

Abstract
Cardiac fibroblasts play critical roles in the maintenance of cardiac structure and the response to cardiac insult. Extracellular matrix deposition by activated resident cardiac fibroblasts, called myofibroblasts, is an essential wound healing response. However, persistent fibroblast activation contributes to pathological fibrosis and cardiac chamber stiffening, which can cause diastolic dysfunction, heart failure, and initiate lethal arrhythmias. The dynamic and phenotypically plastic nature of cardiac fibroblasts is governed in part by the transcriptional regulation of genes encoding extracellular matrix molecules. Understanding how fibroblasts integrate various biomechanical cues into a precise transcriptional response may uncover therapeutic strategies to prevent fibrosis. Here, we provide an overview of the recent literature on transcriptional control of cardiac fibroblast plasticity and fibrosis, with a focus on canonical and non-canonical TGF-β signaling, biomechanical regulation of Hippo/YAP and Rho/MRTF signaling, and metabolic and epigenetic control of fibroblast activation.
Keywords: heart, fibrosis, fibroblast, transcription
Introduction
The heart is a muscular pump responsible for providing oxygenated blood to the entire body. Heart muscle, called myocardium, is composed of a variety of cell types with distinct functions and spatial distributions, including cardiomyocytes, cardiac conduction system cells, vascular endothelial and mural cells, resident immune cells, valvular interstitial cells, and cardiac fibroblasts (CFs). Fibroblasts, which make up ~20% of the non-myocytes in the heart1,2, provide a framework of fibrillar collagen that support cardiac structure and function.3 Perhaps more importantly, resident CFs respond to cardiac insult by proliferating and acquiring a contractile and secretory phenotype. These activated fibroblasts, also called myofibroblasts secrete copious amounts of extracellular matrix (ECM) in an adaptive response that supports cardiac integrity.4,5 However, unchecked CF activation is a primary cause of fibrotic scar formation, which sustains myocardial integrity at the expense of pliability, leading to diastolic dysfunction, heart failure and eventually increasing the risk of lethal arrhythmias.6 A deeper understanding of the mechanisms that control fibroblast plasticity and adverse myocardial remodeling may accelerate the development of anti-fibrotic strategies to treat cardiac pathologies including diastolic heart failure (heart failure with preserved ejection fraction, or HFpEF), a poorly understood condition with limited treatment options. The goal of this review is to provide a short summary of the recent literature related to transcriptional control of the fibroblast phenotype and cardiac fibrosis; we apologize to the authors of studies that were not cited due to limited space.
TGFβ signaling
Transforming growth factor beta (TGFβ) is ubiquitously involved in cell growth, differentiation, migration, and apoptosis during embryonic development and adult cellular pathophysiology and is the cornerstone of fibroblast activation and cardiac fibrosis. The canonical TGFβ signaling pathway is mediated by SMAD family transcription factors (TFs), which include receptor regulated (R)-SMADs (SMAD1/2/3/5/8), a common SMAD (SMAD4) and inhibitory (I)-SMADs (SMAD 6/7). Upon TGFβ binding to type I receptors, R-SMADS such as SMAD2/3 are phosphorylated, stimulating their recruitment of SMAD4 and translocation to the nucleus (see Figure). This complex binds to and activates SMAD-binding elements (SBEs) in the promoter region of target genes to initiate transcription.7–9 Initial studies using global gene deletion in mice described SMAD3 as a critical regulator of the myofibroblast phenotype, which stimulates ECM deposition in pressure overload and myocardial infarction (MI) models.10,11 More recently, CF-specific deletion of Tgfbr1/2 or Smad3 in mice confirmed their roles in ECM deposition during pressure overload and ischemia-induced cardiac remodeling.12 However, these studies also revealed a more nuanced role of SMAD2 and SMAD3. SMAD3 directly induces the expression of genes encoding ECM molecules, suppresses MMP (matrix metalloproteinase)-3 and MMP-8, and induces TIMP (tissue inhibitor of metalloproteinases)-1 to stabilize the collagen network and support cardiac integrity in left ventricle pressure overload without impacting CF proliferation.13 In fact, fibroblast specific Smad3 deletion often leads to lethal left ventricle rupture after MI, which is attributed to disorganized scar formation and insufficient repair.14 In contrast, fibroblasts that lack Smad2 elicit a surprisingly normal fibrotic response to pressure overload and ischemia.12,15 While these studies don’t explain why Smad2 deletion does not impede the development of cardiac fibrosis, one clue may be the unique induction of integrin α2 and α5 by SMAD3, which may mediate important cellular interactions with the ECM.15 Indeed, fibroblasts are reported to play an important structural role in the healing cardiac scar.16
Figure. Summary of the signaling pathways and transcriptional regulation of fibroblast activation.
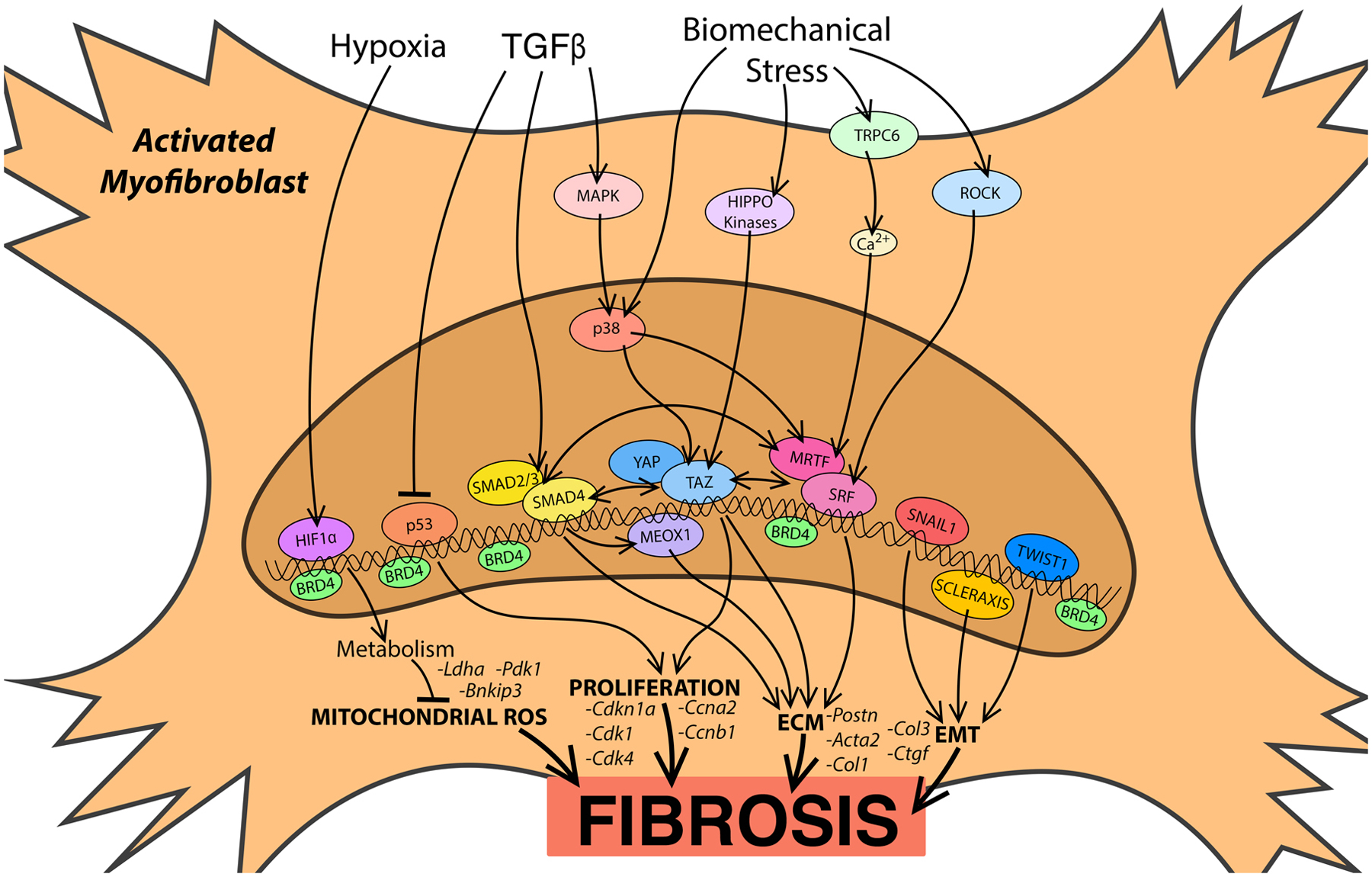
Arrows indicate upstream signaling pathways converging on select transcription factors, or experimentally validated interactions between transcription factors. BRD14 (bromodomain containing protein 4), Ca2+ (calcium), ECM (extracellular matrix), EMT (epithelial-to-mesenchymal transition), HIF1α (hypoxia inducible factor 1 alpha), MAPK (mitogen-activated protein kinase), MRTF (myocardin-related transcription factor), ROCK (Rho-kinase), ROS (reactive oxygen species), SRF (serum response factor), TAZ (transcriptional coactivator with PDZ-binding motif), TGF-β (Transforming growth factor beta), TRPC6 (transient receptor potential cation channel subfamily C member 6), YAP (Yes-associated protein).
The TGFβ−SMAD axis is also influenced by cooperative association with additional TFs related to epithelial to mesenchymal transition (EMT). For example, RAS-responsive element binding protein 1 (RREB1) was identified as a molecular link between RAS and TGFβ/SMAD pathways in carcinoma cells, where RREB1 and SMADs cooperate to promote the expression of Snai1, a TF that drives fibrogenic EMT in intratumoral myofibroblasts.17 Scleraxis, a basic helix-loop-helix TF, has been shown to induce Twist1 and Snai1 expression to stimulate EMT.18 Scleraxis also plays a critical role in CF activation and fibrosis, potentially via synergizing with SMAD3 to induce expression of ECM genes (see Figure).19 The fibroblast phenotype is also supported, in part, by the post transcriptional regulation of genes encoding EMT and fibrosis associated proteins by the RNA-binding protein muscleblind-like 1 (MBNL1)20. It is important to note that while similarities exist between the molecular programs inducing EMT and fibroblast activation, EMT does not appear to influence fibrosis in the heart to the same extent as in other organs; instead EMT is primarily driven by the activation of pre-existing resident CFs4,21.
In contrast to SMAD-dependent signaling, non-canonical TGFβ signaling is mediated by mitogen-activated protein kinases (MAPKs), including p38 isoforms (α, β, γ, and δ), extracellular signal-regulated kinase 1 and 2 (Erk1/2), and c-Jun N-terminal kinases (JNKs). TGFβ-activated kinase 1 (TAK1)-p38 activation is observed in MI and pressure overload models, where it stimulates cardiac fibrogenesis22. Fibroblast-specific deletion of MAPK14 (p38α) prevents fibroblast activation and cardiac fibrosis in mice, often leading to left ventricle rupture after MI (see Figure),23 and salinmoycin, a small molecule that inhibits p38, can block and reverse pathological fibrosis in mouse models of ischemic and non-ischemic cardiac remodeling.24 Cardiac fibrosis can also be influenced by negative regulators of TGFβ-SMAD signaling. In fact, SMAD7 expression is suppressed in the infarcted rat heart, which may allow for the propagation of TGFβ-SMAD signaling and fibroblast activation.25 Overexpression of SMAD7 has been shown to reduce ECM deposition both in vitro and in vivo.26,27 Interestingly, the anti-fibrotic activity of SMAD7 is attributed to the inhibition of SMAD2/3 and SMAD-independent suppression of Erbb2 activation.28 The lysine de-acetylase sirtuin 1 (SIRT1) also plays a cardioprotective role in a mouse pressure overload model, at least partially by inhibiting SMAD2/3 transactivation to alleviate cardiac fibrosis.29 Transcriptional cofactors Ski and SnoN are also negative regulators of SMAD-dependent transcription and myofibroblast activation.30–32 A recent gene delivery approach blocking the STAT3/FOXM1 pathway enhanced SnoN/Ski signaling and suppressed the TGFβ/Smad pathway in pulmonary fibrosis.33
Biomechanical control of transcription and fibroblast activation
Importantly, fibrotic tissue stiffening is both a consequence and a trigger of myofibroblast activation. Therefore, scar formation can stimulate further fibroblast activation to exacerbate pathological fibrosis. Recent studies have begun to elucidate the biomechanical mechanisms that control myofibroblast activation in the injured heart. Indeed, biomechanical regulation of chromatin accessibility is at least partially responsible for stimulating the myofibroblast phenotype.34 Biomechanical regulation of the Rho-Rho kinase (ROCK)-myocardin-related transcription factor (MRTF)-serum response factor (SRF) axis is also particularly important in cardiac fibrosis. MRTFs are transcriptional co-factors for SRF that are sequestered in the cytoplasm via interactions with globular (G) actin. Mechanical tension can trigger Rho-ROCK signaling that initiates actin polymerization, reducing the G-actin pool and allowing MRTFs to translocate to the nucleus where they activate SRF target genes responsible for myofibroblast activation (see Figure).35,36 Global deletion of MRTF-A suppresses myofibroblast activation and fibrosis following MI in mice37, and altering Rho-MRTF signaling with a small molecule reduces the severity of fibrosis in animal models.38,39 In Idiopathic pulmonary fibrosis, the stretch-dependent transient receptor potential vanilloid 4 channel (TRPV4) enhances actomyosin remodeling and increases nuclear translocation of MRTF-A in a SMAD-independent manner.40 In addition, non-canonical TGFβ/p38 signaling stimulates SRF-dependent transient receptor potential cation channel subfamily C member 6 (TRPC6) expression, which induces Ca2+-dependent NFAT/SRF activation and further myofibroblast activation.41 NFAT-dependent fibroblast activation is also facilitated by the non-canonical Ca2+ – calmodulin dependent control of G protein-coupled receptor kinase 5 (GRK5) nuclear accumulation, revealing considerable crosstalk between pro-fibrotic signaling pathways.42
The Hippo pathway is another mechanosensitive gene-regulatory program that impacts the CF phenotype. Cell stretch or loss of contact inhibition disrupts Hippo-pathway kinase cascades, allowing for the nuclear accumulation of Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ).43 In lung and cardiac fibroblasts, YAP/TAZ nuclear translocation directly stimulates TEAD (transcriptional enhanced associate domain)-dependent transcription of genes encoding ECM components and inflammatory mediators, in part through H3K9 methylation (see Figure).44–46 Hippo signaling can also cooperate with canonical and non-canonical TGFβ and Rho signaling pathways. YAP/TAZ binding to SMAD2/3 allows SMAD2/3 sub-cellular localization to be directly regulated by cell stretch47. Mechanical tension also facilitates p38-YAP-TEAD-dependent transcription, linking tissue stiffness to excessive fibroblast activation after MI.48 Interestingly, Caveolin-1 positively modulates mechanosensitive YAP activation through a Hippo-kinase-independent mechanism, which requires Rho-dependent actin-cytoskeleton alterations, revealing a common signaling paradigm upstream of both YAP- and MRTF-dependent gene programs.49 Indeed, MRTF enhances YAP transcriptional activity through TEAD50, and MRTFs and YAP/TAZ facilitate crosstalk between G-protein coupled receptor- and Rho-dependent gene expression.51 A recent study also found that YAP induces the expression of Mrtf-a in cardiac fibroblasts, potentially establishing a pro-fibrotic positive feedback loop52. However, both TGF-β and cell stretch appear to be required for the cooperative activation of target genes by YAP/TAZ, SMAD3 and MRTFs, suggesting a more nuanced and context dependent functional interaction between these gene regulatory programs.53 Of note, fibroblasts reportedly acquire “mechanical memory”, whereby culture on a stiff substrate lowers the threshold for subsequent fibroblast activation; a priming event that is hypothesized to aggravate chronic fibrotic conditions such as idiopathic pulmonary fibrosis54. Hippo/YAP and Rho/MRTF are perfectly positioned to contribute to biomechanical transcriptional memory, which may play a particularly important role later in the remodeling process when ECM deposition increases organ stiffness. Conversely, it is interesting to speculate that an appropriate mechanical intervention may return activated myofibroblasts to a quiescent state, or even shift their phenotype from profibrotic to pro-resolution.
Regulation of fibroblast phenotype by hypoxia and reactive oxygen species
Ischemic heart disease, hypertrophic cardiac remodeling, inflammation, and fibrosis all disrupt perfusion of the heart with oxygenated blood, leading to intermittent or more extended bouts of hypoxia and reactive oxygen species (ROS) dysregulation. A transcriptomic analysis of CFs isolated from mice that were subjected to an exercise regimen, compared to ischemic and non-ischemic models of pathologic cardiac remodeling, revealed surprisingly divergent phenotypic responses; differentially regulated gene programs included ROS scavenger pathways and p53-dependent gene expression.55 Transgenic overexpression of the p53 target gene, Cdkn1a (p21), in CFs restrains their proliferation and attenuates cardiac fibrosis in vivo.56 Control of CF proliferation by the p53-Cdkn1a axis appears to be a physiological characteristic of heart disease, as SPRR2B/MDM2-dependent degradation of p53 accelerates cardiac fibroblast proliferation in vitro and was observed in fibrotic foci of human heart failure tissue (see Figure).57 Indeed, a common characteristic of cardiac insult is the altered expression of cell cycle regulators supporting the transient proliferation of fibroblasts in the diseased heart58. ROS and hypoxia signaling also provide metabolic control of the fibroblast phenotype - fibroblasts in the healthy heart are surprisingly hypoxic, and exhibit elevated hypoxia-inducible factor 1α (HIF-1α) levels59. This study found that elevated HIF-1α dependent gene expression in CFs is important for metabolic buffering that limits mitochondrial ROS production after MI (see Figure).59 This protective mechanism breaks down upon genetic deletion of Hif-1α in fibroblasts, leading to excessive post-MI mitochondrial ROS production, CF proliferation and the generation of a robust fibrotic scar. Metabolic reprogramming may also facilitate fibroblast activation via stimulating the action of histone demethylases that enhance the accessibility of chromatin, particularly in regions that harbor pro-fibrotic genes.60 Although antioxidant therapies have not been successful in clinical trials, these studies suggest that the development of a more targeted approach to metabolic and redox control may still hold promise for treating cardiovascular disease, in part via the prevention of CF activation and pathological cardiac fibrosis.
Targeting the epigenetic control of fibroblast activation
Epigenetic changes in chromatin condensation are associated with gene expression changes that impact cell state and have recently been correlated with the CF phenotype.61 Bromodomain and extraterminal (BET) proteins are a family of epigenetic readers that recruit coregulatory factors and promote transcription of target genes; BRD4 plays a particularly important role in the progression of heart disease.62–64 BRD4 increases innate immune activation, ECM production, and cell adhesion via activation of nuclear factor kappa B (NFκB) and TGFβ signaling pathways (see Figure).64 BRD4 responds to non-canonical TGFβ signaling to facilitate TF binding in enhancer regions, and a small molecule BET inhibitor, JQ1, suppresses fibroblast activation and fibrosis.65 A recent study utilizing JQ1 to suppress the myofibroblast phenotype identified a dynamic and reversible transcriptional switch responsible for the plasticity of the fibroblast phenotype.66 Fibroblast activation in response to left ventricle pressure overload was ameliorated with JQ1, and fibroblast re-activation was observed upon cessation of JQ1. The bi-directional phenotypic switch is tightly correlated with chromatin occupancy of BRD4 at myofibroblast gene enhancers, and especially with chromatin accessibility at the Meox1 gene locus. Meox1 induction by TGF-β was shown to be an important regulator of fibroblast activation and cardiac fibrosis. Further studies are certainly warranted to establish the therapeutic potential of inhibiting the BRD4-dependent pro-fibrotic gene program, or whether a targeted approach that silences a more specific maladaptive switch such as Meox1 is feasible.
Conclusion
The studies described here highlight the importance of CFs in cardiac health and disease. Indeed, CF are emerging as a dynamic and highly malleable cell type that provides structural support during normal cardiac homeostasis and contributes to heart repair and scar formation following cardiac insult. A more complete understanding of the transcriptional control of fibroblast function may lead to unique therapeutic strategies to ameliorate the development of pathological fibrosis that prevent diastolic cardiac dysfunction and heart failure.
Funding:
This work was supported by grants from the National Institutes of Health R01-HL120919, R01-HL133761, R01-HL144867, R01-HL136179 to EMS, and F31-HL158037 to KNBV; by the New York Department of Health NYSTEM-C32566GG to EMS; and by the Harold S. Geneen Charitable Trust Awards Program to EMS.
Footnotes
Declaration of Interests: None
References
- 1.Litvinukova M et al. Cells of the adult human heart. Nature 588, 466–472, doi: 10.1038/s41586-020-2797-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinto AR et al. Revisiting Cardiac Cellular Composition. Circ Res 118, 400–409, doi: 10.1161/CIRCRESAHA.115.307778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber KT Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 13, 1637–1652, doi: 10.1016/0735-1097(89)90360-4 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Quijada P et al. Pre-existing fibroblasts of epicardial origin are the primary source of pathological fibrosis in cardiac ischemia and aging. J Mol Cell Cardiol 129, 92–104, doi: 10.1016/j.yjmcc.2019.01.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanisicak O et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 7, 12260, doi: 10.1038/ncomms12260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plikus MV et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 184, 3852–3872, doi: 10.1016/j.cell.2021.06.024 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Feng XH & Derynck R Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 394, 909–913, doi: 10.1038/29814 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Hoodless PA et al. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell 85, 489–500, doi: 10.1016/s0092-8674(00)81250-7 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Macias-Silva M et al. MADR2 is a substrate of the TGFbeta receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell 87, 1215–1224, doi: 10.1016/s0092-8674(00)81817-6 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Bujak M et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 116, 2127–2138, doi: 10.1161/CIRCULATIONAHA.107.704197 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Divakaran V et al. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ Heart Fail 2, 633–642, doi: 10.1161/CIRCHEARTFAILURE.108.823070 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khalil H et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 127, 3770–3783, doi: 10.1172/JCI94753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russo I et al. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ Res 124, 1214–1227, doi: 10.1161/CIRCRESAHA.118.314438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong P et al. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 137, 707–724, doi: 10.1161/CIRCULATIONAHA.117.029622 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang S et al. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J Mol Cell Cardiol 132, 84–97, doi: 10.1016/j.yjmcc.2019.05.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu X et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest 128, 2127–2143, doi: 10.1172/JCI98215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su J et al. TGF-beta orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 577, 566–571, doi: 10.1038/s41586-019-1897-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Hattab DS et al. Scleraxis regulates Twist1 and Snai1 expression in the epithelial-to-mesenchymal transition. Am J Physiol Heart Circ Physiol 315, H658–H668, doi: 10.1152/ajpheart.00092.2018 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Bagchi RA & Czubryt MP Synergistic roles of scleraxis and Smads in the regulation of collagen 1alpha2 gene expression. Biochim Biophys Acta 1823, 1936–1944, doi: 10.1016/j.bbamcr.2012.07.002 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Bugg D et al. MBNL1 drives dynamic transitions between fibroblasts and myofibroblasts in cardiac wound healing. Cell Stem Cell 29, 419–433 e410, doi: 10.1016/j.stem.2022.01.012 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivey MJ et al. Resident fibroblast expansion during cardiac growth and remodeling. J Mol Cell Cardiol 114, 161–174, doi: 10.1016/j.yjmcc.2017.11.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li CY et al. Dual-specificity phosphatase 14 protects the heart from aortic banding-induced cardiac hypertrophy and dysfunction through inactivation of TAK1-P38MAPK/-JNK1/2 signaling pathway. Basic Res Cardiol 111, 19, doi: 10.1007/s00395-016-0536-7 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Molkentin JD et al. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 136, 549–561, doi: 10.1161/CIRCULATIONAHA.116.026238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burke RM et al. Prevention of Fibrosis and Pathological Cardiac Remodeling by Salinomycin. Circ Res 128, 1663–1678, doi: 10.1161/CIRCRESAHA.120.317791 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study reports the results of a high throughput screen that identified salinomycin as an anti-fibrotic compound. Salinomycin has potent anti-fibrotic activity in mouse models of ischemic and non-ischemic cardiac remodeling and in cardiac fibroblasts obtained from heart failure patients, in part via inhibiting TGF-beta / p38 signaling.
- 25.Wang B et al. Decreased Smad 7 expression contributes to cardiac fibrosis in the infarcted rat heart. Am J Physiol Heart Circ Physiol 282, H1685–1696, doi: 10.1152/ajpheart.00266.2001 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Yu H et al. Overexpression of Smad7 suppressed ROS/MMP9-dependent collagen synthesis through regulation of heme oxygenase-1. Mol Biol Rep 40, 5307–5314, doi: 10.1007/s11033-013-2631-2 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Wei LH et al. Smad7 inhibits angiotensin II-induced hypertensive cardiac remodelling. Cardiovasc Res 99, 665–673, doi: 10.1093/cvr/cvt151 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Humeres C et al. Smad7 effects on TGF-beta and Erbb2 restrain myofibroblast activation, and protect from post-infarction heart failure. J Clin Invest, doi: 10.1172/JCI146926 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study provides further evidence that inhibitory Smads block fibroblast activation. Smad7 blocked canonical TGF-beta signaling as expected, and also prevented Erbb2 activity and fibroblast activation in a TGF-beta indepenedent manner.
- 29.Bugyei-Twum A et al. Sirtuin 1 activation attenuates cardiac fibrosis in a rodent pressure overload model by modifying Smad2/3 transactivation. Cardiovasc Res 114, 1629–1641, doi: 10.1093/cvr/cvy131 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu JW et al. Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-beta signaling. Cell 111, 357–367, doi: 10.1016/s0092-8674(02)01006-1 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Cunnington RH et al. Antifibrotic properties of c-Ski and its regulation of cardiac myofibroblast phenotype and contractility. Am J Physiol Cell Physiol 300, C176–186, doi: 10.1152/ajpcell.00050.2010 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Wang J et al. The Role of c-SKI in Regulation of TGFbeta-Induced Human Cardiac Fibroblast Proliferation and ECM Protein Expression. J Cell Biochem 118, 1911–1920, doi: 10.1002/jcb.25935 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Bisserier M et al. AAV1.SERCA2a Gene Therapy Reverses Pulmonary Fibrosis by Blocking the STAT3/FOXM1 Pathway and Promoting the SNON/SKI Axis. Mol Ther 28, 394–410, doi: 10.1016/j.ymthe.2019.11.027 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker CJ et al. Nuclear mechanosensing drives chromatin remodelling in persistently activated fibroblasts. Nat Biomed Eng, doi: 10.1038/s41551-021-00709-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study describes the role of mechanical tension in controlling aortic valve fibroblast behavior through alterations in nuclear architecture. Persistent strain on the nuclear membrane alters chromatin condensation and facilitates the myofibroblast phenotype.
- 35.Tomasek JJ, McRae J, Owens GK & Haaksma CJ Regulation of alpha-smooth muscle actin expression in granulation tissue myofibroblasts is dependent on the intronic CArG element and the transforming growth factor-beta1 control element. Am J Pathol 166, 1343–1351, doi: 10.1016/s0002-9440(10)62353-x (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomasek JJ et al. Contraction of myofibroblasts in granulation tissue is dependent on Rho/Rho kinase/myosin light chain phosphatase activity. Wound Repair Regen 14, 313–320, doi: 10.1111/j.1743-6109.2006.00126.x (2006). [DOI] [PubMed] [Google Scholar]
- 37.Small EM et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res 107, 294–304, doi: 10.1161/CIRCRESAHA.110.223172 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sisson TH et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am J Pathol 185, 969–986, doi: 10.1016/j.ajpath.2014.12.005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Velasquez LS et al. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc Natl Acad Sci U S A 110, 16850–16855, doi: 10.1073/pnas.1316764110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahaman SO et al. TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis in mice. J Clin Invest 124, 5225–5238, doi: 10.1172/JCI75331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis J, Burr AR, Davis GF, Birnbaumer L & Molkentin JD A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell 23, 705–715, doi: 10.1016/j.devcel.2012.08.017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eguchi A et al. GRK5 is a regulator of fibroblast activation and cardiac fibrosis. Proc Natl Acad Sci U S A 118, doi: 10.1073/pnas.2012854118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dupont S et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183, doi: 10.1038/nature10137 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Liu F et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 308, L344–357, doi: 10.1152/ajplung.00300.2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ligresti G et al. CBX5/G9a/H3K9me-mediated gene repression is essential to fibroblast activation during lung fibrosis. JCI Insight 5, doi: 10.1172/jci.insight.127111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao Y et al. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev 33, 1491–1505, doi: 10.1101/gad.329763.119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szeto SG et al. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J Am Soc Nephrol 27, 3117–3128, doi: 10.1681/ASN.2015050499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study further establishes the role of Hippo signaling in fibroblast activation and cardiac fibrosis. Here, in vitro and in vivo assays linked strain-dependent p38 signaling to YAP-TEAD dependent fibroblast activation.
- 48.Bugg D et al. Infarct Collagen Topography Regulates Fibroblast Fate via p38-Yes-Associated Protein Transcriptional Enhanced Associate Domain Signals. Circ Res 127, 1306–1322, doi: 10.1161/CIRCRESAHA.119.316162 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moreno-Vicente R et al. Caveolin-1 Modulates Mechanotransduction Responses to Substrate Stiffness through Actin-Dependent Control of YAP. Cell Rep 25, 1622–1635 e1626, doi: 10.1016/j.celrep.2018.10.024 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim T et al. MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J 36, 520–535, doi: 10.15252/embj.201695137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu OM, Miyamoto S & Brown JH Myocardin-Related Transcription Factor A and Yes-Associated Protein Exert Dual Control in G Protein-Coupled Receptor- and RhoA-Mediated Transcriptional Regulation and Cell Proliferation. Mol Cell Biol 36, 39–49, doi: 10.1128/MCB.00772-15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Francisco J et al. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl Sci 5, 931–945, doi: 10.1016/j.jacbts.2020.07.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Speight P, Kofler M, Szaszi K & Kapus A Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFbeta-regulated Smad3. Nat Commun 7, 11642, doi: 10.1038/ncomms11642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balestrini JL, Chaudhry S, Sarrazy V, Koehler A & Hinz B The mechanical memory of lung myofibroblasts. Integr Biol (Camb) 4, 410–421, doi: 10.1039/c2ib00149g (2012). [DOI] [PubMed] [Google Scholar]
- 55.Lighthouse JK et al. Exercise promotes a cardioprotective gene program in resident cardiac fibroblasts. JCI Insight 4, doi: 10.1172/jci.insight.92098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pu W et al. A genetic system for tissue-specific inhibition of cell proliferation. Development 147, doi: 10.1242/dev.183830 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Burke RM et al. Small proline-rich protein 2B drives stress-dependent p53 degradation and fibroblast proliferation in heart failure. Proc Natl Acad Sci U S A 115, E3436–E3445, doi: 10.1073/pnas.1717423115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teekakirikul P et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest 120, 3520–3529, doi: 10.1172/JCI42028 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janbandhu V et al. Hif-1a suppresses ROS-induced proliferation of cardiac fibroblasts following myocardial infarction. Cell Stem Cell, doi: 10.1016/j.stem.2021.10.009 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study describes a nuanced role of hypoxia and metabolic reprogramming in fibroblast phenotypic plasticitye. The authors found that fibroblasts are surprisingly hypoxic during normal cardiac homeostasis; HIF1-alpha dependent transcriptional regulation facilitates buffering of mitochondrial ROS, preventing excessive fibroblast activation and the development of pathological scarring after MI.
- 60.Lombardi AA et al. Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat Commun 10, 4509, doi: 10.1038/s41467-019-12103-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li C et al. The landscape of accessible chromatin in quiescent cardiac fibroblasts and cardiac fibroblasts activated after myocardial infarction. Epigenetics, 1–20, doi: 10.1080/15592294.2021.1982158 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anand P et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 154, 569–582, doi: 10.1016/j.cell.2013.07.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Antolic A et al. BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy. JCI Insight 5, doi: 10.1172/jci.insight.138687 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duan Q et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med 9, doi: 10.1126/scitranslmed.aah5084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stratton MS et al. Dynamic Chromatin Targeting of BRD4 Stimulates Cardiac Fibroblast Activation. Circ Res 125, 662–677, doi: 10.1161/CIRCRESAHA.119.315125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alexanian M et al. A transcriptional switch governs fibroblast activation in heart disease. Nature 595, 438–443, doi: 10.1038/s41586-021-03674-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; **This study utilized the BRD4 inhibitor, JQ1, to interrogate fibroblast phenotypic plasticity in a mouse model of left ventricle pressure overload. The authors found that chromatin accessibility at pro-fibrotic gene loci is dynamic and reversible, and identified Meox1 as an important regulator of fibroblast activation.