

Abstract
Millepachine, a bioactive natural product isolated from the seeds of Millettia pachycarpa, is reported to display potential antitumor activity. In this study, novel indole-containing hybrids derived from millepachine were designed, synthesized and evaluated for their antitumor activities. Among all the compounds, compound 14b exhibited the most potent cytotoxic activity against five kinds of human cancer cell lines, with IC50 values ranging from 0.022 to 0.074 μM, making it almost 100 times more active than millepachine. Valuable structure–activity relationships (SARs) were obtained. Furthermore, the mechanism studies showed that compound 14b induced cell-cycle arrest at the G2/M phase by inhibiting tubulin polymerization and further induced cell apoptosis through reactive oxygen species (ROS) accumulation and mitochondrial membrane potential (MMP) collapse. In addition, the low cytotoxicity toward normal human cells and equivalent sensitivity towards drug-resistant cells of compound 14b highlighted its potential for the development of antitumor drugs.
Keywords: tubulin polymerization inhibitors, millepachine, antitumor
1. Introduction
Natural products (NPs) and their structural analogues are historically valuable sources of lead compounds for drug discovery, especially for cancer and infectious diseases [1]. However, the investigation of NPs for drug discovery is hampered by various inevitable difficulties, such as the low amount of NPs available for pharmacological evaluation, the limited availability of natural resources, the difficulty of total chemical synthesis, and the relatively low bioavailability in NPs [2,3]. Therefore, searching for small molecular compounds based on the structures of natural products is an important strategy for drug discovery [4]. The high structural diversity and various bioactivities of NPs provide diverse scaffolds for NP-based drug discovery.
Microtubules, which consist of α- and β-tubulin, are dynamic cytoskeleton components involved in a range of intracellular processes, such as substance transportation, beating of cilia and flagella, cell division and mitosis [5]. Tubulin inhibitors that interfere with the dynamic equilibrium of polymerization and depolymerization are also named antimitotic agents, and they not only perturb mitosis but also arrest cells during the interphase [6]. The prominent role of tubulin in cell division makes it an effective target for antitumor drugs, resulting in tremendous efforts in drug discovery targeting the tubulin–microtubule system [7,8].
Millepachine is a bioactive natural product isolated from the seeds of Millettia pachycarpa that is reported to have potential antitumor activity at the micromole level in cell-based evaluations [9]. On the basis of these reports, various attempts have been undertaken to obtain diverse millepachine-derived compounds, aiming to increase its antitumor activity [10,11,12]. Most mechanism studies demonstrated that millepachine exerted anticancer activity as a tubulin polymerization inhibitor that disturbed mitotic spindle formation and causes cell-cycle arrest at the G2/M phase, followed by apoptosis [13,14].
The indole moiety is one of the most broadly N-heterocyclic frameworks occurring in natural products and in the area of medicinal chemistry [15]. The interesting molecular architecture and wide spectrum of pharmacological activities of indole make it an important pharmacophore present in various drugs. Moreover, indole and its derivatives are well-known, privileged scaffolds that have been used in the discovery and development of many antitumor drug molecules [16,17]. Indole derivatives, such as tubulin inhibitors, comprise a vast area in chemotherapeutic drug discovery [18,19]. They include numerous natural indole-containing derivatives, such as vinca alkaloid, moroidin and diazionamide A, and synthetic indole-based compounds, such as indibulin, rosabulin and MKC-1, have been reported as tubulin inhibitors with prominent positions in clinical trials in anticancer drug development [20].
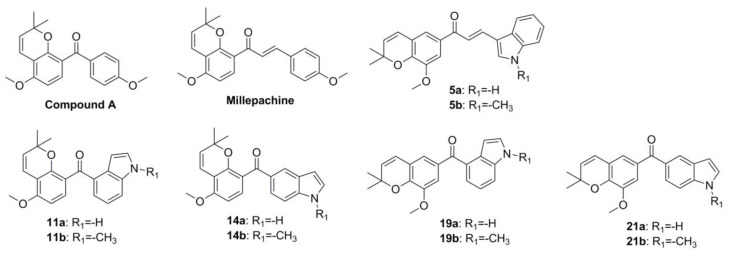
In recent years, our group has been committed to the design and development of indole-containing antitumor agents that act as tubulin polymerization inhibitors [21,22,23,24,25,26]. In this study, as shown in Figure 1, starting from the structure of millepachine, we moved the position of the 2,2-dimethylbenzopyran group and introduced the indole ring to obtain millepachine-indole derivatives. Then, by replacing the α,β-unsaturated ketones with a benzophenone moiety and by simultaneously infusing the indole heterocycle, the target benzophenone-indole derivatives were obtained. The preliminary cytotoxicity screening revealed various structure–activity relationships (SARs). Tubulin polymerization inhibition evaluation, analysis of cell-cycle arrest and ROS production, MMP detection and molecular docking studies were also conducted to elucidate the derivatives’ antitumor mechanism.
Figure 1.

The design strategy for indole-containing hybrids derived from millepachine.
2. Results and Discussion
2.1. Chemistry
The synthesis route for target compounds 5a and 5b is outlined in Scheme 1. The important intermediate 3 was obtained according to the method we previously reported [21]. Then, compound 3 was reacted with different substituted indole aldehydes through aldol condensation to obtain target compound 5.
Scheme 1.

Synthesis route of target compounds 5a-5b. Reagents and conditions: (a) 3-chloro-3-methylbut-1-yne, DBU, CuCl2, CH3CN; (b) pyridine, reflux; (c) EtOH, piperidine, reflux.
The synthesis route of target compounds 11 and 14 is outlined in Scheme 2. Firstly, the important intermediate 8 was obtained completely according to the method we previously reported [27]. Then, compound 8 was reacted with different substituted indole aldehydes to afford diarylmethanol derivative 10 or 13 in the presence of n-BuLi. The oxidation of 10 and 13 with pyridinium chlorochromate (PCC) provided target compounds 11 and 14, respectively.
Scheme 2.

Synthesis route of target compounds 11a-11b and 14a-14b. Reagents and conditions: (a) 3-chloro-3-methylbut-1-yne, CuCl2, DBU, CH3CN, 0 °C; (b) pyridine, 120 °C; (c) n-BuLi, anhydrous THF, −78 °C; (d) PCC, CH2Cl2, rt.
The synthesis routes of target compounds 19 and 21 in Scheme 3 are under the same procedures as described above.
Scheme 3.

Synthesis route of target compounds 19a-19b and 21a-21b. Reagents and conditions: (a) 3-chloro-3-methylbut-1-yne, CuCl2, DBU, CH3CN, 0 °C; (b) pyridine,120 °C; (c) n-BuLi, anhydrous THF, −78 °C; (d) PCC, CH2Cl2, rt.
2.2. In Vitro Antiproliferative Activity Evaluation and SAR Summary
In order to evaluate the antiproliferative activity of the designed compounds and to obtain some valuable structure–activity relationships (SARs), the initial cytotoxicity screening using a cell counting kit-8 (CCK-8) assay was performed. In the CCK-8 assay, a chemical reagent (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium sodium salt), named WST-8, which can be reduced by some dehydrogenase in mitochondria to generate orange formazan in the presence of an electron coupling reagent, was used. Hence, there is a linear relationship between the shades of orange and the number of live cells, which can be used to reflect the proliferation of cells. In our previous studies, the microtubule-targeting agents (MTAs) often displayed extraordinary cytotoxicity towards solid tumors. In this study, we also selected some common cancer cell lines, including A549 (human non-small-cell-lung cancer cell line), Caski (human epithelial cervical cancer cell line), HepG2 (human liver carcinoma cell line), C42B (Human prostate cancer line C42B) and MCF-7 (human breast carcinoma cell line) derived from different tissues for preliminary activity screening. On the base structure of millepachine, we firstly moved the position of the 2,2-dimethylbenzopyran ring of millepachine and replaced the benzene ring with the indole ring to obtain compounds 5a and 5b. Then, we replaced the α,β-unsaturated ketones with a benzophenone moiety to obtain compounds 11, 14, 19 and 21. Overall, the effects of the position of the 2,2-dimethylbenzopyran ring, intermediate linking groups (α,β-unsaturated ketones or benzophenone moiety) and substituents on the indole ring were mainly investigated. As shown in Table 1, the general SARs were summarized as follows: (a) The reference compound millepachine displayed micromolar IC50 values in a range of cancer cell lines (IC50 = 2.566–6.712 μM); compared with millepachine, the antiproliferative activities of all of the compounds in this study have been greatly improved. (b) The introduction of the indole ring greatly improved the activity. Replacing the p-methoxyphenyl moiety of compound A (IC50 = 0.226–0.892 μM, previously reported by our group [21]) with an indole moiety to obtain the corresponding compound 14b (IC50 = 0.022–0.074 μM), the antiproliferative activities were increased 27.3–78.0-fold; comparing the chemical structures of compound 14b and compound A reminded us of the favorable effect of introducing indole moiety. (c) Moving the position of the 2,2-dimethylbenzopyran ring of millepachine was unfavorable to the activity, evidenced by comparing the activities of compounds 11a vs. 19a, 11b vs. 19b, 14a vs. 21a and 14b vs. 21b. (d) N-methylation of the indole ring was favorable for the activity. By comparing the antiproliferative activity results of compounds 5a vs. 5b, 11a vs. 11b, 14a vs. 14b, 19a vs. 19b and 21a vs. 21b, the activities were generally improved by about 1.2–4.4 folds due to N-methylation. (e) The position of aldol condensation has a great effect on the activity. The indole-5-carbaldehyde-derived products displayed better activities than the corresponding indole-4-carbaldehyde products, with the antiproliferative activities improved by 6.13–24.11 folds, for example, comparing the results of compounds 11a vs. 14a, 11b vs. 14b, 19a vs. 21a and 19b vs. 21b. In summary, from the rational design and activity screening, we obtained some valuable SARs and chose compound 14b with as remarkable a cytotoxicity as the optimized compound for further study.
Table 1.
The antiproliferative activities evaluated in five kinds of human cancer cell lines a.
| |||||
|---|---|---|---|---|---|
| Compds | IC50, Mean ± SE (μΜ) b | ||||
| A549 | Caski | HepG2 | C42B | MCF-7 | |
| 5a | 0.812 ± 0.022 | 1.929 ± 0.035 | 0.963 ± 0.032 | 1.377 ± 0.021 | 1.553 ± 0.014 |
| 5b | 0.362 ± 0.017 | 0.729 ± 0.033 | 0.461 ± 0.017 | 0.577 ± 0.021 | 0.350 ± 0.011 |
| 11a | 0.631 ± 0.018 | 0.832 ± 0.011 | 0.998 ± 0.034 | 0.911 ± 0.034 | 0.816 ± 0.045 |
| 11b | 0.208 ± 0.009 | 0.516 ± 0.012 | 0.454 ± 0.021 | 0.455 ± 0.015 | 0.651 ± 0.024 |
| 14a | 0.072 ± 0.004 | 0.086 ± 0.009 | 0.142 ± 0.014 | 0.089 ± 0.021 | 0.082 ± 0.011 |
| 14b | 0.022 ± 0.002 | 0.037 ± 0.003 | 0.074 ± 0.013 | 0.051 ± 0.005 | 0.027 ± 0.006 |
| 19a | 1.321 ± 0.016 | 2.311 ± 0.019 | 3.098 ± 0.021 | 2.411 ± 0.042 | 1.775 ± 0.004 |
| 19b | 0.642 ± 0.081 | 1.098 ± 0.032 | 0.898 ± 0.009 | 1.076 ± 0.034 | 1.021 ± 0.015 |
| 21a | 0.105 ± 0.011 | 0.211 ± 0.012 | 0.441 ± 0.071 | 0.209 ± 0.020 | 0.121 ± 0.021 |
| 21b | 0.082 ± 0.010 | 0.106 ± 0.012 | 0.097 ± 0.031 | 0.107 ± 0.006 | 0.099 ± 0.031 |
| Compd. A | 0.226 ± 0.035 | 0.892 ± 0.024 | 0.758 ± 0.017 | 0.526 ± 0.007 | 0.456 ± 0.021 |
| Millepachine | 2.566 ± 0.131 | 6.712 ± 0.233 | 3.881 ± 0.244 | 3.176 ± 0.384 | 4.528 ± 0.338 |
a Cell lines were treated with different concentrations of the compounds for 48 h. Cell viability was measured by a CCK-8 assay, as described in the Materials and Methods section. b IC50 values are indicated as the mean ± SE (standard error) of at least three independent experiments.
2.3. Cytotoxicity of Compound 14b in Human Normal Cells Activities
High cytotoxicity is one of the most limiting factors in chemotherapeutics. Hence, the cytotoxicity of compound 14b towards three kinds of human non-tumorigenic cell lines including L-02 (normal human hepatocytes), RWPE-1 (human prostate epithelial cells) and MCF-10A (normal human mammary epithelial cells) was also evaluated. As shown in Table 2, compound 14b has much less cytotoxicity against normal cells, which was evidenced by the high selectivity ratio (16.97–231.7) between the human non-tumorigenic cell lines and the corresponding human cancer cell lines.
Table 2.
Cytotoxicity selectivity of compound 14b toward human non-tumorigenic cell lines and cancer cell lines a.
| Cell Lines | IC50, Mean ± SE (μM) b | Selectivity Ratio c |
|---|---|---|
| Hep-G2 | 0.074 ± 0.013 | 16.97 |
| L-02 | 1.256 ± 0.214 | |
| C42B | 0.051 ± 0.005 | 72.51 |
| RWPE-1 | 3.698 ± 0.036 | |
| MCF-7 | 0.027 ± 0.006 | 231.7 |
| MCF-10A | 6.256 ± 0.311 |
a Cell lines were treated with different concentrations of the compounds for 48 h. Cell viability was measured by a CCK-8 assay, as described in the Materials and Methods section. b IC50 values are indicated as the mean ± SE (standard error) of at least three independent experiments. c Selectivity ratio = (IC50 of human normal cells)/(IC50 of corresponding cancer cell lines).
2.4. Cytotoxicity of Compound 14b towards Drug-Resistant Cancer Cell Lines
Besides the high cytotoxicity, drug resistance is the other limiting factor of chemotherapeutics. In this study, the cytotoxicities of compound 14b towards five kinds of drug-resistant cancer cell lines were also evaluated. Except three kinds of cell lines that are resistant to three drugs commonly used in clinical practice, two kinds of cell lines that are resistant to MTAs were also evaluated. As listed in Table 3, the resistance index of compound 14b was 0.82–1.88, which demonstrated that 14b exhibited almost similar potent activities between the parental cells and the drug-resistant cells.
Table 3.
Cytotoxicity of compound 14b toward drug-resistant cancer cell lines a.
| Cell Lines | IC50, Mean ± SE (μM) b | Resistance Index c |
|---|---|---|
| A549 | 0.022 ± 0.002 | 1.59 |
| A549/CDDP d | 0.035 ± 0.021 | |
| C42B | 0.051 ± 0.005 | 0.82 |
| C42B/ENZR e | 0.042 ± 0.011 | |
| MCF-7 | 0.027 ± 0.006 | 1.88 |
| MCF-7/DOX f | 0.051 ± 0.024 | |
| A2780 | 0.036 ± 0.004 | 1.17 |
| A2780/TAX g | 0.042 ± 0.011 | |
| HCT-8 | 0.089 ± 0.015 | 1.37 |
| HCT-8/VCR h | 0.122 ± 0.036 |
a Cell lines were treated with different concentrations of the compounds for 48 h. Cell viability was measured by a CCK-8 assay, as described in the Materials and Methods section. b IC50 values are indicated as the mean ± SE (standard error) of at least three independent experiments. c Resistance index = (IC50 of drug-resistant cell lines)/(IC50 of corresponding parent cancer cell lines); d A549/CDDP: A549 cell line resistant to cisplatin; e C42B/ENZR: C42b cell line resistant to Enzalutamide; f MCF-7/DOX: MCF-7 cell line resistant to doxorubicin; g A2780/TAX: A2780 cell line resistant to taxol; h HCT-8/VCR: HCT-8 cell line resistant to vincristine.
2.5. Compound 14b Inhibited Tubulin Polymerization
Tubulin–microtubule homeostasis is crucial for cell mitosis. In order to investigate the effect of compound 14b on the tubulin–microtubule system, an in vitro tubulin polymerization inhibition assay, intracellular microtubule morphology detection, a molecular docking study and a colchicine competitive inhibition assay were performed. To our knowledge, the agents that interfere with tubulin are classified as microtubule stabilizing agents (MSAs) such as paclitaxel or microtubule destabilizing agents (MDAs) such as colchicine [28]. As shown in Figure 2A and 2B, compound 14b inhibited tubulin polymerization in a colchicine-like manner, which was completely different from that of paclitaxel. Moreover, 14b inhibited tubulin polymerization in a dose-dependent manner (IC50 = 2.07 ± 0.15 μM). As shown in Figure 2C, the immunofluorescence assay demonstrated that the intracellular tubulin-microtubule homeostasis was heavily disturbed by compound 14b, evidenced by the gradually shrunken microtubule to the nucleus, and the disappearance of slim and fibrous filaments. A molecular docking study was adopted to investigate the potential mode of compound 14b to the colchicine binding site of α,β-tubulin. As shown in Figure 2D, 14b could occupy the colchicine binding site of tubulin in agreement with the X-ray structure complex of colchicine-α,β-tubulin (PDB code:1SA0). The 2,2-dimethylbenzopyran moiety was positioned in the binding cavity buried in the β-subunit and formed hydrophobic interactions (σ-π conjugate) with the two key amino acids of β-tubulin (Leuβ248 and Leuβ255). The indole part of 14b increased the hydrophobic interaction with the Met259 and Thr314 residues. In order to rationalize the increase in potency, we performed the superposition of 14b and millepachine using a docking study. As the results show in Figure S1 in the Supplementary Materials, the indole ring of 14b formed a π-H reaction with Ala12 of β-tubulin, which was not observed in millepachine. Additionally, the benzophenone moiety of 14b enables it to better trap the binding cavity buried in the β-subunit. These results might partially explain the increase in potency of 14b. Finally, the results of the colchicine competitive inhibition assay (Figure 2E) show that the IC50 value of compound 14b inhibiting the binding of colchicine to tubulin was 1.04 ± 0.02 μM, which was a little better than that of the reference compound colchicine (IC50 = 1.67 μM), further validating the fact that compound 14b inhibited tubulin polymerization in a colchicine-like manner.
Figure 2.

Effect of compound 14b on tubulin. (A,B) In vitro tubulin polymerization assay of compound 14b (5 μM), paclitaxel (TAX, 5 μM), colchicine (5 μM) and millepachine (5 μM). (C) Immunofluorescence assay of compound 14b interferes with intracellular microtubules with an LSM 570 laser confocal microscope (Carl Zeiss, Jena, Germany). A549 cells were treated with or without compound 14b (10, 20 and 50 nM), taxol (100 nM) and colchicine (5 μM) for 12 h, and followed by immunofluorescence. The experiments were performed at least three times, and representative images are shown. Scale bars are 10 μm. (D) Superimposition of the docked conformation of the compound 14b on top of the X-ray structure of DAMA-colchicine (PDB code: 1SA0). The backbone of tubulin is shown in ribbon representation (α-tubulin, yellow; β-tubulin, green). (E) Competitive combining capacity of compound 14b and colchicine by the colchicine competition binding assay. The treated concentrations of compound 14b and colchicine were 10, 5, 2.5, 1.25, 0.625 and 0.3125 μM. Additionally, the IC50 values of compound 14b and colchicine when inhibiting the binding of colchicine to tubulin were 1.04 ± 0.02 μM and 1.67 ± 0.05 μM, respectively. The data were shown as mean ± SE (standard error) of at least three independent experiments.
2.6. Compound 14b Induced Cell Cycle Arrest at the G2/M Phase
Microtubules play an important role in maintaining the normal process of cell mitosis. Since the above results demonstrated that compound 14b could disturb tubulin–microtubule homeostasis, it is reasonable to speculate that compound 14b may also interfere with the cell cycle. As the results of the flow cytometry analysis show in Figure 3, after the A549 cells were treated with compound 14b for 24 h, the percentage of cells located in the G2/M phase increased from 14.92% in the normal group to 26.88%, 35.02% and 52.67% in the 10 nM-, 20 nM- and 50 nM-treated groups, respectively. These results demonstrated that compound 14b induced cell cycle arrested at the G2/M phase in a dose-dependent manner.
Figure 3.

Compound 14b induced cell cycle arrest at the G2/M phase. (A) The PI-stained DNA content detected by flow cytometry. A549 cells were treated with or without compound 14b at concentrations of 10, 20 and 50 nM for 24 h; then, the cells were harvested for cell cycle analysis by flow cytometry. (B) Quantitative analysis of the percentage of cells in each cell cycle phase by CytExpert analysis software. The experiments were performed at least three times, and the results of the representative experiments are shown.
2.7. Induction of Apoptosis by Compound 14b
To further elucidate the mechanism of cell death caused by the cytotoxicity of compound 14b, the Hoechst 33342 fluorochrome was used to observe the nucleus morphology, Annexin V-FITC double staining was performed to distinguish the apoptotic cells, and the caspase 3 colorimetric assay kit was used to detect the activity of caspase 3. As shown in Figure 4A, after A549 cells treated with compound 14b at concentrations of 10, 20 and 50 nM, an increasing number of cells displayed condensed nuclei, which is a typical morphology of apoptotic cells. At the early stage of apoptosis, different types of cells turn phosphatidylserine (PS) out toward the cell surface, that is, the outside of the cell membrane, which can be selectively marked with Annexin V. Additionally, in the late stage of apoptosis, the integrity of cell membrane is lost and can be stained with PI. The flow cytometry results of Annexin V-FITC double staining in Figure 4B showed that, when A549 cells were treated with compound 14b for 24 h, significant early apoptotic cells (Annexin V+/PI−) were observed. Additionally, when the treatment time was extended to 48 h, a gradually increasing number of late apoptotic (Annexin V+/PI+) cells were observed. These results demonstrated that the compound 14b treatment caused cell apoptosis in a dose- and time-dependent manner. Finally, considering the activation of caspase 3 is a central event in the process of apoptosis, the activity of caspase 3 was also evaluated. As shown in Figure 4C, the activity of caspase 3 was significantly activated and increased in a dose- and time-dependent manner. In summary, the above results indicated that compound 14b caused A549 cell death predominantly through the induction of apoptosis.
Figure 4.

Induction of apoptosis by compound 14b. (A) The Hoechst 33342 staining of A549 cells after treatment with compound 14b at the indicated concentrations for 24 h. (B) The percentages of cells in each apoptosis quantitated by flow cytometry through Annexin V-FITC double staining analysis. Upper left quadrant (Annexin V−/PI+): necrotic cells; upper right quadrant (Annexin V+/PI+): late apoptotic cells; bottom left quadrant (Annexin V−/PI−): live cells; bottom right quadrant (Annexin V+/PI−): early apoptotic cells. (C) The caspase 3 activity detected by the caspase 3 colorimetric assay kit after A549 cells were treated with compound 14b at concentrations of 10, 20 and 50 nM for 24 h. The experiments were performed at least three times, and the results of the representative experiments are shown. Data are presented as mean ± SEM. ** p < 0.01, *** p < 0.001 vs. control group; significant difference as compared with the control group by t test.
2.8. Compound 14b Treatment Leads to ROS Accumulation and MMP Decrease
The intrinsic mitochondrial pathway is one of the most well-studied mechanisms relevant to cell apoptosis [29]. ROS accumulation and mitochondrial membrane potential (MMP) alteration play crucial roles in this pathway. In this study, a fluorescent dye including DCFH-DA and JC-1 was used to monitor the intracellular ROS and MMP, respectively. As shown in Figure 5A, the level of ROS significantly rose abruptly in a dose-dependent manner after the A549 cells was treated with compound 14b at concentrations of 10, 20 and 50 nM. Meanwhile, the MMP, represented by the ratio of JC-1 aggregates to monomers, also remarkably decreased, evidenced by the weakening of red fluorescence and the corresponding enhancement in green fluorescence (Figure 5B). Altogether, these results demonstrated that the treatment of compound 14b resulted in an abnormal accumulation of ROS and a sharp decrease in MMP, which led to the dysfunction of mitochondria, thus ultimately inducing cell apoptosis.
Figure 5.

ROS accumulation and MMP alteration induced by compound 14b. (A,B) The intracellular ROS detection by DCFH-DA after A549 cells treated with compound 14b at indicated concentrations for 12 h. (C,D) The detection of MMP monitored by JC-1 dye after A549 cells treated with compound 14b at the concentrations of 10, 20 and 50 nM for 12 h. The experiments were performed at least three times, and the results of the representative experiments are shown. Data are presented as mean ± SEM *** p < 0.001 vs. Control group; significant difference as compared with the control group by t test. Scar bar: 10 μm.
2.9. The Metabolic Stability of Compound 14b
Metabolic stability is one of the most important properties in drug development. In this study, commercially available human liver microsomes were used to evaluate the metabolic stability of compound 14b. After incubation with human liver microsomes at 37 °C in vitro for an indicated time in a buffer solution containing a NADPH regenerating system, the residue of 14b was monitored by HPLC. CA-4 was used as the reference compound. As shown in Table 4, compound 14b was relatively stable in the liver microsomes. Still, 50.6% of the compound 14b residues could be observed after 3 h of incubation; however, only 20.5% of CA-4 remained after incubation for 1 h, indicating that CA-4 displayed a relatively faster metabolic profile than 14b. As well known, a short half-life means a low bioavailability in vivo and is required for frequent administration with a high dose, which might result in drug accumulation and may produce related toxicity. In comparison with the reference compound CA-4, which had a shorter half-life time (t1/2 < 60 min), the t1/2 of compound 14b (t1/2 = 198 min) was greatly improved, which encouraged us to carry out further detailed studies in vivo.
Table 4.
The metabolic stability of 14b and CA-4.
| Compd. | Time/min, Remaining% | t1/2, min | ||||||
|---|---|---|---|---|---|---|---|---|
| 30 | 60 | 90 | 120 | 150 | 180 | 240 | ||
| 14b | 96.18% | 91.38% | 84.14% | 74.45% | 67.75% | 56.17% | 43.43% | 198 |
| CA-4 | 70.2% | 28.3% | 20.5% | 18.7% | NT a | NT | NT | ND b |
a NT: not tested because of the fast metabolic rate of CA-4; b ND: not determined.
3. Materials and Methods
All the chemical reagents were purchased from Aldrich (Sigma-Aldrich, St. Louis, MO, USA), Innochem. Ltd. (Shanghai, China) and Bide. Ltd. (Shanghai, China) and were used without further purification. Melting points were determined on an X-5 micromelting automated melting point apparatus. NMR spectra were recorded on a Bruker AvanceIII spectrometer with TMS as the internal standard. High-resolution mass spectra (HRMS) data were obtained by ThermoFisher TQ-S. The purity of the target compounds was determined by high-performance liquid chromatography (HPLC) with a TC-C18 column (4.6 mm × 250 mm, 5 μm) using the following conditions: injection volume: 10 μL; flow rate: 1 mL/min; and mobile phase: acetonitrile/water or methanol/water.
The cell lines used in this study were obtained from National Collection of Authenticated Cell Cultures (Shanghai, China). The tubulin polymerization kit was obtained from cytoskeleton, Inc. (cat.#BK011P, Denver, MA, USA). The CCK-8 assay kit, cell cycle analysis kit and Annexin V-FITC double staining kit were purchased from Beyotime Biotechnology (Shanghai, China). The fluorescent dye including Hoechst 33342, DCFH-DA and JC-1 were obtained from Sigma-Aldrich (St. Louis, MO, USA).
3.1. Chemistry
3.1.1. General Procedures for Known Compounds
The preparation of the known compound 3, compound 8 and compound 17 were carried out in complete accordance with our previously reported methods [24,26].
3.1.2. General Procedures for Preparation of Compounds 5a-5b
The preparation of compounds 5a-5c was carried out according to our previously reported method [24]. Briefly, compound 3 (1 mmol, 232 mg) was dissolved in EtOH (4 mL), and piperidine (1.2 mmol, 0.12 mL) and indolal 4a (0.8 mmol, 116 mg) were added in sequence to the solution. After the mixture was stirred at 95 °C for 20 h, the diluted hydrochloric acid was added to quench the reaction and to adjust the reaction system to pH = 6. The mixture was extracted with EtOAc, and the organic layer was subsequently washed with aqueous NaHCO3, water and brine. After drying over anhydrous Na2SO4, the mixture was concentrated under reduced pressure. The residue was purified by column chromatography with petroleum/ethyl acetate (10:1) as an eluent to afford the target compound 5a.
(E)-3-(1H-indol-3-yl)-1-(8-methoxy-2,2-dimethyl-2H-chromen-6-yl)prop-2-en-1-one (5a)
Light yellow solid, yield: 41.2%. 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.04 (d, J = 7.2 Hz, 1H), 7.92 (s, 1H), 7.87 (d, J = 9.8 Hz, 1H), 7.51 (s, 1H), 7.36 (d, J = 7.2 Hz, 1H), 7.32 (m, 2H), 7.17 (d, J = 8.3 Hz, 1H), 6.69 (s, 1H), 6.48 (d, J = 9.8 Hz, 1H), 5.18 (d, J = 9.8 Hz, 1H), 3.90 (s, 3H), 1.62 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 191.11 (CO, C-7), 146.35 (C, C-3), 145.42 (C, C-2), 139.07 (C, C-18), 134.22 (C, C-5), 132.41 (CH, C-9), 131.45 (CH, C-11), 130.31 (CH, C-17), 126.28 (C, C-19), 123.95 (C, C-6), 123.43 (CH, C-8), 122.45 (CH, C-10), 122.33 (CH, C-21), 120.92 (C, C-1), 120.62 (CH, C-23), 120.17 (CH, C-22), 116.71 (C, C-16), 112.77 (CH, C-20), 110.58 (CH, C-4), 77.53 (C, C-12), 56.28 (CH3, C-15), 28.04 (CH3, C-13, 14). HRMS (ESI) (m/z) [M+H]+calcd for C23H22NO3, 360.15942; found, 360.15952. Purity: 97.7% (by HPLC).
(E)-1-(8-methoxy-2,2-dimethyl-2H-chromen-6-yl)-3-(1-methyl-1H-indol-3-yl)prop-2-en-1-one (5b)
Light yellow solid, yield: 43.5%. 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 16.0 Hz, 1H), 7.80 (d, J = 9.8 Hz, 1H), 7.66 (d, J = 8.7 Hz, 1H), 7.55 (s, 1H), 7.46 (s, 1H), 7.13 (d, J = 8.7 Hz, 1H), 7.00 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 8.8 Hz, 1H), 6.61 (d, J = 7.8 Hz, 1H), 6.54 (d, J = 8.8 Hz, 1H), 5.18 (d, J = 9.8 Hz, 1H), 3.92 (s, 3H), 3.89 (s, 3H), 1.62 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 190.21 (CO, C-7), 146.35 (C, C-3), 145.42 (C, C-2), 138.52 (C, C-18), 136.98 (C, C-5), 134.22 (CH, C-9), 132.99 (CH, C-11), 130.51 (CH, C-17), 125.01 (C, C-19), 124.49 (C, C-6), 123.85 (CH, C-8), 122.65 (CH, C-10), 122.35 (CH, C-21), 120.58 (C, C-1), 120.36 (CH, C-23), 118.83 (CH, C-22), 113.41 (C, C-16), 112.87 (CH, C-20), 110.29 (CH, C-4), 76.52 (C, C-12), 56.28 (CH3, C-15), 33.41 (CH3, C-24), 28.27 (CH3, C-13, 14). HRMS (ESI) (m/z) [M+H]+ calcd for C24H24NO3, 374.17507; found, 374.17511 Purity: 99.2% (by HPLC).
3.1.3. General Procedures for Preparation of Compounds 11a-11b, 14a-14b, 19a-19b and 21a-21b
The preparation of compounds 11a-11b, 14a-14b, 19a-19b and 21a-21b was carried out according to our previously reported method [27,30]. To a solution of compound 8 (1.0 mmol, 269 mg) in 20 mL of anhydrous THF in nitrogen atmosphere at −78 °C, n-butyllithium (0.8 mL, 2.5 M in hexane) was added in dropwise. After the solution was stirred for 0.5 h, commercially available compound 9a (1.0 mmol, 145 mg) was added, and then, the reaction mixture was stirred overnight in darkness. A saturated NH4Cl solution was added, and the mixture was extracted with EtOAc, and the organic layer was subsequently washed with aqueous NaHCO3, water and brine. After drying over anhydrous Na2SO4, the mixture was concentrated under reduced pressure to obtain the crude product 10a, which was used directly in the next step of the reaction without further purification.
Compound 10a (1 mmol, 335 mg) was dissolved in dichloromethane; then, PCC (1.2 mmol, 451 mg) and silica gel (451 mg) were added to the mixture and stirred at room temperature for 5–8 h. After the reaction was completed, the suspension was filtered and the solvent was concentrated under a vacuum to provide the crude product, which was subsequently purified by silica gel column chromatography with petroleum/ethyl acetate (10:1–3:1) as an eluent to afford the target compound 11a.
(1H-indol-4-yl)(5-methoxy-2,2-dimethyl-2H-chromen-8-yl)methanone (11a)
White solid, yield: 39.8%. 1H NMR (400 MHz, CDCl3) δ 8.18 (s, 1H), 7.50 (d, J = 9.8 Hz, 1H), 7.46 (d, J = 16.0 Hz, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.17 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 8.7 Hz, 1H), 6.84 (d, J = 11 Hz, 1H), 6.67 (d, J = 7.8 Hz, 1H), 6.59 (d, J = 7.8 Hz, 1H), 5.46 (d, J = 9.8 Hz, 1H), 3.90 (s, 3H), 1.53 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 191.91 (CO, C-7), 155.26 (C, C-2), 152.41 (C, C-6), 135.06 (C, C-12), 130.57 (C, C-13), 127.98 (CH, C-15), 127.56 (CH, C-9), 127.06 (C, C-8), 126.64 (CH, C-4), 125.84 (CH, C-21), 124.81 (CH, C-10), 117.53 (CH, C-16), 116.31 (C, C-5), 107.82 (CH, C-3), 106.72 (CH, C-11), 104.90 (C, C-1), 101.44 (CH, C-20), 76.70 (C, C-14), 55.35 (CH3, C-17), 27.85 (CH3, C-18,C-19). HRMS (ESI) (m/z) [M+H]+calcd for C21H20NO3, 334.14377; found, 334.14430. Purity: 98.9% (by HPLC).
(5-methoxy-2,2-dimethyl-2H-chromen-8-yl)(1-methyl-1H-indol-4-yl)methanone (11b)
White solid, yield: 40.5%. 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 4.8 Hz, 1H), 7.46 (d, J = 11.0 Hz, 1H), 7.17 (d, J = 7.8 Hz, 1H), 7.14 (d, J = 7.8 Hz, 1H), 6.95 (d, J = 9.8 Hz, 1H), 6.80 (d, J = 11 Hz, 1H), 6.64 (d, J = 9.8 Hz, 1H), 6.55 (d, J = 7.8 Hz, 1H), 5.42 (d, J = 9.8 Hz, 1H), 3.91 (s, 3H), 3.85 (s, 3H),1.51 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 195.21 (CO, C-7), 157.58 (C, C-2), 155.45 (C, C-6), 136.55 (C, C-12), 131.47 (C, C-13), 130.48 (CH, C-15), 129.13 (CH, C-9), 128.33 (C, C-8), 127.03 (CH, C-4), 126.10 (CH, C-21), 123.50 (CH, C-10), 117.66 (CH, C-16), 116.58 (C, C-5), 113.42 (CH, C-3), 112.26 (CH, C-11), 107.61 (C, C-1), 100.57 (CH, C-20), 76.48 (C, C-14), 56.01 (CH3, C-17), 32.55 (CH3, C-22), 27.87 (CH3, C-18,19). HRMS (ESI) (m/z) [M+H]+calcd for C22H22NO3, 348.15942; found, 348.15968. Purity: 99.2% (by HPLC).
(1H-indol-5-yl)(5-methoxy-2,2-dimethyl-2H-chromen-8-yl)methanone (14a)
White solid, yield: 46.2%. 1H NMR (400 MHz, CDCl3) δ 8.66 (s, 1H), 8.32 (s, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.53 (d, J = 9.8 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 6.52 (d, J = 79.8 Hz, 1H), 6.45-6.48 (m, 2H), 5.23 (d, J = 9.8 Hz, 1H), 3.85 (s, 3H), 1.52 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 195.58 (CO, C-7), 157.42 (C, C-2), 155.12 (C, C-6), 138.41 (C, C-11), 129.16 (C, C-8), 127.82 (CH, C-12), 127.13 (CH, C-17), 125.80 (CH, C-4), 125.42 (CH, C-14), 125.18 (CH, C-9), 121.50 (CH, C-13), 117.62 (CH, C-18), 117.41 (C, C-5), 111.27 (CH, C-3), 109.37 (CH, C-10), 106.95 (C, C-1), 102.81 (CH, C-15), 77.68 (C, C-16), 55.91 (CH3, C-19), 27.82 (CH3, C-20, 21). HRMS (ESI) (m/z) [M+H]+ calcd for C21H20NO3, 334.14377; found, 334.14382. Purity: 98.7% (by HPLC).
(5-methoxy-2,2-dimethyl-2H-chromen-8-yl)(1-methyl-1H-indol-5-yl)methanone (14b)
White solid, yield: 38.5%. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.78 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 9.8 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.11 (d, J = 7.8 Hz, 1H), 6.62 (d, J = 9.8 Hz, 1H), 6.57–6.55 (m, 2H), 5.28 (d, J = 9.8 Hz, 1H), 3.90 (s, 3H), 3.78 (s, 3H), 1.54 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 195.47 (CO, C-7), 157.50 (C, C-2), 155.22 (C, C-6), 139.27 (C, C-11), 130.17 (C, C-8), 129.02 (CH, C-12), 128.18 (CH, C-17), 127.03 (CH, C-4), 125.80 (CH, C-14), 125.14 (CH, C-9), 122.24 (CH, C-13), 117.56 (CH, C-18), 117.40 (C, C-5), 111.97 (CH, C-3), 109.11 (CH, C-10), 107.19 (C, C-1), 102.88 (CH, C-15), 77.88 (C, C-16), 56.26 (CH3, C-19), 32.81 (CH3, C-22), 27.82 (CH3, C-20, 21). HRMS (ESI) (m/z) [M+H]+calcd for C22H22NO3, 348.15942; found, 348.15921. Purity: 96.6% (by HPLC).
(1H-indol-4-yl)(8-methoxy-2,2-dimethyl-2H-chromen-6-yl)methanone (19a)
Light yellow solid, yield:42.1%. 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.81 (d, J = 9.8 Hz, 1H), 7.67 (d, J = 9.8 Hz, 1H), 7.50 (s, 1H), 7.39 (t, J = 7.8 Hz, 1H), 7.28 (d, J = 7.8 Hz, 1H), 7.19 (s, 1H), 6.43 (d, J = 7.8 Hz, 1H), 6.37 (d, J = 7.8 Hz, 1H), 5.23 (d, J = 11.0 Hz, 1H), 3.90 (s, 3H), 1.61 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 195.10 (CO, C-7), 145.98 (C, C-3), 144.75 (C, C-2), 135.97 (C, C-12), 133.80 (C, C-5), 131.51 (CH, C-15), 130.07 (C, C-13), 127.80 (C, C-8), 126.93 (CH, C-9), 124.94 (CH, C-20), 123.12 (CH, C-10), 122.76 (CH, C-6), 121.98 (CH, C-14), 120.05 (C, C-1), 111.64 (CH, C-11), 108.89 (CH, C-4), 101.31 (CH, C-21), 77.82 (C, C-16), 55.68 (CH3, C-19), 27.97 (CH3, C-17,18). HRMS (ESI) (m/z) [M+H]+ calcd for C21H20NO3, 334.14377; found, 334.14310. Purity: 98.2% (by HPLC).
(8-methoxy-2,2-dimethyl-2H-chromen-6-yl)(1-methyl-1H-indol-4-yl)methanone (19b)
Light yellow solid, yield:39.8%. 1H NMR (400 MHz, CDCl3) δ7.74 (d, J = 8.8 Hz, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.51 (s, 1H), 7.38 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.21 (s, 1H), 6.49 (d, J = 11.0 Hz, 1H), 6.43 (d, J = 7.8 Hz, 1H), 5.23 (d, J = 11.0 Hz, 1H), 3.86 (s, 3H), 3.78 (s, 3H), 1.62 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 195.89 (CO, C-7), 146.28 (C, C-3), 144.75 (C, C-2), 137.06 (C, C-12), 134.63 (C, C-5), 132.81 (CH, C-15), 131.83 (C, C-13), 131.15 (C, C-8), 130.14 (CH, C-9), 129.13 (CH, C-20), 123.44 (CH, C-10), 122.88 (CH, C-6), 122.35 (CH, C-14), 120.26 (C, C-1), 111.44 (CH, C-11), 110.58 (CH, C-4), 100.57 (CH, C-21), 77.88 (C, C-16), 56.28 (CH3, C-19), 33.53 (CH3, C-22), 28.33 (CH3, C-17,18). HRMS (ESI) (m/z) [M+H]+calcd for C22H22NO3, 348.15942; found, 348.15968. Purity: 97.9% (by HPLC).
(1H-indol-5-yl)(8-methoxy-2,2-dimethyl-2H-chromen-6-yl)methanone (21a)
Light yellow solid, yield:36.2%. 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.28 (s, 1H), 7.76 (d, J = 8.8 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.15 (s, 1H), 6.63 (d, J = 8.8 Hz, 1H), 6.47 (d, J = 11.0 Hz, 1H), 5.19 (d, J = 10.8 Hz, 1H), 3.89 (s, 3H), 1.61 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 194.92 (CO, C-7), 146.09 (C, C-3), 144.62 (C, C-2), 138.40 (C, C-11), 133.64 (C, C-5), 131.41 (CH, C-17), 129.03 (C, C-8), 127.19 (C, C-12), 125.80 (CH, C-9), 125.39 (CH, C-14), 123.22 (CH, C-6), 122.35 (CH, C-16), 121.11 (CH, C-13), 120.11 (C, C-1), 109.48 (CH, C-4), 109.36 (CH, C-10), 102.71 (CH, C-15), 77.93 (C, C-18), 56.12 (CH3, C-21), 28.05 (CH3, C-19, 20). HRMS (ESI) (m/z) [M+H]+calcd for C21H20NO3, 334.14377; found, 334.14570. Purity: 98.0% (by HPLC).
(8-methoxy-2,2-dimethyl-2H-chromen-6-yl)(1-methyl-1H-indol-5-yl)methanone (21b)
Light yellow solid, yield:39.1%. 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.48 (s, 1H), 7.23 (s, 1H), 7.19 (d, J = 7.8 Hz, 1H), 6.70 (d, J = 8.8 Hz, 1H), 6.52 (d, J = 11.0 Hz, 1H), 5.19 (d, J = 11.0 Hz, 1H), 3.90 (s, 3H), 3.80 (s, 3H), 1.62 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 195.12 (CO, C-7), 146.22 (C, C-3), 145.02 (C, C-2), 139.82 (C, C-11), 133.94 (C, C-5), 131.71 (CH, C-17), 128.82 (C, C-8), 128.61 (C, C-12), 127.36 (CH, C-9), 125.15 (CH, C-14), 123.42 (CH, C-6), 122.65 (CH, C-16), 121.73 (CH, C-13), 119.91 (C, C-1), 109.72 (CH, C-4), 108.49 (CH, C-10), 102.68 (CH, C-15), 77.89 (C, C-18), 56.38 (CH3, C-21), 32.67 (CH3, C-22), 28.07 (CH3, C-19, 20). HRMS (ESI) (m/z) [M+H]+calcd for C22H22NO3, 348.15942; found, 348.16061. Purity: 98.2% (by HPLC).
3.2. Cell Lines and Cell Culture
A549 (non-small-cell-lung cancer cell line), Caski (human epithelial cervical cancer cell line), HepG2 (human liver carcinoma cell line), MCF-7 (human breast carcinoma cell line), C42B (human prostate cancer line C42B), L-02 (human normal hepatocytes), RWPE-1 (human prostate epithelial cells) and MCF-10A (human normal mammary epithelial cells) were obtained from National Collection of Authenticated Cell Cultures (Shanghai, China). A549/CDDP (A549 cell line resistant to cisplatin), C42B/ENZR (C42b cell line resistant to Enzalutamide), MCF-7/DOX (MCF-7 cell line resistant to doxorubicin), A2780/TAX (human ovarian cancer cell line A2780 resistant to taxol) and HCT-8/VCR (human ileocecum carcinoma cell lines HCT-8 resistant to vincristine) were kindly provided by Professor Junjian Wang and Professor Xingshu Li at Sun Yat-Sen University, China. All cell lines were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI-1640 medium containing 10% (v/v) heat-inactivated FBS, 100 units/mL penicillin and 100 mg/mL streptomycin and cultured in a humidified atmosphere of 5% CO2 at 37 °C.
3.3. Biological Evaluation Assays
The biological evaluation applied in this study, including a CCK-8 assay, an in vitro tubulin polymerization inhibition assay, a molecular docking study, colchicine competitive inhibition assay intracellular microtubule morphology detection using immunofluorescence, a cell cycle and apoptosis assay using flow cytometry, a caspase 3 activity evaluation, and intracellular ROS and MMP detection were performed completely under the guidance of our previously published protocols [24,25,26]. These protocols are briefly described in the Supplementary Materials.
4. Conclusions
Natural-product-based drug discovery provides a strong basis for the development of antitumor drugs. In this study, under the guidance of our previously obtained valuable SARs and starting from the structure of millepachine, a natural product from the seeds of Millettia pachycarpa, a rational drug design strategy that included replacing the α,β-unsaturated ketones with a benzophenone moiety and simultaneously infusing the indole heterocycle was applied. The optimized compound 14b displayed excellent antiproliferative activities towards a panel of human cancer cell lines (0.022–0.074 μM), which was almost 100-fold more active than millepachine (IC50 = 2.566–6.712 μM). For target exploration, some experiments used a tubulin–microtubule system to validate that compound 14b inhibited tubulin polymerization. As with most microtubule target agents, compound 14b inhibited tubulin polymerization by binding to the colchicine binding site of tubulin; disturbed the equilibrium of the tubulin–microtubule system; arrested the cell cycle at the G2/M phase; and therefore, interfered with mitosis. The 14b-induced apoptosis was accompanied by the activation of caspase 3, an abnormal accumulation of ROS and a decrease in MMP, which ultimately led to cancer cell death. Meanwhile, 14b displayed nearly equally potent antiproliferative activity toward drug-resistant cells and parent cancer cells, which provides a potential for 14b co-administration with currently clinically used anticancer drugs. Finally, the extraordinary in vitro anticancer activity and metabolic stability in human liver microsomes of compound 14b encourages us to evaluate its in vivo antitumor activity in a further study. Altogether, these results demonstrate the promising worth of 14b as a potential chemotherapeutic agent.
Acknowledgments
We thank Junjian Wang for providing the cancer cell lines.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules28031481/s1, S I: Figure S1. The superposition of 14b and millepachine using docking study; S II: NMR spectra of target compounds (S2–S11); S III: HPLC chromatograms of target compounds (S12–S15); S IV: High resolution mass spectra of target compounds (S16–S25); S V: Biological evaluation methods (S26–S27).
Author Contributions
Conceptualization, B.H. and B.L.; methodology, B.L.; investigation, Q.Z.; validation, L.Y.; writing—original draft preparation, B.L.; visualization, J.Y. and Y.W.; supervision, B.L.; funding acquisition, B.L. and J.Y. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used in this study are presented in this article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
Funding Statement
This research was funded by the National Natural Science Foundation of China (21907017), Natural Science Foundation of Guangdong Province of China (2018A030310186, 2022A1515012292), Science and Technology Planning Project of Guangzhou (202102010132, 202102010213), Guangzhou Health and Family Planning Commission of Guangdong Province of China (20181A011068, 20201A011080), Foundation of Guangdong Polytechnic of Science and Trade (GDKM2022-90), and Department of Education Characteristic Innovation project of colleges and universities of Guangdong Province (2021KTSCX023).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Azab A., Nassar A., Azab A.N. Anti-Inflammatory Activity of Natural Products. Molecules. 2016;21:1321. doi: 10.3390/molecules21101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinto M.M., Palmeira A., Fernandes C., Resende D.I., Sousa E., Cidade H., Tiritan M.E., Correia-da-Silva M., Cravo S. From Natural Products to New Synthetic Small Molecules: A Journey through the World of Xanthones. Molecules. 2021;26:431. doi: 10.3390/molecules26020431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ekiert H.M., Szopa A. Biological Activities of Natural Products. Molecules. 2020;25:5769. doi: 10.3390/molecules25235769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arbour C.A., Imperiali B. Uridine natural products: Challenging targets and inspiration for novel small molecule inhibitors. Bioorg. Med. Chem. 2020;28:115661. doi: 10.1016/j.bmc.2020.115661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ilan Y. Microtubules: From understanding their dynamics to using them as potential therapeutic targets. J. Cell Physiol. 2019;234:7923–7937. doi: 10.1002/jcp.27978. [DOI] [PubMed] [Google Scholar]
- 6.Xia L., Zhang Y., Yang R., Wang Z., Lu Y., Wang B., Zhu H. Tubulin Inhibitors Binding to Colchicine-Site: A Review from 2015 to 2019. Curr. Med. Chem. 2020;27:6787–6814. doi: 10.2174/0929867326666191003154051. [DOI] [PubMed] [Google Scholar]
- 7.Dong M., Liu F., Zhou H., Zhai S., Yan B. Novel Natural Product- and Privileged Scaffold-Based Tubulin Inhibitors Targeting the Colchicine Binding Site. Molecules. 2016;21:1375. doi: 10.3390/molecules21101375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Y., Di B., Niu M. Structure-Based Pharmacophore Design and Virtual Screening for Novel Tubulin Inhibitors with Potential Anticancer Activity. Molecules. 2019;24:3181. doi: 10.3390/molecules24173181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye H., Fu A., Wu W., Li Y., Wang G., Tang M., Li S., He S., Zhong S., Lai H., et al. Cytotoxic and apoptotic effects of constituents from Millettia pachycarpa Benth. Fitoterapia. 2012;83:1402–1408. doi: 10.1016/j.fitote.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Huang X., Wang M., Wang C., Hu W., You Q., Ma T., Jia Q., Yu C., Liao Z., Wang H. Synthesis and biological evaluation of novel millepachine derivative containing aminophosphonate ester species as novel anti-tubulin agents. Bioorg. Chem. 2020;94:103486. doi: 10.1016/j.bioorg.2019.103486. [DOI] [PubMed] [Google Scholar]
- 11.Huang X., Hua S., Huang R., Liu Z., Gou S., Wang Z., Liao Z., Wang H. Dual-targeting antitumor hybrids derived from Pt(IV) species and millepachine analogues. Eur. J. Med. Chem. 2018;148:1–25. doi: 10.1016/j.ejmech.2018.02.012. [DOI] [PubMed] [Google Scholar]
- 12.Yang Z., Wu W., Wang J., Liu L., Li L., Yang J., Wang G., Cao D., Zhang R., Tang M., et al. Synthesis and Biological Evaluation of Novel Millepachine Derivatives as a New Class of Tubulin Polymerization Inhibitors. J. Med. Chem. 2014;57:7977–7989. doi: 10.1021/jm500849z. [DOI] [PubMed] [Google Scholar]
- 13.Yang J., Yan W., Yu Y., Wang Y., Yang T., Xue L., Yuan X., Long C., Liu Z., Chen X., et al. The compound millepachine and its derivatives inhibit tubulin polymerization by irreversibly binding to the colchicine-binding site in β-tubulin. J. Biol. Chem. 2018;293:9461–9472. doi: 10.1074/jbc.RA117.001658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu W., Liu Y., Ye H., Li Z. Millepachine showed novel antitumor effects in cisplatin-resistant human ovarian cancer through inhibiting drug efflux function of ATP-binding cassette transporters. Phytother. Res. 2018;32:2428–2435. doi: 10.1002/ptr.6180. [DOI] [PubMed] [Google Scholar]
- 15.Kumari A., Singh R.K. Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorg. Chem. 2019;89:103021. doi: 10.1016/j.bioorg.2019.103021. [DOI] [PubMed] [Google Scholar]
- 16.Devi N., Kaur K., Biharee A., Jaitak V. Recent Development in Indole Derivatives as Anticancer Agent: A Mechanistic Approach. Anti-Cancer Agents Med. Chem. 2021;21:1802–1824. doi: 10.2174/1871520621999210104192644. [DOI] [PubMed] [Google Scholar]
- 17.Wan Y., Li Y., Yan C., Yan M., Tang Z. Indole: A privileged scaffold for the design of anti-cancer agents. Euro. J. Med. Chem. 2019;183:111691. doi: 10.1016/j.ejmech.2019.111691. [DOI] [PubMed] [Google Scholar]
- 18.Lu Y., Chen J., Xiao M., Li W., Miller D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012;29:2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang S., Zhou Z., Jiang Z., Zhu W., Qiao D. Indole-Based Tubulin Inhibitors: Binding Modes and SARs Investigations. Molecules. 2022;27:1587. doi: 10.3390/molecules27051587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naaz F., Neha K., Haider M.R., Shafi S. Indole derivatives (2010–2020) as versatile tubulin inhibitors: Synthesis and structure–activity relationships. Future Med. Chem. 2021;13:1795–1828. doi: 10.4155/fmc-2020-0385. [DOI] [PubMed] [Google Scholar]
- 21.An B., Zhang S., Yan J., Huang L., Li X. Synthesis, in vitro and in vivo evaluation of new hybrids of millepachine and phenstatin as potent tubulin polymerization inhibitors. Org. Biomol. Chem. 2017;15:852–862. doi: 10.1039/C6OB02507B. [DOI] [PubMed] [Google Scholar]
- 22.Chen J., Yan J., Hu J., Pang Y., Huang L., Li X. Synthesis, biological evaluation and mechanism study of chalcone analogues as novel anti-cancer agents. RSC Adv. 2015;5:68128–68135. doi: 10.1039/C5RA14888J. [DOI] [Google Scholar]
- 23.Zhang S., An B., Li J., Hu J., Huang L., Li X., Chan A.S.C. Synthesis and evaluation of selenium-containing indole chalcone and diarylketone derivatives as tubulin polymerization inhibition agents. Org. Biomol. Chem. 2017;15:7404–7410. doi: 10.1039/C7OB01655G. [DOI] [PubMed] [Google Scholar]
- 24.Yan J., Chen J., Zhang S., Hu J., Huang L., Li X. Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization in Vitro and in Vivo. J. Med. Chem. 2016;59:5264–5283. doi: 10.1021/acs.jmedchem.6b00021. [DOI] [PubMed] [Google Scholar]
- 25.Yan J., Pang Y., Sheng J., Wang Y., Chen J., Hu J., Huang L., Li X. A novel synthetic compound exerts effective anti-tumour activity in vivo via the inhibition of tubulin polymerisation in A549 cells. Biochem. Pharmacol. 2015;97:51–61. doi: 10.1016/j.bcp.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 26.Zhou J., Pang Y., Zhang W., OuYang F., Lin H., Li X., Yan J. Discovery of a Novel Stilbene Derivative as a Microtubule Targeting Agent Capable of Inducing Cell Ferroptosis. J. Med. Chem. 2022;65:4687–4708. doi: 10.1021/acs.jmedchem.1c01775. [DOI] [PubMed] [Google Scholar]
- 27.Pang Y., Lin H., Ou C., Cao Y., An B., Yan J., Li X. Design, synthesis, and biological evaluation of novel benzodiazepine derivatives as anticancer agents through inhibition of tubulin polymerization in vitro and in vivo. Eur. J. Med. Chem. 2019;182:111670. doi: 10.1016/j.ejmech.2019.111670. [DOI] [PubMed] [Google Scholar]
- 28.Cao Y., Zheng L., Wang D., Liang X., Gao F., Zhou X. Recent advances in microtubule-stabilizing agents. Euro. J. Med. Chem. 2018;143:806–828. doi: 10.1016/j.ejmech.2017.11.062. [DOI] [PubMed] [Google Scholar]
- 29.Singh P., Lim B. Targeting Apoptosis in Cancer. Curr. Oncol. Rep. 2022;24:273–284. doi: 10.1007/s11912-022-01199-y. [DOI] [PubMed] [Google Scholar]
- 30.Zhang S., An B., Yan J., Huang L., Li X. The synthesis and evaluation of new benzophenone derivatives as tubulin polymerization inhibitors. RSC Adv. 2016;6:88453–88462. doi: 10.1039/C6RA16948A. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this study are presented in this article.