

Summary
Viroids and viroid-like covalently closed circular (ccc) RNAs are minimal replicators that typically encode no proteins and hijack cellular enzymes for replication. The extent and diversity of viroid-like agents are poorly understood. We developed a computational pipeline to identify viroid-like cccRNAs and applied it to 5,131 metatranscriptomes and 1,344 plant transcriptomes. The search yielded 11,378 viroid-like cccRNAs spanning 4,409 species-level clusters, a five-fold increase compared to the previously identified viroid-like elements. Within this diverse collection, we discovered numerous putative viroids, satellite RNAs, retrozymes, and ribozy-like viruses. Diverse ribozyme combinations and unusual ribozymes within the cccRNAs were identified. Self-cleaving ribozymes were identified in ambiviruses, some mito-like viruses and capsid-encoding satellite virus-like cccRNAs. The broad presence of viroid-like cccRNAs in diverse transcriptomes and ecosystems implies that their host range is far broader than currently known, and matches to CRISPR spacers suggest that some cccRNAs replicate in prokaryotes.
Graphical Abstract
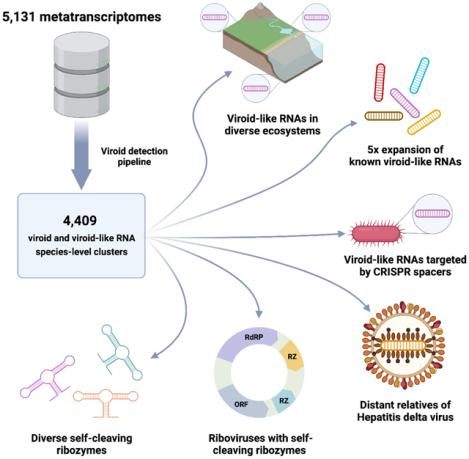
In Brief
A large-scale survey of covalently-closed circular RNA across ecosystems reveals that viroids infect a wide range of host species, extending beyond plants, and identifies additional types of ribozyme activity as well as functional features in these molecules.
Introduction
Viroids, which cause several economically important diseases in agricultural plants, are the smallest and simplest among the known infectious agents1–3. Viroids are small, covalently closed circular (ccc) RNA molecules of 220 to 450 nucleotides that encode no proteins and consist largely of RNA structures that are required for replication or viroid-host interaction. In contrast to viruses, which hijack the host translation system to produce proteins encoded in virus genes, viroids take advantage of the host transcriptional machinery. Specifically, viroids hijack the host plant’s DNA-dependent RNA polymerase II to transcribe their RNA and thus catalyze viroid replication4–6. Viroids utilize the rolling circle replication (RCR) mechanism, producing multimeric intermediates that are cleaved into genome-size monomers by ribozymes that are present in both polarities of viroid RNAs or by recruited host RNases7,8. The resulting linear monomers are then ligated by a host DNA ligase to form the mature cccRNA9,10.
Since the discovery of viroids in 197111, about 50 viroid species were identified in plants and classified into two families, Avsunviroidae and Pospiviroidae. Members of the Avsunviroidae use viroid-encoded autocatalytic hammerhead (HHR) ribozymes to process replication intermediates into unit length viroid genomes12,13. Members of the family Pospiviroidae lack ribozymes and instead rely on conserved sequence motifs that serve as recognition and cleavage sites for host RNase III14. The members of the two viroid families adopt distinct RNA structures: a branched RNA conformation is predominant in avsunviroids15, in contrast to the typically rod-shaped conformation of pospiviroids16.
In addition to viroids, several other groups of infectious agents also possess genomes consisting of cccRNA17. Many plant viruses support the replication of small (about 300 nt) circular satellite RNAs (satRNAs) that closely resemble viroids18 and also replicate via RCR19. The satRNAs differ from viroids in that they are replicated by the RNA-dependent RNA polymerase (RdRP) of the helper virus and are encapsidated in that virus’s capsid20,21. Thus, satRNAs are effectively encapsidated viroids. Unlike viroids, satRNAs encode both HHRs and hairpin ribozymes (HPR)22.
Another viroid-like agent is the retroviroid, carnation small viroid-like RNA (CarSV) which, unlike viroids, does not appear to transmit horizontally among plants23. CarSV, the only currently known retroviroid, is a cccRNA that is similar to viroids in size and contains HHRs in both strands. However, in contrast to the viroids, an extrachromosomal DNA copy of CarSV has been discovered and shown to integrate into the plant genome with the help of a pararetrovirus24,25.
A recently discovered group of cccRNA agents are retrozymes, retrotransposons that propagate via circular RNA intermediates of about 170 to 400 nucleotides. The retrozymes are viroid-like in that they do not encode any proteins but contain self-cleaving HHRs26,27. However, unlike viroids, the retrozymes are neither infectious nor autonomous, but rather, hijack the replication machinery of autonomous retrotransposons. Resembling satRNAs and avsunviroids, retrozyme cccRNAs adopt a branched conformation.
A conspicuous group of viroid-like agents is the viral realm Ribozyviria28 that includes deltaviruses, such as hepatitis delta virus (HDV), an important human pathogen. Similarly to pospiviroids, ribozyviruses possess rod-shaped cccRNA genomes that replicate via the RCR mechanism and encode distinct ribozymes, unrelated to those of viroids, that autocatalytically process multimeric replication intermediates29–31. Ribozyviruses have substantially larger genomes than other viroid-like agents (about 1.7 kb) and encode their own nucleocapsid protein. The reproduction of ribozyviruses relies on a helper virus (hepatitis B virus in the case of HDV), which provides the envelope protein for ribozyvirus virions. For years, HDV remained the only known deltavirus. Recently, however, viruses more distantly related to HDV have been discovered in various vertebrates and invertebrates32–36, suggesting a considerable uncharacterized diversity of ribozyviruses.
Viroids and viroid-like cccRNAs are minimal replicators, or ultimate parasites, that lack genes and effectively consist only of RNA structures required for replication. This extreme simplicity of viroids triggered speculation on their direct descendance from primordial RNA replicators37,38. However, the apparent narrow host range of viroids, which so far have been reported only in plants, is poorly compatible with this evolutionary scenario. Instead, given the similarities between retrozymes and avsunviroids, it has been suggested that avsunviroids descended from retrozymes27,39.
Given the ultimate structural simplicity of viroids and related cccRNAs and the universality of the DNA-dependent RNA polymerases involved in their replication across life forms, the current narrow spread and limited diversity of parasitic cccRNAs appear puzzling. Furthermore, this apparent paucity of viroid-like agents is in stark contrast to the burgeoning diversity of RNA viruses, many thousands of which including numerous, distinct groups have been discovered by metatranscriptome analyses40–43. At present, there are at least three orders of magnitude more known RNA viruses than there are viroids and viroid-like cccRNAs.
We were interested in investigating the global diversity of viroids and viroid-like agents. To this end, we performed an exhaustive search for cccRNAs in a collection of 5,131 diverse metatranscriptomes that have been recently employed for massive RNA virus discovery43, and additionally searched 1,341 plant transcriptomes44. This search yielded more than 10,000 viroid-like cccRNAs that represent an about five-fold increase of the known diversity of viroid-like agents. Further analysis of these cccRNAs led to the identification of numerous putative viroids, satRNAs, retrozymes and ribozy-like viruses.
Results
Computational approach for the discovery of viroid-like cccRNAs
We developed an integrated, scalable computational pipeline for the discovery and analysis of viroids and viroid-like cccRNAs directly from assembled transcriptomes and metatranscriptomes (Figure 1). The pipeline starts with the reference-free and de novo identification of cccRNAs or RCR intermediates, capitalizing on the fact that assemblies of both complete circular monomers and multimeric linear intermediates contain head-to-tail repeats45. The identified sequences are then cleaved in silico to unit length and deduplicated, taking circularity into account. Starting from the set of detected cccRNAs, the pipeline performs both alignment-free and alignment-based searches. The primary approach for the identification of viroid-like agents among the cccRNAs is the prediction of self-cleaving ribozymes using RNA sequence and secondary structure covariance models46. Assuming that the diversity of ribozymes in viroid-like RNAs could be greater than so far uncovered, we curated a database of known self-cleaving ribozyme models from Rfam47, of which only a minority were detected in viroids and viroid-like RNAs. We supplemented this model database with the pospiviroid RY motif48 to enable detection of potential pospiviroids, which lack ribozymes. The pipeline also performs direct sequence similarity searches against reference databases such as ViroidDB49.
Figure 1:
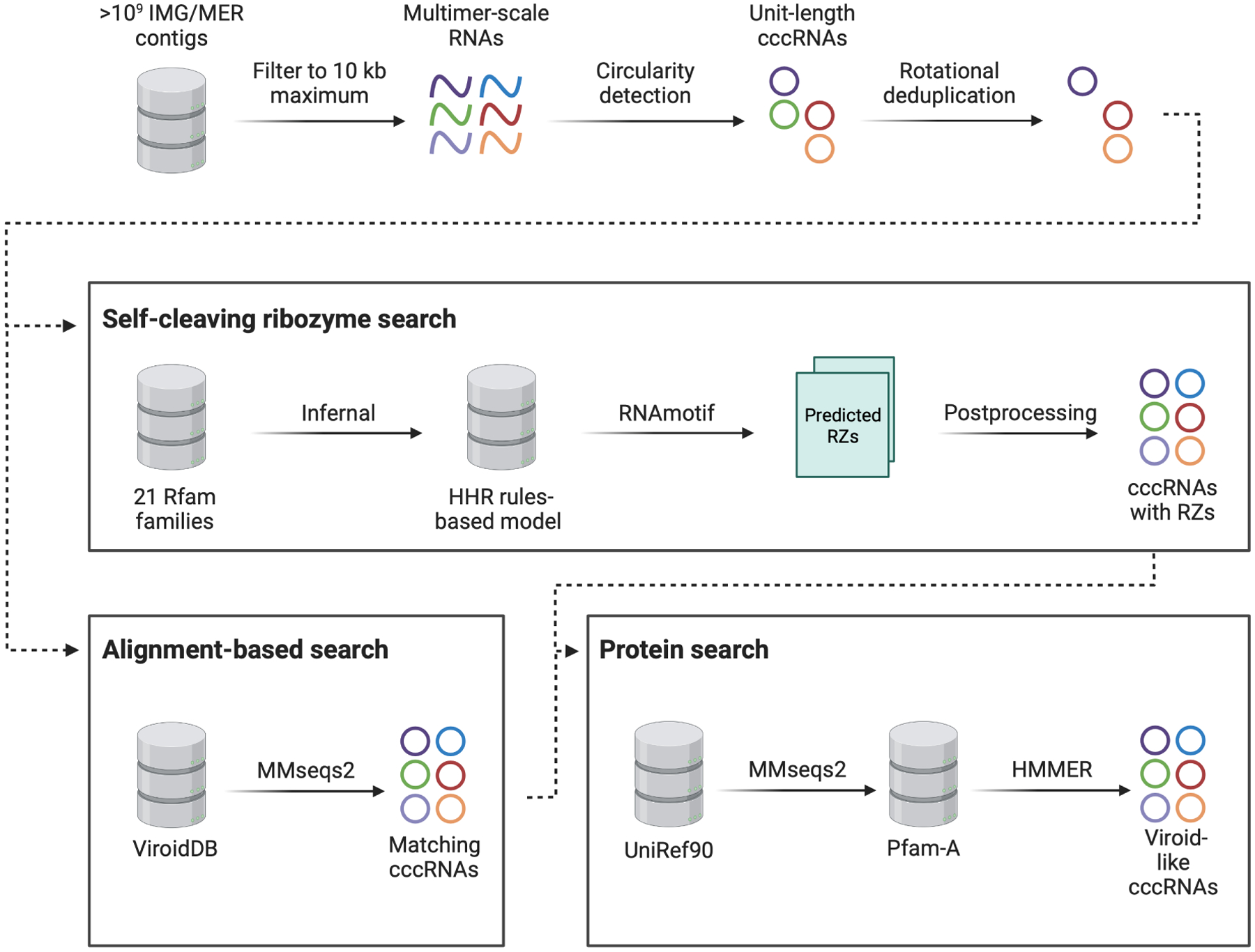
Viroid-like cccRNA detection pipeline.
Ribozyme-containing cccRNA sequences were classified as symmetric or asymmetric depending on whether they contained predicted ribozymes in both or only one RNA polarity, respectively, reflecting the RCR mode these cccRNAs are likely to undergo. However, it cannot be ruled out that some apparently asymmetric cccRNAs actually contain a second ribozyme distinct from the currently known ones.
We validated this method by demonstrating its ability to recover known viroid-like RNAs in transcriptomes and metatranscriptomes (Supplementary Table S1). For the transcriptomic validation, we processed and searched the 1,000 Plant transcriptome (1KP) data set44. We chose this data set due to the known presence of all type of viroid-like cccRNAs except ribozyviruses in plant transcriptomes. Assembling the raw reads of 1,344 transcriptomes resulted in 103,139,086 contigs, of which 163,970 were predicted to be circular. Of these putative cccRNAs, 42 were identified as viroid-like via ribozyme search (15 sequences), sequence search against ViroidDB (33 sequences), or both (6 sequences).
To verify the efficacy of the detection method, we performed a direct search of all contigs against ViroidDB and identified 12 contigs that matched a known viroid sequence with at least 50% target coverage. The detection pipeline found four of these potentially complete viroid contigs. Of the rejected contigs, four were much larger than typical viroids (>1000 nt) and contained major ambiguous regions. The other four were low-coverage fragments that could not be verified as circular being smaller than unit length. Iresine viroid 1, Citrus exocortis viroid, and a Coleus blumei viroid (CbVd) were successfully retrieved. While the former two were nearly identical to the corresponding reference sequences, the CbVd-like sequence was not. At 350 nt, this sequence differed in length from all known coleviroids and in the terminal conserved region, which was identical to that of Dahlia latent viroid, suggesting an origin of this viroid by recombination, as reported for other CbVd species50,51. At 85% identity, this CbVd-like sequence falls below the species membership threshold for coleviroids52. The relatively low number of viroids identified in plant transcriptomes is likely due to selection of healthy plants for RNA isolation and transcriptome analysis.
A five-fold expansion of the known diversity of viroid-like cccRNAs
After testing the pipeline on plant transcriptomes, we applied it to a set of 5,131 diverse metatranscriptomes totalling 1.5 billion metatranscriptomic contigs (708 Gbp) after size filtration. We identified 10,183,455 putative cccRNAs with a median contig length of 269 nt. After removing overlapping regions and eliminating rotationally identical sequences, the median length of the 8,748,001 resultant monomers was 165 nt. Of these, 2,791,251 were within the known size range of viroids (200–400 nt), including 11,378 we classified as viroid-like because they contained a confidently predicted self-cleaving ribozyme in at least one RNA polarity. No metatranscriptomic cccRNAs matched the pospiviroid RY motif. Among the viroid-like cccRNAs, 10,181 were detected by alignment-free methods, that is, showed no detectable sequence similarity to known viroids. The remaining 1,197 sequences shared significant nucleotide sequence similarity with known viroid-like RNAs spanning the entire gamut from viroids to satRNAs to retrozymes (Supplementary Table S2). 907 sequences were identified as viroid-like by both the ribozyme detection and alignment-based search approaches. Among the 10,181 ribozyme-containing viroid-like cccRNAs unrelated to known viroids, 3,434 were symmetric, that is, contained predicted ribozymes in both polarities. Of the 5,131 metatranscriptomes searched, 1,841 contained at least one viroid-like cccRNA.
Of the sequences aligning to viroid-like agents, the majority only contained short (<40 nt) alignable regions, generally localized to the ribozyme motifs. However, 33 sequences yielded long (>100 nt) alignments. These cccRNAs aligned to Tobacco ringspot virus satRNA (satTRSV), Lucerne transient streak virus satRNA (satLTSV), citrus dwarfing viroid (CDVd), and two retrozymes. For satTRSV and satLTSV, the range of identity between the recovered cccRNAs and the reference sequence ranged 80%–98% and 81%–99%, respectively. The match to CDVd was 80% identical to the nearest reference sequence. In all cases, the cccRNAs were similar in length and structure to the reference sequences.
We clustered the viroid-like cccRNAs identified here to estimate the increase in diversity compared to previously known viroid-like RNAs (Figure 2). Aligning cccRNAs poses a challenge due to the variation in the rotation of the sequences. Two identical cccRNAs could appear to have only half the bases aligning if rotated completely out of phase. Therefore, we took special care to compensate for the circularity of the sequences during the postprocessing of the pairwise nucleotide search results (see Methods). To validate our clustering method, we tested it on ViroidDB. Previously, we identified 458 clusters at the average nucleotide identity (ANI) 90% level in ViroidDB using a method that was not circular-aware49. Using the improved method, we identified 50 clusters in ViroidDB, generally corresponding to individual species.
Figure 2: Viroid-like cccRNAs identified in metatranscriptomes.
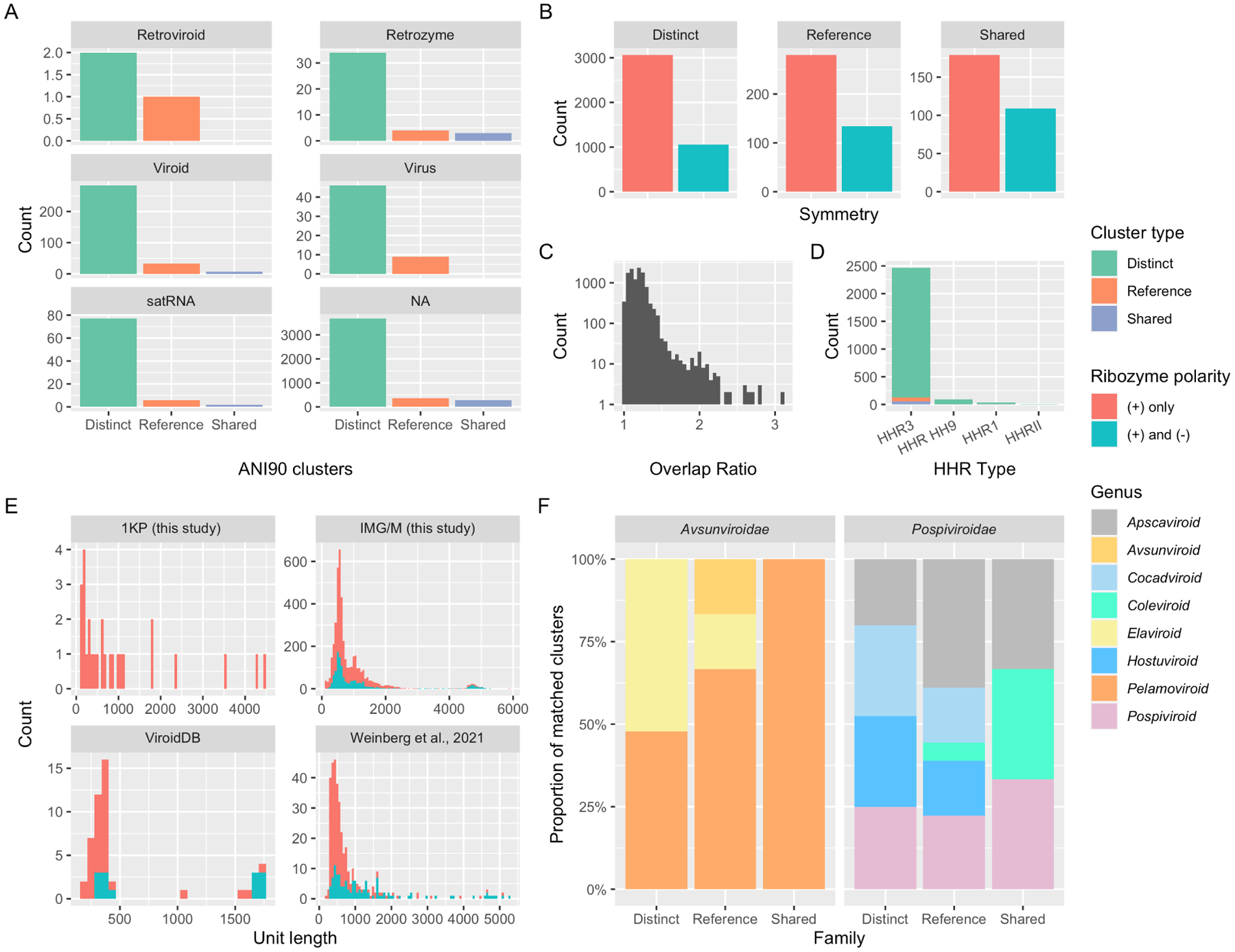
A. Number of ANI90 clusters with most significant matches to given viroid-like cccRNA agent types which are either “distinct” (derived exclusively from transcriptome and metatranscriptome analysis in this work), “reference” (no members distinct from previously identified ones), or “shared” (containing at least one of both types of sequence). B. Comparative distribution of inferred ribozyme architectures by cluster type. C. Plot of overlap ratios in cccRNAs, defined as the assembled length divided by the monomer length, from IMG and 1KP. D. Counts of HHR types in representative clusters. E. Length distributions of cluster representatives in the present analysis (transcriptomes and metatranscriptomes), ViroidDB, and a previous study53. F. Relative abundance of clusters matching different genera within each viroid family by cluster type
In the combined metatranscriptomic, transcriptomic, and reference datasets, we identified 4,823 ANI90 clusters of which 4,121 did not include any sequences from the reference datasets and thus were considered distinct, a 5.9-fold increase in viroid-like RNA diversity. Of the remaining 702 clusters containing at least one known sequence, 288 (41%) were expanded by at least one distinct sequence. Notably, 39 distinct clusters were represented in plant transcriptomes, of which 8 were symmetric.
The relative abundance of HHR types in the cccRNAs varied significantly from what would be expected given the sequence and species count. Within Rfam, HHR1 swamps HHR3 by two orders of magnitude by sequence count (190,679 vs 538 sequences). However, among the cccRNA cluster representatives, HHR3 was found in 94% and was be two orders of magnitude more common than HHR1 (1,952 vs 32). Given the dominance of HHR3 in known viroids8,18, this overabundance of HHR3 is suggestive of the presence of numerous viroid-like cccRNAs.
Putative distinct viroid-like cccRNAs
We briefly describe the 5 largest distinct ANI90 clusters (denoted 1 to 5, in the descending order of the cluster size) derived from the metatranscriptomic data to exemplify the type of findings obtained in this work. All these clusters included members with symmetric, matched ribozymes. The cccRNAs in four of these clusters contained matched HHR3s, whereas those in the fifth cluster contained twister-P1 ribozymes.
The largest, cluster 1, consisted of 149 sequences with a mean length of 562 nt (±9.0 nt). During circularity detection, an average of 18% of the monomer was removed, although one member of the cluster yielded a contig with a 60% (341 nt) overlap. The cccRNAs in this cluster are predicted to adopt a rod-shaped conformation with 74% of the bases (on average) paired in both polarities. Most members of this cluster (n=137) contain symmetric HHR3s, whereas for the remaining 12 members, only one ribozyme was predicted, suggesting the presence of a divergent HHR3. Among the members of this cluster, 38 sequences yielded a short (26–37 nt) alignment to the HHR3 of Eggplant latent viroid or Grapevine hammerhead viroid-like RNA. However, the cccRNAs comprising this cluster are substantially longer than the respective viroids (334 and 375 nt, respectively). The majority of the cluster members (n=133) were found in terrestrial metatranscriptomes from 11 distinct locations, 4 members were identified in 3 distinct freshwater locations, and one member was found in a spruce rhizosphere sample.
The cccRNAs in cluster 2 (68 members) contained twister-P1 ribozymes in both polarities. All but 4 of these cccRNAs are 494 nt in length and form a branched structure with a mean of 70% base complementarity. These cccRNAs were found in 10 locations in terrestrial ecosystems, such as soil and plant litter, with nearly half (n=31) identified in switchgrass phyllosphere samples. Cluster members were repeatedly found during sampling of the switchgrass phyllosphere over the course of a year at a sample site in Michigan, USA.
Like cluster 1, cluster 3 (61 members) consisted of comparatively large (605 nt), rod-shaped cccRNA with symmetric HHR3s. However, unlike the largest cluster, no member shows a detectable direct nucleotide match to any ViroidDB sequence, ribozyme or otherwise. In the majority (n=40) of the members symmetric ribozymes were not detected, but remaining ones were symmetric, again, suggesting the presence of divergent HHRs. Between 73% and 83% of the bases in these cccRNAs were paired in the (+) polarity and between 74% and 82% of bases were paired in the (−) polarity. On average, 28% of the monomer’s length was cleaved during circularity detection although one member was sequenced at 2.78 unit length (almost a complete head-to-tail trimer). The largest two members of the cluster (1,693 and 1,640 nt originally, 1,224 nt after cleavage) were not correctly monomerized by the circularity detection procedure due to mismatches in the seed sequence (see Methods). Manual monomerization of these sequences showed they both were 612 nt, resulting in overlap ratios of 2.76 and 2.67, respectively. Alignment results in 99.0% and 99.5% identity between monomers within each dimer, approximately the error rate of RNA polymerase II when using an RNA template54–56. Almost all members of this cluster were identified in soil samples from 6 locations around the world (including Colombia, Czech Republic, Germany, and USA), and two were found in creek freshwater samples.
Cluster 4 (n=61) is similar to clusters 1 and 3 in that its members consist of 615 nt and are predicted to form rods containing HHR3s in both polarities. The secondary structure results in a high mean self-complementarity of 75% of bases. However, these cccRNAs contain no alignable regions to the other large clusters or to any ViroidDB sequence. As with many other HHR3 rods, the majority (n=39) contain only one HHR3 above the significance threshold. All but two members of the cluster were derived from eight soil locations, and the remaining ones were found in the spruce rhizosphere.
Among the largest clusters we examined, cluster 5 (55 members) is the only one that consists of cccRNAs with predicted quasi-rod shaped structures, with 64% bases paired on average. At 454 nt mean length (±8.9 nt), these cccRNAs are the smallest among the 5 largest clusters. In this case, all members contained two HHR3s, but no nucleotide matches to ViroidDB were detected. Most members were found in terrestrial samples including soil (n=38) and plant litter and peat (n=2). As in the case of cluster 2, 14 members of this cluster were also found over the span of a year in the phyllosphere of switchgrass, and one member was found in a freshwater sample. Altogether, nine distinct locations contained members of this cluster.
In summary, analysis of these 5 largest clusters showed that they consisted of cccRNAs endowed with all the hallmarks of viroids including symmetric ribozymes (HHR3s, with one notable exception), extensive branched or (quasi) rod-shaped secondary structure, and evidence of multimeric intermediates. Furthermore, the members of these clusters were independently identified in diverse samples, primarily, those from soil, indicating they are widespread and are likely to be infectious agents.
Virus-like elements blurring lines between riboviruses, ribozyviruses and viroids
Among the cccRNAs containing symmetric HHR3s, we identified rod-shaped sequences up to 4,705 nt, far outside the length range of viroids and ribozyviruses. We hypothesized that these cccRNAs could be previously uncharacterized ribozy-like viruses. To perform a comprehensive search for potential ribozy-like viruses, all open reading frames (ORFs) longer than 75 codons present in cccRNAs were translated, and the resulting sequences of putative proteins were clustered by amino acid sequence similarity and compared to protein sequence databases (STAR Methods and Supplementary Table S3).
Almost all reliable matches were to virus proteins (Supplementary Table S3). One protein cluster showed significant sequence similarity to the predicted RdRPs of a distinct group of ssRNA viruses, ambiviruses, that were recently discovered in fungal isolates and transcriptomes57–59. Ambiviruses have RNA genomes of approximately 4 kb which encompass bidirectional ORFs, one of which encodes a predicted RdRP. To date, ambiviruses have not been reported to be circular58. In the IMG metatranscriptomes, 163 ANI90 clusters (274 cccRNAs total) were found to encode ambivirus-like RdRP (E-values between 1.3e-229 and 8.7e-04). Notably, these clusters of cccRNAs were also predicted to contain HHR3, HPR-meta1, and CEPB3 ribozymes, including symmetric sequences with different ribozymes in the two RNA polarities. All these sequences were predicted to adopt a rod-like structure in which the two ORFs encoding, respectively, the RdRP and an uncharacterized protein are arranged along the rod in the opposite strands (Figure 4). These sequences showed varying degrees of terminal overlap, with a median trimmed repeat length of 123 nt. Three representative sequences were recovered with >2000 nt overlaps, of which one was an almost-complete dimer, suggestive of RCR. Three of the ambi-like clusters were detected at very low levels in 10 plant transcriptomes.
Figure 4:
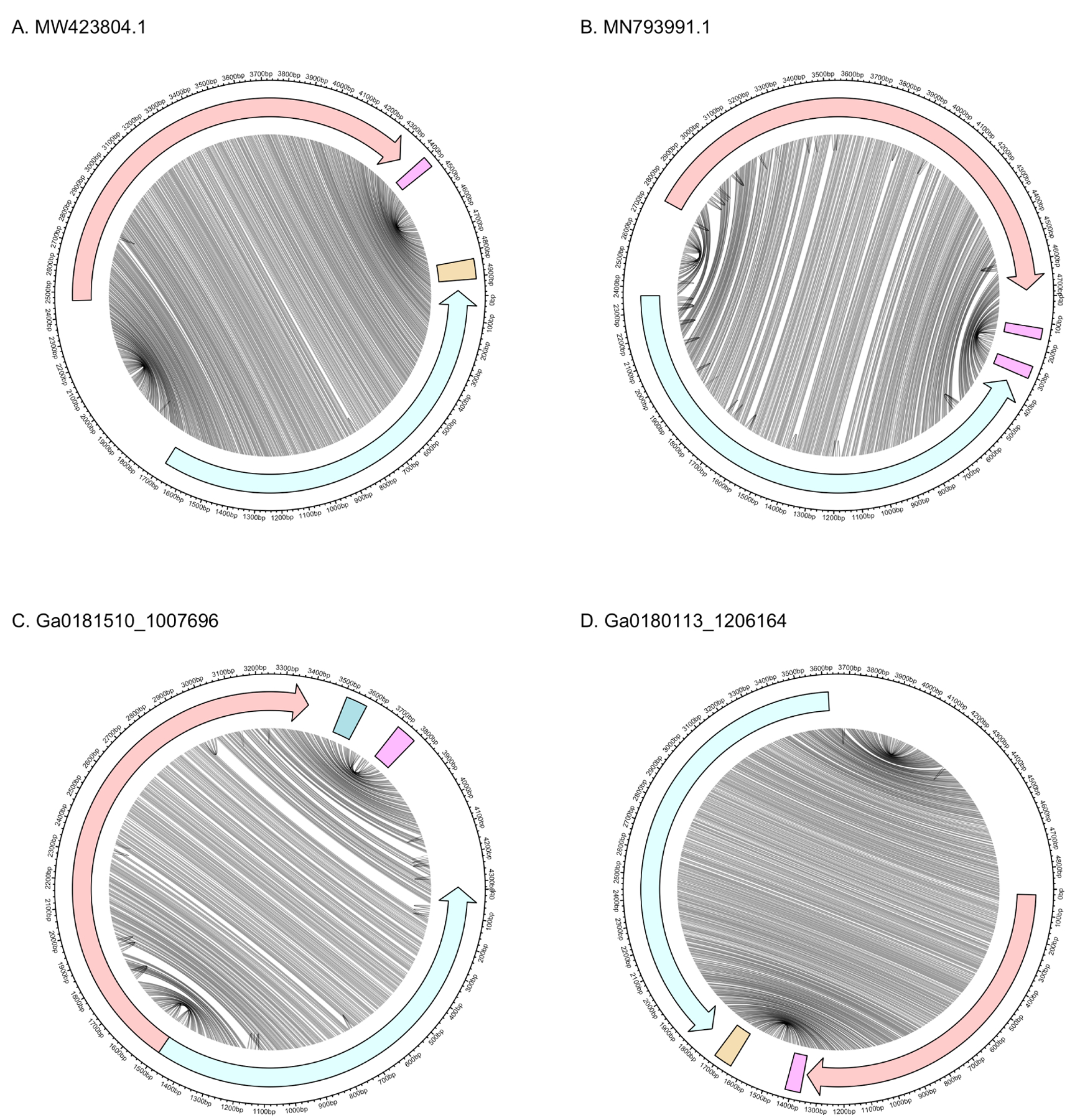
Genomic and secondary structure of Armillaria borealis ambi-like virus 1 (A), Tulasnella ambivirus 1 (B), and ambi-like sequences discovered here (C, D). Red and blue denote (+) and (−) polarities, respectively. Lines connect bases in the genome that are paired in the predicted secondary structure. Arrows represent ORFs and rectangles represent self-cleaving ribozymes. The (+) and (−) ribozymes are HHR3 and HPR-meta1 in (A), HHR3 and HHR3 in (B), CPEB3 and HHR3 in (C), and HHR3 and HPR-meta1 in (D). In all cases, the ribozymes are located outside the ORFs at the end of the rod.
We then ran the detection pipeline on the 30 ambivirus and ambivirus-like sequences from GenBank and found significant ribozyme matches in 15 of these sequences, of which 13 contained two predicted ribozymes. Of the remaining 15 sequences, 11 showed ribozyme matches in the expected locations that failed to pass the significance threshold (Supplementary Table S4). As in the IMG data, the HHR3 and HPR-meta1 ribozymes are present in both matched and mismatched combinations. Similarly, three of the published genomes (MT354566.2, MN793994.1, and MT354567.1) contain terminal overlaps of 160–250 nt, suggestive of circularity. Furthermore, all known ambivirus and ambivirus-like sequences were predicted to adopt a rod-shaped conformation. Taken together, these observations strongly suggest that ambiviruses comprise a distinct group of ribozy-like viruses that encode a RdRP homologous to the RdRPs of riboviruses.
Three cccRNA clusters with significant mitovirus RdRP matches were detected, including two with symmetric ribozymes. The symmetric singletons are 3,283 and 3,058 nt in size and contain matched twister-P1 ribozymes and an HHR3/twister-P1 combination, respectively. The HHR3 aligns to ELVd with 96% identity. A third cccRNA cluster with three members encoding putative mitovirus-like RdRP, of 3,363 nt, contains a similar match to ELVd (including the HHR conserved core) that was not identified as an HHR by either detection method. This cccRNA lacks the HHR core in the opposite polarity but shows weak similarity (E-value = 0.19) to twister-P1. All cccRRNAs were detected with >100 nt overlaps and are predicted to adopt a branched conformation with between 63% and 66% of bases paired in both polarities. These three genomes have a low (36–40%) GC content, a hallmark of mitoviruses60. Searching the predicted RdRPs against the protein sequence databases yielded the most significant matches for the three cccRNAs to Grapevine-associated mitovirus 13, Grapevine-associated mitovirus 14, and Fusarium asiaticum mitovirus 8. Upon closer inspection of Grapevine-associated mitoviruses 11 and 13, we found that they also contained HHR3s in both polarities and a twister-P1/HHR3 combination, respectively. Ribozyme searches of all available Lenarviricota (taxid 2732407) and unclassified Riboviria (taxid 2585030) sequences did not yield other matches besides the ambiviruses and these few mitoviruses.
Apart from the RdRps, we identified 135 sequences comprising 53 ANI90 clusters with significant similarity to capsid proteins of single-stranded (ss) DNA viruses, in particular, CRESS viruses61. The sequences in 50 of these clusters contained predicted ribozymes, and 13 contained HHR3s in both polarities. Two clusters contained paired HHR3 and twister-P1 ribozymes, whereas two other clusters contained symmetric HP-meta1 ribozymes. 26 clusters contained a single HHR3, three a twister-P1, and four a HPR-meta1. 21 clusters, including all three without ribozyme profile matches, produced a nucleotide alignment to a known viroid’s ribozyme, ranging in length from 25 to 50 nt at 83–96% identity. The cccRNAs in these clusters varied between 1,092 and 1,632 nt in length, with a mean of 1,317 nt and GC content with the mean of 44%. Four cccRNAs were sequenced as complete head-to-tail dimers. The secondary structures of these cccRNAs included, on average, 66% paired bases. Given the strong evidence of circularity, extensive self-complementarity resulting in predicted branched structure and confident prediction of ribozymes, these cccRNAs most likely represent a distinct group of ribozy-like satellite viruses.
Diverse deltavirus-like viruses
Apart from the viruses that resembled ribozyviruses conceptually, that is, were identified as protein-coding viroid-like cccRNA but encoded proteins unrelated to HDV antigen (HDVAg), we searched for actual relatives of the deltaviruses. To this end, clusters of ORFs from the identified cccRNAs were compared to the sequences of the HDVAg and its homologs from other ribozyviruses. A total of 12 ORF clusters were identified above the HDVAg Pfam profile’s gathering threshold; additional 21 representative ORFs were significant at the E-value < 1e-03 level, and 34 at E-value < 1e-02. Of these clusters, only one showed a significant nucleotide alignment to a known HDV-like virus. The other clusters were found in a variety of environments ranging from soil to wastewater to coastal wetland sediment. Samples with matching cccRNAs were collected from as far north as Alaska to as far south as Florida. All 69 members of the clusters encompassing ORFs with HDVAg profile matches (E-value < 1e-02) were predicted to adopt a rod shape in both polarities. The genome size of ribozyviruses in ViroidDB ranges from 1,547 to 1,735 nt. However, among the HDV-like clusters identified in metatranscriptomes, the size ranged from 1,019 nt to 1,757 nt, with a median length of 1,317 nt.
Clustering the HDV-like sequences in combination with the known ribozyviruses in ViroidDB produces no ANI80 clusters with both reference and distinct members. Each of the 26 HDV-like clusters falls below the species demarcation criterion for ribozyviruses (80% nucleotide identity)28. Clustering the ORFs from both the detected HDV-like sequences and reference ribozyviruses with 60% minimum identity (the genus demarcation criterion) using CD-HIT resulted in 36 clusters, of which 10 consisted entirely of reference sequences whereas 26 contained only sequences discovered here.
Clustering of the HDVAg sequences and their homologs from other animals and metatranscriptomes with a permissive threshold showed that all previously known HDVAg homologs formed a single tight cluster whereas the metatranscriptome sequences formed multiple smaller clusters and singletons distant from each other and from HDV (Figure 5A). The conservation profile of the multiple alignment of the HDVAg homologs showed that the dimerization region and one of the RNA-binding regions were prominently conserved whereas the second RNA-binding region was not (Figure 5B,C). The sequences of the distant HDVAg homologs from metatranscriptomes showed low sequence similarity to the previously known HDVAgs, far below the similarity among the latter, with the distributions of percent identities almost non-overlapping (Figure 5D). Finally, in the phylogenetic tree of the HDVAg homologs, all previously known sequences formed one compact clade, whereas the homologs from metatranscriptomes identified here comprised several remaining clades, with a much greater phylogenetic depth (Figure 5E).
Figure 5: Diversity of HDV antigen-like proteins in known ribozyviruses and ribozy-like viruses identified in metatranscriptomes.
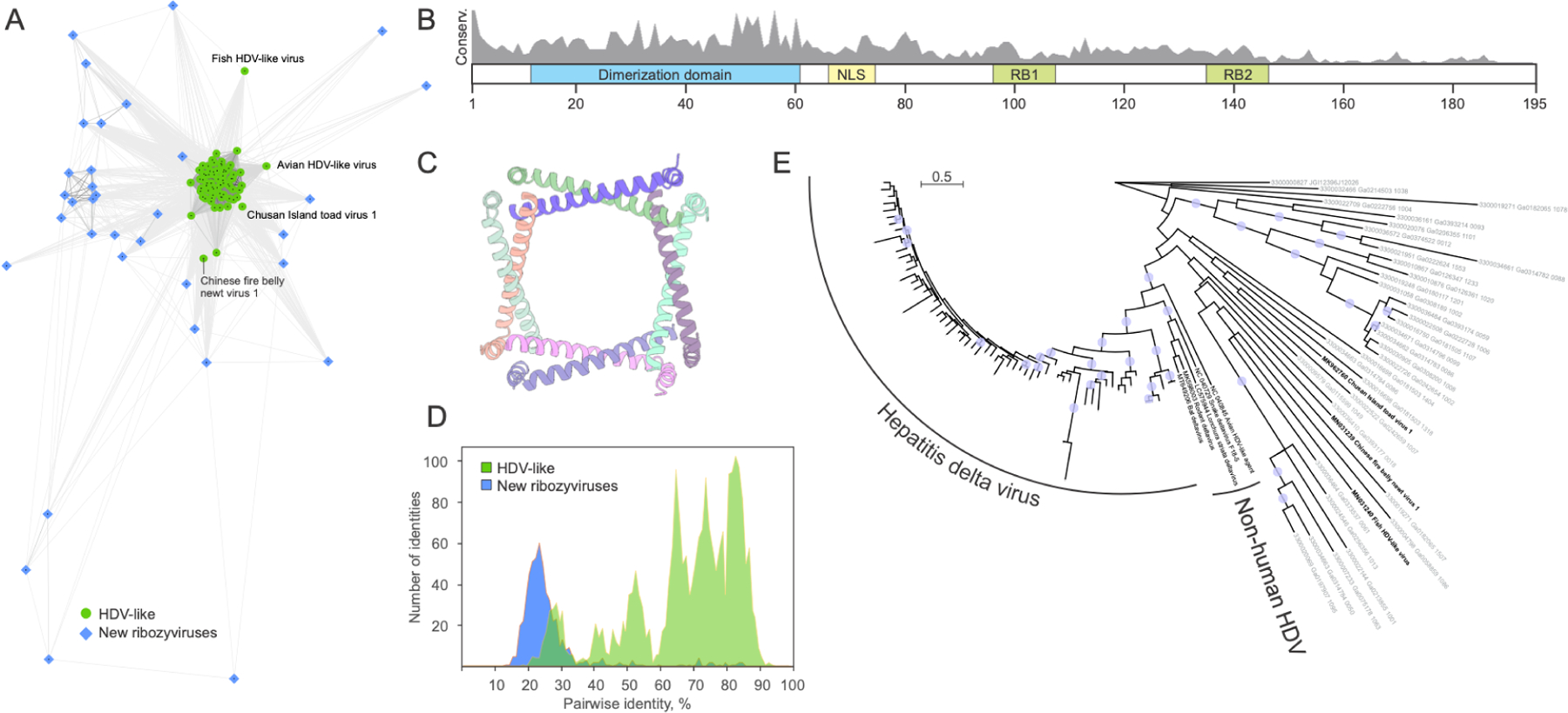
A. Clustering of the HDV antigen (Ag)-like protein homologs based on their sequence similarity. Lines connect nodes (sequences) with P-value < 1e-05. Reference HDVAg-like sequences from GenBank are shown as green circles, whereas those detected in metatranscriptomic datasets as blue diamonds. Some of the divergent reference sequences are labeled. B. Schematic representation of the HDVAg with functionally important regions indicated with colored boxes. RB1 and RB2, RNA-binding sites 1 and 2, respectively; NLS, nuclear localization signal. Gray histogram shows the sequence conservation (percent identity) of HDVAg-like sequences from metatranscriptomic datasets. C. Octameric structure of the conserved dimerization domain of HDVAg. PDB ID: 1A9262. Each protein molecule is shown with a different color. D. Comparison of the sequence conservation among reference HDVAg from GenBank (green) and those from metatranscriptomic datasets (blue). E. Maximum likelihood phylogeny of HDVAg-like sequences. The tree was constructed with IQ-TREE63. Circles at the nodes represent SH-aLRT support higher than 90%. The scale bar represents the number of substitutions per site.
The nucleotide sequences of these HDV-like cccRNAs formed 26 ANI90 clusters, none of which contained confidently predicted self-cleaving ribozymes. However, 13 of these clusters produced weak ribozyme matches (E-value < 1e-01), and 8 of these were symmetric. Both HHR-like (n=36) and HDV-like (n=26) ribozymes were detected although no clusters contained ribozymes of both types. Of the HDV-like ribozymes detected, only five most closely matched the canonical HDV ribozyme. Ten putative ribozymes showed the strongest similarity to the HDV ribozyme (HDVR) found in the genome of Faecalibacterium prausnitzii64, seven were most similar to the HDV-like ribozyme found in the genome of Anopheles gambiae64, and four were most similar to the mammalian CPEB3 ribozyme65,66. The causes of these similarities remain to be investigated; given the small size of the ribozyme, convergence cannot be ruled out.
The limited number of significant ribozyme matches among the HDV-like sequences posed an opportunity for detecting distinct ribozymes or diverged variants of known ones. For example, we examined an HDV-like cccRNA cluster (representative member 3300009579_Ga0115599_1049451) with no predicted ribozymes. However, upon closer examination, sequences from this cluster were shown to contain the conserved HHR core in both polarities in the expected locations, a recently discovered ribozyme configuration67. Some clusters entirely lacked the HHR core in either polarity, suggesting the use of alternative, yet unknown ribozymes.
Diverse ribozymes and ribozyme combinations
Almost all viroid-like RNAs described to date contain the same type of ribozyme in both polarities, with the exception of some satRNAs. Surprisingly, many viroid-like cccRNAs identified here were predicted to contain ribozyme combinations that have not been so far reported in replicating cccRNAs (Figure 6).
Figure 6: Ribozyme diversity in viroid-like cccRNAs.
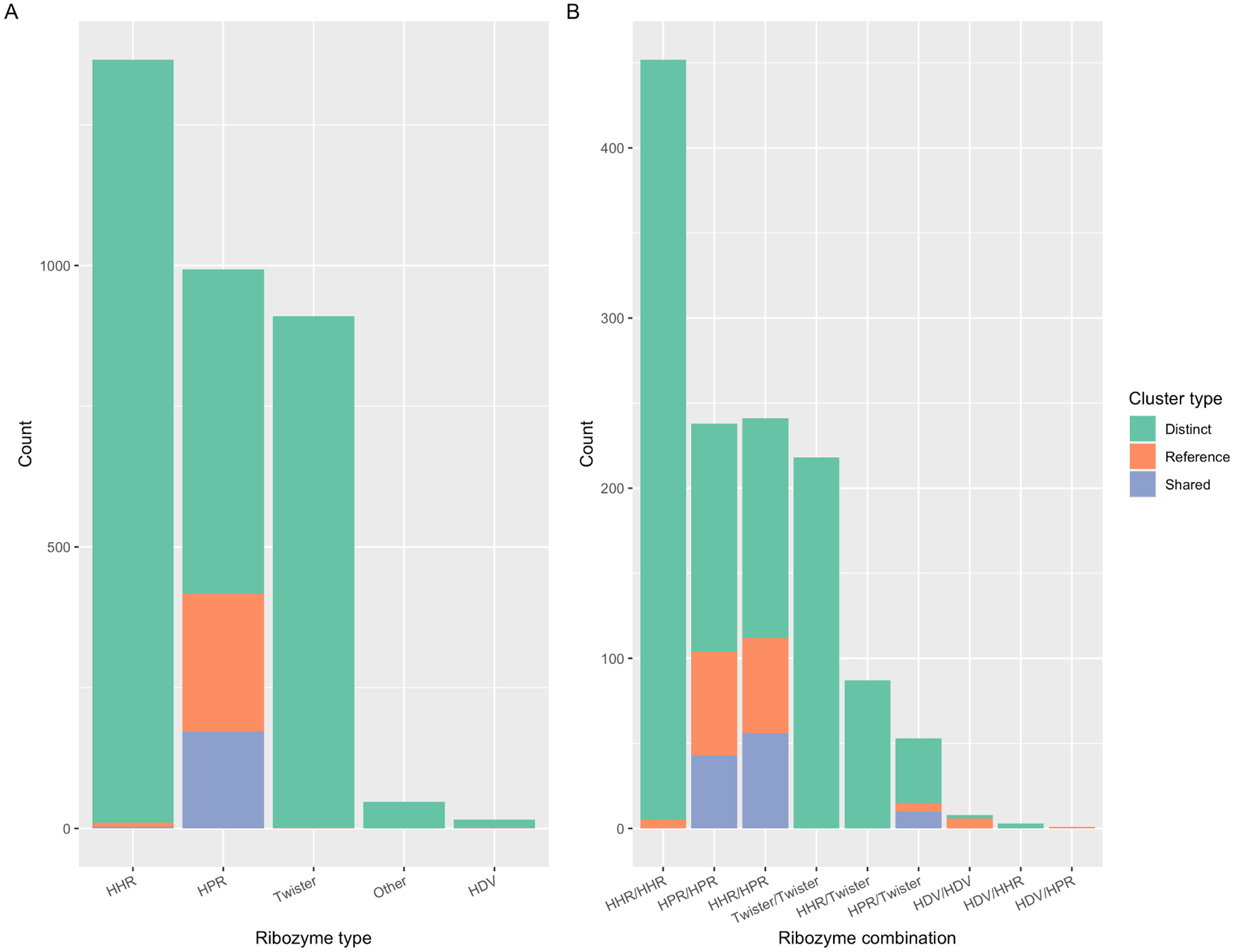
A. Distribution of ribozyme types in asymmetric clusters. B. Ribozyme co-occurrence within the symmetric viroid-like cccRNA cluster representatives derived from metatranscriptomes.
Specifically, we identified numerous cccRNAs containing twister ribozymes, a recently described ribozyme motif that so far has only been found in combination with the HP-meta1 ribozyme. Both symmetric (n=381) and asymmetric (n=930) variants are present in the metatranscriptomic cccRNA clusters. Most symmetric twister clusters contained matched twister ribozymes (218 clusters) in both polarities, an unexpected ribozyme combination. In 87 clusters including mitovirus-like and satellite-like cccRNAs, we found another distinct combination of ribozymes, with HHRs opposite twister ribozymes. The unusual twister ribozyme is widespread in plant transcriptomes, with 59% of the transcriptomes containing reads mapping to a twister-bearing cccRNA. Indeed, we recovered three asymmetric cccRNA clusters from plants that contained a twister-P1 ribozyme.
In addition to the twister combinations, we identified several other unusual ribozyme combinations in symmetric cccRNAs. Previously, HHR3s have been found in conjunction with HDVAg67, but have not been reported to be paired with HDV ribozymes. We identified three clusters in which HHR3s were paired with HDV-type ribozymes, namely, CPEB3 and HDVR F. prausnitzii. The CPEB3-HHR3 combination was found in an ambivirus-like sequence, and the two HDVR F. prausnitzii/-HHR3 clusters were both predicted to adopt rod-shaped structures. One of these, a 1,052 nt singleton, did not match any sequences in ViroidDB, nt, or UniRef90, but in the other cluster (978 nt, two members), the HHR was closely similar to that of Cryphonectria parasitica ambivirus 1 (44/50 nt identical), whereas the HDVR F. prausnitzii motif (32/33 nt and 41/46 nt) aligned to two chromosomes of the Vanessa atalanta butterfly.
Among the asymmetric cccRNAs, we identified two additional types of self-cleaving ribozymes. The hatchet ribozyme was found in 34 ANI90 clusters that ranged from 357 nt to 567 nt in length and came primarily from aquatic metatranscriptomes, in contrast to the general trend among the cccRNAs that derived from soil metatranscriptomes (Figure 7B). For example, the most diverse of these clusters (440 nt) contained 22 members from aquatic (almost all freshwater) metatranscriptomes sampled from around the United States. For the hatchet clusters, the Rfam profile matches were the strongest among all detected ribozymes (median E-value 1.4e-08) and, unusually for viroid-like cccRNAs, the GC content was low (median 35%). The predicted structures of these sequences varied from branched to quasi-rod shaped, with a mean of 62% of the bases paired.
Figure 7: Global distribution and habitats of viroid-like cccRNAs found in metatranscriptomes.
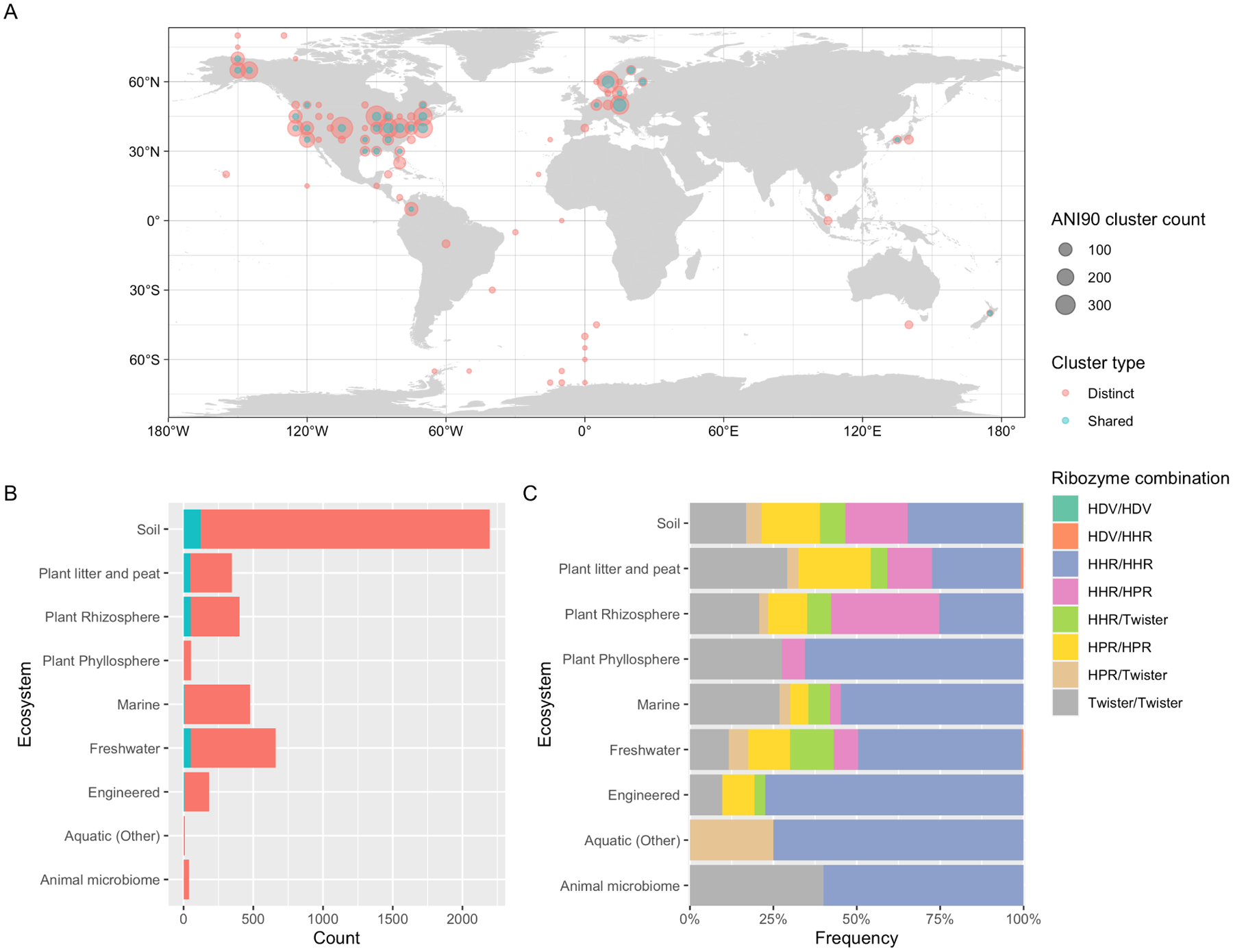
A. Map of sample locations from which viroid-like cccRNAs were detected. The size of each circle corresponds to the number of clusters identified in each location (grouped to the nearest five degrees of latitude and longitude) while the color represents the fraction of distinct clusters (blue shows reference clusters and red, distinct clusters). B. Reference (blue) and distinct clusters of viroid-like cccRNAs in different types of ecosystems. C. Relative frequencies of ribozyme combinations within symmetric cccRNA clusters in each ecosystem type.
The pistol ribozyme was identified in asymmetric cccRNAs. Like the clusters containing the hatchet ribozyme, clusters with the pistol ribozyme were found primarily (9/13) in marine metatranscriptomes ranging from the Antarctic Ocean to the Baltic Sea. The clusters have a slightly lower median profile match E-value (E-value = 6.4e-05) compared to the hatchet ribozyme but, unlike the hatchet ribozyme, have GC content ranging from 33% to 59% (median 49%) more characteristic of viroid-like RNAs. The predicted secondary structures of the pistol-containing cccRNAs were branched, often with several long hairpin structures.
CRISPR spacers matching cccRNAs
CRISPR spacer matches provide one of the most reliable means for assigning hosts to viruses and other mobile genetic elements in prokaryotes, and for differentiating prokaryote-infecting from eukaryote-infecting viruses68,69. Our recent search for riboviruses in the same set of metatranscriptomes that is analyzed here identified multiple spacers matching RNA viruses, resulting in the assignment of several groups of viruses to bacterial hosts including several previously thought to infect eukaryotes43. To identify viroid-like agents that potentially might replicate in prokaryotes, we searched the viroid-like cccRNA sequences identified here against the IMG CRISPR spacer database70, and detected 89 spacers with significant matches to viroid-like cccRNAs from 9 clusters (Supplementary Table S5).
One spacer was an identical match of 37 nt to a member of a cluster of 16 cccRNAs with prominent viroid-like features. The cccRNAs of this cluster are 315 nt long, contain symmetric HHR3s, and are predicted to adopt a rod shape, with 73% of the bases paired. Unusual for viroids, the cccRNAs comprising this cluster were found in Mushroom Spring, a hot spring at Yellowstone National Park. The matching spacer was also detected in a 60° C hot spring, Great Boiling Spring, albeit more than 800 km away71. The repeats in this CRISPR locus were identical to those in the type III CRISPR locus of Roseiflexus sp. RS-1, an anoxygenic filamentous bacterium of the phylum Chloroflexota that was itself identified in Yellowstone hot springs72,73. Searching for spacers matching all 16 cluster members with more relaxed criteria (i.e., more than 1 mismatch but with E-value < 1e-05), we identified a further 13 nearly identical matching spacers from 8 Yellowstone hot springs samples collected between 2007 and 2017. One spacer (35/37 identities, E-value = 3e-06) included a precise match to the HHR core motif. The repeats from this expanded set of loci all matched those from Roseiflexus sp. RS-1. Previously, we identified multiple spacers in Roseiflexus sp. RS-1 that matched a group of partiti-like riboviruses that were accordingly assigned to this bacterial host43. Apparently, the type III CRISPR system of Roseiflexus sp. that encompasses a reverse transcriptase actively incorporates spacers from multiple RNA replicons.
The cluster with the most spacer matches—57 matches spanning 26 metagenomes, largely from sludge and bioreactor samples—includes a single cccRNA of 606 nt (recovered as 841 nt) with an asymmetric twister-P5 ribozyme and a predicted branched conformation with 63% of bases paired. Eight spacers, detected in 7 metagenomes, covered the predicted ribozyme region.
The second most frequently matched cluster, also a singleton, was associated with 13 spacers from 10 metagenomes, all from the same location in the Southern Indian Ocean. This cccRNA is 286 nt long (recovered with a 123 nt overlap) and contains a predicted HHRII in one polarity only. Like the most matched singletons, this sequence also is predicted to adopt a branched conformation, with 57% of bases paired. The spacers collectively covered 33% of the sequence but do not include the HHR region.
Geographic and ecological distribution of viroid-like cccRNAs
We examined the global distribution of the cccRNA clusters. Distinct clusters were found throughout the world (Figure 7A) and in all types of ecosystems (Figure 7B). Soil samples were the primary source of both distinct and shared clusters, reflecting both the greater number of such samples (twice as many as the next most common sample type) and the apparent greater sequence diversity in soils.
The viroid-like cccRNAs displayed non-uniform ribozyme distribution among ecosystems (Figure 7B). Mismatched HPR/HHR ribozymes were especially prevalent among samples from plant rhizospheres, whereas matched HHRs were notably more abundant in engineered ecosystems, such as bioreactors, than in soil environments.
Among the 10 most geographically dispersed, distinct clusters, 8 included symmetric ribozymes, of which 6 were matched HHR3s. The other two symmetric clusters contain HPR-meta1/twister-P1 ribozymes and the two asymmetric clusters contain HHR3s. These widely dispersed clusters ranged in length from 372 nt to 1,039 nt and were predicted to adopt either a rod-like shape (the 6 HHR3 clusters) or branched conformations.
We examined the coverage depth of the viroid-like cccRNAs relative to the other contigs within the same metatranscriptomes. The absolute coverage depth is not directly informative due to the variation between sequencing methods used, but the ranked coverage depth percentile can serve as a proxy for the abundance of a sequence within the sample. In the 747 samples for which coverage information was available, viroid-like cccRNAs ranged from the most covered to least covered contigs. Notably, 150 viroid-like cccRNAs were within the top percentile (Supplementary Table S7). Among these highly covered contigs are known viroid-like cccRNAs, such as satLTSV.
Identifying potential hosts of the viroid-like agents within these ecosystems remains a challenge. Based on the IMG annotation pipeline, the majority of the analyzed metatranscriptomes were dominated by prokaryotic sequences43, but still contained at least 1% of contigs affiliated to eukaryotes (Supplementary Table S6). Nonetheless, 187 metatranscriptomes in which viroid-like cccRNAs were detected contained < 0.1% of eukaryotic contigs, suggesting that these elements replicate in either rare and undetected eukaryotes or in some of the much more abundant prokaryotic hosts. Notable among these datasets were the hot spring metatranscriptomes in which CRISPR spacers targeting viroid-like cccRNAs were identified (see above). The apparent lack of eukaryotic RNA in these samples strengthen the hypothesis of prokaryotic hosts. Additionally, we found 104 symmetric clusters in marine samples which are far beyond the habitation range of the known hosts of viroids and satRNAs. These findings combined with the clusters from prokaryote-dominated samples suggests that the ecological and host ranges of viroid-like agents are far broader than currently appreciated.
Discussion
Viroids and viroid-like cccRNAs, such as satRNAs and ribozyviruses, are the smallest, simplest known replicators that hijack either a host DNA-dependent RNA polymerase or a virus RdRP for their replication. Given the universality of the cellular transcription machinery across all life forms and the enormous diversity and near ubiquity of RdRP-encoding riboviruses, the narrow diversity and host range of the known viroid-like agents appeared puzzling. We suspected that viroid-like agents actually could be far more common than presently known and, with this motivation, searched a collection of more than 5,000 metatranscriptomes for viroid-like cccRNAs.
We identified millions of putative cccRNAs by searching for signatures of circularity or RCR, namely, the presence of head-to-tail repeats in assembled contigs. Because reads spanning the origin cannot be reconciled with a linear sequence, the assembler produces contigs with the same subsequence repeated at both the end and the beginning45. Alternatively, when linear replication intermediates containing head-to-tail repeats are sequenced, the ends of the sequences are also repeated. After compensating for the low-fidelity of RNA polymerase II by allowing for up to 5% mismatches in the repeated regions, testing this method on assembled plant transcriptomes demonstrated that known viroids were reliably recovered in the absence of major assembly errors. However, the extensive secondary structure of many viroid-like cccRNAs and the use of poly-A enrichment during RNA isolation prior to sequencing74 makes it likely that many viroid-like cccRNAs either were not sequenced at all or were grossly misassembled and thus could not be recognized as circular. Even under a conservative approach, where only predicted cccRNAs containing confidently identified ribozymes counted as “viroid-like”, this search resulted in an approximately five-fold increase in the diversity of viroid-like agents. This is most likely a substantial underestimate of the true span of the viroid-like domain of the replicator space because among the millions of the predicted cccRNAs, in which no ribozymes were confidently identified, some, and possibly, many could be viroid-like agents containing unknown ribozymes or lacking ribozymes altogether like pospiviroids. Although perhaps only a coincidence, it is worth noting that a recent analysis of the same collection of metatranscriptomes also yielded an approximately five-fold increase in the diversity of riboviruses43. It is further notable that a substantial number of viroid-like cccRNAs are among the most abundant sequences in the respective metatranscriptomes, emphasizing the prominence of these agents in diverse ecosystems.
The majority of the detected viroid-like cccRNAs possessed characteristic features of viroids including the presence of HHR, often in both polarities, and predicted rod-like or extensive branched conformation. However, the search resulted not only in quantitative expansion of viroid diversity but also in qualitatively distinct findings, in particular, uexpected ribozyme combinations, such as those including twister, and ribozymes not previously found in viroid-like RNAs, such as hatchet and pistol. There is little doubt that additional ribozymes and ribozyme combinations in viroid-like cccRNAs remain to be discovered.
Another key finding is the discovery of diverse groups of ribozy-like viruses. Even if perhaps not unexpected, it is notable that the diversity of the ribozyvirus sequences discovered in metatranscriptomes far exceeds that of the previously known HDV relatives including the recently discovered non-mammalian ones. Moreover, many of the identified ribozyviruses lack the HDV ribozyme or even any known ribozymes, suggesting distinct replication mechanisms. The host range of the discovered ribozyviruses remains to be explored but, probably, includes non-animal hosts (see discussion below).
In contrast, the demonstration that ambiviruses are actually viroid-like agents and the discovery of viroid-like mitoviruses and satellite viruses was surprising. These three groups of viroid-like agents resemble ribozyviruses in that these are relatively large, protein-coding viroid-like cccRNAs. However, unlike HDV and its relatives, these viroid-like agents are clearly linked to riboviruses through the RdRPs encoded by ambiviruses and mitoviruses, and capsid proteins encoded by satellite viruses. These findings show that combinations of viroid-like cccRNA and protein-coding genes emerged on multiple occasions during evolution. These ribozy-like viruses unrelated to deltaviruses likely evolved through recombination between typical riboviruses and viroids. The implications for virus taxonomy, in particular, whether such viruses should be classified into the existing realm Ribozyviria or into the respective divisions of the realm Riboviria75, or perhaps, into a separate realm, remain to be sorted out.
One of the most interesting but also most challenging problems is the host range of the expanded diversity of viroid-like agents. There is currently no direct computational approach for connecting viroid-like RNAs with specific hosts. Nevertheless, it appears exceedingly unlikely that all or even the majority of the viroid-like cccRNAs discovered in metatranscriptomes are parasites of plants. Indeed, we identified orders of magnitude more viroid-like cccRNAs in metatranscriptomes than in plant transcriptomes, and most of the analyzed metatranscriptomes are dominated by bacteria followed by unicellular eukaryotes. Furthermore, ambiviruses were isolated from fungi57–59, and the demonstration that these are viroid-like agents expands the host range of the latter. A potential prokaryotic connection of viroid-like cccRNAs through CRISPR spacer matches is particularly notable. The detected spacer matches were not numerous but reliable, in particular, because multiple spacers matching the same cccRNA were identified in diverse metagenomes. At least, the typical viroid-like cccRNAs that matched spacers from the reverse-transcribing type III CRISPR system of Roseiflexus sp. appear to be strong candidates for distinct bacterial parasites. These viroid-like cccRNAs that likely replicate in bacteria merit further, dedicated metatranscriptome and metagenome searches as well as experimental investigation. These findings echo the recent expansion of the bacterial RNA virome through the search of the same metatranscriptome collection and suggest that bacteria might support a much greater diversity of RNA replicators than previously suspected43.
Limitations of the Study
This work targets low hanging fruits in the search for viroid-like agents, being limited to the cccRNAs that contain reliably identifiable, known ribozymes or align directly to known viroid-like agents. This conservative approach was adopted purposefully, in order to avoid potential artifacts resulting from erroneous identification of cccRNA, contamination with DNA-encoded sequences or other sources. A potential opportunity for the discovery of a far greater diversity of viroid-like agents and a challenge for further research is a comprehensive analysis of the massive set of predicted cccRNAs that lack known ribozymes. Computational methods for de novo prediction of ribozymes need to be developed to advance such analyses. Furthermore, there are numerous additional metatranscriptomes, in particular, those recently analyzed by the Serratus41 and Tara42 teams, as well as numerous animal metatranscriptomes, that should be searched for viroid-like cccRNAs. To make a comprehensive search practicable, more efficient algorithms for circularity detection are required. A technical limitation is that some of the software used to assemble metatranscriptomes (Supplementary table S5) would cut circular molecules to unit length excluding terminal repeats. Although the method we used for cccRNA detection took into account even short repeats, this feature of some assemblers could yield false negatives. Evidently, the computational approaches applied in this work only identify candidates for viroid-like agents. Experimental validation is needed and is especially important in the case of putative cccRNAs lacking known ribozymes.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and data should be directed to and will be fulfilled by the lead contact, Benjamin Lee (benjamin.lee@chch.ox.ac.uk).
Materials Availability
This study did not generate unique reagents. All data used as inputs for the pipeline are listed in the “data acquisition” section. All output of analyses are listed in the “data and code availability” section below.
Data and code availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. Data generated during downstream analysis have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources.
All the code and data generated in this work are freely available through public portals listed below.
Method Details
Data acquisition
The search for cccRNAs was performed on a collection of 5,131 assembled metatranscriptomes sourced from the IMG/MER database. In addition to the IMG metatranscriptomes, we searched the complete set of transcriptomes of the 1,000 Plants (1KP) project, totalling 1,344 paired-end sequencing runs. Before applying the search pipeline to 1KP, we filtered the raw reads for quality using fastp76 and assembled them using rnaSPAdes77 using default parameters. We also included the 2021-09-07 release of ViroidDB and the set of HPR sequences identified by53.
Circularity detection
We identified cccRNAs using a modified and improved version of the reference-free and de novo Cirit algorithm45,78 implemented in the Nim programming language. This method relies upon assembly errors for circular sequences resulting in terminal repeats. Detecting cccRNAs via this method requires searching forward for the last several bases of the sequence (the seed) and, if a match is found, comparing backwards to the start of the sequence. If the start and end of the sequence “overlap”, this repetitive region is then trimmed off. However, the existing implementation was unable to monomerize multimeric transcripts resulting from rolling circle replication among known viroid-like agents due to its single pass design and requirement of exact sequence identity within repetitive regions. Our reimplementation solves these problems by reiteratively attempting to monomerize putative cccRNAs while allowing for a configurable minimum identity within repeats. For this study, we required a minimum of 95% identity with no insertions or deletions within the overlapping region. In addition, the ratio of the length of the contig and computed monomers is reported. Sequences with monomer lengths below a threshold of 100 nt were excluded.
cccRNA deduplication
Standard approaches to sequence deduplication are insufficient for cccRNAs. Most modern approaches rely on hashing for memory efficiency. Such approaches are effective for linear sequences but circular sequences pose a challenge due to the arbitrariness of their start position. To enable deduplication of putative cccRNAs, we define a sequence’s canonical representation as the alphabetically earlier of the lexicographically minimal rotations of the sequence and its reverse complement. This approach, drawn from k-mer counting methods79,80 and, if further optimized, would be able to be computed in linear time and with constant memory for even greater scalability81.
Ribozyme-based filtering
To identify sequences likely to replicate via ribozyme-catalyzed rolling circle replication, we searched the cccRNAs for the presence of known self-cleaving ribozymes using Infernal46. In each polarity, we identified ribozymes above Rfam’s curated gathering cutoff or with E-values < 0.1. Sequences with ribozymes in both polarities that met these criteria were considered viroid-like. Alternatively, we considered sequences as viroid-like with one ribozyme with an E-value < 0.01 or a score above the gathering cutoff. For each polarity, we considered only the most significant (by E-value) ribozyme. To identify more divergent ribozymes that were not detected using Infernal, we searched sequences containing one significant (E-value < 0.01) using RNAmotif82.
Sequence search
We searched all cccRNAs against ViroidDB using MMseqs2 easy-search (version 13.45111)83 with the highest available sensitivity (-s 7.5). For each sequence, we considered only the most significant match as determined by bit score. In addition to searching ViroidDB, we also searched the cccRNAs identified by metatranscriptome mining against the set of cccRNAs recovered from plant transcriptomes using the same method.
RNA secondary structure prediction
We predicted the secondary structures of all viroid-like cccRNAs for both polarities using the ViennaRNA package84. For each predicted structure, we computed the percentage of bases paired and the number of hairpins present. We used a temperature of 25° C and the circular prediction mode.
Clustering
We performed several types of clustering including both alignment-based and alignment-free methods. To produce the alignment-based clustering, we performed an all-versus-all search using MMseqs2 on the sequences of ViroidDB, the HPR dataset of53, and this study. For this method, each sequence was concatenated to itself to compensate for potential variation in the sequence relative to otherwise-similar sequences due to their circular nature. Next, we executed MMseqs2 (v13.45111, easy-search -s 7.5 --min-seq-id 0.40 --search-type 3 -e 0.001 -k 5 --max-seqs 1000000) and computed the pairwise average nucleotide identity (ANI) between sequences by taking the alignment identity of the best hit for each pair. We computed the ANI for two self-concatenated sequences by taking the length of the smaller sequence and dividing by two. We then cap the computed alignment length the length of the now-monomerized smaller sequence. The ANI is then defined as the percent identity within the aligned region times the alignment length divided by the smaller sequence monomer length. Similarly, we defined the alignment fraction as the smaller of the doubled query coverage, doubled target coverage, or one.
To cluster the viroids based on their pairwise ANI, we build a graph by connecting pairs of sequences where the alignment covers at least 25% of the shorter sequence with 40% identity within the alignment. We then weighted the connections between the sequences by the ANI and employed the Leiden algorithm (as implemented in the igraph Python library, version 0.9.10) to delineate communities of similar sequences85. The clustering granularity was optimized by iterating over the resolution parameter space until the difference between average intra-cluster ANI and the target ANI began to increase.
ORF prediction
To find ORFs present within the sequences, we used orfipy86 configured to operate on circular sequences. Specifically, we searched sequences concatenated to themselves to ensure ORFs spanning the origin were detected. Only ORFs longer than 100 amino acids and using the standard genetic code were considered for each cccRNA.
Protein searches
We searched all viroid-like cccRNAs for matches to known proteins. The primary search method we used was by performing translated searches (BLASTX-style) against the UniRef90 protein database87 using MMseqs283. For each cccRNA, we considered only the best match by E-value.
As a second approach, we also searched the ORFs from all cccRNAs, viroid-like or not, using HMMER88. We searched both the full Pfam-A profile database using hmmscan as well as a curated subset (the profiles for RdRP clan combined with the HDVAg profile) using hmmsearch.
HDV antigen analysis
Sequences were clustered using CLANS with BLASTP option (BLOSUM62 matrix, E-value cutoff of 1e-03)89. Sequence similarity among reference HDVAg from GenBank and those from metatranscriptomic datasets was analyzed with the Sequence Demarcation Tool90. For phylogenetic analysis, HDVAg-like sequences were aligned using PROMALS3D91. Due to the short length of the sequences, the alignment was not further processed. Maximum likelihood phylogenetic analysis was performed using IQ-TREE63. The best fitting model was selected by IQ-TREE and was VT+F+R4. The tree was visualized with iTOL92.
Read mapping
We used bowtie2 to perform read mapping from the 1KP transcriptomes to the entire viroid-like cccRNA data set in parallel. We configured bowtie to use its most sensitive setting (--very-sensitive) and ignore unaligned reads.
CRISPR spacer analysis
Viroid-like sequences were compared to predicted CRISPR spacers from prokaryotic (meta)genomes to identify potential cases of spacer acquisition from, and possible defense against, viroids by prokaryotes. The full set of 22,109 viroid and viroid-like sequences, including all reference sequences and sequences identified in this work, was compared to 1,961,109 CRISPR spacers predicted from whole genomes of bacteria and archaea (vJune2022) and 61,658,467 CRISPR spacers predicted from metagenomes in the IMG database70 using BLASTN v2.9.0 with options -dust no -word_size 7. To minimize the number of false-positive hits due to low-complexity and/or repeat sequences, CRISPR spacers were excluded from this analysis if (i) they were encoded in a predicted CRISPR array including 2 spacers or less, (ii) less than 66% of the predicted repeats were 100% identical to each other, (iii) the spacers were at most 20 bp, or (iv) they included a low-complexity or repeat sequence as detected by dustmasker (v1.0.0)93 (options -window 20 -level 10) or a direct repeat of at least 4 bp detected with etandem94 (options -minrepeat 4 -maxrepeat 15 -threshold 2). To initially link viroid-like sequences to CRISPR spacers, only hits with 0 or 1 mismatch over the entire spacer were considered (Table S5). To find additional spacer matches, we searched all members of the clusters with a spacer match against the IMG public metagenomic spacer data (dated 2022-06-18) set using IMG’s workspace BLAST with a minimum E-value of 1e-05. We also extracted the repeats matching loci using MinCED95 and searched them against nt using BLASTN v2.13.096,97.
Quantification and Statistical Analysis
No statistical analysis was involved in this study apart from the sequence similarity searches. For ribozyme detection, significance was defined by a single ribozyme match with E-value < 0.01 or a score above the gathering threshold. Additionally, two ribozymes were considered significant if both had E-values < 0.1. For CRISPR spacer detection, the BLASTN cut-off was set at E <e-05. For protein sequence similarity, the BLASTX cut-off was set at E < 0.01. The rest of the search parameters are indicated in the respective section of STAR Methods
Supplementary Material
Supplementary Table S1: Viroid-like cccRNAs from plant transcriptomes, metatranscriptomes, ViroidDB, and53, related to Figure 1 and Figure 2.
Supplementary Table S2: Metatranscriptomic cccRNAs and clusters with sequence matches to viroid-like agents, related to Figure 2.
Supplementary Table S4: Predicted self-cleaving ribozymes in known ambi- and ambi-like viruses, related to Figure 5.
Supplementary Table S5: CRISPR spacer matches to cccRNAs, related to STAR Methods section “CRISPR spacer analysis.”
Supplementary Table S6: Sample preparation method and taxonomic composition for each metatranscriptome, related to Figure 7.
Supplementary Table S7: Viroid-like cccRNAs coverage depth, related to Figure 7.
Figure 3: Predicted secondary structures of representatives of the five largest clusters of distinct viroid-like cccRNAs.
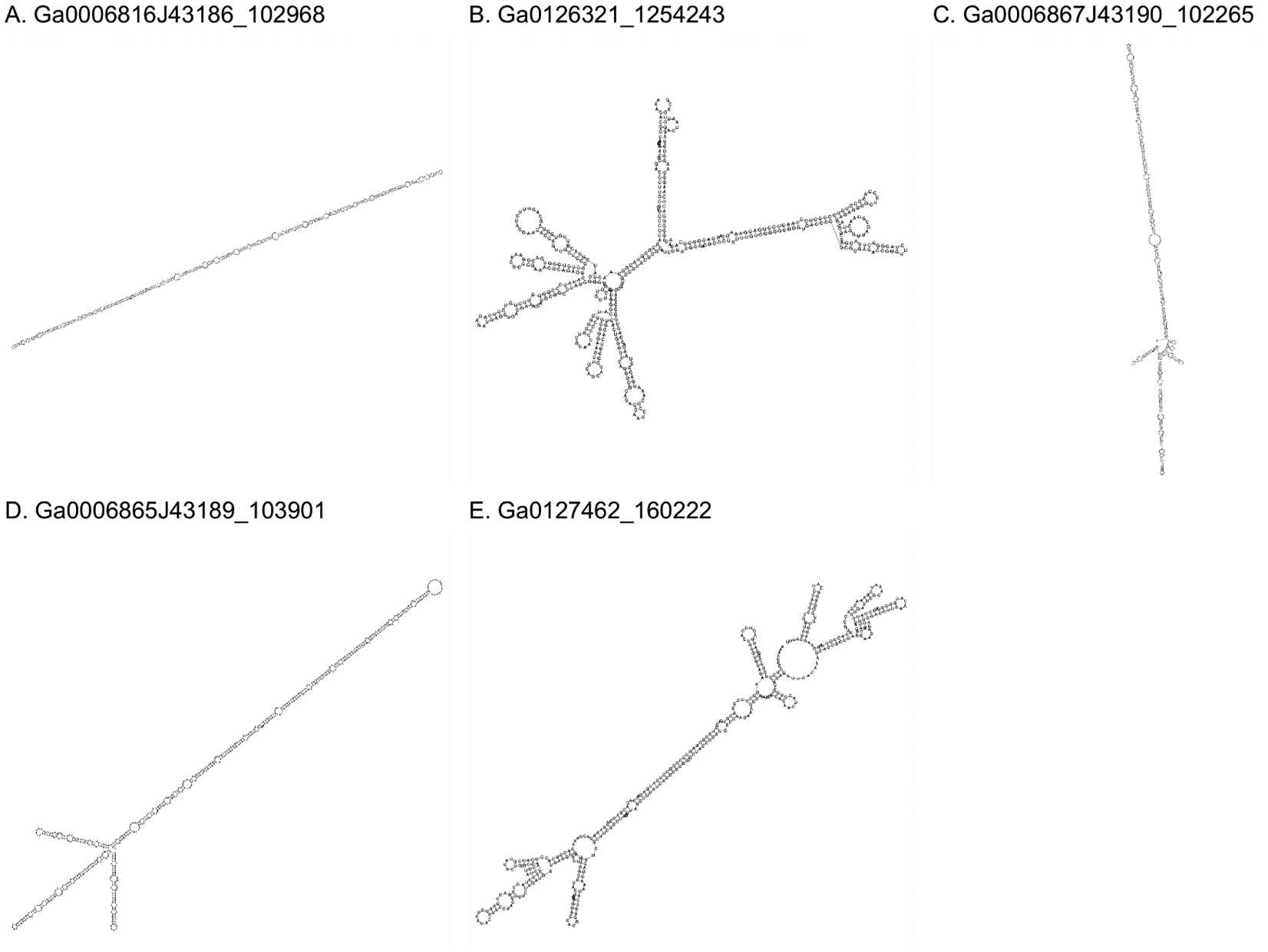
Structures were predicted using ViennaRNA’s RNAfold program configured to operate on circular sequences. Sequence data and metadata are available in Supplementary Table S1.
Highlights.
Metatranscriptome search yields a 5-fold increase in viroid-like circular RNA diversity
Distant relatives of hepatitis delta virus and diverse viroid-like viruses are discovered
Viroid-like circular RNAs are targeted by endogenous CRISPR systems
Diverse ribozymes and ribozyme combinations identified in viroid-like RNAs
Acknowledgements
The authors thank Samuel Wilder for his support while developing the software pipeline and Caleb Oh for advice on software architecture. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). Figure 1 and the graphical abstract were created with BioRender.com. B.D.L. was supported by a fellowship from the National Institutes of Health Oxford-Cambridge Scholars Program. Y.I.W. and E.V.K. are supported through the Intramural Research Program of the US National Institutes of Health (National Library of Medicine). U.G. and U.N. are supported by the European Research Council (ERC-AdG 787514). U.N. is partially supported by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel Aviv University. V.V.D. was partially supported by NIH/NLM/NCBI Visiting Scientist Fellowship. The work of the U.S. Department of Energy Joint Genome Institute (S.R., N.K. and all JGI co-authors), a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. M.K. was supported by l’Agence Nationale de la Recherche grants ANR-20-CE20-009-02 and ANR-21-CE11-0001-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Diener TO (2001). The viroid: Biological oddity or evolutionary fossil? In Advances in Virus Research (Elsevier; ), pp. 137–184. 10.1016/S0065-3527(01)57003-7. [DOI] [PubMed] [Google Scholar]
- 2.Mascia T, and Gallitelli D (2017). Economic Significance of Satellites. In Viroids and Satellites (Elsevier; ), pp. 555–563. 10.1016/B978-0-12-801498-1.00051-6. [DOI] [Google Scholar]
- 3.Daròs J-A, Elena SF, and Flores R (2006). Viroids: An Ariadne’s thread into the RNA labyrinth. EMBO Rep 7, 593–598. 10.1038/sj.embor.7400706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mühlbach H-P, and Sänger HL (1979). Viroid replication is inhibited by α-amanitin. Nature 278, 185–188. 10.1038/278185a0. [DOI] [PubMed] [Google Scholar]
- 5.Schindler I-M, and Mühlbach H-P (1992). Involvement of nuclear DNA-dependent RNA polymerases in potato spindle tuber viroid replication: A reevaluation. Plant Science 84, 221–229. 10.1016/0168-9452(92)90138-C. [DOI] [Google Scholar]
- 6.Navarro J-A, Vera A, and Flores R (2000). A Chloroplastic RNA Polymerase Resistant to Tagetitoxin Is Involved in Replication of Avocado Sunblotch Viroid. Virology 268, 218–225. 10.1006/viro.1999.0161. [DOI] [PubMed] [Google Scholar]
- 7.Branch AD, and Robertson HD (1984). A replication cycle for viroids and other small infectious RNA’s. Science 223, 450–455. 10.1126/science.6197756. [DOI] [PubMed] [Google Scholar]
- 8.Flores R, Minoia S, López-Carrasco A, Delgado S, Martínez de Alba Á-E, and Kalantidis K (2017). Viroid Replication. In Viroids and Satellites (Elsevier; ), pp. 71–81. 10.1016/B978-0-12-801498-1.00007-3. [DOI] [Google Scholar]
- 9.Nohales M-Á, Flores R, and Daròs J-A (2012). Viroid RNA redirects host DNA ligase 1 to act as an RNA ligase. Proc Natl Acad Sci USA 109, 13805–13810. 10.1073/pnas.1206187109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nohales M-Á, Molina-Serrano D, Flores R, and Daròs J-A (2012). Involvement of the Chloroplastic Isoform of tRNA Ligase in the Replication of Viroids Belonging to the Family Avsunviroidae. Journal of Virology 86, 8269–8276. 10.1128/JVI.00629-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diener TO (1971). Potato spindle tuber “virus.” Virology 45, 411–428. 10.1016/0042-6822(71)90342-4. [DOI] [PubMed] [Google Scholar]
- 12.Di Serio F, Li S-F, Pallás V, Owens RA, Randles JW, Sano T, Verhoeven J.Th.J., Vidalakis G, and Flores R (2017). Viroid Taxonomy. In Viroids and Satellites (Elsevier; ), pp. 135–146. 10.1016/B978-0-12-801498-1.00013-9. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y (2021). Current view and perspectives in viroid replication. Curr Opin Virol 47, 32–37. 10.1016/j.coviro.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Branch AD, Benenfeld BJ, and Robertson HD (1988). Evidence for a single rolling circle in the replication of potato spindle tuber viroid. Proc Natl Acad Sci USA 85, 9128–9132. 10.1073/pnas.85.23.9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giguère T, Adkar-Purushothama CR, Bolduc F, and Perreault J-P (2014). Elucidation of the structures of all members of the Avsunviroidae family. Molecular Plant Pathology 15, 767–779. 10.1111/mpp.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giguère T, Adkar-Purushothama CR, and Perreault J-P (2014). Comprehensive secondary structure elucidation of four genera of the family Pospiviroidae. PLoS ONE 9, e98655. 10.1371/journal.pone.0098655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de la Peña M, Ceprián R, and Cervera A (2020). A Singular and Widespread Group of Mobile Genetic Elements: RNA Circles with Autocatalytic Ribozymes. Cells 9, E2555. 10.3390/cells9122555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarro B, Rubino L, and Di Serio F (2017). Small Circular Satellite RNAs. In Viroids and Satellites (Elsevier; ), pp. 659–669. 10.1016/B978-0-12-801498-1.00061-9. [DOI] [Google Scholar]
- 19.Bruening G, Passmore BK, van Tol H, Buzayan JM, and Feldstein PA (1991). Replication of a Plant Virus Satellite RNA: Evidence Favors Transcription of Circular Templates of Both Polarities. MPMI 4, 219–225. 10.1094/MPMI-4-219. [DOI] [PubMed] [Google Scholar]
- 20.Rao ALN, and Kalantidis K (2015). Virus-associated small satellite RNAs and viroids display similarities in their replication strategies. Virology 479–480, 627–636. 10.1016/j.virol.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y-W, Hu C-C, Hsu Y-H, and Lin N-S (2017). Replication of Satellites. In Viroids and Satellites (Elsevier; ), pp. 577–586. 10.1016/B978-0-12-801498-1.00053-X. [DOI] [Google Scholar]
- 22.Ferré-D’Amaré AR, and Scott WG (2010). Small self-cleaving ribozymes. Cold Spring Harb Perspect Biol 2, a003574. 10.1101/cshperspect.a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daròs J-A, and Flores R (1995). Identification of a retroviroid-like element from plants. Proc Natl Acad Sci USA 92, 6856–6860. 10.1073/pnas.92.15.6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vera A, Daròs JA, Flores R, and Hernández C (2000). The DNA of a plant retroviroid-like element is fused to different sites in the genome of a plant pararetrovirus and shows multiple forms with sequence deletions. J Virol 74, 10390–10400. 10.1128/jvi.74.22.10390-10400.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hegedus K, Dallmann G, and Balázs E (2004). The DNA form of a retroviroid-like element is involved in recombination events with itself and with the plant genome. Virology 325, 277–286. 10.1016/j.virol.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 26.Cervera A, Urbina D, and de la Peña M (2016). Retrozymes are a unique family of non-autonomous retrotransposons with hammerhead ribozymes that propagate in plants through circular RNAs. Genome Biol 17, 135. 10.1186/s13059-016-1002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de la Peña M, and Cervera A (2017). Circular RNAs with hammerhead ribozymes encoded in eukaryotic genomes: The enemy at home. RNA Biology 14, 985–991. 10.1080/15476286.2017.1321730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hepojoki J, Hetzel U, Paraskevopoulou S, Drosten C, Harrach B, Zerbini FM, Koonin EV, Krupovic M, Dolja VV, and Kuhn JH (2020). Create one new realm (Ribozyviria) including one new family (Kolmioviridae) including genus Deltavirus and seven new genera for a total of 15 species (International Committee on Taxonomy of Viruses; ). [Google Scholar]
- 29.Kos A, Dijkema R, Arnberg AC, van der Meide PH, and Schellekens H (1986). The hepatitis delta (δ) virus possesses a circular RNA. Nature 323, 558–560. 10.1038/323558a0. [DOI] [PubMed] [Google Scholar]
- 30.Modahl LE, Macnaughton TB, Zhu N, Johnson DL, and Lai MMC (2000). RNA-Dependent Replication and Transcription of Hepatitis Delta Virus RNA Involve Distinct Cellular RNA Polymerases. Mol. Cell. Biol 20, 6030–6039. 10.1128/MCB.20.16.6030-6039.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sureau C, and Negro F (2016). The hepatitis delta virus: Replication and pathogenesis. Journal of Hepatology 64, S102–S116. 10.1016/j.jhep.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Paraskevopoulou S, Pirzer F, Goldmann N, Schmid J, Corman VM, Gottula LT, Schroeder S, Rasche A, Muth D, Drexler JF, et al. (2020). Mammalian deltavirus without hepadnavirus coinfection in the neotropical rodent Proechimys semispinosus. Proc Natl Acad Sci USA 117, 17977–17983. 10.1073/pnas.2006750117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergner LM, Orton RJ, Broos A, Tello C, Becker DJ, Carrera JE, Patel AH, Biek R, and Streicker DG (2021). Diversification of mammalian deltaviruses by host shifting. Proc Natl Acad Sci USA 118, e2019907118. 10.1073/pnas.2019907118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hetzel U, Szirovicza L, Smura T, Prähauser B, Vapalahti O, Kipar A, and Hepojoki J (2019). Identification of a Novel Deltavirus in Boa Constrictors. mBio 10, e00014–19. 10.1128/mBio.00014-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wille M, Netter H, Littlejohn M, Yuen L, Shi M, Eden J-S, Klaassen M, Holmes E, and Hurt A (2018). A Divergent Hepatitis D-Like Agent in Birds. Viruses 10, 720. 10.3390/v10120720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang W-S, Pettersson JH-O, Le Lay C, Shi M, Lo N, Wille M, Eden J-S, and Holmes EC (2019). Novel hepatitis D-like agents in vertebrates and invertebrates. Virus Evol 5, vez021. 10.1093/ve/vez021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diener TO (2016). Viroids: “Living fossils” of primordial RNAs? Biol Direct 11, 15. 10.1186/s13062-016-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flores R, Navarro B, Serra P, and Di Serio F (2022). A scenario for the emergence of protoviroids in the RNA world and for their further evolution into viroids and viroid-like RNAs by modular recombinations and mutations. Virus Evolution 8, veab107. 10.1093/ve/veab107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee BD, and Koonin EV (2022). Viroids and Viroid-like Circular RNAs: Do They Descend from Primordial Replicators? Life 12, 103. 10.3390/life12010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolf YI, Silas S, Wang Y, Wu S, Bocek M, Kazlauskas D, Krupovic M, Fire A, Dolja VV, and Koonin EV (2020). Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat Microbiol. 10.1038/s41564-020-0755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC, Taylor J, Lin V, Altman T, Barbera P, Meleshko D, Lohr D, Novakovsky G, Buchfink B, Al-Shayeb B, et al. (2022). Petabase-scale sequence alignment catalyses viral discovery. Nature 602, 142–147. 10.1038/s41586-021-04332-2. [DOI] [PubMed] [Google Scholar]
- 42.Zayed AA, Wainaina JM, Dominguez-Huerta G, Pelletier E, Guo J, Mohssen M, Tian F, Pratama AA, Bolduc B, Zablocki O, et al. (2022). Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome. Science 376, 156–162. 10.1126/science.abm5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neri U, Wolf YI, Roux S, Camargo AP, Lee B, Kazlauskas D, Chen IM, Ivanova N, Allen LZ, Paez-Espino D, et al. (2022). A five-fold expansion of the global RNA virome reveals multiple new clades of RNA bacteriophages. 10.1101/2022.02.15.480533. [DOI] [PubMed] [Google Scholar]
- 44.One Thousand Plant Transcriptomes Initiative (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685. 10.1038/s41586-019-1693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin Y, Xu T, Lin W, Jia Q, He Q, Liu K, Du J, Chen L, Yang X, Du F, et al. (2020). Reference-free and de novo Identification of Circular RNAs. 10.1101/2020.04.21.050617. [DOI] [Google Scholar]
- 46.Nawrocki EP, and Eddy SR (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalvari I, Nawrocki EP, Ontiveros-Palacios N, Argasinska J, Lamkiewicz K, Marz M, Griffiths-Jones S, Toffano-Nioche C, Gautheret D, Weinberg Z, et al. (2021). Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Research 49, D192–D200. 10.1093/nar/gkaa1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gozmanova M (2003). Characterization of the RNA motif responsible for the specific interaction of potato spindle tuber viroid RNA (PSTVd) and the tomato protein Virp1. Nucleic Acids Research 31, 5534–5543. 10.1093/nar/gkg777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee BD, Neri U, Oh CJ, Simmonds P, and Koonin EV (2021). ViroidDB: A database of viroids and viroid-like circular RNAs. Nucleic Acids Research, gkab974. 10.1093/nar/gkab974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou W-Y, Li S-F, Wu Z-J, Jiang D-M, and Sano T (2009). Coleus blumei viroid 6: A new tentative member of the genus Coleviroid derived from natural genome shuffling. Arch Virol 154, 993–997. 10.1007/s00705-009-0388-7. [DOI] [PubMed] [Google Scholar]
- 51.Nie X, and Singh RP (2017). Coleus Blumei Viroids. In Viroids and Satellites (Elsevier; ), pp. 289–295. 10.1016/B978-0-12-801498-1.00027-9. [DOI] [Google Scholar]
- 52.Chiumenti M, Navarro B, Candresse T, Flores R, and Di Serio F (2021). Reassessing species demarcation criteria in viroid taxonomy by pairwise identity matrices. Virus Evol 7, veab001. 10.1093/ve/veab001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weinberg CE, Olzog VJ, Eckert I, and Weinberg Z (2021). Identification of over 200-fold more hairpin ribozymes than previously known in diverse circular RNAs. Nucleic Acids Research 49, 6375–6388. 10.1093/nar/gkab454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu J, and Bisaro DM (2020). Biased Pol II fidelity contributes to conservation of functional domains in the Potato spindle tuber viroid genome. PLoS Pathog 16, e1009144. 10.1371/journal.ppat.1009144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gago S, Elena SF, Flores R, and Sanjuán R (2009). Extremely High Mutation Rate of a Hammerhead Viroid. Science 323, 1308–1308. 10.1126/science.1169202. [DOI] [PubMed] [Google Scholar]
- 56.López-Carrasco A, Ballesteros C, Sentandreu V, Delgado S, Gago-Zachert S, Flores R, and Sanjuán R (2017). Different rates of spontaneous mutation of chloroplastic and nuclear viroids as determined by high-fidelity ultra-deep sequencing. PLoS Pathog 13, e1006547. 10.1371/journal.ppat.1006547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sutela S, Forgia M, Vainio EJ, Chiapello M, Daghino S, Vallino M, Martino E, Girlanda M, Perotto S, and Turina M (2020). The virome from a collection of endomycorrhizal fungi reveals new viral taxa with unprecedented genome organization. Virus Evolution 6, veaa076. 10.1093/ve/veaa076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forgia M, Isgandarli E, Aghayeva DN, Huseynova I, and Turina M (2021). Virome characterization of Cryphonectria parasitica isolates from Azerbaijan unveiled a new mymonavirus and a putative new RNA virus unrelated to described viral sequences. Virology 553, 51–61. 10.1016/j.virol.2020.10.008. [DOI] [PubMed] [Google Scholar]
- 59.Linnakoski R, Sutela S, Coetzee MPA, Duong TA, Pavlov IN, Litovka YA, Hantula J, Wingfield BD, and Vainio EJ (2021). Armillaria root rot fungi host single-stranded RNA viruses. Sci Rep 11, 7336. 10.1038/s41598-021-86343-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marais A, Nivault A, Faure C, Theil S, Comont G, Candresse T, and Corio-Costet M-F (2017). Determination of the complete genomic sequence of Neofusicoccum luteum mitovirus 1 (NLMV1), a novel mitovirus associated with a phytopathogenic Botryosphaeriaceae. Arch Virol 162, 2477–2480. 10.1007/s00705-017-3338-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krupovic M, Varsani A, Kazlauskas D, Breitbart M, Delwart E, Rosario K, Yutin N, Wolf YI, Harrach B, Zerbini FM, et al. (2020). Cressdnaviricota: A Virus Phylum Unifying Seven Families of Rep-Encoding Viruses with Single-Stranded, Circular DNA Genomes. J Virol 94, e00582–20. 10.1128/JVI.00582-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zuccola HJ, Rozzelle JE, Lemon SM, Erickson BW, and Hogle JM (1998). Structural basis of the oligomerization of hepatitis delta antigen. Structure 6, 821–830. 10.1016/S0969-2126(98)00084-7. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen L-T, Schmidt HA, von Haeseler A, and Minh BQ (2015). IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Molecular Biology and Evolution 32, 268–274. 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Webb C-HT, Riccitelli NJ, Ruminski DJ, and Lupták A (2009). Widespread occurrence of self-cleaving ribozymes. Science 326, 953. 10.1126/science.1178084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chadalavada DM, Gratton EA, and Bevilacqua PC (2010). The human HDV-like CPEB3 ribozyme is intrinsically fast-reacting. Biochemistry 49, 5321–5330. 10.1021/bi100434c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salehi-Ashtiani K, Lupták A, Litovchick A, and Szostak JW (2006). A Genomewide Search for Ribozymes Reveals an HDV-Like Sequence in the Human CPEB3 Gene. Science 313, 1788–1792. 10.1126/science.1129308. [DOI] [PubMed] [Google Scholar]
- 67.de la Peña M, Ceprián R, Casey JL, and Cervera A (2021). Hepatitis delta virus-like circular RNAs from diverse metazoans encode conserved hammerhead ribozymes. Virus Evol 7, veab016. 10.1093/ve/veab016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, Rubin E, Ivanova NN, and Kyrpides NC (2016). Uncovering Earth’s virome. Nature 536, 425–430. 10.1038/nature19094. [DOI] [PubMed] [Google Scholar]
- 69.Munson-McGee JH, Peng S, Dewerff S, Stepanauskas R, Whitaker RJ, Weitz JS, and Young MJ (2018). A virus or more in (nearly) every cell: Ubiquitous networks of virushost interactions in extreme environments. ISME J 12, 1706–1714. 10.1038/s41396-018-0071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen I-MA, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, Hajek P, Ritter S, Varghese N, Seshadri R, et al. (2021). The IMG/M data management and analysis system v.6.0: New tools and advanced capabilities. Nucleic Acids Research 49, D751–D763. 10.1093/nar/gkaa939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thomas SC, Tamadonfar KO, Seymour CO, Lai D, Dodsworth JA, Murugapiran SK, Eloe-Fadrosh EA, Dijkstra P, and Hedlund BP (2019). Position-Specific Metabolic Probing and Metagenomics of Microbial Communities Reveal Conserved Central Carbon Metabolic Network Activities at High Temperatures. Front. Microbiol 10, 1427. 10.3389/fmicb.2019.01427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van der Meer MTJ, Klatt CG, Wood J, Bryant DA, Bateson MM, Lammerts L, Schouten S, Sinninghe Damsté JS, Madigan MT, and Ward DM (2010). Cultivation and Genomic, Nutritional, and Lipid Biomarker Characterization of Roseiflexus Strains Closely Related to Predominant In Situ Populations Inhabiting Yellowstone Hot Spring Microbial Mats. J Bacteriol 192, 3033–3042. 10.1128/JB.01610-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Madigan MT, Jung DO, Karr EA, Sattley WM, Achenbach LA, and van der Meer MTJ (2005). Diversity of anoxygenic phototrophs in contrasting extreme environments. In Geothermal Biology and Geochemistry in Yellowstone National Park, Inskeep WP and McDermott TR, eds. (Thermal Biology Institute; ), pp. 203–219. [Google Scholar]
- 74.Johnson MTJ, Carpenter EJ, Tian Z, Bruskiewich R, Burris JN, Carrigan CT, Chase MW, Clarke ND, Covshoff S, dePamphilis CW, et al. (2012). Evaluating Methods for Isolating Total RNA and Predicting the Success of Sequencing Phylogenetically Diverse Plant Transcriptomes. PLoS ONE 7, e50226. 10.1371/journal.pone.0050226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koonin EV, Dolja VV, Krupovic M, Varsani A, Wolf YI, Yutin N, Zerbini FM, and Kuhn JH (2020). Global Organization and Proposed Megataxonomy of the Virus World. Microbiol Mol Biol Rev 84, e00061–19. 10.1128/MMBR.00061-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen S, Zhou Y, Chen Y, and Gu J (2018). Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bushmanova E, Antipov D, Lapidus A, and Prjibelski AD (2019). rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8, giz100. 10.1093/gigascience/giz100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gao Y, Wang J, and Zhao F (2015). CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16, 4. 10.1186/s13059-014-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Melsted P, and Pritchard JK (2011). Efficient counting of k-mers in DNA sequences using a bloom filter. BMC Bioinformatics 12, 333. 10.1186/1471-2105-12-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marçais G, and Kingsford C (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Duval JP (1983). Factorizing words over an ordered alphabet. Journal of Algorithms 4, 363–381. 10.1016/0196-6774(83)90017-2. [DOI] [Google Scholar]
- 82.Macke TJ (2001). RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Research 29, 4724–4735. 10.1093/nar/29.22.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Steinegger M, and Söding J (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 35, 1026–1028. 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 84.Lorenz R, Bernhart SH, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol Biol 6, 26. 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: Guaranteeing well-connected communities. Sci Rep 9, 5233. 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Singh U, and Wurtele ES (2021). Orfipy: A fast and flexible tool for extracting ORFs. Bioinformatics 37, 3019–3020. 10.1093/bioinformatics/btab090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, and the UniProt Consortium (2015). UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eddy SR (2011). Accelerated Profile HMM Searches. PLoS Comput Biol 7, e1002195. 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frickey T, and Lupas A (2004). CLANS: A Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20, 3702–3704. 10.1093/bioinformatics/bth444. [DOI] [PubMed] [Google Scholar]
- 90.Muhire BM, Varsani A, and Martin DP (2014). SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 9, e108277. 10.1371/journal.pone.0108277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pei J, and Grishin NV (2014). PROMALS3D: Multiple Protein Sequence Alignment Enhanced with Evolutionary and Three-Dimensional Structural Information. In Multiple Sequence Alignment Methods Methods in Molecular Biology., Russell DJ, ed. (Humana Press; ), pp. 263–271. 10.1007/978-1-62703-646-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Letunic I, and Bork P (2021). Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Research 49, W293–W296. 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morgulis A, Gertz EM, Schäffer AA, and Agarwala R (2006). A Fast and Symmetric DUST Implementation to Mask Low-Complexity DNA Sequences. Journal of Computational Biology 13, 1028–1040. 10.1089/cmb.2006.13.1028. [DOI] [PubMed] [Google Scholar]
- 94.Rice P, Longden I, and Bleasby A (2000). EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet 16, 276–277. 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 95.Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, and Hugenholtz P (2007). CRISPR Recognition Tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8, 209. 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J Mol Biol 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 97.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: Architecture and applications. BMC Bioinformatics 10, 421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li W, and Godzik A (2006). Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 99.Gábor C, and Nepusz T (2005). The igraph software package for complex network research. InterJournal Complex Systems, 1695. [Google Scholar]
- 100.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Knight R, Maxwell P, Birmingham A, Carnes J, Caporaso JG, Easton BC, Eaton M, Hamady M, Lindsay H, Liu Z, et al. (2007). PyCogent: A toolkit for making sense from sequence. Genome Biol 8, R171. 10.1186/gb-2007-8-8-r171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rumpf A (2022). Mastering Nim: A complete guide to the programming language. [Google Scholar]
- 103.Shen W, Le S, Li Y, and Hu F (2016). SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 11, e0163962. 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McKinney W (2010). Data Structures for Statistical Computing in Python. In, pp. 56–61. 10.25080/Majora-92bf1922-00a. [DOI] [Google Scholar]
- 105.Wickham H (2016). Ggplot2 (Springer International Publishing; ) 10.1007/978-3-319-24277-4. [DOI] [Google Scholar]
- 106.Mölder F, Jablonski KP, Letcher B, Hall MB, Tomkins-Tinch CH, Sochat V, Forster J, Lee S, Twardziok SO, Kanitz A, et al. (2021). Sustainable data analysis with Snakemake. F1000Res 10, 33. 10.12688/f1000research.29032.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gu Z, Gu L, Eils R, Schlesner M, and Brors B (2014). Circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 108.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: Viroid-like cccRNAs from plant transcriptomes, metatranscriptomes, ViroidDB, and53, related to Figure 1 and Figure 2.
Supplementary Table S2: Metatranscriptomic cccRNAs and clusters with sequence matches to viroid-like agents, related to Figure 2.
Supplementary Table S4: Predicted self-cleaving ribozymes in known ambi- and ambi-like viruses, related to Figure 5.
Supplementary Table S5: CRISPR spacer matches to cccRNAs, related to STAR Methods section “CRISPR spacer analysis.”
Supplementary Table S6: Sample preparation method and taxonomic composition for each metatranscriptome, related to Figure 7.
Supplementary Table S7: Viroid-like cccRNAs coverage depth, related to Figure 7.
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. Data generated during downstream analysis have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources.
All the code and data generated in this work are freely available through public portals listed below.