

Abstract
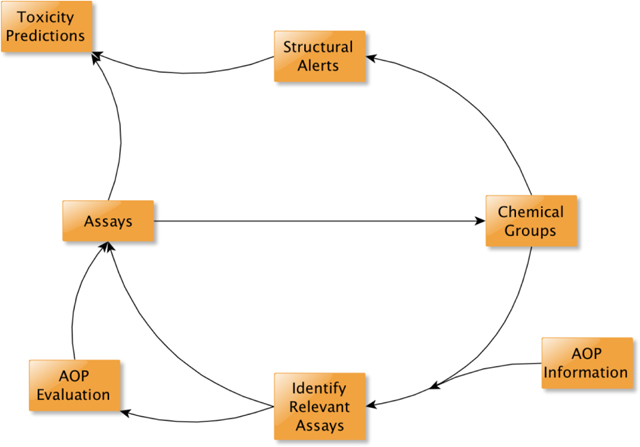
Adverse Outcome Pathways (AOPs) establish a connection between a molecular initiating event (MIE) and an adverse outcome. Detailed understanding of the MIE provides the ideal data for determining chemical properties required to elicit the MIE. This study utilized high-throughput screening data from the ToxCast program, coupled with chemical structural information, to generate chemical clusters using three similarity methods pertaining to nine MIEs within an AOP network for hepatic steatosis. Three case studies demonstrate the utility of the mechanistic information held by the MIE for integrating biological and chemical data. Evaluation of the chemical clusters activating the glucocorticoid receptor identified activity differences in chemicals within a cluster. Comparison of the estrogen receptor results with previous work showed that bioactivity data and structural alerts can be combined to improve predictions in a customizable way where bioactivity data are limited. The aryl hydrocarbon receptor (AHR) highlighted that while structural data can be used to offset limited data for new screening efforts, not all ToxCast targets have sufficient data to define robust chemical clusters. In this context, an alternative to additional receptor assays is proposed where assays for proximal key events downstream of AHR activation could be used to enhance confidence in active calls. These case studies illustrate how the AOP framework can support an iterative process whereby in vitro toxicity testing and chemical structure can be combined to improve toxicity predictions. In vitro assays can inform the development of structural alerts linking chemical structure to toxicity. Consequently, structurally related chemical groups can facilitate identification of assays that would be informative for a specific MIE. Together, these activities form a virtuous cycle where the mechanistic basis for the in vitro results and the breadth of the structural alerts continually improve over time to better predict activity of chemicals for which limited toxicity data exist.
Keywords: Molecular Initiating Event (MIE), Structure Activity Relationship (SAR), ToxCast, Adverse Outcome Pathway (AOP), Chemical Grouping, Structural Alert
Graphical Abstract
1. Introduction
In 2007, the National Research Council recommended increased use of alternative approaches to toxicity testing such as in vitro, in chemico, in silico, and –omics techniques that report on toxicity pathways at the molecular and cellular level (NRC, 2007). To make full use of the information generated by these alternative approaches, a framework that integrates these different data streams and connects the toxicity pathways with apical endpoints typically used for evaluation of toxicity was needed. The Adverse Outcome Pathway (AOP) concept provides such a framework that links mechanistic information derived from these alternative approaches to a biological target and, subsequently, to an apical adverse outcome (Ankley et al., 2010, Villeneuve et al., 2014a). This mechanistic focus enables the use of AOPs for organizing and integrating information from multiple methods for toxicity assessment. As such, the assays associated with these approaches may measure changes to the same or different biological processes along the pathway. Additionally, the structure of AOPs provides a scaffold on which mechanistic data from various sources including in silico models, in chemico, in vitro and/or in vivo assays, and literature information can be organized.
The molecular initiating event (MIE) is the initial key event (KE) within an AOP and its elicitation is essential for initiation of an AOP. The MIE is unique compared to other KEs as it is the stage where the chemical directly interacts with the biological system (Villeneuve et al., 2014b). As such, the MIE can provide mechanistic understanding of the interaction between the chemical and the biological system at the molecular level. Insight into these underlying mechanisms can help discern the chemical properties that are responsible for the interaction with the biological system (Enoch et al., 2013, Enoch and Roberts, 2013, Przybylak and Schultz, 2013). Once structural features have been identified as being associated with the elicitation of a particular MIE, they can facilitate the development of structural alerts, i.e. structural fragments associated with a specific MIE (Allen et al., 2014, Allen et al., 2016, Gutsell and Russell, 2013, Przybylak and Schultz, 2013). Collections of structural alerts associated with a given MIE have been recently termed in silico profilers especially in the context of their practical implementation into software tools such as the OECD Toolbox (Dimitrov et al., 2016). The information held by these profilers can be used to generate chemical clusters centered on the ability to elicit the same MIE (Allen et al., 2016, Enoch et al., 2011, Enoch et al., 2012, Enoch et al., 2013, Enoch and Roberts, 2013, Naven et al., 2013, Nelms et al., 2015a, Nelms et al., 2015b, Sakuratani et al., 2013a, Sakuratani et al., 2013b).
Alternatively, in vitro data related to a MIE can be utilized as a primary filter to identify chemicals active against the target of interest. Subsequently, the active chemicals can be clustered based upon structural similarity (Enoch and Roberts, 2013). To develop chemical clusters in this manner requires: 1) a representation of the chemical structure and, 2) a method to calculate the similarity between two chemicals (Leach and Gillet, 2007). There are a multitude of options available for both components, leading to an overwhelming number of possible combinations (Cereto-Massague et al., 2015). However, encoding chemical structure as a binary fingerprint (i.e. a string of vectors indicating the presence (1) or absence (0) of particular substructural features) and calculating similarity using the Tanimoto coefficient are most commonly used (Leach and Gillet, 2007, Willett, 2009, Willett, 2006). The outcome of this provides a measure of the similarity between two chemicals ranging from zero to one, where zero indicates a complete dissimilarity and one indicates the chemicals are identical.
As noted by Carrió et al. (2016), since there is no general agreement about which method is the most appropriate for quantifying chemical similarity, as this will be dependent on the characteristics of both the chemicals and the endpoint being evaluated, in this study we investigated three different similarity approaches that were chosen arbitrarily: 1) atom-pair descriptors and Tanimoto similarity, 2) ToxPrint chemotypes and Tanimoto similarity, and 3) atom environment descriptors and Hellinger distance (Carhart et al., 1984, Yang et al., 2015, Jeliazkova et al., 2009). For more discussion about the issues of characterizing chemical similarity, the reader is referred to other works by Todeschini et al. (2012), Floris et al. (2014), and Floris and Olla (2018).
A practical number of chemical clusters can be identified by using a similarity threshold. An index of 0.6 (i.e. 60% similarity) or greater, has been found to be useful in developing chemical clusters in previous works (Enoch et al., 2009). Investigation of the resultant chemical clusters in conjunction with the associated bioactivity data can, in certain circumstances, also enable the identification of structural alerts that may be incorporated into in silico profilers. It should be noted that the chemicals present within clusters can vary depending upon the chemical representation and similarity calculation used (Cereto-Massague et al., 2015). As such, just because chemicals cluster based upon one method does not mean they will necessarily cluster based upon a different method.
Recently, Angrish et al. (2016) outlined an AOP network (i.e. individual AOPs that share at least one common KE) for hepatic steatosis focusing on the key events of chemical mediated non-alcoholic fatty liver disease (Angrish et al., 2016). Fatty liver disease is currently the most common liver disease in the United States, affecting between 20–30% of the population (Noureddin and Rinella, 2015). Hepatic steatosis can be induced either by excessive alcohol consumption (alcoholic fatty liver disease) or by a variety of other stressors, including both therapeutic drugs and environmental chemicals (non-alcoholic fatty liver disease) (Al-Eryani et al., 2015). Within their AOP network Angrish and colleagues identified seven MIEs that may induce non-alcoholic fatty liver disease, five of these MIEs related to nuclear receptor interactions. Work performed by Mellor et al. expanded the number of MIEs present in the AOP network developed by Angrish et al. by identifying five additional nuclear receptors (NR) associated with hepatic steatosis (Mellor et al., 2016a). Consequently, a total of ten NRs have been identified as MIEs associated with the potential to induce hepatic steatosis, namely: Aryl hydrocarbon receptor (AHR), Constitutive androstane receptor (CAR), Estrogen receptor (ER), Farnesoid X receptor (FXR), Glucocorticoid receptor (GR), Liver X receptor (LXR), Peroxisome proliferator-activated receptor (PPAR), Pregnane X receptor (PXR), Retinoic acid receptor (RAR), and Retinoid X receptor (RXR).
In a follow-up paper, Mellor et al. identified two hundred and fourteen structural features and eight physicochemical descriptors that can be used as structural alerts to screen for chemicals with the potential to bind to one, or more, of these NRs (Mellor et al., 2016b). It should be noted that due to a lack of data in the database used to construct these alerts (only 40 chemical structures could be found in ChEMBL and none of these structures had an associated pChEMBL value), no structural alerts could be developed for the constitutive androstane receptor (CAR). Therefore, in this study we will only consider the nine NRs for which structural alerts were developed.
Both the AOP network developed by Angrish et al., and the work undertaken by Mellor et al., identify interactions between chemical ligands and various NRs as important MIEs that have the potential to induce hepatic steatosis. In order to discern chemicals with the ability to elicit the same MIE, biological information pertaining to the targets of interest is needed. The U.S. EPA’s Toxicity Forecasting (ToxCast) project, and the U.S. federal cross-agency Tox21 program have utilized high-throughput screening methods to generate large quantities of in vitro assay data (Dix et al., 2007, Judson et al., 2010, Kavlock et al., 2012, Attene-Ramos et al., 2013, Collins et al., 2008, Tice et al., 2013). At present, across both projects, a total of more than 9000 chemicals have undergone some level of assay screening (Richard et al., 2016, ToxCast). Of these 9000 chemicals, the vast majority have been tested in approximately 100 assays as part of the Tox21 program; whilst a subsection (~1060 ToxCast Phases I and II chemicals) have the broadest assay coverage, having been tested in more than 1000 assays across both the Tox21 and ToxCast projects. These chemicals cover a broad range of uses including food additives, cosmetic/personal care product ingredients, pesticides, pharmaceutical drugs, and industrial chemicals (Richard et al., 2016). As such, not only the chemical-use space, but the structure-feature space associated with the ToxCast and Tox21 data is highly diverse. The ToxCast database (which contains all the data from both ToxCast and Tox21 projects), contains at least two assay endpoints associated with each of the NRs identified as MIEs for hepatic steatosis.
The aim of this study was to highlight the power of combining biological activity and chemical structure in an iterative manner to better leverage both when attempting to predict chemical toxicity in data limited situations. To illustrate this process, chemical clusters were formed based on in vitro data corresponding to a set of MIE(s) within an AOP network for hepatic steatosis. Given the importance of hepatic steatosis as an AO, and interactions with NRs as MIEs within the AOP network, it provided an ideal set of case studies for demonstrating some of the applications for these chemical clusters. However, as we are only utilizing data pertaining to the MIEs any predictions of downstream events are, therefore, beyond the scope of the current study.
2. Materials and Methods
2.1. ToxCast high-throughput screening data
High-throughput screening data for the 9076 chemicals and 1192 assay endpoints, contained within the October 2015 data released by the ToxCast project, were used for this analysis (Judson et al., 2010, Kavlock et al., 2012, Richard et al., 2016). Further information regarding the chemicals and assays within ToxCast can be obtained from the data download page (https://www.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data, accessed on July 16th 2018), the interactive webpage of the iCSS ToxCast Dashboard (https://actor.epa.gov/dashboard/, accessed on July 16th 2018), or the CompTox Chemistry Dashboard (https://comptox.epa.gov/dashboard/, accessed on July 16th 2018) (Williams et al., 2017). The DSSTox Substance Identifiers (DTXSIDs) provided in this manuscript can be utilized to access highly curated chemical information, with associated chemical properties, present in the CompTox Chemistry Dashboard. Three files were utilized for this analysis, the first containing AC50 values (i.e. the concentration at 50% of maximum activity) for each chemical-assay pair (oldstyle_ac50_Matrix_151020.csv, provided by ToxCast), the second containing a list of chemical-assay combinations with a flag for quality control (QC) (AllResults_flags_151020.csv, provided by ToxCast), and the third containing the chemical specific cytotoxicity point (Supplementary Table 1). All ToxCast files referenced throughout this manuscript are available for download from the ToxCast data download page at ftp://newftp.epa.gov/comptox/High_Throughput_Screening_Data/Summary_Files (accessed July 16th 2018).
The AC50 file comprises a matrix containing an AC50 value (in μM) for each chemical-assay pair that has been tested and identified as being active within ToxCast. A value of 1,000,000μM (1M) is used to represent a chemical-assay combination that has been tested but was identified as being inactive, and “NA” values represent chemical-assay combinations that have not currently been tested (as of the October 2015 data release) (Judson et al., 2016, Documentation, 2015). Meanwhile, the QC flag file identifies chemical-assay pairs where there may be false positive/negative results; alternatively, the associated flag(s) may help to explain potentially anomalous data (Filer et al., 2015, Filer et al., 2017(ToxCast, 2018)).
The cytotoxicity point file contains a list of the chemicals within ToxCast and the associated concentration (in log units and μM) at which the chemical exhibited cytotoxicity. It has been shown that chemicals can cause a large number of non-specific hits (the “burst”) in in vitro assays at concentrations near or above where cell stress and cytotoxicity occur (Judson et al., 2016). The burst region is defined by taking activity data from a range of cytotoxicity assays (46 total in the current analysis) in the form of hits (i.e. was the chemical active or not) and AC50 values (concentrations at 50% of maximal activity). A chemical is defined as being cytotoxic if at least 2 of the cytotoxicity assays were active below the typical upper limit of testing of 100μM. For each chemical, we take the median(log10(AC50)) as the center of the cytotoxicity region, and we additionally calculate the median absolute deviation (MAD) of the log10(AC50) values. Since the individual chemical MAD values are sensitive to the number of cytotoxicity assays that are positive, we then take the median of the cytotoxicity MAD values across all chemicals, which is termed the global cytotoxicity MAD. In this instance, the global cytotoxicity MAD value is 0.289. Finally, the lower bound of cytotoxicity for each chemical is defined as the median(log10(AC50)) – (3*global cytotoxicity MAD) (Judson et al., 2016). Thus, any chemical-assay pair concentration above this lower bound value would be considered within the cytotoxicity region. Additionally, because this lower bound value takes into consideration the variability of the cytotoxicity point across the entire ToxCast dataset, it acts as a more conservative estimate of the cytotoxicity point. For chemicals with 0 or 1 active cytotoxicity assays, the center of the cytotoxicity region is set to a default value of 1000μM. More information regarding how the values contained within this file were generated can be found in Judson et al. (2016).
2.2. Data Analysis
Unless otherwise stated all analyses in this study were performed using R, v3.3.2 (Team, 2016).
2.2.1. Generation of filtered hit-call matrix
2.2.1.1. Use of cytotoxicity burst phenomenon information
The AC50 and cytotoxicity point ToxCast data files, discussed above, were utilized to develop a discretized hit-call matrix that takes into account the cytotoxicity-associated burst phenomenon. To generate the burst hit-call matrix, we first identified, in a chemical-specific manner, the lower value of either 1) the μM value of the lower bound of cytotoxicity, or 2) the lowest reported AC50 value for the chemical within the 46 cytotoxicity assays. These criteria were used to provide us with the most conservative estimate of the cytotoxicity burst threshold. For this study, the lower of these two values will be termed the lower cytotoxicity value. Next, for each chemical, the lower cytotoxicity value was compared against the AC50 values present in the ToxCast AC50 matrix. Those chemical-assay combinations with an AC50 below the lower cytotoxicity value for the chemical are considered active (i.e. assigned a “1”) and those combinations with an AC50 above the lower cytotoxicity value are considered inactive (i.e. assigned a “0”) (Supplementary Figure 1). It should be noted that some of the activity present above this lower cytotoxicity value may be driven by the target specific mechanism of the assay. However, where AC50 values fall within this region it is more difficult to ascertain if those chemical-assay pairs are driven by a target specific mechanism or by more generalized cytotoxicity or cell stress effects (Judson et al., 2016). For this work, it was more important to enrich for chemicals that specifically act via the MIE rather than to identify all chemicals that may act via the MIE.
2.2.1.2. Use of quality control flag information
To evaluate the burst-filtered hit call matrix, the cytotoxicity assay AC50s were compared with the AC50 for the target assay across the chemicals. To perform this examination, the original –log10AC50 data matrix (old_style_neg_log_ac50_Matrix_151020.csv, provided by ToxCast) was integrated with the burst hit-call matrix developed above through scalar multiplication. This was done so that only the –log10AC50 data for those chemical-assay combinations active below the lower cytotoxicity value remained. Next, a scatter graph of the results from the cytotoxicity assays was plotted against the results for the corresponding specific target assay for each chemical (Supplementary Figure 1, inset). For example, the –log10AC50 values in the TOX21_AR_BLA_Antagonist_viability assay (a cytotoxicity assay) were plotted against the results from the TOX21_AR_BLA_Antagonist_ratio assay (the corresponding target-specific assay).
The scatter graphs showed that the majority of the points fell on either the x-axis (i.e. the chemical exhibited a target-specific effect prior to cytotoxicity) or the y-axis (i.e. the chemical exhibited cytotoxicity prior to the observation of a target-specific effect). However, a number of points fell along the diagonal. These points are more difficult to decipher because the target-specific effect occurs at approximately the same concentration at which the chemical exhibits cytotoxicity. Therefore, we are less certain about the mechanism behind these chemicals because one of two scenarios may be occurring: 1) a target-specific mechanism may be inducing cytotoxicity at approximately the same concentration, or 2) cytotoxicity may be occurring via a secondary mechanism and the target-specific effect coincidentally occurs at the same concentration. Investigation of the points along the diagonal led us to identify four quality control (QC) flags that were associated with a large proportion of these chemical-assay pairs. These four flags are: “Only one concentration above baseline, active”, “Borderline active”, “Gain AC50 < lowest concentration and loss AC50 < mean concentration”, and “Hit-call potentially confounded by overfitting”. The four QC flags utilized within this study represent a subset of the nine flags present within the quality control flag file released by ToxCast (AllResults_flags_151020.csv, provided by ToxCast). More information regarding each of these flags can be found in the ToxCast data analysis pipeline (tcpl, V1.4.3, accessed July 16th 2018) R package or the associated vignette (Filer et al., 2018). These four flags were added to the filtering criteria when creating the final hit-call matrix for this work to remove potentially confounding chemicals and increase confidence in target-specific active calls.
2.2.1.3. Combining burst and QC flag information
To generate the final hit-call matrix (the burst flag hit-call matrix) that was used for the remainder of this study, a chemical was considered active in an assay only when it passed two criteria: 1) the AC50 was below the lower cytotoxicity value, and 2) it did not have an associated QC flag. Unless otherwise noted, these criteria were used for determining chemical activity whenever a positive assay result is referenced throughout the manuscript. A copy of this final burst flag hit-call matrix is available as a comma separate value (csv) file within the Supplementary Information (Supplementary Table S2).
2.2.2. Extraction of chemicals active for nuclear receptors associated with hepatic steatosis
Since at least two cytotoxicity assays were required to establish a burst point, chemicals that were tested in fewer than two of the 46 cytotoxicity assays were not considered for this study. This information was gathered from the “tested_Matrix_151020.csv” file available in the ToxCast data release. Of the chemicals with two or more cytotoxicity assays, active chemicals for each NR from Mellor et al. (2016a) were defined based on activity in at least one human-relevant assay from the final hit-call matrix. Therefore, a separate data frame was generated for each of the nine NRs that contained 1) the list of active chemicals for the specific NR; 2) the assay results for the NR in question; 3) SMILES notation, where available, for each chemical and; 4) a number of chemical identifiers. Only use those chemical with a defined structure and corresponding simplified molecular-input line entry system (SMILES) notation are used in the present study. The relatively small number of compounds without a defined SMILES string in ToxCast are typically macromolecules, mixtures/formulations, or polymers. Only the chemicals with a SMILES string were used because the SMILES strings provide the necessary chemical structure information to create structure-based chemical clusters and profile against the previously developed structural alerts. Therefore, unless otherwise stated, numbers referenced will relate to active chemicals that have associated SMILES notation. All of the SMILES strings were neutralized, and salt counter ions were removed using ChemAxon’s Standardizer program.
2.2.3. Clustering of active chemicals
Three separate clustering methods were used to generate chemical clusters for each chemical list identified above based upon structural similarity. The first approach used the ChemmineR package (Cao et al., 2011, Cao et al., 2008) to generate atom-pair descriptors for each chemical within the separate chemical lists and then calculated the Tanimoto index between chemicals (AP_Tani) (Carhart et al., 1984). The second approach utilized the more than 700 freely available ToxPrint chemotypes implemented within the ChemoTyper software to generate a molecular fingerprint for each chemical (Yang et al., 2015) (https://chemotyper.org, accessed July 16th 2018). Subsequently, the ChemmineR package was used to calculate the Tanimoto index between chemicals (TP_Tani). For the final approach, we used Toxmatch (v1.07) to generate a similarity matrix identifying the Hellinger distance between atom environments within each chemical list (AE_Hell) (Gallegos Saliner et al., 2008, Jeliazkova et al., 2009, Patlewicz et al., 2008).
The chemical cluster generation for each individual method was performed using the single-linkage (nearest neighbor) binning clustering function implemented within the ChemmineR package (Cao et al., 2011). It should be noted that for the first two approaches a distance matrix was calculated on the fly by the ChemmineR package. For the final approach, a distance matrix calculated as 1-similarity was generated prior and subsequently utilized by the clustering function within ChemmineR. We used an initial similarity threshold of 0.6 (60%) and increased in 5% increments. Our initial threshold was based upon previous work that demonstrated a cut-off of 0.6 works well for chemicals clustering for teratogenicity (Enoch et al., 2009) The final similarity thresholds were chosen based upon visual inspection of the clusters after each iteration to keep the cluster sizes generally consistent across the different chemicals/metrics. Neither chemical structure, toxicity information, nor any other information regarding expected clustering of the chemicals were used as criteria. As such, the chosen similarity thresholds varied between chemical list and similarity approach used.
2.3. Profiling active chemicals against previously developed structural alerts
Each chemical list was profiled against a set of previously developed structural alerts associated with NRs that have the potential to induce hepatic steatosis (Mellor et al., 2016b). The majority of these alerts are present in the AOP network developed by Angrish et al. (2016). The KNIME workflow developed by Mellor et al. (2016b) was utilized within KNIME (v3.2) to perform the profiling of each of the chemical lists to identify those chemicals that contain a structural fragment(s) associated with NR binding. Chemicals triggering an alert for a NR, therefore, suggest chemicals that have the potential to bind to the specific NR.
2.4. Use of Judson et al. estrogen receptor model to evaluate our approach
The results from the full ER model developed by Judson et al. (2015) were used to evaluate our approach. These results were utilized because the Judson et al. (2015) ER model has been shown to perform on par with other, more traditional, lower-throughput tests (Browne et al., 2015, Kleinstreuer et al., 2016). To evaluate our approach, chemicals not given a designation of “agonist”, “antagonist”, or “none” within the supplemental table provided by Judson et al. (2015) were removed. The chemicals were then split into two groups based upon their classification by the Judson et al. (2015) ER model: those classified as either “agonist” or “antagonist” were consider active, and those classified as “none” were considered inactive. Subsequently, we varied the conditions under which a chemical would be considered active by our approach. The conditions utilized were as follows: 1) an active call in at least one ER assay; 2) an active call in at least one ER assay AND the presence of a structural alert for ER binding; 3) an active call in at least one ER assay OR the presence of a structural alert for ER binding; and 4) the presence of a structural alert only. For each of the first three conditions we iterated through every combination of sets of four ER assays (a total of 3060 different combinations per condition) based on the work of Judson et al. (2017). Finally, we calculated the mean sensitivity, specificity, accuracy, and Matthews’ correlation coefficient and the corresponding 95% confidence interval for each condition.
Additionally, the same calculations for the same three conditions were made when iterating through every combination of between five and eighteen ER assays to estimate the minimum number of assays needed to better define chemical clusters for cases like the AHR case study.
3. Results and Discussion
This study demonstrates the use of chemical structure and bioactivity information to improve chemical toxicity prediction in situations where data are limited. To ensure that chemical activity used for developing the chemical clusters represented target-mediated effects, stringent filters removed assay results that were potentially confounded by non-specific cytotoxicity. These filters include explicit filtering on cytotoxicity and more general filtering via quality control flags identified as informative based on preliminary analysis. While the stringency of the filters likely resulted in a high false negative rate, it increases our confidence that an active call was due to a target-mediated mechanism. This, in turn, makes the structural clusters more reliably focused on target-mediated mechanisms.
Using the target-specific hit-calls, active chemicals were defined for each NR acting as an MIE within the liver steatosis AOP network (Angrish et al., 2016, Mellor et al., 2016a). Active chemicals were subsequently profiled against previously developed structural alerts associated with the potential to bind to NRs linked to inducing hepatic steatosis (Mellor et al., 2016b). It should be noted that different numbers of assays are associated with the different NRs. For example, there are twenty human-relevant assays associated with ER but only two assays associated with AHR (Table 1). Furthermore, the total number of chemicals active below the cytotoxicity burst also varied across the NRs with the liver X receptor (LXR) having the fewest number of chemicals active below the burst (20) and ER having the highest number of chemicals active below the burst (1449) (Table 1). Additionally, there is no apparent correlation between the number of active chemicals identified and the total number of assays associated with a NR; although, increased sensitivity to actives with increasing number of assays cannot be ruled out. For example, there are 680 total chemicals active within at least one of the three assays associated with pregnane X receptor (PXR); in comparison, there are only 285 total chemicals active within at least one of the eight assays associated with farnesoid X receptor (FXR) (Table 1).
Table 1.
Data extracted from the burst flag hit-call matrix showing: 1) the number of human relevant assays present in ToxCast for each nuclear receptor identified as being associated with liver steatosis, 2) the total number of chemicals tested in at least one of the NR-related assays, 3) the total number of chemicals active below the lower cytotoxicity value (numbers within parentheses provide number of chemicals with associated SMILES strings), 4) the number of chemicals that contain any of the structural alerts developed by Mellor et al (2016), and 5) the number of chemicals that contain at least one structural alert for the specific nuclear receptor (numbers within parentheses are the percentage of chemicals with SMILES strings that contain a nuclear receptor specific structural alert).
| Nuclear Receptor | Number of human relevant assays in ToxCast | Number of chemicals tested in at least 1 assay | Number of chemicals active in at least 1 assay | Number of chemicals triggering any alert | Number of chemicals triggering nuclear receptor specific structural alert |
|---|---|---|---|---|---|
| AHR | 2 | 8415 | 461 (448) | 349 | 161 (36%) |
| ER | 20 | 8415 | 1449 (1341) | 905 | 576 (43%) |
| FXR | 8 | 7639 | 285 (278) | 206 | 38 (14%) |
| GR | 5 | 8415 | 340 (284) | 250 | 170 (60%) |
| LXR | 3 | 3282 | 20 (20) | 15 | 1 (5%) |
| PPAR | 12 | 8707 | 541 (513) | 360 | 222 (43%) |
| PXR | 3 | 3282 | 680 (639) | 436 | 117 (18%) |
| RAR | 6 | 3282 | 72 (65) | 51 | 0 (0%) |
| RXR | 5 | 3284 | 192 (175) | 87 | 3 (1%) |
3.1. Coverage of previously developed structural alerts
After identifying active chemicals for each NR, each chemical list was profiled against the structural alerts developed by Mellor et al. (2016b). Upon inspecting the profiling results, it became clear that the structural alerts associated with certain NRs performed better than others at identifying potential binders (Table 1). The four best performing suites of structural alerts were able to identify between 36–60% of chemicals active for a particular NR as having the potential to bind to that specific NR (Table 1). For example, of the 448 chemicals active in at least one AHR assay, 161 (36%) were profiled as having a structural alert for AHR. Meanwhile, the suites of structural alerts perform poorly for other NRs, with the structural alerts for LXR, the retinoic acid receptor (RAR), and the retinoid X receptor (RXR) identifying between 0–5% of chemicals active for one of these NRs as having the potential to bind to that specific NR. The varying success of suites of structural alerts will depend, in part, on the size of the active NR sets from which the alerts were derived, and to which the alerts are being applied. As discussed below, the iterative nature of evaluating structural alerts based on functional assay information and then refining chemical activity predictions using the structural alerts holds the promise to both refine the structural alerts, and their predictive ability, as well as provide the additional support of the structural predictions when evaluating assay activity.
In using the more stringent threshold for defining our active chemicals, we may have removed false positives that resulted in aberrant structural alerts that were more influenced by general cytotoxicity previously or we may have removed true positives that fell above the cytotoxicity burst threshold the would have been informative for developing structural alerts. To determine the impact of our stringent threshold on the lack of coverage by the structural alerts, we investigated how the suites of structural alerts performed when the chemical lists were generated using the original ToxCast hit-call matrix. As would be expected a larger number of chemicals were identified as being active for each NR (Table 2). However, for the vast majority of the NRs there was either no change or only a modest decline in the percentage of active chemicals containing a structural alert for the specific NR (Table 2): the exception being those chemicals active in at least one GR assay that had a decline of 15% compared to the results using the burst flag hit-call matrix.
Table 2.
Data extracted from the original ToxCast hit-call matrix showing: 1) the number of chemicals that were active (numbers within parentheses give the number of chemicals with associated SMILES strings), 2) the number of those chemicals that contain any of the structural alerts developed by Mellor et al. (2016b), and 3) the number of chemicals that contain at least one structural alert for the specific nuclear receptor (numbers within parentheses are the percentage of chemicals with SMILES strings that contain a nuclear receptor specific structural alert).
| Nuclear Receptor | Number of chemicals active in at least 1 assay | Number of chemicals triggering any alert | Number of chemicals triggering nuclear receptor specific structural alert |
|---|---|---|---|
| AHR | 974 (954) | 740 | 302 (32%) |
| ER | 2840 (2677) | 1837 | 1154 (43%) |
| FXR | 1177 (1127) | 904 | 134 (12%) |
| GR | 941 (858) | 710 | 383 (45%) |
| LXR | 148 (142) | 96 | 8 (6%) |
| PPAR | 1584 (1517) | 1119 | 557 (38%) |
| PXR | 1533 (1467) | 961 | 244 (17%) |
| RAR | 339 (318) | 242 | 1 (0%) |
| RXR | 632 (596) | 364 | 14 (2%) |
Given that the results seem to be at least as good with the more stringent cutoff, utilizing the lower cytotoxicity value to remove chemicals that are (potential) false positives is likely to be a less significant contributing factor to the lack of coverage by the structural alerts when compared to the chemical space that the structural alerts cover. This may be explained, at least in part, by examining the two datasets used as the training and test set. To develop the structural alerts Mellor et al. (2016b) used ChEMBL as the training set, a data set containing mainly drug-like bioactive molecules. In comparison, the test set chemicals, i.e. ToxCast, contains mainly environmentally-relevant chemicals with few drug-like molecules. Therefore, it is likely that a difference in chemical space between the chemicals in the training and test sets contributed to our not observing more active chemicals associated with an alert for the specific NR. In other words, many ToxCast chemicals lie outside the applicability domain of the existing alerts, which were derived based on drug-like chemicals.
Chemicals with biological activity for each NR were clustered based on structural similarity using three separate clustering methods to generate chemical clusters. We chose to employ three different fingerprinting and similarity approaches to investigate what differences, if any, may be observed. It should be noted that detailed analysis as to the distinctions between the methodologies employed by the different approaches is beyond the scope of this study. Whilst investigating these chemical clusters, we were able to hypothesize refinements to some of the existing alerts whilst also positing additional alerts. One example of a cluster that may lead to a possible refinement to an existing alert can be seen in Figure 1, whereby seven chemicals active in at least one AHR assay were clustered together using the AP_Tani similarity approach. The original alert developed by Mellor et al. (2016b) (center structure, Figure 1) enables either a fluorine, chlorine, or hydrogen atom to be at the meta and/or para position on the benzene ring. However, this existing alert does not cover two of the chemicals present within this cluster because they contain a bromine atom para to the amino substituent. After examination of the chemicals within the cluster it would make sense to expand the existing alert to cover bromine substituted chemicals, especially given that bromine is also a halogen and, therefore, has the potential to act in a similar manner to the fluorine and chlorine substituted chemicals.
Figure 1.

The chemicals around the perimeter are those contained within AP_Tani Cluster 26 of the AHR active chemicals. The structure in the center is the previously identified structural alert generated by Mellor et al. (2016b). It is hypothesized that this structural alert could be expanded to cover brominated chemicals as well as chlorinated and fluorinated chemicals. As a structural alert is either present or absent from a chemical there is no relative similarity that can be calculated between the alert and the chemical(s) being profiled.
One example of a hypothesized additional alert regarding the potential to bind to the peroxisome proliferator-activated receptor (PPAR) can be seen in Figure 2 (center structure). Each of the four chemicals used to derive this potential alert were active in at least one PPAR assay and were clustered together using the TP_Tani similarity approach. As can be seen in Figure 2, each of the four chemicals contain the same backbone that has been utilized to generate the posited alert. Three of the chemicals present within this cluster (mefenamic acid (DTXSID5023243), tolfenamic acid (DTXSID1045409), and meclofenamate sodium (DTXSID8045567)) were also clustered when using the AP_Tani approach. However, both putative alerts used as examples here should not yet be considered “finished”. This is because additional data are required to confirm (or refute) the mechanistic information as to how these chemicals bind AHR or PPAR. Additionally, for the hypothesized PPAR alert more data from further studies would likely be needed for other, similar chemicals to further define/refine the alert.
Figure 2.

The chemical fragment in the center is a hypothesized additional alert for chemicals with the potential to bind to PPAR. The chemicals around the perimeter are those contained within TP_Tani Cluster 124 of the PPAR active chemicals form the basis upon which this hypothesized alert was developed.
3.2. Case Studies
Three nuclear receptors will be considered in turn to evaluate the findings and to demonstrate the value of integrating in vitro and in silico data using the AOP framework. As the data used within this study only relate to the MIE, and not to any downstream KEs, we will be limiting discussion to those applications that could be performed using the data included in this study. Thus, predictions regarding the likelihood that a chemical induces hepatic steatosis will not be made.
3.2.1. Glucocorticoid Receptor
A total of 340 chemicals were active below the cytotoxicity burst in at least one of the five GR related assays (Table 1). Comparing the structure-based chemical clusters for GR active chemicals highlights some of the main differences in results between the different clustering methods.
As can be seen in Table 3, irrespective of the clustering approach used, approximately two thirds of the active chemicals were assigned to “clusters” with either one or two members and the vast majority (at least 85%) of “clusters” contain only a single chemical. This is perhaps expected given the diversity of chemical space in the ToxCast chemical library (Richard et al., 2016). Within this study, any cluster with fewer than three members was not investigated further. Overall, there is modest variability across the three clustering approaches in terms of the number of clusters produced containing three or more cluster members: the AE_Hell (i.e. Hellinger distance between atom environments) approach generated the most clusters (14) covering 99 active chemicals; the TP_Tani (i.e. Tanimoto similarity between ToxPrint chemotypes) approach generated the fewest clusters (7) covering 97 active chemicals; and the AP_Tani Tani (i.e. Tanimoto similarity between atom pair descriptors) approach generated 10 clusters covering the fewest active chemicals (88) (Table 3). Even though the number of clusters generated across the approaches differ, the chemicals that comprise the clusters essentially remain the same, i.e. the extra clusters generated by the AP_Tani and AE_Hell approaches are primarily subsets of the clusters containing larger numbers of chemicals generated by the TP_Tani approach. For example, the 32 chemicals that form Cluster 44 generated by the TP_Tani approach are, in large part, spread over two separate clusters (Clusters 95 and 136) when using the AP_Tani approach, whilst these chemicals are split into four clusters (Clusters 7, 27, 29, and 77) when using the AE_Hell approach. There are cases where novel clusters are formed, however, such as two chemical clusters (Clusters 76 and 98) defined by AE_Hell that were not identified by either the AP_Tani or TP_Tani.
Table 3.
Clustering data that shows the similarity threshold chosen, the number of clusters that contain three or more chemicals, and the number of active chemicals that are contained within a cluster with three or more members for each similarity measure. AE_Hell - atom environment descriptors and Hellinger distance, AP_Tani – atom pair descriptors and Tanimoto similarity, and TP_Tani – ToxPrint chemotypes and Tanimoto similarity.
| Nuclear Receptor | Similarity Approach | Similarity Threshold (%) | Number of clusters with 3 or more members | Number of active chemicals covered |
|---|---|---|---|---|
| AHR | AE_Hell | 70 | 19 | 92 |
| AP_Tani | 60 | 17 | 90 | |
| TP_Tani | 75 | 24 | 121 | |
| ER | AE_Hell | 85 | 27 | 173 |
| AP_Tani | 65 | 42 | 237 | |
| TP_Tani | 80 | 59 | 324 | |
| FXR | AE_Hell | 70 | 14 | 61 |
| AP_Tani | 60 | 10 | 32 | |
| TP_Tani | 75 | 10 | 41 | |
| GR | AE_Hell | 80 | 14 | 99 |
| AP_Tani | 70 | 10 | 88 | |
| TP_Tani | 85 | 7 | 97 | |
| LXR | AE_Hell | 60 | 0 | 0 |
| AP_Tani | 60 | 0 | 0 | |
| TP_Tani | 70 | 0 | 0 | |
| PPAR | AE_Hell | 80 | 11 | 76 |
| AP_Tani | 65 | 14 | 62 | |
| TP_Tani | 75 | 26 | 102 | |
| PXR | AE_Hell | 75 | 22 | 91 |
| AP_Tani | 60 | 14 | 61 | |
| TP_Tani | 80 | 19 | 110 | |
| RAR | AE_Hell | 75 | 1 | 5 |
| AP_Tani | 60 | 1 | 4 | |
| TP_Tani | 80 | 1 | 5 | |
| RXR | AE_Hell | 75 | 8 | 44 |
| AP_Tani | 60 | 4 | 14 | |
| TP_Tani | 80 | 4 | 29 |
The AP_Tani approach was, in most cases, able to cluster chemicals that were active in either the TOX21_GR_BLA_Agonist_ratio or TOX21_GR_BLA_Antagonist_ratio (Supplemental Figure 12). This is also borne out for the other two similarity approaches (Supplemental Figures 11 and 13). One example where this is not the case is Cluster 10 generated by the AP_Tani approach that contains 25 chemicals, of which 22 are active in the TOX21_GR_BLA_Agonist_ratio assay, with three (prednisone (DTXSID4021185), meprednisone (DTXSID8023260), and cortisone (DTXSID5022857)) active in the TOX21_GR_BLA_Antagonist_ratio assay. Upon closer inspection, the three chemicals active within the antagonist assay contain a carbonyl substituent at the 11-carbon position within the steroid ring system (Figure 3A). In contrast, the chemicals active in the Tox21 GR β-lactamase agonist assay all contain a hydroxyl substituent at the 11-carbon position (Figure 3B). This is further corroborated by Cluster 5 generated by the TP_Tani approach, of the 48 chemicals contained within this cluster only the two chemicals (prednisone and meprednisone) active in the Tox21 GR β-lactamase antagonist assay did not have a hydroxyl substituent present at the 11-carbon position within the steroid ring. The results from these clusters suggest that the change in substituent at the 11-carbon position from a ketone to a hydroxyl may play a role in conferring agonist activity. Information within the literature further supports this hypothesis: prednisone is a known pro-drug that must be metabolized in order to be converted to the GR active form, prednisolone (Becker, 2013). In the conversion from prednisone to prednisolone the only difference between the two chemical structures is the change from a ketone moiety at the 11-carbon position to a hydroxyl moiety. Additionally, in terms of GR activity, cortisone can be considered the inactive form of cortisol (Rask et al., 2001). In this instance, the 11β-hydroxysteroid dehydrogenase enzyme is responsible for metabolizing cortisone to cortisol. Therefore, the information we have gained by taking into consideration both the assay and chemical structure data can be utilized to help inform the development and/or refinement of structural alerts pertaining to chemicals that contain this steroid backbone.
Figure 3.

A) Structures of the three chemicals present within Cluster 10 using the AP_Tani approach that are active within the TOX21_GR_BLA_Antagonist_ratio assay. B) General structure of the majority of chemicals present within AP_Tani Cluster 10 and TP_Tani Cluster 5 that are active in the TOX21_GR_BLA_Agonist_ratio assay. The main differences in structure occur at the R1 and R2 substituents. As can be seen, each of the chemicals active only in the antagonist assay contain a carbonyl moiety at the 11-carbon position of the steroid structure whilst the chemicals active in only the agonist assay contain a hydroxyl moiety at the same position.
3.2.2. Estrogen Receptor
A total of 1449 chemicals, were active below the cytotoxicity burst in at least one of the 20 ER related assays (Table 1). As discussed above, there is a moderate level of variability in terms of the number of clusters containing three or more cluster members generated by the different clustering approaches: the AE_Hell approach generated 27 clusters covering 173 active chemicals (12%), the AP_Tani approach generated 42 clusters covering 237 active chemicals (16%), and the TP_Tani approach generated 59 clusters covering 325 active chemicals (22%) (Table 3 ). Whilst there was overlap between the chemicals contained within clusters with three or more members, together the three clustering approaches covered a total of 443 active chemicals (31%).
Work recently published by Judson and colleagues demonstrated that for certain combinations of in vitro assays, as few as four assays were sufficient to predict ER agonist activity at a level comparable to the 16-assay ER agonist model (Judson et al., 2017). In this work, Judson et al. generated subset models for each combination of 1–16 in vitro assays that comprise the ER agonist pathway model. For each subset model, a variety of performance metrics were calculated (i.e. sensitivity, specificity, and balanced accuracy) for different contexts, namely: using all chemicals against the previously developed full ER model (Judson et al., 2015); using only the reference chemicals against the full ER model, using only in vitro literature-based reference chemicals (Judson et al., 2015); and using only in vivo literature-based reference chemicals (Kleinstreuer et al., 2016). The results of certain combinations of four ER agonist-related assays were observed to have a minimum balanced accuracy (across the four different situations named above) of 0.94, which was on par with the minimum balanced accuracy for the model containing all 16 ER agonist-related assays. As a consequence of this, and the fact that few targets within ToxCast contain as many assays as ER, we decided to use the minimum number of assays identified by Judson et al. (2017) to generate two distinct models.
The models were created by iterating through every combination of four ER-relevant assays from the burst flag hit-call matrix and combining this information with the ER-specific structural alerts developed by Mellor et al. (2016b). The first model (the higher confidence model) aimed to provide a higher confidence in a hazard call; this was achieved by requiring the presence of 1) activity within at least one ER assay in the burst flag hit-call matrix and 2) an ER structural alert. In comparison, the second model (the wider coverage model) was designed to capture the maximal number of positive chemicals; this was achieved by requiring only the presence of activity in at least one ER assay or an ER structural alert. Due to the relative simplicity of our models, we wanted to evaluate them against the much more detailed ER model developed by Judson et al. (2015) (Table 4). The reason for this was not to attempt to replace the Judson et al. (2015) ER model, but rather to investigate how reliable our approach would be in instances where only limited bioactivity data are available.
Table 4.
Mean performance data of every variation of four ER assays for the two ER models and how they compare to using the burst flag hit-call data or the presence of a structural alert in isolation. Values in parentheses are 95% confidence intervals (no confidence interval could be calculated for the presence of a structural alert as there were no parameters to change). MCC – Matthews Correlation Coefficient.
| Active call in burst hit-call matrix | Presence of structural alert only | Active call in burst hit-call matrix AND presence of structural alert | Active call in burst hit-call matrix OR presence of structural alert | |
|---|---|---|---|---|
| Sensitivity | 67% (48–80%) | 69% | 52% (40–62%) | 84% (77–89%) |
| Specificity | 92% (85–97%) | 73% | 98% (96–99%) | 67% (61–71%) |
| Accuracy | 90% (84–94%) | 72% | 94% (93–95%) | 68% (63–71%) |
| MCC | 0.466 (0.38–0.575) | 0.234 | 0.546 (0.484–0.617) | 0.27 (0.244–0.292) |
As can be seen in Table 4, the higher confidence model has a higher specificity, accuracy, and Matthew’s Correlation Coefficient (MCC) than the wider coverage model or bioactivity data in isolation; thus, demonstrating that the higher confidence model is able to utilize the combination of bioactivity data from only four assays and structural alert information to correctly identify chemicals with the ability to bind to the ER (Table 4). Therefore, if the higher confidence model predicts a chemical as being active, there is a greater likelihood the chemical is a true active. In comparison, the wider coverage model has a higher sensitivity and lower specificity, i.e. it identifies a larger number of chemicals as being active at the expense of identifying a larger number of false positives. This is to be expected as the aim of the wider coverage model is to identify chemicals that have the potential to bind to the ER with further, confirmatory, toxicity tests subsequently being performed.
Based on how the models performed against the Judson et al. (2015) ER model we believe our approach of integrating structural information into predictions could be of use in instances where bioactivity data are limited. This approach will be useful to limit the amount of testing required for novel chemicals that contain either a structural alert or are structurally similar to other chemicals that have already undergone testing.
3.2.3. Aryl Hydrocarbon Receptor
Overall, a total of 461 chemicals were active in at least one of the two AHR assays present in ToxCast (Table 1). The AP_Tani approach generated the fewest clusters with at least three cluster members (17) covering the fewest chemicals (90), the TP_Tani approach generated the most clusters (24) covering the largest number of chemicals (121), and the AE_Hell approach generated an intermediate number of clusters (19) covering 92 chemicals (Table 3).
Unlike ER, the majority of targets within ToxCast have many fewer assays in which a chemical can be tested to verify bioactivity. For example, considering the two assays related to AHR binding, over half (55%) of the active chemicals were tested in only one assay. Therefore, it is difficult to determine a chemical’s activity with a high degree of certainty. Determining true activity is especially difficult in instances where a chemical has been tested in both AHR assays but is active in only one. Even taking cytotoxicity into consideration, certain chemicals may be active in a specific assay or technology due to: 1) assay interference (e.g. chemicals used as dyes interfering with fluorescence readouts), or 2) real activity that can only be captured in a specific assay (e.g. the assay may have metabolic capabilities not present in other assays within the battery). With data from fewer assays it is also more difficult to define robust chemical clusters. Thus, while four assays are sufficient once the pathway is known and mechanistic models exist for integrating the data, the number of assays needed to define this information initially is likely to be higher.
To estimate the minimum number of assays required when nothing is known about the chemical clusters or mechanistic linkage among the assays, we again used the Judson et al. (2015) model results. However, this time we varied the number of assays used between five and eighteen and iterated through every combination to calculate the same statistics (Table 5 and Figure 4). The results suggest that chemical activity in any one (or more) of 10 assays provides a good balance between bringing the statistics in-line with using the entire 18 assay battery and the number of assays used to generate the statistics. Therefore, it is likely that information from up to 10 assays may be required to enable the development of robust chemical clusters and allow development of more sophisticated models for integrating the assay results. There are two means by which we could acquire the additional assay data. The first is by using additional assays specific for the biological target (e.g. AHR) or MIE. Alternatively, the AHR AOP could be utilized to identify appropriate assays that measure changes in downstream KEs and the information from these assays could be used. This second approach may be more desirable, especially where data for downstream KEs is available such as in ToxCast, as it would allow for data from proximal KEs to be used not only for determining the chemical clusters but also to provide orthogonal assays to evaluate active calls at the MIE in the context of the broader AOP.
Table 5.
Mean performance data of every variation of between 5 and 18 ER assays for the two ER models and how they compare to using the burst flag hit-call data in isolation. Values in parentheses are 95% confidence intervals. MCC – Matthews Correlation Coefficient.
| No. Assays | Burst Only | Burst AND Alert | Burst OR Alert | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sensitivity | Specificity | Accuracy | MCC | Sensitivity | Specificity | Accuracy | MCC | Sensitivity | Specificity | Accuracy | MCC | |
| 5 | 71 (54–81%) | 90 (84–96%) | 89 (83–94%) | 0.457 (0.379–0.554) | 55 (43–62%) | 97 (96–99%) | 94 (93–95%) | 0.545 (0.493–0.607) | 85 (79–90%) | 65 (61–70%) | 67 (63–71%) | 0.269 (0.245–0.291) |
| 6 | 73 (57–82%) | 89 (82–95%) | 88 (82–93%) | 0.447 (0.377–0.537) | 56 (46–63%) | 97 (95–99%) | 94 (93–95%) | 0.542 (0.496–0.597) | 86 (80–90%) | 64 (60–69%) | 66 (62–70%) | 0.268 (0.245–0.289) |
| 7 | 75 (60–83%) | 87 (82–95%) | 87 (81–92%) | 0.438 (0.374–0.523) | 58 (48–63%) | 97 (95–98%) | 94 (92–95%) | 0.538 (0.495–0.586) | 87 (81–90%) | 64 (59–69%) | 65 (61–70%) | 0.266 (0.245–0.288) |
| 8 | 77 (64–84%) | 86 (81–94%) | 86 (81–92%) | 0.429 (0.37–0.509) | 59 (50–63%) | 96 (95–98%) | 94 (92–95%) | 0.534 (0.495–0.577) | 88 (82–91%) | 63 (58–68%) | 64 (60–69%) | 0.265 (0.243–0.286) |
| 9 | 79 (66–84%) | 85 (80–93%) | 85 (80–91%) | 0.421 (0.368–0.494) | 60 (52–63%) | 96 (95–98%) | 93 (92–95%) | 0.530 (0.493–0.572) | 88 (84–91%) | 62 (58–68%) | 64 (60–69%) | 0.263 (0.241–0.285) |
| 10 | 80 (69–85%) | 84 (79–91%) | 84 (80–89%) | 0.414 (0.366–0.483) | 60 (53–63%) | 96 (95–97%) | 93 (92–94%) | 0.525 (0.493–0.567) | 89 (85–91%) | 61 (57–66%) | 63 (60–67%) | 0.260 (0.240–0.282) |
| 11 | 81 (72–85%) | 83 (79–89%) | 83 (79–88%) | 0.407 (0.364–0.472) | 61 (53–63%) | 95 (94–97%) | 93 (92–94%) | 0.521 (0.491–0.563) | 89 (85–91%) | 60 (57–65%) | 63 (60–67%) | 0.258 (0.239–0.280) |
| 12 | 82 (73–85%) | 82 (79–88%) | 82 (79–88%) | 0.401 (0.363–0.459) | 61 (55–63%) | 95 (94–97%) | 93 (92–94%) | 0.517 (0.491–0.557) | 89 (86–91%) | 60 (57–64%) | 62 (59–66%) | 0.256 (0.239–0.277) |
| 13 | 82 (75–85%) | 82 (79–87%) | 82 (79–87%) | 0.394 (0.362–0.446) | 62 (56–63%) | 95 (94–96%) | 93 (92–94%) | 0.513 (0.491–0.552) | 90 (88–91%) | 59 (57–64%) | 61 (59–65%) | 0.254 (0.238–0.275) |
| 14 | 83 (76–85%) | 81 (78–85%) | 81 (79–85%) | 0.389 (0.361–0.439) | 62 (56–63%) | 95 (94–96%) | 93 (92–94%) | 0.509 (0.489–0.544) | 90 (88–91%) | 59 (57–62%) | 61 (59–64%) | 0.253 (0.237–0.273) |
| 15 | 83 (80–85%) | 80 (78–85%) | 80 (79–84%) | 0.383 (0.361–0.431) | 62 (60–63%) | 95 (94–96%) | 92 (92–93%) | 0.504 (0.49–0.534) | 90 (88–91%) | 58 (57–61%) | 60 (59–63%) | 0.251 (0.238–0.268) |
| 16 | 84 (82–85%) | 79 (78–83%) | 80 (79–83%) | 0.378 (0.361–0.406) | 62 (62–63%) | 95 (94–95%) | 92 (92–93%) | 0.500 (0.49–0.529) | 90 (89–91%) | 58 (57–60%) | 60 (59–62%) | 0.249 (0.238–0.261) |
| 17 | 84 (83–85%) | 79 (78–81%) | 79 (79–81%) | 0.372 (0.362–0.4) | 63 (62–63%) | 94 (94–95%) | 92 (92–93%) | 0.500 (0.49–0.517) | 91 (89–91%) | 57 (57–59%) | 60 (59–61%) | 0.248 (0.241–0.258) |
| 18 | 85 | 78 | 79 | 0.368 | 63 | 94 | 92 | 0.491 | 91 | 57 | 59 | 0.246 |
Figure 4.

Four line graphs depicting the mean change in sensitivity, specificity, accuracy, and Matthew’s correlation coefficient (MCC) when adjusting the minimum number of assays required to initially provide a reliable determination of activity. The 95% confidence interval is represented by the ribbon plot surrounding each line graph. The different colors represent a different testing method: blue represents instances where activity required an active call in the burst hit-call matrix and the presence of a structural alert; orange represents instances where only an active call in the burst hit-call matrix was required, and; green represents instances where either an active call in the burst hit-call matrix or the presence of a structural alert was required.
4. Conclusion
ToxCast data has shown promise for providing cost-effective bioactivity information for a large number of chemicals, especially in cases where many related or orthogonal assays exist for a common target-mediated mechanism (Judson et al., 2015, Browne et al., 2015). Cheminformatics approaches are also providing valuable information for chemicals where little or no toxicity data exist (Allen et al., 2016, Enoch et al., 2011, Enoch et al., 2012, Enoch et al., 2013, Enoch and Roberts, 2013, Nelms et al., 2015a, Nelms et al., 2015b, Sakuratani et al., 2013b, Sakuratani et al., 2013a, Naven et al., 2013). The present work focused on how in vitro and in silico data can be leveraged to provide the maximal information in data-limited situations. Our results have shown the importance of the applicability domain and how this can be difficult to overcome with the wide array of structural diversity seen with environmental chemicals.
The ultimate goal for this work would be an iterative process whereby solutions can be provided to maximize the utility of existing in vitro and in silico data while providing concrete information for use in improving the confidence in those predictions when needed. The AHR example (Figure 2) showed how biological activity could be used to refine existing structural alerts. The PPAR example (Figure 3) showed how biological activity could be used to identify new structural alerts. The glucocorticoid receptor case study highlighted how small molecular changes that are critical for agonist activity can be identified in a data-driven way. The estrogen receptor showed how existing structural alerts can be combined with biological activity readouts to provide fit-for-purpose toxicity predictions making optimal use of the available data for data-poor chemicals. With the aryl-hydrocarbon receptor case study, however, we saw that in many cases the biological activity data associated with many MIEs is insufficient to support many applications. While structural information can improve confidence in those situations as well, it can be combined with AOP information to assist in improving the suite of biological assays for these MIEs. As data-driven approaches to AOP development are established (Oki and Edwards, 2016, Oki et al., 2016, Bell et al., 2016), these efforts can be integrated to continually improve 21st century toxicity predictions.
Evaluation of the wealth of data associated with estrogen receptor activity (Judson et al., 2015) suggests that up to ten assays may be required to define the optimal model for integrating the toxicity data and create high confidence chemical clusters to support the development of structural alerts. This does not mean that the methods described herein cannot be employed until after a major data gathering effort. Chemical clusters could be defined based on as little as two assays as shown with the AHR example, and these could be used to increase confidence in predictions with the two assays alone. As chemical clusters are defined that are highly likely to work through a given MIE, the biological activity of the chemicals in those clusters could be used to identify additional assays related to that mechanism and potentially expand the number of assays without required additional screening. This, in turn, could improve the clustering of chemicals to establish higher quality structural groups with broader coverage of the chemical space, which then could be used to further expand the number of assays considered for the toxicity assessment. As high throughput toxicogenomics measurements are generated, this method of iterative development should prove extremely valuable.
It is also important to note that data from the full set of assays covering any one MIE is only required for a subset of the chemical universe to evaluate the assays and determine the optimal subset for screening. Results from Judson, et al. (2017) suggest that the remaining chemicals following this initial phase can be screened with as few as four assays. By employing an iterative process where chemical structure as well as mechanistic information is used as the initial assay battery is defined and evaluated, we would expect that the process can be optimized to minimize the up-front screening required to establish the biological assay panel and maximize the domain of applicability for the structural alerts identified.
Supplementary Material
Supplementary Table 2. Complete final burst flag hit-call matrix that takes into consideration both cytotoxicity and quality control flag information.
Supplementary Table 3. Shows the similarity threshold chosen for each nuclear receptor and similarity method, the resulting number of clusters with three or more cluster members, and the number of active chemicals covered by these clusters. Each tab labelled with the abbreviated nuclear receptor name contains the chemical identified from ToxCast (chid), the cluster size at the chosen similarity threshold, and the cluster identifier for each similarity method.
Supplementary Table 1. Cytotoxicity point, and associated lower bound of cytotoxicity point, data calculated using information from the 46 cytotoxicity assays. Each row present in the table contains a chemical-specific cytotoxicity point, lower bound of the cytotoxicity point, and number of “hits” within the 46 cytotoxicity assays.
Supplementary Figures 2-19 contain a vectorized image (PDF) of the negative log10AC50 values for each nuclear receptor and clustering method utilized for which chemical groups were identified. The colored side bar identifies the chemical cluster the chemical belongs to. Each column corresponds to a ToxCast assay. Blue represents activity within the assays, with darker blues depicting activity at a lower concentration and lighter blues depicting activity at higher concentrations. White depicts chemical-assay pairs that were tested but not activity was observed, and grey depicts, as yet, untested chemical-assay pairs.
Supplementary Figure 1 contains a scatterplot that depicts, using an example, how the cytotoxicity burst point was used to increase confidence in active calls.
Acknowledgements
The authors would like to acknowledge the support of Brian Chorley, Michelle Angrish for their help and expertise and David Hines and Antony Williams for constructive comments on the manuscript. M.D.N. was supported by an appointment to the Research Participation Program of the U.S. Environmental Protection Agency, Office of Research and Development, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. EPA.
Funding Sources
This project was supported in part by an appointment to the Internship/Research Participation Program at the Office of Research and Development, U.S. Environmental Protection Agency, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and EPA.
Disclaimer
The information in this document has been funded wholly (or in part) by the U.S. Environmental Protection Agency. It has been subjected to review by the National Health and Environmental Effects Research Laboratory and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
Abbreviations
- AE_Tani
Atom Environment and Hellinger distance approach
- AHR
Aryl Hydrocarbon Receptor
- AOP
Adverse Outcome Pathway
- AO
Adverse Outcome
- AP_Tani
Atom Pair and Tanimoto similarity approach
- ER
Estrogen Receptor
- GR
Glucocorticoid Receptor
- MAD
Median Absolute Deviation
- MIE
Molecular Initiating Event
- NR
Nuclear Receptor
- TP_Tani
ToxPrint chemotypes and Tanimoto similarity approach
Footnotes
The authors declare no competing financial interests. The views expressed in this paper are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
Data Statement
All the data used in this manuscript is available either in the paper, in the Supplementary Information, or from the URLs provided within the manuscript.
References
- AL-ERYANI L, WAHLANG B, FALKNER KC, GUARDIOLA JJ, CLAIR HB, PROGUH RA & CAVE M. 2015. Identification of environmental chemicals associated with the develoment of toxicant associated fatty liver disease in rodents. Toxicol. Pathol, 43, 482–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALLEN TE, GOODMAN JM, GUTSELL S. & RUSSELL PJ 2014. Defining molecular initiating events in the adverse outcome pathway framework for risk assessment. Chem Res Toxicol, 27, 2100–12. [DOI] [PubMed] [Google Scholar]
- ALLEN TE, LIGGI S, GOODMAN JM, GUTSELL S. & RUSSELL PJ 2016. Using Molecular Initiating Events To Generate 2D Structure-Activity Relationships for Toxicity Screening. Chem Res Toxicol, 29, 1611–1627. [DOI] [PubMed] [Google Scholar]
- ANGRISH MM, KAISER JP, MCQUEEN CA & CHORLEY BN 2016. Tipping the Balance: Hepatotoxicity and the 4 Apical Key Events of Hepatic Steatosis. Toxicol Sci, 150, 261–8. [DOI] [PubMed] [Google Scholar]
- ANKLEY GT, BENNETT RS, ERICKSON RJ, HOFF DJ, HORNUNG MW, JOHNSON RD, MOUNT DR, NICHOLS JW, RUSSOM CL, SCHMIEDER PK, SERRRANO JA, TIETGE JE & VILLENEUVE DL 2010. Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem, 29, 730–41. [DOI] [PubMed] [Google Scholar]
- ATTENE-RAMOS MS, MILLER N, HUANG R, MICHAEL S, ITKIN M, KAVLOCK RJ, AUSTIN CP, SHINN P, SIMEONOV A, TICE RR & XIA M. 2013. The Tox21 robotic platform for the assessment of environmental chemicals - From vision to reality. Drug Discov Today, 18, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BECKER DE 2013. Basic and clinical pharmacology of glucocorticosteroids. Anesth Prog, 60, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELL SM, ANGRISH MM, WOOD CE & EDWARDS SW 2016. Intergrating publically available data to generate computationally predictied adverse outcome pathways for fatty liver. Toxicol Sci, 150, 510–520. [DOI] [PubMed] [Google Scholar]
- BROWNE P, JUDSON RS, CASEY WM, KLEINSTREUER NC & THOMAS RS 2015. Screening chemicals for estrogen receptor bioactivity using a computational model. Environ Sci Technol, 49, 8804–8814. [DOI] [PubMed] [Google Scholar]
- CAO Y, BACKMAN T, WANG Y. & GIRKE T. 2011. ChemmineR - V2: Analysis of small molecule and screening data. ChemMineR Manual. [Google Scholar]
- CAO Y, CHARISI A, CHENG LC, JIANG T. & GIRKE T. 2008. ChemmineR: a compound mining framework for R. Bioinformatics, 24, 1733–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARHART RE, SMITH DH & VENKATARAGHAVAN R. 1984. Atom Pairs as Molecular Features in Structure-Activity Studies: Definition and Applications. J Chem Inf Comput Sci, 25, 64–73. [Google Scholar]
- CARRIÓ P, SANZ F. & PASTOR M. 2016. Toward a unifying strategy for the structure-based prediction of toxicological endpoints. Arch Toxicol, 90, 2445–2460. [DOI] [PubMed] [Google Scholar]
- CERETO-MASSAGUE A, OJEDA MJ, VALLS C, MULERO M, GARCIA-VALLVE S. & PUJADAS G. 2015. Molecular fingerprint similarity search in vitrual screening. Methods, 71, 58–63. [DOI] [PubMed] [Google Scholar]
- COLLINS FS, GRAY GM & BUCHER JR 2008. Transforming environmental health protection. Science, 319, 906–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMITROV SD, DIDERICH R, SOBANSKI T, PAVLOV TS, CHANKOV GV, CHAPKANOV AS, KARAKOLEV YH, TEMELKOV SG, VASILEV RA, GEROVA KD, KUSEVA CD, TODOROVA ND, MEHMED AM, RASENBERG M. & MEKENYAN OG 2016. QSAR Toolbox - workflow and major functionalities. SAR QSAR Environ Res, 1–17. [DOI] [PubMed] [Google Scholar]
- DIX DJ, HOUCK KA, MARTIN MT, RICHARD AM, SETZER W. & KAVLOCK RJ 2007. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci, 95, 5–12. [DOI] [PubMed] [Google Scholar]
- DOCUMENTATION TR 2015. US EPA ToxCast data release October 2015 - Summary Files. [Google Scholar]
- ENOCH SJ, CRONIN MTD & ELLISON CM 2011. The use of a chemistry based profiler for covalent DNA binding in the development of chemical categories for read-across for genotoxicity. ATLA, 39, 131–145. [DOI] [PubMed] [Google Scholar]
- ENOCH SJ, CRONIN MTD, MADDEN JC & HEWITT M. 2009. Formation of Structural Categories to Allow for Read-Across for Teratogenicity. QSAR & Combinatorial Science, 28, 696–708. [Google Scholar]
- ENOCH SJ, PRZYBYLAK KR & CRONIN MTD 2013. Category Formation Case Studies. In: CRONIN MTD, MADDEN JC, ENOCH SJ, ROBERTS DW (ed.) Chemical Toxicity Prediction: Category Formation and Read-Across. Cambridge, United Kingdom: Royal Society of Chemistry. [Google Scholar]
- ENOCH SJ & ROBERTS DW 2013. Approaches for Grouping Chemicals into Categories. In: CRONIN MTD, MADDEN JC, ENOCH SJ, ROBERTS DW (ed.) Chemical Toxicity Prediction: Category Formation and Read-Across. Cambridge, United Kingdom: Royal Society of Chemistry. [Google Scholar]
- ENOCH SJ, SEED MJ, ROBERTS DW, CRONIN MT, STOCKS SJ & AGIUS RM 2012. Development of mechanism-based structural alerts for respiratory sensitization hazard identification. Chem Res Toxicol, 25, 2490–8. [DOI] [PubMed] [Google Scholar]
- FILER D, KOTHIYA P, SETZER RW, JUDSON RS & MARTIN MT 2018. ToxCast Data Analysis Pipeline (tcpl) V1.4.3. R Package. [DOI] [PubMed] [Google Scholar]
- FILER DL, KOTHIYA P, SETZER RW, JUDSON RS & MARTIN MT 2017. tcpl: the ToxCast pipeline for high-throughput screening data. Bioinformatics, 33, 618–620. [DOI] [PubMed] [Google Scholar]
- FILER DL, KOTHIYA P, SETZER WR, JUDSON RS & MARTIN MT 2015. The ToxCast Analysis Pipeline: An R package for processing and modeling chemical screening data. Available: https://www.epa.gov/sites/production/files/2015-08/documents/pipeline_overview.pdf. [Google Scholar]
- FLORIS M, MANANARO A, NICOLOTTI O, MEDDA R, MANGIATORDI GF & BENFENATI E. 2014. A generalizable definition of chemical similarity for read-across. J Cheminform, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLORIS M. & OLLA S. 2018. Molecular Similarity in Computational Toxicology. In: NICOLOTTI O. (ed.) Methods Mol Biol. New York: Springer. [DOI] [PubMed] [Google Scholar]
- GALLEGOS SALINER A, POATER A, JELIAZKOVA N, PATLEWICZ G. & WORTH AP 2008. Toxmatch - A Chemical Classification and Activity Prediction Tool based on Similarity. Regul Toxicol Pharmacol, 52, 77–84. [DOI] [PubMed] [Google Scholar]
- GUTSELL S. & RUSSELL P. 2013. The role of chemistry in developing understanding of adverse outcome pathways and their application in risk assessment. Toxicology Research, 2, 299. [Google Scholar]
- JELIAZKOVA N, PATLEWICZ G. & GALLEGOS SALINER A. 2009. IdeaConsult Toxmatch User Manual. [Google Scholar]
- JUDSON R, HOUCK K, MARTIN M, RICHARD AM, KNUDSEN TB, SHAH I, LITTLE S, WAMBAUGH J, WOODROW SETZER R, KOTHYA P, PHUONG J, FILER D, SMITH D, REIF D, ROTROFF D, KLEINSTREUER N, SIPES N, XIA M, HUANG R, CROFTON K. & THOMAS RS 2016. Editor’s Highlight: Analysis of the Effects of Cell Stress and Cytotoxicity on In Vitro Assay Activity Across a Diverse Chemical and Assay Space. Toxicol Sci, 152, 323–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUDSON RS, HOUCK KA, KAVLOCK RJ, KNUDSEN TB, MARTIN MT, MORTENSEN HM, REIF DM, ROTROFF DM, SHAH I, RICHARD AM & DIX DJ 2010. In vitro screening of environmental chemicals for targeted testing prioritization: The ToxCast project. Environ Health Perspect, 118, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUDSON RS, HOUCK KA, WATT ED & THOMAS RS 2017. On selecting a minimal set of in vitro assays to reliably determine estrogen agonist activity. Regul Toxicol Pharmacol, 91, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUDSON RS, MAGPANTY FM, CHICKARMANE V, HASKELL C, TANIA N, TAYLOR J, XIA M, HUANG R, ROTROFF DM, FILER DL, HOUCK KA, MARTIN MT, SIPES N, RICHARD AM, MANSOURI K, SETZER RW, KNUDSEN TB, CROFTON KM & THOMAS RS 2015. Integrated model of chemical perturbations of a bioloigcal pathway using 18 in vitro high-throughput screening assays for the estrogen receptor. Toxicol Sci, 148, 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAVLOCK RJ, CHANDLER K, HOUCK KA, HUNTER S, JUDSON RS, KLEINSTREUER N, KNUDSEN TB, MARTIN MT, PADILLA S, REIF DM, RICHARD AM, ROTROFF D, SIPES N. & DIX DJ 2012. Update on EPA’s ToxCast program: Providing high throughput decision support tools for chemical risk management. Chem Res Toxicol, 25, 1287–1302. [DOI] [PubMed] [Google Scholar]
- KLEINSTREUER NC, CEGER PC, ALLEN DG, STRICKLAND J, CHANG X, HAMM JT & CASEY WM 2016. A curated database of rodent uterotrophic bioactivity. Environ Health Perspect, 124, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEACH AR & GILLET VJ 2007. An Introduction to Chemoinformatics, Netherlands, Springer. [Google Scholar]
- MELLOR CL, STEINMETZ FP & CRONIN MT 2016a. The identification of nuclear receptors associated with hepatic steatosis to develop and extend adverse outcome pathways. Crit Rev Toxicol, 46, 138–52. [DOI] [PubMed] [Google Scholar]
- MELLOR CL, STEINMETZ FP & CRONIN MT 2016b. Using Molecular Initiating Events to Develop a Structural Alert Based Screening Workflow for Nuclear Receptor Ligands Associated with Hepatic Steatosis. Chem Res Toxicol, 29, 203–12. [DOI] [PubMed] [Google Scholar]
- NAVEN RT, SWISS R, KLUG-MCLEOD J, WILL Y. & GREENE N. 2013. The Development of Structure-Activity Relationships for Mitochondrial Dysfunction: Uncoupling of Oxidative Phosphorylation. Toxicol Sci, 131, 271–278. [DOI] [PubMed] [Google Scholar]
- NELMS MD, ATES G, MADDEN JC, VINKEN M, CRONIN MT, ROGIERS V. & ENOCH SJ 2015a. Proposal of an in silico profiler for categorisation of repeat dose toxicity data of hair dyes. Arch Toxicol, 89, 733–41. [DOI] [PubMed] [Google Scholar]
- NELMS MD, MELLOR CL, CRONIN MT, MADDEN JC & ENOCH SJ 2015b. Development of an in Silico Profiler for Mitochondrial Toxicity. Chem Res Toxicol, 28, 1891–902. [DOI] [PubMed] [Google Scholar]
- NOUREDDIN M. & RINELLA ME 2015. Nonalcoholic fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clin Liver Dis, 19, 361–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NRC 2007. Toxicity testing in the 21st Century: A vision and a strategy, Washington, DC. USA, The National Academies Press. [Google Scholar]
- OKI NO & EDWARDS SW 2016. An integrative data mining approach to identifying adverse outcome pathway signatures. Toxicol, 350–352, 49–61. [DOI] [PubMed] [Google Scholar]
- OKI NO, NELMS MD, BELL SM, MORTENSEN HM & EDWARDS SW 2016. Accelerating Adverse Outcome Pathway development using publicly available data sources. Current Environmental Health Reports. [DOI] [PubMed] [Google Scholar]
- PATLEWICZ G, JELIAZKOVA N, GALLEGOS SALINER A. & WORTH AP 2008. Toxmatch-a new software tool to aid in the development and evaluation of chemically similar groups. SAR QSAR Environ Res, 19, 397–412. [DOI] [PubMed] [Google Scholar]
- PRZYBYLAK KR & SCHULTZ TW 2013. Informing chemical categories through the development of Adverse Outcome Pathways. In: CRONIN MTD, MADDEN JC, ENOCH SJ, ROBERTS DW (ed.) Chemical Toxicity Prediction: Category Formation and Read-Across. Cambridge, United Kingdom: Royal Society of Chemistry. [Google Scholar]
- RASK E, OLSSON T, SODERBERG S, ANDREW R, LIVINGSTONE DEW, JOHNSON O. & WALKER BR 2001. Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab, 86, 1418–1421. [DOI] [PubMed] [Google Scholar]
- RICHARD AM, JUDSON RS, HOUCK KA, GRULKE CM, VOLARATH P, THILLAINADARAJAH I, YANG C, RATHMAN J, MARTIN MT, WAMBAUGH JF, KNUDSEN TB, KANCHERLA J, MANSOURI K, PATLEWICZ G, WILLIAMS AJ, LITTLE SB, CROFTON KM & THOMAS RS 2016. ToxCast chemical landscape: Paving the road to 21st Century toxicology. Chem Res Toxicol, 29, 1225–1251. [DOI] [PubMed] [Google Scholar]
- SAKURATANI Y, ZHANG HQ, NISHIKAWA S, YAMAZAKI K, YAMADA T, YAMADA J, GEROVA K, CHANKOV G, MEKENYAN O. & HAYASHI M. 2013a. Hazard Evaluation Support System (HESS) for predicting repeated dose toxicity using toxicological categories. SAR QSAR Environ Res, 24, 351–63. [DOI] [PubMed] [Google Scholar]
- SAKURATANI Y, ZHANG HQ, NISHIKAWA S, YAMAZAKI T, YAMADA J. & HAYASHI M. 2013b. Categorization of nitrobenzenes for repeated dose toxicity based on adverse outcome pathways. SAR QSAR Environ Res, 24, 35–46. [DOI] [PubMed] [Google Scholar]
- TEAM, R. C. 2016. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- TICE RR, AUSTIN CP, KAVLOCK RJ & BUCHER JR 2013. Improving the human hazard characterization of chemicals: a Tox21 update. Environ Health Perspect, 121, 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TODESCHINI R, CONSONNI V, XIANG H, HOLLIDAY J, BUSCEMA M. & WILLETT P. 2012. Similarity Coefficients for Binary Chemoinformatics Data: Overview and Extended Comparison Using Simulated and Real Data Sets. J Chem Inf Model, 52, 2884–2901. [DOI] [PubMed] [Google Scholar]
- TOXCAST. Available: https://www.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data.
- TOXCAST 2018. ToxCast Owner’s Manual - Guidance for Exploring Data. [Google Scholar]
- VILLENEUVE DL, CRUMP D, GARCIA-REYERO N, HECKER M, HUTCHINSON TH, LALONE CA, LANDESMANN B, LETTIERI T, MUNN S, NEPELSKA M, OTTINGER MA, VERGAUWEN L. & WHELAN M. 2014a. Adverse outcome pathway (AOP) development I: strategies and principles. Toxicol Sci, 142, 312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VILLENEUVE DL, CRUMP D, GARCIA-REYERO N, HECKER M, HUTCHINSON TH, LALONE CA, LANDESMANN B, LETTIERI T, MUNN S, NEPELSKA M, OTTINGER MA, VERGAUWEN L. & WHELAN M. 2014b. Adverse outcome pathway development II: best practices. Toxicol Sci, 142, 321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLETT P. 2006. Similarity-based virtual screening using 2D fingerprints. Drug Discov Today, 11, 1046–1053. [DOI] [PubMed] [Google Scholar]
- WILLETT P. 2009. Similarity methods in chemoinformatics. Ann Rev Inform Sci, 43, 3–71. [Google Scholar]
- WILLIAMS AJ, GRULKE CM, EDWARDS J, MCEACHRAN AD, MANSOURI K, BAKER NC, PATLEWICZ G, SHAH I, WAMBAUGH JF, JUDSON RS & RICHARD AM 2017. The CompTox Chemistry Dashboard: a community data resource for environmental chemistry. J Cheminform, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG C, TARKHOV A, MARUSCZYK J, BIENFAIT B, GASTEIGER J, KLEINOEDER T, MAGDZIARZ T, SACHER O, SCHWAB CH, SCHWOEBEL J, TERFLOTH L, ARVIDSON K, RICHARD A, WORTH A, RATHMAN J. 2015. New publically available chemical query language, CSRML, to support chemotype representations for application to data mining and modeling. J Chem Inf Model, 55, 510–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 2. Complete final burst flag hit-call matrix that takes into consideration both cytotoxicity and quality control flag information.
Supplementary Table 3. Shows the similarity threshold chosen for each nuclear receptor and similarity method, the resulting number of clusters with three or more cluster members, and the number of active chemicals covered by these clusters. Each tab labelled with the abbreviated nuclear receptor name contains the chemical identified from ToxCast (chid), the cluster size at the chosen similarity threshold, and the cluster identifier for each similarity method.
Supplementary Table 1. Cytotoxicity point, and associated lower bound of cytotoxicity point, data calculated using information from the 46 cytotoxicity assays. Each row present in the table contains a chemical-specific cytotoxicity point, lower bound of the cytotoxicity point, and number of “hits” within the 46 cytotoxicity assays.
Supplementary Figures 2-19 contain a vectorized image (PDF) of the negative log10AC50 values for each nuclear receptor and clustering method utilized for which chemical groups were identified. The colored side bar identifies the chemical cluster the chemical belongs to. Each column corresponds to a ToxCast assay. Blue represents activity within the assays, with darker blues depicting activity at a lower concentration and lighter blues depicting activity at higher concentrations. White depicts chemical-assay pairs that were tested but not activity was observed, and grey depicts, as yet, untested chemical-assay pairs.
Supplementary Figure 1 contains a scatterplot that depicts, using an example, how the cytotoxicity burst point was used to increase confidence in active calls.