one variant ranked worse than the fifth best. On this basis, FPAs with scores below 0.35 and ranking worse than fifth were removed, eliminating 1765 alignments; further inspection of conflicting alignments removed an additional 400–450 rows, reducing to under 1000 of the original 3247 FPAs.
DISCUSSION
The field of LC-MS metabolomics has long been constrained by the incompatibility of measurements acquired under disparate analytical conditions. metabCombiner provides an opportunity to bridge the gap between some previously incomparable metabolomics data and to increase the utility of multiple studies beyond their initial uses. metabCombiner is a versatile method designed to be widely applicable to metabolomics data sets, with and without prior knowledge of shared coverage.
Unlike many previously discussed alignment approaches, metabCombiner uses traditionally peak-picked and aligned metabolomics data rather than raw MS files as input. This allows for identically acquired spectral data to be peak-picked and aligned using any method; we have examined data analyzed using several different software tools in this study. It is important to acknowledge that important spectral information may be lost in translation from raw MS data to the tabular format. Different preprocessing software tools were used in this study to determine potential sources of error resulting from data set generation. We encountered errors similar to those reported previously,35 such as incomplete or multipeak integration, low signal-to-noise features, and missed peaks. Such errors may complicate the one-to-one alignment of features, as illustrated in Figure 5. In addition, factors affecting the accurate estimation of m/z, retention time location, and peak area as well as overall quantity of features have important ramifications for this analysis. Therefore, the preprocessing method and choice of parameters should be considered carefully for each data set. Another important distinction is that metabCombiner focuses on determining the intersection, as opposed to the union, of input data set features. While information from nonoverlapped features may be missed, the final output table consists of an expanded set of observations over a smaller set of observed compounds, providing increased statistical power to detect significant biological changes. The output must be corrected for batch effects before carrying out further statistical and bioinformatics analyses.
Figure 5.
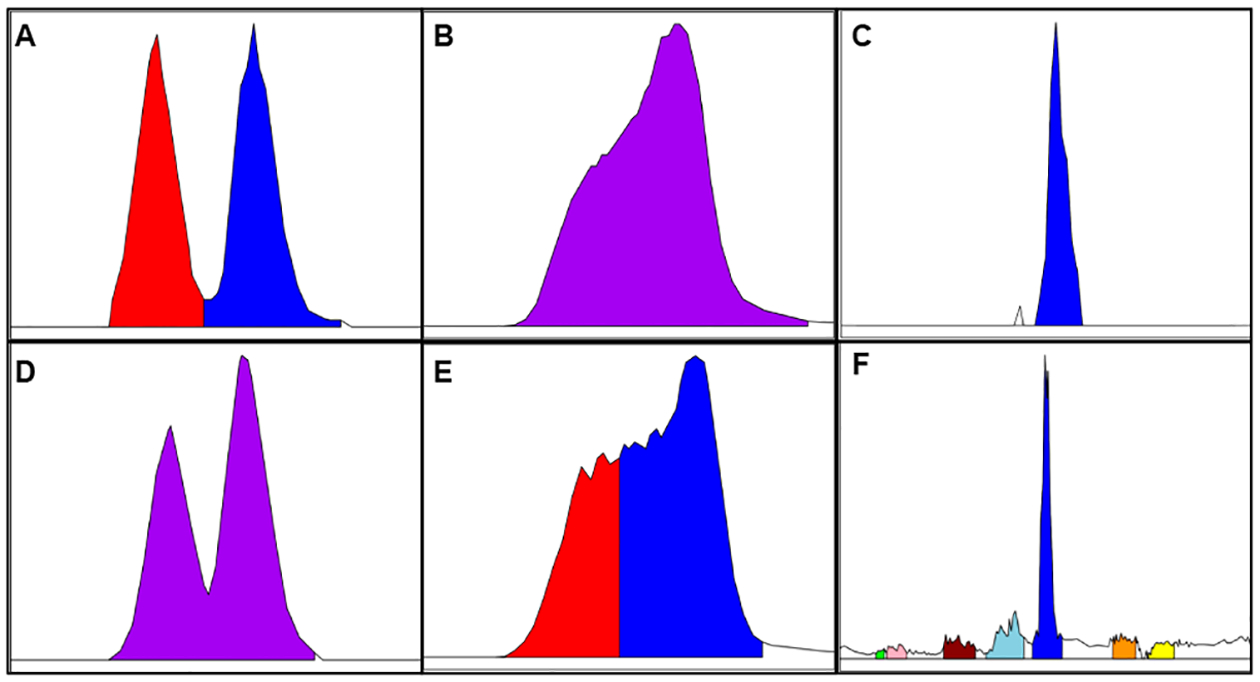
Illustration of common processing errors. The top row (A–C) displays correctly integrated peaks, whereas the second row (D–F) displays errors. In (A), two isomers are integrated as separate peaks, but in (D), they are fused as one feature. In (B), a wide peak is integrated as one feature, but in (E), it is split into multiple features. In (C) and (E), one peak is matched to numerous low signal/noise features, with only the abundant peak representing the true compound match. Processing errors such as these complicate accurate 1–1 feature matching between data sets.
To our knowledge, few methods for aligning metabolomics features make extensive use of relative abundance (Q). In metabCombiner, it is first used to select order pair anchors for RT mapping, and then it is incorporated alongside RT and m/z in pairwise alignment scoring. While useful for contrasting high- and low-abundance compounds, disparities in relative abundance may occur due to experimental factors, such as sample preparation and in-source ionization. We occasionally observe that the formation and relative abundances of in-source adducts and fragments may differ for the same compounds between data sets. Therefore, metabCombiner can be configured to give relative abundance a less prominent role than RT and m/z for compound matching by selecting appropriate weight parameters. metabCombiner is not the first tool to use a GAM for the purposes of RT mapping between chromatograms. GAMs have distinct advantages over local regression (LOESS) and regression tree ensemble approaches. Their simplicity, versatility, and robustness to overfitting have been noted.18 In particular, setting the default “family” argument to scat (scaled t-family for heavy-tailed data) helps to eliminate the influence of outlier points, which may cause other overfitting in other approaches.
There are some important limitations to the current work that we hope to address in future iterations of this tool. The first is that metabCombiner is currently designed for combining two data sets. In order to align more data sets, many of the package constructs may need to be appropriately generalized or sequentially applied to additional data sets. For the current implementation, it is possible to iteratively merge combined data set outputs with an additional data set in a stepwise manner, using one set of m/z, rt, and sample measurements for comparison and the others as “extra”, nonanalyzed columns, as detailed in S6. A second limitation is that our RT mapping approach does not yet provide for prediction intervals as only the point estimate of the mapped RTs is used to determine the pairwise alignment score; however, we recognize that intervals may provide great utility due to the nonuniformity of prediction errors throughout the chromatogram. Cases presented in this study vary in key chromatographic variables, such as gradients, column types and dimensions, and mobile phase solvents, yet numerous other variables have yet to be fully explored. In general, we observe that data sets acquired from HILIC methods are more difficult to align than those by RPLC methods, a difficulty shared with previous studies attempting to predict compound retention in separate HILIC assays.36 Finally, achieving optimal matching of features between two data sets using metabCombiner benefits significantly from careful refinement of weighting parameters and other factors. We attempted to provide guidance to enable users to achieve good results, but future developments may focus on automating this process to a greater extent, allowing for a more hands-off data alignment approach more compatible with nonexpert users.
In conclusion, we have developed a computational pipeline for comprehensive mapping of features detected in two distinct untargeted metabolomics experiments to generate an aligned data set in a semiautomated manner. This tool has numerous applications, such as allowing for comparisons of experimental protocols and reproducibility assessments, facilitating collaborations in compound identification efforts across institutions, and generating expanded data sets suitable for performing meta-analyses. While this study focused on the alignment of metabolomics data, the methods described here may be adapted to other untargeted LC-MS analyses of complex mixtures, as long as the input data sets meet the main assumptions described in this manuscript. This workflow is implemented in the metabCombiner R package which is available on Github at https://github.com/hhabra/metabCombiner.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge support of NIH grants U2CES030164 and U2CES026553 to the University of Michigan. The authors thank Dr. Ivana Blaženović for her assistance with the urine data set analysis. The authors also thank Dr. George Michailidis, Dr. Veerabhadran Baladandayuthapani, and Dr. Joshua Errickson for their technical assistance in developing the metabCombiner R package and methods.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03693.
Initial table size by m/z binGap; guide to weight coefficient selection; guide to FPA table reduction and conflicting FPA examples; experimental and computational analysis details for plasma, urine, and muscle data sets; evaluation data set RT mapping accuracy by observed RT; unsupervised vs semisupervised urine map curve image; and muscle data set RT mapping curve image, with and without RT restriction (PDF)
Supplementary Sheet S1, plasma metabolites (adduct/in-source fragment/multimers) used for evaluation (XLSX)
Supplementary Sheet S2, shared identified muscle metabolite feature pair alignments (XLSX)
Supplementary Sheet S3, consistent, inconsistent, and hypothesized named aligned urine metabolites (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c03693
The authors declare no competing financial interest.
Contributor Information
Hani Habra, Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Arbor, Michigan 48109, United States.
Maureen Kachman, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan, Ann Arbor, Michigan 48105, United States.
Kevin Bullock, Metabolomics Platform, Broad Institute, Cambridge, Massachusetts 02142, United States.
Clary Clish, Metabolomics Platform, Broad Institute, Cambridge, Massachusetts 02142, United States.
Charles R. Evans, Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan, Ann Arbor, Michigan 48105, United States
Alla Karnovsky, Department of Computational Medicine and Bioinformatics, University of Michigan Medical School, Arbor, Michigan 48109, United States; Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan, Ann Arbor, Michigan 48105, United States.
REFERENCES
- (1).Patti GJ J. Sep Sci 2011, 34 (24), 3460–9. [DOI] [PubMed] [Google Scholar]
- (2).Sindelar M; Patti GJ J. Am. Chem. Soc 2020, 142 (20), 9097–9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Smith R; Ventura D; Prince JT Briefings Bioinf 2015, 16 (1), 104–17. [DOI] [PubMed] [Google Scholar]
- (4).Smith CA; Want EJ; O’Maille G; Abagyan R; Siuzdak G Anal. Chem 2006, 78 (3), 779–87. [DOI] [PubMed] [Google Scholar]
- (5).Pluskal T; Castillo S; Villar-Briones A; Oresic M BMC Bioinf 2010, 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Brunius C; Shi L; Landberg R Metabolomics 2016, 12 (11), 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Koch S; Bueschl C; Doppler M; Simader A; Meng-Reiterer J; Lemmens M; Schuhmacher R Metabolites 2016, 6 (4), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mak TD; Goudarzi M; Laiakis EC; Stein SE Anal. Chem 2020, 92 (7), 5231–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tautenhahn R; Patti GJ; Kalisiak E; Miyamoto T; Schmidt M; Lo FY; McBee J; Baliga NS; Siuzdak G Anal. Chem 2011, 83 (3), 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cajka T; Smilowitz JT; Fiehn O Anal. Chem 2017, 89 (22), 12360–12368. [DOI] [PubMed] [Google Scholar]
- (11).Martin JC; Maillot M; Mazerolles G; Verdu A; Lyan B; Migné C; Defoort C; Canlet C; Junot C; Guillou C; Manach C; Jabob D; Bouveresse DJ; Paris E; Pujos-Guillot E; Jourdan F; Giacomoni F; Courant F; Favé G; Le Gall G; Chassaigne H; Tabet JC; Martin JF; Antignac JP; Shintu L; Defernez M; Philo M; Alexandre-Gouaubau MC; Amiot-Carlin MJ; Bossis M; Triba MN; Stojilkovic N; Banzet N; Molinié R; Bott R; Goulitquer S; Caldarelli S; Rutledge DN Metabolomics 2015, 11 (4), 807–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Benton HP; Want E; Keun HC; Amberg A; Plumb RS; Goldfain-Blanc F; Walther B; Reily MD; Lindon JC; Holmes E; Nicholson JK; Ebbels TM Anal. Chem 2012, 84 (5), 2424–32. [DOI] [PubMed] [Google Scholar]
- (13).Baker JK; Ma C-Y Journal of Chromatography A 1979, 169, 107–115. [Google Scholar]
- (14).Bogusz M; Aderjan RJ Anal. Toxicol 1988, 12 (2), 67–72. [DOI] [PubMed] [Google Scholar]
- (15).Hill DW; Kelley TR; Langner KJ; Miller KW Anal. Chem 1984, 56 (13), 2576–9. [DOI] [PubMed] [Google Scholar]
- (16).Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Proteomics 2012, 12 (8), 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Abate-Pella D; Freund DM; Ma Y; Simon-Manso Y; Hollender J; Broeckling CD; Huhman DV; Krokhin OV; Stoll DR; Hegeman AD; Kind T; Fiehn O; Schymanski EL; Prenni JE; Sumner LW; Boswell PG Journal of chromatography. A 2015, 1412, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Boswell PG; Abate-Pella D; Hewitt JT Journal of chromatography. A 2015, 1412, 52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Starczewski A; Krzyżak A In A Modification of the Silhouette Index for the Improvement of Cluster Validity Assessment; Cham, Springer International Publishing: Cham, 2016; pp 114–124, DOI: 10.1007/978-3-319-39384-1_10. [DOI] [Google Scholar]
- (20).Bouwmeester R; Martens L; Degroeve S Anal. Chem 2020, 92 (9), 6571–6578. [DOI] [PubMed] [Google Scholar]
- (21).Hsu YH; Churchhouse C; Pers TH; Mercader JM; Metspalu A; Fischer K; Fortney K; Morgen EK; Gonzalez C; Gonzalez ME; Esko T; Hirschhorn JN PLoS Comput. Biol 2019, 15 (1), e1006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jaitly N; Monroe ME; Petyuk VA; Clauss TR; Adkins JN; Smith RD Anal. Chem 2006, 78 (21), 7397–409. [DOI] [PubMed] [Google Scholar]
- (23).Palmblad M; Mills DJ; Bindschedler LV; Cramer RJ Am. Soc. Mass Spectrom 2007, 18 (10), 1835–43. [DOI] [PubMed] [Google Scholar]
- (24).Tsou C-C; Tsai C-F; Tsui Y-H; Sudhir P-R; Wang Y-T; Chen Y-J; Chen J-Y; Sung T-Y; Hsu W-L Molecular & Cellular Proteomics 2010, 9 (1), 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tsou CC; Avtonomov D; Larsen B; Tucholska M; Choi H; Gingras AC; Nesvizhskii AI Nat. Methods 2015, 12 (3), 258–64. 7 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mitra V; Smilde A; Hoefsloot H; Suits F; Bischoff R; Horvatovich P Journal of chromatography. A 2014, 1373, 61–72. [DOI] [PubMed] [Google Scholar]
- (27).Eilers PHC; Marx BD Statistical Science 1996, 11 (2), 89–102. [Google Scholar]
- (28).Wood SN Statistics and Computing 2017, 27 (4), 985–989. [Google Scholar]
- (29).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TW; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD; Thaden JJ; Viant MR Metabolomics 2007, 3 (3), 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kachman M; Habra H; Duren W; Wigginton J; Sajjakulnukit P; Michailidis G; Burant C; Karnovsky A Bioinformatics 2020, 36 (6), 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Blaženović I; Kind T; Sa MR; Ji J; Vaniya A; Wancewicz B; Roberts BS; Torbašinović H; Lee T; Mehta SS; Showalter MR; Song H; Kwok J; Jahn D; Kim J; Fiehn O Anal. Chem 2019, 91 (3), 2155–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sud M; Fahy E; Cotter D; Azam K; Vadivelu I; Burant C; Edison A; Fiehn O; Higashi R; Nair KS; Sumner S; Subramaniam S Nucleic Acids Res 2016, 44 (D1), D463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Myers OD; Sumner SJ; Li S; Barnes S; Du X Anal. Chem 2017, 89 (17), 8696–8703. [DOI] [PubMed] [Google Scholar]
- (34).Stein SE; Scott DR J. Am. Soc. Mass Spectrom 1994, 5 (9), 859–66. [DOI] [PubMed] [Google Scholar]
- (35).Myers OD; Sumner SJ; Li S; Barnes S; Du X Anal. Chem 2017, 89 (17), 8689–8695. [DOI] [PubMed] [Google Scholar]
- (36).Wang N; Boswell PG Journal of chromatography. A 2017, 1520, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.