

SUMMARY
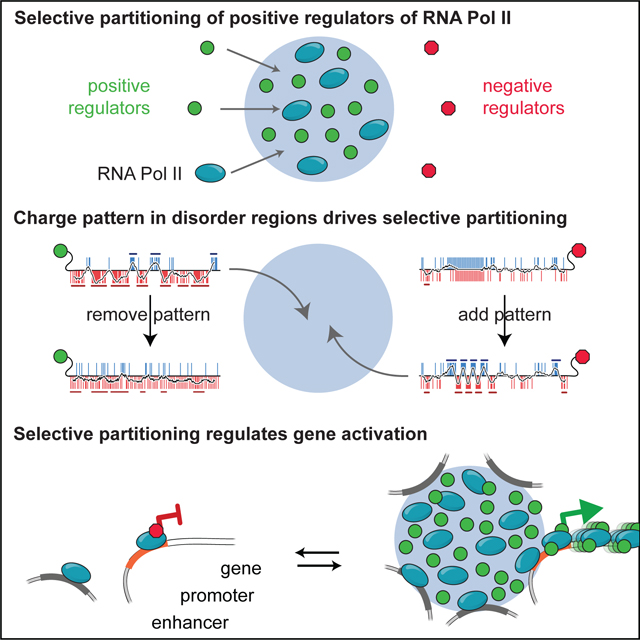
Components of the transcriptional machinery are selectively partitioned into specific condensates, often mediated by protein disorder, yet we know little about how this specificity is achieved. Here we show that condensates composed of the intrinsically disordered region (IDR) of MED1 selectively partition RNA Pol II together with its positive allosteric regulators while excluding negative regulators. This selective compartmentalization is sufficient to activate transcription and is required for gene activation during a cell-state transition. The IDRs of partitioned proteins are necessary and sufficient for selective compartmentalization and require alternating blocks of charged amino acids. Disrupting this charge pattern prevents partitioning whereas adding the pattern to proteins promotes partitioning with functional consequences for gene activation. IDRs with similar patterned charge blocks show similar partitioning and function. These findings demonstrate that disorder-mediated interactions can selectively compartmentalize specific functionally related proteins from a complex mixture of biomolecules, leading to regulation of a biochemical pathway.
eTOC Blurb
Charge patterning in disordered regions of transcriptional regulators mediate selective partitioning into MED1IDR condensates for gene activation.
Graphical Abstract
INTRODUCTION
Transcription is a multi-step process requiring the recruitment of dozens of proteins to specific genomic loci1,2. Many of these components form dynamic local concentrations correlating with transcriptional activity3–17, yet we know little about how the multiple components required for transcription are selectively concentrated. Here we refer to these high local concentrations of the transcription machinery as condensates18. While the exact physicochemical process underlying transcriptional condensate formation is unresolved, there is general agreement that weak multivalent interactions drive their assembly19–21. How specificity in condensate composition is achieved by weak multivalent interactions remains a major unanswered question.
Weak multivalent interactions among components of the transcriptional machinery, in particular those mediated by intrinsically disordered regions (IDRs), have long been understood to be important for transcription22,23. Recently, specific pairwise interactions among IDRs have been documented for candidate factors in the context of condensates3,4,6,24. Significant work has also been done on dissecting the molecular underpinnings of IDR self-association, with RNA-binding proteins being among the most comprehensively characterized25–32. These studies conclude that multivalency and patterning of diverse types of interactions along the sequence of these IDRs are important for condensate formation. Because most studies focused on self-association or partitioning of candidate factors, general principles for selective partitioning in the context of a complex cellular environment remain elusive. In this study, we aim to perform unbiased identification of partitioned proteins to uncover sequence features that engender specificity. Towards that goal, we focus on identifying and characterizing proteins that selectively partition into condensates composed of the largest IDR within the Mediator complex: MED1IDR.
We focus on MED1IDR as a case study to investigate selective partitioning mediated by protein disorder and its functional consequences for two key reasons. First, the Mediator coactivator complex is a central hub of interactions critical for gene activation33–36 and disordered portions of several Mediator subunits are thought to play a role in these interactions37,38. The largest IDR in the complex is on subunit MED1 and has been implicated in the formation of condensates involved in gene activation in both cell-based and in vitro systems3,4,24,39,40, providing us with biochemical and cell-based assays to investigate selective partitioning. Second, while Med1 knockout mice are embryonic lethal41, knockout cells grow in culture42,43 but are unable to undergo certain cell-state transitions including adipogenesis42, providing us with a cell-state transition model to examine the functional role of selective partitioning. Here we investigate the interactions and function of this case study IDR in cell-free reconstitutions, an engineered cell reporter, and the adipocyte cell-state transition.
Here we show that MED1IDR condensates reconstituted in a cell-free lysate selectively partition positive regulators of transcription while excluding negative regulators. High local concentration of MED1IDR at a specific genomic locus partition these positive regulators and activate transcription in cells. The IDRs of positive regulators of transcription are necessary and sufficient for selective partitioning, which is driven by the patterning of charged residues into alternating blocks of charge in these IDRs. Manipulating the charge pattern of IDRs or swapping IDRs can retarget partitioning in cells, leading to changes in function. IDRs with similar charge patterning to MED1IDR exhibit similar selective partitioning and function, demonstrating that these findings are applicable beyond MED1 and represent general principles for how proteins are sorted into different condensates. While we identify specific feature patterns for a transcriptional pathway, it is likely that other biochemical pathways where IDRs play a role in compartmentalization will have evolved their own set of feature patterns that facilitate the sorting of proteins into the various cellular condensates.
RESULTS
MED1IDR condensates selectively partition positive regulators of transcription
To investigate selective partitioning of IDR-mediated condensates, we focused on MED1IDR (Fig 1A, Table S1) which forms condensates when added to a soluble nuclear extract (Fig 1B, methods). These condensates were isolated by centrifugation, which fractionates the extract into a pellet, containing proteins partitioned into MED1IDR condensates, and a supernatant (sup), containing proteins not partitioned (Fig 1C). By proteomic analysis of pellet and supernatant fractions, we calculated a partition ratio, defined as pellet over supernatant (pellet/sup) for every identified protein in the extract (Table S2). Gene ontology analysis of highly partitioned proteins showed enrichment for positive regulation of RNA Pol II transcription (Fig S1A). Among these positive regulators of RNA Pol II transcription are SPT6, IWS1, and CTR9 (along with all subunits of the PAF1 complex), as well as subunits of RNA Pol II (Fig 1D,S1B). Among the least partitioned proteins were proteins known to form distinct nuclear condensates (e.g., HP1α in heterochromatin, NPM1 in nucleoli, etc.), demonstrating the selectivity of partitioning in this assay (Fig 1D,S1B). Proteomics data were corroborated by immunoblot of an independent fractionation experiment with an additional control of not adding MED1IDR to ensure pelleting was due to MED1IDR. RPB1 (largest subunit of RNA Pol II), CTR9, SPT6, and IWS1 were only in the pellet fraction in the presence of MED1IDR while FUS and PTBP1, RNA-binding proteins implicated in nuclear condensates, remained in the supernatant (Fig 1E), confirming that specific positive regulators of transcription are selectively partitioned by MED1IDR condensates in vitro.
Figure 1. MED1IDR condensates selectively partition positive regulators of transcription from a nuclear extract.
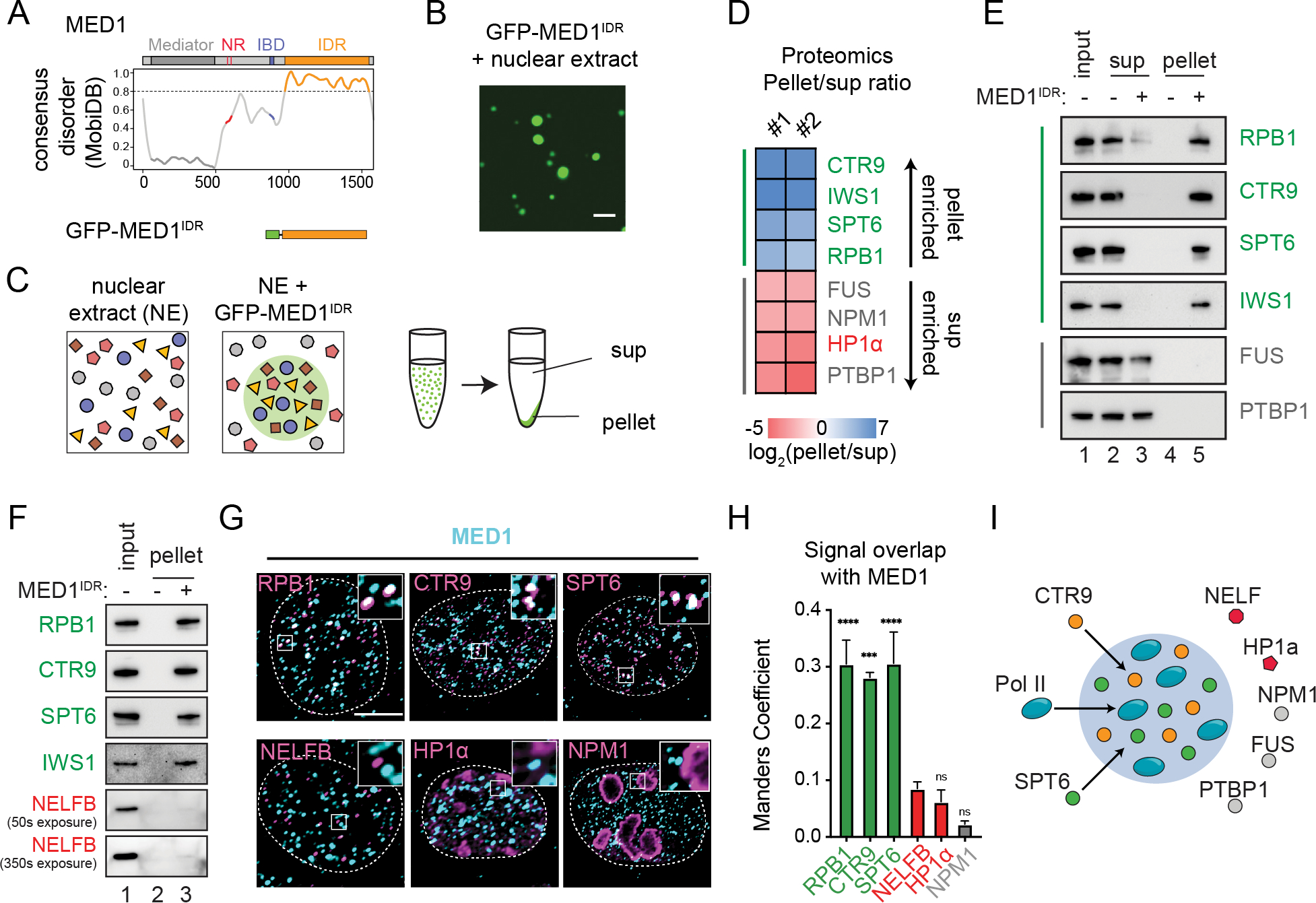
A) Consensus MobiDB disorder prediction102 for MED1. The MED1IDR (orange) was purified as an mEGFP fusion (methods).
B) Purified mEGFP-MED1IDR forms droplets in nuclear extract (methods). Scale, 5μm.
C) Illustration of pelleting experiment.
D) Heatmap of log2(pellet/sup) values from proteomics analysis. See also Figure S1B.
E) Immunoblot of input, supernatant or pellet fractions in the presence or absence of MED1IDR using indicated antibodies.
F) Immunoblot of input and pellet fraction in presence or absence of MED1IDR using indicated antibodies.
G) Co-immunofluorescence against MED1 (cyan) and indicated proteins (magenta). Scale, 5μm.
H) Manders overlap coefficient per cell from co-IF shown in 1G. Mean±SEM. P-values, one-way ANOVA.
I) Illustration summarizing observed selective partitioning by MED1IDR condensates.
See also Figure S1.
To investigate whether the identified proteins were specific to MED1IDR condensates, we analyzed data44 from a similar fractionation experiment with NPM1. For the 200 most partitioned proteins by either NPM1 or MED1IDR condensates 92% of proteins are unique (Fig S1C). Importantly, no positive regulators of RNA Pol II were in the NPM1 condensate dataset nor were the NPM1-partitioned nucleolar proteins found in our MED1IDR dataset (Fig S1D), providing evidence that the proteins partitioned by MED1IDR condensates are specific.
Given that MED1IDR condensates selectively partition positive allosteric regulators of RNA Pol II which function in opposition to the negative elongation factor (NELF) complex45, we next investigated the partitioning of NELF. The interactions of RNA Pol II with CTR9 and SPT6 or with the NELF complex are mutually exclusive, and each provides distinct conformations leading to active or inactive states for RNA Pol II46,47. In our proteomics dataset, NELF subunits were neutral or depleted from the MED1IDR pellet fraction (Fig S1B). Immunoblots of an independent experiment confirmed that nearly all RPB1, CTR9, SPT6, or IWS1 were in the MED1IDR pellet fraction (relative to the input signal) whereas the NELFB subunit was only modestly detected (Fig 1F). While NELF is capable of forming condensates48, our data suggest MED1IDR condensates have a strong bias for partitioning positive regulators over NELF.
To investigate whether endogenous MED1 colocalizes with partitioned proteins in cells, we performed co-immunofluorescence (co-IF). Co-IF of MED1 with partitioned proteins RPB1, CTR9, or SPT6 revealed significant signal overlap with MED1 foci, while excluded factors showed relatively lower signal overlap (Fig 1G–1H). These data show that at endogenous protein concentrations, MED1 foci colocalize with RPB1, CTR9, and SPT6 (Fig 1I).
High levels of MED1 occupancy at enhancers correlate with increased RPB1, CTR9, SPT6, and transcription at associated genes
To investigate the relative chromatin occupancy of MED1 to RNA Pol II, CTR9, and SPT6, we analyzed published ChIP-seq datasets. Clusters of enhancers with high MED1 occupancy are termed “super-enhancers” and represent condensates in mouse embryonic stem cells (mESCs)3,49. Using ChIP-seq datasets from mESCs3,50,51 and annotations for super-enhancers52, the gene body occupancy of RPB1, CTR9, or SPT6 was highest for genes near enhancers with the largest MED1 occupancy (Fig 2A,S2A). NELFA ChIP-seq data51 did not follow this trend (Fig 2A,S2A). Furthermore, genes with highest MED1 occupancy at their enhancers had highest RPB1, CTR9, and SPT6 occupancy at their gene bodies with an apparent threshold level of MED1 (Fig 2B), supporting a model where genomic loci with the highest MED1 occupancy, above an apparent threshold, partition proteins identified as constituents of MED1IDR condensates. We next asked whether this increase in occupancy of positive regulators of transcription was associated with increased RNA Pol II activity and rates of transcription. Phosphorylation of RNA Pol II at S5 or S2 is associated with RNA Pol II activity53. Genes associated with enhancers with high MED1 occupancy had higher levels of gene body S5ph and S2ph (Fig S2B–S2D). RNA Pol II is regulated by NELF-mediated promoter-proximal pausing which can be assessed by the traveling ratio54. The lower the ratio, the less often RNA Pol II is found paused at promoters relative to elongating within gene bodies. Strikingly, genes with the highest MED1 occupancy at their enhancers had lower traveling ratios (Fig S2E). Consistent with having higher occupancy of positive regulators and lower pausing, genes with the highest MED1 occupancy at their enhancers had higher rates of transcription, as measured by GRO-seq55 (Fig 2C), with the same apparent threshold observed for ChIP signal (Fig 2B). These data support a model where high local concentration of MED1 partitions RNA Pol II and its positive allosteric regulators, leading to a greater frequency of assembling elongation-competent RNA Pol II and, therefore, higher rates of transcription.
Figure 2. High local concentration of MED1IDR is sufficient to partition positive regulators and activate gene transcription in cells.
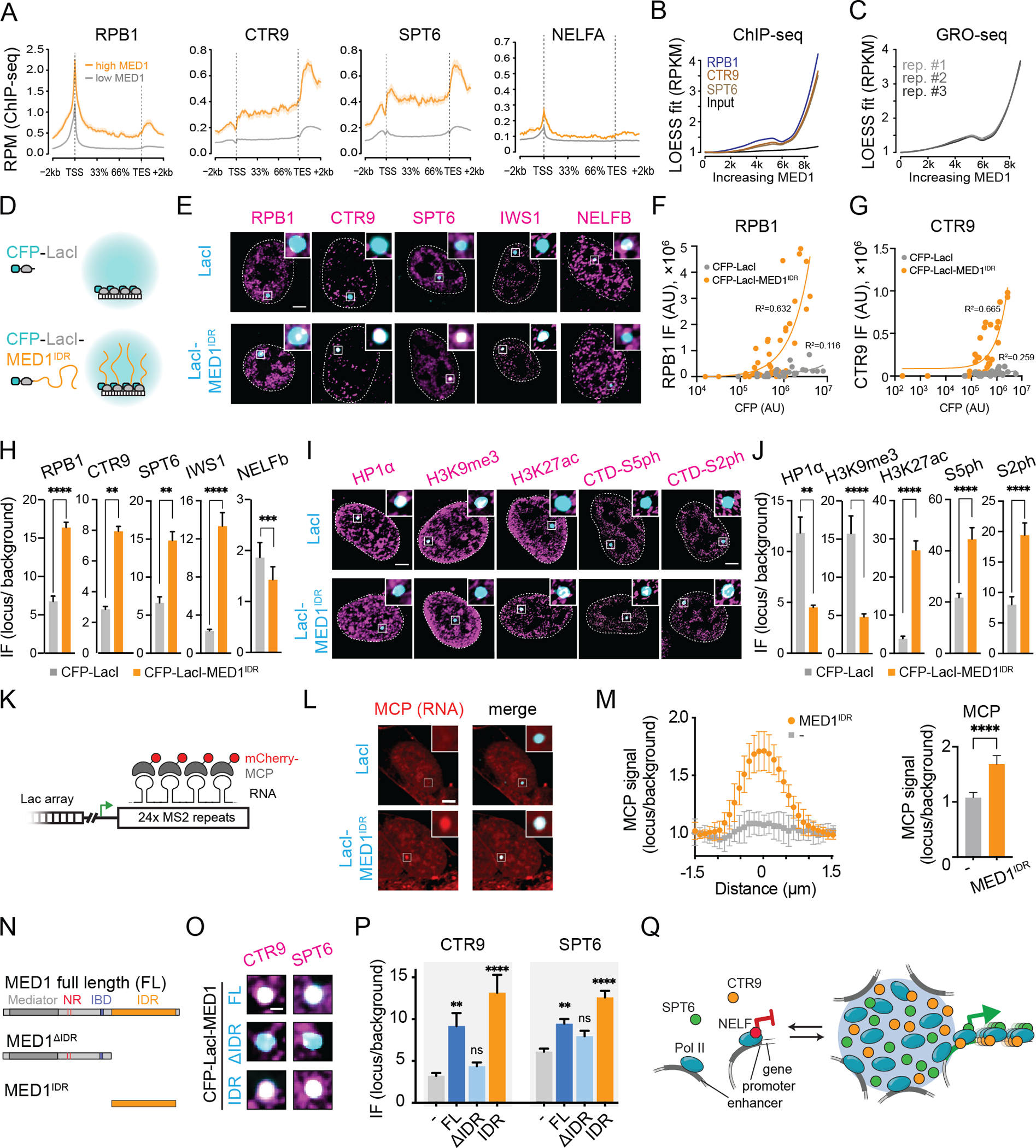
A) Metagene plots for indicated ChIP-seq datasets for genes proximal to annotated enhancers in mESCs that either have high (orange) or low (gray) MED1 occupancy.
B) LOESS polynomial fit curve for scatter plots of ChIP-seq read densities at gene bodies ranked by MED1 occupancy at each gene’s annotated enhancer.
C) Same as 2B, but for GRO-seq data for mESCs.
D) Schematic of Lac array cells.
E) IF for indicated proteins (magenta) in Lac array cells expressing indicated LacI fusion. Scale, 5μm.
F) Scatter plots of RPB1 IF (Y-axis) against CFP (X-axis) for foci of CFP-LacI (gray) or CFP-LacI-MED1IDR (orange).
G) Same as 2F for CTR9.
H) IF quantification of indicated protein at CFP-LacI (gray) or CFP-LacI-MED1IDR (orange). Mean±SEM. p-values, t-test.
I) Same as 2E for indicated factors.
J) Same as 2H for indicated factors.
K) Schematic of MS2 repeats in Lac array cells.
L) Cells co-transfected with mCherry-MCP and CFP-LacI or CFP-LacI-MED1IDR. Scale, 5μm.
M) Left: mCherry-MCP relative signal across foci for CFP-LacI (gray) or CFP-LacI-MED1IDR (orange). Right: mean±SD of central MCP data points (methods). P-value, t-test.
N) Diagram of MED1 truncations tested as CFP-LacI fusions.
O) IF for indicated proteins (magenta) in Lac array cells expressing indicated LacI fusion. Zoom in on Lac focus. Scale, 1μm.
P) IF quantification for 2O. Mean±SEM. p-values, one-way ANOVA vs CFP-LacI.
Q) Model for function of MED1IDR condensate-mediated partitioning.
See also Figure S2.
High local concentration of MED1IDR is sufficient to partition positive regulators and activate gene transcription in cells
To investigate whether MED1IDR is sufficient to selectively partition positive regulators of transcription when recruited to a specific genomic locus, we used the U2OS 2-6-3 cell line56 which contains an integrated LacO array (hereafter referred to as Lac array cells) and compared cells transfected with either CFP-LacI or CFP-LacI-MED1IDR (Fig 2D). In Lac array cells, immunofluorescence (IF) signal for the positive regulators of transcription identified in the extract fractionation was enriched at CFP-LacI-MED1IDR foci relative to control (Fig 2E–2H). NELFB showed the opposite result (Fig 2E–2H). Interestingly, IF signal for the positive regulators was observed only in the brightest CFP-LacI-MED1IDR foci, with an apparent threshold amount of MED1IDR required for partitioning (Fig 2F–2G). These results demonstrate that high concentrations of MED1IDR at a defined genomic locus recapitulate its selective partitioning observed in nuclear extract.
We next investigated whether recruitment of high local concentrations of MED1IDR is sufficient to change the chromatin landscape and activate transcription. Under basal conditions, the Lac array locus is within heterochromatin and transcriptionally silent56. IF for HP1α or H3K9me3, markers of heterochromatin, showed enrichment at CFP-LacI foci, but not at CFP-LacI-MED1IDR foci (Fig 2I–2J). Conversely, IF signal for H3K27ac, a marker for active chromatin, showed enrichment at CFP-LacI-MED1IDR foci (Fig 2I–2J). IF signal for RNA Pol II S5ph or S2ph was also enriched at CFP-LacI-MED1IDR foci (Fig 2I–2J). There is a reporter gene with MS2 RNA loop repeats downstream of the LacO array, which can be visualized by mCherry-MCP (Fig 2K)56. We observed enrichment of mCherry-MCP at CFP-LacI-MED1IDR foci compared to control (Fig 2L–2M). These results demonstrate that high local concentration of MED1IDR at a specific genomic locus is sufficient to create an active chromatin environment.
MED1IDR is necessary for selective partitioning
To test whether MED1IDR is necessary for partitioning CTR9 and SPT6, we performed IF for these factors in Lac array cells containing CFP-LacI fusions with either full-length MED1 (FL), MED1 with the IDR region deleted (ΔIDR), or just MED1IDR (IDR) (Fig 2N). IF signal for CTR9 and SPT6 was depleted in foci lacking MED1IDR (Fig 2O–2P), demonstrating that MED1IDR is necessary for partitioning. To investigate whether the Mediator complex was recruited by MED1IDR, we performed IF for MED14, a core subunit of the complex. In agreement with studies showing that MED1 interacts with Mediator via its N-terminus37,57, MED14 was only found in FL and ΔIDR foci (Fig S2F–S2G). In summary, high local concentrations of MED1IDR partition RNA Pol II with its positive regulators, exclude negative regulators, alter chromatin environment, and activate gene transcription (Fig 2Q).
MED1IDR and its partitioned proteins are required for gene activation during a cell-state transition
To investigate whether MED1IDR is required for gene activation events during a cell-state transition, we used the 3T3-L1 adipogenesis model58 which requires MED1 for differentiation59. During differentiation, MED1 is redistributed into new super-enhancers60 (Fig 3A–3C), which activate genes critical for adipogenesis (Fig 3D–3E). In agreement with these ChIP-seq data, co-IF for MED1 and RPB1 or CTR9 showed an increase in overlap and focus size during differentiation (Fig S3A–S3B) while protein levels were unchanged or reduced (Fig S3C). These data suggest a redistribution of RPB1 and CTR9 into newly formed MED1 foci during differentiation.
Figure 3. MED1IDR is required for gene activation during a cell-state transition.
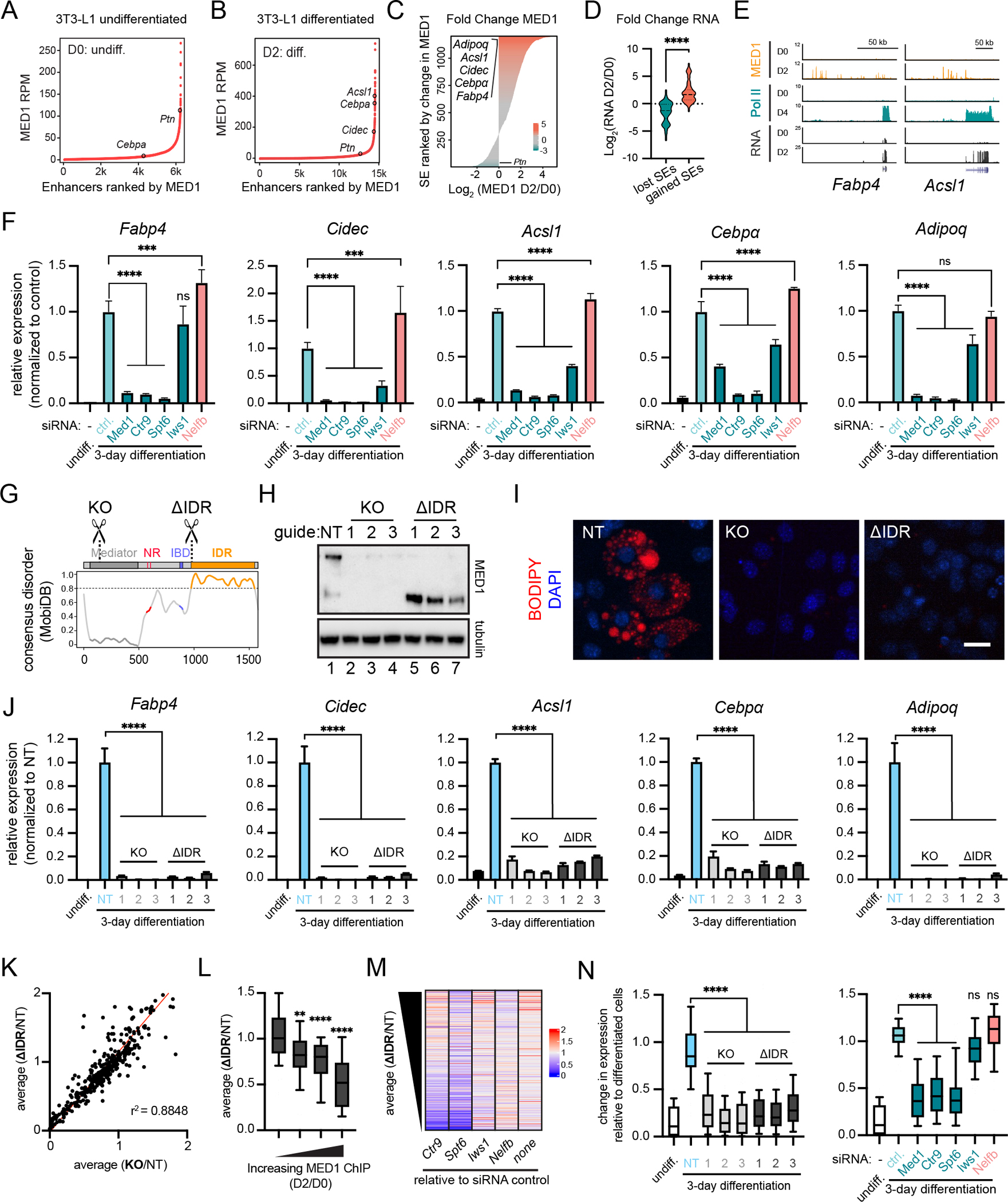
A) Rank ordered plot of enhancers in untreated 3T3-L1 cells (Day 0, D0) ranked by increasing MED1 ChIP-seq signal (RPM).
B) Same as 3A, but for 3T3-L1 cells after 2 days (D2) of differentiation.
C) Horizontal bar plot of all genomic regions containing a super-enhancer at D0 or D2 ranked by log2 (MED1_D2/MED1_D0).
D) Violin plots of the log2 fold change in RNA expression for the genes proximal to top 100 and bottom 100 super-enhancers from Figure 3C. p-values, t-test.
E) Gene tracks of MED1 and RNA Pol II ChIP-seq, and RNA-seq from 3T3-L1 cells.
F) Relative expression of adipocyte genes after knockdown of the indicated transcriptional regulators. Mean±SD. p-values, one-way ANOVA vs control.
G) Location of sgRNAs for MED1 knockout (KO) and MED1ΔIDR (ΔIDR) cell lines. See also Figure S3H.
H) Immunoblot for N-terminal epitope of MED1 in 3T3-L1 CRISPR-Cas9 engineered cells.
I) Images of CRISPR-Cas9 engineered cell lines stained with BODIPY and DAPI (nuclei). Scale, 20μm.
J) Relative expression of adipocyte genes. Mean±SD. p-values, one-way ANOVA vs NT.
K) Scatter plot of average FPKM from either the three ΔIDR cells (y-axis) or the three KO cells (x-axis) each divided by FPKM from the NT cells for all adipocyte-activated genes. r2 = goodness-of-fit for a linear regression.
L) Adipocyte-activated genes with annotated MED1-occupied enhancers were ranked by the fold increase from D0 to D2 and split into 4 equal bins. Boxplot(10–90%) of average(ΔIDR/NT) for each bin. p-values, one-way ANOVA vs first bin.
M) Heatmap of fold change in FPKM for the indicated knockdowns (X-axis labels) relative to the siRNA control (“none”) ranked by decreasing average(ΔIDR/NT). See also Fig S3O.
N) Boxplot(10–90%) showing FPKM of KO or ΔIDR samples (left) or siRNA experiments (right) relative to FPKM of control day-3 differentiated cells for adipocyte-activated genes sensitive to deletion of MED1IDR (ΔIDR/NT ≤ 0.5). p-values, one-way ANOVA vs NT (left) or ctrl (right).
See also Figure S3.
To test the necessity of MED1 and proteins partitioned by MED1IDR, we performed siRNA knockdown of Med1, Ctr9, Spt6, Iws1, and Nelfb followed by an adipogenesis assay. These transcription regulators are expressed at the same level before and after adipogenesis (Fig S3D), but their expression is significantly reduced by siRNA knockdown (Fig S3F). Knockdown of Med1 led to a reduction in the activation of key adipocyte genes regulated by the largest accumulation of MED1 at newly formed super-enhancers (Fig 3F). Similarly, knockdown of Ctr9 and Spt6 led to a comparable or greater decrease, while knockdown of Iws1 led to a modest decrease (Fig 3F). Interestingly, knockdown of Nelfb led to a modest increase in expression (Fig 3F). These effects were corroborated by BODIPY staining (Fig S3G).
To test whether MED1IDR is required for 3T3-L1 differentiation, we used CRISPR/Cas9 to delete MED1IDR. We designed three guide RNAs to target either early exons or the last exon of the Med1 gene (Fig 3G,S3H), leading to frameshift-induced knockout (KO) or deletion of the IDR (ΔIDR), respectively (Fig 3H). We confirmed by co-immunoprecipitation that, as expected57, the MED1-ΔIDR still interacts with the Mediator complex (Fig S3I). These KO and ΔIDR cell lines were compared to a non-targeting control (NT) under differentiation conditions (methods). While the NT cells produced large fat deposits, KO and ΔIDR cells lacked fat deposits (Fig 3I,S3J). Similarly, expression of key adipocyte genes was activated during differentiation in NT cells but had decrease expression in all KO and ΔIDR cells (Fig 3J). To ensure that our engineered cells respond to signals in the differentiation media, we tested the expression of genes activated during early adipogenesis (4h) independently of MED1 and found that all cell populations showed increased expression (Fig S3K). We also performed a time course and found that KO and ΔIDR cells failed to activate these genes even at later time points (Fig S3L). Taken together, MED1IDR and proteins partitioned into MED1IDR condensates are required for the activation of genes necessary for adipocyte cell-state transition.
We next performed RNA-seq of the siRNA and CRISPR cell lines described above (Table S3). By focusing our analysis on genes activated during adipogenesis (methods), we find that the average effect on expression for the 3 KO and 3 ΔIDR cell lines show a linear correlation (r2 = 0.88) (Fig 3K), demonstrating that deletion of the MED1IDR phenocopies MED1 KO in our assay conditions. As expected59,61, only a subset of adipocyte-activated genes is downregulated by Med1 KO or ΔIDR. By comparing the change in MED1 ChIP-seq to the change in expression for ΔIDR cells relative to NT cells, we observed that genes most sensitive to loss of MED1IDR are those genes with the largest increase in MED1 occupancy at their associated enhancers (Fig 3L). The same trend is observed for Med1 KO cells (Fig S3M), for Ctr9 and Spt6 knockdown, to a lesser degree for Iws1 knockdown, and in the opposite direction for Nelfb knockdown (S3N). This trend is not observed for the respective control differentiated cell lines (Fig S3M–S3N). Additionally, heatmaps plotting change in expression due to siRNA knockdowns ranked by ΔIDR/NT, show that the most ΔIDR-sensitive genes are also most sensitive to Ctr9 and Spt6 knockdown (Fig 3M,S3O). Furthermore, ΔIDR-sensitive genes (average ΔIDR/NT<0.5) are also downregulated in Med1 KO and siRNA knockdown of Med1, Ctr9, and Spt6 (Fig 3N). These RNA-seq results further support the conclusion that MED1IDR, CTR9, and SPT6 are operating through a similar pathway with the greatest effects for genes regulated by enhancers with highest MED1 occupancy.
IDRs of partitioned factors are sufficient and necessary to recapitulate selective partitioning
Analysis of proteomics data revealed that proteins with the highest partition ratio were enriched for IDRs (Fig 4A), which led us to investigate the sufficiency of IDRs for selective compartmentalization into MED1IDR condensates. We purified the terminal IDRs of CTR9 and SPT6 as recombinant mCherry fusions (Fig 4B, Table S1). As negative controls, we purified previously studied IDRs of NELFA, HP1a, and FUS (Fig 4B, Table S1), which are implicated in nuclear condensates48,62–64, and were all neutral or excluded from MED1IDR condensates (Fig 4C). We tested the partitioning of these mCherry-IDRs into GFP-MED1IDR droplets in vitro (Fig 4D, methods) either in the presence of total cellular RNA, which has been shown to stimulate MED1IDR droplet formation65 (Fig 4D–4F), or in the absence of RNA where MED1IDR will not form droplets on its own (Fig S4A–S4C). No nuclear extract was added. In both experiments, CTR9IDR or SPT6IDR partition with GFP-MED1IDR droplets (Fig 4E–4F, S4B–S4C). Consistent with our proteomics analysis, NELFAtentacle, HP1aIDR, and FUSIDR do not partition into MED1IDR droplets (Fig 4E–4F, S4B–S4C). In these conditions, none of the mCherry fusions form condensates on their own (Fig S4D–S4F). These results demonstrate that CTR9IDR and SPT6IDR are sufficient for selective partitioning into MED1IDR condensates without any other proteins or annotated interaction domains.
Figure 4. IDRs of partitioned factors are sufficient and necessary to recapitulate selective partitioning.
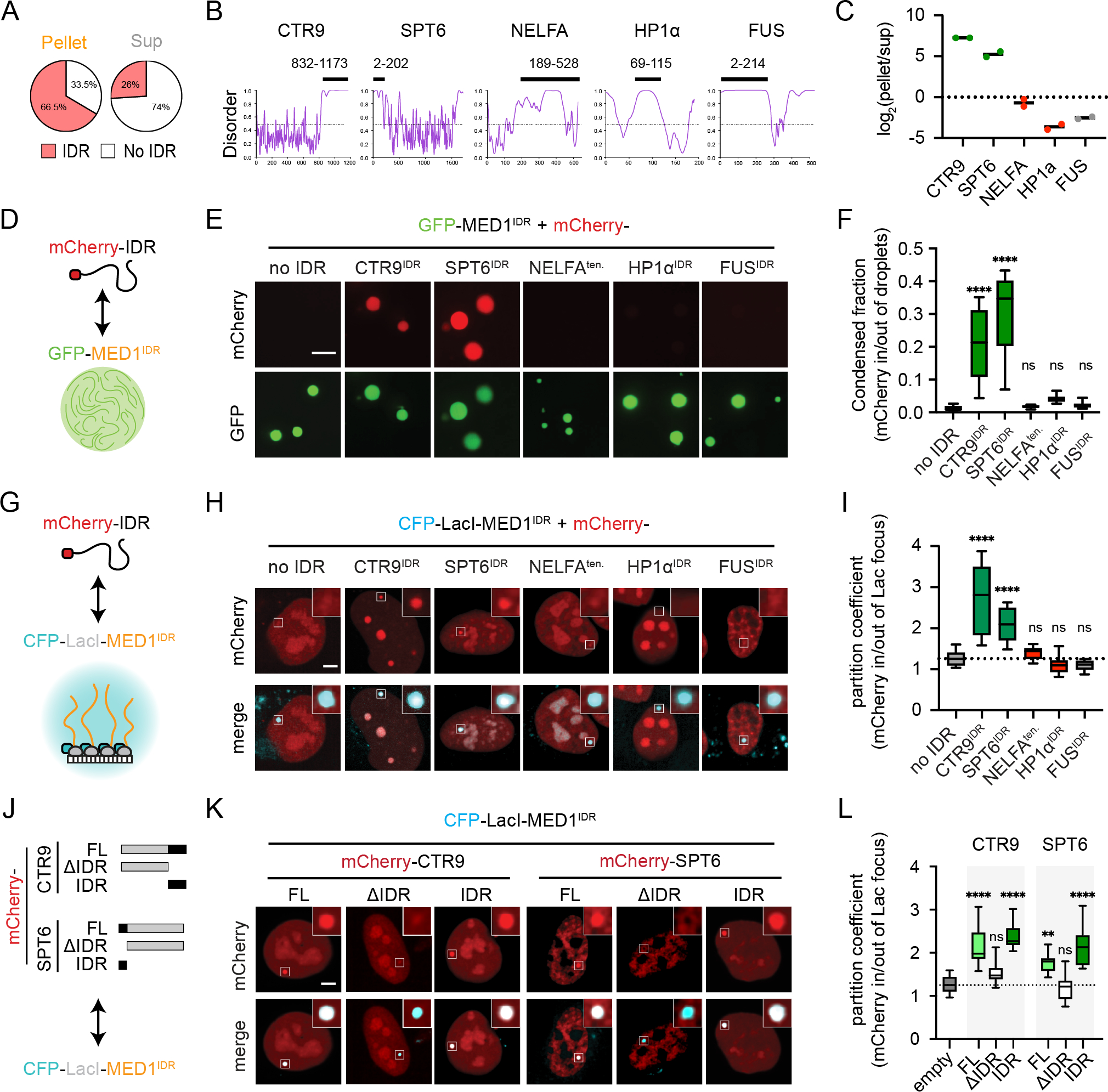
A) MED1IDR partitioned proteins are themselves enriched for IDRs.
B) PONDR plots (VSL2). Black bar highlights IDR purified as an mCherry fusion.
C) Proteomics data from MEDIDR pelleting experiments showing replicate log2(pellet/sup) values for each protein.
D) Cartoon depiction of purified mCherry-IDRs tested. See also Figure S4A.
E) Images of indicated mCherry-IDR partitioning into mEGFP-MED1IDR droplets in total RNA. Scale, 5μm. See also Figure S4B.
F) Boxplot(10–90%) of condensed fraction. p-values, one-way ANOVA vs no IDR control.
G) Cartoon depiction of experimental setup.
H) Images of mCherry-IDRs (top row) tested for partitioning into CFP-LacI-MED1IDR foci (bottom row). Scale, 5μm.
I) Boxplot(min-max) of partition coefficients for experiments in 4H. p-values, one-way ANOVA vs no IDR control.
J) Diagram of CTR9 and SPT6 truncations as mCherry fusions tested for partitioning into CFP-LacI-MED1IDR in Lac array cells.
K) Same as 4H, but for indicated mCherry fusions.
L) Same as 4I, but for indicated mCherry fusions.
See also Figure S4.
To corroborate these findings in Lac array cells, we co-transfected CFP-LacI-MED1IDR with mCherry fusions of the same IDRs (Fig 4G). Matching in vitro data, CTR9IDR and SPT6IDR partitioned into MED1IDR foci, while NELFAtentacle, HP1aIDR, and FUSIDR were not partitioned (Fig 4H–4I). The results were similar when the fusion fluorescent proteins were flipped (Fig S4G–S4I). No partitioning of CTR9IDR or SPT6IDR in CFP-LacI without MED1IDR was observed (Fig S4J–S4L). To test whether the IDRs of CTR9 and SPT6 are necessary for partitioning into MED1IDR foci, we cloned full-length (FL) and IDR deletion (ΔIDR) (Fig 4J) as mCherry-fusions and tested their partitioning into CFP-LacI-MED1IDR. FL and IDRs of CTR9 and SPT6 partitioned, but ΔIDR proteins did not (Fig 4K–4L). These results corroborate in vitro experiments and demonstrate that IDRs are necessary for partitioning.
We next tested seven additional IDRs from two classes: 1) IDRs from other highly partitioned proteins from our proteomics data (IWS1, LEO1, and PAF1) and 2) other IDRs implicated in the formation of nuclear condensates (DDX4, NELFE, EWSR1, and p300) (Table S1)6,31,48,66. These IDRs were co-transfected with CFP-LacI-MED1IDR into Lac array cells (Fig S4M). Only the IDRs from highly partitioned proteins (IWS1IDR, LEO1IDR, and PAF1IDR) significantly partitioned into MED1IDR foci above the control (Fig S4N–S4O). DDX4IDR and NELFEtentacle were not different from the control, while EWSR1IDR and p300IDR had lower partitioning than the control. Interestingly, EWSR1IDR and p300IDR are both implicated in positive regulation of transcription6,66 yet are effectively excluded, suggesting specialization among transcriptional condensates (discussion). These results show that even among disordered regions there is a wide range of partitioning.
Selectively partitioned IDRs are enriched for blocks of both positive and negative amino acids
To understand the IDR sequence features responsible for the observed selectivity, we compared sequence parameters of IDRs from the most partitioned proteins (partitioned IDRs), IDRs from the least partitioned proteins (excluded IDRs), all IDRs in the proteome (All IDRs) (based on consensus IDR predictions, methods, Table S4), and all IDRs from proteins identified in the nuclear extract used in our pelleting assays (extract IDRs). As expected, all groups had a similar degree of disorder-promoting amino acids (Fig S5A). Although aromatic residues play a well-documented role in phase separation26,67, the distributions for aromatic residue fraction or patterning of all groups were similar (Fig S5B–S5C). Partitioned IDRs had a significantly higher fraction of charged amino acids (E, D, R, and K) and a significantly lower fraction of A, G, Q, and P relative to excluded IDRs (Fig S5D). In agreement with these results, partitioned IDRs had a significantly higher fraction of charged residues (FCR) (Fig 5A), fraction of acidic residues (Fig S5E), and fraction of basic residues (Fig S5F), but partitioned IDRs as a group did not show a difference in net charge per residue (NCPR) (Fig S5G).
Figure 5. Patterned blocks of acidic and basic residues in IDRs are necessary and sufficient for selective partitioning.
A) Partitioned IDRs have a higher FCR than other IDR classes tested. Boxplots(10–90%). p-values, one-way ANOVA vs partitioned IDRs.
B) NCPR for 10-residue sliding window (black line) across the indicated disordered regions. Vertical red and blue lines represent acidic or basic residues, respectively. Y-axis, charge index. The position of acidic or basic blocks (10-residue window NCPR ≥ abs (5)) is shown as dark red bars or dark blue bars, respectively.
C) Same as 5A, but for number of charged blocks.
D) Correlation analysis of partition coefficient and FCR for all IDRs tested in Lac array cells (Fig 4I, S4O). r2 = goodness-of-fit for a linear regression.
E) Same as 5D but for number of charged blocks. Red line, linear fit.
F) NCPR plots as in 5B are shown for CTR9IDR and the CTR9IDR mutants tested.
G) Left: images of indicated mCherry fusions and CFP-LacI-MED1IDR. Scale, 1μm. Right: Boxplot(10–90%) of partition coefficients for mCherry-CTR9IDR wildtype and mutants shown on left. p-value, one-way ANOVA vs WT.
H) Same as 5F for SPT6IDR.
I) Same as 5G for SPT6IDR.
J) NCPR plots as in 5B for SPT6IDR charge and non-charge scramble mutants.
K) Same as 5G for indicated SPT6IDR mutants.
L) NCPR plots as in 5B for the middle region of SPT6IDR (ABABA pattern) and block pattern rearrangements of this middle region.
M) Same as 5G but for block pattern mutants.
N) Schematic of synthetic IDR design. See methods.
O) Illustration of synthetic IDRs tested for partitioning into CFP-LacI-MED1IDR foci in cells and GFP-MED1IDR droplets in vitro.
P) Same as 5G but for synthetic IDRs.
Q) Left: images and quantification of synthetic IDRs partitioning into GFP-MED1IDR droplets in the presence of RNA. Scale, 2μm. Right: Boxplot(10–90%) of condensed fraction of mCherry signal. p-values, one-way ANOVA vs no IDR control.
R) NCPR plots as in 5B of NELFEtentacle (WT) and NELFE blocky tentacle (blocky).
S) Same as 5G, but with WT and blocky NELFEtentacle.
T) Same as 5Q, but with WT and blocky NELFEtentacle.
U) Illustration summarizing observed sufficiency and necessity of alternating charge block pattern for partitioning into MED1IDR condensates.
See also Figure S5.
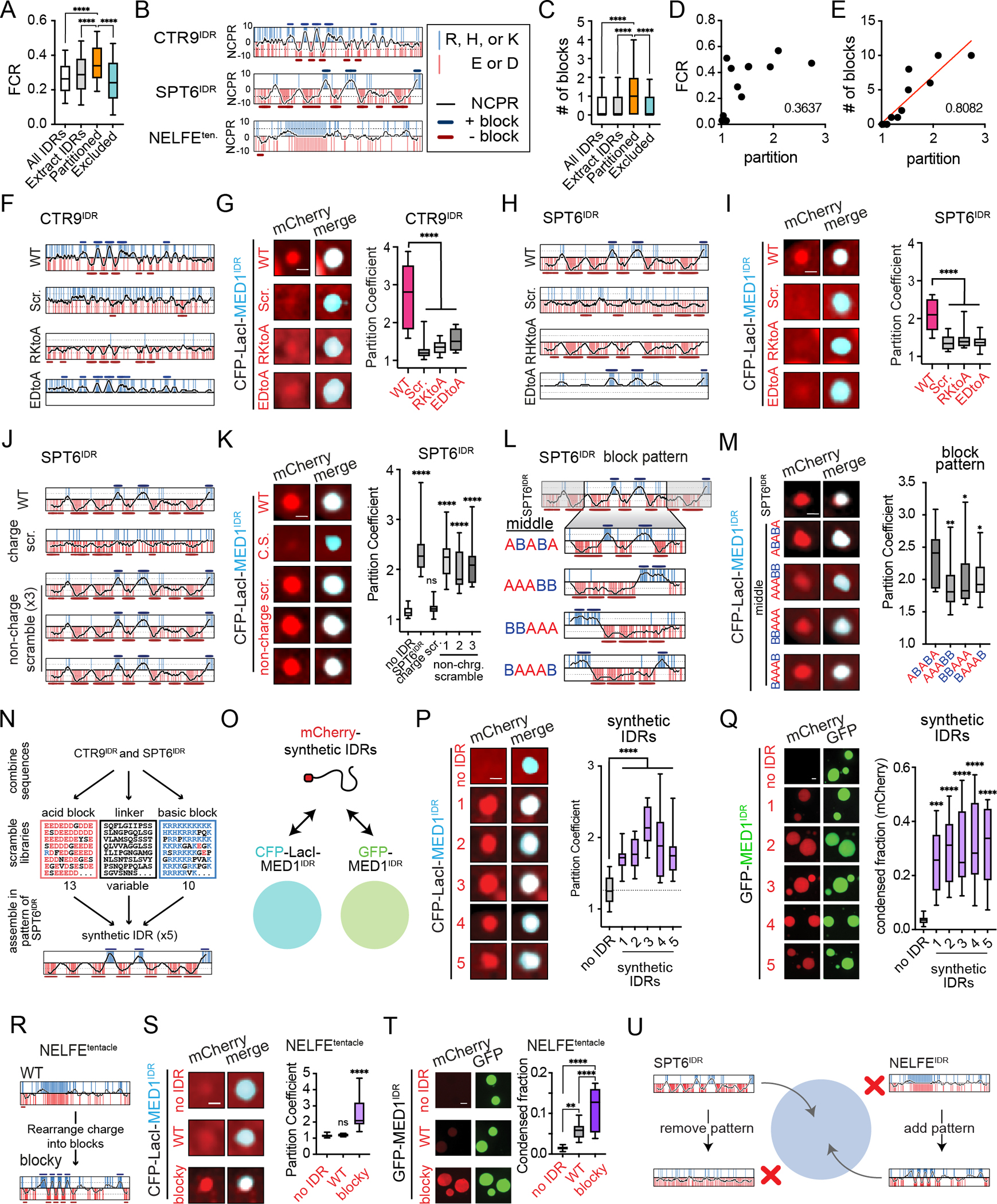
Many of the most partitioned IDRs (e.g., CTR9IDR and SPT6IDR) had striking patterns of local charge density or charge blocks (Fig 5B). Some of these sequences had high kappa values, a measure of charge segregation68, yet as a group, the partitioned IDRs did not have higher kappa (Fig 5SH) but did have a higher value for sequence charge decoration (SCD)69,70, another measure of charge patterning (Fig S5I). To annotate charge blocks, we plotted NCPR for 10-residue sliding windows and defined blocks as any window with NCPR>0.5 for basic and <−0.5 for acidic, followed by merging overlapping windows of the same charge (methods). These plots (Fig 5B) for CTR9IDR or SPT6IDR highlight the striking pattern of alternating charge blocks present throughout these sequences. As a comparison, the NEFLEtentacle, a protein region not partitioned by MED1IDR (Fig S4N), has a similar charge content but lacks the blocky pattern (Fig 5B). By this NCPR block definition, partitioned IDRs have more charge blocks relative to the other groups (Fig 5C). Correlation analysis of partition coefficients for twelve IDRs tested (Fig 4H–4I, S4N–S4O) with FCR or the two parameters for charge patterning (kappa and SCD) gave poor fits in a linear regression (r2 < 0.5), but number of blocks gave the best fit (r2 = 0.81) (Fig 5D–5E, S5J–S5K). These data led us to the hypothesis that multivalent charge blocks are responsible for preferential partitioning into MED1IDR condensates.
Patterning and local density of charged amino acids on CTR9IDR and SPT6IDR are required for selective partitioning
To determine whether the pattern of charge is required for partitioning, we modified specific sequence features in CTR9IDR and SPT6IDR and tested their effect on partitioning. Disrupting overall sequence pattern by scrambling the sequence or disrupting acidic and basic content by alanine substitutions for either IDR diminished partitioning relative to the wildtype sequence (Fig 5F–5I). We next made SPT6IDR sequences where 1) only charged residues were scrambled (“charge scramble”) or 2) only non-charged residues were scrambled (“non-charge scramble”) in three different versions (Fig 5J). The charge scramble SPT6IDR failed to partition into CFP-LacI-MED1IDR foci (Fig 5J–5K). In agreement with charge pattern driving partitioning, all three non-charge scramble mutants partitioned with MED1IDR (Fig 5J–5K). Matching the cell-based data, the charge scramble SPT6IDR had diminished partitioning into GFP-MED1IDR droplets while a non-charge scramble mutant (#3) partitioned like the WT SPT6IDR (Fig S5L). The region within SPT6IDR containing the most striking alternating block pattern (“middle”) was sufficient for partitioning. Changing the pattern of charge blocks within this middle sequence diminished partitioning (Fig 5L–5M). These results demonstrate that patterning and local density of charged residues are important for IDR-mediated partitioning.
We next investigated the conservation of charge block features in CTR9IDR and SPT6IDR. As expected for disordered regions71,72, the positional sequence identity is poorly conserved, but fraction of disordered amino acids, FCR, kappa values, and number of charge blocks are conserved (Fig S5M–S5O). Examining the local sequence alignments for individual blocks revealed insertions, deletions, and substitutions which disrupt positional sequence identity, but maintain local charge density (Fig S5N–S5P). These results suggest that charge blocks within CTR9IDR and SPT6IDR are conserved even as positional sequence identity diverges across the species analyzed.
To understand what types of proteins in the human proteome contain IDRs with charge blocks, we calculated the number of charge blocks in our consensus IDR table (Table S4) and performed gene ontology analysis (methods). IDRs with the highest number of blocks were enriched for positive regulation of RNA Pol II transcription, while highly charged IDRs without blocks were enriched for cytoskeletal protein binding (Fig S5Q).
Charge content and patterning within IDRs are known to be important for phase separation31,73–75. We tested seven of these IDRs from 1) a set of IDRs from transcriptional repressors (MeCP2, CBX2, KDM1A, and RCOR3) (Fig S5R–S5T) and 2) a set of IDRs known to phase separate by charge-mediated interactions (Ki67, NICD, and NPM1)73,74 (Fig S5U–S5W) and none of these IDRs partitioned into MED1IDR foci in Lac array cells. While each of these IDRs contains their own charge pattern, they do not have the charge pattern features described for IDRs partitioned into MED1IDR condensates. These results highlight that even among charged disordered regions there is specificity for partitioning.
Engineered IDRs containing alternating charge blocks selectively partition
Having shown that patterned charge blocks are necessary for selective partitioning, we next engineered these features into proteins to confer gain-of-function partitioning. In the first approach, we designed 5 synthetic IDRs based on CTR9 and SPT6 IDRs (Fig 5N, Table S1, methods). These synthetic IDRs (Fig 5O) partition into MED1IDR foci in cells (Fig 5P) and into MED1IDR droplets in vitro (Fig 5Q). In the second approach, we rearranged the position of charged residues in NELFEtentacle to form an alternating charge block pattern (Fig 5R, Table S1, methods); no changes were made to the position of non-charged residues. This “blocky” NELFEtentacle partitioned into MED1IDR foci in cells (Fig 5S) and into MED1IDR droplets in vitro (Fig 5T) to a greater extent than the wildtype sequence. These results demonstrate that the patterning of charged residues into alternating blocks allows for selective partitioning into MED1IDR condensates (Fig 5U).
Introducing IDRs with alternating charge blocks into NELFE leads to NELF complex partitioning and a decrease in gene activation
We next tested whether replacing the disordered region of NELFE would lead to aberrant partitioning of the NELF complex. We made NELFE chimeras that contain the structured N-terminal region of NELFE, which engages with the complex47, fused to IDRs with alternating charge blocks or IDRs without this pattern and tested their partitioning into MED1IDR foci in cells (Fig 6A). These NELFE chimeras behaved as expected, with those containing patterned IDRs partitioning and those containing non-patterned IDRs not partitioning (Fig 6B–6C). These NELFE chimeras did not partition in the absence of MED1IDR (Fig S6A–B). IF for NELFB showed that the NELF complex was recruited to CFP-LacI-MED1IDR foci only by patterned NELFE chimeras (Fig 6E–6F). This aberrant partitioning of NELF by IDR swaps (Fig 6G) diminished reporter gene expression, as measured by a decrease in MCP signal (Fig 6H–6I). Partitioning of CTR9IDR or SPT6IDR without NELFE does not decrease MCP signal (Fig S6C–S6E). These results demonstrate that protein complexes can be spatially organized by sequence features in their IDRs with functional consequences for gene activity.
Figure 6. Introducing IDRs with alternating charge blocks in NELFE leads to NELF complex partitioning and a decrease in gene activation.
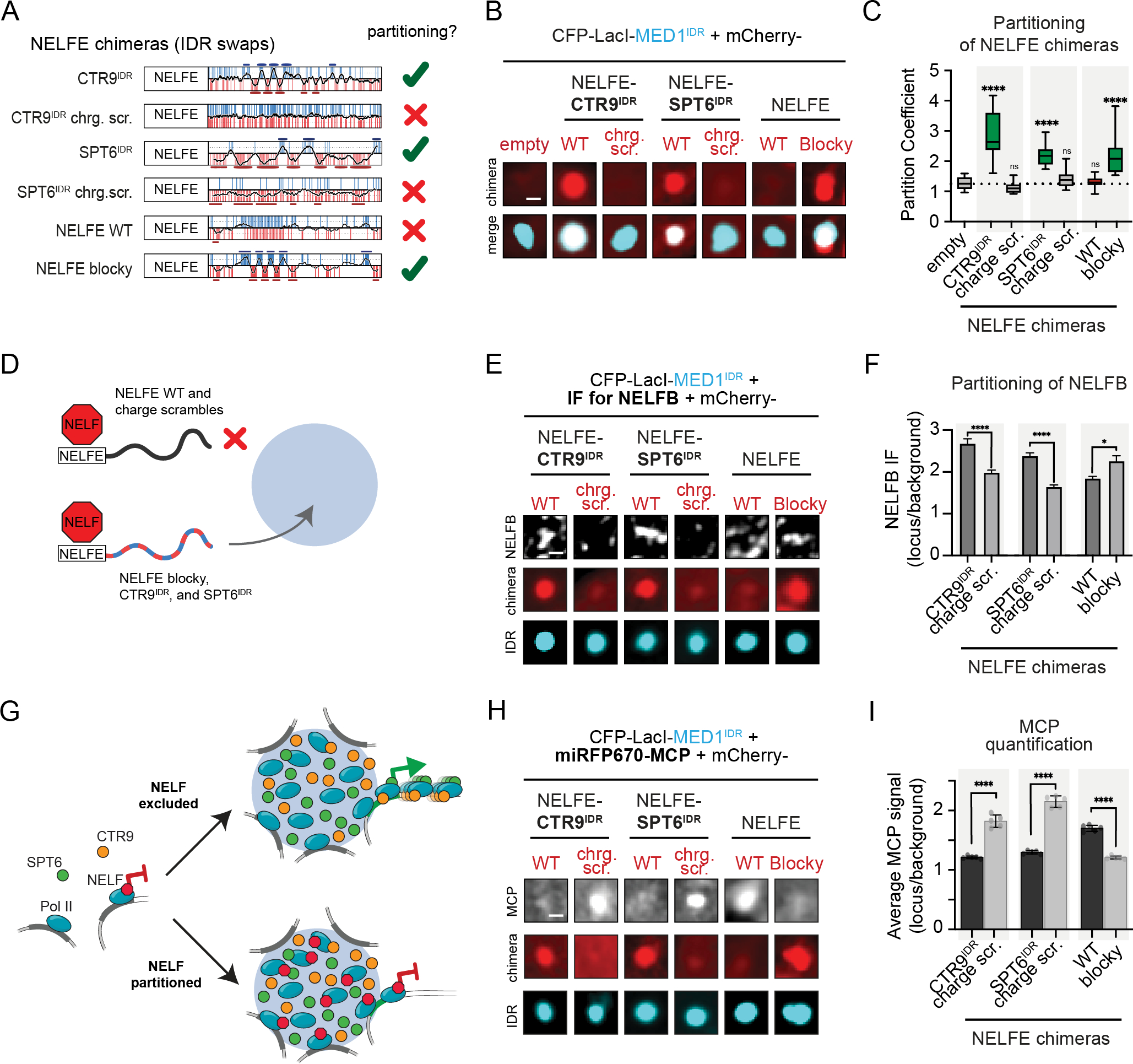
A) Schematic showing NELFE chimeras. Checkmark or X on the right indicates whether the fused IDR partitions into MED1IDR condensates.
B) Representative images of NELFE chimeras as mCherry fusions partitioning into CFP-LacI-MED1IDR foci. Scale, 1μm.
C) Boxplot(10–90%) of partition coefficients for experiments shown in 6B. p-values, one-way ANOVA vs no IDR control.
D) Illustration showing the partitioning or not of NELFE chimeras and consequently the recruitment or not of other NELF subunits.
E) Representative images of NELFB immunofluorescence. Scale, 1μm.
F) NELFB IF quantification for experiments in 6E. Mean±SEM. p-values, t-test for each pair of NELFE chimeras.
G) Illustration of how NELF is expected to be aberrantly partitioned into MED1IDR condensates when its wildtype tentacle is replaced by IDRs with alternating charge blocks, leading to a decrease in gene activation.
H) Representative images showing MCP signal. Scale, 1μm.
I) MCP signal quantification (line profile method as Fig 2M) in CFP-LacI-MED1IDR foci from experiments in 6H. Mean±SEM. p-values, t-test for each pair of NELFE chimeras.
See also Figure S6
Patterning and local density of charged amino acids on MED1IDR are required for selective partitioning, reporter gene activation, and cell-state transition
Having demonstrated the importance of charge blocks for the partitioned IDRs, we next investigated whether the same features were required for MED1IDR function. Similar to CTR9IDR and SPT6IDR, MED1IDR positional sequence identity is not conserved, but disorder, FCR, and the number of charged blocks are conserved (Fig S7A–S7B). While WT MED1IDR formed condensates in extract, scrambled and charge-to-alanine-substitution mutants of MED1IDR (Fig 7A) disrupted droplet formation (Fig 7B–7C). The WT MED1IDR was able to pellet RPB1, CTR9, SPT6, and IWS1 from the extract, but none of the MED1IDR mutants could partition these factors to the same extent (Fig 7D). In Lac array cells, these MED1IDR mutations disrupted partitioning of CTR9IDR and SPT6IDR (Fig 7F–7G, S7E), prevented exclusion of HP1αIDR (Fig S7C–S7D), and prevented reporter gene activation (Fig 7F–7G, S7F).
Figure 7. IDRs with similar alternating charge blocks are required and sufficient for selective partitioning, reporter gene activation, and cell-state transition.
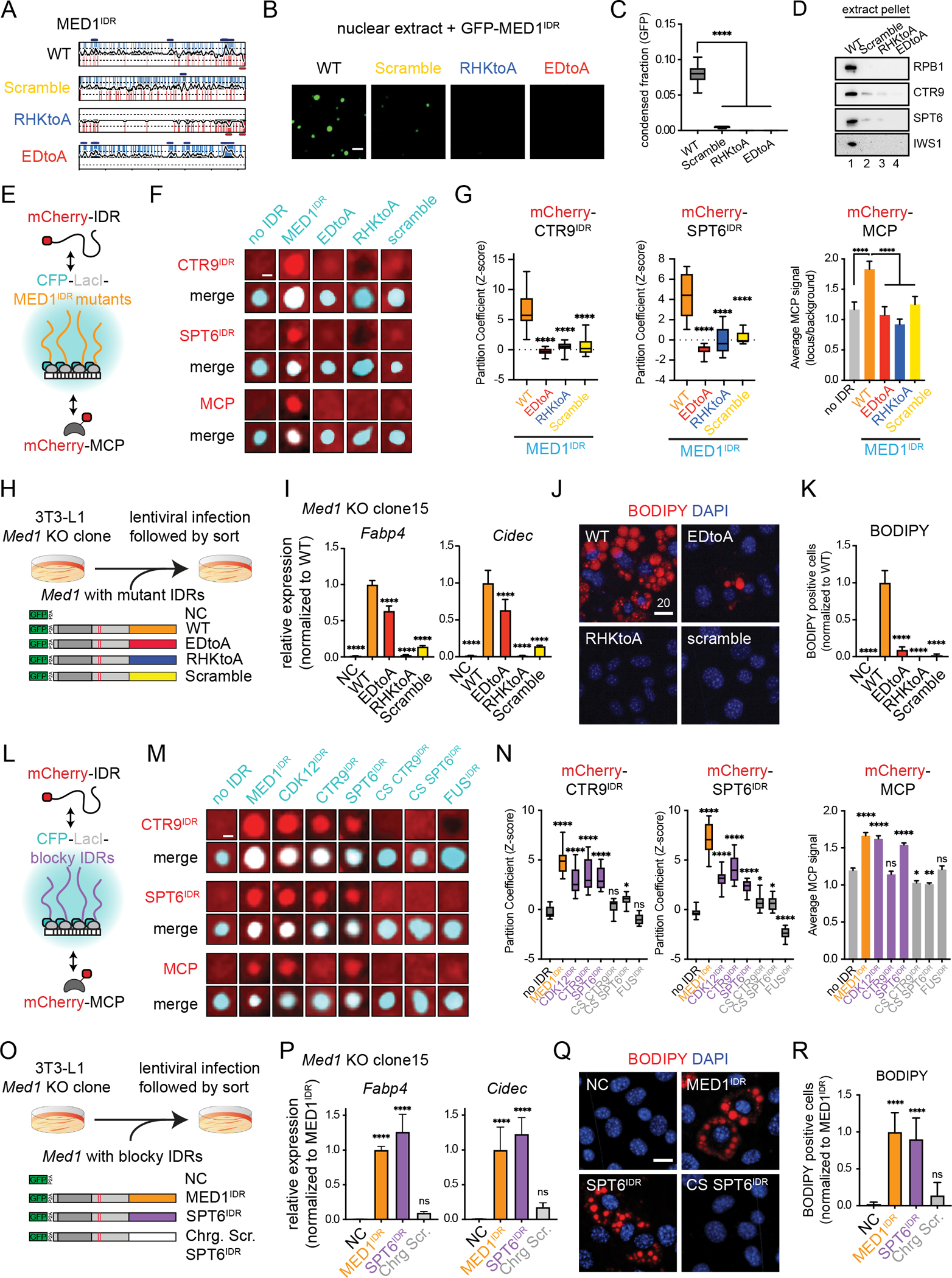
A) NCPR plots for MED1IDR and the mutants tested in 7B-7K.
B) Representative images of mEGFP-MED1IDR, scramble, or charge mutants tested for droplet formation in nuclear extract. Scale, 2μm.
C) Quantification of 7B. Boxplot(10–90%) of condensed fraction of GFP signal. p-values, one-way ANOVA vs WT.
D) Immunoblot of pellet fractions in the presence of recombinant mEGFP-MED1IDR, scramble, or charge mutants using indicated antibodies.
E) Cartoon depicting experiments to test mCherry-IDR partitioning and reporter gene activation by MED1IDR mutants.
F) Representative images of partitioning of mCherry fused to CTR9IDR (top set), SPT6IDR (middle set), or MCP (bottom set) into either wildtype or mutant MED1IDR foci (listed across top in cyan) in Lac array cells. Scale, 1μm.
G) Quantification of 7F. Left and middle: Boxplot of partition coefficients for CTR9IDR and SPT6IDR as Z-scores relative to no IDR mCherry control (Fig S7E). Right: Bar chart of MCP signal at indicated LacI focus normalized to background signal, mean±SD. p-values, one-way ANOVA vs wildtype MED1IDR.
H) Cartoon depiction of 3T3-L1 MED1 KO clone addback experiments with indicated lentiviral constructs.
I) Relative expression of adipocyte genes after induction (day 3) for the indicated addback experiments in Med1 KO clone #15. Mean±SD. Values are normalized to WT. p-values, one-way ANOVA vs WT. See also Figure S7H.
J) Images of Med1 KO clone #15 + indicated addback construct stained with BODIPY and DAPI. Scale, 20μm. See also Figure S7I.
K) Percentage of BODIPY-stained cells for indicated addback constructs. Mean±SD. p-values, one-way ANOVA vs WT.
L) Cartoon depicting experiment to test mCherry-IDR partitioning and gene activation by other blocky IDRs and controls.
M) Same as 7F for indicated CFP-LacI and mCherry fusions
N) Same as 7G for experiments in 7M. See also Fig S7M–S7P.
O) Same as 7H for indicated constructs.
P) Same as 7I, but with indicated addback constructs.
Q) Same as 7J-K, but with indicated addback constructs.
See also Figure S7.
Using a published strategy of addback complementation in 3T3-L1 cells59, we expressed MED1 with either WT, RHKtoA, EDtoA, or scramble IDR, or a non-complement control (NC) in Med1 knockout clones (Fig 7H,S7G). While MED1 with a wildtype IDR rescued both gene activation (Fig 7I) and adipogenesis (Fig 7J–7K, S7I), cells containing MED1 with either RHKtoA, EDtoA, or scramble IDRs were unable to fully rescue either process in two independent Med1 KO clones (Fig S7J–S7L). While the EDtoA mutant exhibited some degree of rescue for the adipocyte genes tested, this construct had only a modest rescue of adipogenesis (Fig 7K,S7L). These data demonstrate that the patterning and local density of charged amino acids on MED1IDR are required for selective partitioning, reporter gene activation, and cell-state transition.
IDRs with similar block patterns have similar partitioning and function
To test whether other IDRs with charge blocks had similar function to MED1IDR, we tested three IDRs containing alternating charge blocks (CDK12IDR, CTR9IDR, SPT6IDR), two control IDRs where the charged residues were scrambled (charge scramble of CTR9IDR and SPT6IDR), and an IDR depleted of charge blocks (FUSIDR) (Fig S7M). Strikingly, in Lac array cells, the blocky IDRs partitioned CTR9IDR, SPT6IDR, and NELFE blocky tentacle while the charge scramble controls and FUSIDR did not (Fig 7M–7N, S7N–S7O). Exhibiting the same selectivity as MED1IDR, the blocky IDRs did not partition NELFE tentacle (Fig S7N–S7L). Like MED1IDR, CDK12IDR and SPT6IDR were both sufficient to activate the reporter gene, but CTR9IDR failed to do so (Fig 7M–N, S7P).
We next performed addback experiments with MED1 having its IDR swapped for 1) a blocky IDR (SPT6IDR), or 2) a control IDR without charge blocks (charge scramble of SPT6IDR) (Fig 7O,S7Q). Surprisingly, the MED1-SPT6IDR chimera rescued adipocyte gene activation (Fig 7P), and adipogenesis like the wildtype MED1 (Fig 7Q–R). In contrast, the MED1 chimera containing the charge scramble SPT6IDR did not rescue adipocyte gene activation (Fig 7P) or adipogenesis (Fig 7Q–R). Notably, SPT6IDR does not share positional sequence identity or linear motifs with MED1IDR but recapitulates the same function. We attempted to test MED1-CDK12IDR chimeras, but they did not express in 3T3-L1 cells. These results show that IDRs with no shared positional sequence identity exhibit similar partitioning and function by nature of their shared charge patterning and demonstrate that the patterning and local charge density on IDRs in general are required for selective partitioning, reporter gene activation, and cell-state transition.
DISCUSSION
Our results demonstrate that IDR-mediated partitioning can be specific, highlighting the degree of functional specificity encoded within IDRs. We propose a model where this functional selectivity facilitates a local environment in which the elongation-competent RNA Pol II complex can be assembled without being challenged by NELF, thereby increasing the rate of RNA Pol II transcription. The formation of these environments will be highly dynamic and their presence and stability at specific genomic loci may relate to the different patterns of frequency and amplitude of transcriptional bursts observed in cells17. The regulatory element landscape (i.e. accessible DNA sequence and chromatin state) will determine the local concentration of transcription activators and coactivators locally seeding and scaffolding condensate assembly76. In support of this model, the amount of H3K27ac at a gene’s associated enhancer positively correlates with burst frequency77. Multivalent interactions among disordered regions, described here, work together with site-specific structure-mediated interactions (e.g., DNA/RNA-binding domains, specific protein-protein interaction domains, etc.) to regulate transcription by creating functionally specialized local proteomes at specific loci21,78,79.
While we focus on MED1IDR and a set of other blocky IDRs from transcriptional regulators, other disordered regions will likely partition functionally related proteins with varying degrees of overlap. Interestingly, we find that the IDR of p300, a coactivator known to form condensates66, does not partition into MED1IDR condensates, suggesting that there are likely different types of transcriptional condensates. Recent data demonstrate that different regulatory elements have specific dependencies for different factors defined by the underlying regulatory element sequence80–83. Whether these different classes of regulatory elements seed and scaffold distinct types of transcriptional condensates which would compete for the available RNA Pol II is an exciting topic for future study.
Our study supports a model wherein different condensates partition biomolecules in part by utilizing specific patterns and types of interactions among IDRs. In the context of transcription, several studies have demonstrated that IDRs are sufficient to target proteins to specific genomic loci40,84,85. There is a great diversity of compositional complexity within IDR sequences that form condensates25,26,31,73,86, suggesting that there are mechanisms of partitioning yet to be discovered. Even among charged IDRs, the specific patterning and density of charge can be enough to change specificity of partitioning70,87–92. For example, while condensate formation of DDX4IDR, NICD, MeCP2IDR, Ki67IDR, and NPM1IDR require patterning of charged residues31,73–75, these IDRs do not partition into MED1IDR condensates (Fig S4M–S4O, S5R–S5W). This suggests that selective compartmentalization by MED1IDR condensates could be achieved by “statistical pattern matching”, where matched pairs of charge pattern distributions along a disordered heteropolymer enable specificity90,93,94. Electrostatic pattern matching among disordered proteins in the formation of binary complexes has been experimentally demonstrated95,96. Interestingly, a theoretical study97 found that even when interactions between components are random, multicomponent fluids segregate into many coexisting phases with distinct compositions, suggesting that the differences in patterns may not need to be stark to ensure selectivity. Given that these functional yet weak interactions are likely dependent on context and on concentration of all components in the complex mixture of the cell, it is likely that most have been missed by standard discovery methods.
While we focus on IDRs, it should be noted that condensate formation does not require protein disorder98,99. Seminal work on condensate formation and composition control has demonstrated a role for multivalent modular structured domain-ligand pairs98,100. The transcriptional and chromatin machinery contain many domain-ligand pairs where a structured domain of a protein binds to a short linear motif (SLiM) on a partner protein. A recent study discovered a new type of domain-SLiM interaction enriched in transcriptional elongation machinery101, suggesting that there are others to be discovered. The IDR-IDR interactions described here likely work together with domain-SLiM interactions to create networks of interactions, enabling selective compartmentalization.
Our study demonstrates that multivalent interactions mediated by IDRs can selectively partition functionally related proteins from a complex mixture of proteins. The sequence features required for this selectivity are also required for transcriptional activity. Our findings demonstrate that selective partitioning can be driven by interactions among disordered regions. It remains unknown whether these disordered regions adopt transient structures upon interaction or remain disordered. Our ability to reconstitute selective partitioning with protein regions lacking annotated structure suggests that site-specific structured domain-mediated interactions are not required for specificity. Our work opens the possibility that there are other specific disorder-mediated interactions overlooked by conventional discovery methods and that these types of interactions play a functional role in the spatial organization of biochemical pathways in the cell.
Limitations of the study
Our study focused on the partitioning of proteins into a condensate composed of an IDR which enabled us to identify previously undescribed interactions but introduced a limitation in that we miss the potential contributions of other regions of MED1 or other IDRs within the Mediator complex. While many proteins partitioned by MED1IDR condensates contain IDRs with multiple patterned charge blocks, some do not have these features, suggesting that there are other ways proteins can be partitioned. While we demonstrate necessity and sufficiency, we do not uncover how alternating charge blocks enable specificity. Biophysical characterization of these interactions is needed to address how multivalent charge blocks interact to enable selective partitioning.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Benjamin R. Sabari (benjamin.sabari@utsouthwestern.edu)
Materials availability
All plasmids generated in this study are available upon request.
Data and code availability
The RNA sequence data and processed files are available at GEO under accession GSE210875. The raw proteomics data are available at MassIVE under accession MSV000089301. Microscopy data reported in this paper will be shared by the lead contact upon request.
Code is available from lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
U2OS cells
Cells were cultured as previously described56. Briefly, U2OS 2-6-3 cells (Lac array cells) were grown in DMEM supplemented with 10% Tet system approved FBS (Takara Bio USA 631105), 1% Penicillin Streptomycin (Fisher 15-140-122), 1% GlutaMAX Supplement (Fisher Scientific 35050061). Cells were grown at 37°C with 5% CO2 in a humidified sterile incubator. For U2OS cells without the Lac array system (used in Figure 1), cells were grown in regular FBS (Sigma F0926).
3T3-L1 and 293T cells
3T3-L1 cells (ATCC CL-173) were cultured in full medium (DMEM (FisherScientific 11995) supplemented with 10% FBS (Sigma F0926), 1x GlutaMAX (FisherScientific 35050), 1x Penicillin-Streptomycin (FisherScientific 15140). 293T cells were grown in full medium on plates coated with 0.001% poly-L-lysine (Sigma P4832).
3T3-L1 differentiation
Cells were grown to 100% confluence and then maintained for two days prior to initiating differentiation. Differentiation was induced by treatment with full medium supplemented with 0.5mM IBMX (Sigma I7018), 1μM dexamethasone (Sigma D1881), 5μg/ml insulin (Sigma I5500), 5μM troglitazone (Sigma T2573). Cells were treated with this differentiation medium for three days before being switched to full medium containing 5μg/ml insulin.
METHOD DETAILS
Nuclear extract preparation
1–2 × 108 U2OS cells were grown in DMEM (Fisher Scientific 11995073) supplemented with 10% FBS, penicillin-streptomycin (Thermo Fisher 15140122) and GlutaMAX (Fisher Scientific 35050061) at 37 °C under 5% CO2. Cells were collected by trypsinization, pelleted at 500xg, and resuspended in 10 mL of buffer CE+NP40 (cytoplasmic extract) (20 mM HEPES, 10 mM KCl, 5 mM MgCl2, 1 mM EDTA, 0.1% NP40, 1 mM DTT, cOmplete protease inhibitor cocktail (Sigma 11873580001)). After 5 min incubation on ice, the sample was centrifuged at 500×g, 4°C. Pellet was washed three times in 4 mL of buffer CE (20 mM HEPES, 10 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, cOmplete protease inhibitor cocktail (Sigma 11873580001)) and resuspended in 1 mL of buffer NE (nuclear extract) (20 mM Tris-HCl, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM PMSF, 25% glycerol, 1 mM DTT, cOmplete protease inhibitor cocktail (Sigma 11873580001)). This sample was rotated at 4°C for 1 hour and clarified by centrifugation at 20,000×g. Clarified lysate was dialyzed in dialysis buffer (20 mM Tris-HCl, 75 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM PMSF, 10% glycerol, 1 mM DTT, cOmplete protease inhibitor cocktail (Sigma 11873580001)) using Slide-A-Lyzer™ MINI Dialysis Device (Fisher Scientific 88404). Extracts were clarified and protein concentration was measured by Qubit™ Protein Assay Kit (Thermo Fisher Q33211).
Protein expression and purification
The DNA fragments encoding the protein region of interest were cloned into a modified T7 pET expression vector, resulting in the protein having N terminal 6xHis mEGFP or mCherry (see Key Resource Table and Table S1) followed by a 14 amino acid linker sequence “GAPGSAGSAAGGSG” before the protein region of interest. NEBuilder® HiFi DNA Assembly Master Mix (NEB E2621L) was used to insert the DNA sequence in-frame with the linker sequence. The generated construct was transformed into NEB 5-alpha Competent E. coli (C2987H) and plasmids were isolated from a selected bacterial colony. Sequence identity was confirmed using sanger sequencing.
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Recombinant DNA | ||
| pET_6His_TEV_mEGFP_MED1IDR | Sabari et al. 3 | N/A |
| pET_6His_TEV_mEGFP_MED1IDR scramble | This paper | N/A |
| pET_6His_TEV_mEGFP_ MED1IDR EDtoA | This paper | N/A |
| pET_6His_TEV_mEGFP_ MED1IDR RHKtoA | This paper | N/A |
| pET_6His_TEV_mCherry | Sabari et al. 3 | N/A |
| pET_6His_TEV_mCherry_CTR9 IDR | This paper | N/A |
| pET_6His_TEV_mCherry_SPT6 IDR | This paper | N/A |
| pET_6His_TEV_mCherry_NELFA tentacle | This paper | N/A |
| pET_6His_TEV_mCherry_HP1A IDR | This paper | cDNA: Addgene 17652 |
| pET_6His_TEV_mCherry_FUS IDR | This paper | cDNA: Addgene 26374 |
| pET_6His_TEV_mCherry_NELFE tentacle | This paper | cDNA: gift from D’Orso Lab |
| pET_6His_TEV_mCherry_NELFE Blocky_tentacle | This paper | N/A |
| pET_6His_TEV_mCherry_synthetic_IDR_1 | This paper | N/A |
| pET_6His_TEV_mCherry_synthetic_IDR_2 | This paper | N/A |
| pET_6His_TEV_mCherry_synthetic_IDR_3 | This paper | N/A |
| pET_6His_TEV_mCherry_synthetic_IDR_4 | This paper | N/A |
| pET_6His_TEV_mCherry_synthetic_IDR_5 | This paper | N/A |
| pET_6His_TEV_mCherry_SPT6IDR-charge_scramble | This paper | N/A |
| pET_6His_TEV_mCherry_SPT6IDR-non-charge_scramble_3 | This paper | N/A |
| CFP-LacI-MED1IDR_WT | Zamudio et al. 40 | N/A |
| CFP-LacI-MED1 FL | This paper | N/A |
| CFP-LacI-MED1 ΔIDR | This paper | N/A |
| CFP-LacI-MED1IDR_EDtoA | This paper | N/A |
| CFP-LacI-MED1IDR_RHKtoA | This paper | N/A |
| CFP-LacI-MED1IDR-Scramble | This paper | N/A |
| CFP-LacI-CTR9IDR | This paper | N/A |
| CFP-LacI-SPT6IDR | This paper | N/A |
| CFP-LacI-NELFAtentacle | This paper | N/A |
| CFP-LacI-HP1aIDR | This paper | N/A |
| CFP-LacI-FUSIDR | This paper | N/A |
| CFP-LacI-CDK12IDR | This paper | cDNA: Addgene #116723 |
| CFP-LacI-FUSIDR | This paper | N/A |
| CFP-LacI-CTR9IDR-charge_scramble | This paper | N/A |
| CFP-LacI-SPT6IDR-charge_scramble | This paper | N/A |
| pCDNA3.1_mCherry | This paper | N/A |
| pCDNA3.1_mCherry_CTR9IDR | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR | This paper | N/A |
| pCDNA3.1_mCherry_LEO1IDR | This paper | N/A |
| pCDNA3.1_mCherry_PAF1IDR | This paper | N/A |
| pCDNA3.1_mCherry_CTR9IDR_EDtoA | This paper | N/A |
| pCDNA3.1_mCherry_CTR9IDR_KRtoA | This paper | N/A |
| pCDNA3.1_mCherry_NELFAtentacle | This paper | N/A |
| pCDNA3.1_mCherry_CTR9IDR_scramble | This paper | N/A |
| mCherry-MS2coatProtein (MCP) | Addgene | #103832 |
| miRFP670-MS2coatProtein (MCP) | This paper | N/A |
| pCDNA3.1_mCherry_CTR9_FL | This paper | N/A |
| pCDNA3.1_mCherry_CTR9_ΔIDR | This paper | N/A |
| pCDNA3.1_mCherry_SPT6_FL | This paper | cDNA: gift from Vos Lab |
| pCDNA3.1_mCherry_SPT6_ΔIDR | This paper | N/A |
| pCDNA3.1_mCherry_HP1aIDR | This paper | N/A |
| pCDNA3.1_mCherry_FUSIDR | This paper | N/A |
| pCDNA3.1_mCherry_NELFEtentacle | This paper | N/A |
| pCDNA3.1_mCherry_p300IDR | This paper | N/A |
| pCDNA3.1_mCherry_DDX4IDR | This paper | cDNA: Addgene 101225 |
| pCDNA3.1_mCherry_EWSR1IDR | This paper | cDNA: Addgene 26377 |
| pCDNA3.1_mCherry_IWS1IDR | This paper | N/A |
| pCDNA3.1_mCherry_MeCP2IDR | This paper | cDNA: Li et al. 103 |
| pCDNA3.1_mCherry_CBX2IDR | This paper | cDNA: Addgene 82510 |
| pCDNA3.1_mCherry_ NICD | This paper | cDNA: gift from the Rosen Lab |
| pCDNA3.1_mCherry_ NPM1IDR | This paper | cDNA: Addgene 17578 |
| pCDNA3.1_mCherry_ KDM1AIDR | This paper | N/A |
| pCDNA3.1_mCherry_ RCOR3IDR | This paper | N/A |
| pCDNA3.1_mCherry_ Ki67R12_pm9×2 | cDNA: design from Yamazaki et al. 74 | N/A |
| pCDNA3.1_mCherry_SPT6IDR_EDtoA | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_RHKtoA | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_Scramble | This paper | N/A |
| pCDNA3.1_mCherry_MED1IDR | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR-charge_scramble | This paper | N/A |
| pCDNA3.1_mCherry_CTR9IDR-charge_scramble | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_non-charge_scramble_1 | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_non-charge_scramble_2 | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_non-charge_scramble_3 | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_middle_WT(ABABA) | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_middle_AAABB | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_middle_BBAAA | This paper | N/A |
| pCDNA3.1_mCherry_SPT6IDR_middle_BAAAB | This paper | N/A |
| pCDNA3.1_mCherry_synthetic_IDR_1 | This paper | N/A |
| pCDNA3.1_mCherry_synthetic_IDR_2 | This paper | N/A |
| pCDNA3.1_mCherry_synthetic_IDR_3 | This paper | N/A |
| pCDNA3.1_mCherry_synthetic_IDR_4 | This paper | N/A |
| pCDNA3.1_mCherry_synthetic_IDR_5 | This paper | N/A |
| pCDNA3.1_mCherry_NELFEtentacle_blocky | This paper | N/A |
| pCDNA3.1_mCherry_NELFE-CTR9IDR chimera | This paper | N/A |
| pCDNA3.1_mCherry_NELFE-CTR9IDR_charge_scramble chimera | This paper | N/A |
| pCDNA3.1_mCherry_NELFE-SPT6IDR chimera | This paper | N/A |
| pCDNA3.1_mCherry_NELFE-SPT6IDR_charge_scramble chimera | This paper | N/A |
| pCDNA3.1_mCherry_NELFE FL (WT tentacle) | This paper | N/A |
| pCDNA3.1_mCherry_NELFE-blockytentacle chimera | This paper | N/A |
| LentiCRISPR V2 | Addgene | #52961 |
| psPAX2-GAG-Pol-Rev | Addgene | #12260 |
| pAdVAntage | Promega | E1711 |
| pCMV-VSV-G | Addgene | #8454 |
| pUltra_GFP-P2A | Ito et al. 59 | |
| pUltra_GFP-P2A_mMed1_FL | Ito et al. 59 | |
| pUltra_GFP-P2A_mMed1-IDR_EDtoA | This paper | N/A |
| pUltra_GFP-P2A_mMed1-IDR_RHKtoA | This paper | N/A |
| pUltra_GFP-P2A_mMed1-IDR_Scramble | This paper | N/A |
| pUltra_GFP-P2A_mMed1-SPT6IDR chimera | This paper | N/A |
| pUltra_GFP-P2A_mMed1-SPT6IDR_charge_scramble chimera | This paper | N/A |
| Antibodies for IF | ||
| NELF-B | Abcam | ab167401 |
| MED1 | Bethyl | A303-876A |
| CTR9 | Bethyl | A301-395A |
| RNAP II Subunit B1 (Phospho-CTD Ser-5) Clone 3E8 | Sigma | 04-1572 |
| RNAP II Subunit B1 (Phospho-CTD Ser-2) Clone 3E10 | Sigma | 04-1571 |
| POLII CTD | Sigma Aldrich (Millipore) | 05-952-I-25UG |
| H3K27ac | Active Motif | 39133 |
| SPT6 | Cell Signaling | 15616S |
| IWS1 | Cell Signaling | 5681S |
| HP1alpha | Abcam | ab109028 |
| NPM1 | Invitrogen | 32-5200 |
| H3K9me3 | Active Motif | 39062 |
| Anti-goat IgG-Alexa Fluor 488 | Thermo Fisher | A-11055 |
| Anti-rabbit IgG-Alexa Fluor 555 | Thermo Fisher | A32732 |
| Anti-mouse IgG-Alexa Fluor 555 | Thermo Fisher | A48270 |
| Anti-rat IgG-Alexa Fluor 555 | Thermo Fisher | A48270 |
| Anti-rabbit IgG-Alexa Fluor 635 | Thermo Fisher | A-31577 |
| Antibodies for Western Blot | ||
| POLII CTD | Millipore | 05-952-I-25UG |
| CTR9 | Bethyl | A301-395A |
| SPT6 | Cell Signaling | 15616S |
| IWS1 | Cell Signaling | 5681S |
| NELF-B | Abcam | ab167401 |
| FUS/TLS (E3O8I) | Cell Signaling | 67840S |
| PTBP1 (E5O2S) | Cell Signaling | 72669S |
| GFP (B-2) | Santa Cruz | sc-9996 |
| MED1 | Bethyl | A300-793A |
| A.v. GFP (JL-8) | Takara | 632381 |
| MED1 | Invitrogen | PA5-36200 |
| MED4 | Abcam | ab129170 |
| Beta Tubulin | Abcam | ab6046 |
| Lamin A | Bethyl | A303-432A |
| horseradish peroxidase (HRP)-conjugated anti-Mouse IgG | Cytiva | NA931V |
| horseradish peroxidase (HRP)-conjugated anti-Rabbit IgG | Cytiva | NA934V |
| Oligonucleotides | ||
| Primers for qPCR | This paper | See table in methods section |
| sgRNA for Med1 KO and IDR deletion | This paper | See table in methods section |
| Experimental models: Cell lines | ||
| 293T | ATCC | CRL-3216 |
| 3T3-L1 | ATCC | CL-173 |
| 3T3-L1_CRISPR KO and ΔIDR cells | This paper | |
| 3T3-L1_MED1 KO clones #2 and #15 | Ito et al. 59 | |
| U2OS 2-6-3 | Janicki et al. 56 | |
| U2OS | Sabari Lab | |
| siRNA | ||
| ON-TARGETplus Non-targeting Pool | Horizon Discovery | D-001810-10-05 |
| ON-TARGETplus Mouse Med1 (19014) siRNA - Individual 040964-15 | Horizon Discovery | J-040964-15-0005 |
| ON-TARGETplus Mouse CTR9 siRNA - Individual 047267-09 | Horizon Discovery | J-047267-09-0002 |
| ON-TARGETplus Mouse IWS1 siRNA - Individual 055676-09 | Horizon Discovery | J-055676-09-0002 |
| ON-TARGETplus Mouse SUPT6 siRNA - Individual 62143-09 | Horizon Discovery | J-062143-09-0002 |
| ON-TARGETplus Mouse NELFB siRNA - Individual 048011-09 | Horizon Discovery | J-048011-09-0002 |
| Chemicals and Commercial Assays | ||
| DMEM | FisherScientific | 11995 |
| FBS | Sigma | F0926 |
| GlutaMAX | FisherScientific | 35050 |
| Penicillin-Streptomycin | FisherScientific | 15140 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma | 11873580001 |
| Slide-A-Lyzer™ MINI Dialysis Device | FisherScientific | 88404 |
| Qubit™ Protein Assay Kit | Thermo Fisher | Q33211 |
| Eppendorf™ LoBind Microcentrifuge Tubes: Protein | Eppendorf | 22431081 |
| S-Trap Micro | Protifi | C02-micro-80 |
| Oasis HLB solid-phase extraction plate | Waters | 186001828BA |
| EasySpray column | Thermo | ES905 |
| 3 to 8% Tris-Acetate 1.0 mm polyacrylamide gel | Thermo Fisher | EA03752BOX |
| Tris-Acetate SDS Running Buffer | FisherScientific | LA0041 |
| Invitrogen Mini Gel Tank and Blot Module set | Thermo Fisher | NW2000 |
| 0.45-um PVDF membrane | Millipore | IPVH00010 |
| Novex Tris-Glycine Transfer Buffer | FisherScientific | LC3675 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher | 34577 |
| Tet system approved FBS | Takara Bio USA | 631105 |
| coverslips | VWR | 48366-067 |
| paraformaldehyde | VWR | BT140770 |
| triton X100 | Sigma | T9284 |
| IgG-free bovine serum albumin, BSA | VWR | 102643-516 |
| Hoechst 33342 | Thermo Fisher | 62249 |
| slides | VWR | 10144-820 |
| Vectashield | VWR | 101098-042 |
| nail polish | VWR | 100491-940 |
| Lipofectamine 3000 transfection reagent | FisherScientific | L3000015 |
| poly-L-lysine | Sigma | P4832 |
| IBMX | Sigma | I7018 |
| dexamethasone | Sigma | D1881 |
| insulin | Sigma | I5500 |
| troglitazone | Sigma | T2573 |
| Lipofectamine RNAiMAX transfection reagent | FisherScientific | 13-778-075 |
| Polybrene Infection/Transfection Reagent | Sigma | TR-1003 |
| puromycin | FisherScientific | A11138 |
| IGEPAL CA-630 | Sigma | I8896 |
| DynaBeads Protein G | Invitrogen | 10004D |
| RNeasy Plus Mini Kit | Qiagen | 74136 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | A25778 |
| BODIPY 493/503 | Thermo Fisher | D3922 |
| Hoechst 33342 Solution | Thermo Fisher | 62249 |
| NEBuilder® HiFi DNA Assembly Master Mix | NEB | E2621L |
| Isopropyl β-D-1-thiogalactopyranoside, IPTG | Sigma | 70527 |
| HisPur Ni-NTA resin | FisherScientific | PI88222 |
| Poly-Prep® Chromatography Columns | Bio-Rad | 7311550 |
| ENrich SEC 650 10 × 300 column | Bio-Rad | 7801650 |
| UNO S6 column | Bio-Rad | 7200023 |
| Bacterial and virus strains | ||
| 5-alpha Competent E. coli | NEB | C2987H |
| NiCo21(DE3) Competent E. coli | NEB | C2529H |
| Software and algorithms | ||
| Proteome Discoverer v2.4 | Thermo | Version 2.4 |
| GraphPad Prism 9 | GraphPad Software, LLC. | Version 9.3.1 https://www.graphpad.com/ |
| CellProfiler | CellProfiler | Version 4.1.3 https://cellprofiler.org/ |
| ImageJ (FIJI) | Open Source | Version 2.3.0/1.53f https://imagej.net/software/fiji/ |
| Nikon Elements | Nikon instruments | https://www.microscope.healthcare.nikon.com/products/software |
| ngsplot | Shen et al. 104 | Version 2.63/ https://github.com/shenlab-sinai/ngsplot |
| SRA tool kit | NCBI | Version 2.8.2/ https://github.com/ncbi/sra-tools |
| Bedtools | Quinlan and Hall 105 | Version 2.29.2/ http://bedtools.readthedocs.io/en/latest/ |
| ROSE Package | Whyte et al. 52,Lovén et al. 106 | http://younglab.wi.mit.edu/super_enhancer_code.html |
| STAR aligner | Dobin et al. 107 | Version 2.7.3/ https://github.com/alexdobin/STAR |
| DEseq2 | Love et al. 108 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Samtools | Li et al. 109 | Version 1.6/ http://samtools.sourceforge.net |
| bowtie | Langmead et al. 110 | Version 1.0.0/ http://bowtie.cbcb.umd.edu. |
| Macs | Zhang et al. 111 | Version 14.2 |
| Panther | Mi et al. 112 | Version 17.0/ http://pantherdb.org/ |
| MobiDB lite | Necci et al. 113 | Version 3.9.0/ http://old.protein.bio.unipd.it/mobidblite/ |
| D2P2 | Oates et al. 114 | |
| metapredict | Emenecker et al. 115 | Version 1.31/ https://github.com/idptools/metapredict |
| localCIDER | Holehouse et al. 116 | Version 0.1.17/ http://pappulab.github.io/localCIDER/ |
| GEO Accession Numbers of previously published data used in this study | ||
| MED1 and RNA Pol II ChIP-seq (mESC) | Sabari et al. 3 | GEO: GSE112806 |
| SPT6 ChIP-seq (mESC) | Wang et al. 50 | GSE103180 |
| CTR9, NELFA, Pol2Ser2P and Pol2Ser5P ChIP-seq (mESC) | Ding et al. 51 | GSE149999 |
| GRO-seq data (mESC) | Min et al. 55 | GSE27037 |
| RNA Pol II ChIP-seq (3T3-L1) | Nielsen et al. 117 | GSE13511 |
| MED1 ChIP-seq (3T3-L1 cells) | Siersbæk et al. 60 | GSE95533 |
| RNA-seq (3T3L1 cells) | Siersbæk et al. 60 | GSE95533 |
NiCo21(DE3) Competent E. coli cells (NEB C2529H) were transformed with the bacterial expression plasmids described above. Transformed colonies were grown in LB media at 37 °C on a rotating shaker (250 rpm) until OD600 of 0.6 was reached. Following 1mM of Isopropyl ß-D-1-thiogalactopyranoside (IPTG, Sigma 70527) induction for protein expression, the bacterial culture was incubated at room temperature for 16 hours. For Synthetic_IDR_1 and NELFEtentacle_blocky protein production cells were incubated at 21°C at 135 rpm for 20 hours after induction. For SPT6IDR_non-charge_scramble protein production, cells were grown in LB media at 37°C at 250 rpm until OD600 of 0.6 reached and further incubated at 21°C at 135 rpm for 20 hours without induction. All bacterial pellets were collected by spinning the culture at 14000 rpm at 4 °C for 30 minutes in a Sorvall Rc6+ centrifuge. The pellet was lysed and dissolved using 30 mL of lysis buffer (50mM Tris, 500mM NaCl, 5mM β-mercaptoethanol) supplemented with cOmplete EDTA free 1x protease inhibitor cocktail (Sigma 11873580001). Dissolved pellets were subjected to sonication on ice using a Branson Digital Sonifier 250 at 50% amplitude with 5 second bursts followed by 10 second cooling period until homogenous lysate was obtained. The lysate was centrifuged at 18000 rpm at 4 °C for 30 minutes. During the lysate centrifugation step, HisPur Ni-NTA resin (FisherScientific PI88222) was equilibrated with lysis buffer. The supernatant from the bacterial lysate was collected and incubated with the equilibrated resin for 1 hour at 4 °C, after which the mixture was poured into empty polypropylene columns (Bio-Rad 7311550). Wash buffer (50mM Tris, 2.5 M NaCl, 5mM BME, 1x cOmplete protease inhibitor cocktail (Sigma 11873580001)) was applied to the column prior to elution with elution buffer (50 mM Tris, 500 mM NaCl, 5 mM BME, 500 mM imidazole, and 1x cOmplete protease inhibitor cocktail). mEGFP-MED1IDR was purified by cation exchange chromatography on UNO S6 column (Bio-Rad 7200023) using Buffer A (50 mM Tris, 5 mM BME) and Buffer B (50 mM Tris, 5 mM BME, 1M NaCl). Fractions (0.5 mL each) containing purified protein were collected. For all other proteins mentioned in the manuscript, size exclusion chromatography was performed by FPLC (Bio-Rad NGC quest) with an ENrich SEC 650 10 × 300 column (Bio-Rad 7801650) in buffer containing 50 mM Tris, 75 mM NaCl and 5mM BME. Next, all proteins were dialyzed against protein storage buffer (50 mM Tris, 75 mM NaCl, 5% glycerol, 1mM PMSF, 1 mM DTT) overnight at 4 °C. The concentration of dialyzed protein was calculated by reading A280 on a spectrophotometer (DeNovix, DS-11 FX+). The purified proteins were either used immediately or stored at −80 °C.
DNA synthesis of IDR cDNA
Custom DNA fragments for natural, mutated, or artificial IDR sequences were ordered from Twist Bioscience as gene fragments without adapter sequences. Gene fragments were directly inserted using HiFi (NEB) into either bacterial expression vectors for protein purification or mammalian expression vectors for cell-based experiments. All synthetic DNA were sequence verified by sanger sequencing. See table S1 for the amino acid sequences of all IDRs used in this study.
Pelleting assay
Purified recombinant mEGFP-MED1IDR and U2OS nuclear extract were pre-clarified at 22,000 × g for at least 30 minutes to remove any insoluble material. Purified recombinant mEGFP-MED1IDR (final 15mM) and nuclear extract (final 0.6 mg/mL) were mixed in Protein LoBind tubes (Eppendorf 22431081) and incubated for 15 minutes. The mixture was then centrifuged at 10,000 × g for 10 minutes in fixed angle rotor. The supernatant fraction was carefully removed leaving behind the pellet fraction. Both the pellet and supernatant fractions were brought to equal volume and resuspended in either 1x Laemmli sample buffer (58mM Tris-HCl pH 6.8, 5% glycerol, 1.6% SDS, 3.3% beta-mercaptoethanol, 0.007% bromophenol blue) for western blot analysis or in a resuspension buffer for downstream proteomic analysis.
Proteomics
Two independent pelleting assays were performed as described above. Pellets and supernatants were brought up to equal volumes in a resuspension buffer (25mM Tris, 500mM NaCl, 5% glycerol, 1mM DTT). Samples were dried in a SpeedVac to reduce the volume to 10–20 uL, after which an equal volume of 10% SDS with 100 mM triethylammonium bicarbonate (TEAB) was added to each. Reduction and alkylation were performed with tris(2-carboxyethyl) phosphine (TCEP) and iodoacetamide. Next, trypsin digestion was performed overnight at 37°C on an S-Trap Micro (Protifi) following the manufacturer’s directions. After digestion, the peptide eluate was cleaned using an Oasis HLB solid-phase extraction plate (Waters), dried in a SpeedVac, and reconstituted in a 2% acetonitrile, 0.1% TFA buffer.
The resulting samples were injected onto an Orbitrap Fusion Lumos mass spectrometer coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system. Samples were injected onto a 75 um i.d., 75-cm long EasySpray column (Thermo) and eluted with a gradient from 0–28% Buffer B over 90 min at 250 nL/min. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and Buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 1.5 kV and an ion transfer tube temperature of 275 °C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Dynamic exclusion was set for 25s after an ion was selected for fragmentation.
Raw MS data files were analyzed using Proteome Discoverer v2.4 (Thermo), with peptide identification performed using Sequest HT searching against the human protein database from UniProt along with the sequence for mEGFP-MED1IDR. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with oxidation of Met set as a variable modification. The false-discovery rate (FDR) cutoff was 1% for all peptides.
Proteomic data analysis
We used the Mass spectrometry data for MED1IDR fractions to calculate the fold enrichment between the pellet fraction(P) and the supernatant fraction(S) (n=2). Average pellet to supernatant fraction (P/S) values were used to determine the top 40 pellet-enriched proteins. Gene ontology enrichment analysis was performed for highly enriched pellet fraction proteins using PANTHER Overrepresentation Test (v16.0) with slim molecular function categories and Fisher’s exact test. The list of all proteins identified from the nuclear extract was used as background.
Data for the protein composition of NPM1 induced cell lysate granules were obtained from a published work 118 and a list of the top 200 highly enriched proteins was created based on the NPM1 enrichment score mentioned in this dataset. Using the UniProt IDs, we compared the overlap between the MED1 and NPM1 pellet enriched proteins.
Western blot
Samples were run on a 3% to 8% Tris-Acetate 1.0 mm polyacrylamide gel (Thermo Fisher EA03752BOX) using Tris-Acetate SDS Running Buffer (Fisher Scientific LA0041), in an Invitrogen Mini Gel Tank and Blot Module set (Thermo Fisher NW2000) at 150V for 75 min. Protein was wet transferred to a 0.45-um PVDF membrane (Millipore IPVH00010) in cold 10% Methanol Novex Tris-Glycine Transfer Buffer (Fisher Scientific LC3675) at 200 mA for 105 min at room temperature. After transfer, the membrane was blocked with 5% non-fat milk in TBST for 1 h at room temperature (RT), then incubated in primary antibody in 5% non-fat milk in TBST overnight at 4°C. After three subsequent washes in TBST at room temperature, the membrane was incubated with 1:10,000 horseradish peroxidase (HRP)-conjugated anti-Mouse IgG (Cytiva NA931V) or anti-Rabbit IgG (Cytiva NA934V) secondary antibody diluted in 5% non-fat-milk in TBST for 1h at RT. After washing with TBST, the membrane was developed with chemiluminescent HRP substrate (Thermo Fisher 34577) and imaged using Chemidoc MP system from Bio-Rad.
Microscopy
All micrographs shown in figures are representative for each experimental sample. These images were acquired using a Hamamatsu ORCA-Fusion C14440 digital camera and a CSU-W1 Yokogawa Spinning Disk Field Scanning Confocal System equipped with a Super Resolution by Optical Pixel Reassignment (SoRa) module. Data in Figure 1 were acquired using a 60x Plan Apo Lambda Oil Immersion objective (NA, 1.40) with SoRa engaged. Unless otherwise noted, all other images were acquired with the same objective without SoRa. Thickness of Z-slices were 0.15μm (SoRa) or 0.2 μm otherwise. SoRa images were processed using Nikon Instruments 3D deconvolution. Images of BODIPY stained 3T3-L1 cells were acquired using a 20x Plan Apo Lambda objective (NA, 0.75) and a Z stack step size of 5.6μm. Exposure time and laser intensity were the same for samples imaged in parallel. Laser lines used: 405nm, 488 nm, 561 nm, and 640 nm.
Immunofluorescence
Immunofluorescence was performed as formerly described3 with certain modifications. In brief, cells were grown on coverslips (VWR 48366–067) in a 6-well plate and fixed in 4% paraformaldehyde (VWR BT140770) in PBS for 10 min at room temperature (RT). After three washes in PBS for 10 min, samples were permeabilized with 0.5% triton X100 (Sigma T9284) in PBS for 10 min at RT. After washing three times in PBS for 5 min, cells were blocked with 4% IgG-free bovine serum albumin, BSA, (VWR 102643–516) for 1 hour at RT and incubated overnight at RT with primary antibodies in 4% IgG-free BSA (samples were kept in the dark if they contained transfected fluorophores). Following three washes in PBS for 10 min each, samples were incubated with secondary antibodies for 2h in the dark. For co-immunofluorescence, cells were incubated with secondary antibodies, one species at a time, to avoid cross-reactivity (donkey against goat followed by PBS washing and then an incubation with goat against rabbit or mouse). Next, cells were incubated with 1:5000 Hoechst 33342 (Thermo Fisher 62249) in double-distilled water at RT in the dark (5 min). After washing once more with water, coverslips were mounted on slides (VWR 10144–820) with Vectashield (VWR 101098–042). Coverslips were sealed with nail polish (VWR 100491–940) and stored at 4°C in the dark. All primary antibodies for immunofluorescence were diluted 1:500. All secondary antibodies were diluted 1:10,000.
Manders coefficients were calculated using CellProfiler 119. The analysis pipeline included image segmentation to identify individual cells based on Hoechst staining in a 60X magnification image. The channels for immunofluorescence (MED1 and other proteins of interest) were analyzed per identified nuclei and a Manders coefficient was calculated per cell considering MED1 as the channel of reference. Thresholding was performed for the highest range of fluorescence using FIJI as a reference and then CellProfiler’s Robust background thresholding. A single Z-slice per image sample was used (focused on best plane).
Analysis of mESC sequencing data
ChIP-seq and GRO-seq alignment and processing
ChIP-seq and GRO-seq data collected from mESCs were downloaded using SRA tool kit (v2.8.2) from the following GEO datasets: GSE112806 (RNA Polymerase II and MED1 ChIP-seq and Input), GSE103180 (SPT6 ChIP-seq), GSE149999 (CTR9, NELFA, S2ph, and S5Ph ChIP-seq) and GSE27037 (GRO-seq). The sequenced reads (FASTQ) were aligned to the mm10 version of the mouse reference genome using bowtie (v1.0.0) with parameters –k 1 –m 1 –best and –strata. The aligned reads were processed using samtools (v1.6) to convert into its binary counterpart (.bam) as well as to sort and remove potential PCR duplicates. Processed bam files were converted to BED files and sorted using BEDtools (v 2.29.2).
Ranking of MED1 enhancer genes and LOESS fit
The list of enhancers and their nearest Refseq gene IDs in mESCs were obtained from a previously published dataset 120. The list was sorted based on the MED1 signal and the chromosomal positions were added to the mm10 version of the genome. The average profile plots for all typical enhancer genes and super-enhancer genes (annotated in original dataset as super-enhancer YES/NO) were generated using ngsplot (v2.63) with parameters -G mm10 -R gene body -GO none.
RPKM values were calculated for MED1 sorted enhancer genes for the gene body region (TSS-TES) from different ChIP samples and the LOESS polynomial regression fit for RPKM values was plotted against MED1 sorted and ranked enhancer regions using custom R scripts. Predicted fit values were normalized by the minimum predicted value before plotting.
RNA polymerase II travel ratio calculation
RNA Pol II travel ratio was calculated as previously 54 by comparing the ratio between Pol II density in the promoter region and in the gene region. The promoter region is defined from −30 to +300 relative to the TSS, and the gene body as the TSS-TES region. The ratio was calculated from RPKM values for RNA Pol II ChIP-seq data at the promoter and gene body regions. LOESS polynomial regression fit curve was plotted for ratio values against MED1 sorted and ranked enhancer regions (defined in previous method section) using custom R scripts. Predicted fit values were normalized by the first predicted value (lowest MED1 rank ratio fit value) before plotting.
Lac array cell methods
Culture
Cells were cultured as previously described56. Briefly, U2OS 2-6-3 cells (Lac array cells) were grown in DMEM supplemented with 10% Tet system approved FBS (Takara Bio USA 631105), 1% Penicillin Streptomycin (Fisher 15-140-122), 1% GlutaMAX Supplement (Fisher Scientific 35050061). Cells were grown at 37°C with 5% CO2 in a humidified sterile incubator. For U2OS cells without the Lac array system (used in Figure 1), cells were grown in regular FBS (Sigma F0926).
Transfections
Lac array cells were seeded in 6-well plates at 50% confluency and transfected on the same day using Lipofectamine™ 3000 Transfection Reagent (ThermoFisher L3000015). All transfections for CFP-LacI containing plasmids (see Key Resource Table) were performed at approximately equimolar concentrations following manufacturer’s guidelines (≈3000 ng). mCherry-containing plasmids were co-transfected at a lower concentration to avoid non-specific interactions (≈2000 ng). For transfections with three plasmids (NELFE chimera experiments), the CFP and mCherry plasmids were used at the concentrations described above. The miRFP670-MS2coatProtein plasmid was co-transfected at a concentration of 2000 ng. Media was changed in the morning after transfection and cells were allowed to recover and then fixed the next day. Imaging was performed 1–2 days after fixing cells.
Processing coverslips of co-transfected cells
After cells were transfected and allowed to recover, cells were fixed in 4% paraformaldehyde (VWR BT140770) in PBS for 10 min at room temperature (RT) in the dark (plate wrapped with foil). After three washes in PBS for 10 min, samples were permeabilized with 0.5% triton X100 (Sigma T9284) in PBS for 10 min at RT. Then cells were incubated with 1:5000 Hoechst 33342 (Thermo Fisher, 62249) in double-distilled water at RT in the dark (5 min). After washing once more with water, coverslips were mounted on slides (VWR 10144–820) with Vectashield (VWR 101098–042). Coverslips were sealed with nail polish (VWR 100491–940) and stored at 4°C.
Two-dimensional analysis for IF at CFP foci
The relative fraction of IF intensity described in Figure 2 and Figure S2 was performed considering the integrated background fluorescence in the cytoplasm and the integrated fluorescence at the LacO locus core (highest fluorescence intensity range on the Z-slice) using FIJI 2D measurement tools and ROI tools. The same ROI area was used to measure the fluorescence at both background and CFP foci. For this analysis, only high intensity CFP foci were considered given the results in Figure 2F–G. These “high intensity” foci were on a different histogram distribution as shown by the Brightness/Contrast tool in FIJI. The quotient used in this analysis (arbitrary units) was calculated as raw intensity at CFP foci divided by raw intensity at background (cytoplasm) using the color channel for the IF of interest. Despite this internal control, it must be noted that images were processed together using the same IF treatment, exposure time, and laser intensity.
Three-dimensional analysis for IF at CFP foci
The “3D Objects Counter” analysis tool in FIJI was used to quantify the integrated fluorescence intensity of CFP foci and IF signal at the LacO locus. Using the ROI manager, the CFP region was defined after automatically resetting the min and max intensity of the CFP channel using the Brightness/Contrast tool and defining this region at the brightest Z-slice. Next, this region was duplicated across channels and Z-slices containing CFP signal. Then the channels were split using the Channels Tool, and for every channel of interest (IF or CFP) a threshold was set based on a mid-range value observed in control and MED1IDR conditions. Based on this threshold value, the 3D Objects Counter tool can then determine 3D objects and provide their volume and integrated intensity (arbitrary units). For each channel, the integrated intensity data for the largest object was recorded and plotted on the XY graph shown in Figure 2F–G. Control and experimental images were processed together using the same IF treatment, exposure time, and laser intensity.
Calculation of Goodness of Fit
Data from the three-dimensional analysis of IF for CTR9 and RPB1 shown in Figure 2 was plotted on an XY sheet in Prism. These data were analyzed using a “Logistic growth” model in Prism. This analysis provided the goodness of fit parameter R2. The growth curves shown in Figure 2 represent the early phase of this logistic growth model. The carrying capacity of this model is not shown as saturation of this system was not fully achieved.
Partition coefficient calculation for mCherry-IDR in Lac array cells
The partition coefficient was calculated by measuring the mCherry integrated fluorescence at the LacO locus core (highest intensity range on the Z-slice) and at a proximal spot in the nucleus (background) using FIJI 2D measurement tools and ROI tools. The same ROI area was used to measure the fluorescence at both background and CFP foci. For this analysis, all CFP foci were equally considered. The quotient of these values (arbitrary units) was calculated as raw intensity at CFP locus divided by raw intensity in the surrounding background. Despite this internal control, it must be noted that images were processed together using the same treatment, exposure time, and laser intensity as their mCherry no IDR control or CFP-LacI no fusion control.
MCP analysis
Intensity line profiles of mCherry-MCP or miRFP670-MCP on a line across the CFP focus were drawn using FIJI and saved in the ROI manager for use in all samples (≈ 3 microns). The values outside the CFP foci were taken as reference to normalize the rest of the data by averaging the first and last 4 data points and using a quotient between the rest of the data points and this average. Control and experiment cells containing low and comparable levels of mCherry- or miRFP670-MCP were considered for analysis. The 5 data points in the center of this line profile were averaged per cell and considered for statistical analysis against a control group.
Z score calculation
Z scores were calculated using the STANDARDIZE function on Microsoft Excel. The formula was given individual partition coefficient values, and the mean and standard deviation of the mCherry no IDR distribution. This normalization score was used for CFP-LacI MED1IDR mutants because their mCherry no IDR baseline partition level was different, so partition coefficients for other mCherry fusions across samples were not directly comparable.
Analysis of 3T3-L1 sequencing data
Alignment and processing
Publicly available MED1 (GSE95533) and RNA polymerase II (GSE13511) ChIP-seq data from 3T3-L1 at different stages of differentiation were used in this study.
Reads were aligned to the mm10 genome assembly using bowtie (v1.0.0) with parameters –k 1 –m 1 –best and –strata. Subsequent analyses were performed using a combination of samtools (v1.6) and BEDtools (v 2.29.2). Peak calling was done with MACS (v1.4.2). Counts Per Million normalized bigwig files were displayed on the UCSC genome browser for the visualization of ChIP-seq tracks.
Mapping MED1 enhancers and analysis
Enhancers and super-enhancers were mapped using the ROSE software package available at younglab.wi.mit.edu/super_enhancer_code.html for individual macs peak file by considering their respective bam files. Enhancers are defined as stitched regions of MED1 ChIP-seq binding not contained in promoters (t=2000). The stitched enhancer regions were annotated by considering the distance from the center of the enhancer to nearby TSS for genes with FPKM > 10. RPM values were calculated for enhancer regions of respective days of differentiation and the list was sorted based on increasing MED1 RPM signal. RPM values were plotted against ranked MED1 enhancer regions in Figure 3A and 3B.
Super-enhancer regions defined by the ROSE algorithm were annotated with the previous strategy for D0 (416 SE) and D2 (785 SE) ChIP-seq samples. These regions were combined and calculated a log2 fold change values (using RPM values) in comparison to Day 0 sample. These regions were sorted and ranked based on their log2FC values (lower to higher), and a bar plot was created with colors representing the loss and gain of super-enhancers (log2FC>=1.5 for gain and log2FC =<1.5 for loss).
3T3-L1 RNA-seq data analysis
Published datasets from GEO (MED1: GSE95533) were downloaded for RNA sequencing data for 3T3-L1 cells at different time points of adipocyte differentiation. mRNA-seq reads were mapped to the genome using STAR(v2.7.3a) and subsequent analyses were performed using samtools (v1.6) and BEDtools (v 2.29.2). Counts Per Million normalized bigwig files were displayed in the UCSC genome browser.
Read count quantification for genes was performed with htseq-count (v0.6.1) with parameters -i gene_id –stranded = reverse -f bam -m intersection-strict and a GTF file containing transcript positions was downloaded for the mm10 version of the genome from UCSC. Differential analysis was performed using DESeq2108 for RNA samples from different days of adipocyte differentiation in comparison to Day 0 replicates using the standard workflow and both replicates of each condition. Heatmaps and boxplots were plotted for selected candidate genes using Log2FC values in comparison to Day 0 RNA sequencing data.
3T3-L1 model
Differentiation
3T3-L1 cells (ATCC CL-173) were cultured in full medium (DMEM (FisherScientific 11995) supplemented with 10% FBS (Sigma F0926), 1x GlutaMAX (FisherScientific 35050), 1x Penicillin-Streptomycin (FisherScientific 15140). Cells were grown to 100% confluence and then maintained for two days prior to initiating differentiation. Differentiation was induced by treatment with full medium supplemented with 0.5mM IBMX (Sigma I7018), 1μM dexamethasone (Sigma D1881), 5μg/ml insulin (Sigma I5500), 5μM troglitazone (Sigma T2573). Cells were treated with this differentiation medium for three days before being switched to full medium containing 5μg/ml insulin.
siRNA knockdowns
6 wells of 3T3-L1 cells at approximately 60% confluency were transfected with 30pmol of the indicated Dharmacon siRNAs using Lipofectamine RNAiMAX transfection reagent (FisherScientific 13-778-075) according to the manufacturer’s instructions. 48h post transfection, cells were treated with differentiation medium as described above for three days prior to harvesting for RT-qPCR analysis.
CRISPR/Cas9 Med1 knockout and Med1ΔIDR in 3T3-L1 cells
6 wells of 293T cells (ATTC CRL-3216) grown in full medium on plates coated with 0.001% poly-L-lysine (Sigma P4832) at approximately 50% confluency were transfected with 3.3μg LentiCRISPR V2 (Addgene plasmid #52961) containing an sgRNA targeting exon 2, exon 3, or exon 17 of Med1, together with 2μg psPAX2-GAG-Pol-Rev (Addgene plasmid #12260), 0.85μg pAdVAntage (Promega E1711), and 1.3μg pCMV-VSV-G (Addgene plasmid #8454) using Lipofectamine 3000 transfection reagent (FisherScientific L3000015) according to the manufacturer’s instructions. A LentiCRISPR V2 plasmid containing a non-targeting sgRNA was transfected as a control. See table below for sgRNA sequences. At 48- and 72-hours post-transfection, culture medium containing virus was collected and passed through a 45μm PVDF filter onto 6 wells of 3T3-L1 cells at approximately 50% confluency. 50μg Polybrene (Sigma TR-1003) was added per well following each addition of virus. 48h after the second addition of virus, infected 3T3-L1 cells were selected using 2μg/ml puromycin (FisherScientific A11138), expanded, and frozen for future use. Med1 knockout and Med1ΔIDR cell lines were validated by probing cell extracts for MED1 protein expression using an antibody (Invitrogen, PA5-36200) that binds to an epitope located in the N-terminal domain of MED1.
List of sgRNA. Related to STAR Methods.
| sgRNA | Sequence |
|---|---|
| Med1_exon2_sgRNA1 | AATTTAACCAGAACAGACCT |
| Med1_exon2_sgRNA2 | TTGCATGGAGCCGTTCCAGG |
| Med1_exon3_sgRNA1 | GGGTCGTAATGAGTTCTGGA |
| Med1_exon17_sgRNA1 | TTTGGGTGCTGCAGATCTGA |
| Med1_exon17_sgRNA2 | ATCTGATGGAGCACCACAGT |
| Med1_exon17_sgRNA3 | AAGGACTCTGACTCCCACTG |
| Mouse_Non-Targeting_sgRNA1 | GCGAGGTATTCGGCTCCGCG |
MED1 Co-IP
40 million cells per line were resuspended in 3.5mL cold lysis buffer (50mM Tris-HCl pH 8, 150mM NaCl, 0.5% IGEPAL CA-630 (Sigma I8896), cOmplete protease inhibitor cocktail (Sigma 11873580001)). Samples were sonicated at 20% amplitude for five pulses of 5seconds on, 30seconds off using a Branson Digital Sonifier 250, and subsequently clarified via centrifugation for 15minutes at 16,000×g and 4°C. Protein concentrations of the cleared lysates were measured using Qubit Protein Assay Kit (Invitrogen Q33211) according to the manufacturer’s specifications. 50uL Protein G DynaBeads (Invitrogen 10004D) were washed three times with 0.5% BSA (w/v) and twice with PBS before being resuspended in 40uL cold lysis buffer. The beads were then incubated with 5ug of MED1 antibody (Invitrogen, PA5-36200) at 4°C rotating for 4hours. After washing three times in cold lysis buffer, antibody conjugated beads were incubated with cleared lysate (~3,000ug protein) overnight at 4°C. Beads were washed three times with cold lysis buffer, resuspended in 60uL 2x Laemmli buffer, and boiled for 10minutes to release bound proteins. Samples were probed with an antibody that recognizes the N-terminus of Med1 (Invitrogen, PA5-36200) and an antibody that recognizes Med4 (Abcam, ab129170).
RT-qPCR
3T3-L1 cell lines were seeded in 6 wells and differentiated as described above. Samples were taken at the indicated timepoints, and RNA was isolated using the RNeasy Plus Mini Kit (Qiagen 74136) according to the manufacturer’s instructions. cDNA was generated using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems 4368814), according to the manufacturer’s protocol, without RNase inhibitors. qPCR reactions were setup and standard cycling mode (primer Tm ≥60°C) and melt curve settings were used as described in the PowerUp SYBR Green Master Mix (Applied Biosystems A25778) user guide. Amplification cycles and melt curve analyses were carried out using a Roche LightCycler 480 384-well qPCR system. For each cell line, target gene expression was normalized to the expression of GAPDH. These values were further normalized to the non-targeting control.
The sequences of primers used for qPCR are listed below.
List of primers used for RT-qPCR. Related to STAR Methods.
| RT-qPCR | Forward primer | Reverse primer |
|---|---|---|
| Gapdh | TGGTGAAGGTCGGTGTGAAC | CCATGTAGTTGAGGTCAATGAAGG |
| Fabp4 | GCTGGTGGTGGAATGTGTTATG | ATTTCCATCCAGGCCTCTTCC |
| Cidec | GCCACGCGGTATTGCC | GATTGTGTTGGACCTCCCCCT |
| Acsl1 | CTCCTTAAATAGCATCGCAACCC | TTCTCTATGCAGAATTCTCCTCC |
| Cebpa | AATGGCAGTGTGCACGTCTA | CCCCAGCCGTTAGTGAAGAG |
| Adipoq | TGACGACACCAAAAGGGCTC | ACGTCATCTTCGGCATGACT |
| Cebpb | AAGCTGAGCGACGAGTACAAGA | GTCAGCTCCAGCACCTTGTG |
| Vdr | GCATCCAAAAGGTCATCGGC | TCAGAGGTGAGGTCCCTGAA |
| Med1 | TGAAAAAGAACCTGCCCCCG | CCACTCATTGGGTTGTTGCC |
| Ctr9 | CCGGATGCCTGGTCTTTGAT | TTTTCTCGGTCTCGGGTTGG |
| Spt6 | AGAGACGCCAGAGACCTTCT | TCATTTCGGATGGCCTGGTC |
| Iws1 | GAGTCATCCAGGTTTCAGGCA | ATTGCCCTCGATGCTGATTTTC |
| Nelfb | ATGGCGAGGACCTGAAAGAG | GCCATTCTCTGTCTGAAATTGTTC |
BODIPY staining
3T3-L1 cell lines were seeded in 6-well plates and differentiated as described above. At the indicated timepoints, cells were prepared for microscopy and imaged on plates. Briefly, cells were washed with PBS and fixed with 4% paraformaldehyde for 10 minutes at room temperature. All subsequent steps were carried out at 37°C. Following fixation, cells were washed with PBS three times for 5minutes each. Cells were permeabilized with 0.5% Triton-X-100 (Sigma T9284) for 10 minutes and then washed three times as before. Next, cells were stained with 3.8μM BODIPY 493/503 (Thermo Fisher D3922) for 30 minutes and then washed three times. Finally, cells were counterstained with 600nM DAPI for 10 minutes, washed with PBS, and left in PBS for the duration of imaging. Images presented in figures are false colored with Red LUT.
BODIPY image analysis
BODIPY images were processed and analyzed using FIJI121. Max intensity projections were generated from Z-stacks, and subsequently split into constituent channels. A Gaussian Blur filter with a radius of 4.00 was applied to the DAPI channel prior to using the StarDist Plugin122 to identify and count nuclei. The StarDist 2D feature was implemented using the “versatile (fluorescent nuclei)” neural network prediction model and the option to “set optimized post-processing thresholds (for selected model).” The Cell Counter Plugin (Kurt De Vos, University of Sheffield, Academic Neurology) was used to manually count cells that contained BODIPY-stained fat droplets. The number of BODIPY-stained cells was divided by the number of nuclei within the field of view, with the quotient multiplied by 100 to obtain the percentage of differentiating cells.
RNA-seq library construction protocol
Samples were run on the Agilent 2100 Bioanalyzer to determine level of degradation thus ensuring only high-quality RNA is used (RIN Score 8 or higher). The Qubit fluorometer is used to determine the concentration prior to staring library prep. 4 μg of total DNAse treated RNA are then prepared with the TruSeq Stranded Total RNA LT Sample Prep Kit from Illumina. Poly-A RNA is purified and fragmented before strand specific cDNA synthesis. cDNA are then a-tailed and indexed adapters are ligated. After adapter ligation, samples are PCR amplified and purified with Ampure XP beads, then validated again on the Agilent 2100 Bioanalyzer. Samples are quantified by Qubit before being normalized and pooled, then run on the Illumina HiSeq 2500 using SBS v3 reagents
3T3-L1 RNA-seq data analysis for CRISPR lines and siRNA knockdowns
Fifteen 3T3-L1 samples were analyzed by mRNA-seq. One sample of undifferentiated parental 3T3-L1 cells (WT_undiff). One sample of 3-day differentiated parental 3T3-L1 cells (WT_diff). Thirteen samples of 3-day differentiated 3T3-L1 cells with various genetic perturbations: three independent CRISPR MED1 KO cell lines (Med1_KO_1-3), three independent CRISPR MED1 ΔIDR cell lines (Med1_ΔIDR_1-3), CRISPR non-targeting control cell line (NT), Med1 siRNA knockdown, Ctr9 siRNA knockdown, Spt6 siRNA knockdown, Iws1 siRNA knockdown, Nelfb siRNA knockdown, and control siRNA. Raw and processed data are available on GEO (GSE210875).
The quality of the Fastq files obtained for RNA seq samples were confirmed and Demultiplexed mRNA seq reads were mapped to the mm10 version of the mouse reference genome using STAR methods (v2.7.3a) and subsequent analyses were performed using samtools (v1.6) and BEDtools (v 2.29.2). Bigwig files were generated using deeptools (v.3.5.0) with the following parameters -bs 50 –normalize Using CPM and were displayed in the UCSC genome browser
Per-transcript expression was quantified as FPKMs using featureCounts from the subread (v.1.6.3) package and annotated to the genes.gtf file of mm10 version of the mouse reference genome. ChIP RPM values for the region of TSS+/−2kb for each gene was calculated from the 3T3L1 ChIP seq samples (GSE95533) for Day 0, Day2, and Day 7. Also, RPM values of predicted and stitched enhancer genes (highest RPM) from the ChIP data is added into the RNA seq FPKM table.
Analysis in Figure 3 and S3 focused on genes activated by adipogenesis which was defined by two filters on the FPKM values of processed data 1) WT_diff ≥ 10 and 2) WT_diff/WT_undiff > 2. These two filters remove genes not well-expressed at 3-days of differentiation and focus on those genes that exhibit at least a 2-fold increase in expression at 3 days of differentiation. These two filters give 369 genes as activated during adipogenesis. Using this list of 369 genes, the subset of MED1 ΔIDR-sensitive genes is defined as average(ΔIDR)/NT ≤ 0.5. This filter gives a list of 63 ΔIDR-sensitive genes.
In vitro droplet assay
Components of the droplet assay were mixed together in protein storage buffer (50 mM Tris, 75 mM NaCl, 5% glycerol, 1mM PMSF, 1 mM DTT) and immediately loaded onto a homemade chamber comprising coverslip attached to a glass slide by parallel strips of double-sided tape. Droplets that settled on the coverslip were imaged using a Hamamatsu ORCA-Fusion C14440 digital camera and a CSU-W1 Yokogawa Spinning Disk Field Scanning Confocal System equipped with a Super Resolution by Optical Pixel Reassignment (SoRa) module. Images were acquired using a 60x Plan Apo Lambda Oil Immersion objective (NA, 1.40) with SoRa engaged. To test the partitioning of mCherry fusion proteins into mEGFP-MED1IDR droplets pre-formed by addition of total cellular RNA, 25 μM mEGFP-MED1IDR was mixed with 42.5 ng/uL total RNA extracted from U2OS cells using Qiagen RNeasy kit and then mixed with 4μM of mCherry-fusion as shown in Figure 4E. Similar experiments were performed in the absence of RNA where 8μM mEGFP-MED1IDR was with 1.75 μM mCherry fusions as shown in Figure S4B. To test whether mCherry fusions formed droplets on their own 42.5ng/uL was mixed with 4μM mCherry fusion protein as shown in Figure S4E. To test the partitioning of mutated IDRs and synthetic IDRs, 20uM mEGFP-MED1IDR was mixed with 40ng/uL total RNA and then mixed with 4μM mCherry fusion protein as indicated in Figures 5Q and S5L or 7μM mCherry fusion protein as indicated in Figure 5T. To test the ability of MED1IDR substitution and scramble mutants to form droplets in extract, 0.54mg/mL nuclear extract was mixed with 5μM of mEGFP-MED1IDR of indicated mutants as shown in Figure 7B.
Analysis of in vitro droplet experiments
To analyze in vitro droplet experiments, we used a Python script obtained from https://github.com/jehenninger/in_vitro_droplet_assay. The script was written to identify droplets and characterize their size, shape, and intensity. Beside using default parameters to identify droplets, parameters of circ, max_a and min_a were adjusted to identify the maximum number of droplets. The output file obtained for individual droplets (individual_output.xlsx) with unique IDs for each droplet was further analyzed and plotted using GraphPad prism version 9.3.1(350). Briefly, condensed fraction (C.F.) of channel (mCherry or GFP) in each field of view was plotted and the difference between group of a family were statistically tested using ordinary one-way ANOVA. As described previously 76, condensed fraction is defined as:
Where, Ichannel,droplets is total intensity in the condensed droplet phase and Ichannel,bulk is total background intensity outside droplets.
Analysis of protein sequence features
Generating consensus IDR table with sequence parameters
Consensus IDRs are defined as the overlapping ranges of disordered regions predicted by three different predictive methods: MobiDB 102, Metapredict 115 and D2P2 114. For D2P2 disorder predictions, we used consensus sequence ranges for which 75% of predictors agree, and these regions were stitched if the difference between two IDRs was less than 10 amino acids. For MobiDB and Metapredict, we directly used the predicted ranges of disordered regions. Further, we generated a table of consensus IDR ranges for all UniProt-reviewed proteins by considering the overlapping predicted ranges from these three different disorder predictors. The minimum length cutoff used to define an IDR was 30 amino acids (Table S3). For each predicted consensus IDR sequence, we obtained the following sequence parameters: IDR length, NCPR, FCR, disorder promoting fraction, kappa, molecular weight, acidic fraction, basic fraction, aromatic patterning (Ω_aro), SCD, and fraction of all amino acids using the localCIDER algorithm. The fraction of aromatic residues was calculated using custom Perl scripts by counting the number of F, Y, W residues and dividing it by the total length of the IDR or protein in respective cases. The number of acidic, basic, and charged blocks were defined using a threshold of greater than or equal to the absolute value of 5 on NCPR sliding window (10 amino acid) using in-house Python and R scripts. The number of blocks were calculated by merging the adjacent overlapping sliding windows that satisfy previously mentioned threshold criteria (NCPR>=abs(5)). The charged blocks per residue was calculated by taking the sum of acidic and basic blocks predicted for each IDR and dividing it with the length of the sequence.
Analysis of protein and IDR features from proteomics data
Average pellet to supernatant fraction (P/S) values were used to determine the top 200 pellet enriched proteins and bottom 200 supernatant enriched proteins from the proteomics data. Using the Consensus IDR definition (>=30AA) we defined whether a pellet enriched protein or supernatant enriched protein contains at least one IDR.
Average pellet to supernatant fraction (P/S) values were used to determine the top 200 pellet enriched IDRs (>=30AA) and bottom 200 supernatant enriched IDRs (>=30AA) from consensus ranges of the IDR dataset defined previously (Table S3). The proteins which do not have an IDR>=30 AA were removed from pellet and supernatant fractions (top and bottom fractions) to keep the IDR number uniform (200) for both categories. The P/S score for the top 200 IDRs ranges from 1325 to 30, whereas the P/S values for the bottom 200 IDRs ranges from 0.05 to 0.003. All IDRs (>=30AA) predicted by consensus IDR table for input proteins from the mass spectrometry data were considered as Input IDRs.
Defining acidic and basic blocks
The Net Charge Per Residue (NCPR) values were calculated for 10 amino acid sliding windows, and these values were used to define acidic and basic blocks on IDRs. If the sliding window NCPR values were greater than or equal to the absolute value of 5, then the respective window is defined as an acidic block or basic block based on the charge (negative: acidic, positive: basic). The number of blocks were calculated by merging the adjacent overlapping sliding windows that satisfy previously mentioned threshold criteria (NCPR>=abs(5)). The charged blocks per residue value was calculated by taking the sum of these blocks predicted for a particular protein or IDR and by dividing it by the total length.
IDR conservation analysis
IDRs of different organisms used for the conservation analysis and heatmap representation were defined using MobiDB positions obtained from UniProt IDs and the parameters for these IDRs were calculated by using localCIDER or by calculating the charged blocks per residue using the above-mentioned method. The sequence identity values compared to human IDR were obtained using UniProt aligner with default parameters.
GO analysis for blocky IDRs
We sorted the consensus IDR table based on the number of merged blocks (both acidic and basic) and selected the top 500 IDRs in this ranking as the blockiest IDRs. Gene ontology enrichment analysis was performed for their UniProt protein IDs using PANTHER Overrepresentation Test (v16.0) with slim molecular function categories and Fisher’s exact test. UniProt IDs of all consensus IDRs (>=30 amino acid) was used as background. The same analysis was also performed for sorted top 500 IDRs containing higher number of charge residues in their sequence with no patterning of charge into blocks.
Design strategy for artificial IDR sequences
Design of synthetic blocky IDRs
The five synthetic IDR sequences were designed to each have the same length and have the same pattern of charge blocks and linker sequences. Using the sequences of CTR9IDR and SPT6IDR, we built three libraries of sequences: 1) amino acids found in acidic blocks, 2) amino acids found in basic blocks, and 3) amino acids found in intervening (linker) sequence. To avoid potential disruption to charge pattern, we removed all charge amino acids from the linker library. To ensure that each synthetic IDR had the same length, we fixed the length of acidic blocks and basic blocks to the average length found in CTR9IDR and SPT6IDR: 13 for acidic and 10 for basic. We fixed the linker length to the observed linker length in SPT6IDR and used the pattern of block type found in SPT6IDR. The scaffold for the synthetic IDR was therefore (each number indicates the length of the linker sequence, “A” is acidic block and “B” is basic block): 1-A-10-A-16-A-2-B-3-A-4-B-2-A-12-A-6-A-4-B-1. The three libraries were scrambled and then for each synthetic IDR sequences were pulled from each library and stitched together to fit the scaffold. This was repeated 5 times with each new IDR stitched together from randomly scrambled libraries. The amino acid sequences were ordered as codon optimized gene fragments from Twist Bioscience. See Table S1 for the sequences of all five synthetic IDRs.
Design of NELFE blocky tentacle
To make the NELFEtentacle blocky sequence, charged amino acids were rearranged without changing the position of non-charge amino acids. NELFEtentacle contains a perfectly mixed alternating acid and basic residues continuously for 48 amino acids (RD domain). The design centered around creating 6 blocks of 8 amino acids from this RD region and then charged amino acids from elsewhere in the sequence were rearranged to segregate charge into as close to 8 amino acid regions as possible. The amino acid sequence was ordered as a codon-optimized gene fragment from Twist Bioscience. See Table S1 for the sequence.
3T3-L1 addback experiments
Lentivirus preparation for addback experiments of Med1IDR Mutants
Lentiviral expression constructs were generated using pUltra plasmids (Addgene #24129). For lentivirus packaging, psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) plasmids were used. 6 μg amounts of lentiviral plasmids carrying mouse Med1 cDNA with mutation in the IDR (pUltra-MED1, pUltra-MED1(EDtoA) pUltra-MED1(RHKtoA)) were transfected together with 3 μg of GAG-Pol and VSVG-Rev plasmids into 293T cells in 10 cm plates using polyethyleneimine (MW25000, Polyscience Inc. #23966). 10μM forskolin (SIGMA #F3917) was added to the culture 16 hours after transfection, and supernatant was collected at 48 and 72 hours after transfection. Pooled viral supernatant was centrifuged and concentrated.
Lentivirus preparation for addback experiments of Med1 Chimeras
6 wells of 293T cells (ATTC CRL-3216) at approximately 50% confluency were transfected with 3.3μg pUltra plasmids (Addgene #24129) containing cDNA for mouse Med1 chimeras, together with 2μg psPAX2-GAG-Pol-Rev (Addgene plasmid #12260), 0.85μg pAdVAntage (Promega E1711), and 1.3μg pCMV-VSV-G (Addgene plasmid #8454) using Lipofectamine 3000 transfection reagent (FisherScientific L3000015) according to the manufacturer’s instructions. At 48- and 72-hours post-transfection, culture medium containing virus was collected and passed through a 45μm PVDF filter onto 6 wells of Med1-KO 3T3-L1 cells at approximately 50% confluency. 50μg Polybrene (Sigma TR-1003) was added per well following each addition of virus. Infected 3T3-L1 cells were sorted for GFP signal, expanded, and frozen for future use.
Med1IDR mutant and Med1 chimera addback experiments in Med1-KO 3T3-L1
The ability of MED1IDR mutants and Med1 chimeras to restore adipogenesis capacity in Med1-KO 3T3-L1 preadipocytes was assessed following the protocol described previously59. Med1-KO 3T3-L1 clones #2 and #15 were infected with concentrated lentiviral particles and sorted for GFP-expressing cells. Differentiation was induced as described earlier in the methods section and monitored by qPCR to measure expression levels of key adipocyte genes and BODIPY staining to measure fat accumulation.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Software was used for statistical analysis. Parametric or non-parametric tests were determined after performing a normality test on samples. GraphPad notation for p values was used on our reports: (ns p > 0.05, * p ≤ 0.05, **p ≤ 0.01 ***, p ≤ 0.001, **** p ≤ 0.0001). Multiple comparisons tests were Dunnett’s test or Dunn’s test for non-parametric samples against a control group.
| Figure Panel | Statistical Test | Post-hoc Test | Planned Comparison | Sample Size |
|---|---|---|---|---|
| 1H | one-way ANOVA | Dunnett’s test | vs NELFB | ≥9 |
| 2H | two-tailed unpaired t-test | N/A | N/A | ≥10 |
| 2J | two-tailed unpaired t-test | N/A | N/A | ≥10 |
| 2M | two-tailed unpaired t-test | N/A | N/A | 10 |
| 2P | one-way ANOVA | Dunnett’s test | vs CFP-LacI (“-”) | ≥13 |
| 3D | two-tailed unpaired t-test | N/A | N/A | 100 |
| 3F | one-way ANOVA | Dunnett’s test | vs control siRNA (ctrl.) | 3 |
| 3J | one-way ANOVA | Dunnett’s test | vs NT control | 3 |
| 3L | one-way ANOVA | Dunnett’s test | vs First bin | 207 |
| 3N | one-way ANOVA | Dunnett’s test | vs non-targeting gRNA (NT) (left plot) or vs siRNA control (ctrl) (right plot) | 63 |
| 4F | one-way ANOVA | Dunnett’s test | vs no IDR control | ≥9 |
| 4I | one-way ANOVA | Dunnett’s test | vs no IDR control | 10 |
| 4L | one-way ANOVA | Dunnett’s test | vs mCherry only (no IDR control) | 20 |
| 5A | one-way ANOVA | Dunnett’s test | vs partitioned IDRs | All IDRs, 14088; Extract IDRs, 3955; partitioned IDRs, 200; and Excluded IDRs, 200 |
| 5G | one-way ANOVA | Dunnett’s test | vs CTR9 IDR wildtype | 10 |
| 5I | one-way ANOVA | Dunnett’s test | vs SPT6 IDR wildtype | 10 |
| 5K | one-way ANOVA | Dunnett’s test | vs no IDR control | 20 |
| 5M | one-way ANOVA | Dunnett’s test | vs ABABA | ≥12 |
| 5P | one-way ANOVA | Dunnett’s test | vs no IDR control | ≥12 |
| 5Q | one-way ANOVA | Dunnett’s test | vs no IDR control | ≥10 |
| 5S | one-way ANOVA | Dunn’s test | vs no IDR control | no IDR= 20; WT= 20; blocky= 16 |
| 5T | one-way ANOVA | Dunnett’s test | vs WT or vs no IDR | no IDR= 14; WT= 16; Blocky= 16 |
| 6C | one-way ANOVA | Dunnett’s test | vs no IDR control | 15 |
| 6F | two-tailed unpaired t-test | N/A | charge pattern vs no pattern | 13 |
| 6I | two-tailed unpaired t-test | N/A | charge pattern vs no pattern | 8 |
| 7C | one-way ANOVA | Dunnett’s test | vs WT MED1 IDR | ≥10 |
| 7G left | one-way ANOVA | Dunnett’s test | vs WT MED1 IDR | 20 |
| 7G middle | one-way ANOVA | Dunnett’s test | vs WT MED1 IDR | 20 |
| 7G right | one-way ANOVA | Dunnett’s test | vs WT MED1 IDR | 10 |
| 7I | one-way ANOVA | Dunnett’s test | vs WT MED1 IDR | 3 |
| 7K | one-way ANOVA | Dunnett’s test | vs WT MED1 IDR | 5 |
| 7N left | one-way ANOVA | Dunnett’s test | vs no IDR control | 20 |
| 7N middle | one-way ANOVA | Dunnett’s test | vs no IDR control | 20 |
| 7N right | one-way ANOVA | Dunnett’s test | vs no IDR control | 10 |
| 7P | one-way ANOVA | Dunnett’s test | vs NC | 3 |
| 7R | one-way ANOVA | Dunnett’s test | vs NC | 5 |
Statistical details for supplemental figures are given in the supplemental figure legends.
Supplementary Material
Table S1. Sequence of IDRs used in this study. Related to STAR Methods
Table S2. Proteomics data from MED1IDR condensate pelleting. Related to Figure 1
Table S3: FPKM values of 3T3-L1 differentiation RNA-seq. Related to Figure 3
Table S4. List of consensus IDRs. Related to Figure 5
HIGHLIGHTS.
Disorder-mediated selective partitioning regulates gene activation
Disordered regions are necessary and sufficient for selective partitioning
Charge patterning in disordered regions is required for specificity of partitioning
Disordered regions with similar patterning have similar partitioning and function
ACKNOWLEDGMENTS
The authors thank the UTSW Proteomics Core facility for assistance with mass spectrometry experiments, UTSW Genome Core for assistance in RNA-seq experiments, D’Orso Lab for providing cDNA for NELFE, Vos Lab for providing cDNA for SPT6, Rosen Lab for providing cDNA for NICD, and Banaszynski Lab for an aliquot of H3K9me3 antibody. This work was supported by Cancer Prevention and Research Institute of Texas (CPRIT) grant RR190090 to B.R.S and National Institutes of Health (NIH) grants GM147583 to B.R.S. and DK071900 and CA234575 to R.G.R. K.I. was supported by National Cancer Institute T32 grant CA009673 and by a Japan Society for the Promotion of Science postdoctoral fellowship for research abroad.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Roeder RG (2019). 50+ years of eukaryotic transcription: an expanding universe of factors and mechanisms. Nature Structural & Molecular Biology 26, 783 – 791. 10.1038/s41594-019-0287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cramer P (2019). Organization and regulation of gene transcription. Nature 573, 45 – 54. 10.1038/s41586-019-1517-4 [DOI] [PubMed] [Google Scholar]
- 3.Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. 10.1126/science.aar3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842 – 1855.e1816. 10.1016/j.cell.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho W-K, Spille J-H, Hecht M, Lee C, Li C, Grube V, and Cissé II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412 – 415. 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X, and Tjian R (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361. 10.1126/science.aar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cissé II, Izeddin I, Causse SZ, Boudarene L, Senecal A, Muresan L, Dugast-Darzacq C, Hajj B, Dahan M, and Darzacq X (2013). Real-time dynamics of RNA polymerase II clustering in live human cells. Science 341, 664 – 667. 10.1126/science.1239053 [DOI] [PubMed] [Google Scholar]
- 8.Iborra FJ, Pombo A, Jackson DA, and Cook PR (1996). Active RNA polymerases are localized within discrete transcription “factories’ in human nuclei. Journal of cell science 109 (Pt 6), 1427 – 1436. [DOI] [PubMed] [Google Scholar]
- 9.Izeddin I, Récamier V, Bosanac L, Cissé II, Boudarene L, Dugast-Darzacq C, Proux F, Bénichou O, Voituriez R, Bensaude O, et al. (2014). Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife 3, 23352. 10.7554/elife.02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson DA, Hassan AB, Errington RJ, and Cook PR (1993). Visualization of focal sites of transcription within human nuclei. The EMBO journal 12, 1059 – 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Zhang Z, Li L, Chen B-C, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, et al. (2014). Single-Molecule Dynamics of Enhanceosome Assembly in Embryonic Stem Cells. Cell 156, 1274 – 1285. 10.1016/j.cell.2014.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mir M, Reimer A, Haines JE, Li X-Y, Stadler M, Garcia H, Eisen MB, and Darzacq X (2017). Dense Bicoid hubs accentuate binding along the morphogen gradient. Gene Dev 31, 1784 – 1794. 10.1101/gad.305078.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mir M, Stadler MR, Ortiz SA, Hannon CE, Harrison MM, Darzacq X, and Eisen MB (2018). Dynamic multifactor hubs interact transiently with sites of active transcription in Drosophila embryos. Elife 7. 10.7554/elife.40497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papantonis A, and Cook PR (2013). Transcription Factories: Genome Organization and Gene Regulation. Chem Rev 113, 8683 – 8705. 10.1021/cr300513p. [DOI] [PubMed] [Google Scholar]
- 15.Tantale K, Mueller F, Kozulic-Pirher A, Lesne A, Victor J-M, Robert M-C, Capozi S, Chouaib R, Bäcker V, Mateos-Langerak J, et al. (2016). A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nat Commun 7, 12248. 10.1038/ncomms12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai A, Muthusamy AK, Alves MRP, Lavis LD, Singer RH, Stern DL, and Crocker J (2017). Nuclear microenvironments modulate transcription from low-affinity enhancers. Elife 6, e28975. 10.7554/elife.28975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho W-K, Jayanth N, English BP, Inoue T, Andrews JO, Conway W, Grimm JB, Spille J-H, Lavis LD, Lionnet T, and Cissé II (2016). RNA Polymerase II cluster dynamics predict mRNA output in living cells. Elife 5, 1123. 10.7554/elife.13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nature Reviews Molecular Cell Biology 18, 285 – 298. 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woringer M, and Darzacq X (2018). Protein motion in the nucleus: from anomalous diffusion to weak interactions. Biochemical Society Transactions 46, 945 – 956. 10.1042/bst20170310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rippe K (2021). Liquid–Liquid Phase Separation in Chromatin. Cold Spring Harbor Perspectives in Biology, a040683. 10.1101/cshperspect.a040683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabari BR (2020). Biomolecular Condensates and Gene Activation in Development and Disease. Developmental Cell 55, 84 – 96. 10.1016/j.devcel.2020.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Sigler PB (1988). Acid blobs and negative noodles. Nature 333, 210 – 212. 10.1038/333210a0. [DOI] [PubMed] [Google Scholar]
- 23.Fuxreiter M, Tompa P, Simon I, Uversky VN, Hansen JC, and Asturias FJ (2008). Malleable machines take shape in eukaryotic transcriptional regulation. Nature Chemical Biology 4, 728 – 737. 10.1038/nchembio.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille J-H, Afeyan LK, Zamudio AV, Shrinivas K, et al. (2019). Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543 – 548. 10.1038/s41586-019-1464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Choi J-M, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D, et al. (2018). A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 174, 688 – 699.e616. 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin EW, Holehouse AS, Peran I, Farag M, Incicco JJ, Bremer A, Grace CR, Soranno A, Pappu RV, and Mittag T (2020). Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 367, 694 – 699. 10.1126/science.aaw8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das S, Lin Y-H, Vernon RM, Forman-Kay JD, and Chan HS (2020). Comparative roles of charge, π, and hydrophobic interactions in sequence-dependent phase separation of intrinsically disordered proteins. Proc National Acad Sci 117, 28795–28805. 10.1073/pnas.2008122117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fawzi NL, Parekh SH, and Mittal J (2021). Biophysical studies of phase separation integrating experimental and computational methods. Current Opinion in Structural Biology 70, 78–86. 10.1016/j.sbi.2021.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim TH, Payliss BJ, Nosella ML, Lee ITW, Toyama Y, Forman-Kay JD, and Kay LE (2021). Interaction hot spots for phase separation revealed by NMR studies of a CAPRIN1 condensed phase. Proc National Acad Sci 118, e2104897118. 10.1073/pnas.2104897118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Y-H, and Chan HS (2017). Phase Separation and Single-Chain Compactness of Charged Disordered Proteins Are Strongly Correlated. Biophysical Journal 112, 2043–2046. 10.1016/j.bpj.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, and Baldwin AJ (2015). Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 57, 936 – 947. 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou X, Sumrow L, Tashiro K, Sutherland L, Liu D, Qin T, Kato M, Liszczak G, and McKnight SL (2022). Mutations linked to neurological disease enhance self-association of low-complexity protein sequences. Science 377, eabn5582. 10.1126/science.abn5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malik S, and Roeder RG (2010). The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet 11, 761 – 772. 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soutourina J (2017). Transcription regulation by the Mediator complex. Nature Reviews Molecular Cell Biology 34, 77. 10.1038/nrm.2017.115. [DOI] [PubMed] [Google Scholar]
- 35.Richter WF, Nayak S, Iwasa J, and Taatjes DJ (2022). The Mediator complex as a master regulator of transcription by RNA polymerase II. Nature Reviews Molecular Cell Biology, 1–18. 10.1038/s41580-022-00498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin J. w., and Wang G (2014). The Mediator complex: a master coordinator of transcription and cell lineage development. Development 141, 977 – 987. 10.1242/dev.098392. [DOI] [PubMed] [Google Scholar]
- 37.Abdella R, Talyzina A, Chen S, Inouye CJ, Tjian R, and He Y (2021). Structure of the human Mediator-bound transcription preinitiation complex. Science 372, 52–56. 10.1126/science.abg3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen BL, and Taatjes DJ (2015). The Mediator complex: a central integrator of transcription. Nature Reviews Molecular Cell Biology 16, 155 – 166. 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nair SJ, Yang L, Meluzzi D, Oh S, Yang F, Friedman MJ, Wang S, Suter T, Alshareedah I, Gamliel A, et al. (2019). Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat Struct Mol Biol 26, 193–203. 10.1038/s41594-019-0190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zamudio AV, Dall’Agnese A, Henninger JE, Manteiga JC, Afeyan LK, Hannett NM, Coffey EL, Li CH, Oksuz O, Sabari BR, et al. (2019). Mediator Condensates Localize Signaling Factors to Key Cell Identity Genes. Mol Cell 76, 753 – 766.e756. 10.1016/j.molcel.2019.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito M, Yuan C-X, Okano HJ, Darnell RB, and Roeder RG (2000). Involvement of the TRAP220 Component of the TRAP/SMCC Coactivator Complex in Embryonic Development and Thyroid Hormone Action. Mol Cell 5, 683 – 693. 10.1016/s1097-2765(00)80247-6. [DOI] [PubMed] [Google Scholar]
- 42.Ge K, Guermah M, Yuan C-X, Ito M, Wallberg AE, Spiegelman BM, and Roeder RG (2002). Transcription coactivator TRAP220 is required for PPARγ2-stimulated adipogenesis. Nature 417, 563 – 567. 10.1038/417563a. [DOI] [PubMed] [Google Scholar]
- 43.Khattabi LE, Zhao H, Kalchschmidt J, Young N, Jung S, Blerkom PV, Kieffer-Kwon P, Kieffer-Kwon K-R, Park S, Wang X, et al. (2019). A Pliable Mediator Acts as a Functional Rather Than an Architectural Bridge between Promoters and Enhancers. Cell 0. 10.1016/j.cell.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freibaum BD, Messing J, Yang P, Kim HJ, and Taylor JP (2021). High-fidelity reconstitution of stress granules and nucleoli in mammalian cellular lysate. J Cell Biol 220, e202009079. 10.1083/jcb.202009079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwak H, and Lis JT (2013). Control of Transcriptional Elongation. Genetics 47, 483–508. 10.1146/annurev-genet-110711-155440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, and Cramer P (2018). Structure of activated transcription complex Pol II–DSIF–PAF–SPT6. Nature 560, 607 – 612. 10.1038/s41586-018-0440-4. [DOI] [PubMed] [Google Scholar]
- 47.Vos SM, Farnung L, Urlaub H, and Cramer P (2018). Structure of paused transcription complex Pol II–DSIF–NELF. Nature 560, 601 – 606. 10.1038/s41586-018-0442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rawat P, Boehning M, Hummel B, Aprile-Garcia F, Pandit AS, Eisenhardt N, Khavaran A, Niskanen E, Vos SM, Palvimo JJ, et al. (2021). Stress-induced nuclear condensation of NELF drives transcriptional downregulation. Mol Cell 81, 1013–1026.e1011. 10.1016/j.molcel.2021.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13 – 23. 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang AH, Juan AH, Ko KD, Tsai P-F, Zare H, Dell’Orso S, and Sartorelli V (2017). The Elongation Factor Spt6 Maintains ESC Pluripotency by Controlling Super-Enhancers and Counteracting Polycomb Proteins. Mol Cell 68, 398–413.e396. 10.1016/j.molcel.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding L, Paszkowski-Rogacz M, Mircetic J, Chakraborty D, and Buchholz F (2021). The Paf1 complex positively regulates enhancer activity in mouse embryonic stem cells. Life Sci Alliance 4, e202000792. 10.26508/lsa.202000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 153, 307 – 319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phatnani HP, and Greenleaf AL (2006). Phosphorylation and functions of the RNA polymerase II CTD. Gene Dev 20, 2922–2936. 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 54.Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc Regulates Transcriptional Pause Release. Cell 141, 432 – 445. 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, and Lis JT (2011). Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Gene Dev 25, 742–754. 10.1101/gad.2005511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, and Spector DL (2004). From Silencing to Gene Expression: Real-Time Analysis in Single Cells. Cell 116, 683 – 698. 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malik S, Guermah M, Yuan C-X, Wu W, Yamamura S, and Roeder RG (2004). Structural and functional organization of TRAP220, the TRAP/mediator subunit that is targeted by nuclear receptors. Mol Cell Biol 24, 8244 – 8254. 10.1128/mcb.24.18.8244-8254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Green H, and Kehinde O (1975). An established preadipose cell line and its differentiation in culture II. Factors affecting the adipose conversion. Cell 5, 19–27. 10.1016/0092-8674(75)90087-2. [DOI] [PubMed] [Google Scholar]
- 59.Ito K, Schneeberger M, Gerber A, Jishage M, Marchildon F, Maganti AV, Cohen P, Friedman JM, and Roeder RG (2021). Critical roles of transcriptional coactivator MED1 in the formation and function of mouse adipose tissues. Genes & Development 35, 729–748. 10.1101/gad.346791.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Siersbæk R, Madsen JGS, Javierre B-M, Nielsen R, Bagge EK, Cairns J, Wingett SW, Traynor S, Spivakov M, Fraser P, and Mandrup S (2017). Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation. Mol Cell 66, 420 – 435.e425. 10.1016/j.molcel.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 61.Siersbæk R, Rabiee A, Nielsen R, Sidoli S, Traynor S, Loft A, Poulsen LLC, Rogowska-Wrzesinska A, Jensen ON, and Mandrup S (2014). Transcription Factor Cooperativity in Early Adipogenic Hotspots and Super-Enhancers. Cell Reports 7, 1443 – 1455. 10.1016/j.celrep.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 62.Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236 – 240. 10.1038/nature22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241 – 245. 10.1038/nature22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. (2015). A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162, 1066 – 1077. 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 65.Henninger JE, Oksuz O, Shrinivas K, Sagi I, LeRoy G, Zheng MM, Andrews JO, Zamudio AV, Lazaris C, Hannett NM, et al. (2021). RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 184, 207–225.e224. 10.1016/j.cell.2020.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ma L, Gao Z, Wu J, Zhong B, Xie Y, Huang W, and Lin Y (2021). Co-condensation between transcription factor and coactivator p300 modulates transcriptional bursting kinetics. Mol Cell 81, 1682–1697.e1687. 10.1016/j.molcel.2021.01.031. [DOI] [PubMed] [Google Scholar]
- 67.Vernon RM, Chong PA, Tsang B, Kim TH, Bah A, Farber P, Lin H, and Forman-Kay JD (2018). Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife 7, e31486. 10.7554/elife.31486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Das RK, and Pappu RV (2013). Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc National Acad Sci 110, 13392 – 13397. 10.1073/pnas.1304749110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Firman T, and Ghosh K (2018). Sequence charge decoration dictates coil-globule transition in intrinsically disordered proteins. The Journal of Chemical Physics 148, 123305. 10.1063/1.5005821. [DOI] [PubMed] [Google Scholar]
- 70.Lin Y-H, Brady JP, Chan HS, and Ghosh K (2020). A unified analytical theory of heteropolymers for sequence-specific phase behaviors of polyelectrolytes and polyampholytes. The Journal of Chemical Physics 152, 045102. 10.1063/1.5139661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, Williams CJ, and Dunker AK (2002). Evolutionary Rate Heterogeneity in Proteins with Long Disordered Regions. J Mol Evol 55, 104–110. 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 72.Brown CJ, Johnson AK, and Daughdrill GW (2010). Comparing Models of Evolution for Ordered and Disordered Proteins. Mol Biol Evol 27, 609–621. 10.1093/molbev/msp277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin Y, Protter DSW, Rosen MK, and Parker R (2015). Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell 60, 208 – 219. 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamazaki H, Takagi M, Kosako H, Hirano T, and Yoshimura SH (2022). Cell cycle-specific phase separation regulated by protein charge blockiness. Nature Cell Biology 24, 625–632. 10.1038/s41556-022-00903-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Plys AJ, Davis CP, Kim J, Rizki G, Keenen MM, Marr SK, and Kingston RE (2019). Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Gene Dev 33, 799 – 813. 10.1101/gad.326488.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shrinivas K, Sabari BR, Coffey EL, Klein IA, Boija A, Zamudio AV, Schuijers J, Hannett NM, Sharp PA, Young RA, and Chakraborty AK (2019). Enhancer Features that Drive Formation of Transcriptional Condensates. Mol Cell 75, 549 – 561.e547. 10.1016/j.molcel.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Larsson AJM, Johnsson P, Hagemann-Jensen M, Hartmanis L, Faridani OR, Reinius B, Segerstolpe Å, Rivera CM, Ren B, and Sandberg R (2019). Genomic encoding of transcriptional burst kinetics. Nature 565, 251 – 254. 10.1038/s41586-018-0836-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sabari BR, Dall’Agnese A, and Young RA (2020). Biomolecular Condensates in the Nucleus. Trends in Biochemical Sciences 45, 961 – 977. 10.1016/j.tibs.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Strom AR, and Brangwynne CP (2019). The liquid nucleome - phase transitions in the nucleus at a glance. Journal of cell science 132, jcs235093. 10.1242/jcs.235093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haberle V, Arnold CD, Pagani M, Rath M, Schernhuber K, and Stark A (2019). Transcriptional cofactors display specificity for distinct types of core promoters. Nature 570, 122 – 126. 10.1038/s41586-019-1210-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Serebreni L, and Stark A (2021). Insights into gene regulation: From regulatory genomic elements to DNA-protein and protein-protein interactions. Curr Opin Cell Biol 70, 58–66. 10.1016/j.ceb.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 82.Alerasool N, Leng H, Lin Z-Y, Gingras A-C, and Taipale M (2022). Identification and functional characterization of transcriptional activators in human cells. Mol Cell 82, 677–695.e677. 10.1016/j.molcel.2021.12.008. [DOI] [PubMed] [Google Scholar]
- 83.Neumayr C, Haberle V, Serebreni L, Karner K, Hendy O, Boija A, Henninger JE, Li CH, Stejskal K, Lin G, et al. (2022). Differential cofactor dependencies define distinct types of human enhancers. Nature, 1–8. 10.1038/s41586-022-04779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu F, Portz B, and Gilmour DS (2019). The C-Terminal Domain of RNA Polymerase II Is a Multivalent Targeting Sequence that Supports Drosophila Development with Only Consensus Heptads. Mol Cell 73, 1232 – 1242.e1234. 10.1016/j.molcel.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brodsky S, Jana T, Mittelman K, Chapal M, Kumar DK, Carmi M, and Barkai N (2020). Intrinsically Disordered Regions Direct Transcription Factor In Vivo Binding Specificity. Mol Cell. 10.1016/j.molcel.2020.05.032. [DOI] [PubMed] [Google Scholar]
- 86.Parker MW, Kao JA, Huang A, Berger JM, and Botchan MR (2021). Molecular determinants of phase separation for Drosophila DNA replication licensing factors. Elife 10. 10.7554/elife.70535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dinic J, Marciel AB, and Tirrell MV (2021). Polyampholyte Physics: Liquid-Liquid Phase Separation and Biological Condensates. Curr Opin Colloid In 54, 101457. 10.1016/j.cocis.2021.101457. [DOI] [Google Scholar]
- 88.Kapelner RA, Yeong V, and Obermeyer AC (2020). Molecular determinants of protein-based coacervates. Curr Opin Colloid In 52, 101407. 10.1016/j.cocis.2020.101407. [DOI] [Google Scholar]
- 89.Lin Y-H, Forman-Kay JD, and Chan HS (2018). Theories for Sequence-Dependent Phase Behaviors of Biomolecular Condensates. Biochemistry 57, 2499 – 2508. 10.1021/acs.biochem.8b00058. [DOI] [PubMed] [Google Scholar]
- 90.Lin Y-H, Brady JP, Forman-Kay JD, and Chan HS (2017). Charge pattern matching as a ‘fuzzy’ mode of molecular recognition for the functional phase separations of intrinsically disordered proteins. New Journal of Physics 19, 115003. 10.1088/1367-2630/aa9369. [DOI] [Google Scholar]
- 91.Das S, Amin AN, Lin Y-H, and Chan HS (2018). Coarse-grained residue-based models of disordered protein condensates: utility and limitations of simple charge pattern parameters. Physical Chemistry Chemical Physics 20, 28558–28574. 10.1039/c8cp05095c. [DOI] [PubMed] [Google Scholar]
- 92.Hazra MK, and Levy Y (2020). Charge pattern affects the structure and dynamics of polyampholyte condensates. Physical Chemistry Chemical Physics 22, 19368–19375. 10.1039/d0cp02764b. [DOI] [PubMed] [Google Scholar]
- 93.Chakraborty AK (2001). Disordered heteropolymers: models for biomimetic polymers and polymers with frustrating quenched disorder. Phys Reports 342, 1–61. 10.1016/s0370-1573(00)00006-5. [DOI] [Google Scholar]
- 94.Golumbfskie AJ, Pande VS, and Chakraborty AK (1999). Simulation of biomimetic recognition between polymers and surfaces. Proceedings of the National Academy of Sciences of the United States of America 96, 11707 – 11712. 10.1073/pnas.96.21.11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schuler B, Borgia A, Borgia MB, Heidarsson PO, Holmstrom ED, Nettels D, and Sottini A (2020). Binding without folding – the biomolecular function of disordered polyelectrolyte complexes. Current Opinion in Structural Biology 60, 66 – 76. 10.1016/j.sbi.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 96.Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, et al. (2018). Extreme disorder in an ultrahigh-affinity protein complex. Nature 19, 31. 10.1038/nature25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shrinivas K, and Brenner MP (2021). Phase separation in fluids with many interacting components. Proc National Acad Sci 118, e2108551118. 10.1073/pnas.2108551118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li P, Banjade S, Cheng H-C, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. (2012). Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336 – 340. 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Choi J-M, Holehouse AS, and Pappu RV (2020). Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annual Review of Biophysics 49, 107 – 133. 10.1146/annurev-biophys-121219-081629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, and Rosen MK (2016). Compositional Control of Phase-Separated Cellular Bodies. Cell 166, 651 – 663. 10.1016/j.cell.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cermakova K, Demeulemeester J, Lux V, Nedomova M, Goldman SR, Smith EA, Srb P, Hexnerova R, Fabry M, Madlikova M, et al. (2021). A ubiquitous disordered protein interaction module orchestrates transcription elongation. Science 374, 1113–1121. 10.1126/science.abe2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Piovesan D, Necci M, Escobedo N, Monzon AM, Hatos A, Mičetić I, Quaglia F, Paladin L, Ramasamy P, Dosztányi Z, et al. (2020). MobiDB: intrinsically disordered proteins in 2021. Nucleic Acids Res 49, D361–D367. 10.1093/nar/gkaa1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li CH, Coffey EL, Dall’Agnese A, Hannett NM, Tang X, Henninger JE, Platt JM, Oksuz O, Zamudio AV, Afeyan LK, et al. (2020). MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature 586, 440–444. 10.1038/s41586-020-2574-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. Bmc Genomics 15, 284. 10.1186/1471-2164-15-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 153, 320 – 334. 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup GPDP (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078 – 2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10, R25–R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mi H, Ebert D, Muruganujan A, Mills C, Albou L-P, Mushayamaha T, and Thomas PD (2020). PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res 49, gkaa1106–. 10.1093/nar/gkaa1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Necci M, Piovesan D, Clementel D, Dosztányi Z, and Tosatto SCE (2020). MobiDB-lite 3.0: fast consensus annotation of intrinsic disorder flavors in proteins. Bioinformatics 36, 5533–5534. 10.1093/bioinformatics/btaa1045. [DOI] [PubMed] [Google Scholar]
- 114.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, et al. (2013). D2P2: database of disordered protein predictions. Nucleic Acids Res 41, D508–D516. 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Emenecker RJ, Griffith D, and Holehouse AS (2021). metapredict: a fast, accurate, and easy-to-use predictor of consensus disorder and structure. Biorxiv, 2021.2005.2030.446349. 10.1101/2021.05.30.446349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Holehouse AS, Das RK, Ahad JN, Richardson MOG, and Pappu RV (2017). CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophysical Journal 112, 16 – 21. 10.1016/j.bpj.2016.11.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nielsen R, Pedersen TÅ, Hagenbeek D, Moulos P, Siersbæk R, Megens E, Denissov S, Børgesen M, Francoijs K-J, Mandrup S, and Stunnenberg HG (2008). Genome-wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Gene Dev 22, 2953–2967. 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Freibaum BD, Messing J, Yang P, Kim H, and Taylor JP (2021). High-fidelity reconstitution of stress granules and nucleoli in mammalian cellular lysate. The Journal of Cell Biology 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100. 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Whyte A, Warren, Orlando A, David, Hnisz D., Abraham J, Brian, Lin Y, Charles, Kagey H, Michael, Rahl B, Peter, Lee I, Tong, and Young A, Richard (2013). Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 153, 307–319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schmidt U, Weigert M, Broaddus C, and Myers G (2018). Cell Detection with Star-Convex Polygons. In Medical Image Computing and Computer Assisted Intervention – MICCAI 2018, (Springer International Publishing; ), pp. 265–273. 10.1007/978-3-030-00934-2_30. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequence of IDRs used in this study. Related to STAR Methods
Table S2. Proteomics data from MED1IDR condensate pelleting. Related to Figure 1
Table S3: FPKM values of 3T3-L1 differentiation RNA-seq. Related to Figure 3
Table S4. List of consensus IDRs. Related to Figure 5
Data Availability Statement
The RNA sequence data and processed files are available at GEO under accession GSE210875. The raw proteomics data are available at MassIVE under accession MSV000089301. Microscopy data reported in this paper will be shared by the lead contact upon request.
Code is available from lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.