

Abstract
Biofilm infection has a high prevalence in chronic wounds and can delay wound healing. Current treatments using repeated debridement and long-term antibiotic administration impose a significant burden on patients and healthcare systems. To address their limitations, we describe a highly efficacious electrical anti-biofilm system in this paper. Our system uses high-intensity current (75 mA cm−2) to completely debride biofilm above the wound surface and enhance antibiotic delivery and penetration into biofilm-infected wounds simultaneously. Combining these two effects, our system uses short treatments (≤2 h) to reduce bacterial count of MRSA biofilm-infected ex vivo skin wounds from over 1010 CFU g−1 to 105.2 CFU g−1. Taking advantage of the hydrogel ionic circuit design, our system enhances the in vivo safety of high-intensity current application compared to conventional devices. The in vivo anti-biofilm efficacy of our system is tested using a diabetic mouse-based wound infection model. MRSA biofilm bacterial count decreases from 109.0 CFU g−1 to 104.6 CFU g−1 at 1-day post-treatment and to 103.3 CFU g−1 at 7-day post-treatment, both of which are below the clinical threshold for infection. Overall, our novel technology provides a quick, safe, yet highly efficacious treatment to chronic wound biofilm infection and will facilitate wound healing process.
Keywords: biofilm, chronic wound infection, hydrogel ionic circuit, electrical debridement, iontophoretic antibiotic delivery
Graphical Abstract
1. Introduction
Chronic, non-healing wounds are currently affecting 1% of the world’s total population[1]. Chronic wounds can significantly affect patients’ quality of life and lead to a high rate of lower-extremity amputation[2]. The current clinical care for chronic wounds imposes a huge burden on both patients and healthcare systems[3]. One of the key contributing factors that lead to chronic wounds is bacterial biofilm infection[4], which exists in 78.2% of all chronic wounds[5]. These biofilms arrest wounds in a prolonged inflammatory phase and inhibit skin tissue regeneration[4, 6]. Due to the devastating effects of biofilm, reducing bacterial bioburden is considered a critical component in chronic wound care[1]. The latest consensus considers a bacterial count of 105 colony-forming units (CFU) per gram wound tissue a critical threshold for clinical infection and healing inhibition[7, 8]. If the bacterial bioburden is below this threshold, a wound can generally heal in otherwise healthy patients[8]. Above this threshold, the infection overwhelms the host immune system and stalls the wound healing process[7].
The current clinical standard of care for chronic wound biofilm infections includes debridement and antimicrobial treatment. Debridement physically reduces biofilms on chronic wounds[9]. However, bacterial burden is quickly restored within 48 hours after debridement[10, 11]. As a result, repeated debridement followed by long-term treatment with antimicrobial agents is currently practiced in clinics[1, 9, 12]. Besides limited efficacy, conventional debridement methods can cause several safety-related issues, including dispersing bacteria to deeper tissues[10], overaggressive resection of healthy tissues[8], and pain to patients[13]. Topical antibiotics are commonly administered for clinical wound care[14]. However, its efficacy against chronic wound biofilm infections is found to be very limited [15]. One reason is the high tolerance of biofilm bacteria to antibiotics. The antibiotic concentration required to kill biofilm bacteria can be 10 to 1,000 times higher than the concentration required to inhibit planktonic bacterial growth[16]. The second reason is that bacterial communities in chronic wound biofilms are encapsulated in a matrix of protective extracellular polymeric substances (EPS)[17], which presents a high resistance to antibiotic penetration. Multiple studies have demonstrated the decreased diffusion rate of antibiotics in various biofilms compare to their free aqueous diffusion[18]. A 2012 study showed the diffusion coefficient of vancomycin in S. aureus biofilms was as low as 0.2 μm2 s−1, which was more than 1,000 times slower than its diffusion in water[19].
In vivo chronic wound biofilms can be very thick. The thickness of mature P. aeruginosa biofilms can reach 200 μm above the wound surface in a rat model[20]. S. aureus biofilms of 150 μm thick have been observed on the wound surface in mouse[21] and pig[22] models. In vivo biofilms can also infiltrate the underlying wound tissues. Sarker et al. reported that P. aeruginosa biofilms penetrated up to 1,400 μm below the surface of rat burn wounds[20]. S. aureus biofilm was reported to penetrate up to 190 μm below the surface of an ex vivo human skin wound infection model[23]. Low antibiotic diffusion rates combined with high biofilm thicknesses lead to a long antibiotic diffusion time required to penetrate the entire thickness of biofilm-infected wound tissues. For example, 80.3 hours is required for vancomycin to diffuse through a S. aureus biofilm that has a thickness of 150 μm above the wound surface and infiltrates 190 μm below wound surface[19]. As a result of such a slow antibiotic penetration, some biofilm bacteria (e.g. those in deeper layers) may be exposed to a sub-lethal concentration of antibiotics for a long period of time, which give bacteria a chance to develop antibiotic tolerance[24].
Several new technologies have been explored to enhance antibiotic delivery into biofilm-infected wound tissues, but they all have their limitations. Although microneedle array can physically penetrates the EPS and reduce the diffusion distance of antibiotics [25, 26], it does not increase the antibiotic diffusion speed. As a result, long-term continuous application of microneedle array on wounds (24 to 48 hours[26, 27]) is required to achieve a good biofilm treatment efficacy. Both pharmacological and physical disruptions of biofilm can improve antibiotic penetration in biofilms[28]. However, the specificity of pharmacological biofilm-disruption compounds and the heterogeneity of clinical biofilms limit the applicability of these compounds[29]. Physical disruption (e.g. laser and ultrasound), on the other hand, can cause dissemination of biofilm bacteria and damage of host tissues[30].
Due to the limited efficacy of the current care of chronic wound biofilm infections, a next-generation biofilm treatment strategy is critically needed.
Electric current as a biofilm treatment modality has attracted a lot of interest due to its easy application, non-invasiveness and easy dose control. In the last 20 years, many studies have demonstrated that low-intensity direct current (DC) applied for a long duration (12 – 48 hours) was able to enhance the anti-biofilm efficacy of antibiotics in in vitro experiments, which was termed the “bioelectric effect”[31–33]. Combining the low-intensity current with antibiotics, the anti-biofilm efficacy was enhanced by 1–3 log10 scales[31–33], compared to antibiotic treatment alone. Pozo et al. applied a 2 mA current (0.17 mA cm−2) to S. aureus biofilms for 24 h in the presence of 32 μg mL−1 vancomycin. This treatment resulted in a biofilm bacterial density reduction of 1 log10 scale, while no antibacterial effect was observed when using vancomycin alone[34]. Similar bioelectric effect was observed when treating in vitro S. gordonii biofilms with a 0.4 mA cm−2 current in the presence of 2 μg mL−1 gentamicin for 24 h. After the treatment, the biofilm bacterial count was reduce by 4.3 log10 scales. When antibiotic was used alone, the bacteria count was only reduced by 0.8 log10 scale[31]. Although the working mechanism of bioelectric effect is still under investigation, enhanced antibiotic delivery by iontophoresis has been proposed as a key contributing factor[35]. Several studies have demonstrated that iontophoresis could increase the permeation rate of antibiotics in skin tissues[36, 37]. Datta et al. evaluated the transdermal iontophoresis of vancomycin. Their results showed that the amount of vancomycin delivered into the epidermis by iontophoresis (0.3 mA cm−2 current applied for 4 h) was 2.1 times higher than that delivered by passive diffusion[37]. In a 2006 study, Nicoli et al. demonstrated that transdermal iontophoresis (0.5 mA cm−2 current applied for 2 h) was able to increase the amount of amikacin delivered into the epidermis by 3 times compared to the amount delivered by passive diffusion[38]. Although the bioelectric effect is able to enhance the efficacy of antibiotics and reduce biofilm bacterial densities, a very long treatment duration (≥24 hours) is required due to the low current intensities (≤0.5 mA cm−2) used[31, 35]. Such a long treatment duration is not practical for clinical use and will negatively affect patients’ compliance. Increasing current intensity leads to a higher iontophoretic antibiotic delivery efficiency. So, the treatment duration can be reduced[32, 39, 40]. However, applying high-intensity currents using conventional electrical devices can induce significant pH changes and temperature increases at the device/tissue interface due to electrochemical reactions. These side effects can lead to severe tissue damage[41]. Besides the bioelectric effect, high-intensity pulsed electrical field has been reported to have an antibacterial effect. This effect is mainly attributed to the electroporation-induced irreversible damage to bacterial cell membranes. Electroporation also enhances the permeation of antimicrobial agents into bacterial cells. However, the high electrical energy used to treat biofilms has also been reported to cause mammalian cell membrane damage[40, 42], skin tissue injuries[43], and neuromuscular damage[44]. Joule heating induced by high electrical energy can also result in severe thermal damage to host tissues[45].
In this paper, we developed a novel high-intensity DC electric current-based biofilm treatment system. Our system tackled biofilm infections through two mechanisms that worked simultaneously and demonstrated enhanced safety compared to conventional devices. The working mechanisms of our system included: 1) efficacious electrical debridement to rapidly remove the biofilm above the wound surface; and 2) fast iontophoretic delivery of high-concentration antibiotics into biofilm and underlying wound tissues to minimize the chance of bacterial cells to adapt tolerance and rapidly reduce bacterial count. The high-intensity electric current for electrical debridement and antibiotic delivery were applied by our well-established hydrogel ionic circuit (HIC) principle[46, 47].
In this work, we first demonstrated that our biofilm treatment system was able to apply up to 75 mA cm−2 current intensity (150 times higher than the maximal safe current intensity used by conventional transdermal iontophoresis) to ex vivo (porcine) skin tissues without causing significant pH and temperature changes. The efficacy of high-intensity electrical debridement and iontophoretic antibiotic delivery of our biofilm treatment system were then characterized separately using a porcine skin-based wound model infected with methicillin-resistant S. aureus (MRSA) biofilms. By combining these two mechanisms, we demonstrated that a 65-minute to 2-hour treatment using our system reduced the bacterial count of MRSA biofilm-infected porcine skin wounds from over 1010 CFU g−1 to 105 CFU g−1 at 24 hours post treatment and then to 103.8 CFU g−1 at 4 days post treatment. 103.8 CFU g−1 is below the threshold for clinical wound infection and is considered not inhibitory to wound healing. Afterwards, the in vivo safety of high-intensity currents applied by our system was studied using a healthy mouse model. The highly efficacious biofilm treatment of our system was finally studied in vivo using a diabetic mouse-based wound model infected with MRSA biofilms. The in vivo anti-biofilm efficacy was consistent with the ex vivo test results. Overall, by rapidly reducing biofilm bioburden to below the clinical infection threshold, our novel technology will be able to resume the normal healing process in chronic wounds.
2. Design and working mechanism of an HIC-based electrical biofilm treatment system
One challenge of conventional electrical biofilm treatment devices is the significant pH change at device/tissue interface when high-intensity current is applied. Current electrical devices typically conduct electron currents. They have to be converted to ion currents at the device/tissue interface via electrochemical (EC) reactions, which decompose water molecules. The water electrolysis generates hydrogen ions on anode and hydroxide ions on cathode. When high-intensity currents are applied, these ions cannot be sufficiently buffered by conventional electrical devices due to the limited pH buffering capacity of their buffering systems (e.g. PBS, which contains only 12 mM phosphate ions). The accumulated acidic and alkaline concentrations result in chemical burns of skin tissues. Non-polarizable electrodes (e.g. silver/silver chloride) are capable of transferring charges without splitting water molecules[48]. However, if the current intensity exceeds their charge transfer capacity, water decomposition can still be triggered to cause significant pH changes[49]. Another problem of conventional electrical devices is the thermal effect during high-intensity current application. Water molecule decomposition requires 1.48 V DC potential, but the voltage across the electrode/media interface is usually higher than 1.48 V and it increases with increasing current intensity. The excess voltage (i.e. electrode over-potential) can results in temperature increase on tissue surface[50]. In addition, the Joule heating generated by electric current conduction can also contribute to thermal damage to skin tissues.
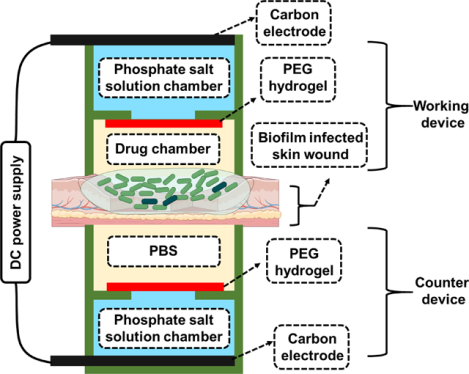
To overcome the problems induced by conventional electrical devices when applying high-intensity currents, we developed a hydrogel ionic circuit (HIC)-based system for combating bacterial infection. Figure 1 showed the design of our HIC-based electrical biofilm treatment system. Our biofilm treatment system consisted of a working device attached to the biofilm-infected wound and a counter device attached to the back side of the skin tissue. The working device could be either anode or cathode depending on the polarity of the iontophoresis required to deliver the antibiotics. Each device had four components: carbon electrode, phosphate salt solution chamber, polyethylene glycol (PEG) hydrogel membrane and drug chamber/phosphate-buffered saline (PBS) chamber. The carbon electrode was inserted in the phosphate salt solution chamber, which was filled with a high-concentration mixture of sodium dihydrogen phosphate and disodium phosphate solutions. The phsophate salt solution chamber was separated by the PEG hydrogel membrane from the drug chamber in the working device or the PBS chamber in the counter device. The drug chamber was filled with antibiotic solution in the working device. The drug/PBS chamber was in direct contact with the skin or the wound to allow current conduction and drug delivery. The carbon electrode of the working and the counter device was connected to the positive and negative output of a DC power supply, respectively, to complete the circuit.
Figure 1.
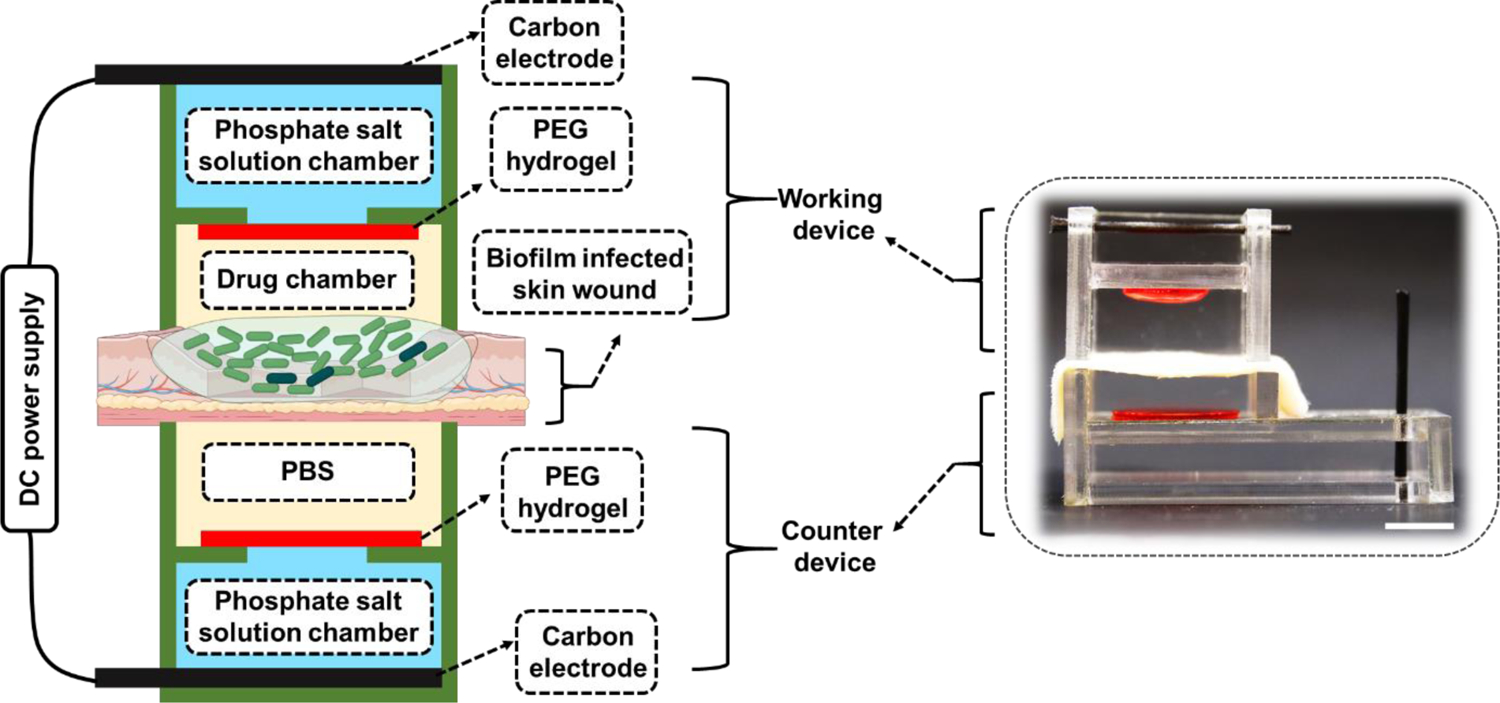
General design of HIC-based electrical biofilm treatment system for combating chronic wound bacterial infections (Created with BioRender.com). The inset shows a digital camera image of the actual system (scale bar: 10 mm). The actual counter device shown here had a slightly different design than the working device to allow easy and secure placement of the skin/wound tissue and easy insertion of the carbon electrode.
During biofilm treatment, direct currents (DC) was applied to our working and counter devices through carbon electrodes. The hydrogen/hydroxide ions generated by EC reaction were neutralized in our system by the high-concentration phosphate salt ions in the phosphate salt solution chambers (contained 600 to 1080 mM phosphate salt ions)[46, 51]. The EC reaction-induced heat was absorbed by the high water content in our devices[51]. In addition, the Joule heat produced by current conduction was minimized due to the high electrical conductivity of our high-concentration phosphate salt solutions[51]. These allowed us to apply current intensities that were significantly higher than the safe current intensity used by conventional electrical devices (e.g. 0.5 mA cm−2 is typically applied by conventional transdermal iontophoresis[52]) without causing tissue damage. The PEG hydrogel formed a unique aqueous two-phase separation (ATPS) with the high-concentration phosphate salt solutions[46]. The ATPS minimized the diffusion of phosphate salt ions to the drug chamber to avoid osmolarity changes in the drug chamber and tissues[46]. The ion currents were transmitted to the drug chamber through the PEG hydrogel and then to the biofilm-infected wound tissue. In the drug chamber, the high-intensity ion current mobilized the antibiotics and iontophoretically delivered them into the biofilm and the underlying wound tissue with a high permeation rate. In the wound tissue, the high-intensity ion current electrically debrided the biofilm.
3. In vitro and ex vivo safety evaluation of high-intensity current applied by HIC-based electrical biofilm treatment system
In this section, we evaluated the in vitro and ex vivo safety of our HIC-based biofilm treatment system when applying a high current intensity of 75 mA cm−2 for 1 h. This current intensity is 150 times higher than the maximal safe current (0.5 mA cm−2) used by conventional transdermal iontophoresis[52]. We first confirmed that the PEG hydrogel in our HIC-based system retained its ability to maintain ATPS and to minimize the diffusion of high-concentration phosphate salt ions out from the phosphate salt solution chamber after 75 mA cm−2 current application for 1 h (Figure S1 and S2 in Supporting information, method described in Section 12.4). We then measured the temperature and pH changes induced by 75 mA cm−2 current application using fetal porcine skins (Figure 2A). In this test, the drug chamber of our HIC-based devices was filled with PBS. A conventional electrical device, constructed by directly inserting a carbon electrode in a PBS-filled drug chamber, was tested as a comparison (Figure 2B). This conventional working device used the same carbon rod electrode, and drug chamber design and dimension as our HIC-based device. This allowed us to determine the effect of our HIC design only on safety as the single variable. This conventional device design was consistent with the transdermal iontophoresis and electrical stimulation device design used in the most recently published literature[53]. During the 1 h current application, we monitored the surface temperature of the skin in contact with the working device, which was anode in our test. Based on our previous experience, HIC-based anode device always generated more heat than HIC-based cathode device[46], so it represented the worst case. The average peak skin surface temperature treated by our system was 42.5±0.32 °C, which was lower than the maximal safe temperature (43 °C) that skin can tolerate[54]. In contrast, the conventional device increased skin surface temperature up to 67.0±5.29 °C (Figure 2C), which was considerably higher than the safe temperature threshold. We also measured the pH in the drug chambers and on the surface of the skin in contact with the device immediately after current application. When HIC-based devices were used, the pH in both locations remained between 6.5 and 8.5, which were considered safe for skin tissues[55]. The conventional device, however, changed the pH to around 2 on the anode side and around 12 on the cathode side (Figure 2D–E). The significant pH changes were owing to the hydrogen ions and hydroxide ions generated on anode and cathode, respectively, from EC reactions. These ions cannot be sufficiently neutralized by the pH-buffering system used in the conventional device (i.e. PBS) due to its low phosphate ion concentration (12 mM). Hence, these results demonstrated the ability of our HIC-based system to maintain a safe pH and temperature on skin surface during high-intensity current application to avoid thermal and chemical damage to skin tissues.
Figure 2.
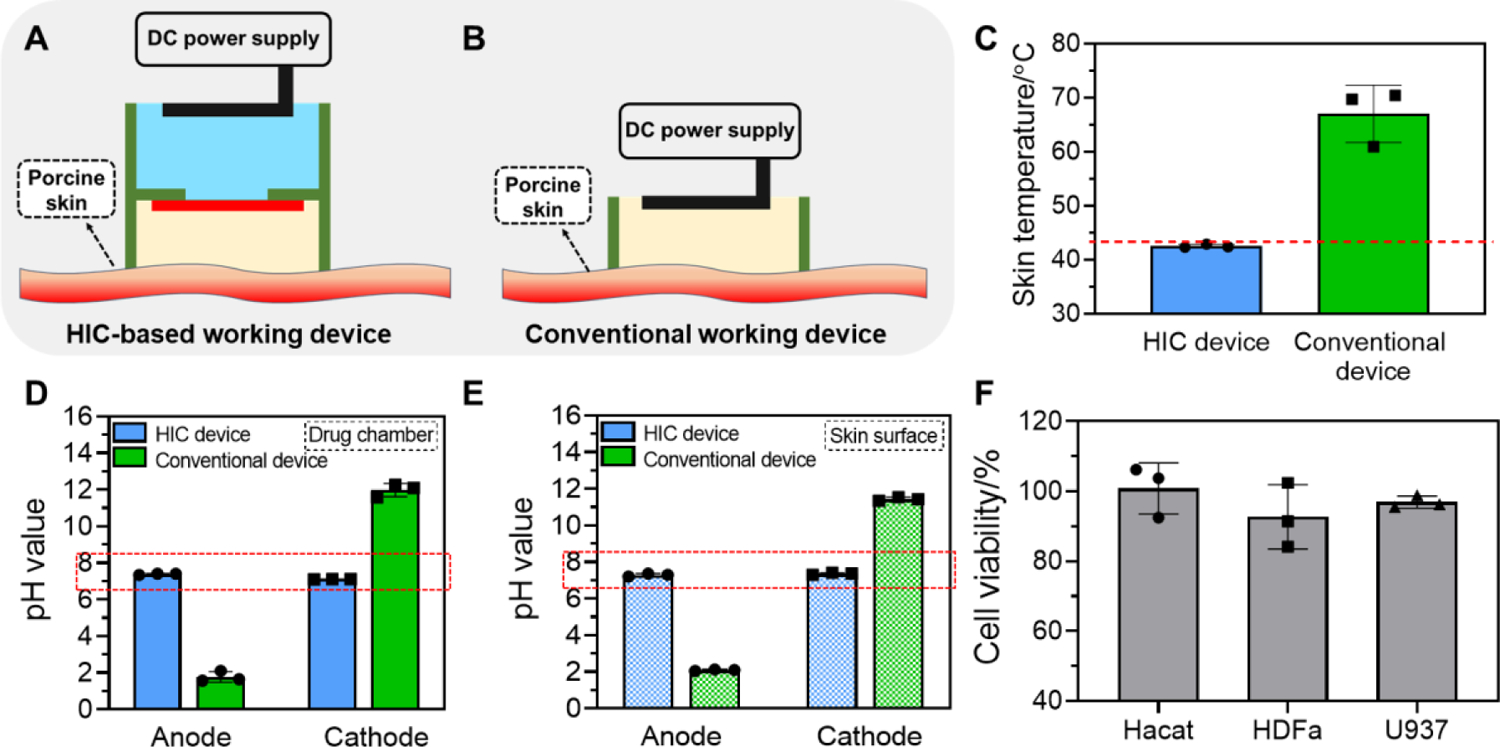
In vitro and ex vivo safety tests of our biofilm treatment system applying a high-intensity current. (A-B) Schematic of the ex vivo safety test setup. (A) HIC-based biofilm treatment working device. (B) Conventional electrical working device. (C) The peak temperature on the skin surface treated with 75 mA cm−2 current for 1 h applied by our HIC-based system or a conventional device. Red dashed line showed 43°C. (D-E) pH changes after 75 mA cm−2 current application for 1 h by our HIC-based device or a conventional device. (D) pH in drug chambers (filled with PBS). (E) pH on skin surface. Red dashed box in (D) and (E) showed pH range of 6.5 to 8.5, which was considered safe for skin tissues. (F) The viability (normalized to untreated control) of human keratinocyte (HaCaT), human primary dermal fibroblast cells (HDFa), and human monocyte (U937) after 75 mA cm−2 current treatment for 1 h applied by HIC-based system.
Since our system did not cause significant pH and temperature changes when applying high-intensity current to tissues, we then evaluated the cytotoxicity of high-intensity current itself applied by our system. Here, we tested the viability of in vitro cultured wound healing-related cells after being exposed to 75 mA cm−2 current applied by our HIC-based system for 1 h. Human keratinocyte (HaCaT), human primary dermal fibroblast cells (HDFa), and human monocyte (U937) were tested. Our result showed that treatment with 75 mA cm−2 current for 1 h had minimal impact on the viability of these cells (Figure 2F), which provided further evidence for the safety of our HIC-based biofilm treatment system.
4. Ex vivo electrical debridement of biofilm induced by the high-intensity electrical biofilm treatment system
Our goal in this section was to determine the high-intensity electrical biofilm debridement efficacy of our system using an ex vivo MRSA biofilm-infected porcine skin wound. Porcine skin has a high similarity to human skin and is recommended by the Wound Healing Society for pre-clinical studies[6, 56]. Biofilm cultured on ex vivo porcine skin wound is expected to provide a more accurate assessment of the biofilm treatment efficacy of our system than conventional in vitro biofilm models cultured on abiotic substrates. An excisional skin wound was created by a 6-mm biopsy punch down to the dermis layer. The wound was then inoculated with 20 μL of 1×108 CFU mL−1 MRSA bacteria solution. The bacteria were allowed to grow for 48 h to form a mature biofilm[57]. Our HIC-based biofilm treatment system was then used to apply current to the infected wound for 1 h. The drug chamber of our system was loaded with PBS in this test. Different current intensities, including 0 mA cm−2 (untreated control), 0.5 mA cm−2, 19 mA cm−2, 38 mA cm−2, and 75 mA cm−2, were tested. Only electric current was applied and no antibiotic was used in this study.
After current application, we first evaluated the biofilm structure. As showed in Figure 3A, when no current was applied, a thick biofilm could be seen on the surface of the wound. The thickness of this biofilm was 149.8±27.81 μm (Figure 3B), which was similar to the reported thickness of in vivo chronic wound MRSA biofilms[20–22, 58], indicating the formation of a mature biofilm. The thickness of the MRSA biofilm was reduced to 55.3±18.96 μm and 58.6±12.72 μm after low current intensity treatments at 0.5 mA cm−2 and 19 mA cm−2, respectively. When the current intensity further increased to 38 mA cm−2 and 75 mA cm−2, more biofilm thickness reduction was observed. The biofilm thickness was reduced to 14.6±12.72 μm after 38 mA cm−2 treatment, and no discernible biofilm could be observed on wound surface under microscope after 75 mA cm−2 treatment. These results highlighted the structural damage to biofilm induced by high-intensity current application.
Figure 3.
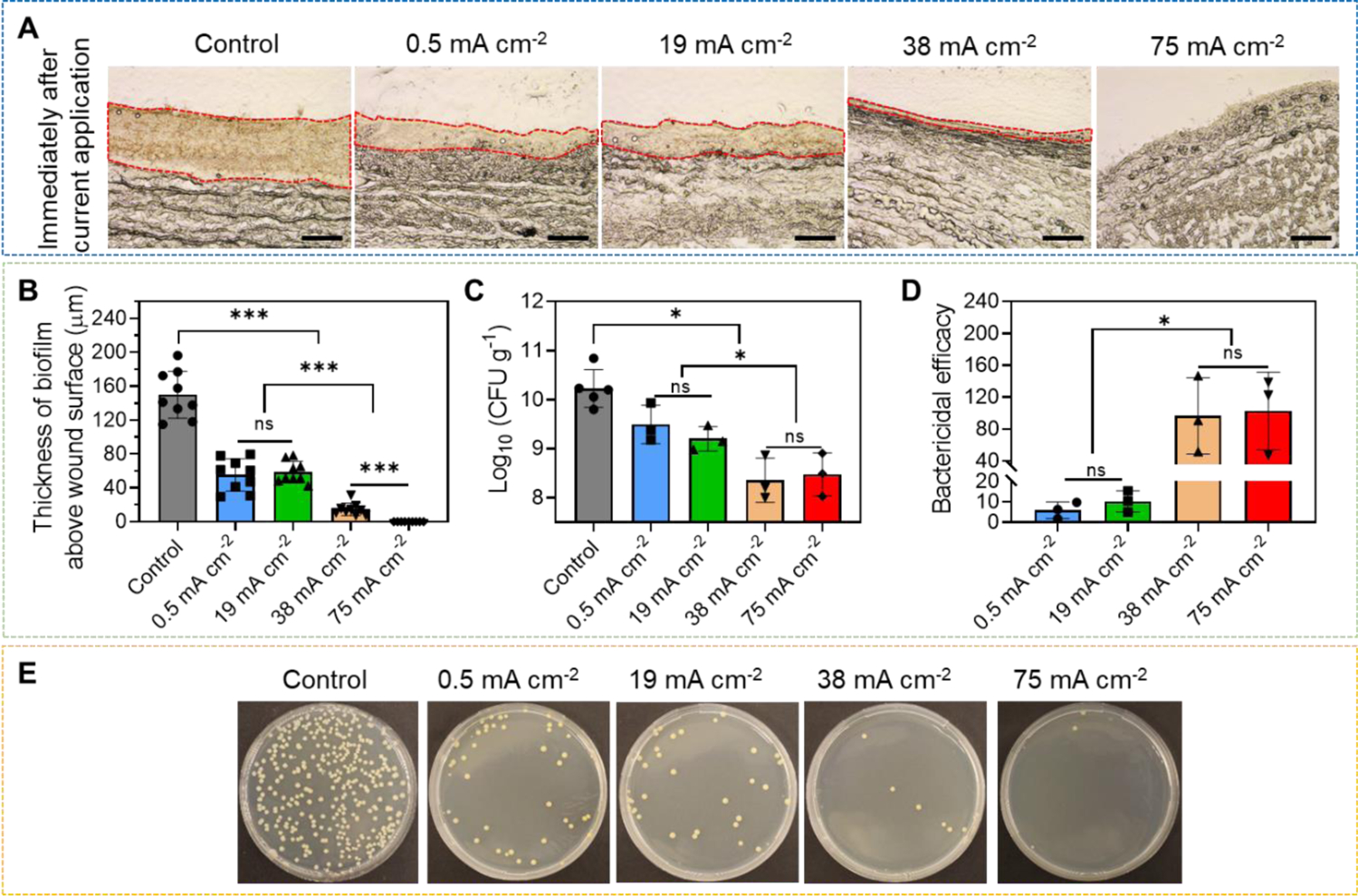
Ex vivo electrical biofilm debridement by high-intensity current applied by our HIC-based biofilm treatment system. (A) Representative cryo-section images of MRSA biofilm-infected porcine skin wounds after current application at different intensities for 1 h. All tissue samples were collected and embedded immediately after treatment. Red dashed area showed the biofilm above wound surface. Scale bar: 100 μm. (B) Biofilm thickness above wound surface measured immediately after treatment. (C) MRSA bacterial count (log10 scale) in infected skin wounds immediately after treatment measured by standard plate counting assay. (D) Bactericidal efficacy immediately after treatment. Bactericidal efficacy was calculated as initial bacterial count divided by bacterial count after treatment. (E) Representative photographs of bacterial culture from MRSA biofilm-infected porcine skin wound tissues immediately after treatment. All tissue homogenates were diluted 106 times with PBS before plating.
The biofilm debridement effect of low-intensity electrical current treatment has been reported previously[59]. The main mechanism of electrical debridement was attributed to physical biofilm disruption and detachment caused by the current-induced electrostatic force that pulled biofilm bacteria away from their substrate[60]. For example, Hong et al. demonstrated that 15 μA cm−2 current applied for 60 min induced 80% detachment of P. aeruginosa biofilm from a glass surface[61]. In another study, van der Borden et al. observed 78% detachment of S. epidermidis biofilm and 54% detachment of S. aureus biofilm from surgical stainless steel after applying 15–125 μA current for 2.5 h[60]. In addition to physical biofilm disruption and detachment, it has also been proposed that the adverse effects of electrochemical reactions may cause bacterial cell death[62]. However, there is no consensus on the contribution of this effect, as some studies reported no bacterial killing induced by electric current alone[35, 63]. To investigate this, we collected the solution in the drug chamber of our working device, which contained the debrided materials, immediately after the high-intensity electrical debridement and measured the bacterial density. We found the density of viable bacteria in the collected solution was similar to the bacterial count in the untreated control biofilm (Figure S3, Supporting information). This result suggested that the high-intensity current applied by our system had no bacterial killing effect. To further verify this result, we also performed an in vitro experiment by applying 75 mA cm−2 current to planktonic MRSA bacteria for 60 min using our system. The number of viable bacteria was counted before, immediately after and at 24 h after our treatment (Figure S4, Supporting information). No significant difference in viable bacterial count was observed among these three time-points, further confirming the lack of bacterial killing by the high-intensity current applied by our system. This result was also consistent with our in vitro and ex vivo safety study which showed that our system could effectively minimize pH and temperature changes induced by high-intensity current application and damage to mammalian cells.
Next, we quantitatively measured the bacterial count remaining in the wound tissue immediately after current application at different intensities using the standard plate counting assay[64] (Figure 3C and E). The untreated wound sample had a MRSA bacterial count of 1010.3 CFU g−1. The bacterial count was reduced to 109.3 CFU g−1 and 109.2 CFU g−1 after low-intensity current application at 0.5 mA cm−2 and 19 mA cm−2, respectively. When high current intensities of 38 mA cm−2 and 75 mA cm−2 were used, the bacterial count was further reduced to 108.4 CFU g−1 and 108.5 CFU g−1, respectively.
The bactericidal efficacy, defined as initial bacterial count divided by remaining bacterial count after treatment, was 5.9±4.05, 10.2±5.2, 96.6±47.84 and 102.7±48.82 times for 0.5 mA cm−2, 19 mA cm−2, 38 mA cm−2 and 75 mA cm−2 treatment, respectively (Figure 3D). The bactericidal efficacy at 38 mA cm−2 and 75 mA cm−2 was equivalent to a 99% bactericidal rate. Our biofilm thickness and bacterial count measurements showed that 75 mA cm−2 applied for 1 h by our biofilm treatment system effectively destroyed mature MRSA biofilm above wound surface and removed a majority (>99%) of the bacteria.
Although our system achieved a biofilm debridement efficacy that was better than the conventional low current intensity method[32], our system did not prevent biofilm reformation similar to conventional debridement methods (Figure S5A, Supporting information). The biofilm thickness recovered to 79.0±20.54 μm and 91.1±14.66 μm at 24 h after electrical debridement at 0.5 mA cm−2 and 19 mA cm−2, respectively (Figure S5B, Supporting information). The restoration of biofilm was also observed in high current intensity treatment groups. The biofilm thickness was recovered to 43.4±13.19 μm and 28.9±7.99 μm at 24 h after electrical debridement at 38 mA cm−2 and 75 mA cm−2, respectively. The restoration of biofilm was further evidenced by bacterial count, which was above 109.1 CFU g−1 in all groups at 24 h after electrical debridement (Figure S5C, supporting information). Accordingly, the bactericidal efficacy was decreased (Figure S5D, Supporting information) compared to that immediately after electrical debridement. These results suggested that although high-intensity currents alone was able to quickly debride mature biofilm, it did not prevent biofilm recovery after treatment.
5. High-intensity iontophoretic delivery of vancomycin (VAN)
In this section, we aimed to determine the iontophoretic antibiotic delivery efficiency of our electrical biofilm treatment system using the high-intensity current. We performed ex vivo antibiotic delivery study using the same MRSA biofilm-infected porcine skin wound model as in the previous section. Vancomycin (VAN) was used here as a model drug, since it is the FDA-approved antibiotic and is the clinical gold standard for combating S. aureus infections[65].
We first tested the VAN delivery efficiency at different current intensities (from 0 mA cm−2 (passive diffusion) to 75 mA cm−2) applied by our biofilm treatment system for 1 h. The drug chamber of the working device was loaded with 1 mg mL−1 VAN in this study. The anode side was used as the working device because VAN required anodal iontophoresis. The accumulated concentration of VAN in biofilm infected skin wound were measured immediately after iontophoresis using high performance liquid chromatography (HPLC). As shown in Figure 4A, when 38 mA cm−2 was used, the accumulated concentration of VAN in wound tissue reached 2.5±0.32 mg g−1. When 75 mA cm−2 was used, the VAN concentration in wound tissue increased to 4.5±0.52 mg g−1. The VAN concentration in wound tissue after passive diffusion, 0.5 mA cm−2, and 19 mA cm−2 iontophoresis were also measured. However, they could not be distinguished from the background interference of the residual biofilm components, so no quantitative results were obtained (data not shown). Importantly, the VAN concentration delivered by 75 mA cm−2 iontophoresis for 1 h exceeded the minimal biofilm eradication concentration (MBEC) of VAN reported for MRSA biofilm, which was 4 mg g−1[66]. The ability to deliver a lethal antibiotic concentration into biofilm-infected wounds within a short period of time was critical for biofilm treatment as it would minimize the chance for bacteria to develop adaptive tolerance.
Figure 4.
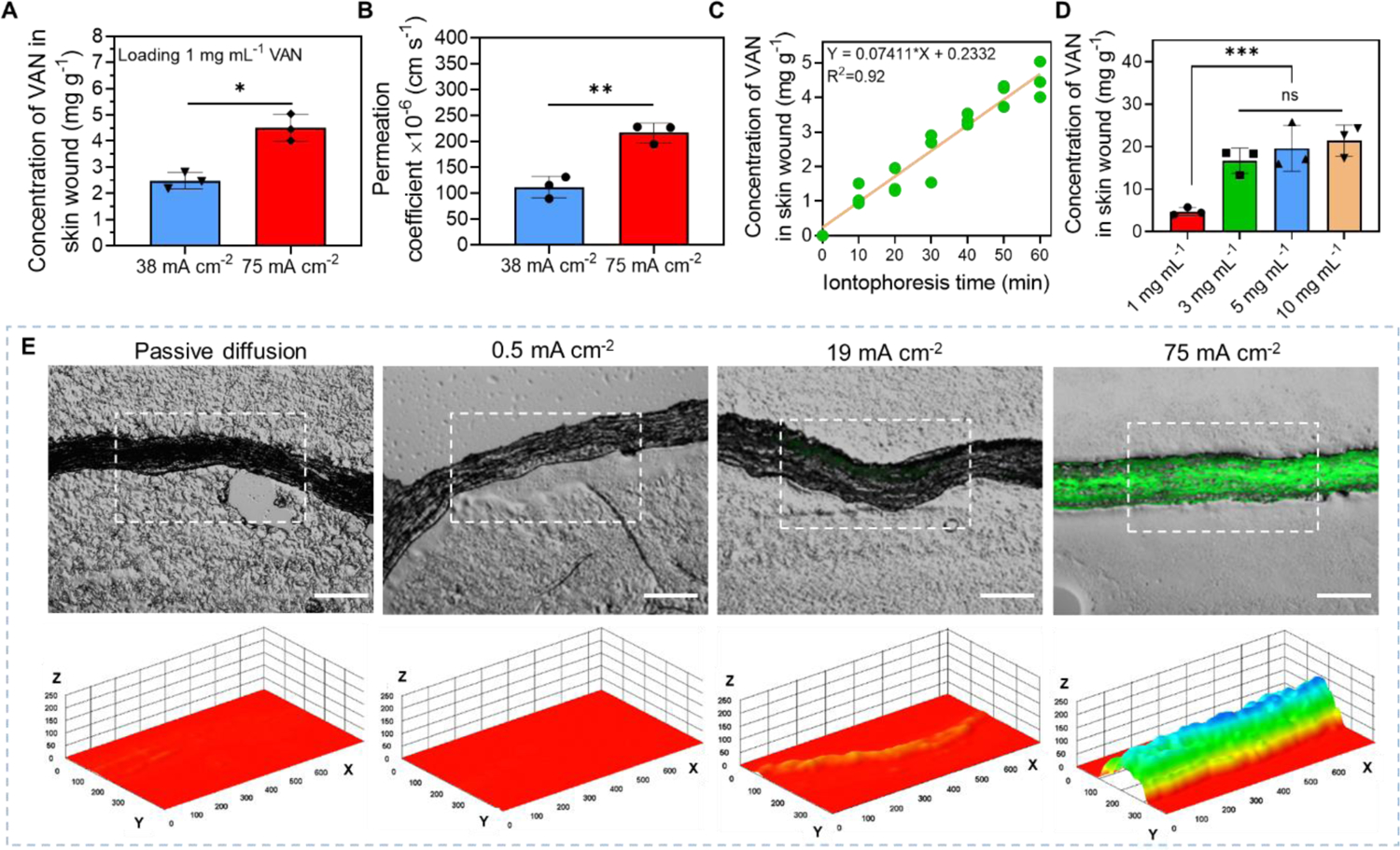
High-intensity iontophoretic delivery of vancomycin (VAN) into an ex vivo MRSA biofilm-infected porcine skin wound model using our electrical biofilm treatment system. (A) Accumulated concentration of VAN collected from skin wound tissues immediately after 1-h iontophoresis delivery at different current intensities. The drug chamber of our working device (anode) was loaded with 1 mg mL−1 VAN. (B) Permeation coefficient of VAN for 1-h iontophoretic delivery at different current intensities. (C) Accumulated concentration of VAN in biofilm-infected skin wound tissues as a function of iontophoresis time. The drug chamber of the working device (anode) was loaded with 1 mg mL−1 VAN and 75 mA cm−2 was applied. (D) Accumulated concentrations of VAN collected from biofilm-infected skin wound tissues with different loading concentrations of VAN in the drug chamber of the working device after 75 mA cm−2 current applied for 1 h. (E) Upper panel: Representative fluorescent images of cryo-sectioned skin wound samples after iontophoretic delivery of a fluorescently labeled dextran with a molecular weight of 4,000 Da (FD-4) at different current intensities for 1 h using our system (Scale bar: 200 μm). Lower panel: A three-dimensional illustration of the fluorescent intensity distribution in the skin tissue sample (in white dashed region) showed in the upper panel. Z-axis showed fluorescent intensity in random unit. X- and Y-axis had a unit of μm.
The VAN permeation coefficient (Pc) achieved by 38 mA cm−2 and 75 mA cm−2 iontophoresis were calculated (Figure 4B). At 38 mA cm−2, the Pc was 112.0±20.97 × 10−6 cm s−1. It increased to 216.5±18.96 × 10−6 cm s−1 when 75 mA cm−2 was used. This was a significant improvement from conventional low-intensity transdermal iontophoresis. A 2021 paper reported that a conventional transdermal iontophoresis using 0.31 mA cm−2 achieved a VAN Pc of 0.221 × 10−6 cm s−1 in excised porcine skin tissues[67]. This was 980 times lower than the VAN Pc achieved by our 75 mA cm−2 iontophoresis. This substantial increase in VAN Pc achieved by our system was attributed to the higher current intensity used[68] and the enhanced wound tissue permeability induced by the moderate wound temperature increase from our electric current application[69].
Next, we evaluated the progression of VAN accumulation in wound tissues at different time points during the 75 mA cm−2 1-h iontophoresis application (Figure 4C, Figure S6, Supporting information). The drug chamber of the working device was loaded with 1 mg mL−1 VAN in this test. Our results showed that VAN concentration increased almost linearly with time (R2=0.92).
Since iontophoretic drug delivery efficiency was highly dependent on drug loading concentration, we next tested the effect of VAN loading concentration on its iontophoretic delivery efficacy (using 75 mA cm−2 1 h) by loading the drug chamber with 1 mg mL−1, 3 mg mL−1, 5 mg mL−1, and 10 mg mL−1 VAN. The results showed that the VAN concentration in wound tissue was significantly increased to 16.7±2.96 mg g−1 when 3 mg mL−1 VAN was used (Figure 4D). However, there was no further increase in VAN tissue concentration that was statistically significant when higher VAN loading concentrations (5 mg mL−1, and 10 mg mL−1) were used. As a result, the calculated Pc for 5 mg mL−1, and 10 mg mL−1 loading concentrations was decreased compared to the Pc achieved by 1 mg mL−1, and 3 mg mL−1 loading concentrations (Figure S7, Supporting information).
Finally, we determined if our high-intensity iontophoresis was able to enhance the VAN penetration depth in wound tissues. A high penetration depth would allow effective treatment of bacteria in deeper layers of clinical biofilm infections to minimize biofilm reformation. We used a fluorescently labeled dextran (M.W. = 4 kDa, FD-4) to visualize drug distribution in wound tissue immediately after iontophoresis in this test. The hydrodynamic radius and net charge of FD-4 were similar to VAN, so FD-4 was expected to exhibit a similar iontophoretic behavior as VAN[70]. Figure 4E upper panel showed the distribution of FD-4 in wound tissue cross-sections after passive diffusion, 0.5 mA cm−2, 19 mA cm−2, and 75 mA cm−2 iontophoresis applied by our biofilm treatment system (loaded with 1 mg mL−1 FD-4 in drug chamber) for 1 h. All wound tissues were dehydrated by acetone before sectioning to minimize FD-4 mobility during sample processing. When passive diffusion and 0.5 mA cm−2 were applied, the fluorescence was indiscernible under microscope. When 19 mA cm−2 was applied, fluorescence could only be seen near the upper boundary of the wound tissue facing the working device during iontophoresis. In contrast, after 75 mA cm−2 iontophoresis for 1 h, FD-4 penetrated the entire thickness of the tissue sample, which was 955.7±192.56 μm thick before dehydration and 182.4±12.62 μm thick after dehydration. This penetration depth was higher than the typical thickness of in vivo S. aureus biofilm infections in skin wounds (150 μm above wound surface[21, 22] and 190 μm below wound surface[23]). Figure 4E lower panel showed the distribution of FD-4 reconstructed in a three-dimensional format with the Z-axis showing the fluorescent intensity. Our results in this section provided both quantitative and qualitative evidence to show that high-intensity iontophoresis applied by our biofilm treatment system not only increased drug permeation rate, but also enhanced drug penetration depth in wound tissues.
6. The combined anti-biofilm efficacy of high-intensity electrical debridement and iontophoretic VAN delivery
In previous sections, we demonstrated that high-intensity current application physically removed biofilm biomass and high-intensity iontophoresis significantly enhanced VAN delivery into wound tissues. In this section, our goal was to evaluate the anti-biofilm efficacy of our biofilm treatment system when combining these two effects.
Same as previous tests, an ex vivo MRSA biofilm-infected porcine skin wound model was used here. We first tested the biofilm treatment efficacy when different current intensities were used with a fixed VAN loading concentration at 1 mg mL−1. After a single treatment for 1 h, wound tissues were incubated for 24 h in a 37°C incubator to allow VAN to take effect[71]. After incubation, tissue samples were collected and evaluated for bacterial count using the standard plate counting assay for MRSA colony. As shown in Figure 5A and 5F, 1010.2 CFU g−1 MRSA were measured in control wound sample receiving no treatment, which indicated the formation of a mature biofilm. Treating the biofilm with no current for 1 h (only VAN diffusion) or a low current of 0.5 mA cm−2 for 1 h induced a minimal MRSA count reduction to 109.8 CFU g−1 and 109.6 CFU g−1, respectively. When 19 mA cm−2, 38 mA cm−2, and 75 mA cm−2 were applied, MRSA count was further reduced to 108.6 CFU g−1, 108.3 CFU g−1 and 107.6 CFU g−1, respectively (Figure 5A and 5F). We calculated the bactericidal efficacy of these treatments using different current intensities, which was shown in Figure 5B. A bactericidal efficacy of 2.7±2.06 times, 4.4±2.76 times, 57.8±50.30 times, and 81.4±13.34 times was achieved for passive diffusion, 0.5 mA cm−2, 19 mA cm−2, and 38 mA cm−2, respectively. 75 mA cm−2 significantly increased the bactericidal efficacy to 452.8±115.32 times, which was equivalent to a 99.8% bactericidal rate. Because 75 mA cm−2 had better anti-biofilm efficacy than other conditions, it was selected for our following tests.
Figure 5.
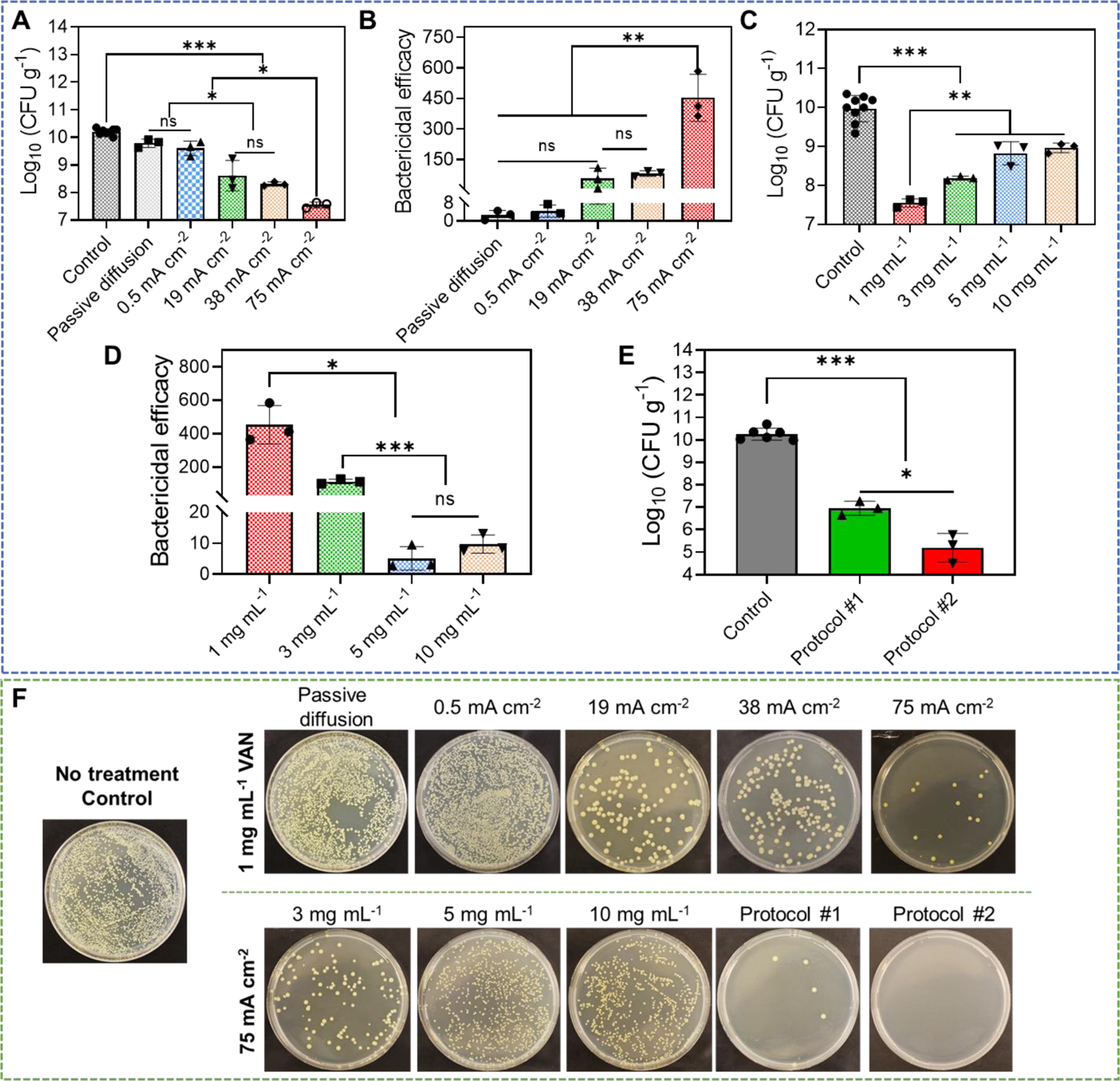
Anti-biofilm efficacy of our electrical biofilm treatment system combining the effects of high-intensity electrical debridement and iontophoretic VAN delivery. An ex vivo MRSA biofilm-infected porcine skin wound model was used in this study. (A) MRSA bacterial count (CFU g−1) in wound tissues measured at 24 h after biofilm treatment using our system. Treatment duration was 1 h. The drug chamber of the working device (anode) was loaded with 1 mg mL−1 VAN. Different current intensities were tested. Control received no treatment. Passive diffusion used 0 mA cm−2. (B) Bactericidal efficacy at 24 h after biofilm treatment calculated using data in (A). (C) MRSA bacterial count (CFU g−1) in wound tissues measured at 24 h after biofilm treatment using our system. Treatment duration was 1 h. The drug chamber of the working device (anode) was loaded with different concentrations of VAN. 75 mA cm−2 current intensity was tested. Control received no treatment. (D) Bactericidal efficacy at 24 h after biofilm treatment calculated using data in (C). (E) Anti-biofilm efficacy of repeated treatment protocols. Protocol #1 applied two treatments separated by 6 h. Protocol #2 applied two treatments separated by 24 h. Each treatment applied 75 mA cm−2 for 1 h using our biofilm treatment system. The drug chamber of the working device (anode) was loaded with 1 mg mL−1 VAN for all treatments. MRSA bacterial count (CFU g−1) in wound tissues was measured at 24 h after the last treatment. Control received no treatment. (F) Representative photographs of bacterial culture from wound tissues at 24 h after treatment (for single-treatment protocols) or after the last treatment (for repeated treatment protocols). All tissue homogenates were diluted 104 times with PBS before plating.
Next, we tried to study the biofilm treatment efficacy of our system using 75 mA cm−2 as the current intensity while varying the VAN loading concentration. A single treatment of 1 h was applied. At 24 h after the completion of the treatment, MRSA bacterial counts of wound tissues were measured. As shown in Figure 5C and 5F, the bacterial count for 3 mg mL−1, 5 mg mL−1, and 10 mg mL−1 loading concentrations were 108.2 CFU g−1, 108.8 CFU g−1, and 109.0 CFU g−1, respectively. They were significantly higher than the bacterial count in tissue treated with 1 mg mL−1 VAN loading concentration, which was 107.6 CFU g−1. The bactericidal efficacy was calculated and was found to decrease from 452.8±115.32 times (1 mg mL−1) to 112.6±13.58 times (3 mg mL−1), 5.1±3.80 times (5 mg mL−1) and 9.7±2.92 times (10 mg mL−1) (Figure 5D). Our results here showed that although a higher VAN loading concentration in our system increased the concentration of VAN delivered into the wound tissue (Figure 4D), it did not lead to a better biofilm treatment efficacy. This phenomenon was known as a paradoxical effect[72], defined as a decrease in antibacterial activity of an antibiotic at higher concentrations compared to lower concentrations. Jarrad et al. reported that VAN displayed the paradoxical effect when treating C.difficle (a Gram-positive bacteria), where the antibacterial activity started to decrease at 64 × MIC[73]. Our results in Figure 4D showed that VAN loading concentrations of 3 mg mL−1, 5 mg mL−1 and 10 mg mL−1 all delivered more than 16 mg g−1 VAN into the wound tissue when 75 mA cm−2 was applied. We therefore speculated that the concentration threshold for the paradoxical effect of VAN pertaining to MRSA biofilm treatment happened somewhere between 4.5 mg g−1 (VAN concentration delivered by 1 mg mL−1 loading concentration) and 16.7 mg g−1 (VAN concentration delivered by 3 mg mL−1 loading concentration). To test this hypothesis, we performed a study to investigate the anti-biofilm efficacy of vancomycin at different concentrations from 1 mg mL−1 to 20 mg mL−1 after passive incubation on MRSA biofilm-infected porcine skin wound for 24 h. Immediately after treatment, the bacterial count in skin wound was evaluated (Figure S8, Supporting information). The bacterial count was 108.7 and 108.2 CFU g−1 when 1 mg mL−1 and 4.5 mg mL−1 vancomycin were used, respectively. This result suggested that better anti-biofilm efficacy was achieved when increasing vancomycin concentration from 1 to 4.5 mg mL−1. However, further increasing vancomycin concentration to 10 mg mL−1 and 20 mg mL−1 led to worse anti-biofilm efficacies. The bacterial count was 108.9 CFU g−1 and 109.2 CFU g−1, for 10 mg mL−1 and 20 mg mL−1 vancomycin, respectively. These results provided further evidence to support to our threshold concentration hypothesis. Because 1 mg mL−1 VAN loading concentration had better anti-biofilm efficacy than other loading concentrations, it was selected for our following tests.
Although a single 1 h treatment using 75 mA cm−2 and 1 mg mL−1 VAN loading concentration reduced the MRSA bacterial count in wound tissue by 2.6 log10 scales, the final bacterial count (107.6 CFU g−1) was above the clinical threshold for infection (105 CFU g−1). Here, we sought to further improve the bactericidal efficacy by applying multiple treatments. Two protocols were tested. Protocol #1 applied two treatments separated by 6 h. Protocol #2 applied two treatments separated by 24 h. Each treatment applied 75 mA cm−2 for 1 h and used 1 mg mL−1 VAN loading concentration. MRSA bacterial count in wound tissues was measured at 24 h after the last treatment. As shown in Figure 5E and 5F, the bacterial count reduced to 107.0 CFU g−1 using protocol #1. When protocol #2 was used, bacterial count decreased to 105.2 CFU g−1. We also tested the biofilm treatment efficacy of 24 h passive diffusion of 1 mg mL−1 VAN, which mimicked the conventional long-term topical antibiotic administration used to treat chronic wound biofilm infections. 24 h VAN diffusion only reduced bacterial count to 108.7 CFU g−1 measured at 24 h after treatment (Figure S9, Supporting information), which was inferior to both of our single and repeated treatment protocols. When we incubated protocol #2-treated wound samples for 4 days after treatment (instead of 24 h), the number of MRSA colonies was further reduced to 103.8 CFU g−1 (Figure S10, Supporting information). This was below the threshold for clinical infection and thus was considered not inhibitory to normal wound healing[7, 8]. It is worth noting that the viability of skin cells was not affected by 4.5 mg mL−1 VAN (the VAN concentration delivered to wound tissues by our system) after 48-h incubation (Figure S11, Supporting information), suggesting a good cytocompatibility of the high VAN concentrations delivered by our system.
Our findings in this section demonstrated the therapeutic efficacy of our high-current intensity biofilm treatment system that used a 2-hour treatment in total (protocol #2) to reduce the bacterial count in MRSA biofilm-infected porcine skin wound tissues from 1010.2 CFU g−1 before treatment to 105.2 CFU g−1 at 24 hour after treatment and 103.8 CFU g−1 at 4-day after treatment.
7. Further enhancement of anti-biofilm efficacy by combining high-intensity current with daptomycin (DAP)
Although VAN is the standard treatment for clinical S. aureus infections, newly developed antibiotics have shown better treatment efficacy. Daptomycin (DAP), a cyclic lipopeptide antibiotic, was recently approved by FDA in 2003 to treat serious infections caused by Gram-positive bacteria[74]. Because the mode of action of DAP is less reliant on the metabolic activities of bacteria, it is considered more effective in killing metabolically inactive bacteria than conventional antibiotics, such as VAN[75]. This was corroborated by our bacteria killing studies using planktonic MRSA in stationary phase (Figure S12, Supporting information). Our study showed that 10 mg mL−1 VAN reduced bacterial count from 108 CFU g−1 to 106 CFU g−1 after a 4-day incubation (Figure S12A, Supporting information), while 5 mg mL−1 DAP reduced bacterial count from 108 CFU g−1 to 0 CFU g−1 within 24 h (Figure S12B, Supporting information).
In light of the superb MRSA killing efficacy of DAP, in this section, we sought to use DAP to further enhance the anti-biofilm efficacy of our biofilm treatment system. Since DAP is negatively charged at physiological pH[76], it was loaded in the drug chamber of the cathode device, which was used as the working device in all our tests in this section. An ex vivo MRSA biofilm-infected porcine skin wound model was used in all tests in this section.
We first evaluated the iontophoretic delivery efficiency of DAP at different current intensities and DAP loading concentrations. As shown in Figure 6A, the concentration of DAP delivered to wound tissues after 1 h iontophoresis was significantly increased with increasing current intensity, from 1.7±0.59 mg g−1 by passive diffusion to 19.9±1.45 mg g−1 by 75 mA cm−2. The Pc of DAP for 75 mA cm−2 iontophoresis reached 158.3±11.18 ×10−6 cm s−1, which was 12.0 times higher than passive diffusion (Pc = 13.2±3.15 ×10−6 cm s−1) and 10.1 times higher than 0.5 mA cm−2 iontophoresis (Pc = 15.7±3.37 ×10−6 cm s−1) (Figure 6B). The concentration of DAP delivered to wound samples after 1 h iontophoresis also had a linear relationship with the DAP loading concentration. As shown in Figure 6C, when 1 mg mL−1 DAP was loaded in our working device, 75 mA cm−2 iontophoresis delivered 3.3±1.49 mg g−1 DAP into the wound tissue. When 5 mg mL−1 and 10 mg mL−1 DAP was loaded, the DAP concentration delivered into wound tissue increased to 19.9±1.45 mg g−1 and 32.8±5.68 mg g−1, respectively. The Pc of DAP at different loading concentrations was calculated and was found to be independent of loading concentration (Figure S13, Supporting information). This result suggested that unlike VAN, DAP did not exhibit concentration saturation effect.
Figure 6.
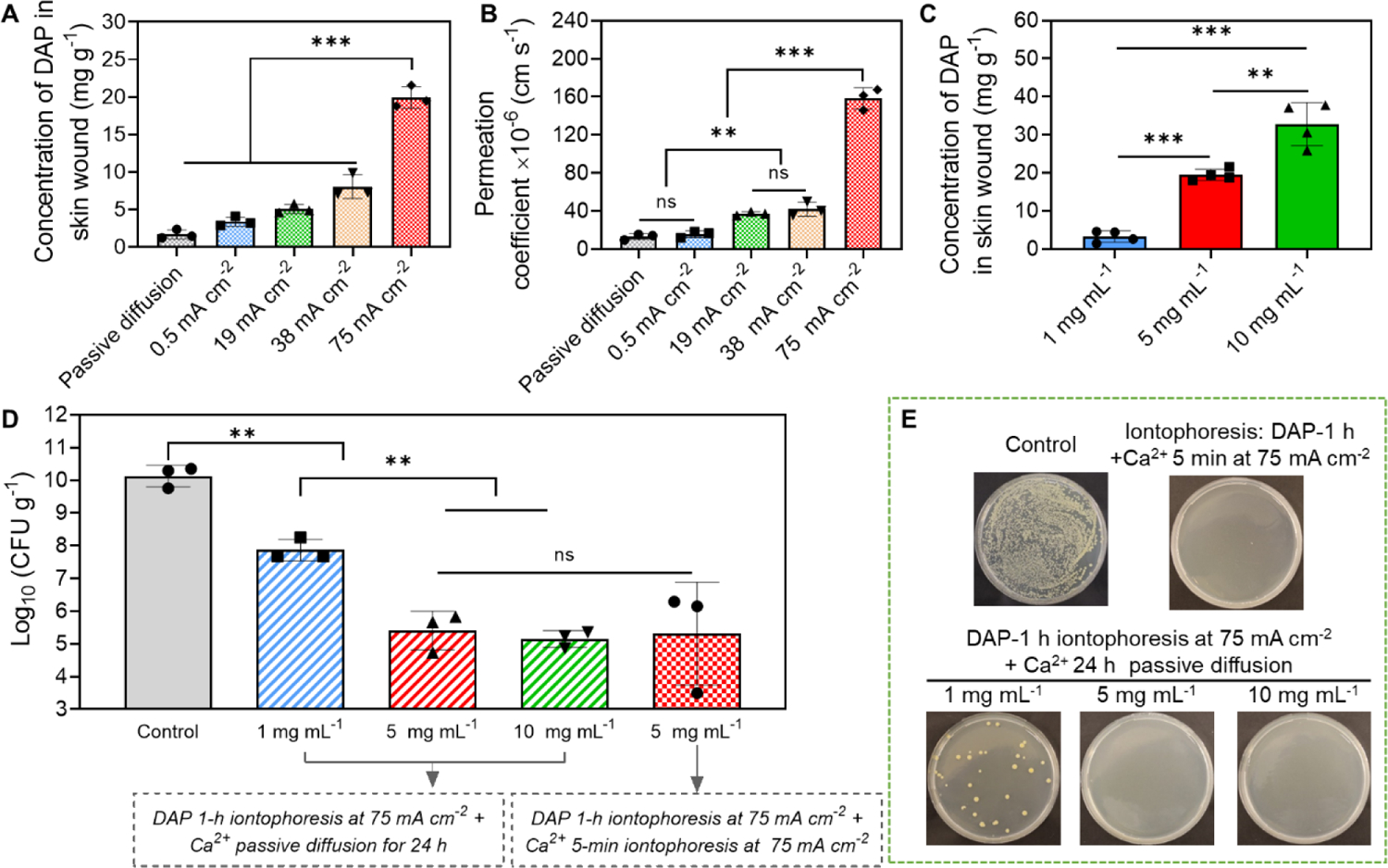
Anti-biofilm efficacy of our electrical biofilm treatment system using DAP. (A-C) Iontophoretic delivery of DAP using our system into an ex vivo MRSA biofilm-infected porcine skin wound model. (A) Accumulated concentration of DAP in wound tissues measured immediately after 1 h iontophoresis at different current intensities. The drug chamber of the working device (cathode) was loaded with 5 mg mL−1 DAP. (B) Permeation coefficient (Pc) of DAP as a function of applied current intensity. (C) Accumulated concentration of DAP in wound tissues measured immediately after 1 h iontophoresis with different DAP loading concentrations. The current intensity was 75 mA cm−2. (D) MRSA bacterial count in wound tissues measured at 24 h after treatment with our system (75 mA cm−2 1-h) using different DAP loading concentrations. Detailed treatment protocols were noted in the figure. (E) Representative photographs of bacterial colony cultured from ex vivo biofilm infected skin wounds after different treatments. All tissue homogenates were diluted 104 times with PBS before plating.
Since 75 mA cm−2 produced the highest iontophoretic delivery efficiency for DAP, it was used as the current intensity in the following anti-biofilm efficacy study. Three DAP loading concentrations (1, 5 and 10 mg mL−1) were tested in the anti-biofilm efficacy study. After a single 1 h treatment, wound tissues were incubated in a 37°C incubator for 24 h. During incubation, calcium chloride solution was topically applied to the wound, because DAP required calcium ions to function[77]. After incubation, bacterial count in wound tissues was measured. As shown in Figure 6D stripped columns and Figure 6E, MRSA bacterial count was reduced from 1010.1 CFU g−1 (untreated control) to 107.9 CFU g−1, 105.4 CFU g−1 and 105.2 CFU g−1 when 1 mg mL−1, 5 mg mL−1 and 10 mg mL−1 DAP was loaded in the drug chamber, respectively. The final bacterial count achieved by 5 mg mL−1 DAP loading concentration reached the clinical threshold for infection and was same as the bacterial count achieved by VAN treatment protocol #2 (measured at 24 h post treatment, Figure 5E). Similar to VAN, 20 mg mL−1 DAP (the DAP concentration delivered to wound tissues by 5 mg mL−1 loading concentration) did not have significant impact on the viability of skin cells after a 24-h incubation (Figure S11, Supporting information), suggesting a good cytocompatibility of the high DAP concentrations delivered by our system.
Although efficacious, the current protocol required a long total treatment duration consisting of a 1 h electrical treatment by our system followed by a 24 h topical application of calcium chloride solution. To further reduce the total treatment duration, we tried to use a 5 min anodal iontophoresis at 75 mA cm−2 to introduce calcium ions into the wound tissue immediately after the 1 h treatment with 5 mg mL−1 DAP loading concentration. The anodal iontophoresis of calcium ions was applied by our system. Wound samples were then incubated for 24 h without topical calcium chloride solution before bacterial counting. As shown in Figure 6D checkered pattern column and Figure 6E, this new protocol achieved a similar bacterial count reduction to 105.3 CFU g−1 as the previous protocol, while only requiring a 65 min total treatment duration.
As a comparison, when we treated the same biofilm-infected wound tissue with topically applied 5 mg mL−1 DAP mixed with calcium ions (passive diffusion) for 65-min, the bacterial count was only reduced to 107.7 CFU g−1 at 24-hour post treatment, and no further bacterial reduction was obtained when the topical application time was prolonged to 24 h (Figure S14, Supporting information). In summary, our results in this section showed that by using DAP, a 65 min high-intensity electrical treatment could achieve the same anti-biofilm efficacy as a 2 h treatment using VAN.
8. In vivo safety of our high-intensity electrical biofilm treatment system
In previous section, we demonstrated the in vitro and ex vivo safety of our high-intensity biofilm treatment system focusing on pH and temperature stability and cell viability. Here, we took a step further to evaluate the in vivo safety of our system, which is a critical prerequisite for therapeutic efficacy evaluation of our system using animal models and eventually in human patients. Healthy mice were used in this test. The working device (anode) was attached to the lower back of the mouse along the midline (Figure 7A). The counter device was also attached to the back located 1 cm away from the working device. The effective treating area with skin was 0.125 cm2. The drug chamber of both working and counter devices was filled with 1×PBS. Since anode device always represents a worst-case scenario in terms of tissue damage[46], we only evaluated skin tissue samples that were in contact with the anode device.
Figure 7.
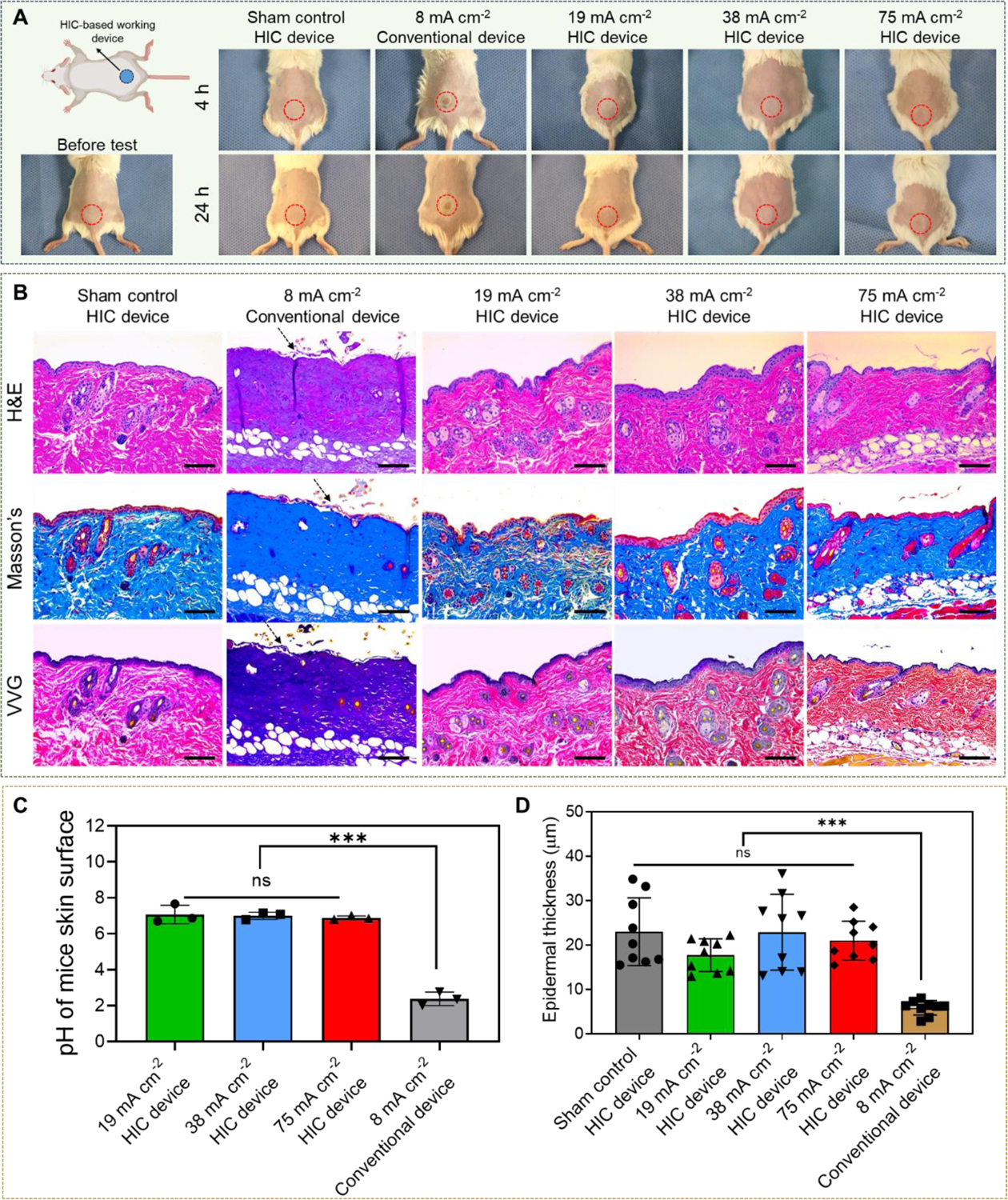
In vivo safety of our high-intensity electrical biofilm treatment system. (A) Representative photographs of mouse in five different treatment groups (0 mA cm−2 (sham control), 19 mAcm−2, 38 mA cm−2, and 75 mA cm−2 1 h treatments applied by our system, and 8 mA cm−2 1 h treatment applied by a conventional electrical device (Figure 2B). Photographs were taken at 4 h and 24 h after treatment. (The upper left schematic was created with BioRender.com) (B) Representative histological sections of skin tissues that were in direct contact with the working device using three stains (hematoxylin and eosin (H&E), Masson’s trichrome, and Verhoeff-Van Gieson (VVG)). Skin samples were collected at 24 h after treatment. Arrows showed the detachment of epidermal from the dermis layer in 8 mA cm−2 conventional device group. Scale bar: 100 μm. (C) Skin surface pH measured immediately after treatment. (D) Thickness of epidermal layer measured from skin sections collected at 24 h after treatment.
Different current intensities, including 0 mA cm−2 (sham control), 19 mA cm−2, 38 mA cm−2, and 75 mA cm−2, were applied by our system for 1 h. A conventional electrical device was constructed by inserting a carbon electrode in a PBS-filled drug chamber with the same volume as our biofilm treatment system (Figure 2B). We gradually increased the current intensity from 0.5 mA cm−2 to identify the minimum current that induced significant skin tissue damage after 1 h application when the conventional device was used to apply the current. Human clinical trials showed that conventional low-intensity iontophoresis typically induced mild skin irritation and erythema that rarely lasted more than 3 h after the removal of iontophoresis[78]. Therefore, we chose to follow up at 4 h and 24 h after treatment for skin damage evaluation.
As shown in Figure 7A, no signs of skin irritation, redness or blisters were observed in mice treated with our electrical biofilm treatment system for all current intensities tested at both 4 h and 24 h checkpoints. However, the skin tissue treated with the conventional device started to show obvious redness and irritation in device contact area at 8 mA cm−2, which did not recover by 24 h. We measured the skin surface pH in current treating area immediately after different treatments. Figure 7C showed that the pH of the skin treated by our system remained in the safe range for all current intensities tested (pH = 7.1±0.52, 7.0±0.20, and 6.9±0.11 for 19 mA cm−2, 38 mA cm−2, and 75 mA cm−2 treatments, respectively). However, the pH of the skin treated by conventional device at 8 mA cm−2 decreased to 2.38±0.38. This was mainly due to the accumulation of hydrogen ions generated by EC reactions, which was not sufficiently neutralized by the pH buffering system used in the conventional device (i.e. PBS). This acidic environment caused the skin irritation as seen in Figure 7A. Similar skin tissue damage caused by pH-induced chemical burn was also reported by other studies[79]. We also measured the skin surface temperature immediately after treatment, which remained below 43°C in all groups (Figure S15, Supporting information). This result suggested that pH change was the major cause for skin damage in conventional device-treated animals. Histological sections of the skin tissues in direct contact with our system or the conventional device were collected at 24 h after treatment (Figure 7B). In tissues treated by conventional device at 8 mA cm−2, fluid accumulation and swelling of collagen bundles in the dermis layer were observed. These resulted in missing cleft spaces between collagen bundles, homogenization and full-thickness hyalinized necrosis of the dermal collagens. The epidermis in conventional device-treated samples demonstrated significant thinning with keratinocytes necrosis and focal detachment as compared to the sham control. In contrast, although mild neutrophil infiltration was observed, there was no significant epidermal or dermal damage in skin tissues treated by our system. We measured the epidermal thickness for all samples and found that treatment with our system did not significantly alter the epidermal thickness in all current intensity treatment groups (Figure 7D). However, conventional device treatment at 8 mA cm−2 significantly reduced the average epidermal thickness by more than 50% compared to sham control. This result was consistent with our histological observation. The outcome of this study, for the first time, demonstrated that our HIC-based system significantly enhanced the in vivo safety of high-intensity current application to mouse skin tissues compared to the conventional electrical device.
9. In vivo anti-biofilm efficacy of our high-intensity electrical biofilm treatment system
We finally investigated the in vivo anti-biofilm efficacy of our high-intensity electrical biofilm treatment system using a type II diabetic mouse-based skin wound model infected with the MRSA biofilm. The wound infection model was established following previously published protocols with modifications[57, 80]. In brief, full-thickness wounds were created with 4 mm biopsy punch on the back of the diabetic mouse. MRSA was cultured for 4 h to reach 1×108 CFU mL−1 in vitro, and then inoculated into the wound site. Two days after wound inoculation, mature biofilms were formed on the wound[57]. Daptomycin was used in this in vivo anti-biofilm efficacy study because it required a shorter treatment duration to achieve the same efficacy as vancomycin in our ex vivo studies. The working device was attached to the wound site on the back of the mouse. The counter device was attached to the belly directly below the working device. The effective treating area with skin wound was 0.125 cm2, which can entirely cover the wound site. Five different treatments were tested, including untreated negative control, DAP-alone (topical application of DAP at 5 mg mL−1 with calcium ions for 65 min), high-intensity current alone (75 mA cm−2 was applied for 60 min, drug chamber was loaded with PBS), low-intensity current (0.5 mA cm−2 was applied for 60 min) with DAP (5 mg mL−1 was loaded in the drug chamber), and high-intensity current (75 mA cm−2 was applied for 60 min) with DAP (5 mg mL−1 was loaded in the drug chamber). For all electrical treatment groups, iontophoresis of calcium ions was applied for 5 min at 75 mA cm−2 after the 60 min treatment. Our experimental procedure was illustrated in Figure 8A. The biofilm bacterial count was measured at 24 h or 7 days after the treatment by using standard plate counting assay. Figure 8B showed representative photographs of the biofilm infected diabetic wound immediately before and at 24 h after the treatment. Figure 8C showed the bacterial colony cultured from wound samples collected at 24 h after treatment. The quantitative measurement of bacterial count in wound samples collected at 24 h after treatment was presented in Fig. 8D. The bacterial count in no-treatment control group reached 109.0 CFU g−1 at Day 3. The high-intensity electrical treatment alone did not reduce the bacteria density, which remained at 109.1 CFU g−1. This result was consistent with our ex vivo electrical debridement study, which showed that biofilm recovery happened within 24 h after the treatment. Low current intensity combined with DAP and DAP-alone only reduced the bacterial density from 109.0 CFU g−1 to 107.6 CFU g−1 and 106.5 CFU g−1, respectively, which were both above the clinical threshold for wound infection. Although the final bacterial count of low-intensity current combined with DAP was higher than that of DAP-alone treatment, there was no statistically significant difference between these two treatments (Figure 8D). In contrast, when high-intensity current combined with DAP was applied, the bacterial count in the wound at 24 h after treatment reduced to 104.6 CFU g−1, which was below the clinic threshold for wound infection. Our results here showed that the high-intensity electrical treatment with DAP applied by our HIC-based system achieved a significantly higher anti-biofilm efficacy compared to low-intensity electrical treatment and conventional topical application using the same DAP concentration. Moreover, the bacterial count at 7-day after the treatment of high-intensity current with DAP further decreased to 103.3 CFU g−1 (Figure 8E), which suggested that the high DAP concentration delivered by our system exhibited a sustained anti-biofilm effect. The outcome of this study was significant because it showed that in vivo wound biofilm infections can be effectively reduced by our one-time, short-duration electrical treatment. This treatment efficacy achieved by our system was better than the efficacy achieved by existing technologies based on low-intensity iontophoresis[81], co-delivery of biofilm-destabilizing and antimicrobial agents, microneedle arrays[82], antimicrobial nanoparticles[81, 83], and drug-loaded wound dressings[84] using long treatment durations of 24 h or longer. These existing technologies typically achieved a biofilm bacterial reduction of up to 3 log10 scales. A much shorter treatment duration would be particularly advantageous for management of chronic wound infections as it would reduce patient discomfort and enhance patient compliance.
Figure 8.
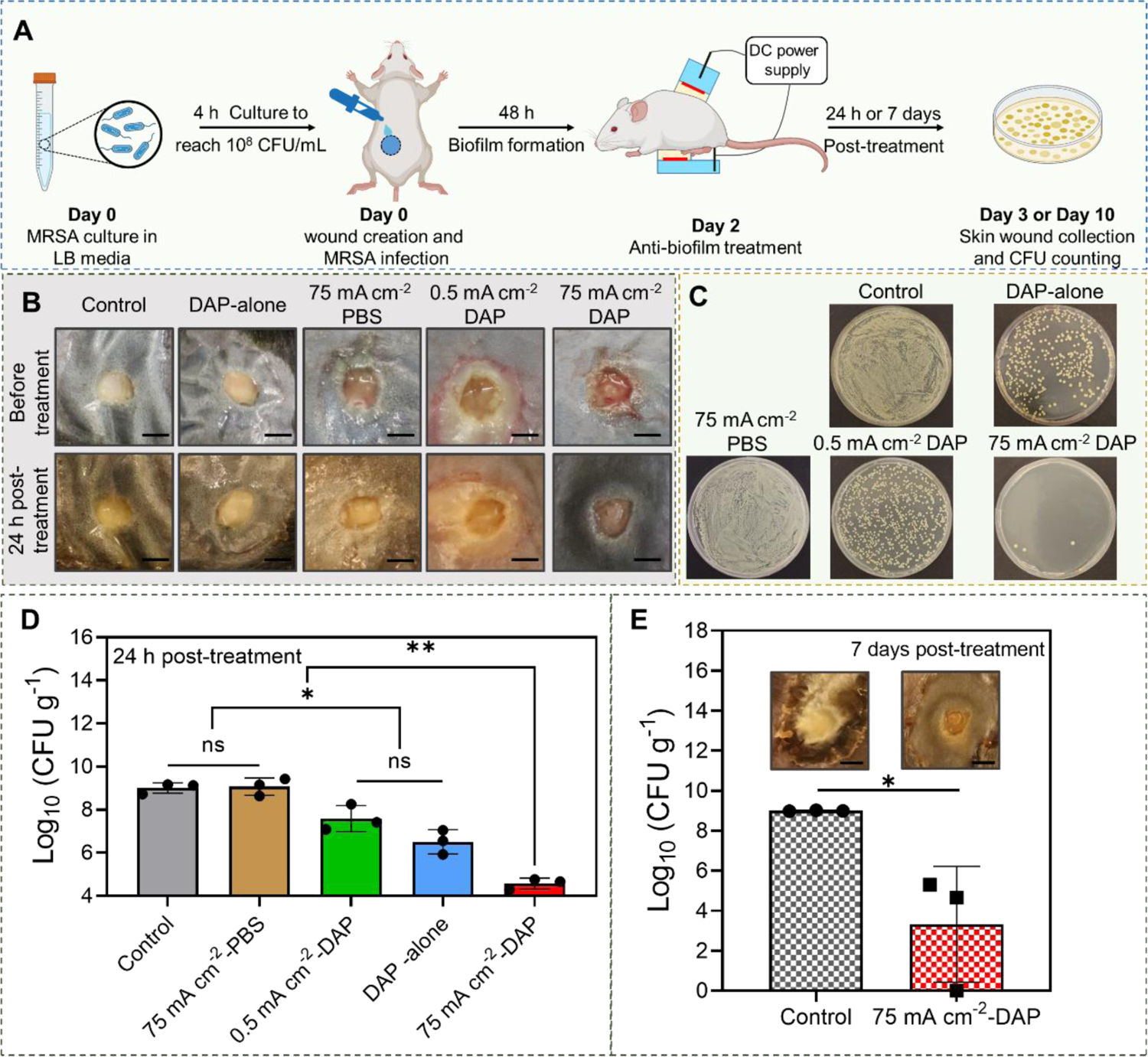
In vivo efficacy of high-intensity electrical biofilm treatment system using a type II diabetic mouse-based wound model infected with MRSA biofilms (n=3). (A) Experimental timeline and schematic illustration of our system setup on diabetic mouse (Created with BioRender.com). (B) Representative photographs of MRSA biofilm-infected wounds in different treatment groups. (C) Representative photographs of bacterial colony cultured from skin wounds at 24 h after different treatments. All tissue homogenates were diluted 102 times with PBS before plating. (D) MRSA bacterial count in skin wounds measured at 24 h after different treatments. (E) MRSA bacterial count in skin wounds measured at 7 days after treatment. (Scale bar: 3 mm)
10. Limitations and outlook
The limitation of our high-intensity electrical biofilm treatment system is that it did not completely eradicate the biofilm (i.e. the biofilm bacterial count was not reduced to 0 CFU g−1). The biofilm treatment efficacy of our technology is likely limited by the antibiotics used in our system. It has been reported that VAN and DAP are not capable of killing S. aureus persister cells even at 100 × MIC. The development of new anti-biofilm agents that are more effective against persister cells is an active research area[85]. More efficacious anti-biofilm drugs will undoubtedly enhance the biofilm treatment efficacy of our technology. Delivering a combination of anti-biofilm agents instead of a single one may further enhance the efficacy of our technology. EPS degrading agents and metabolic adjuvants have been shown to enhance biofilm bacterial killing of antibiotics[86, 87]. Anti-inflammatory agents can reduce tissue damage induced by biofilm infections[86]. By applying high-intensity current and combining with multi-drugs using our novel electrical system, we expect greater anti-biofilm treatment efficacy can be achieved. Although we demonstrated that our system had enhanced safety compared to the conventional electrical device, a more comprehensive study will need to be performed in the future to further and fully characterize the safety of the high-intensity current applied by our system, including the evaluation of integrity and function of skin cells and tissues, pain sensation, and neuromuscular functions.
Our study demonstrated that high-intensity currents could significantly enhance the efficiency of transdermal iontophoresis. Transdermal iontophoresis, in both forward and reverse modes, have been widely used in many different areas of biomedical and clinical application, including drug delivery and disease diagnosis. The high iontophoretic efficiency enabled by our HIC technology can be potentially applied to enhance the efficacy of these applications. Our study also showed that our HIC-based system could improve the safety of high-intensity current application to biological tissues compared to conventional electrical stimulation devices. Electrical stimulation is an important physical treatment modality. A high current intensity is required by many therapeutic applications of electrical stimulation to achieve good efficacy, such as transcutaneous electrical nerve stimulation for pain management[88], denervated muscle stimulation for prevention of muscle atrophy[89], and electrical current-induced cell migration (i.e. electrotaxis) for accelerated wound closure[90]. The safety enhancement enabled by our HIC design could potentially lead to safer and more efficacious treatment system design in these application areas.
11. Conclusion
In this paper, we described a novel electrical current-based biofilm treatment system for combating chronic wound biofilm infections. We demonstrated the safety, the electrical biofilm debridement efficacy and the iontophoretic antibiotic delivery efficacy of our system using high current intensities of up to 75 mA cm−2. By combining high-intensity electrical debridement and high-efficacy iontophoretic antibiotic delivery, our system used a short treatment (≤2 h) to successfully reduce the bacterial count of mature biofilm-infected skin wounds ex vivo and in vivo to below the clinical threshold for wound infection. Our innovative technology provides a simple, quick, safe, yet highly efficacious means to manage biofilm infections in chronic wounds. The reduction of bacterial bioburden will help resume the normal healing process in chronic wounds. Ultimately, this will reduce the amputation rate related to chronic wounds, enhance patients’ quality of life, and reduce the overall healthcare cost.
12. Materials and Methods
12.1. Materials
Polyethylene glycol dimethacrylate (PEGMDA, molecular weight = 8000) was purchased from Polysciences (Warrington, PA, USA). Poly(ethylene glycol) diacrylate (PEGDA, molecular weight = 700), IRGACURE 2959, Benzophenone, Fluorescein isothiocyanate (FITC) labeled dextran-4 kDa (FD-4), water with 0.1% (v:v) trifluoroacetic acid (TFA), acetonitrile, acetonitrile with 0.1% (v:v) TFA, Agar, sodium phosphate monobasic (NaH2PO4), and sodium phosphate dibasic (Na2HPO4) were purchased from Sigma-Aldrich (St. Louis, MO, USA). LB Broth (Miller) was purchased from Fisher Bioreagents (Fair Lawn, NJ, USA). Acrylic sheets and very-high-bond (VHB) foam tape were purchased from Mcmaster-Carr (Robbinsville, NJ, USA). Fetus porcine skin was purchased from Nebraska Scientific (Omaha, NE, USA). Human keratinocyte cell (HaCaT), human primary dermal fibroblast cells (HDFa), and human monocytic cells (U937) were kind gifts from Dr. Jingwei Xie at the University of Nebraska Medical Center (Omaha, NE, USA).
12.2. Bacterial strain, and antibiotics
Methicillin-resistant S. aureus (MRSA) USA300 strain was used in this study. The planktonic bacteria were cultured in LB medium. Vancomycin hydrochloride (Apexbio Technology LLC, Houston, TX, USA) and daptomycin (Combi-Blocks, San Diago CA, USA) were used to treat MRSA biofilm. The minimum inhibitory concentration (MIC) of vancomycin hydrochloride to planktonic MRSA cells was determined as 1.2 μg mL−1 (Figure S16, Supporting information), which was in consistent with the results reported previously[91].
12.3. Fabrication of HIC-based electrical biofilm treatment system
Our HIC-based system, including working device and counter device, was fabricated by laser micromachining (Trotec Speedy 300, Trotec., MI, USA). The design and fabrication process of our HIC-based devices has been previously reported[51]. Briefly, drug chamber and phosphate salt solution chamber were fabricated using acrylic plastic. To assemble different components of device, a double-adhesive VHB tape was used. Polyethylene glycol (PEG) hydrogel was composed of 10% PEGDMA, 5% PEGDA and 1% Irgacure 2959 and 84% deionized water. It was bond to the phosphate salt solution chamber to form a two-phase separation system (ATPS) by using UV lamp. Benzophenone (10% w/v) was used to allow the binding. To obtain phosphate salt solutions with high conductivities, saturated Na2HPO4 solution (0.6 mol L−1, pH = 9.0, 46.1±2.40 ms cm−1) was used in anode HIC-based device. And a mixture solution containing NaH2PO4 (0.6 mol L−1) and Na2HPO4 (0.48 mol L−1) (pH = 6.4, 51.0±0.51 ms cm−1) was used in cathode HIC-based device. To connect our system with DC power supply, carbon electrode was used.
12.4. In vitro evaluation of ATPS stability during high-intensity current treatment.
In this experiment, we first applied 75 mA cm−2 current for 60 minutes using our HIC-based anode and cathode devices (same design, both had no drug/PBS chamber) to PBS (1st PBS, see Figure S1A, Supporting information). After current application, the anode and cathode devices were collected and refilled with new high-concentration phosphate salt solutions. Then each of them was half-way dipped in fresh PBS (2nd PBS, 4 mL) for 60 minutes (Figure S1B, Supporting information). As a negative control, HIC-based anode and cathode devices (without drug/PBS chamber) that were not used for high-intensity current application were also dipped in fresh PBS (2nd PBS, 4 mL) for 60 minutes. After incubation, the conductivity and pH of the 2nd PBS solutions were measured. A change in the conductivity or pH of the 2nd PBS serves as a good indicator of high-concentration phosphate salt ion diffusion out from the phosphate salt solution chamber. As can be seen in Figure S2 (Supporting information), the pH (Figure S2A) and conductivity (Figure S2B) of the 2nd PBS incubated with our HIC devices that had gone through high-intensity current application were not significantly different than that of the 2nd PBS incubated with negative control HIC devices. These results suggested that after high-intensity current application, the PEG hydrogel in our HIC devices retained its ability to minimize the diffusion of high-concentration phosphate salt ions out from the phosphate salt solution chamber.12.4
12.5. In vitro safety test of high-intensity current application and high concentration of antibiotics
The in vitro cell viability test setup of high-intensity ion current was described in our previous work[51]. Briefly, two adhering cells, HaCaT cells (human keratinocyte cell line) and HDFa cells (human primary dermal fibroblast cells), were seeded in 8 mm×8 mm areas in cell culture dishes defined by a PDMS stencil, and cultured overnight. After removing the PDMS stencils the next day, the test device with a rectangular fluidic chamber was amounted on the cell culture dish, so the current density adhering cells experienced can be precisely defined. After adding complete growth media, 75 mA cm−2 DC current intensity was conducted for 1 h. After test, cell counting assay was used to count the live cells. Human monocytic cells (U937) was also used to evaluate the cell viability after high current intensity treatment. They were cultured overnight in complete growth media and transferred to the test device chamber the next day. Cells with no electrical current treatment was used as control group for cell viability comparison.
To evaluate the cell toxicity of the high concentration of antibiotics, HaCaT cells and HDFa cells were used. 2×104 HaCat cells and 2×103 HDFa cells were seeded in each well of a 96-well plate. After cultured in an incubator under 37°C and 5% CO2 for 24 h, 4.5 mg mL−1 VAN and 20 mg mL−1 DAP were added to the culture media, respectively. Both HaCat cells and HDFa cells were treated by VAN and DAP for two days and one day, respectively. The cell toxicity of the two cells at the end time points were evaluated by LIVE/DEAD stain and visualized by a confocal laser scanning microscope (CLSM, LSM 710, Zeiss, Germany).
12.6. In vitro bactericidal effect of MRSA planktonic bacteria by high-intensity current
MRSA bacteria was cultured with LB media in a 37°C water shaking bath for 4 h to reach the density of 108 cells mL−1. Then MRSA solution was added to a rectangular fluidic chamber. After immersing our HIC-based anode device and cathode device in the bacteria solution located at two sides of the chamber, 75 mA cm−2 DC current intensity was conducted for 1 h. The viable bacteria densities before, immediately after treatment, and 24 h after treatment were measured by standard plate counting assay.
12.7. Ex vivo safety test of high-intensity current application
Freshly preserved fetal pig skin samples purchased from Nebraska Scientific (Omaha, NE, USA) was used for the ex vivo safety test of high current intensity applied by HIC-based biofilm treatment system. Skin samples were stored at −20°C upon arrival and used within 1 month. Skin samples were sandwiched between drug chamber and PBS chamber, and the test setup was demonstrated in Fig. 2A. The counter device was attached to the back of the skin sample. Before testing, drug chamber was filled with PBS. The high concentration phosphate salt solutions were added into phosphate salt solution chambers. 75 mA cm−2 current intensity was conducted from DC power supply and applied on the skin sample for 1 h continuously. By using a K-type thermocouple (Digi-Sense™ Standard Precalibrated Thermocouple, Vernon Hills, IL, USA), the peak temperature of device/tissue interface during current application was monitored. After the test, the pH of drug solution and drug chamber contacting-skin area were measured using a flat pH probe (Sensorex Corporation, Stanton, CA). For comparison, carbon electrode directly contacted with the drug chamber and applied same current conditions was used as the conventional control (Figure 2B).
12.8. Establishment of ex vivo MRSA biofilm infected skin wound
The fetal pig skin samples (approximately 30×30 mm) were immersed in 10% (v/v) bleach for 5 min after hair removal. Then, skin samples were sterilized in 70% ethanol for 30 min. The surface of skin sample was further cleaned by 10% povidone-iodine and 70% isopropanol to prevent contamination. A 6-mm disposable biopsy punch was used to create an excisional wound. Samples were put into sterile 10-cm petri-dishes filled with sterile PBS (6–10 mL). The wound was then inoculated with 1×108 CFU mL−1 MRSA bacteria solution (20 μL). The bacteria were allowed to grow in a 37°C Heratherm™ compact microbiological incubator (ThermoFischer Scientific™, Waltham, MA, USA) for at least 48 h to form mature biofilms.
12.9. Electrical debridement treatment of high-intensity current on ex vivo MRSA biofilm infected skin wounds
The test setup was shown in Figure S17 (Supporting information). MRSA biofilm infected skin wound was sandwiched between anode and cathode HIC-based devices. To evaluate the anti-biofilm effect of high current intensity only, PBS (5 mL) was filled in both drug chamber and PBS chamber and then 0.5 mA cm−2, 19 mA cm−2, 38 mA cm−2, and 75 mA cm−2 was applied for 1 h, respectively. The device/skin wound contact area was 0.502 cm2. Skin samples were collected by 8-mm biopsy punch immediately after treatment or at 24 h after treatment. To evaluate the biofilm thickness before and after different treatments, samples were embedded in tissue freezing medium (OCT) and sectioned at 8 μm. After imaged in microscope, the thickness of biofilms after different treatments were measured using ImageJ software. To evaluate the colony-forming unit (CFU) counts per gram of MRSA from infected skin wounds, samples were homogenized in PBS (10 mL) and diluted with PBS, and then transferred to standard plate counting assay. The bactericidal efficacy was calculated as follow:
| (1) |
12.10. Iontophoresis of antibiotics induced by HIC-based biofilm treatment system to ex vivo MRSA biofilm infected skin wound
To evaluate the antibiotic delivery efficacy of VAN by high-intensity current, biofilm infected fetal pig skin wound was used. The test setup was demonstrated in Figure S17 (Supporting information). The DAP test setup was same as that of VAN, except no PBS chamber was used. Instead, the inner surface of the skin wound contacted directly to the hydrogel of the counter device. To evaluate the effect of electrical current intensity on the drug delivery efficacy, different current intensities from 0 to 75 mA cm−2 were applied. To evaluate the effect of loading concentration of antibiotics, different concentrations of VAN and DAP from 1 mg mL−1 to 10 mg mL−1 were used. Since VAN is neutral and DAP is negatively charged under physiological environment, anode HIC-based device was placed facing biofilm for VAN iontophoresis, and cathode HIC-based device was placed facing biofilm for DAP iontophoresis. Samples were collected immediately after different treatments. The concentrations of antibiotics delivered into skin wound samples were measured by high-performance liquid chromatography (HPLC). The permeation coefficient (Pc, cm s−1) was then calculated (Pc=J/C, where J is antibiotic delivery flux in tissue samples (μg (cm2s)−1, C is the loading concentration of antibiotic solution (μg mL−1)).
12.11. Quantitative measurement of antibiotic concentrations in infected skin wounds using high-performance liquid chromatography (HPLC)
12.11.1. Sample preparation
Immediately after iontophoresis, the infected skin wounds were collected by an 8-mm disposable biopsy punch and washed in PBS three times to remove surface antibiotics, and then weighted before use. Samples were homogenized in 4–10 mL PBS and then centrifuged (4000 rpm, 20 min). The supernatant (200 μL) was collected and mixed with methanol or acetonitrile to remove potential tissue proteins. After solvent entirely evaporated by a hot plate (85 °C, 4 h), samples were reconstituted in optimized solution (200 μL) to restore original volume. Centrifuge (10,000 rpm) was performed for 5 min and the supernatant was injected for analysis. For VAN, the optimized solution to re-dissolve samples was the mixture of water with 0.1% trifluoroacetic acid (TFA) and acetonitrile with 0.1% TFA (50:50). For DAP, samples were re-dissolved in the mixture of buffered solution (Monosodium phosphate: 188.8 mmol L−1, Disodium phosphate: 11.16 mmol L−1, pH=5.5) and acetonitrile (50:50).
12.11.2. HPLC test procedure
The concentrations of antibiotics accumulated in infected skin wounds were detected by HPLC on an Agilent 1260 Infinity system (Agilent, Santa Clara, CA, USA) using a EC-C18 column (150 mm×4 mm, particle size 2.7 μm). The injection volume was 20 μL with a flow rate of 0.6 mL min−1. For VAN concentration detection, a gradient procedure was used for precisely detection. The mobile phase was water with 0.1% TFA (Buffer A) and acetonitrile with 0.1% TFA (Buffer B). The gradient procedure was developed as follow:
The detection was performed at 284 nm. Retention time of VAN was around 4 min (Figure S18A, Supporting information). For DAP concentration detection, the mobile phase was buffered solution (Monosodium phosphate: 188.8 mmol L−1, Disodium phosphate: 11.16 mmol L−1, pH=5.5) and acetonitrile with ratio of 50:50 (v/v). The detection was performed at 223 nm. The retention time of DAP was around 2.3 min (Figure S18B, Supporting information). Accumulated concentration of VAN and DAP was calculated via a standard calibration curve of VAN and DAP ranging from 25 μg mL−1 to 100 μg mL−1, respectively (Figure S19, Supporting information).
12.12. Cryo-section and fluorescent microscopy
FD-4 penetration and distribution in the MRSA biofilm infected skin wound was evaluated by cryo-section and fluorescent microscopy. Briefly, samples were collected by 8-mm disposable biopsy punch and dehydrated in acetone for 10 to 30 min. After air dried for another 10 to 30 min, samples were embedded in tissue freezing medium (OCT) and sectioned to obtain 10 μm section under −20 °C. Section samples were then observed under a fluorescent microscope (DMI 6000 B, Leica, Bannockburn, IL, USA).
12.13. Minimum inhibitory concentration (MIC) and time-killing tests of antibiotics to planktonic MRSA
MRSA bacteria was cultured in LB media in a 37 °C water shaking bath overnight to reach the concentration of 1×108 CFU mL−1. Then, they were diluted to 105 CFU mL−1 in 96-well plates by adding different concentrations of VAN solutions. After incubation for 24 hours without shaking, MIC of VAN to inhibit MRSA growth was measured by a Biotek Synergy H1 hybrid multi-mode microplate reader (BioTek, Winooski, VT, USA) at OD600. To evaluate the killing time of VAN to bacteria in stationary phase (1×108 CFU mL−1), VAN powder was added to bacteria solution to obtain concentration of 1 mg mL−1, 3 mg mL−1, 5 mg mL−1, and 10 mg mL−1, respectively, while remaining MRSA concentration of 1×108 CFU mL−1. After incubation in a 37 °C water bath for 1 day, 2 days, 3 days and 4 days, the CFU of MRSA cells was tested by the standard plate counting assay, respectively. The time-killing tests of DAP with different concentrations (0.1 mg mL−1, 0.5 mg mL−1, 1 mg mL−1, and 5 mg mL−1) on MRSA planktonic cells were also performed, with the incubation time up to 24 h.
12.14. Combined biofilm treatment efficacy of electrical debridement and high-concentration antibiotics delivery
The test setup using VAN was described in Figure S17 (Supporting information). The combined biofilm treatment efficacy of electrical debridement and VAN delivery was first evaluated. To evaluate the anti-biofilm treatment efficacy of current intensity, 0 mA cm−2 (passive diffusion) to 75 mA cm−2 was applied on the test system with 1 mg mL−1 VAN loaded in HIC-based working device, respectively. To evaluate the anti-biofilm treatment efficacy of loading concentration of VAN, 1 mg mL−1, 5 mg mL−1, and 10 mg mL−1 VAN was loaded in the drug chamber of the HIC-based working device, respectively, and applied 75 mA cm−2. To further enhance the anti-biofilm efficacy of electrical debridement and high concentration delivery of VAN, two treatments were applied, named protocol #1 and protocol #2. Protocol #1 applied two treatments (treatment: 75 mA cm−2 for 1 h, 1 mg mL−1 VAN loaded in HIC-based working device) separated by 6 h. Protocol #2 applied two treatments (treatment same to Protocol #1) separated by 24 h. CFU was counted 24 h after last treatment using the standard plate counting assay. The bactericidal efficacy was calculated accordingly, as described in Section 12.9.
The combined biofilm treatment efficacy of electrical debridement and DAP delivery was also evaluated. The test setup was same as that of VAN, except no PBS chamber was used. Instead, the inner surface of the skin wound contacted directly to the hydrogel of the counter device. The 0 mA cm−2 (passive diffusion) to 75 mA cm−2 were applied on the test system with 5 mg mL−1 DAP loaded in HIC-based working device to measure the effect of current intensity on the anti-biofilm efficacy. 1 mg mL−1, 5 mg mL−1, and 10 mg mL−1 DAP was loaded in HIC-based working device, respectively, and applied 75 mA cm−2 to evaluate the anti-biofilm treatment efficacy of DAP loading concentration. Calcium chloride solution (1 mL, 100 mg mL−1) was supplied on the infected wound and allowed for 24-h passive diffusion after DAP treatment, since DAP require calcium ion to have anti-bacteria function. CFU was counted immediately after calcium ions 24-h diffusion using the standard plate counting assay. We further developed a protocol with two steps to reduce the treatment time. For step 1, biofilm-infected skin wound was treated with 75 mA cm−2 for 1 h induced by HIC-based cathode device, loaded with 5 mg mL−1 DAP. Immediately after step 1, HIC-based anode device was applied on the wound loaded with calcium chloride solution (100 mg mL−1) and applied 75 mA cm−2 for 5 min (step 2). CFU was counted 24 h after the treatment of step 2. The bactericidal efficacy was calculated accordingly, as described in Section 12.9.
To evaluate the paradoxical effect of VAN, the ex vivo mature MRSA biofilm established on the porcine skin wound was incubated in 1 mg mL−1, 4.5 mg mL−1, 10 mg mL−1, and 20 mg mL−1 VAN for passive diffusion of VAN, respectively. After 24 h incubation, the CFU of biofilm infected wounds under different VAN treating concentrations were measured.
12.15. In vivo safety test of high-intensity current application
BALB/c mouse (6–10 weeks, ~20 g) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The animal test was incompliance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Nebraska Medical Center (protocol #21-049-06-FC). To evaluate the safety of high current intensity on the mouse skin, the drug chamber (diameter: 4 mm) of anode HIC-based device was amounted on the back of mouse with the help of 3M Tegaderm transparent film after mouse were anesthetized and hair removal. After adding PBS (0.5 mL) in the drug chamber, the HIC-based hydrogel and phosphate salt solution chamber were attached on the drug chamber. Different current intensities (0 mA cm−2 (sham control), 19 mA cm−2, 38 mA cm−2, and 75 mA cm−2) were applied from a DC power supply and route to the mouse skin. The counter device had the same design as the working device and was also attached to the back of mouse skin to complete the circuit. The HIC-based counter device had slightly larger skin contact area (diameter: 7 mm) to minimize the pH/temperature impact of the counter device. A conventional electrical device with the carbon electrode contacted with drug chamber directly was used as a comparison. 8 mA cm−2 was induced by the conventional device and last for 1 h. The pH of skin surface immediately after test was measured by using a flat pH probe. Digital images of the skin were taken to inspect for signs of redness, blisters, and tissue damage at 4 h and 24 h after different treatments. At 24 hour post treatment, mouse were sacrificed and the skin tissues were collected for histological evaluation (Hematoxylin and Eosin (H&E) staining, Masson’s trichrome stain, and Verhoeff-Van Gieson (VVG) staining)
12.16. In vivo anti-biofilm efficacy test of our high-intensity electrical biofilm treatment system
The in vivo anti-biofilm efficacy test was incompliance with the protocol approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center (protocol #21-049-06-FC). MRSA was cultured in LB media in water shaking bath (37°C) overnight. Then 100 μL bacteria was transferred to LB media (4 mL) and cultured for another 4 h to reach the density of 1×108 mL−1 and stored in ice before use. In this study, homozygous B6.BKS(D)-Leprdb/J mouse (5–6 weeks, around 30 g, Jackson Laboratory 000697) was used to establish the biofilm infected skin wound model. Specifically, after mouse anesthesia and hair removal, one full-thickness wound (4 mm in diameter) was created on the back of the mouse by using 4-mm biopsy punch. For negative control group, two wounds were created on the back of the mouse. Bacteria solution (10 μL) was carefully dropped in the wound site. Then the infected wound was covered and sealed by 3M Tegederm transparent film. After mouse was infected for 2 days, different anti-biofilm treatments were applied on the wound respectively. The HIC-based working device used in this study had slightly smaller dimension than the ex vivo working device due to the smaller wound used. Working device was attached on the wound site with the help of 3M tegaderm transparent film. Cathode phosphate salt solution (2 mL) was loaded in phosphate salt solution chamber and PBS/daptomycin solution (0.5 mL) was added in the drug chamber. The effective treatment area was 0.125 cm2 to fully cover the infected wound site. The counter device attached to the belly of the mouse directly down to the working device to complete the circuit. To evaluate the anti-biofilm efficacy of high-intensity current alone, 75 mA cm−2 ionic current was applied in the system for 60 min with PBS loaded in the drug chamber. To evaluate the anti-biofilm efficacy of low-intensity current combined with DAP, 0.5 mA cm−2 ionic current was applied in the system for 60 min with DAP (5 mg mL−1) loaded in the drug chamber. To evaluate the efficacy of our high-intensity electrical biofilm treatment system, 75 mA cm−2 ionic current was applied in the system for 60 min with DAP (5 mg mL−1) loaded in the drug chamber. To perform the antibacterial function of DAP, immediately after all electrical treatment of each group, HIC-anode device was applied on the wound loaded with calcium chloride solution (100 mg mL−1) and applied 75 mA cm−2 for 5 min. To evaluate the efficacy of conventional topical treatment of DAP alone, the mixture of DAP (5 mg mL−1) and calcium chloride (0.5 mg mL−1) was loaded in the drug chamber for 65 min, with no electrical current applied. The infected wound without treatment was used as the negative control. Then, 24 h or 7 days after treatment, the infected skin wounds and surrounding tissues were collected by 6-mm biopsy punch. Bacterial count per gram was evaluated using the standard plate counting assay. Each group has three wound samples to perform statistical analysis.
12.17. Statistical Analysis
The statistical difference between samples were determined by unpaired student’s t-tests (GraphPad Software, SanDiego, CA, USA). The statistical difference in the figures were present by * for p < 0.05, ** for p < 0.01, and *** for p < 0.005. At least 3 replicates were performed for all statistics.
Supplementary Material
Table 1.
Gradient procedure of HPLC for vancomycin chloride detection
| Time (min) | Buffer A (%) | Buffer B (%) | Flow (mL min−1) |
|---|---|---|---|
| 0 | 100 | 0 | 0.6 |
| 5 | 100 | 0 | 0.6 |
| 8 | 70 | 30 | 0.6 |
| 10 | 100 | 0 | 0.6 |
Acknowledgements
This research was supported by Departmental Start-Up Fund provided by the Holland Regenerative Medicine Program at the University of Nebraska Medical Center. This research was also supported by the COBRE Nebraska Center for Nanomedicine pilot grant (funded by NIGMS P30GM127200); NIAMS R21 AR078439; NIAMS R21 AR080906; and NIGMS R01 GM138552.The content in this publication was solely the responsibility of the authors and did not necessarily represent the official views of the NIH. The authors acknowledge Dr. Wen Xue for her assistance in in vitro cell viability test; Dr. Donghee Lee for his assistance in fluorescent microscopy; Dr. Bin Xue for paraffin section and histological staining of skin tissues; Dr. Wen Shi for his assistance in high-performance liquid chromatography (HPLC); and Dr. Noel Johnson for his assistance in in vivo safety study.
Contributor Information
Fan Zhao, Mary & Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, NE 68198, USA; Department of Surgery, University of Nebraska Medical Center, Omaha, NE 68198 USA.
Yajuan Su, Mary & Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, NE 68198, USA; Department of Surgery, University of Nebraska Medical Center, Omaha, NE 68198 USA.
Junying Wang, Mary & Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, NE 68198, USA; Department of Surgery, University of Nebraska Medical Center, Omaha, NE 68198 USA.
Svetlana Romanova, Department of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE 68198, USA.
Dominick J. DiMaio, Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198, USA
Jingwei Xie, Mary & Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, NE 68198, USA; Department of Surgery, University of Nebraska Medical Center, Omaha, NE 68198 USA.
Siwei Zhao, Mary & Dick Holland Regenerative Medicine Program, University of Nebraska Medical Center, Omaha, NE 68198, USA; Department of Surgery, University of Nebraska Medical Center, Omaha, NE 68198 USA.
Data availability
All relevant data are included in the main manuscript and the Supporting Information. Additional data are available from the corresponding authors upon reasonable request.
Reference
- [1].Eriksson E, Liu PY, Schultz GS, Martins-Green MM, Tanaka R, Weir D, Gould LJ, Armstrong DG, Gibbons GW, Wolcott R, Olutoye OO, Kirsner RS, Gurtner GC, Wound Repair Regen 2022, 30, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].C. D. C. o. D. F. W. Care, Diabetes Care 1999, 22, 1354. [DOI] [PubMed] [Google Scholar]
- [3].Graves N, Zheng H, Wound Practice & Research: Journal of the Australian Wound Management Association 2014, 22. [Google Scholar]
- [4].Clinton A, Carter T, Laboratory medicine 2015, 46, 277; [DOI] [PubMed] [Google Scholar]; Gajula B, Munnamgi S, Basu S, IJS Global Health 2020, 3, e16. [Google Scholar]
- [5].Malone M, Bjarnsholt T, McBain AJ, James GA, Stoodley P, Leaper D, Tachi M, Schultz G, Swanson T, Wolcott RD, Journal of wound care 2017, 26, 20. [DOI] [PubMed] [Google Scholar]
- [6].Ganesh K, Sinha M, Mathew-Steiner SS, Das A, Roy S, Sen CK, Advances in wound care 2015, 4, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Alves PJ, Barreto RT, Barrois BM, Gryson LG, Meaume S, Monstrey SJ, Int Wound J 2021, 18, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Anghel EL, DeFazio MV, Barker JC, Janis JE, Attinger CE, Plast Reconstr Surg 2016, 138, 82S. [DOI] [PubMed] [Google Scholar]
- [9].Wolcott R, Kennedy J, Dowd S, Journal of wound care 2009, 18, 54. [DOI] [PubMed] [Google Scholar]
- [10].Roy S, Elgharably H, Sinha M, Ganesh K, Chaney S, Mann E, Miller C, Khanna S, Bergdall VK, Powell HM, The Journal of pathology 2014, 233, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wolcott RD, Rumbaugh KP, James G, Schultz G, Phillips P, Yang Q, Watters C, Stewart PS, Dowd SE, Journal of wound care 2010, 19, 320. [DOI] [PubMed] [Google Scholar]
- [12].Schultz G, Bjarnsholt T, James GA, Leaper DJ, McBain AJ, Malone M, Stoodley P, Swanson T, Tachi M, Wolcott RD, Wound Repair and Regeneration 2017, 25, 744; [DOI] [PubMed] [Google Scholar]; Falcone M, De Angelis B, Pea F, Scalise A, Stefani S, Tasinato R, Zanetti O, Dalla Paola L, J Glob Antimicrob Resist 2021, 26, 140. [DOI] [PubMed] [Google Scholar]
- [13].Leren L, Johansen E, Eide H, Falk RS, Juvet LK, Ljoså TM, International wound journal 2020, 17, 466; [DOI] [PMC free article] [PubMed] [Google Scholar]; Eusen M, Brenaut E, Schoenlaub P, Saliou P, Misery L, Journal of the European Academy of Dermatology and Venereology 2016, 30, 1603; [DOI] [PubMed] [Google Scholar]; Dhar Y, Han Y, Engineered Regeneration 2020, 1, 64. [Google Scholar]
- [14].Lipsky BA, Hoey C, Clinical infectious diseases 2009, 49, 1541. [DOI] [PubMed] [Google Scholar]
- [15].Uçkay I, Kressmann B, Malacarne S, Toumanova A, Jaafar J, Lew D, Lipsky BA, BMC infectious diseases 2018, 18, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; Lipsky BA, Hoey C, Clin Infect Dis 2009, 49, 1541. [DOI] [PubMed] [Google Scholar]
- [16].Castaneda P, McLaren A, Tavaziva G, Overstreet D, Clinical Orthopaedics and Related Research® 2016, 474, 1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Flemming H-C, Neu TR, Wozniak DJ, Journal of bacteriology 2007, 189, 7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Singh R, Ray P, Das A, Sharma M, J Antimicrob Chemother 2010, 65, 1955; [DOI] [PubMed] [Google Scholar]; Nabb DL, Song S, Kluthe KE, Daubert TA, Luedtke BE, Nuxoll AS, Front Microbiol 2019, 10, 2803; [DOI] [PMC free article] [PubMed] [Google Scholar]; Stewart PS, Davison WM, Steenbergen JN, Antimicrob Agents Chemother 2009, 53, 3505; [DOI] [PMC free article] [PubMed] [Google Scholar]; Walters MC 3rd, Roe F, Bugnicourt A, Franklin MJ, Stewart PS, Antimicrob Agents Chemother 2003, 47, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Daddi Oubekka S, Briandet R, Fontaine-Aupart MP, Steenkeste K, Antimicrob Agents Chemother 2012, 56, 3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sarker RR, Tsunoi Y, Haruyama Y, Sato S, Nishidate I, J Biomed Opt 2022, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang H, Zhang H, Wang J, Yu J, Wei H, Sci Rep 2017, 7, 40182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Roy S, Santra S, Das A, Dixith S, Sinha M, Ghatak S, Ghosh N, Banerjee P, Khanna S, Mathew-Steiner S, Ghatak PD, Blackstone BN, Powell HM, Bergdall VK, Wozniak DJ, Sen CK, Ann Surg 2020, 271, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bullock AJ, Garcia M, Shepherd J, Rehman I, Sheila M, Applied Spectroscopy Reviews 2020, 55, 158. [Google Scholar]
- [24].Tseng BS, Zhang W, Harrison JJ, Quach TP, Song JL, Penterman J, Singh PK, Chopp DL, Packman AI, Parsek MR, Environ Microbiol 2013, 15, 2865; [DOI] [PMC free article] [PubMed] [Google Scholar]; Jefferson KK, Goldmann DA, Pier GB, Antimicrob Agents Chemother 2005, 49, 2467; [DOI] [PMC free article] [PubMed] [Google Scholar]; Doroshenko N, Tseng BS, Howlin RP, Deacon J, Wharton JA, Thurner PJ, Gilmore BF, Parsek MR, Stoodley P, Antimicrob Agents Chemother 2014, 58, 7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guillot AJ, Cordeiro AS, Donnelly RF, Montesinos MC, Garrigues TM, Melero A, Pharmaceutics 2020, 12, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Su Y, Wang H, Mishra B, Lakshmaiah Narayana J, Jiang J, Reilly DA, Hollins RR, Carlson MA, Wang G, Xie J, Molecular Pharmaceutics 2019, 16, 2011. [DOI] [PubMed] [Google Scholar]
- [27].Permana AD, Mir M, Utomo E, Donnelly RF, International journal of pharmaceutics: X 2020, 2, 100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Baelo A, Levato R, Julián E, Crespo A, Astola J, Gavaldà J, Engel E, Mateos-Timoneda MA, Torrents E, Journal of Controlled Release 2015, 209, 150; [DOI] [PubMed] [Google Scholar]; Teirlinck E, Samal SK, Coenye T, Braeckmans K, in Functionalized nanomaterials for the management of microbial infection, Elsevier, 2017, 49; [Google Scholar]; Marshall JS, The Journal of the Acoustical Society of America 2016, 139, EL228. [DOI] [PubMed] [Google Scholar]
- [29].Bridier A, Briandet R, Thomas V, Dubois-Brissonnet F, Biofouling 2011, 27, 1017; [DOI] [PubMed] [Google Scholar]; Stoodley P, Sauer K, Davies DG, Costerton JW, Annual review of microbiology 2002, 56, 187. [DOI] [PubMed] [Google Scholar]
- [30].Bjarnsholt T, Ciofu O, Molin S, Givskov M, Høiby N, Nature Reviews Drug Discovery 2013, 12, 791. [DOI] [PubMed] [Google Scholar]
- [31].Wattanakaroon W, Stewart PS, Archives of Oral Biology 2000, 45, 167. [DOI] [PubMed] [Google Scholar]
- [32].Del Pozo JL, Rouse MS, Mandrekar JN, Sampedro MF, Steckelberg JM, Patel R, Antimicrobial agents and chemotherapy 2009, 53, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Froughreyhani M, Salemmilani A, Mozafari A, Hosein-Soroush M, Journal of Clinical and Experimental Dentistry 2018, 10, e1223; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zou P, Li P, Liu J, Cao P, Luan Q, Journal of Microbiology 2022, 60, 70. [DOI] [PubMed] [Google Scholar]
- [34].del Pozo JL, Rouse MS, Mandrekar JN, Sampedro MF, Steckelberg JM, Patel R, Antimicrob Agents Chemother 2009, 53, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Costerton JW, Ellis B, Lam K, Johnson F, Khoury AE, Antimicrobial agents and chemotherapy 1994, 38, 2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaweski S, Baldwin RC, Wong RK, Manders EK, Plastic and reconstructive surgery 1993, 92, 1342; [PubMed] [Google Scholar]; Nicoli S, Santi P, Journal of controlled release 2006, 111, 89. [DOI] [PubMed] [Google Scholar]
- [37].Datta D, Panchal DS, Venuganti VVK, International Journal of Pharmaceutics 2021, 602, 120663. [DOI] [PubMed] [Google Scholar]
- [38].Nicoli S, Santi P, J Control Release 2006, 111, 89. [DOI] [PubMed] [Google Scholar]
- [39].Kim YW, Subramanian S, Gerasopoulos K, Ben-Yoav H, Wu H-C, Quan D, Carter K, Meyer MT, Bentley WE, Ghodssi R, npj Biofilms and Microbiomes 2015, 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Aycock KN, Davalos RV, Bioelectricity 2019, 1, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Schulze C, Peters M, Baumgartner W, Wohlsein P, Vet Pathol 2016, 53, 1018; [DOI] [PubMed] [Google Scholar]; Lateef Z, Stuart G, Jones N, Mercer A, Fleming S, Wise L, Int J Mol Sci 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Novickij V, Svediene J, Paskevicius A, Markovskaja S, Lastauskiene E, Zinkeviciene A, Girkontaite I, Novickij J, Molecules 2018, 23; [DOI] [PMC free article] [PubMed] [Google Scholar]; Tirono M, Pertanika Journal of Science & Technology 2021, 29; [Google Scholar]; Geboers B, Scheffer HJ, Graybill PM, Ruarus AH, Nieuwenhuizen S, Puijk RS, van den Tol PM, Davalos RV, Rubinsky B, de Gruijl TD, Radiology 2020, 295, 254. [DOI] [PubMed] [Google Scholar]
- [43].Novickij V, Zinkeviciene A, Perminaite E, Cesna R, Lastauskiene E, Paskevicius A, Svediene J, Markovskaja S, Novickij J, Girkontaite I, Sci Rep 2018, 8, 14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Golberg A, Broelsch GF, Vecchio D, Khan S, Hamblin MR, Austen WG Jr, Sheridan RL, Yarmush ML, Journal of Burn Care & Research 2015, 36, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Davalos RV, Rubinsky B, International journal of heat and mass transfer 2008, 51, 5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhao S, Tseng P, Grasman J, Wang Y, Li W, Napier B, Yavuz B, Chen Y, Howell L, Rincon J, Advanced Materials 2018, 30, 1800598. [DOI] [PubMed] [Google Scholar]
- [47].Zhao S, Mehta AS, Zhao M, Cellular and molecular life sciences 2020, 77, 2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sieg A, Wascotte V, Journal of drug targeting 2009, 17, 690. [DOI] [PubMed] [Google Scholar]
- [49].Chopra P, Hao J, Li SK, International journal of pharmaceutics 2010, 388, 107; [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu H, Liu C, Jiang L, Liu J, Yang Q, Guo Z, Cai X, Electroanalysis: An International Journal Devoted to Fundamental and Practical Aspects of Electroanalysis 2008, 20, 170. [Google Scholar]
- [50].Abe I, Energy carriers and conversion systems 2009, 1. [Google Scholar]
- [51].Zhao F, Fan S, Ghate D, Romanova S, Bronich TK, Zhao S, Advanced Materials 2022, 34, 2107315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Banga AK, Electrically assisted transdermal and topical drug delivery, CRC Press, 1998; [Google Scholar]; Dixit N, Bali V, Baboota S, Ahuja A, Ali J, Current drug delivery 2007, 4, 1. [DOI] [PubMed] [Google Scholar]
- [53].Wang M, Yang Y, Min J, Song Y, Tu J, Mukasa D, Ye C, Xu C, Heflin N, McCune JS, Nature Biomedical Engineering 2022, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; Chaudon M-J, Hulea O, Yakoub A, Monnier P, Saadaoui M, Medical Engineering & Physics 2022, 107, 103861; [DOI] [PubMed] [Google Scholar]; Bolat G, De la Paz E, Azeredo NF, Kartolo M, Kim J, de Loyola e Silva AN, Rueda R, Brown C, Angnes L, Wang J, Analytical and Bioanalytical Chemistry 2022, 1; [DOI] [PubMed] [Google Scholar]; Wu C, Jiang P, Li W, Guo H, Wang J, Chen J, Prausnitz MR, Wang ZL, Advanced Functional Materials 2020, 30, 1907378; [Google Scholar]; Lim C, Hong YJ, Jung J, Shin Y, Sunwoo S-H, Baik S, Park OK, Choi SH, Hyeon T, Kim JH, Science Advances 2021, 7, eabd3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yarmolenko PS, Moon EJ, Landon C, Manzoor A, Hochman DW, Viglianti BL, Dewhirst MW, International Journal of Hyperthermia 2011, 27, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].von Euler H, Söderstedt A, Thörne A, Olsson JM, Yongqing G, Bioelectrochemistry 2002, 58, 163. [DOI] [PubMed] [Google Scholar]
- [56].Alves DR, Booth SP, Scavone P, Schellenberger P, Salvage J, Dedi C, Thet N-T, Jenkins ATA, Waters R, Ng KW, Frontiers in cellular and infection microbiology 2018, 8, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Su Y, Mainardi VL, Wang H, McCarthy A, Zhang YS, Chen S, John JV, Wong SL, Hollins RR, Wang G, ACS nano 2020, 14, 11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bullock AJ, Garcia M, Shepherd J, Rehman I, Sheila M, Applied Spectroscopy Reviews 2019, 55, 158. [Google Scholar]
- [59].Del Pozo J, Rouse M, Patel R, The International journal of artificial organs 2008, 31, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Van der Borden A, Van der Mei H, Busscher H, Journal of Biomedical Materials Research Part B: Applied Biomaterials: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials 2004, 68, 160. [DOI] [PubMed] [Google Scholar]
- [61].Hong SH, Jeong J, Shim S, Kang H, Kwon S, Ahn KH, Yoon J, Biotechnol Bioeng 2008, 100, 379. [DOI] [PubMed] [Google Scholar]
- [62].Van der Borden A, Van der Mei H, Busscher H, Biomaterials 2005, 26, 6731; [DOI] [PubMed] [Google Scholar]; Van Der Borden AJ, Van Der Werf H, Van Der Mei HC, Busscher HJ, Applied and environmental microbiology 2004, 70, 6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jass J, Lappin-Scott HM, Journal of Antimicrobial Chemotherapy 1996, 38, 987; [DOI] [PubMed] [Google Scholar]; Jass J, Costerton JW, Lappin-Scott H, Journal of industrial microbiology and biotechnology 1995, 15, 234. [DOI] [PubMed] [Google Scholar]
- [64].Yuan Z, Lin C, He Y, Tao B, Chen M, Zhang J, Liu P, Cai K, Acs Nano 2020, 14, 3546. [DOI] [PubMed] [Google Scholar]
- [65].Pumerantz A, Muppidi K, Agnihotri S, Guerra C, Venketaraman V, Wang J, Betageri G, International journal of antimicrobial agents 2011, 37, 140; [DOI] [PubMed] [Google Scholar]; Van Hal SJ, Fowler VG Jr, Clinical infectious diseases 2013, 56, 1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Castaneda P, 2020.
- [67].Datta D, Panchal DS, Venuganti VVK, Int J Pharm 2021, 602, 120663. [DOI] [PubMed] [Google Scholar]
- [68].Ita K, Journal of Drug Targeting 2016, 24, 386. [DOI] [PubMed] [Google Scholar]
- [69].Hull W, J Appl Res 2002, 2, 1. [Google Scholar]
- [70].Ottink KD, Strahm C, Muller-Kobold A, Sendi P, Wouthuyzen-Bakker M, Journal of bone and joint infection 2019, 4, 167; [DOI] [PMC free article] [PubMed] [Google Scholar]; Facchetti G, Rimoldi I, “New hybrid imine reductases based on Vancomycin for the asymmetric reduction of cyclic imines in aqueous buffer”, presented at New Trends in Organic Synthesis, 2019; [Google Scholar]; Kanikkannan N, BioDrugs 2002, 16, 339. [DOI] [PubMed] [Google Scholar]
- [71].Nicasio AM, Bulitta JB, Lodise TP, D’Hondt RE, Kulawy R, Louie A, Drusano GL, Antimicrobial agents and chemotherapy 2012, 56, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Eagle H, Musselman A, The Journal of experimental medicine 1948, 88, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jarrad AM, Blaskovich MA, Prasetyoputri A, Karoli T, Hansford KA, Cooper MA, Frontiers in Microbiology 2018, 9, 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].LaPlante KL, Woodmansee S, Antimicrobial agents and chemotherapy 2009, 53, 3880; [DOI] [PMC free article] [PubMed] [Google Scholar]; Kirkham S, Castelletto V, Hamley IW, Inoue K, Rambo R, Reza M, Ruokolainen J, Chemphyschem 2016, 17, 2118; [DOI] [PubMed] [Google Scholar]; Wood TM, Martin NI, Medchemcomm 2019, 10, 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Craft KM, Nguyen JM, Berg LJ, Townsend SD, MedChemComm 2019, 10, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zupančič O, Partenhauser A, Lam HT, Rohrer J, Bernkop-Schnürch A, European Journal of Pharmaceutical Sciences 2016, 81, 129. [DOI] [PubMed] [Google Scholar]
- [77].Ho SW, Jung D, Calhoun JR, Lear JD, Okon M, Scott WR, Hancock RE, Straus SK, European biophysics journal 2008, 37, 421. [DOI] [PubMed] [Google Scholar]
- [78].Roustit M, Blaise S, Cracowski JL, British journal of clinical pharmacology 2014, 77, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kim M, Kim H, Kang HW, Lasers in surgery and medicine 2018, 50, 661; [DOI] [PubMed] [Google Scholar]; Macêdo J, Ferreira Júnior JA, Nascimento KA, Lacerda MS, Pereira NE, Pedroso PM, Pesquisa Veterinária Brasileira 2018, 38, 2088. [Google Scholar]
- [80].Sun J, Li M, Lin M, Zhang B, Chen X, Advanced Materials 2021, 33, 2104402. [DOI] [PubMed] [Google Scholar]
- [81].Mohammed MI, Makky AM, Teaima MH, Abdellatif MM, Hamzawy MA, Khalil MA, Drug delivery 2016, 23, 1558. [DOI] [PubMed] [Google Scholar]
- [82].Ziesmer J, Tajpara P, Hempel NJ, Ehrström M, Melican K, Eidsmo L, Sotiriou GA, Advanced Materials Technologies 2021, 6, 2001307; [DOI] [PMC free article] [PubMed] [Google Scholar]; Woodhouse I, Nejati S, Selvamani V, Jiang H, Chittiboyina S, Grant J, Mutlu Z, Waimin J, Abutaleb NS, Seleem MN, ACS Applied Bio Materials 2021, 4, 5405. [DOI] [PubMed] [Google Scholar]
- [83].Scriboni AB, Couto VM, L. N. d. M. Ribeiro, I. A. Freires, F. C. Groppo, E. De Paula, M. Franz-Montan, K. Cogo-Müller, Frontiers in Pharmacology 2019, 10, 1401; [DOI] [PMC free article] [PubMed] [Google Scholar]; Li C-H, Landis RF, Makabenta JM, Nabawy A, Tronchet T, Archambault D, Liu Y, Huang R, Golan M, Cui W, Materials Horizons 2021, 8, 1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Choi M, Hasan N, Cao J, Lee J, Hlaing SP, Yoo J-W, International journal of biological macromolecules 2020, 142, 680. [DOI] [PubMed] [Google Scholar]
- [85].Wood TK, Microbial biotechnology 2017, 10, 1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Høiby N, Bjarnsholt T, Moser C, Bassi G, Coenye T, Donelli G, Hall-Stoodley L, Holá V, Imbert C, Kirketerp-Møller K, Clinical microbiology and infection 2015, 21, S1. [DOI] [PubMed] [Google Scholar]
- [87].Balaban NQ, Gerdes K, Lewis K, McKinney JD, Nat Rev Microbiol 2013, 11, 587. [DOI] [PubMed] [Google Scholar]
- [88].Moran F, Leonard T, Hawthorne S, Hughes CM, McCrum-Gardner E, Johnson MI, Rakel BA, Sluka KA, Walsh DM, The Journal of Pain 2011, 12, 929. [DOI] [PubMed] [Google Scholar]
- [89].Tamaki H, Tomori K, Yotani K, Ogita F, Sugawara K, Kirimto H, Onishi H, Yamamoto N, Kasuga N, Journal of musculoskeletal & neuronal interactions 2014, 14, 220. [PubMed] [Google Scholar]
- [90].Zhao Z, Zhu K, Li Y, Zhu Z, Pan L, Pan T, Borgens RB, Zhao M, Bioelectricity 2020, 2, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lenhard JR, Brown T, Rybak MJ, Meaney CJ, Norgard NB, Bulman ZP, Brazeau DA, Gill SR, Tsuji BT, Antimicrobial Agents and Chemotherapy 2015, 60, 1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are included in the main manuscript and the Supporting Information. Additional data are available from the corresponding authors upon reasonable request.